
Submitted:
03 June 2023
Posted:
05 June 2023
You are already at the latest version
Abstract
Antibody-drug conjugates (ADCs) have provided new therapeutic options and significant promise for patients with cancers, particularly where existing treatments are limited. Substantial effort in ADC development is underway globally, with 13 ADCs currently approved and many more in development [1]. Therapeutic benefits of ADCs leverage the ability to selectively target cancer cells through antibody binding, resultant relative sparing of non-malignant tissues, and the targeted delivery of a cytotoxic payload. Consequently, this drug class has demonstrated activity in multiple malignancies refractory to standard therapeutic options [1-4]. Despite this, limitations exist, including narrow therapeutic windows, unique toxicity profiles, development of therapeutic resistance, and appropriate biomarker selection [5-7]. This review will describe the development of ADCs, their mechanisms of action, pivotal trials, and approved indications and identify common themes. Current challenges and opportunities will be discussed for this drug class in cancer therapeutics at a time when significant developments in antibody therapies, immunotherapy and targeted agents are occurring.
Keywords:
Antibody-drug conjugate
; cancer
; clinical trials
Introduction
The search for directed and effective cytotoxic therapy has been the Holy Grail of cancer treatment since being hypothesised as a treatment for cancer over a century ago [8]. Despite this, the mainstay of treatment for over half a century and up until today remains as chemotherapy. While effective in many cancers, chemotherapy is associated with frequent off-target effects resulting in significant toxicities [9]. Antibody-drug conjugates (ADCs) are compounds comprising three distinct components to effectively target cancer cells; a monoclonal antibody, a cytotoxic payload, which are connected via a linker. It thus leverages on the ability to selectively target cancer cells through antibody binding to specific receptors, the resultant relative sparing of non-malignant tissues, and the targeted delivery of a smaller but ultimately more impactful cytotoxic payload [4,10].
The theory behind ADCs was first developed in the 1960s, with the first animal studies conducted in the 1980s [11]. Challenges with the first generation of ADCs included the unstable linker component, resulting in premature drug release into the circulation [1]. Stabilising the drug molecule, determining appropriate ratios of drug to antibody, and half-lives which influence efficacy and potency, have been major developmental challenges [12]. In addition, conventional chemotherapies such as anthracyclines were used as payloads but were ineffective due to a lack of relative lack of potency [1]. Early antibodies used were predominantly chimeric humanised or mouse-derived, which are associated with higher immunogenicity and drug reactions compared to next-generation humanised antibodies [1]. Another barrier to the utility of ADCs were the limited number of antigen targets.
Significant progress has been made since and a growing number of ADCs have now been FDA approved for clinical use following positive results from phase 3 trials when compared against current standards of care. In this review, we will discuss the structure and mechanisms of actions of ADCs, present the seminal clinical ADC trials, and provide an overview of the challenges facing the ongoing clinical development of ADCs.
ADC Structure, Pharmacokinetics and Mechanism of Action:
ADCs have a unique formulation consisting of three primary components: an antibody, cytotoxic payload bound by a chemical linker, as illustrated in Figure 1 and Figure 2. Each plays its own role in targeting cancer, efficacious delivery and a desired cytotoxic outcome [13].
Monoclonal antibodies, while used for cancer treatment for many years, have alone not provided the desired outcome regarding therapeutic benefit in most cancers. As such, the vast majority of cancer treatment protocols which include antibodies, are given in combination with chemotherapy. ADCs represent a rational approach to harnessing the characteristics of both therapeutic classes [14]. The first step of any ADC is to identify a molecular target that is over-expressed exclusively on the tumour site [1]. This minimises delivery to non-tumoral tissue, given that ADCs are created to deliver the cytotoxic payload only to the nominated antigen [6]. The internalisation of the ADC requires effective binding between the antibody and surface antigen. Antigens are required to be extracellular or on the cell surface, such as Trop-2 and HER-2, as intracellular antigens may not be recognised by the antibody [1]. In addition, the antigen should be expressed rather than secreted, as the latter can lead to ADC binding in the systemic circulation, increasing the risk of systemic toxicity [1,15].
Current ADCs involve antibodies that are fully humanised, reducing immunogenicity [1]. Most ADCs are based on immunoglobulin G (IgG) antibodies, which has four known subtypes. The benefits of IgG1, the most common subtype used, include its ability to induce antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity and antibody-dependent phagocytosis [1].
The second part of any ADC is the cytotoxic payload, commonly and dramatically termed the missile or warhead in the existing literature. With only 2% of ADCs reaching target tumour cells, potency is essential for these compounds to be efficacious [1,16]. Most agents approved today have employed two categories of payloads, DNA damaging agents and microtubule inhibitors [1]. DNA damaging agents include agents which lead to DNA double stranded breaks (e.g. calicheamicins), DNA intercalation (e.g. topoisomerase inhibitors), DNA alkylation (e.g. duocarmycins), and DNA cross-linking (e.g. pyrrolobenzodiazepines) [1]. These potent DNA damaging agents have an IC50 in the picomolar range [1,16]. Microtubules are a key cytoskeletal element that play an important role in cell division [1]. Microtubule inhibitors include auristatin derivatives, such as monomethyl auristatin E and F (MMAE and MMAF), and maytansinoid derivatives, such as DM1 and DM4 [1]. They typically possess IC50 values in the nanomolar range [1].
The drug antibody ratio (DAR), which is the number of payload molecules that can be attached to the antibody, is an important consideration in ADC development [17,18]. It plays a pivotal role in the potency and influence the therapeutic index of ADCs [1,19]. The DAR also impacts the physiological properties of binding, the drug's pharmacokinetics and half-life [20]. Most currently approved ADCs have a DAR ranging from 2-8. The ADC manufacturing process can impact the DAR, resulting in substantial heterogeneity between drug batches in some ADCs [21,22].
Linkers play a critical role in ensuring the stability of the ADC and the delivery of the cytotoxic payload to the tumour cells. Unstable linkers are associated with the premature release of the payload, resulting in systemic toxicities [23]. The optimal ADC has a stable linker in systemic circulation before binding to cancer cells. The linker can be modified by varying the linker length, which impacts accessibility to the payload, and by the type of linkage cleavage, cleavable and non-cleavable, which influence pharmacokinetic properties and modulates the kinetics of the payload release [24,25]. Non-cleavable linkers work when the antibody is catabolized in the lysosome or via proteases. After internalisation, the degradation of the monoclonal antibody via enzymatic hydrolysis leads to the release of the cytotoxic drug and subsequent cell death [26]. The main advantage of non-cleavable linkers (e.g., thioether or maleimidocaproyl group) is that they remain intact in the circulation and have less off-target toxicity. The catabolite is made whereby the payload retains an amino acid or peptide from the linker in its chemical structure [23,27]. An example of non-cleavable linkers used in clinically used ADCs is trastuzumab emtansine (Trastuzumab-DM1, Kadcyla) [28]. In contrast, cleavable linkers rely on the microenvironment and tumour to release their payload. These include chemically cleavable, enzymatically cleavable, and acid-sensitive linkers. An example of an enzymatically cleavable linker is those sensitive to lysosomal proteases such as cathepsin B, known to be expressed in certain cancer cells. These cleavable linkers enable drug release close to the tumour and are stable in blood [29–31]. Most currently approved ADCs have enzymatically cleavable linkers, examples being brentuximab vedotin and inotuzumab ozogamicin [1,32].
Optimising linker and conjugation chemistry is vital in improving ADC efficacy. Traditional methods for conjugation (connecting the linker plus payload to an antibody) relied upon using lysine and cysteine residues on the antibody as a site for the conjugation reaction to occur [33], producing heterogeneous compounds with variable DARs and conjugation sites. The most common method was amide coupling, where an active carboxylic acid ester connects the lysine residue on the antibody to the payload. An example of this is Trastuzumab-DM1. Coupling via this method was random, resulting in an uneven DAR distribution, ranging from 0 to 8 [34]. Another conjugation method uses a cysteine residue, which has less heterogeneity compared to lysine coupling and is used in trastuzumab deruxtecan, inotuzumab ozogamicin and gemtuzumab ozogamicin. [1,35,36]. Site-specific conjugation has allowed for more homogenous drugs with better therapeutic windows [37]. Improvements in linker technology, such as utilising a hydrolysable carbonate moiety, spontaneous linker hydrolysis or an enzymatically cleavable tetrapeptide linker, have also enabled higher DARs to be achieved so that ADCs containing less potent topoisomerase 1 inhibitor payloads, such as trastuzumab deruxtecan (T-DXD) and sacituzumab govitecan, have been effective [16,32,38,39].
Pharmacokinetics and Pharmacodynamics of ADCs:
ADCs are administered intravenously, and can travel through the systemic circulation without being metabolised, essentially remaining inactive [40]. Its metabolism and elimination are crucial in ensuring drug delivery and excretion. Metabolism in the circulation can influence the pharmacokinetics, efficacy and toxicity profile [41]. The distribution of the ADC initially relies on “volume of distribution” due to vascular and interstitial space. It can also be impacted by antigen expression and subsequent internalisation of the ADC once the target is reached. Distribution can result in off-target toxicities if interacting with non-target tissues [14]. The metabolism of ADCs is complex and involves conjugation sites, linkers, and the payload. Chemical uncoupling through deconjugation or linker cleaving via enzymes are the commonest ways to release the drug into the plasma [41,42]. The internalisation or mobilisation of ADC leads to fusion with lysosomes, where cleavage of the linker occurs. This represents a mechanism of ADC elimination and clearance from circulation by receptor-mediated endocytosis with subsequent lysosomal compartment degradation. The cytotoxic payload is then free to reach the target, binding and triggering the death of the target cell. The free drug, if released into the cytosol, has been reported to travel across plasma membranes leading to a bystander effect where surrounding cells are also exposed to the cytotoxic payload. One upside of this is that a lower antigen expression on tumour cells is required allowing for the targeting of a larger population of cancerous cells, not only being limited to tumour cells with the highest target antigen expression [43].
Proteolytic degradation or catabolism assists in eliminating the monoclonal antibody portion of the ADC, which is repurposed as a protein or new carbon source. They cannot be excreted by the liver or exit the systemic circulation through glomerular filtration. In contrast, the excretion of the payload occurs through the renal and hepatic organs and varies with the ADCs [44]. For example, the payload of T-DM1 is predominantly excreted via the hepatic system with minimal renal excretion, while brentuximab vedotin is predominantly excreted in faeces via the hepatic route [26]. There is limited published data with newer ADCs like sacituzumab govitecan on the elimination process and its impact on hepatic or renal function [45].
