
Submitted:
06 June 2023
Posted:
06 June 2023
You are already at the latest version
Abstract
The growing demands of material longevity in marine environments necessitate the development of highly efficient, low-cost and durable corrosion protective coatings. Although magnesium alloys are widely used in the automotive and aerospace industries, severe corrosion issues still hinder their long-term service in naval architecture. In present work, an epoxy composite coating containing sericite nanosheets is prepared on the AZ31B Mg alloy by one-step electrophoretic deposition method to improve corrosion resistance. Due to the PEI modification, positively-charged sericite nanosheets can be highly orientated in an epoxy coating under the influence of an electric field. The sericite-incorporated epoxy coating prepared in the emulsion with 4 wt.% sericite exhibits the highest corrosion resistance in 3.5 wt.% NaCl solution, as manifested by electrochemical corrosion and long-term immersion tests. The highly-orientated sericite nanosheets in epoxy coating provide excellent barrier effect against corrosive media, thereby significantly improving the long-term anti-corrosion performance of the epoxy coating. This work provides new insight toward the design of lamellar filler/epoxy coatings with superior anticorrosion performance and shows promise in the corrosion protection of magnesium alloys.
Keywords:
Magnesium
; sericite
; epoxy
; corrosion
; electrochemical
1. Introduction
Global efforts are underway to reduce transportation-related greenhouse gas emissions, which can be achieved through increased use of lightweight materials in transportation industry [1,2]. Due to their low density, high elastic modulus, high specific strength, good heat conductivity, good shock resistance, and good castability, magnesium and its alloys are widely used as structural metal materials in terrestrial vehicles and aircrafts [3,4]. However, magnesium alloys experience severe corrosion issues in marine environments due to magnesium’s high chemical reactivity, which impedes their further application in naval architecture [5,6]. In recent decades, numerous efforts have been made to reduce magnesium's susceptibility to corrosion. In addition to improving alloy composition and microstructure, coating technology is the most effective method for preventing and mitigating corrosion of Mg alloys [7,8]. Among anti-corrosion coatings, organic coatings such as polyurethane, epoxy resin, and acrylic resin have been widely used for the corrosion protection of magnesium alloys [9,10,11]. In addition to being environmentally friendly due to their low emission of volatile organic compounds, waterborne epoxy coatings offer superior chemical resistance and adhesion, making them ideal for the automotive and marine industries [11,12]. Nevertheless, during the drying process and film formation, gas bubbles and pinholes are unavoidably formed within the epoxy coating. These inherent defects in pure epoxy coatings allow H2O, O2, and ions to permeate in corrosive media, resulting in poor long-term corrosion resistance [13,14]. Consequently, many works have focused on enhancing the impermeability of organic coatings in order to improve their corrosion resistance [15].
To address the aforementioned issue, lamellar fillers (such as graphene, MXenes, and clays) with high aspect ratio and good barrier capability were added into organic coatings [14,16,17,18]. For example, Yan et al. [14] prepared Ti3C2Tx/epoxy composite coatings on the aluminum alloy to achieve good anti-corrosion and wear-resistance properties. Zhu et al. [16] provided a facile strategy to design a composite coating of cationic dopamine-reduced graphene oxide (DRGO+) nanosheets and epoxy with good corrosion resistance. Hosseini et al. [17] also demonstrated that the addition of 5% polyaniline–montmorillonite clay to an epoxy coating on aluminum alloy can significantly improve corrosion resistance. However, lamellar materials, such as Ti3C2Tx and graphene, currently have drawbacks, such as difficult preparation and high cost, that limit their industrial applications on a large scale. Moreover, their potential electrical conductivity can result in a galvanic effect with the metal substrate, thereby increasing the likelihood of coating failure due to corrosion [19,20].
