
Submitted:
05 June 2023
Posted:
05 June 2023
You are already at the latest version
Abstract
Blood-based circulating cell free DNA(cfDNA) detection offers a non-invasive and easily accessible way for early cancer detection. Despite the extensive utility of cfDNA, there are still many challenges to develop the clinical biomarkers. For example, cfDNA with genetic alterations often compose a small portion of the DNA circulating in plasma, which can be confounded by cfDNA contributed by normal cells. Therefore, filtering out the potential false-positive cfDNA mutations from health population will be important for the cancer-based biomarkers. Additionally, many low-frequency genetic alterations are easily overlooked in small amount of cfDNA-based cancer test. We hypothesize that, the combination of diverse types of cancer studies on cfDNA can provide us a new insight to identify low-frequency genetic variant across cancer types for early clinical detection of cancers. By building a standardized computational pipeline for 1358 cfDNA samples across seven cancer types, we prioritize 129 shard genetic variants in the major cancer types. Further functional analysis of the 129 variants found that they are mainly enriched in ribosome pathways such as cotranslational protein targeting to membrane, some of which are tumor suppressor, oncogene and related to cancer initiation. In summary, our integrative analysis revealed the important roles of ribosome proteins as the common biomarkers in early cancer diagnosis.
Keywords:
Early cancer diagnosis
; cell free DNA
; biomarker
; integrative biology
; pan cancer
1. Introduction
In cancers, apoptotic and necrotic cells release cfDNA into the blood, and cancer patients have elevated levels of cell-free DNA (cfDNA). Therefore has been a great deal of interest in the use of circulating cfDNA as a "liquid biopsy" for noninvasive early cancer detection [1]. In general, cfDNA with genetic alterations constitute a small proportion of the DNA circulating in plasma, which can be confused with cfDNA from normal cells [1]. Therefore, it will be essential for cancer-based biomarkers to eliminate potential false-positive cfDNA mutations from the healthy population. In addition, many low-frequency genetic alterations are easily missed in cancer tests based on a small amount of cfDNA.
Cancer genome project advancements and new applications of next-generation sequencing (NGS) technology have facilitated groundbreaking research on cfDNA over the past decade. However, the development of clinical biomarkers continues to face significant obstacles. Firstly, current cfDNA profiling strategies are insufficiently sensitive for the concurrent detection of multiple cancers. This can be improved in a number of ways, such as by optimizing the pre-analytical steps, collecting samples from body fluids with higher mutation allele fractions, and enriching tumor-derived cfDNA after extraction. The sensitivity and specificity of cfDNA tests can be significantly enhanced by combining multiple biomarkers into a single evaluation. Secondly, the quantitative and qualitative fluctuations of cfDNA in a person's blood hinder the reproducibility of measurements, interpretations, and comparisons. A better comprehension of cfDNA release rate may be able to resolve this problem [2]. Thirdly, it is necessary to validate the quantification of cfDNA, subsequent mutation analysis, and other analytical steps, including the sequencing platform itself, in order to simulate the clinical environment [3]. Lastly, the use of diverse high-throughput sequencing platforms frequently makes it challenging to reproduce results and highlights the need for standardization and analytical validation of liquid biopsy methods [4].
Normally, mutations in oncogenes and tumor suppressor genes play a crucial role in cancer initiation [5,6]. There are fewer than 2,000 genes, despite the fact that these important driver genes are essential for cancer diagnosis. On the other hand, cancer cells typically contain thousands of mutations that do not directly drive cancer initiation and progression, and these mutations can also be found in healthy populations. To concentrate on key cancer progression events, we collect data on 1,358 experiments with original sequences from 14 projects, involving 7 major types of head and neck cancer, lung cancer, breast cancer, prostate cancer, gastric cancer, colon cancer, and liver cancer. We hypothesize that the combination of diverse types of cancer studies on cfDNA will allow us to identify high-quality genetic variants across cancer types for early clinical cancer detection [5].
2. Materials and Methods
2.1. Data sources and the data filtering pipeline
As shown in Figure 1A, our analysis pipeline were started by downloading data from the NCBI SRA database (http://www.ncbi.nlm.nih.gov/sra) using the SRA Toolkit (https://www.ncbi.nlm.nih.gov/sra/docs/toolkitsoft/). SRA database is a public repository for millions of publicly available data related to genomics sequencing. For our project, we only focused on the cell free DNA data in cancers. Therefore, we searched from the SRA database by the following expression: “cell free DNA [title] or single cell DNA [title] or single cell RNA [title]” and “cancer or tumor” on 2019 Apr 10. Then we downloaded 1358 experiments with raw sequence from a total of fourteen projects involving seven major cancer types including breast, colorectal, head and neck, liver, lung, prostate, and stomach cancers.
2.2. Sequence data alignment and pre-processing
The raw data downloaded from SRA database are the short reads in the Fastq format. To translate the raw data to the meaningful information, we adopted the best practices of Genome Analysis Toolkit’s (GATK) for the overall data pre-processing and genome mapping. Firstly, we aligned the raw fastq reads to the Human reference assembly HG19 by using the genome aligner BWA v0.7.13 [7] with default settings. The resulted binary alignment map (BAM) files were used as the input for the tools used in the GATK best practice [8]. In brief, we removed the potential duplicated short reads with Picard’s MarkDuplicates command. We also corrected the local alignment around indels based on GATK’s Indel-Realigner module. The recalibration of the quality score and reduction of machine-read error was further conducted by using GATK’s base quality score recalibration (BQSR) module (Figure 1A).
2.3. Variant calling, filtration and annotation
The pre-processed BAM files with recalibrated quality scores were further analyzed by the somatic mutation calling tools of MuTect2 [9] and Monovar [10] for single-cell DNA data. In detail, the variant calling format (VCF) files were generated from two variant calling tools for each sample. Then the VCF files were used as input to eliminate potential sequencing and germline artifacts. For example, we removed those non-functional variants, and we focused on those somatic variants detected in three or more cancer types. In addition, we also removed the false-positive genetic mutations that may be present in the VCFs (Figure 1B). Then, functional annotations for variants were added to each mutation using the ANNOVAR software v. 2019Oct24 [11]. And the pathogenicity of missense variants was predicted in silico using scores from dbNSFP [12] based on 12 different algorithms such as SIFT and CADD [13].