Bystander Effect:
The bystander effect is an increasingly recognised therapeutic mechanism of ADCs. This occurs where cells within close proximity of the targeted cancer cells is exposed to the antitumour effects of ADCs, irrespective of antigen expression [46]. It relies on the relationship between the linker, payload, and ADC design. Cleavable linkers were shown in preclinical models to diffuse through cell membranes when released from the antibody, and with hydrophobic payloads [22]. This feature is intrinsically linked to both the payload and cleaver, with differences seen when comparing the mechanism of two similar ADCs with a trastuzumab antibody, T-DXd and T-DM1 [5,47]. T-DM1’s payload is released after lysosomal degradation of the complex, leading to reduced cell permeability. In contrast, T-DXd is internalised, and then the payload is released but does not affect membrane permeability, thus neighbouring cells can still be affected by diffusion of the cytotoxic drug. Furthermore, T-DXd is highly membrane permeable compared to T-DM1. T-DXd uses cysteine residues to connect the linker and antibody, whereas T-DM1 uses a non-cleavable thioether linker. The resultant breakdown of T-DM1 results in charged amino acids of the payload that do not diffuse across the cell membrane, while this does not occur with T-DXd [5,47]. Like T-DXd, trastuzumab duocarmazine is another ADC demonstrating a significant bystander effect compared to T-DM1. Trastuzumab duocarmazine also uses a cleavable linker with a duocarmycin payload. Other approved ADCs have also demonstrated the bystander effect in preclinical models including enfortumab vedotin (EV), tisotumab vedotin (TV) and sacituzumab govitecan (SG) [22]. MMAE and MMAF which is extracted from sea hare, are commonly used cytotoxic payloads used across ADCs. MMAE exhibits bystander effect properties as it is not bound by cell membranes, while MMAF does not, and thus found to be less efficient and less toxic due to this pivotal difference [48,49]. While specific research in the bystander effect in the haematological space is limited, MMAE conjugates brentuximab vedotin and polatuzumab vedotin have been approved, respectively, for use in Hodgkin lymphoma and refractory diffuse large B-cell lymphoma. Brentuximab vedotin has also demonstrated the bystander effect in germ cell tumours expressing CD 30 positive and negative cells [50]. As MMAE-based ADCs often utilise cleavable linkers known to retain membrane permeability, these characteristics underpin their ability to create a bystander effect [51].
Seminal Phase II/III Trials of Antibody Drug Conjugates in Cancer
There are currently 13 ADCs approved by the US Food and Drug Administration (FDA) for various malignancies, with approvals occurring particularly rapidly since 2017 [1]. Here we summarise the pivotal trials of ADCs in solid and haematologic malignancies (illustrated in Table 1 and Table 2).

Table 1.
Positive Phase II/III Trials of Antibody Drug Conjugates in Solid Tumours Leading to FDA Approval.
Table 1.
Positive Phase II/III Trials of Antibody Drug Conjugates in Solid Tumours Leading to FDA Approval.
| Drug | FDA approval | Pivotal trial(s) | Population | Number of patients | Antibody Target, Linker and Payload | Results with Intervention vs Comparator |
|---|---|---|---|---|---|---|
|
Trastuzumab emtansine (T-DM1) |
2013 | EMILIA [52] (phase III) |
Advanced HER2+ breast cancer with PD after trastuzumab + taxane. | T-DM1: 495 Capecitabine + lapatinib: 496 |
Ab target: HER2 Linker: SMCC (non-cleavable) Payload: DM1 |
ORR 43.6% vs 30.8%, mPFS 9.6 vs 6.4 mths, mOS 30.9 vs 25.1 mths. |
| 2019 | KATHERINE [53] (phase III) |
Early stage HER2+ breast cancer with residual disease after NACT. | T-DM1: 743 Trastuzumab: 743 |
3 yr iDFS 88.3% vs 77.0%. | ||
|
Trastuzumab deruxtecan (T-DXd) |
2022 | DESTINY-Breast03 [54] (phase III) |
Advanced HER2+ breast cancer with PD after trastuzumab + taxane. | T-DXd: 261 T-DM1: 263 |
Ab target: HER2 Linker: GGFG tetrapeptide (cleavable) Payload: Deruxtecan |
ORR 79.7% vs 34.2%, mPFS not reached vs 6.8 mths with T-DM1, mOS both not reached. |
| 2022 | DESTINY-Breast02 [55] (phase III) |
Advanced HER2+ breast cancer with PD after T-DM1. | T-DXd: 406 TPC: 202 |
ORR 70% vs 29%, mPFS 17.8 vs 6.9 mths, mOS 39.2 vs 26.5 mths. |
||
| 2022 | DESTINY-Breast04 [56] (phase III) |
Advanced HER2 low breast cancer with PD after 1-2 lines of chemotherapy. | T-DXd: 373 TPC: 184 |
ORR 52.3% vs 16.3%, mPFS 9.9 vs 5.1 mths, mOS 23.4 vs 16.8 mths. |
||
| 2021 | DESTINY-Gastric01 [57] (phase II) |
Advanced HER2+ gastric/GOJ cancers after ≥2 lines of therapy. | T-DXd: 125 TPC: 62 |
ORR 51% vs 14%, mPFS 5.6 vs 3.5 mths, mOS 12.5 vs 8.4 mths. |
||
| 2022 | DESTINY-Lung01 [58] (phase II) | Advanced HER2+ NSCLC refractory to standard therapy. | T-DXd: 91 (single arm) | ORR 55%, mPFS 8.2 mths, mOS 17.8 mths. |
||
|
Sacituzumab govitecan (SG) |
2023 | TROPiCS-02 [59] (phase III) | Advanced HR+ breast cancer, HER2- or low with PD after ET and ≥2 systemic therapies. | SG: 272 TPC: 271 |
Ab target: Trop-2 Linker: CL2A (cleavable) Payload: SN-38 |
ORR 21% vs 14%, mPFS 5.5 vs 4.0 mths, mOS 13.9 vs 12.3 mths. |
| 2020 | ASCENT [60] (phase III) |
Advanced TNBC with PD after ≥2 lines of chemotherapy. | SG: 235 TPC: 233 |
ORR 35% vs 5%, mPFS 5.6 vs 1.7 mths, mOS 12.1 vs 6.7 mths. |
||
| 2021 | TROPHY [61] (phase II) |
Advanced urothelial cancer with PD after platinum and immunotherapy. | SG: 113 (single arm) |
ORR 27%, mPFS 5.4 mths, mOS 10.9 mths. |
||
| 2020 | IMMU-132-01 [62] (phase I/II) | Advanced TNBC after ≥2 lines of chemotherapy. | SG: 108 (single arm) |
ORR 33.3%, mPFS 5.5 mths, mOS 13.0 mths. |
||
|
Enfortumab vedotin (EV) |
2019 | EV-201 [63,64] (phase II) | Advanced urothelial carcinoma. Cohort 1: PD after platinum + immunotherapy. Cohort 2: PD after immunotherapy, no prior platinum. |
Cohort 1: 125 Cohort 2: 89 (single arm) |
Ab target: Nectin-4 Linker: mc-VC-PABC (cleavable) Payload: MMAE |
Cohort 1: ORR 44%, mPFS 5.8 mths, mOS 11.7 mths Cohort 2: ORR 52%, mPFS 5∙8 mths, mOS 14.7 mths. |
| 2019 | EV-301 [65] (phase III) | Advanced urothelial carcinoma with PD after platinum and immunotherapy. | EV: 301 TPC: 307 |
ORR 40.6% vs 17.9%, mPFS 5.6 vs 3.7 mths, mOS 12.9 vs 9.0 mths. |
||
|
Disitamab vedotin* (DV) |
2021 | [66] (phase II) |
Advanced HER2+ urothelial carcinoma with PD after ≥1 prior therapy. | DV: 43 (single arm) |
Ab target: HER2 Linker: mc-VC-PABC (cleavable) Payload: MMAE |
ORR 51.2%, mPFS 6.9 mths, mOS 13.9 mths. |
|
Tisotumab vedotin (TV) |
2021 | InnovaTV 204 [67] (phase II) | Recurrent/advanced cervical cancer with PD after ≤2 lines of chemotherapy. | TV: 102 (single arm) |
Ab target: tissue factor Linker: mc-VC-PABC (cleavable) Payload: MMAE |
ORR 24%, mPFS 4.2 mths, mOS 12.1 mths. |
| Mirvetuximab soravtansine (MIRV) | 2022 | SORAYA [68] (phase II) |
FRα high platinum- resistant ovarian cancer with ≤3 prior systemic therapies, including bevacizumab. | MIRV: 106 (single arm) |
Ab target: FRα Linker: disulfide hydrophilic sulfo-SPDB (cleavable) Payload: DM4 |
ORR 32.4%, mPFS 4.3 mths, mOS 13.8 mths. |
Abbreviations: Ab, Antibody; ABVD, doxorubicin, bleomycin, vinblastine and dacarbazine; ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; AVD, doxorubicin, vinblastine and dacarbazine; BCMA, B-cell maturation antigen; BG, bendamustine and obinutuzumab; BR, bendamustine and rituximab; BSC, best supportive care; CHOP, cyclophosphamide, doxorubicin, vincristine and prednisone; CHP, cyclophosphamide, doxorubicin, and prednisone; CL2A, cross-linked 2A; CR, complete response; DLBCL, diffuse large B cell lymphoma; EFS, event-free survival; FRα, folate receptor α; GGFG, Gly-Gly-Phe-Gly; HR, hormone receptor; mc-VC-PABC, maleimidocaproyl-valyl-citrullinyl-p-aminobenzyloxycarbonyl; iDFS, invasive disease free survival; MMAE/F, monomethyl auristatin-E/F; mth, months; mPFS, median progression free survival; mOS, median overall survival; NACT, neoadjuvant chemotherapy; NMPA, National Medical Products Administration of China; ORR, objective response rate; PBD, pyrrolobenzodiazepine; PD, progressive disease; RFS, relapse-free survival; SG, Sacituzumab govitecan; SMCC, succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate; SPDB, N-succinimydl 4-(2-pyridyldithio)−2-sulfobutanoate); TPC, treatment of physician’s choice. * Approved by NMPA.
Table 2.
Positive Phase II/III Trials of Antibody Drug Conjugates in Haematological Malignancies Leading to FDA Approval.
Table 2.
Positive Phase II/III Trials of Antibody Drug Conjugates in Haematological Malignancies Leading to FDA Approval.