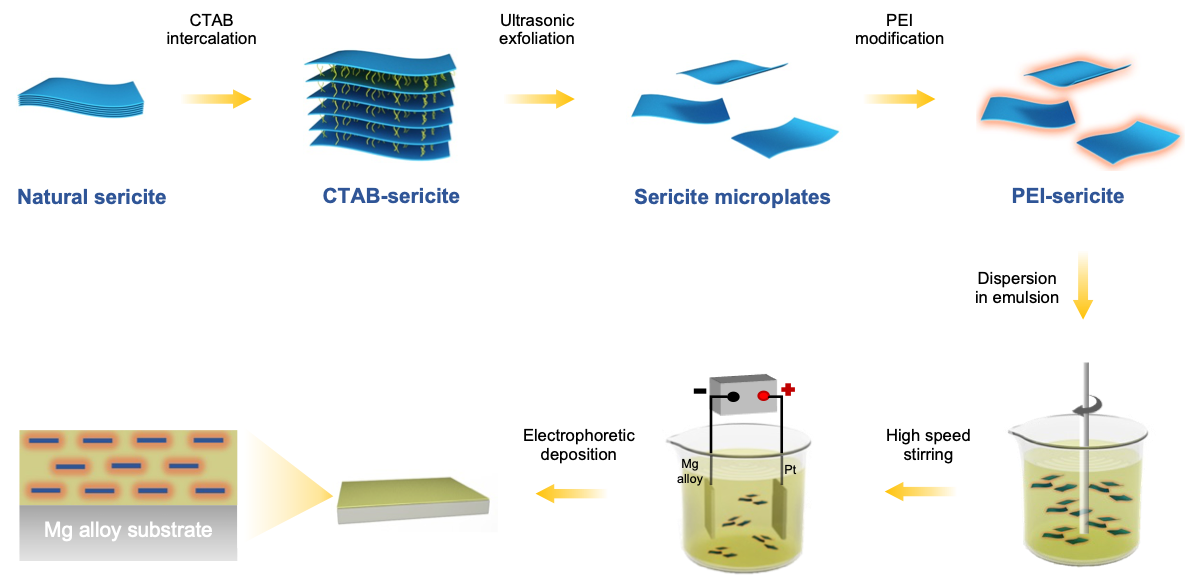
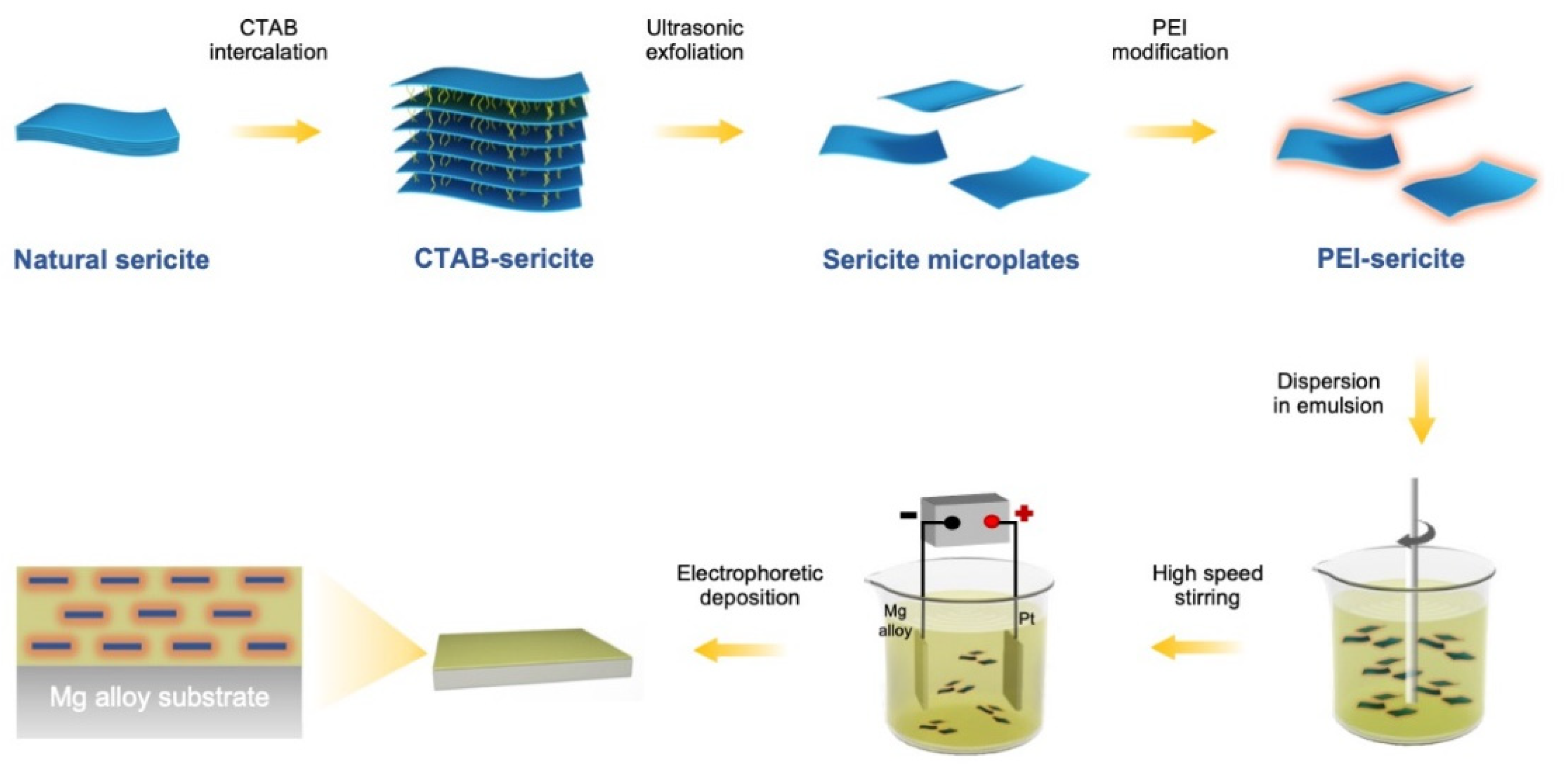
Natural clays have attracted interest in many fields due to their availability, abundance, environmental-friendliness, and low cost [21]. Sericite, a macroscopic transparent phyllosilicate clay, possesses impressive properties including high moduli, electric insulation, ultraviolet ray resistance, temperature stability, and chemical durability [22,23]. Sericite mineral raw materials can be processed into nanosheets or nanoplates via purification, intercalation, and exfoliation [24,25,26]. The high ratio of lateral to axial dimension makes them excellent lamellar fillers for use in coatings, as they provide an outstanding physical barrier. Our recent work [21] demonstrated that the addition of sericite nanosheets can significantly improve the corrosion resistance of micro-arc oxidation coating on aluminum alloy. According to the theoretic study by Lu and Mai [27], the geometric factors of clay fillers in coating can significantly affect the barrier property of coating. To minimize the permeability of coating, the orientation of the clay fillers should be aligned parallel to the surface plane, and their size and aspect ratio should also be optimized. In addition, the bond strength at the filler/polymer interface can also significantly impact the structural stability of composite coating [28]. Herein, we aim to fabricate a highly-orientated sericite/polymer coating by electrophoretic deposition of cationic modified sericite nanosheets and waterborne epoxy. By intercalating cetyltrimethyl ammonium bromide (CTAB) and modifying sericite with branched polyethyleneimine (PEI), cationic modified sericite nanosheets are successfully produced. The PEI modified sericite (PEI-SER) nanosheets can be dispersed steadily in aqueous solution and are used as a filler in a water-based cationic epoxy emulsion. In addition, the composite epoxy coating with aligned PEI-SER is fabricated on the surface of AZ31B magnesium alloy using electrophoretic deposition technology under the influence of electric field (as shown in Schematic 1). The anticorrosion property of composite epoxy coatings incorporating sericite nanosheets is then evaluated, and the relevant mechanisms are also investigated and discussed.