2.4. High-quality variants prediction
It is crucial to accurately predict the deleteriousness of nonsynonymous variants in order to distinguish pathogenic mutations from background polymorphisms [13]. Although numerous methods for predicting deleteriousness have been developed, their prediction results are sometimes inconsistent [14]. The computational algorithms utilised by these prediction methods (Markov model, evolutionary conservation, random forest, neural network, etc.) vary. Therefore, it is recommended to use multiple prediction algorithms for variant evaluation to eliminate algorithm bias [15]. We chose Combined Annotation-Dependent Depletion (CADD) [16] and Functional Analysis through Hidden Markov Models with an eXtended Feature set (FATHMM-XF) [17] as our prediction algorithms based on their relative merits. In brief, CADD assesses the deleterious nature of SNVs based on a variety of genomic characteristics, including the surrounding sequence context, epigenetic measurements, evolutionary constraints, and functional predictions [16]. CADD's ability to prioritise functional, deleterious, and pathogenic variants is unmatched by any single-annotation method currently in use [18]. Compared to traditional procedures (such as SIFT), CADD was determined to be the most effective in silico algorithm in previous SNV pathogenicity analyses [19]. However, the disadvantage of CADD is limited accuracy for predicting variants in the non-coding regions [20]. To add non-coding information, we utilised FATHMM-XF, one of the most efficient tools for non-coding regions [17].
2.5. Functional and pathway enrichment analysis
To investigate the functional patterns of the genes associated with the identified somatic mutations, we conducted a comprehensive functional annotation. In brief, significant gene ontology (GO) biological process terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were performed to analyse the identified biomarkers at the functional level. GO provides a general framework to characterize the gene function shared in multiple species [21]. According to the adjusted statistical P values, the terms were arranged in ascending order, making it simple to focus on the most significant GO terms associated with the biomarker genes. To supplement the missing information in GO annotation, we also consulted the KEGG database for pathway information. KEGG assigns specific gene set pathways to key data containing higher-order functional information and can be used for the functional interpretation and practical application of genomic data [22]. In practice, all human genes as the background and the identified biomarkers as the input were used to perform GO function and KEGG pathway enrichment analysis, and FDR 0.05 was considered statistically significant using Toppfun [23].
2.6. Protein-protein interaction and hub gene analysis
To understand the metabolic and molecular mechanisms related to the identified biomarkers shared in multiple cancers, we utilized the existent protein-protein interaction data. In brief, the Search Tool for the Retrieval of Interacting Genes (STRING) database (version 10.0) [24] provides a comprehensive analysis and integration of protein-protein interactions, including direct physical connections and indirect functional associations such as co-expression in multiple dataset. The output from the STRING results were further visualized by using Cytoscape 3.7.1, which is easy to depict the genes from different functional groups [25]. In addition, the plug-in app cytohubba in Cytoscape was downloaded and installed to explore the hub genes [26]. Using the top scores of the Maximal Clique Centrality (MCC) algorithm, the hub genes with high connectivity in the gene expression network were eliminated and clustered.
2.7. Survival and mutational analysis of the top module genes in TCGA database
Using 10967 samples from 33 TCGA pan-cancer study, we further explored the potential clinical application of those key genes identified in the network modules. For instance, mutational analysis was performed to investigate the single-nucleotide somatic mutation and copy number variation pattern of the genes from the top module at a pan-cancer level [27]. The frequency of genetic alteration was further plotted based on the number of tumor samples containing the somatic mutation, copy number alteration associated with the key network genes. Additionally, we associate the genes to patient overall survival data from TCGA by classification of all patients into altered and unaltered groups using cBioportal [28]. To focus on the reliable result, the log-rank analysis and Kaplan–Meier plots were generated.
3. Results
3.1. Identification of potential biomarkers in cfDNA
To collect the high-quality genetic variations in cfDNA for liquid biopsy biomarker, we searched SRA and downloaded raw sequence data from 14 projects involving seven major cancer types. Firstly, we performed a gene-based annotation of all called variants to remove non-functional variants and identified a total of 896193 exonic SNVs or indels (Figure 2A). Secondly, to further minimize the rate of false-positive calls, variants from different cancer types were combined and duplicate variants were removed, leaving a total of 858,176 variants. Figure 2B,C show how the variants were distributed and shared across different cancer types. Thirdly, variants present in at least 3 cancer types were selected to refine the list to 6981 for downstream analysis. A total of 129 variants were predicted to be deleterious by a combination of two pan-genome prediction scores (CADD and FATHMM-XF). The 116 corresponding potential biomarkers then were used for further analysis (Table S1).
3.2. KEGG pathway analysis confirmed the close relationship between ribosome and cancer
As shown in Figure 3A, the top GO terms of cellular component, molecular function and biological process includes co-translational protein targeting to membrane (adjusted P-value = 1.326E-21), protein targeting to ER (adjusted P-value = 2.894E-19), translational initiation (adjusted P-value = 8.101E-17), mRNA catabolic process (adjusted P-value = 2.941E-16), establishment of protein localization to endoplasmic reticulum (adjusted P-value = 5.164E-19), and cytosolic ribosome (adjusted P-value = 4.468E-20) (Table S2).
Additional signaling pathway analysis (Table S3) were conducted. For instance, the KEGG analysis showed that the biomarkers were mainly enriched in Ribosome, Oxidative phosphorylation, Proteasome, and other signaling pathways (Figure 3B). Ribosomes, for instance, are important for the translation of mRNA-contained information into functional proteins, which is align well with the enriched GO function “co-translational protein targeting to membrane” [29]. More interesting, hyperactivation of ribosome biogenesis, which can be triggered by oncogenes or the loss of tumour suppressor genes, plays an essential role in the initiation and progression of cancer [30]. Recent studies suggest that both increased numbers and altered modifications of ribosomes may contribute to the cancer development. For instance, multiple cancers, including endometrial cancer, high-grade gliomas, colorectal cancer, acute and chronic lymphocytic leukaemia, have been found to contain ribosomal genetic mutations [31].