| Drug | FDA approval | Pivotal trial(s) | Population | Number of patients | Antibody Target, Linker and Payload | Results with Intervention vs Comparator |
|---|---|---|---|---|---|---|
| Gemtuzumab ozogamicin (GO) | 2017 | ALFA-0701 [69–71] (phase III) | Newly diagnosed, CD33+ AML, age 50-70. | GO + standard therapy: 140 SOC: 140 |
Ab target: CD33 Linker: hydrazone (cleavable) Payload: calicheamicin |
2 year EFS 40·8% vs 17.1%, RFS 50.3% vs 22.7%. |
| 2017 | AAML0531 [72] (phase III) |
Newly diagnosed AML age 0-29 years. | GO + standard therapy: 511 SOC: 511 |
3 yr EFS 53.1% vs 46.9%, 3 yr OS 69.4% vs 65.4%. | ||
| 2017 | AML-19 [73] (phase III) | Newly diagnosed AML, >75 yrs or 61-75 yrs and unfit for intensive chemotherapy. | GO: 118 BSC: 119 |
mOS 4.9 vs 3.6 mths. | ||
| 2017 | MyloFrance-1 [74] (phase II) |
CD33+ AML in first relapse. | GO: 57 (single arm) |
ORR 33.3%, mOS 8.4 mths, mRFS 11.0 mths. | ||
|
Brentuximab vedotin (BV) |
2018 | ECHELON-2 [75] (phase III) |
Untreated CD30+ peripheral T cell lymphomas. | BV + CHP: 226 CHOP: 226 |
Ab target: CD30 Linker: mc-VC-PABC (cleavable) Payload: MMAE |
5 yr PFS 51.4% vs 43.0%, 5 yr OS 70.1% vs 61.0%. |
| 2018 | ECHELON-1 [76] (phase III) |
Untreated stage III-IV classical Hodgkin lymphoma. | BV + AVD: 664 ABVD: 670 |
5 yr PFS 82.2% vs 75.3%, OS immature. | ||
| 2017 | ALCANZA [77] (phase III) |
Relapsed primary cutaneous anaplastic large cell lymphoma or CD30+ mycosis fungoides. | BV: 64 TPC: 64 |
ORR 54.7% vs 12.5%, mPFS 16.7 vs 3.5 mths, 3 year OS 64.4% vs 61.9%. | ||
| Polatuzumab vedotin (PV) | 2019 | Study GO29365 [78] (phase Ib/II) | Relapsed or refractory DLBCL with ≥2 prior therapies. | 1. PV + BG: 20 2. PV + BR: 40 3. BR: 40 |
Ab target: CD79b Linker: mc-VC-PABC (cleavable) Payload: MMAE |
Phase I: PV + BG mOS 10.8 mths. Phase II: PV + BR vs BR mPFS 12.4 vs 4.7 mths. |
| Belantamab mafodotin (BM) | 2020 | DREAMM-2 [79] (phase II) |
Relapsed or refractory multiple myeloma with ≥4 prior therapies. |
Cohort 1 (BM 2.5 mg/kg): 97 Cohort 2 (BM 3.4 mg/kg): 99 |
Ab target: BCMA Linker: mc (non-cleavable) Payload: MMAF |
Cohort 1: ORR 31%, mPFS 2.9 mths. Cohort 2: ORR 34%, mPFS 4.9 mths. |
| Inotuzumab ozogamicin (InO) | 2017 | INO-VATE [80] (phase III) |
Relapsed or refractory B-cell precursor ALL. | InO: 164 TPC: 162 |
Ab target: CD22 Linker: hydrazone (cleavable) Payload: calicheamicin |
mOS: 7.7 vs 6.2 mths, 2 yr OS: 22.8% vs 10.0%. |
|
Moxetumomab pasudotox (MP) |
2018 | Study 1503 [81] (phase II) |
Relapsed or refractory hairy cell leukaemia. | MP: 80 (single arm) |
Ab target: CD22 Linker: hydrazone (cleavable) Payload: pasudotox |
Durable CR rate of 36%, median CR duration 62.8 mths, mPFS 41.5 mths. |
|
Loncastuximab tesirine (LT) |
2021 | LOTIS-2 [82] (phase II) | Relapsed or refractory DLBCL after ≥2 therapies. | LT: 145 (single arm) |
Ab target: CD19 Linker: valine-alanine (cleavable) Payload: PBD dimer |
ORR 48.3%, mPFS 4.9 mths, mOS 9.9 mths. |
Abbreviations: Ab, Antibody; ABVD, doxorubicin, bleomycin, vinblastine and dacarbazine; ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; AVD, doxorubicin, vinblastine and dacarbazine; BCMA, B-cell maturation antigen; BG, bendamustine and obinutuzumab; BR, bendamustine and rituximab; BSC, best supportive care; CHOP, cyclophosphamide, doxorubicin, vincristine and prednisone; CHP, cyclophosphamide, doxorubicin, and prednisone; CL2A, cross-linked 2A; CR, complete response; DLBCL, diffuse large B cell lymphoma; EFS, event-free survival; FRα, folate receptor α; GGFG, Gly-Gly-Phe-Gly; HR, hormone receptor; mc-VC-PABC, maleimidocaproyl-valyl-citrullinyl-p-aminobenzyloxycarbonyl; MMAE/F, monomethyl auristatin-E/F; mth, months; mPFS, median progression free survival; mOS, median overall survival; NACT, neoadjuvant chemotherapy; NMPA, National Medical Products Administration of China; ORR, objective response rate; PBD, pyrrolobenzodiazepine; PD, progressive disease; RFS, relapse-free survival; SOC, Standard of Care; SG, Sacituzumab govitecan; SMCC, succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate; SPDB, N-succinimydl 4-(2-pyridyldithio)−2-sulfobutanoate); TPC, treatment of physician’s choice. * Approved by NMPA.
Trials of ADCs in Solid Organ Malignancies
Three ADCs have been FDA-approved for the treatment of breast cancer. This is not surprising as therapeutic antibodies are well established in treating this disease, and ADCs represent an extension of this approach. T-DM1 was the first ADC approved for breast cancer and consists of a humanised HER2-directed monoclonal antibody trastuzumab, linked via a non-cleavable linker to DM1, a microtubule inhibitor, with a DAR of approximately 3.5 [1]. It has been shown to improve median overall survival in patients with metastatic HER2 positive breast cancer treated in the second line setting compared to capecitabine plus lapatinib, with a hazard ratio of 0.68 (95% CI 0.55 to 0.85, p <0.001) [52]. It has also been approved for use in patients with residual HER2-amplified breast cancer after neoadjuvant HER2-directed therapy and chemotherapy, where it has been shown to improve invasive disease-free survival by 50% compared to adjuvant trastuzumab (HR 0.50, 95% CI 0.39 to 0.64; P<0.001) [53].
T-DXd has subsequently been shown to improve outcomes compared to T-DM1. T-DXd is a newer ADC that consists of the same HER2 directed monoclonal antibody trastuzumab, linked via a cleavable tetrapeptide linker to the payload deruxtecan, which is a topoisomerase 1 inhibitor [1]. In the DESTINY-Breast03 trial, which compared to T-DXd to T-DM1 in patients with metastatic HER2 positive breast cancer in the second line setting, reported an impressive hazard ratio of 0.28 for disease progression or death (95% CI 0.22 to 0.35, p<0.001) [54]. Furthermore, the DESTINY-Breast02 trial is the first and only trial exploring ADC use in patients who have previously progressed on another ADC, T-DM1. It showed that T-DXd was superior to treatment of physician’s choice in patients with metastatic HER2 positive breast cancer previously treated with T-DM1, with hazard ratio of 0·36 for progression-free survival (PFS) (95% CI 0·28 to 0·45; p<0·0001) [55].
Historically, HER2-positive breast cancer has been defined in a binary fashion as either HER2 positive, defined as a score of 3+ on immunohistochemistry (IHC) or 2+ on IHC and positive in situ hybridisation (ISH), or HER2 negative, defined by a score of 0-1 on IHC or 2+ on IHC and negative on ISH [56]. However, in the phase III randomised controlled DESTINY-Breast04 trial, T-DXd was shown to have benefit in patients with ‘HER2 low’ breast cancer, defined as a score of 1+ on IHC or as 2+ on IHC with negative ISH, with a hazard ratio of 0.64 for overall survival (95% CI 0.49 to 0.84) [56]. This pivotal study has redefined the treatment algorithm and classification of breast cancer. In terms of toxicity, drug-related interstitial lung disease or pneumonitis occurred in 12.1% of patients receiving T-DXd on the DESTINY-Breast 04 trial, including 3 (0.8%) with a fatal event. In the DESTINY-Breast 03 trial, 10.5% developed drug-related pneumonitis with no fatalities [54,56].
More recently, there has been an expansion of drug indications across tumour types based on shared receptor biology between different tumour types. For example, T-DXd is now approved for HER2-positive metastatic gastric and lung cancers [57,58]. In patients with metastatic gastric or gastro-oesophageal junction cancers with disease progression after two or more lines of previous therapy, T-DXd was associated with a 41% improvement in median overall survival compared to chemotherapy (HR 0.59, 95% CI 0.39 to 0.88) [57]. Multiple trials are ongoing using T-DXd in a wide variety of other HER2 positive malignancies. Another similar ADC is disitamab vedotin, which consists of a HER2 directed monoclonal antibody, cleavable linker and MMAE payload [66]. There have been promising results with disitamab vedotin in a single arm phase II trial of patients with HER2-positive advanced urothelial carcinoma, with an objective response rate of 51.2% [66].
Sacituzumab govitecan consists of a Trop-2 directed antibody linked via a cleavable linker to SN-38, a topoisomerase 1 inhibitor payload [60,62]. Trop-2 is an antigen that has been found to be over-expressed in triple negative breast cancer and many other solid malignancies, and is associated with cancer progression and poor prognosis [83]. Sacituzumab govitecan has been shown to improve overall survival for patients with late line metastatic triple negative breast cancer compared to treatment of physicians choice (HR 0.48, 95% CI 0.38 to 0.59; P<0.001) [60]. More recently, it has also been demonstrated to improve PFS in patients with metastatic hormone receptor-positive breast cancer compared to treatment of physicians choice (HR 0.66, 95% CI 0.53-0.83; P= 0.0003) [59]. Furthermore, sacituzumab govitecan has shown clinical benefit in a single-arm phase II trial of patients with metastatic urothelial cancer with progressive disease after platinum and immunotherapy [61,84,85]. There are currently at least 19 trials underway studying the use of sacituzumab govitecan in a range of malignancies, including glioblastoma, refractory metastatic epithelial cancers, and breast, non-small cell lung, urothelial, prostate, head and neck, endometrial, and ovarian cancers [86].
Enfortumab vedotin combines a nectin-4 directed monoclonal antibody linked via a cleavable linker to MMAE [63–65]. Despite having a different antibody target, linker, and payload to sacitizumab govitecan, it has shown similar benefit in patients with metastatic urothelial cancer who have disease progression after platinum and immunotherapy. In the phase III trial in patients with progressive disease after platinum and immunotherapy, enfortumab vedotin was shown to improve overall survival compared with chemotherapy (HR 0.70; 95% CI 0.56 to 0.89; P = 0.001) [65].
Two ADCs have been recently approved for refractory advanced gynaecological cancers based on data from phase II trials. Mirvetuximab soravtansine is composed of a folate receptor alpha (FRα) antibody, cleavable linker and DM4 payload, another microtubule inhibitor [68]. FRα is a cell surface glycoprotein that mediates various cellular processes such as cell division, proliferation and tissue growth [87]. It is over-expressed in over 90% of ovarian cancers, as well as in uterine, lung and breast cancers [87]. A phase II single-arm trial evaluated mirvetuximab soravtansine in patients with platinum-resistant ovarian cancer with high FRα expression who had disease progression after 1-3 lines of chemotherapy plus bevacizumab. ORR was 32.4%, median PFS of 5.5 months, and median overall survival of 13.8 months [68]. In comparison, the standard of care for platinum-resistant recurrent ovarian cancer of chemotherapy plus bevacizumab is associated with response rates of 27.3% and a median PFS of 6.7 months [88]. Similarly, outcomes are poor for those with metastatic cervical cancer who have progressive disease after first-line therapy. Tisotumab vedotin is an ADC directed against tissue factor, with a cleavable linker and DM4 payload [67]. Tissue factor is physiologically expressed on adventitial cells and released after endothelial injury, however is also pathologically over-expressed on the surface of tumour cells and endothelial cells in various cancers, including pancreatic cancer, cervical cancer, sarcoma, lung cancer, triple negative breast cancer, and acute lymphocytic leukaemia [89]. A phase II single-arm trial of tisotumab vedotin in patients with disease progression on or after doublet chemotherapy with bevacizumab demonstrated an ORR of 24%, median PFS of 4·2 months, and median overall survival of 12.1 months [67]. The confirmatory phase III trials of mirvetuximab soravtansine (clinicaltrials.gov identifier NCT04209855) and tisotumab vedotin (clinicaltrials.gov identifier NCT04697628) are ongoing [90].