2. Materials and Methods
2.1. Materials
Water-based Cathodic epoxy resin electrophoresis emulsion (solid content: 35%) was supplied by Jiangxi Gaojie Technology Co., Ltd and natural sericite (800 mesh) was obtained from Wanqiao Mica Inc. (Chuzhou, Anhui, China). PEI (Mw= 25,000), CTAB, nitric acid (HNO3), silver nitrate (AgNO3), ethanol and sodium chloride (NaCl) were purchased from Aladdin Industrial Corporation. AZ31B magnesium alloy specimens (40×25×2 mm) were used as the substrates.
2.2. Intercalation and modification of sericite
Natural sericite was intercalated via the following procedures. First, the natural sericite was heated at 800°C in a muffle furnace for 1 h, and then the sericite powder was stirred with 5.0 mol/L HNO3 at 95 °C for 5 h, filtered and washed with deionized water until the pH was 7, and dried at 80 °C. Then the powder was dispersed into NaCl supersaturated solution at 95 °C, stirred for 3 h, filtered and washed with deionized water to remove redundant NaCl (tested by AgNO3). Subsequently, the resulting powder and a certain amount of CTAB (mass ratio: 15/46) were dispersed into deionized water at 80 °C, stirred for 24 h, filtered and washed with deionized water to remove redundant CTAB (tested by AgNO3), and then dried at 80 °C. For the sericite intercalated by CTAB, the product was named CTAB-SER.
After intercalation, a certain amount of CTAB-SER was dispersed into 500 mL ethanol and stirred in ultrasonication for 2 h. Then the exfoliated CTAB-SER micro plates were obtained by centrifuging and drying at 80 °C. Then the exfoliated CTAB-SER nanosheets were modified with PEI using a typical method. Specifically, 5g of CTAB-SER nanosheets, 100 mg of PEI and 292 mg of NaCl were dissolved in 500 mL of deionized water, followed by stirring for 1 h. The resultant powder was centrifuged, washed with deionized water several times, and then dried at 80 °C for further use. For the sericite modified by PEI, the product was named PEI-SER.
2.3. Preparation of composite coatings
In this study, AZ31B magnesium alloy specimens (40 × 25 × 2 mm) were used as the substrates. They were ground and polished with 400, 1200, 2000 grit silicon carbide paper, cleaned ultrasonically in ethanol, rinsed with distilled water, and dried in the air before coating.
Prior to electrophoretic deposition, the composite electrophoretic emulsion was prepared as follows. First, a given amount of modified sericite was ultrasonically dispersed in 10 g deionized water for 1 h to obtain a homogeneous suspension. Then the homogeneous suspension was slowly added into 90.0 g of cathodic epoxy resin electrophoresis emulsion, and then the mixture was stirred under 3000 rpm for 30 min to achieve the uniform dispersion of sericite and epoxy. After stirring, it was dispersed by ultrasound for 1 h to achieve better dispersion and then degassed in a vacuum oven for 30 min to remove the air bubbles. For comparison, mixed emulsions with different modified sericite contents (0 wt.%, 1.0 wt.%, 2.0 wt.%, 4.0 wt.%, and 6.0 wt.%) were prepared by the same method.
Then, the electrophoretic deposition was conducted in a beaker with the platinum plate electrode as the anode, pretreated AZ31B magnesium alloy as the cathode, and the mixed electrophoretic emulsion placed in. The coating was deposited using electrophoresis at 60 V for 5 min. After coating, the sample was taken out from the electrophoretic emulsion, washed with deionized water and dried at 150 °C for 30 min to obtain epoxy composite coating. For convenience, the coatings obtained by emulsion deposition with different sericite contents were named as E, E-S1, E-S2, E-S4 and E-S6, respectively.