The OXPHOS (oxidative phosphorylation) metabolic pathway is another significant pathway that deserves mention. It produces ATP by transporting electrons to the electron transport chain, a series of transmembrane protein complexes in the mitochondrial inner membrane (ETC) [32,33]. Cancer cells require OXPHOS, and cancer stem cells are frequently characterized by an increased reliance on OXPHOS [34]. Downregulation of OXPHOS is frequently correlated with poor clinical outcomes and metastasis [35]. Inhibition of OXPHOS has also been shown to reduce oxygen consumption rate (OCR) and alleviate hypoxia in tumors [32].
3.3. Network analysis revealed hub genes associated with cancer development
To evaluate the interactive relationships among identified biomarkers, we mapped them to the STRING database. The final interactome contains 115 genes and 477 connections. In the network, the average node degree is 8.3 and the average local clustering coefficient is 0.506. The Protein-protein interaction (PPI) enrichment P < 1.0E-16 (Figure 4A). In sum, these topological characteristics of the network indicate that the genes within it can exchange information efficiently.
Then, we utilized the Molecular COmplex DEtection (MCODE) application to identify clustered modules throughout the entire network. The network consisted of 5 modules, and with the top module containing 25 nodes and 262 edges. The 25 genes in the top module were selected for alteration frequency and survival analysis (Figure 4B). Based on the MCC (Maximal Clique Centrality) scores, we prioritize the most stable hub genes in the network including RPS15A, RPS23, RPS9, RPS21, RPS14, RPS25, RPS6, RPL27, RPL35A, and UBA52 (Figure 4C). Among these genes, RPS15A (Ribosomal protein s15a) has been shown related to many cancers in previous studies. As a component of the 40S subunit, increased RPS15A expression is closely correlated with poor prognosis in gastric cancer (GC) patients and promotes epithelial-mesenchymal transition (EMT) and GC progression, as demonstrated [36].
3.4. Overlapping with OCGs, TSGs, and CIGs revealed multiple roles played by identified biomarkers
In order to evaluate the roles of the potential biomarkers in cancer progression, we mapped the genes to known oncogenes (OCGs) [37], tumor suppressor genes (TSGs) [5], and cancer initiation genes (CIGs) [38]. This analysis identified 10 biomarker genes reported as either CIGs, OCGs, or TSGs (Figure 4D) (Table S4). These genes included DUSP12, VIM, FOS, UBE2C, MIEN1, HINT1, LITAF, GABARAP, PFN1, and MLF2. As a member of the E2 ubiquitin-conjugating enzyme family, UBE2C is overexpressed in all 27 cancers and patients with higher UBE2C expression levels exhibited a shorter overall survival duration [39]. Another interesting gene is LITAF (Lipopolysaccharide-induced tumor necrosis factor-α factor). It possesses transcription factor activity and is involved in the regulation of protein quality. Previous research has suggested that LITAF functions as a TSG and is frequently underrepresented in prostate, pancreatic, and stomach cancers [40]. Taken together, these findings confirmed the significance of these biomarkers in the development of cancer, indicating their potential use in clinical diagnosis.
3.5. Patients with altered genes in the top functional module has a significantly worse overall survival rate
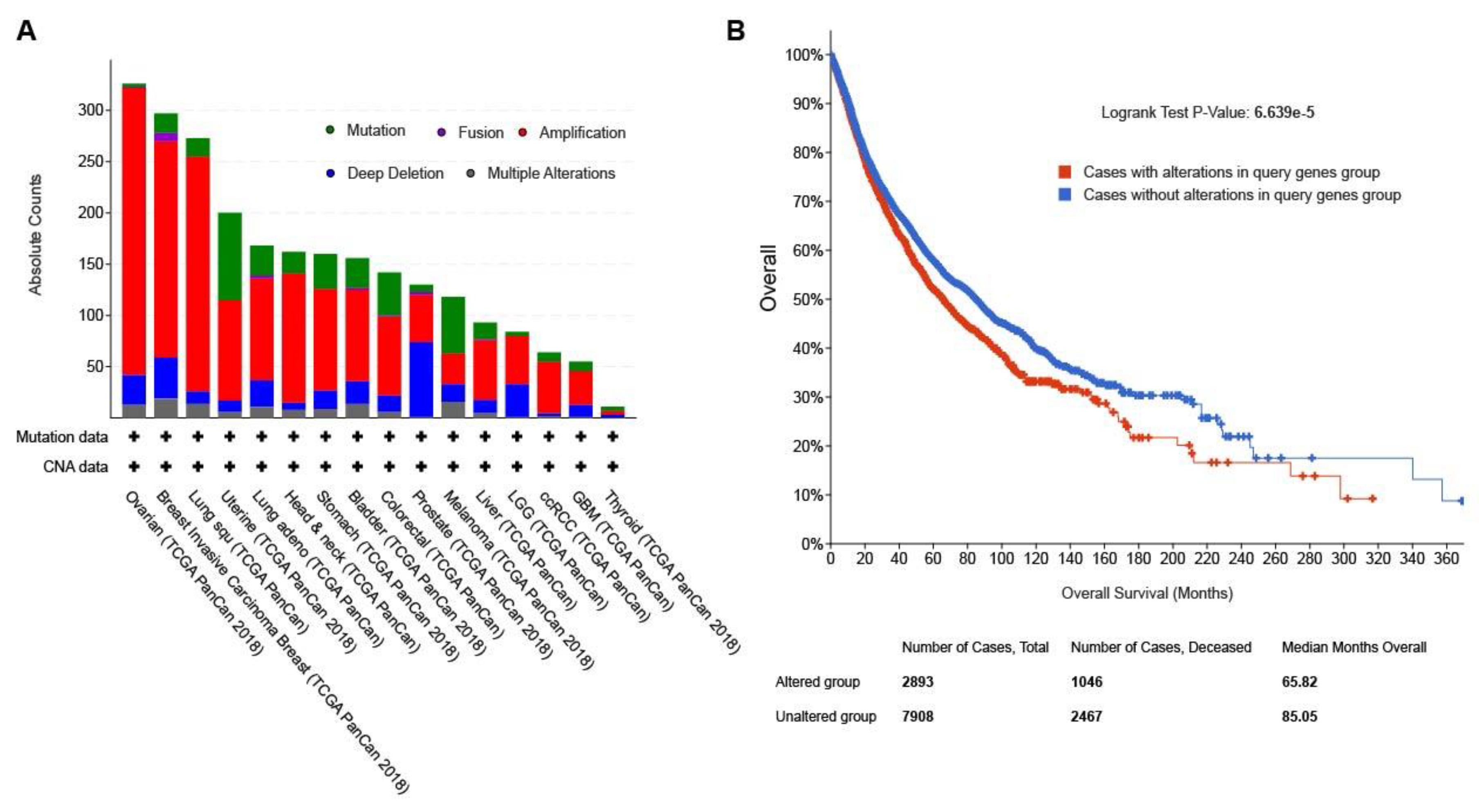
The frequencies of genetic alterations of the 25 genes in the top module were evaluated using the cBioPortal database. Approximately 27% of clinical cases from 32 different cancer studies exhibited significant alterations in the 25 genes (Figure 5A). Kaplan-Meier plots were used to compare Overall survival in 10953 patients with or without alterations in the 25 hub genes (Figure 5B). It was revealed that cases with altered genes exhibited significantly worse OS compared to those with unaltered genes (P value=6.639E-5).