ADCs in Haematological Malignancies
There are multiple ADCs approved in haematological malignancies, as illustrated in Table 2. The first ADC to be approved by the FDA was gemtuzumab ozogamicin for the treatment of adults with relapsed CD33+ acute myeloid leukaemia [1]. Gemtuzumab ozogamicin is a CD33 targeted ADC with a cleavable linker and calicheamicin payload [73]. Approval was subsequently withdrawn after the phase III SWOG S0106 trial demonstrated a higher mortality rate of 5.5% and high rates of hepatic toxicity, with gemtuzumab ozogamicin (6mg/ m2) plus standard chemotherapy compared to 1.4% with standard chemotherapy alone [91]. Later randomised phase III clinical trials using a lower dose of gemtuzumab ozogamicin of 3 mg/m2 showed clinical benefit and improved safety, which led to its re-approval by the FDA in 2017 [69–74]. Inotuzumab ozogamicin, which targets CD22 with a cleavable linker and calicheamicin payload, has been associated with improved overall survival (HR 0.74, 97.5% CI 0.57-0.99, p=0.01) in patients with relapsed or refractory B-cell precursor acute lymphoblastic leukaemia compared to treatment of physician’s choice [80]. Moxetumomab pasudotox which also targets CD22 with the same cleavable linker as inotuzumab ozogamicin, but with a different payload (pasudotox), has shown benefit in a small single-arm phase II trial in patients with relapsed or refractory hairy cell leukaemia, with a median PFS of 41.5 months [81].
Brentuximab vedotin, which targets CD30 with a cleavable linker and MMAE payload, has been approved for several haematological malignancies [50,75–77]. In the phase III ECHELON-1 trial of untreated stage III-IV classical Hodgkin lymphoma, brentuximab vedotin with doxorubicin, vinblastine, dacarbazine was compared to doxorubicin, bleomycin, vinblastine and dacarbazine. 5 year PFS was improved with the addition of brentuximab vedotin (HR 0·68, 95% CI 0·53–0·87, p=0.0017) [76]. It has also shown to improve 5-year overall survival in CD30 expressing peripheral T cell lymphoma when added to cyclophosphamide, doxorubicin, and prednisone compared to cyclophosphamide, doxorubicin, vincristine and prednisone (HR 0.72, 95% CI 0.53-0.99) [75]. Similarly, in a small phase III trial of relapsed primary cutaneous anaplastic large cell lymphoma or CD30 expressing mycosis fungoides, brentuximab has shown improved response rates and PFS [77].
Polatuzumab vedotin targets CD79b, possesses a cleavable linker and MMAE payload [78]. In a small phase Ib/II trial of patients with relapsed or refractory diffuse large B cell lymphoma, when combined with bendamustine and rituximab, it was shown to improve OS, compared to bendamustin and rituximab alone (HR 0.42, 95% CI 0.24 to 0.75, p=0.002) [78]. Similarly, loncastuximab tesirine, which targets CD19, has a cleavable linker and pyrrolobenzodiazepine dimer payload, has been shown in a small single-arm phase II trial of patients with relapsed or refractory large B cell lymphoma to be associated with a high response rate of 48.3% and median overall survival of 9.9 months [82]. In addition, belantamab mafodotin which targets B cell maturation antigen, and possesses a non-cleavable linker and MMAF payload, has been shown to be active in a small phase II trial in a heavily pre-treated population of patients with multiple myeloma, with a 31-34% response rate depending on the dose used [79].
Challenges in the Clinical Development of ADCs and Limitations of Current ADCs
ADCs continue to gain popularity owing to heightened efficacy compared to conventional chemotherapy. Despite this, limitations exist, with over 50 potential ADCs having ceased development due to barriers such as limited efficacy or toxicity [92]. Early-phase research into ADCs poses distinct challenges and requires different approaches to trials of conventional cytotoxic therapy. For instance, phase I trials have traditionally been designed to find the maximum tolerated dose (MTD) of drugs on the basis that toxicity and response are positively correlated. However, in trials of targeted therapies and immunotherapies, it has been shown that there is not a predictable linear correlation between dose and efficacy [93]. This relationship has not been well studied in ADC trials. Still, it is likely to be distinct from traditional cytotoxic chemotherapy since various factors other than dose can influence ADC efficacy and therapeutic windows. These factors include the homogeneity or heterogeneity and level of expression of the target antigen on the tumour tissue, the degree of expression of target antigen on normal tissues, premature extracellular deconjugation of ADCs due to linker instability, and the permeability of the payload across cell membranes, and hence the degree to which the bystander effect occurs [94]. A threshold effect can exist for ADCs, whereby exceeding a particular dose of an ADC does not increase exposure or efficacy. There are various strategies that have been suggested to improve the optimal dose delivery of ADCs. These include body weight dose capping, treatment duration capping, altering the dose frequency, response-guided dosing, and randomised dose-finding studies [94]. Further improvements in the design and dose-finding of early-phase trials using ADCs are greatly needed.
Toxicities resulting from off-target effects where the payload is released to other tissues represent another obstacle to the development and adoption of ADCs. These are typically hepatic, neurologic, haematologic, respiratory or ophthalmic in nature [95]. Key examples of toxicities include the expression of HER2 and Nectin-2 on cardiomyocytes and skin, causing cardiotoxicity and skin toxicity, respectively [96]. Due to the unique makeup of different ADCs, adverse effect profiles vary and may be unique to the ADC, highlighting the importance of being able to provide reference guides for the individual drugs [97]. Early in ADC development, premature release of ADC payloads causing higher toxicity was associated with linker instability. To reduce this, the half-life of the ADC needed to be 10 times that of the payload itself [98]. Another key parameter to minimise premature drug release and toxicity is the polarity of the linker. This balance enhances payload coupling and reduces immunogenicity while maintaining an appropriate payload delivery [99,100]. If the cytotoxic agent is too hydrophobic, this can change the antibody properties leading to aggregation or conjugation. Conversely, the bystander effect can result from hydrophilic payloads. This can lead to increased stability and safety of ADC on non-target cells, allowing for increased cytotoxicity through increased payload delivery. These balances can influence drug efficacy and tolerability [5]. Another potential limitation of ADCs is in cancers that possess a dense tumour stroma. High-molecular weight drugs, including ADCs, have limited ability to penetrate dense tumour stroma to reach the required target, representing another limitation of ADCs [26].
Mechanisms of Resistance to ADCs
Understanding resistance mechanisms to ADCs is an emerging area that needs further research. The areas of weakness that cancer can exploit include the internalisation process, payload mechanism and the interaction between the antigen and antibody. Despite the potency of the payload, resistance to this can occur [101]. Contributing factors can be the ABC transporters (drug efflux pumps), historically known to impact and decrease the effectiveness of chemotherapeutic agents [101]. Preclinical in vitro models of breast cancer cells exhibiting ABCC1 (multidrug resistance protein 1) expression exhibited a 256-fold increased resistance to T-DM1 after three months of cyclical treatment [102]. Cancer cells exposed to chronic unconjugated tubulin inhibitors administration can also induce the drug transporter MDR1 which is hypothesised to play a role in DM1 resistance [101]. This transporter, along with MRP1 can be upregulated from chronic exposure leading to acquired resistance, efflux upregulation and drug deposition [101]. Acquired or intrinsic resistance is a challenge of ADCs, including antibody resistance, inability to traffic the required drug, lysosomal dysfunction and payload inefficiency [101]. Currently, no resistance models correlating loss of ADC activity and conjugate internalisation exist. Proteomic profiling has shown utility in identifying protein alteration involving different aspects of the internalisation process, including lysosome biogenesis, vehicle transport, the cytoskeleton and trafficking of the antibody [102]. Loss of lysosomal transporters could also decrease ADC efficacy [102,103].
ADC payload resistance and challenges are seen in a variety of cancer subtypes. An example includes a patient with long exposure to sacituzumab govitecan who underwent biopsies after death [104]. Tumour subclones with the mutation TOP1 known to encode topoisomerase-1 and the mutation TACSTD2, the encoder for TROP2, were found [104]. Parallel resistance mechanisms could occur affecting payload and antibody concurrently [104]. HER-2 receptor kinase or kinase signalling pathway alteration is a resistance mechanism after persistent ADC exposure. The T-DM1 resistant preclinical model KPL-4-T-DM1-R demonstrated decreased levels of HER2 and HER3 while other kinases such as EGFR increased [105]. Antigen expression and heterogeneity has been shown to be a mechanism of resistance in preclinical studies as described in the JIMT1 lines (resistant cell lines), where xenograft tumours treated with T-DM1 exhibited lower HER2 expression, which is associated with higher relapse rates and lower survival rates [106]. Changed target expression, before or during treatment, was associated with potentially worse outcomes in haematological malignancies, including myeloid leukaemia with low CD33 expression [101,107].
Future Directions
There is a substantial amount of research being conducted into ADCs, with over one hundred ADCs in preclinical and early-stage clinical research [1]. While historically targeted therapies have targeted oncogenic driver mutations, given how efficacious ADCs have been, we are seeing a shift in designs of ADCs so that the antigenic target is not necessarily an oncogenic driver but rather simply a target that is preferentially over-expressed in malignant cells. Haematological malignancies have more identifiable targets due to lineage-specific antigens making them the perfect candidates to target, while antigenic targets are often more heterogeneously expressed and less specific in solid organ tumours [108,109].
Developing Novel Antigenic Targets and Antibodies
Some of these new antigenic targets that are being explored in solid tumours include prostate-specific membrane antigen (PSMA), six-transmembrane epithelial antigen of prostate-1 (STEAP-1), tissue factor, deltalike protein 3 (DLL-3), mesothelin, ENPP3 and B7-H3 family of proteins [110]. DLL-3 is an inhibitory Notch pathway ligand that mediates oncogenesis in melanoma, bladder, endometrial, ovarian, pancreatic and lung cancer via multiple mechanisms, including angiogenesis, tumour stromal remodelling, and effects on immune cells in the tumour stroma [111]. PSMA is a membrane glycoprotein that is highly and selectively expressed in prostate cancer, and a PSMA ADC using an MMAE payload and valine-citrulline dipeptide linker has shown safety and activity in phase I trials [112]. STEAP-1 is a cell membrane protein that acts as an ion channel or transporter protein, and is highly expressed in prostate, breast, pancreas, bladder, gastrointestinal tract, testicular, ovarian, and cervical cancers, Ewing sarcoma and melanoma [113]. DSTP3086S is an ADC which consists of a humanized IgG1 linked through a protease cleavable linker to MMAE, and has demonstrated in a phase I trial to have acceptable safety and evidence of activity in metastatic castrate-resistant prostate cancer [114].
A phase I trial of an ADC targeted against ENPP3, a protein expressed by most clear cell renal cell carcinomas, has reported tolerable toxicity and efficacy [115]. B7-H3 is an immune checkpoint protein that is overexpressed in many paediatric cancers, as well as non-small cell lung cancer and prostate cancer [116]. B7-H3 ADCs are currently being studied in medulloblastoma, peritoneal cancer, neuroblastoma, glioma, prostate cancer, head and neck cancer, non-small cell lung cancer, urothelial cancer, rhabdomyosarcoma, osteosarcoma, Ewing sarcoma, and Wilms’ tumour [116]. For instance, AbBV-155 (mirzotamab clezutoclax), an anti-B7-H3 ADC, has been evaluated in non-small cell lung cancer and breast cancer. No significant dose-limiting toxicities were reported in the single agent phase one cohort, with a partial response occurring in 21% of patients [117]. Mesothelin is a cell membrane glycoprotein that is expressed in mesothelioma, lung adenocarcinoma, pancreatic adenocarcinoma, colorectal adenocarcinoma, serous ovarian cancer, gastric adenocarcinoma and breast cancer [118]. Various other ADCs targeting mesothelin are currently in development [118].
Various other targets are being explored for haematological malignancies, including CD37 for patients with relapsed and refractory diffuse large B cell lymphoma with naratuximab emtansine, CD138 with indatuximab ravtansine for multiple myeloma, CD19 with coltuximab mertansine for diffuse large B cell lymphoma and acute lymphocytic leukaemia, CD56 with lorvotuzumab mertansine for CD56 expressing haematological malignancies, and CD22 with pinatuzumab vedotin for diffuse large B cell lymphoma and follicular non-Hodgkin lymphoma [119].