2.4. Materials characterization
Field-emission scanning electron microscopy (FE-SEM, Carl Zeiss, SUPRA® 55, Germany) was used to observe the surface morphology of sericite powders and the surface and cross-sectional morphologies of the coatings. The surface and lateral elemental distributions were determined by energy-dispersive X-ray spectroscopy (EDS). The X-ray diffraction patterns of untreated and as-treated sericite powders were characterized on an X-ray powder diffractometer (XRD, PANalytical X'pert Powder, Netherlands) with Cu K𝛼 radiation (λ = 0.15418 nm). The XRD data was collected in the 2𝜃 range from 10° to 80° at a scanning rate of 12°/min and from 1° to 10° at a scanning rate of 3°/min The Fourier transform infrared (FTIR) spectra were obtained on the FTIR spectrometer (Nicolet IS50, Thermo Electron Corporation, US) in the wavenumber range from 4000 to 500 cm−1 with a resolution of 1 cm-1. The composition of coating was determined by grazing-incidence X-ray diffraction (GIXRD, Rigaku SmartLab, Japan) with Cu Kα radiation (λ = 0.15418 nm) at an incidence angle of 1°. The XRD data were collected in the 2θ range of 10° ~ 90° at a scanning rate of 2°/min and step size of 0.02°.
2.5. Corrosion evaluation
The corrosion characteristics of the samples were evaluated at room temperature in a 3.5 wt.% NaCl solution. The electrochemical tests were conducted on the electrochemical workstation (Reference 600+, Gamry, USA) using a three-electrode cell with the platinum mesh electrode as the counter electrode, the saturated calomel electrode (SCE) as the reference electrode, and the sample as the working electrode (exposed area is 1 cm2). Electrochemical impedance spectroscopy (EIS) plots were obtained with an interference potential at 5 mV from 105 Hz to 10-1 Hz. The equivalent circuits of EIS were fitted with the Gamry Echem Analyst software. Potentiodynamic polarization (POL) was performed between −0.25 V to 0.5 V (vs. OCP) at a scanning rate of 1 mV/s. In order to obtain the polarization curves, the potential was swept from the cathodic to the anodic regions. Tafel extrapolation was used to derive the corresponding corrosion current densities (icorr) and corrosion potentials (Ecorr). All electrochemical tests were performed at least 3 times to ensure repeatability. Immersion tests were carried out to observe the corrosion behavior of the samples. After immersion in a 3.5 wt% NaCl solution at room temperature for 0, 1, 3, 5, 7, and 14 days, all samples were quickly rinsed and dried in the air. FE-SEM and 3D optical profilometry (UP Dual Model, Rtec, USA) were used to examine the surface and cross-sectional state of composite coatings after 14 days of immersion.
Figure 1.
A schematic showing the fabricating procedure of highly-orientated sericite/epoxy coating on AZ31B Mg alloy.
Figure 1.
A schematic showing the fabricating procedure of highly-orientated sericite/epoxy coating on AZ31B Mg alloy.
3. Results
3.1. Characterization of sericite before and after pretreatment
The wide-angle (Figure 2a) and the small-angle (Figure 2b) XRD spectra of original and modified sericite reveal the evolution of interlaminar structure during the intercalation and modification process of sericite. The diffraction peaks in Figure 2a are consistent with muscovite (JCPDS 07-0032) and quartz (JCPDS 01-079-1910). After CTAB intercalation, the majority of peaks persist, indicating the absence of phase transformation and the preservation of the lamellar structure [22]. However, the intensity of peak 𝑑002 = 2.4 nm (2𝜃 = 8∘) is weaker than that of natural sericite, shifting to a lower diffraction angle than before CTAB intercalation. Concurrently, a new basal reflection corresponding to 𝑑002 = 5.2 nm (2𝜃 = 2∘) was observed, indicating that CTAB molecules enter the interlayer spaces in a regular arrangement and the interlamellar spacing is partly expanded [24,25,29]. After ultrasonication and PEI treatment, the intensity of 𝑑002 = 2.4 nm (2𝜃 = 8∘) further decreases and the broad peak 𝑑002 = 5.2 nm(2𝜃 = 2∘) are disappeared, suggesting further dissociation of the layered lattice along the vertical direction of (002) plane. The XRD results indicate the layered structure of sericite is separated after pretreatment and modification.
FT-IR was used to characterize the functional groups of SER, CTAB, CTAB-SER, PEI, PEI-SER. As depicted in Figure 2c, the 3600 cm-1 stretching band is attributed to the presence of –OH groups on the surface of sericite [26,30]. After CTAB intercalation, the peak of CTAB-SER at 3600 cm-1 is significantly diminished compared to SER. Besides, the spectrum of CTAB-SER displays two weak absorption bands at 2926 cm-1 and 2852 cm-1, which can be attributed to the asymmetric and symmetric stretching vibrations of methylene (−CH2) [31]. The Si–O stretching vibration band (at 1077 cm−1) in CTAB-SER is broader than that of SER, which can be due to the appearance of the C–N and C–C bending vibration bands at 960 cm-1 and 909 cm-1, respectively [25]. According to the above XRD and FT-IR results, it can be concluded that the intercalation was successful because CTA+ entered the interlayer space of sericite. The characteristic peaks of PEI appear at ~3300 cm-1 (-NH and -NH2 stretching) and 1630 cm-1 (-NH2 scissoring vibration) [32]. The typical peaks of CTAB-SER are still present in the PEI-SER spectrum, and two new peaks appear at 3355 cm-1 and 1630 cm-1. The FT-IR results confirm that the PEI is successfully attached onto the surface of the sericite nanosheets.