4. Discussion
Numerous studies have demonstrated the potential of cfDNA as a biomarker for the early detection of cancer. However, the accuracy of cfDNA-based tests faces significant obstacles [2]. Previous cfDNA studies focused on a single tumor type or the results from a single cohort study [41,42], but there is no systematic examination of high-quality variants in different cancer types. In order to collect high-quality biomarkers in cfDNA, we constructed a computational pipeline to screen genetic variants shared by multiple tumor types based on the raw sequence data in the SRA databases.
In total, we identified 116 potential biomarkers following variant calling and filtering pipeline. As was suggested by functional enrichment analysis, these biomarker genes were mainly involved in the ribosome pathway confirming the close relationship between ribosome and cancer development, which contradicts the view held until recently that ribosomes played a rather passive role as the only molecular factory in the translation process [43]. Recent studies have linked the altered ribosome and dysregulated expression of specific ribosomal proteins to cancer initiation, evolution, and progression (RP) [44]. As an example, the correlation between accelerated colorectal cancer (CRC) cell growth and alterations in particular steps of ribosome biogenesis is cited as a key factor in cancer initiation [45]. Erica Buoso et al. provided an analysis on how ribosomes translate cancer progression in breast cancer through the ribosomal protein RACK1 [46]. Amandine et al. provided evidence supporting the role of altered ribosome components in the development of cancer and argued that ribosomes may play a crucial role in the acquisition and maintenance of the cancer stem cell phenotype [43]. Our study confirmed the association between ribosomes and cancer by statistical analysis of large-scale genomic data from multiple cancer types. It also indicated targeting ribosome pathway is another promising possibility for developing a cancer therapeutic strategy.
The ctDNA, which is a portion of the cfDNA released from the blood of cancer patients by tumour cells via apoptosis, necrosis, or active release, is another intriguing aspect of our data. As a new type of cancer biomarker, tumor-specific mutations in the ctDNA sequence can be used to identify cancer patients. To evaluate tumour heterogeneity, cfDNA-based liquid biopsy is less invasive, more feasible, and more comprehensive than tissue biopsy due to the rapid development of next-generation sequencing (NGS) technology. However, the use of ctDNA sequencing for cancer screening and early diagnosis is hindered by a low concentration of ctDNA in the blood and an increase in false positives resulting from normal healthy cells. This study developed a systematic pipeline that integrated a combination of prediction algorithms with optimised parameters to analyse raw sequencing data of cfDNA from various cancer types and identify high-quality variants in order to identify reliable biomarkers for cfDNA tests.
Raw sequence data in FASTQ format were downloaded from public available SRA database, these data come from 14 projects involving seven major cancer types. By applying the systematic pipeline, 116 biomarker genes shared by different cancer types were screened out.from a total of 896,193 exonic SNVs or indels. Functional enrichment analysis shows that these biomarker genes are mainly involved in the ribosomal pathway, implying the close relationship between ribosomes and cancer development. By cross-referencing these 116 biomarker genes with known oncogenes, tumor suppressor genes, and cancer initiation genes, 10 genes were identified with multiple roles in cancer development. Then the importance of these biomarkers in cancer development were confirmed implying their potential application for clinic diagnosis. In summary, this study provided new insight to identify high-quality genetic variants in cfDNA across different cancer types enabling a better application of cfDNA as a non-invasive diagnostic clinical biomarker for early detection of cancers.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1. The 14 bioprojects related to circulating cell-free DNA integrated in this study. Table S2. 116 cfDNA-based common diagnostic genes in multiple cancers. Table S3. The gene ontology enrichment of the 116 genes. Table S4: The KEGG analysis for the 116 genes.
Author Contributions
Conceptualization, M.Z.; methodology, M.L.; formal analysis, M.L.; writing—original draft preparation, M.L.; writing—review and editing, M.Z. and Y.L.; visualization, M.L.; supervision, M.Z.; project administration, M.Z.; All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Our study is based public Gene Expression datasets from Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/sra). Therefore, this study is exempt from ethics approval.
Informed Consent Statement
Patient consent was waived due to the use of public data.
Data Availability Statement
The raw data used were from public Sequence Read Archive (SRA) databases.