ADCs employing bispecific antibodies are being explored as a potential means of improving efficacy in tumours with heterogeneous antigen expression. Bispecific ADCs can target two different antigens or different sites on the same antigen. Targeting two different sites on the same antigen is thought to enable more efficient internalisation of the compound and improve receptor aggregation [1]. For example, ADCs with bispecific antibodies targeting HER-2 and the prolactin receptor have been shown to improve ADC internalisation and have higher anti-tumour activity in vitro compared to a conventional HER2-directed ADC [120].
There is also increasing research into targeting cells in the tumour microenvironment. Cancer-associated fibroblasts are thought to promote therapeutic resistance and promote cancer cell survival. Two novel ADCs, fibroblast activation protein α monoclonal antibody conjugated to DM1 and fibroblast activation protein α conjugated to pseudomonas exotoxin 38, have been shown to be highly effective in xenograft models of lung, head and neck, pancreatic and breast cancers [121,122].
Improving Linker Technology
Several strategies to improve linker technology have been well reviewed elsewhere and hence will not be discussed in detail in this review [32]. Broadly, these include efforts to create highly selective linkers, improve linker-antibody attachments to improve linker stability and homogeneity, the development of additional linker-payload attachments, and various means to optimise their pharmacokinetics [32].
Development of Improved Cytotoxic and Other Payloads
Novel payloads and payload structures are also being explored. Dual payloads are being explored to improve responsiveness in solid tumours with heterogeneous target expression. Newer payloads in development include pyrrolobenzodiazepine monomers or dimers, indolino-benzodiazepines and cyclopropabenzindolone monomers and dimers, with IC50 values in the picomolar range. Some have been hindered by high rates of toxicity [16].
For instance, a HER2-targeted ADC containing dual payloads of MMAE and MMAF was designed and tested in a xenograft model of HER2+ breast cancer. The dual payload containing ADC was highly effective at killing tumour cells in vivo, more so than when both single payload-based ADCs were used together [123]. Other payloads in development include BCL-XL inhibitors that can induce apoptosis selectively in tumours that are BCL-XL dependent [124,125]. Overexpression of BCL-XL is often seen in cancers such as melanoma and glioblastoma [126].
Tyrosine kinase inhibitors have also been studied as payloads, as kinase families are known to be heavily involved in cell cycle progression, proliferation, angiogenesis, and movement of cells around the body. At present, tyrosine kinase inhibitor-based ADCs have not been as efficacious as hoped [16]. Tyrosine kinase inhibitor plus ADC combinations are being investigated to offset tumour heterogeneity and resistance. For instance, T-DM1 and tucatinib, a HER2 selective tyrosine kinase inhibitor, have been used in combination. This demonstrated an objective response rate of 47% in patients previously treated with trastuzumab and a prior taxane, along with a brain-specific response of 36% [127]. Recruitment into this specific combination is ongoing [127]. Photoimmunotherapy is another emerging treatment whereby monoclonal antibodies are conjugated with a light-activated dye, which when activated, disrupts tumour cells leading to cell death. For example, cetuximab sarotalocan combines an EGFR monoclonal antibody with the light activatable dye, IR700, and in a phase I trial of three patients with recurrent head and neck squamous cell carcinoma, two of three patients experienced a response with a manageable safety profile [128].
Immunotherapy and ADCs
There is increasing research into developing ADCs that have a heightened ability to stimulate the immune system, which is particularly relevant as ADCs work partly through Antibody-dependent cellular cytotoxicity. Research is ongoing into two approaches: first, using immunotherapy in combination with ADCs, and second, incorporating immunotherapy into ADCs. ADCs interact with the local tumour immune microenvironment via activation of dendritic cells, activation of T cells, and upregulation of damage-associated molecular patterns (DAMPs) and have been shown to enhance the anti-tumour effect of immunotherapy in preclinical models [129,130]. Combining HER2-directed ADCs and immunotherapy was trialled in the phase II KATE2 study, where T-DM1 and atezolizumab were compared to T-DM1 and placebo in pre-treated patients with HER-2-positive breast cancer. The combined immunotherapy and HER2 treatment failed to improve PFS, but a trend was noted for benefit in those with PDL-1 expression [131]. Despite this combination not having the desired outcome, combining ADC and immunotherapy in early-phase studies is promising in a plethora of cancers, including small cell lung, ovarian, triple-negative breast cancer and urothelial cancer [13].
In the second approach, ADCs are being designed to stimulate the immune system. The two main categories of these immune-stimulating ADCs in development at present are ADCs containing STING agonists and TLR agonists [16]. Conventional STING and TLR agonists have been unsuccessful to date owing to high rates of toxicity, particularly characterised by cytokine release syndrome. The first immunostimulatory ADC to reach clinical trials, NJH395, combines a small molecule TLR7/8 agonist with an anti-HER2 monoclonal antibody. However, results were disappointing in a phase I clinical trial of 18 patients with non-breast HER2-positive malignancies, characterised by high rates of cytokine release syndrome and limited efficacy [132,133].
In summary, ADCs represent a new class of therapies that combines the strengths that therapeutic antibodies and potent chemotherapy delivers. While there has been significant success with ADCs recently, the field remains in its infancy. Multiple areas have not yet been thoroughly studied, such as resistance mechanisms, the optimal dosing of ADCs, and the interplay between the immune system and ADCs. As more ADCs come into clinical use, recurrent themes on its mechanisms of action and toxicities are likely to emerge, although each ADC is likely to be unique in its own right due to its combination of antibody, payload and linker, with significant opportunities existing to improve upon each component.
References
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduction and Targeted Therapy 2022, 7, 93. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2023. MAbs 2023, 15, 2153410. [Google Scholar] [CrossRef]
- Theocharopoulos, C.; Lialios, P.P.; Samarkos, M.; Gogas, H.; Ziogas, D.C. Antibody-Drug Conjugates: Functional Principles and Applications in Oncology and Beyond. Vaccines (Basel) 2021, 9. [Google Scholar] [CrossRef]
- McKertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Yap, T.A.; Heymach, J.V.; Meric-Bernstam, F.; Le, X. Antibody-drug conjugates in lung cancer: dawn of a new era? npj Precision Oncology 2023, 7, 5. [Google Scholar] [CrossRef]
- Marei, H.E.; Cenciarelli, C.; Hasan, A. Potential of antibody–drug conjugates (ADCs) for cancer therapy. Cancer Cell International 2022, 22, 255. [Google Scholar] [CrossRef] [PubMed]
- Valent, P.; Groner, B.; Schumacher, U.; Superti-Furga, G.; Busslinger, M.; Kralovics, R.; Zielinski, C.; Penninger, J.M.; Kerjaschki, D.; Stingl, G.; et al. Paul Ehrlich (1854-1915) and His Contributions to the Foundation and Birth of Translational Medicine. J Innate Immun 2016, 8, 111–120. [Google Scholar] [CrossRef]
- Loadman, P. Anticancer Drug Development. British Journal of Cancer 2002, 86, 1665–1666. [Google Scholar] [CrossRef]
- Firer, M.A.; Luboshits, G. Antibody-Drug-Conjugate Therapy for Hematological Cancers: Matching Cell Biology with Clinical Benefit. Advanced Functional Materials 2021, 31, 2100032. [Google Scholar] [CrossRef]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody–drug conjugates: current status and future directions. Drug Discovery Today 2014, 19, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Jerjian, T.V.; Glode, A.E.; Thompson, L.A.; O'Bryant, C.L. Antibody-Drug Conjugates: A Clinical Pharmacy Perspective on an Emerging Cancer Therapy. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 2016, 36, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Antrás, J.; Genta, S.; Vijenthira, A.; Siu, L.L. Antibody–drug conjugates: in search of partners of choice. Trends in Cancer 2023, 9, 339–354. [Google Scholar] [CrossRef]
- Mahmood, I. Clinical Pharmacology of Antibody-Drug Conjugates. Antibodies (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Stepan, L.P.; Trueblood, E.S.; Hale, K.; Babcook, J.; Borges, L.; Sutherland, C.L. Expression of Trop2 Cell Surface Glycoprotein in Normal and Tumor Tissues:Potential Implications as a Cancer Therapeutic Target. Journal of Histochemistry & Cytochemistry 2011, 59, 701–710. [Google Scholar] [CrossRef]
- Conilh, L.; Sadilkova, L.; Viricel, W.; Dumontet, C. Payload diversification: a key step in the development of antibody–drug conjugates. Journal of Hematology & Oncology 2023, 16, 3. [Google Scholar] [CrossRef]
- Gébleux, R.; Casi, G. Antibody-drug conjugates: Current status and future perspectives. Pharmacol Ther 2016, 167, 48–59. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nature reviews Drug discovery 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res 2001, 7, 1490–1496. [Google Scholar]
- Choi-Sledeski, Y.M.; Wermuth, C.G. Chapter 28 - Designing Prodrugs and Bioprecursors. In The Practice of Medicinal Chemistry (Fourth Edition), Wermuth, C.G., Aldous, D., Raboisson, P., Rognan, D., Eds.; Academic Press: San Diego, 2015; pp. 657–696. [Google Scholar]
- Agarwal, P.; Bertozzi, C.R. Site-specific antibody–drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate chemistry 2015, 26, 176–192. [Google Scholar] [CrossRef]
- Giugliano, F.; Corti, C.; Tarantino, P.; Michelini, F.; Curigliano, G. Bystander effect of antibody–drug conjugates: fact or fiction? Current Oncology Reports 2022, 24, 809–817. [Google Scholar] [CrossRef]
- Su, D.; Zhang, D. Linker Design Impacts Antibody-Drug Conjugate Pharmacokinetics and Efficacy via Modulating the Stability and Payload Release Efficiency. Frontiers in Pharmacology 2021, 12. [Google Scholar] [CrossRef]
- Zhang, D.; Yu, S.-F.; Ma, Y.; Xu, K.; Dragovich, P.S.; Pillow, T.H.; Liu, L.; Del Rosario, G.; He, J.; Pei, Z.; et al. Chemical Structure and Concentration of Intratumor Catabolites Determine Efficacy of Antibody Drug Conjugates. Drug Metabolism and Disposition 2016, 44, 1517. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.V.; Kaur, S.; Saad, O.M. Conjugation Site Influences Antibody-Conjugated Drug PK Assays: Case Studies for Disulfide-Linked, Self-Immolating Next-Generation Antibody Drug Conjugates. Analytical Chemistry 2020, 92, 12168–12175. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Molecular Cancer Research 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Kinneer, K.; Meekin, J.; Tiberghien, A.C.; Tai, Y.T.; Phipps, S.; Kiefer, C.M.; Rebelatto, M.C.; Dimasi, N.; Moriarty, A.; Papadopoulos, K.P.; et al. SLC46A3 as a Potential Predictive Biomarker for Antibody-Drug Conjugates Bearing Noncleavable Linked Maytansinoid and Pyrrolobenzodiazepine Warheads. Clin Cancer Res 2018, 24, 6570–6582. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein & Cell 2016, 9, 33–46. [Google Scholar] [CrossRef]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opinion on Therapeutic Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel Peptide Linkers for Highly Potent Antibody−Auristatin Conjugate. Bioconjugate Chemistry 2008, 19, 1960–1963. [Google Scholar] [CrossRef]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody–drug conjugates—A tutorial review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody-drug conjugates: Recent advances in linker chemistry. Acta Pharm Sin B 2021, 11, 3889–3907. [Google Scholar] [CrossRef] [PubMed]