Figure 2d and e depict the zeta potential and size distribution of SER, CTAB-SER, and PEI-SER dispersed in deionized water. PEI-SER appears to be positively charged with a zeta potential of 45.3 mV, whereas SER and CTAB-SER exhibit negative zeta potentials of 23.6 mV and weak positive zeta potentials of 8.9 mV, respectively. As a result of their positive charge, the sericite nanosheets move toward the cathode during electrophoretic deposition in order to form a composite coating. Although the measured particle size is not the actual lamellar diameter but the equivalent particle size due to sericite's two-dimensional layered structure, it can still approximate the lamellae size. The measured particle size of raw SER ranges from approximately 2000~7000 nm, with an average size of 3585.9 nm. CTAB-SER and PEI-SER display a sharper Gaussian-like size distribution than raw SER, with an average size of 1990.2 nm and 1718.2 nm, respectively. Figure 2f compares SER and PEI-SER at a concentration of 0.5% by weight dispersed in deionized water after one hour of standing. Due to the agglomeration of sericite, the natural sericite solution exhibited distinct stratification and precipitation after 1 h of standing, whereas the PEI-SER solution did not exhibit any discernible precipitation. The surface morphology of SER and PEI-SER are observed by SEM. Figure 2g demonstrates that raw sericite possessed a typical compact and layered structure, whereas PEI-SER exhibited a structure with thinner layers. The PEI-SER lamellae were obtained by exfoliated and stripped from the multi-layered CTAB-SER by ultrasonic and stirring treatment. Consequently, based on the aforementioned results, it can be concluded that CTAB intercalation dissociated the multilayer structure of the original sericite, whereas PEI modification successfully modulated the surface charge properties of the sericite nanosheets.
3.2. Surface morphology and cross-section of coatings
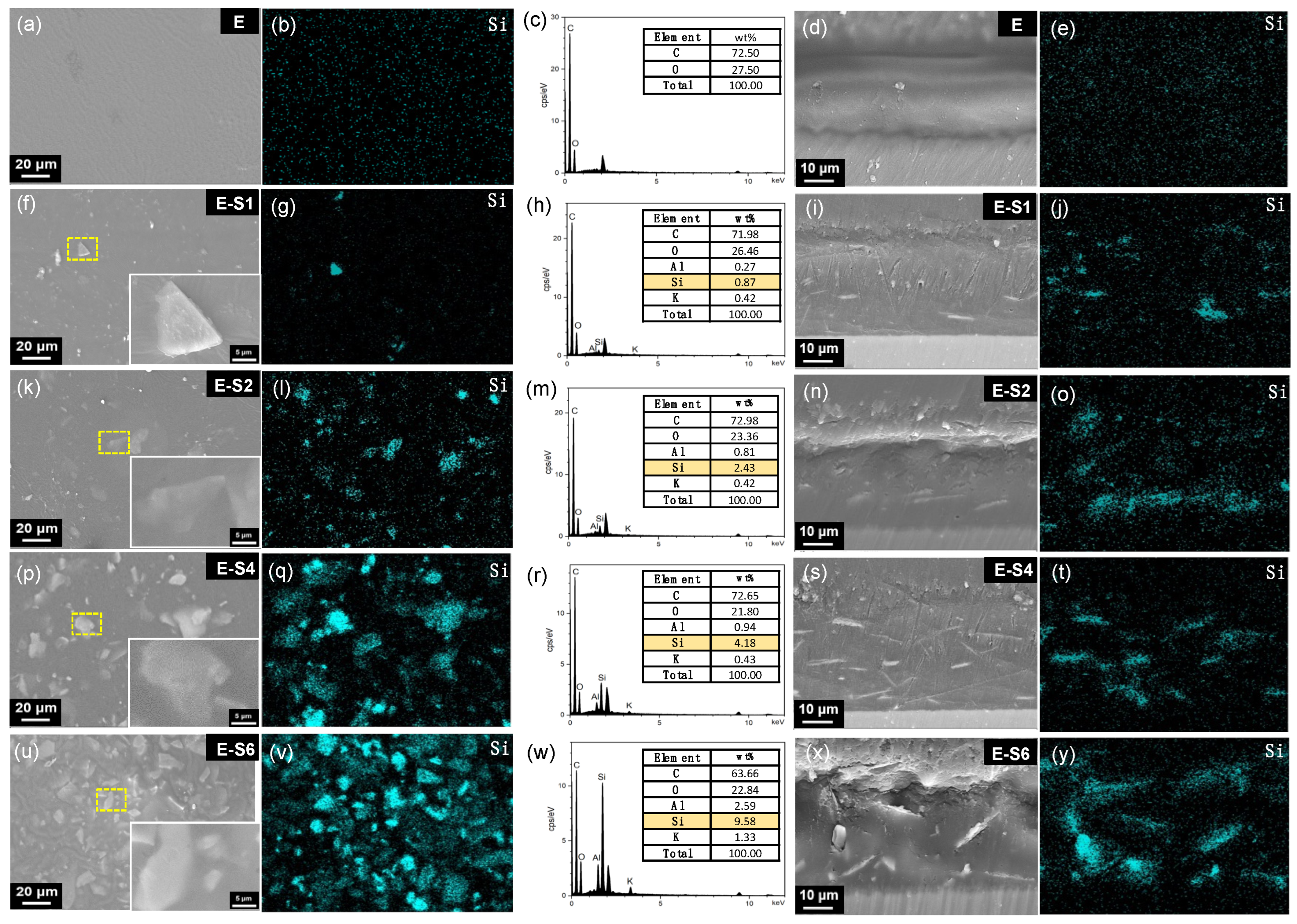
Figure 3 depicts the surface morphology and cross-sections of coating surfaces prepared with emulsions containing varying amounts of PEI-SER. As the sericite concentration in the electrolyte increases, so does the number of nanosheets observed on the coating surface. The sericite nanosheets are nearly uniformly dispersed in the coatings and closely combined with the epoxy resin, demonstrating excellent interfacial compatibility. SEM images and Si element distribution on the surface of different samples reveal that the addition of PEI-modified SER to the electrophoretic emulsion increased the number of sericite nanosheets and the Si element content on the coating surface. The average thickness of the pure epoxy coating is approximately 26 μm, whereas the average thickness of four coatings with sericite is approximately 30 μm, indicating that the addition of PEI-SER increases the electrophoretic deposition rate of epoxy coating. This can be attributed to the molecular brush effect of positively charged sericite, which facilitates the movement of the emulsion towards the cathode during electrophoresis, resulting in denser and thicker coatings [16]. The majority of sericite nanosheets are aligned parallel to the sample surface in the PEI-SER coating. Throughout electrophoretic deposition, a perpendicular DC electric field is applied to the metal surface. Therefore, the positively charged PEI-SER nanosheets are propelled towards the cathode by the electric field. Due to the uniformly applied electric field force and electrostatic repulsion force between nanosheets, the nanosheets tend to align themselves parallel to the surface of the sample during movement. As shown in Figure 3j,n,s, sericite nanosheets are uniform dispersed in parallel in the E-S1, E-S2 and E-S4 coatings. In addition, the sericite nanosheets are compatible