Acknowledgments
This work was supported by computational resources provided by the Australian Government through QCIF under the National Computational Merit Allocation Scheme. This research was also supported by the use of the Nectar Research Cloud, a collaborative Australian research platform supported by the NCRIS-funded Australian Research Data Commons (ARDC).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pantel, K. Blood-Based Analysis of Circulating Cell-Free DNA and Tumor Cells for Early Cancer Detection. PLOS Medicine 2016, 13, e1002205. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol Detect Quantif 2019, 17, 100087–100087. [Google Scholar] [CrossRef] [PubMed]
- Perakis, S.; Speicher, M.R. Emerging concepts in liquid biopsies. BMC Med 2017, 15, 75–75. [Google Scholar] [CrossRef] [PubMed]
- Verlingue, L.; Alt, M.; Kamal, M.; Sablin, M.P.; Zoubir, M.; Bousetta, N.; Pierga, J.Y.; Servant, N.; Paoletti, X.; Le Tourneau, C. Challenges for the implementation of high-throughput testing and liquid biopsies in personalized medicine cancer trials. Personalized medicine 2014, 11, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Kim, P.; Mitra, R.; Zhao, J.; Zhao, Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic acids research 2016, 44, D1023–D1031. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, J.; Zhao, M. ONGene: A literature-based database for human oncogenes. Journal of Genetics and Genomics 2017, 44, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Zafar, H.; Wang, Y.; Nakhleh, L.; Navin, N.; Chen, K. Monovar: single-nucleotide variant detection in single cells. Nature methods 2016, 13, 505–507. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Human mutation 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human molecular genetics 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine : official journal of the American College of Medical Genetics 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019, 47, D886–d894. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics (Oxford, England) 2018, 34, 511–513. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O'Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014, 46, 310–315. [Google Scholar] [CrossRef]
- Ganakammal, S.R.; Alexov, E. Evaluation of performance of leading algorithms for variant pathogenicity predictions and designing a combinatory predictor method: application to Rett syndrome variants. PeerJ 2019, 7, e8106. [Google Scholar] [CrossRef]
- Mather, C.A.; Mooney, S.D.; Salipante, S.J.; Scroggins, S.; Wu, D.; Pritchard, C.C.; Shirts, B.H. CADD score has limited clinical validity for the identification of pathogenic variants in noncoding regions in a hereditary cancer panel. Genetics in medicine : official journal of the American College of Medical Genetics 2016, 18, 1269–1275. [Google Scholar] [CrossRef]
- Thomas, P.D. The Gene Ontology and the Meaning of Biological Function. Methods in molecular biology (Clifton, N.J.) 2017, 1446, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017, 45, D353–d361. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res 2009, 37, W305–311. [Google Scholar] [CrossRef] [PubMed]
- Praneenararat, T.; Takagi, T.; Iwasaki, W. Integration of interactive, multi-scale network navigation approach with Cytoscape for functional genomics in the big data era. BMC Genomics 2012, 13 Suppl 7, S24. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC systems biology 2014, 8 Suppl 4, S11. [Google Scholar] [CrossRef]
- Zhao, M.; Zhao, Z. CNVannotator: a comprehensive annotation server for copy number variation in the human genome. PLoS One 2013, 8, e80170. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013, 6, pl1. [Google Scholar] [CrossRef]
- Beall, G.N. Immunologic aspects of endocrine diseases. Jama 1987, 258, 2952–2956. [Google Scholar] [CrossRef]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat Rev Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef]
- Goudarzi, K.M.; Lindström, M.S. Role of ribosomal protein mutations in tumor development (Review). International journal of oncology 2016, 48, 1313–1324. [Google Scholar] [CrossRef]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clinical Cancer Research 2018, 24, 2482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Chen, Y.; Qu, D.; Qu, H. TSdb: a database of transporter substrates linking metabolic pathways and transporter systems on a genome scale via their shared substrates. Sci China Life Sci 2011, 54, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Sica, V.; Bravo-San Pedro, J.M.; Stoll, G.; Kroemer, G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. 2020, 146, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Gaude, E.; Frezza, C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nature Communications 2016, 7, 13041. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, X.; Liu, X.; Yu, J.; Zhang, M.; Yu, F.; Wang, Y. RPS15A promotes gastric cancer progression via activation of the Akt/IKK-β/NF-κB signalling pathway. Journal of cellular and molecular medicine 2019, 23, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, J.; Zhao, M. ONGene: A literature-based database for human oncogenes. J Genet Genomics 2017, 44, 119–121. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, M.; Li, Q.; Lu, J.; Zhao, M.; Qu, H. CIGene: a literature-based online resource for cancer initiation genes. BMC Genomics 2018, 19, 552–552. [Google Scholar] [CrossRef]
- Dastsooz, H.; Cereda, M.; Donna, D.; Oliviero, S. A Comprehensive Bioinformatics Analysis of UBE2C in Cancers. International journal of molecular sciences 2019, 20. [Google Scholar] [CrossRef]
- Zou, J.; Guo, P.; Lv, N.; Huang, D. Lipopolysaccharide-induced tumor necrosis factor-α factor enhances inflammation and is associated with cancer (Review). Molecular medicine reports 2015, 12, 6399–6404. [Google Scholar] [CrossRef]
- Choudhury, A.D.; Werner, L.; Francini, E.; Wei, X.X.; Ha, G.; Freeman, S.S.; Rhoades, J.; Reed, S.C.; Gydush, G.; Rotem, D.; et al. Tumor fraction in cell-free DNA as a biomarker in prostate cancer. JCI insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Vymetalkova, V.; Cervena, K.; Bartu, L.; Vodicka, P. Circulating Cell-Free DNA and Colorectal Cancer: A Systematic Review. International journal of molecular sciences 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Bastide, A.; David, A. The ribosome, (slow) beating heart of cancer (stem) cell. Oncogenesis 2018, 7, 34. [Google Scholar] [CrossRef] [PubMed]
- Guimaraes, J.C.; Zavolan, M. Patterns of ribosomal protein expression specify normal and malignant human cells. Genome biology 2016, 17, 236. [Google Scholar] [CrossRef]
- Slimane, S.N.; Marcel, V.; Fenouil, T.; Catez, F.; Saurin, J.C.; Bouvet, P.; Diaz, J.J.; Mertani, H.C. Ribosome Biogenesis Alterations in Colorectal Cancer. Cells 2020, 9. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Long, A.; Chiappini, C.; Travelli, C.; Govoni, S.; Racchi, M. Ribosomes as a nexus between translation and cancer progression: Focus on ribosomal Receptor for Activated C Kinase 1 (RACK1) in breast cancer. Br J Pharmacol 2020. [Google Scholar] [CrossRef]
Figure 1.
Flow chart of cfDNA data processing and filtering. (A) A three-stage workflow to identify the cancer driver genes in cfDNA sequence read, and 5 detailed steps for variant calling process on the left. (B) Flowchart for variants filtering, annotation, and deleterious gene prediction.