- Brun, M.-P.; Gauzy-Lazo, L. Protocols for Lysine Conjugation. In Antibody-Drug Conjugates, Ducry, L., Ed.; Humana Press: Totowa, NJ, 2013; pp. 173–187. [Google Scholar]
- Matsuda, Y.; Mendelsohn, B.A. An overview of process development for antibody-drug conjugates produced by chemical conjugation technology. Expert Opinion on Biological Therapy 2021, 21, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Azar, I.; Alkassis, S.; Fukui, J.; Alsawah, F.; Fedak, K.; Al Hallak, M.N.; Sukari, A.; Nagasaka, M. Spotlight on Trastuzumab Deruxtecan (DS-8201,T-DXd) for HER2 Mutation Positive Non-Small Cell Lung Cancer. Lung Cancer (Auckl) 2021, 12, 103–114. [Google Scholar] [CrossRef]
- Nadkarni, D.V. Conjugations to Endogenous Cysteine Residues. In Antibody-Drug Conjugates: Methods and Protocols, Tumey, L.N., Ed.; Springer US: New York, NY, 2020; pp. 37–49. [Google Scholar]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nature biotechnology 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.V.; Cabel, L.; Bidard, F.C. Does sacituzumab-govitecan act as a conventional antibody drug conjugate (ADC), a prodrug of SN-38 or both? Ann Transl Med 2021, 9, 1113. [Google Scholar] [CrossRef]
- Singh, D.; Dheer, D.; Samykutty, A.; Shankar, R. Antibody drug conjugates in gastrointestinal cancer: From lab to clinical development. J Control Release 2021, 340, 1–34. [Google Scholar] [CrossRef]
- Zhao, L.; Ji, P.; Li, Z.; Roy, P.; Sahajwalla, C.G. The antibody drug absorption following subcutaneous or intramuscular administration and its mathematical description by coupling physiologically based absorption process with the conventional compartment pharmacokinetic model. J Clin Pharmacol 2013, 53, 314–325. [Google Scholar] [CrossRef]
- Su, D.; Kozak, K.R.; Sadowsky, J.; Yu, S.-F.; Fourie-O’Donohue, A.; Nelson, C.; Vandlen, R.; Ohri, R.; Liu, L.; Ng, C.; et al. Modulating Antibody–Drug Conjugate Payload Metabolism by Conjugation Site and Linker Modification. Bioconjugate Chemistry 2018, 29, 1155–1167. [Google Scholar] [CrossRef]
- Pillow, T.H.; Sadowsky, J.D.; Zhang, D.; Yu, S.F.; Del Rosario, G.; Xu, K.; He, J.; Bhakta, S.; Ohri, R.; Kozak, K.R.; et al. Decoupling stability and release in disulfide bonds with antibody-small molecule conjugates. Chem Sci 2017, 8, 366–370. [Google Scholar] [CrossRef]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. Journal of Experimental & Clinical Cancer Research 2018, 37, 20. [Google Scholar] [CrossRef]
- Vezina, H.E.; Cotreau, M.; Han, T.H.; Gupta, M. Antibody–Drug Conjugates as Cancer Therapeutics: Past, Present, and Future. The Journal of Clinical Pharmacology 2017, 57, S11–S25. [Google Scholar] [CrossRef]
- Seligson, J.M.; Patron, A.M.; Berger, M.J.; Harvey, R.D.; Seligson, N.D. Sacituzumab Govitecan-hziy: An Antibody-Drug Conjugate for the Treatment of Refractory, Metastatic, Triple-Negative Breast Cancer. Annals of Pharmacotherapy 2020, 55, 921–931. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nature Reviews Clinical Oncology 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Dan, N.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, S.C. Antibody-drug conjugates for cancer therapy: chemistry to clinical implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, Y.; Li, W.; Jeanty, C.; Xiang, G.; Dong, Y. Recent advances of antibody drug conjugates for clinical applications. Acta Pharmaceutica Sinica B 2020, 10, 1589–1600. [Google Scholar] [CrossRef]
- Schönberger, S.; van Beekum, C.; Götz, B.; Nettersheim, D.; Schorle, H.; Schneider, D.T.; Casati, A.; Craveiro, R.B.; Calaminus, G.; Dilloo, D. Brentuximab vedotin exerts profound antiproliferative and pro-apoptotic efficacy in CD30-positive as well as cocultured CD30-negative germ cell tumour cell lines. J Cell Mol Med 2018, 22, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, L.; Fan, S.; Xiao, D.; Xie, F.; Li, W.; Zhong, W.; Zhou, X. Antibody-Drug Conjugate Using Ionized Cys-Linker-MMAE as the Potent Payload Shows Optimal Therapeutic Safety. Cancers 2020, 12, 744. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. New England Journal of Medicine 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. New England Journal of Medicine 2018, 380, 617–628. [Google Scholar] [CrossRef]
- Cortés, J.; Kim, S.-B.; Chung, W.-P.; Im, S.-A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.-M.; Petry, V.; Chung, C.-F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. New England Journal of Medicine 2022, 386, 1143–1154. [Google Scholar] [CrossRef]
- André, F.; Hee Park, Y.; Kim, S.B.; Takano, T.; Im, S.A.; Borges, G.; Lima, J.P.; Aksoy, S.; Gavila Gregori, J.; De Laurentiis, M.; et al. Trastuzumab deruxtecan versus treatment of physician's choice in patients with HER2-positive metastatic breast cancer (DESTINY-Breast02): a randomised, open-label, multicentre, phase 3 trial. Lancet 2023. [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. New England Journal of Medicine 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Shitara, K.; Bang, Y.J.; Iwasa, S.; Sugimoto, N.; Ryu, M.H.; Sakai, D.; Chung, H.C.; Kawakami, H.; Yabusaki, H.; Lee, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Gastric Cancer. N Engl J Med 2020, 382, 2419–2430. [Google Scholar] [CrossRef]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazières, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2-Mutant Non–Small-Cell Lung Cancer. New England Journal of Medicine 2021, 386, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Bardia, A.; Marmé, F.; Cortes, J.; Schmid, P.; Loirat, D.; Tredan, O.; Ciruelos, E.; Dalenc, F.; Pardo, P.G.; et al. Primary results from TROPiCS-02: A randomized phase 3 study of sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) in patients (Pts) with hormone receptor–positive/HER2-negative (HR+/HER2-) advanced breast cancer. Journal of Clinical Oncology 2022, 40, LBA1001–LBA1001. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. New England Journal of Medicine 2021, 384, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Balar, A.V.; Petrylak, D.P.; Kalebasty, A.R.; Loriot, Y.; Fléchon, A.; Jain, R.K.; Agarwal, N.; Bupathi, M.; Barthelemy, P.; et al. TROPHY-U-01: A Phase II Open-Label Study of Sacituzumab Govitecan in Patients With Metastatic Urothelial Carcinoma Progressing After Platinum-Based Chemotherapy and Checkpoint Inhibitors. Journal of Clinical Oncology 2021, 39, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. New England Journal of Medicine 2019, 380, 741–751. [Google Scholar] [CrossRef]
- Yu, E.Y.; Petrylak, D.P.; O'Donnell, P.H.; Lee, J.-L.; van der Heijden, M.S.; Loriot, Y.; Stein, M.N.; Necchi, A.; Kojima, T.; Harrison, M.R.; et al. Enfortumab vedotin after PD-1 or PD-L1 inhibitors in cisplatin-ineligible patients with advanced urothelial carcinoma (EV‑201): a multicentre, single-arm, phase 2 trial. The Lancet Oncology 2021, 22, 872–882. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O'Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J Clin Oncol 2019, 37, 2592–2600. [Google Scholar] [CrossRef]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. New England Journal of Medicine 2021, 384, 1125–1135. [Google Scholar] [CrossRef]
- Sheng, X.; Yan, X.; Wang, L.; Shi, Y.; Yao, X.; Luo, H.; Shi, B.; Liu, J.; He, Z.; Yu, G.; et al. Open-label, Multicenter, Phase II Study of RC48-ADC, a HER2-Targeting Antibody-Drug Conjugate, in Patients with Locally Advanced or Metastatic Urothelial Carcinoma. Clin Cancer Res 2021, 27, 43–51. [Google Scholar] [CrossRef]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): a multicentre, open-label, single-arm, phase 2 study. The Lancet Oncology 2021, 22, 609–619. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Lorusso, D.; Oaknin, A.; Pignata, S.; Dean, A.; Denys, H.; Colombo, N.; Van Gorp, T.; Konner, J.A.; Marin, M.R.; et al. Efficacy and Safety of Mirvetuximab Soravtansine in Patients With Platinum-Resistant Ovarian Cancer With High Folate Receptor Alpha Expression: Results From the SORAYA Study. Journal of clinical oncology 2023, JCO2201900–JCO2201900. [Google Scholar] [CrossRef]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Pautas, C.; Terré, C.; Renneville, A.; Gardin, C.; Suarez, F.; Caillot, D.; Berthon, C.; Rousselot, P.; Preudhomme, C.; et al. Final Analysis of the ALFA 0701 Study. Blood 2014, 124, 376. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. The Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Gamis, A.S.; Alonzo, T.A.; Meshinchi, S.; Sung, L.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Kahwash, S.B.; Heerema-McKenney, A.; Winter, L.; et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol 2014, 32, 3021–3032. [Google Scholar] [CrossRef] [PubMed]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients With Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J Clin Oncol 2016, 34, 972–979. [Google Scholar] [CrossRef] [PubMed]
- Taksin, A.L.; Legrand, O.; Raffoux, E.; de Revel, T.; Thomas, X.; Contentin, N.; Bouabdallah, R.; Pautas, C.; Turlure, P.; Reman, O.; et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: a prospective study of the alfa group. Leukemia 2007, 21, 66–71. [Google Scholar] [CrossRef]
- Horwitz, S.; O'Connor, O.A.; Pro, B.; Trümper, L.; Iyer, S.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. The ECHELON-2 Trial: 5-year results of a randomized, phase III study of brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma. Ann Oncol 2022, 33, 288–298. [Google Scholar] [CrossRef]
- Straus, D.J.; Długosz-Danecka, M.; Connors, J.M.; Alekseev, S.; Illés, Á.; Picardi, M.; Lech-Maranda, E.; Feldman, T.; Smolewski, P.; Savage, K.J.; et al. Brentuximab vedotin with chemotherapy for stage III or IV classical Hodgkin lymphoma (ECHELON-1): 5-year update of an international, open-label, randomised, phase 3 trial. The Lancet Haematology 2021, 8, e410–e421. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Scarisbrick, J.J.; Dummer, R.; Whittaker, S.; Duvic, M.; Kim, Y.H.; Quaglino, P.; Zinzani, P.L.; Bechter, O.; Eradat, H.; et al. Randomized phase 3 ALCANZA study of brentuximab vedotin vs physician's choice in cutaneous T-cell lymphoma: final data. Blood Adv 2021, 5, 5098–5106. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J Clin Oncol 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. The lancet oncology 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.M.; Jabbour, E.; Wang, T.; Liang White, J.; et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: Final report and long-term survival follow-up from the randomized, phase 3 INO-VATE study. Cancer 2019, 125, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Robak, T.; le Coutre, P.D.; Gjertsen, B.T.; Troussard, X.; Roboz, G.J.; Karlin, L.; et al. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): long-term follow-up from the pivotal trial. J Hematol Oncol 2021, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): a multicentre, open-label, single-arm, phase 2 trial. The Lancet Oncology 2021, 22, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Zeng, P.; Chen, M.B.; Zhou, L.N.; Tang, M.; Liu, C.Y.; Lu, P.H. Impact of TROP2 expression on prognosis in solid tumors: A Systematic Review and Meta-analysis. Sci Rep 2016, 6, 33658. [Google Scholar] [CrossRef] [PubMed]
- Fradet, Y.; Bellmunt, J.; Vaughn, D.J.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; Necchi, A.; et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: results of >2 years of follow-up. Ann Oncol 2019, 30, 970–976. [Google Scholar] [CrossRef]