with the epoxy matrix at the interface. The parallel arrangement of sericite nanosheets in epoxy coatings becomes more pronounced and denser as sericite content is continuously added. The parallel distribution and high density of sericite nanosheets are advantageous for enhancing the coating's physical barrier effect and resistance to corrosion. However, when the concentration of PEI-SER nanosheets reaches a certain threshold, disordered arrangement and agglomeration begin to emerge. As seen in Figure 3x, the sericite nanosheets in the E-S6 coating have become disordered and agglomerated, which may lead to the formation of micropores and defects that diminish the impermeability of composite coatings.
Figure 2.
SEM images showing surface morphology and cross-sections of different samples. From the first row to the fifth row are related to samples of E, E-S1, E-S2, E-S4, E-S6, respectively. The first column of images (a, f, k, p, u) depicts surface morphology of different samples with insets showing magnified selected area. The second column images show the Si elemental EDS maps and the third column shows EDS spectra of the related area in the first row: E (b, c), E-S1 (g, h), E-S2 (l, m), E-S4 (q, r), and E-S6 (v, w). The fourth column of images depicts the cross-sections of different samples and the fifth row shows the Si elemental of the related cross-sectional area: E (d, e), E-S1 (i, j), E-S2 (n, o), E-S4 (s, t), and E-S6 (x, y).
Figure 2.
SEM images showing surface morphology and cross-sections of different samples. From the first row to the fifth row are related to samples of E, E-S1, E-S2, E-S4, E-S6, respectively. The first column of images (a, f, k, p, u) depicts surface morphology of different samples with insets showing magnified selected area. The second column images show the Si elemental EDS maps and the third column shows EDS spectra of the related area in the first row: E (b, c), E-S1 (g, h), E-S2 (l, m), E-S4 (q, r), and E-S6 (v, w). The fourth column of images depicts the cross-sections of different samples and the fifth row shows the Si elemental of the related cross-sectional area: E (d, e), E-S1 (i, j), E-S2 (n, o), E-S4 (s, t), and E-S6 (x, y).
3.3. Electrochemical corrosion tests
Figure 4a depicts the potentiodynamic polarization (POL) curves of different samples after immersion in 3.5 wt.% NaCl for 1h. The corrosion potentials (Ecorr) and corrosion current densities (icorr) otained directly from the Tafel region in the cathodic polarization curves by Tafel extrapolation are listed in Table 1 and shown in Figure 4b, respectively. As the concentration of sericite increased, the polarization curve gradually shifted to the left and upward in the order of E1, E2, and E4, with E-S4 occupying the most extreme left and highest position on the POL curves plot. Among all samples, E-S4 shows the smallest icorr of 2.412×10−11 A∗cm−2, which is approximately 2 orders of magnitude lower than that of E and 6 orders of magnitude lower than the AZ31B Mg substrate. Moreover, E-S4 has the highest Ecorr of -1.214 V. More positive Ecorr and lower icorr are generally indicative of greater corrosion resistance [13]. Consequently, the anti-corrosion performance of epoxy samples is significantly enhanced by the addition of sericite. This can be attributed to the self-aligned parallel arrangement of sericite nanosheets, which can significantly affect the barrier performance of epoxy coating and enhance its corrosion resistance. In comparison to E-S4, E-S6 demonstrates a lower Ecorr and a higher icorr, indicating that the disordered arrangement and aggregation of sericite nanosheets negatively impacted the coating's corrosion resistance performance. Nevertheless, due to its relatively high sericite concentration, its performance is still superior to all other samples except E-S4.