Figure 1.
Flow chart of cfDNA data processing and filtering. (A) A three-stage workflow to identify the cancer driver genes in cfDNA sequence read, and 5 detailed steps for variant calling process on the left. (B) Flowchart for variants filtering, annotation, and deleterious gene prediction.
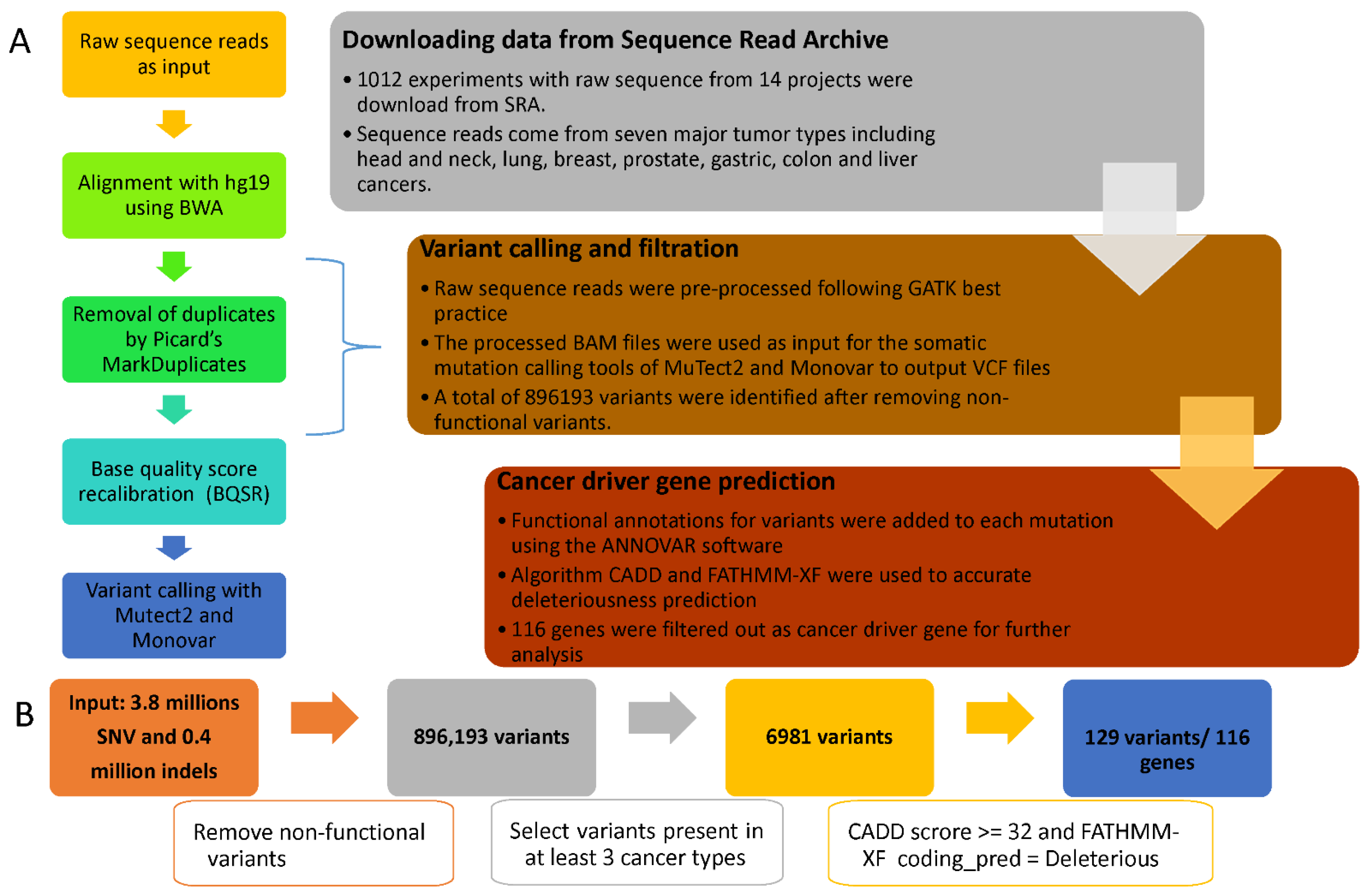
Figure 2.
Summary of cfDNA sequence data processing results. (A) Detailed steps for variant calling, filtration, and biomarkers prediction. (B) A bar chart indicates the number of variants called for different cancer types. (C) Venn diagram depict the overlap of somatic variants detected in various cancer types.
Figure 2.
Summary of cfDNA sequence data processing results. (A) Detailed steps for variant calling, filtration, and biomarkers prediction. (B) A bar chart indicates the number of variants called for different cancer types. (C) Venn diagram depict the overlap of somatic variants detected in various cancer types.