- Raggi, D.; Miceli, R.; Sonpavde, G.; Giannatempo, P.; Mariani, L.; Galsky, M.D.; Bellmunt, J.; Necchi, A. Second-line single-agent versus doublet chemotherapy as salvage therapy for metastatic urothelial cancer: a systematic review and meta-analysis. Ann Oncol 2016, 27, 49–61. [Google Scholar] [CrossRef]
- Wen, Y.; Ouyang, D.; Zou, Q.; Chen, Q.; Luo, N.; He, H.; Anwar, M.; Yi, W. A literature review of the promising future of TROP2 : a potential drug therapy target. Annals of Translational Medicine 2022, 10, 1403. [Google Scholar] [CrossRef]
- Bax, H.J.; Chauhan, J.; Stavraka, C.; Santaolalla, A.; Osborn, G.; Khiabany, A.; Grandits, M.; López-Abente, J.; Palhares, L.C.G.F.; Chan Wah Hak, C.; et al. Folate receptor alpha in ovarian cancer tissue and patient serum is associated with disease burden and treatment outcomes. British Journal of Cancer 2023, 128, 342–353. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Hilpert, F.; Pereira, D.; Wimberger, P.; Oaknin, A.; Mirza, M.R.; Follana, P.; Bollag, D.; Ray-Coquard, I.; Weber, B.; et al. Bevacizumab Combined With Chemotherapy for Platinum-Resistant Recurrent Ovarian Cancer: The AURELIA Open-Label Randomized Phase III Trial. Journal of clinical oncology 2014, 32, 1302–1308. [Google Scholar] [CrossRef]
- Li, X.; Cao, D.; Zheng, X.; Wang, G.; Liu, M. Tissue factor as a new target for tumor therapy—killing two birds with one stone: a narrative review. Annals of Translational Medicine 2022, 10. [Google Scholar] [CrossRef]
- Chelariu-Raicu, A.; Mahner, S.; Moore, K.N.; Lorusso, D.; Coleman, R.L. Integrating antibody drug conjugates in the management of gynecologic cancers. Int J Gynecol Cancer 2023, 33, 420–429. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Brock, K.; Homer, V.; Soul, G.; Potter, C.; Chiuzan, C.; Lee, S. Is more better? An analysis of toxicity and response outcomes from dose-finding clinical trials in cancer. BMC Cancer 2021, 21, 777. [Google Scholar] [CrossRef]
- Liao, M.Z.; Lu, D.; Kågedal, M.; Miles, D.; Samineni, D.; Liu, S.N.; Li, C. Model-Informed Therapeutic Dose Optimization Strategies for Antibody-Drug Conjugates in Oncology: What Can We Learn From US Food and Drug Administration-Approved Antibody-Drug Conjugates? Clin Pharmacol Ther 2021, 110, 1216–1230. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.C.; Nickens, D.J.; Xuan, D.; Shazer, R.L.; Amantea, M. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Investigational New Drugs 2018, 36, 121–135. [Google Scholar] [CrossRef]
- Venetis, K.; Crimini, E.; Sajjadi, E.; Corti, C.; Guerini-Rocco, E.; Viale, G.; Curigliano, G.; Criscitiello, C.; Fusco, N. HER2 low, ultra-low, and novel complementary biomarkers: Expanding the spectrum of HER2 positivity in breast cancer. Frontiers in Molecular Biosciences 2022, 9. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, K.; Wang, K.; Zhu, H. Treatment-related adverse events of antibody–drug conjugates in clinical trials: A systematic review and meta-analysis. Cancer 2023, 129, 283–295. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—an emerging class of cancer treatment. British journal of cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Erickson, H.K.; Widdison, W.C.; Mayo, M.F.; Whiteman, K.; Audette, C.; Wilhelm, S.D.; Singh, R. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem 2010, 21, 84–92. [Google Scholar] [CrossRef]
- Sheyi, R.; de la Torre, B.G.; Albericio, F. Linkers: An Assurance for Controlled Delivery of Antibody-Drug Conjugate. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Xu, Y.-y.; Shao, Z.-M.; Yu, K.-D. Resistance to antibody-drug conjugates in breast cancer: mechanisms and solutions. Cancer Communications 2023, 43, 297–337. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody-drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol Cancer Ther 2015, 14, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.; Tan, X.; Lu, B.; Golas, J.; Hosselet, C.; Wang, F.; Tylaska, L.; King, L.; Zhou, D.; Dushin, R. Caveolae-mediated endocytosis as a novel mechanism of resistance to trastuzumab emtansine (T-DM1) altered trafficking as mechanism of ADC resistance. Molecular cancer therapeutics 2018, 17, 243–253. [Google Scholar] [CrossRef]
- Coates, J.T.; Sun, S.; Leshchiner, I.; Thimmiah, N.; Martin, E.E.; McLoughlin, D.; Danysh, B.P.; Slowik, K.; Jacobs, R.A.; Rhrissorrakrai, K. Parallel genomic alterations of antigen and payload targets mediate polyclonal acquired clinical resistance to sacituzumab govitecan in triple-negative breast cancer. Cancer discovery 2021, 11, 2436. [Google Scholar] [CrossRef]
- Li, G.; Guo, J.; Shen, B.-Q.; Yadav, D.B.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Phillips, G.D.L. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer CellsMechanisms of Resistance to Trastuzumab Emtansine (T-DM1). Molecular cancer therapeutics 2018, 17, 1441–1453. [Google Scholar] [CrossRef] [PubMed]
- Abelman, R.O.; Wu, B.; Spring, L.M.; Ellisen, L.W.; Bardia, A. Mechanisms of Resistance to Antibody-Drug Conjugates. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A.; Rugo, H.S.; Vukelja, S.J.; Vogel, C.L.; Borson, R.A.; Limentani, S.; Tan-Chiu, E.; Krop, I.E.; Michaelson, R.A.; Girish, S. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)–positive breast cancer after prior HER2-directed therapy. Journal of Clinical Oncology 2011, 29, 398–405. [Google Scholar] [CrossRef]
- Criscitiello, C.; Morganti, S.; Curigliano, G. Antibody–drug conjugates in solid tumors: a look into novel targets. Journal of Hematology & Oncology 2021, 14, 20. [Google Scholar] [CrossRef]
- Matsumura, Y. Barriers to antibody therapy in solid tumors, and their solutions. Cancer Science 2021, 112, 2939–2947. [Google Scholar] [CrossRef]
- Sardinha, M.; Palma Dos Reis, A.F.; Barreira, J.V.; Fontes Sousa, M.; Pacey, S.; Luz, R. Antibody-Drug Conjugates in Prostate Cancer: A Systematic Review. Cureus 2023, 15, e34490. [Google Scholar] [CrossRef]
- You, W.-K.; Schuetz, T.J.; Lee, S.H. Targeting the DLL/Notch Signaling Pathway in Cancer: Challenges and Advances in Clinical Development. Molecular Cancer Therapeutics 2023, 22, 3–11. [Google Scholar] [CrossRef]
- Petrylak, D.P.; Kantoff, P.; Vogelzang, N.J.; Mega, A.; Fleming, M.T.; Stephenson, J.J.; Frank, R.; Shore, N.D.; Dreicer, R.; McClay, E.F.; et al. Phase 1 study of PSMA ADC, an antibody-drug conjugate targeting prostate-specific membrane antigen, in chemotherapy-refractory prostate cancer. The Prostate 2019, 79, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Kassambara, A.; Hose, D.; Klein, B. STEAP1 is overexpressed in cancers: a promising therapeutic target. Biochem Biophys Res Commun 2012, 429, 148–155. [Google Scholar] [CrossRef]
- Danila, D.C.; Szmulewitz, R.Z.; Vaishampayan, U.; Higano, C.S.; Baron, A.D.; Gilbert, H.N.; Brunstein, F.; Milojic-Blair, M.; Wang, B.; Kabbarah, O.; et al. Phase I Study of DSTP3086S, an Antibody-Drug Conjugate Targeting Six-Transmembrane Epithelial Antigen of Prostate 1, in Metastatic Castration-Resistant Prostate Cancer. Journal of Clinical Oncology 2019, 37, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.A.; Motzer, R.J.; Molina, A.M.; Choueiri, T.K.; Heath, E.I.; Redman, B.G.; Sangha, R.S.; Ernst, D.S.; Pili, R.; Kim, S.K.; et al. Phase I Trials of Anti-ENPP3 Antibody-Drug Conjugates in Advanced Refractory Renal Cell Carcinomas. Clin Cancer Res 2018, 24, 4399–4406. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int J Biol Sci 2020, 16, 1767–1773. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Carneiro, B.A.; Dowlati, A.; Razak, A.R.A.; Chae, Y.K.; Villella, J.A.; Coppola, S.; Englert, S.; Phillips, A.C.; Souers, A.J.; et al. A first-in-human study of mirzotamab clezutoclax as monotherapy and in combination with taxane therapy in relapsed/refractory solid tumors: Dose escalation results. Journal of Clinical Oncology 2021, 39, 3015–3015. [Google Scholar] [CrossRef]
- Chu, Q. Targeting Mesothelin in Solid Tumours: Anti-mesothelin Antibody and Drug Conjugates. Curr Oncol Rep 2023, 25, 309–323. [Google Scholar] [CrossRef]
- Hong, Y.; Nam, S.M.; Moon, A. Antibody-drug conjugates and bispecific antibodies targeting cancers: applications of click chemistry. Arch Pharm Res 2023. [CrossRef]
- Zong, H.-F.; Zhang, B.-H.; Zhu, J.-W. Generating a Bispecific Antibody Drug Conjugate Targeting PRLR and HER2 with Improving the Internalization. Pharmaceutical Fronts 2022, 04, e113–e120. [Google Scholar] [CrossRef]
- Fang, J.; Xiao, L.; Joo, K.-I.; Liu, Y.; Zhang, C.; Liu, S.; Conti, P.S.; Li, Z.; Wang, P. A potent immunotoxin targeting fibroblast activation protein for treatment of breast cancer in mice. International Journal of Cancer 2016, 138, 1013–1023. [Google Scholar] [CrossRef]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective Immunoconjugate Therapy in Cancer Models Targeting a Serine Protease of Tumor Fibroblasts. Clinical Cancer Research 2008, 14, 4584–4592. [Google Scholar] [CrossRef]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nature Communications 2021, 12, 3528. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: changing partners in the dance towards death. Cell Death & Differentiation 2018, 25, 65–80. [Google Scholar]
- Wang, L.; Doherty, G.A.; Judd, A.S.; Tao, Z.-F.; Hansen, T.M.; Frey, R.R.; Song, X.; Bruncko, M.; Kunzer, A.R.; Wang, X.; et al. Discovery of A-1331852, a First-in-Class, Potent, and Orally-Bioavailable BCL-XL Inhibitor. ACS Medicinal Chemistry Letters 2020, 11, 1829–1836. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Tupone, M.G.; Desideri, M.; Di Martile, M.; Gabellini, C.; Buglioni, S.; Pallocca, M.; Alessandrini, G.; D’Aguanno, S.; Del Bufalo, D. BCL-XL overexpression promotes tumor progression-associated properties. Cell Death & Disease 2017, 8, 3216. [Google Scholar] [CrossRef]
- Borges, V.F.; Ferrario, C.; Aucoin, N.; Falkson, C.; Khan, Q.; Krop, I.; Welch, S.; Conlin, A.; Chaves, J.; Bedard, P.L.; et al. Tucatinib Combined With Ado-Trastuzumab Emtansine in Advanced ERBB2/HER2-Positive Metastatic Breast Cancer: A Phase 1b Clinical Trial. JAMA Oncology 2018, 4, 1214–1220. [Google Scholar] [CrossRef]
- Tahara, M.; Okano, S.; Enokida, T.; Ueda, Y.; Fujisawa, T.; Shinozaki, T.; Tomioka, T.; Okano, W.; Biel, M.A.; Ishida, K.; et al. A phase I, single-center, open-label study of RM-1929 photoimmunotherapy in Japanese patients with recurrent head and neck squamous cell carcinoma. Int J Clin Oncol 2021, 26, 1812–1821. [Google Scholar] [CrossRef]
- Nicolò, E.; Giugliano, F.; Ascione, L.; Tarantino, P.; Corti, C.; Tolaney, S.M.; Cristofanilli, M.; Curigliano, G. Combining antibody-drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives. Cancer Treatment Reviews 2022, 106, 102395. [Google Scholar] [CrossRef]
- Bauzon, M.; Drake, P.M.; Barfield, R.M.; Cornali, B.M.; Rupniewski, I.; Rabuka, D. Maytansine-bearing antibody-drug conjugates induce in vitro hallmarks of immunogenic cell death selectively in antigen-positive target cells. OncoImmunology 2019, 8, e1565859. [Google Scholar] [CrossRef]
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.-B.; Im, S.-A.; Wang, Y.; Salgado, R.; Mani, A.; et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomised, double-blind trial. The Lancet Oncology 2020, 21, 1283–1295. [Google Scholar] [CrossRef]
- Janku, F.; Han, S.-W.; Doi, T.; Ajani, J.; Kuboki, Y.; Mahling, P.; Subramanian, K.; Pelletier, M.; Askoxylakis, V.; Siena, S. 378 A first in-human, multicenter, open-label, dose-finding phase 1 study of the immune stimulator antibody conjugate NJH395 in patients with nonbreast HER2+ advanced malignancies. Journal for ImmunoTherapy of Cancer 2020, 8, A230–A230. [Google Scholar] [CrossRef]
- Janku, F.; Han, S.-W.; Doi, T.; Amatu, A.; Ajani, J.A.; Kuboki, Y.; Cortez, A.; Cellitti, S.E.; Mahling, P.C.; Subramanian, K.; et al. Preclinical Characterization and Phase I Study of an Anti–HER2-TLR7 Immune-Stimulator Antibody Conjugate in Patients with HER2+ Malignancies. Cancer Immunology Research 2022, 10, 1441–1461. [Google Scholar] [CrossRef]
Figure 1.