Figure 5a-c depict the Nyquist, Bode-impedance and Bode-Phase plots of samples after immersion for 1h. For Nyquist curves, a larger diameter of capacitive loops indicates a coating with superior anticorrosion properties [13]. Notably, as shown by Figure 5a, the capacitive loops enlarge after electrophoretic deposition, indicating improved corrosion resistance compared to the substrate. In the case of Bode impedance plots, the impedance modulus at the lower frequency (|Z|0.1Hz) is frequently utilized as a crucial indicator for evaluating the anticorrosion performance of coatings. Usually, the higher |Z|0.1Hz represents better corrosion protection [33,34]. The bode impedance plot in Figure 5b demonstrates that the E-S4 has the highest |Z|0.1Hz value, suggesting better corrosion protection performance than other samples.
According to the characteristics of Bode and Nyquist plots, equivalent circuits (Figure 5d) are used to fit the EIS data of different samples. In this work, constant phase element (CPE) is used to represent the non-ideal capacitors, which is expressed by:
which Y0 and n are the admittance constant and empirical exponent, respectively [35,36]. Based on the characteristics of EIS data and previous literatures [4,37], Rs(CPEf (Rpore(((CPEdlRct)(CPEdiffRdiff))))) is proposed to fit the EIS data of E-S1, E-S2, and E-S6. The EIS data curves of AZ31B show obvious inductive characteristics in the low frequency impedance region, which can be attributable to pit formation during corrosion [38]. Consequently, Rs (((CPEdlRct)(CPEdiffRdiff))(LRL)))) is proposed to fit the EIS data of AZ31B. Since the EIS data curves of E-S4 does not exhibit the full low frequency diffusion impedance region, Rs(CPEf (Rpore (CPEdl Rct ))) is proposed to fit E-S4. In these three circuits, Rs refers to the solution resistance. CPEf represents the capacitance of the coating and Rpore stands for the total resistance of pore and defects in the coating. CPEdl represent the capacitance of the electric double layer at the low-frequency area and and Rct refers to the charge transfer resistance in the Faradic process. CPEdiff represents the capacitance pertaining to the diffusion process and Rdiff denotes the relevant resistance. L refers to the inductance associate with the relaxation process of adsorbed species and RL is the relevant inductance resistance. The fitted data are shown in Table 2. Generally, the capacitive curves at the high frequency region stands for the surface film and charge transfer, while the lower frequency behavior is associated with mass transfer [38,39,40]. For these epoxy-coated samples, the Rpore can be used to evaluate the barrier effect of coating and the Rct can be used to evaluate the ability of coating to maintain the coating/metal interface corrosion [41]. Both of these two parameters indicate a general trend of reducing corrosion resistance: E-S4 > E-S6 > E-S2 > E-S1 > E.
Y = Y0 (jω)n,
In order to evaluate the long-term corrosion behavior and mechanism, EIS is utilized after exposing samples to 3.5 wt. % NaCl for varying time periods. The EIS data are represented in Figure 6d-u as Nyquist, Bode-impedance, and Bode-Phase plots. The appearance of an inductive loop can be used to determine the onset of localized corrosion. Figure 7 depicts the variation between the coating impedance (Rpore) and the impedance modulus at low frequencies (|Z|0.1Hz). The Rpore and |Z|0.1 Hz of the samples decreased as immersion time increased, indicating that the barrier capability of coatings and overall corrosion resistance of the samples would gradually decrease during immersion. The addition of sericite effectively slowed the deterioration of the coating's corrosion resistance, and the E-S4 sample demonstrates the best long-term corrosion resistance.
3.4. Immersion tests
The immersion test was carried out for 14 days in 3.5 wt.% NaCl to investigate the corrosion process. The digital pictures of immersed specimens were shown in Figure 8. After 1 day of immersion, the AZ31B substrate began to show signs of discoloration, filiform corrosion, and cracking. The corrosion gradually intensified and spread across the entire surface as immersion time was increased. For the sample with an epoxy coating, corrosion was inhibited significantly. However, due to flaws and pinholes in the coating, corrosive media can easily penetrate the pure epoxy coating. After 1 day of immersion, some tiny blisters and pores began to appear on the surface of the E sample. With prolonged immersion, surface blistering and localized corrosion in edge regions continued to increase. The corrosion damage of samples with sericite is less severe than that of sample E. Preliminary coating damage starts on the 3rd day of E-S1 and E-S6, and the 5th day of E-S2. In contrast, the corroded areas of these samples are significantly smaller than those of sample E. Notably, after 14 days, only a few bubbles and edge corrosion areas appeared on the surface of E-S4, indicating that the coating still retains good corrosion resistance. CLSM and 3D topography images are used to observe the microscopic corrosion of samples after immersion for 14 days. As shown in Figure 9, the AZ31B substrate and E exhibit a surface that is heavily corroded and covered with corrosion products. Comparatively, the surfaces of samples containing sericite are considerably more intact and refined, indicating a milder corrosion behavior. Nevertheless, after 14 days of immersion, these samples still display shallow pits and corrosion products. Among them, the E-S4 sample has the lowest Ra, indicating that the E-S4 coating has the most effective corrosion barrier effect.