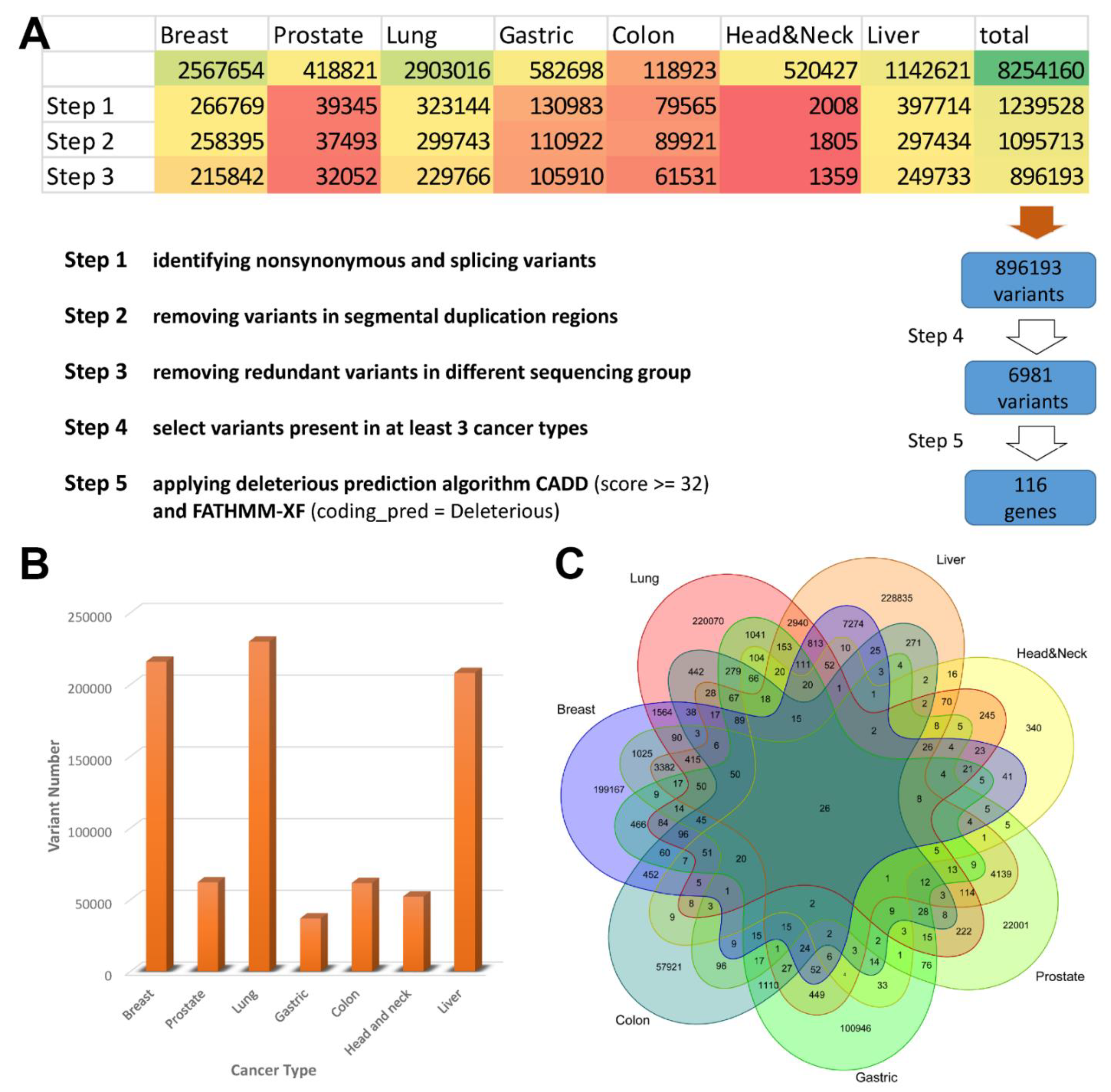
Figure 3.
The functional enrichment analysis of potential cancer biomarkers in cfDNA. (A) The significant enriched gene ontology (GO) terms. (B) All enriched KEGG pathways with statistical significance. (C) The overlapping of the top GO terms and the most frequent mutated genes.
Figure 3.
The functional enrichment analysis of potential cancer biomarkers in cfDNA. (A) The significant enriched gene ontology (GO) terms. (B) All enriched KEGG pathways with statistical significance. (C) The overlapping of the top GO terms and the most frequent mutated genes.
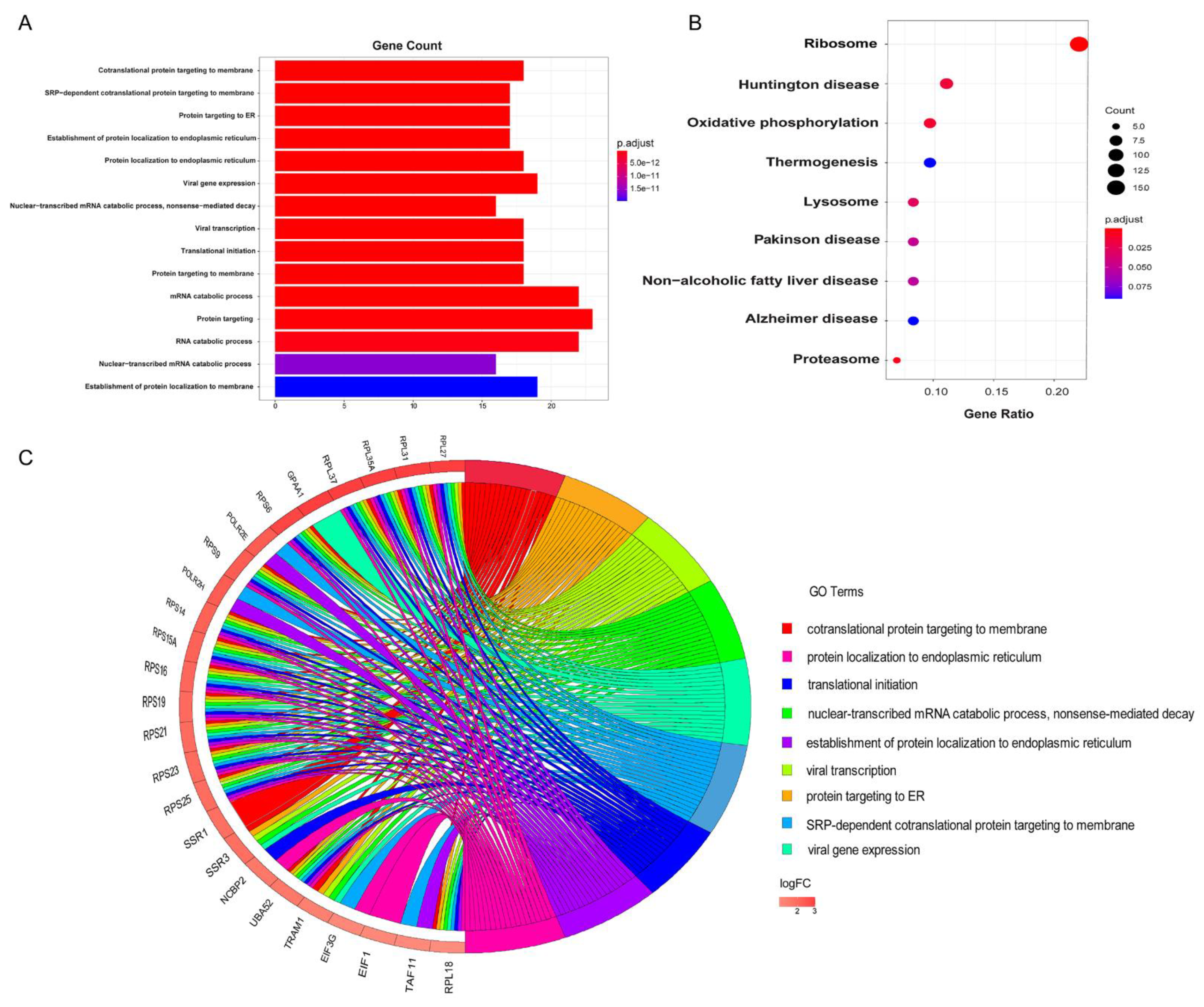
Figure 4.