Mechanism of Action of Action of Antibody Drug Conjugates.Created with BioRender.com.
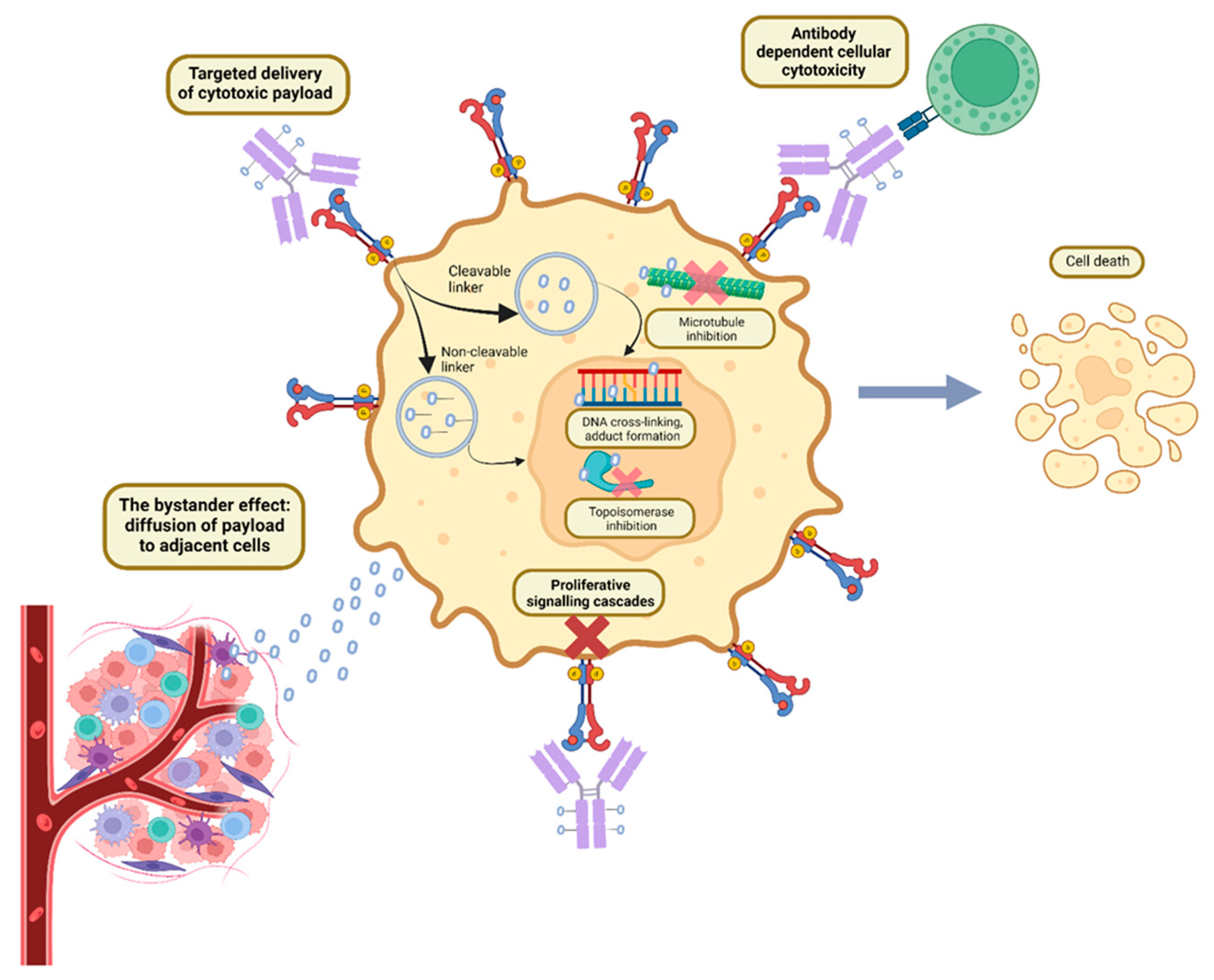
Figure 2.
Structure of an Antibody Drug Conjugate.‚Abbreviations: BCMA; B-cell maturation antigen; B7-H3, B7 homolog 3 protein; DLL3, deltalike protein 3; ENPP3, ectonucleotide pyrophosphatase/phosphodiesterase family member 3; FRα, folate receptor α; MMAE/F, monomethyl auristatin E/F; SMCC, succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate; PBD, pyrrolobenzodiazepine; PSMA, prostate specific membrane antigen. Created using BioRender.com.
Figure 2.
Structure of an Antibody Drug Conjugate.‚Abbreviations: BCMA; B-cell maturation antigen; B7-H3, B7 homolog 3 protein; DLL3, deltalike protein 3; ENPP3, ectonucleotide pyrophosphatase/phosphodiesterase family member 3; FRα, folate receptor α; MMAE/F, monomethyl auristatin E/F; SMCC, succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate; PBD, pyrrolobenzodiazepine; PSMA, prostate specific membrane antigen. Created using BioRender.com.
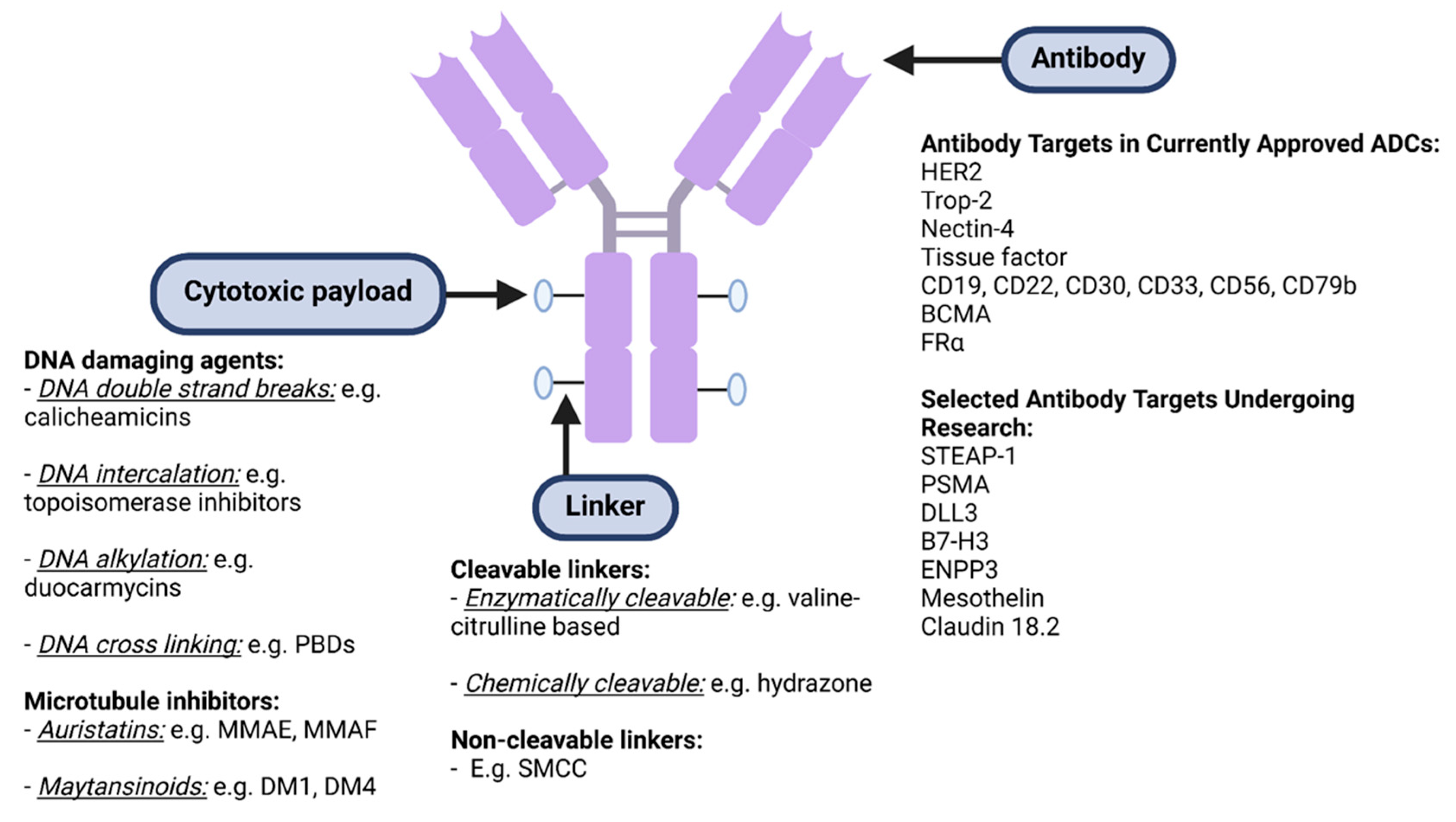
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