The network analysis of potential cancer biomarkers in cfDNA. (A) Visualized PPI analysis of biomarker genes. (B) 25 genes in module 1 with the highest Maximal Clique Centrality (MCC) scores. (C) Interconnection of 10 hub genes; the color represents MCC scores, darker is higher. (D) The overlap of identified biomarkers (CDGs) with CIGs (cancer initiation genes), OCGs (oncogenes), and TSGs (tumor suppressor genes).
Figure 4.
The network analysis of potential cancer biomarkers in cfDNA. (A) Visualized PPI analysis of biomarker genes. (B) 25 genes in module 1 with the highest Maximal Clique Centrality (MCC) scores. (C) Interconnection of 10 hub genes; the color represents MCC scores, darker is higher. (D) The overlap of identified biomarkers (CDGs) with CIGs (cancer initiation genes), OCGs (oncogenes), and TSGs (tumor suppressor genes).
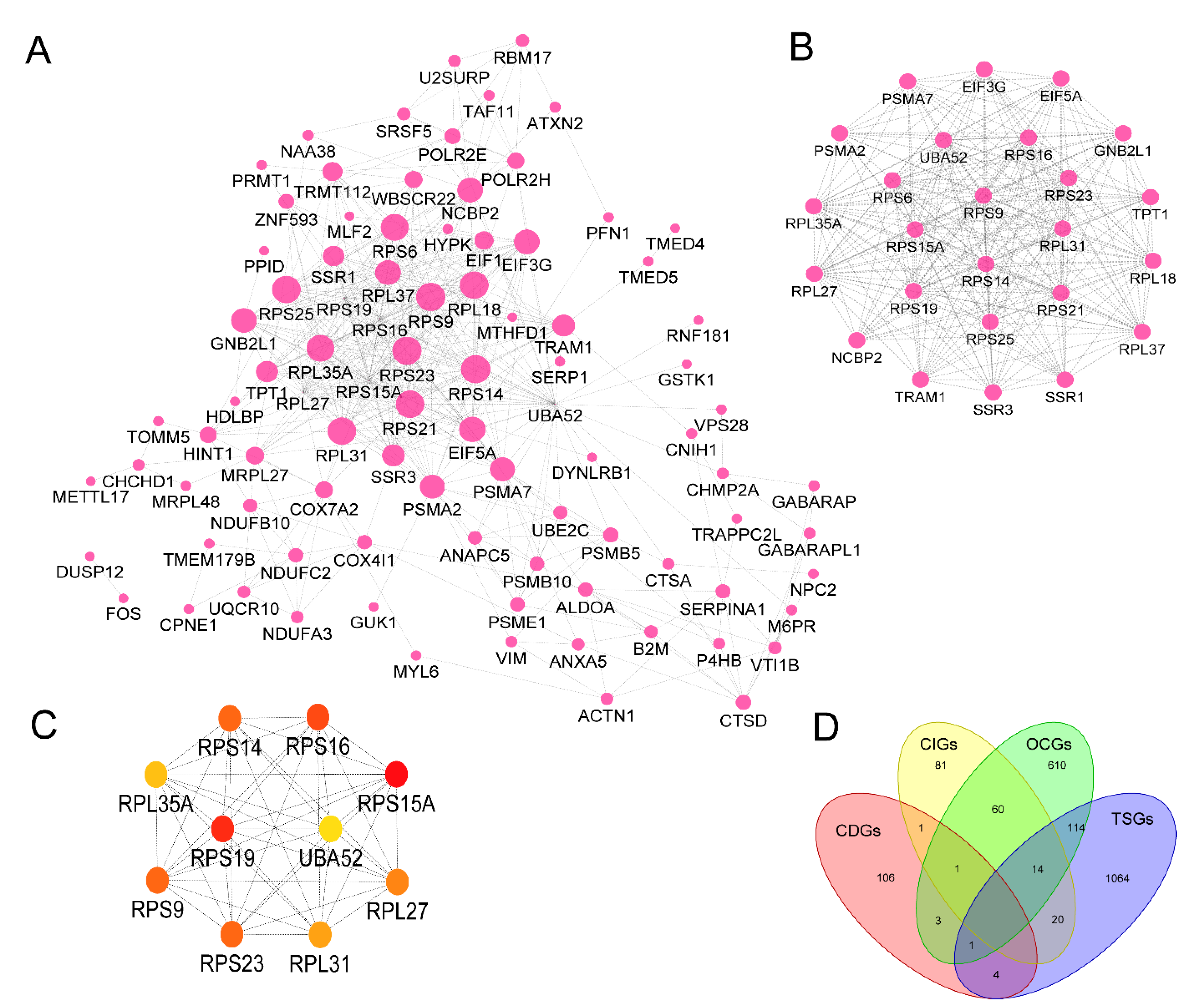
Figure 5.
The mutational and clinical feature of the 25 genes in the top functional module. (A) The mutational frequency across multiple cancer types; the percentages of cases with the 25 altered genes was depicted in the y axis. (B) The overall survival analysis of patients with altered (red) and unaltered (blue) 25 genes from the top functional module.
Figure 5.
The mutational and clinical feature of the 25 genes in the top functional module. (A) The mutational frequency across multiple cancer types; the percentages of cases with the 25 altered genes was depicted in the y axis. (B) The overall survival analysis of patients with altered (red) and unaltered (blue) 25 genes from the top functional module.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