3.5. Corrosion products analysis
The GIXRD patterns of all samples in 3.5 wt.% NaCl solutions after immersing 14 days are shown in Figure 10. In the sericite/epoxy composite coatings, the broad peak between 2θ of 10°~30° indicates epoxy, while the diffraction peaks at 17.5°, 26.6°, and 27.8° provide evidence of sericite (JSPDS NO. 47-1144). Corrosion products such as Mg(OH)2 (Brucite, JCPDS NO. 82-2455) and MgO (JCPDS NO. 89-7746), MgAl-LDH (Hydrotalcite, JCPDS NO. 89-0460) and MgCl2 (JCPDS NO. 80-1752) can also be observed as diffraction peaks on the surface of the sample. Furthermore, E sample has the highest relative intensity of the Mg (JCPDS NO. 89-7195) peak at 34.4° compared to the epoxy peak, indicating that the E sample has the lowest coating integrity and the largest area of substrate exposure.
SEM and EDS were used to determine the corrosion morphology and elemental composition of samples following an immersion test. Figure 11 shows typical regions of samples after immersion in 3.5 wt.% NaCl for 14 days. As shown in Figure 11a,b, the surface of AZ31B is fully covered by cotton-like corrosion products. The elemental composition of the corroded area indicates that the corrosion products consist primarily of MgO and Mg(OH)2, with a small amount of MgAl-LDH and chlorides [42]. On the surface of the E sample, many cracks are distributed and the coating is partially peeled off. On the exposed substrate, corrosion products can be observed in continuous quantities. This is due to the fact that when the corrosive medium penetrates into the coating, the substrate will be severely corroded to produce hydrogen gas and corrosion products, resulting in local cracks and defects in the coating [43,44]. Sericite nanosheets can be observed from the surfaces of sericite-containing samples, with Si element also detected from related area. Instead of cracks and massive corrosion products, these sericite-containing samples typically exhibit corrosion pits and sheet-like corrosion products. The surface corrosion products consist primarily of sheet-like Mg(OH)2 and Mg-Al LDH, which are formed by continuous exudation of Mg and Al ions from the AZ31 substrate[37].
To analyze the internal condition of coatings after 14 days of immersion in 3.5% NaCl, cross-sectional SEM images and EDS maps are acquired. As shown in Figure 12a, the distribution of C and O elements reveals that after immersion, the epoxy coating on E has been replaced by a thick layer of corrosion products. The coatings of E-S1 and E-S2 are also severely corroded, as evidenced by the presence of deep cracks within and beneath the epoxy coating. The distribution of Mg in Figure 12b,c also indicate the formation of corrosion products on and in these coatings. In contrast, Figure 12d demonstrates that E-coating S4's is relatively intact after 14 days of immersion. It is almost difficult to observe the aggregation of Mg elements on its surface, indicating that less corrosion products are generated. However, the SEM image and EDS maps in Figure 12e indicate that the agglomeration of sericite in the E-S6 coating may cause localized defects in the surrounding area and ultimately lead to corrosion failure.
4. Discussion
In order to discuss the anticorrosion mechanism of the highly-oriented epoxy-sericite composite coating, the electrochemical or chemical reaction changes at the substrate-coating interface were analyzed. The main anodic and cathodic reactions occurring at the interface during the electrochemical corrosion of magnesium and magnesium alloys are as follow:
Anodic dissolution:
The M in equation (2) is mainly Mg and Al for the AZ31 alloy. The cathodic reaction can be oxygen reduction reaction (ORR) or hydrogen evolution reaction (HER):
The dissolved metallic ions in (2) can diffuse and react with OH- to form corrosion products such as Mg(OH)2, MgO and Mg-Al LDH:
These corrosion products can form at or around the active site where corrosion occurs. Due to the Pilling-Bedworth ratio (PBR) of Mg, these corrosion products may form defective passivation layers and provide partial protection. However, in chloride solutions, Cl- can destroy the passivation layer and accelerate the corrosion of magnesium, which can be expressed by (8) and (9):
Therefore, it can be inferred that the availability of H2O, O2, and Cl- at the interface are the critical factors affecting the entire corrosion process.
M→ Mx+ + xe-
O2 + 2H2O + 4e- → 4OH-
2H2O + 2e- → H2 + 2OH-
Mg2+ + 2OH- → Mg(OH)2
Mg(OH)2 → MgO + H2O
Mg2+ + Al3+ + OH → MgxAly(OH)z
Mg2++ 2Cl- → MgCl2
MgCl2+ 2OH-→Mg(OH)2+ 2Cl-
As can be seen from the SEM images in Figure 11 and Figure 12, the corrosion failure mode of epoxy coatings on magnesium alloys can be divided into several stages. In the early stage of corrosion of the E sample, the epoxy coating possesses a corrosion retardation effect by preventing the corrosive species from the substrate. However, as the immersion time increases, the corrosive medium can diffuse through the coating via micro-pores and defects to reach the substrate/coating interface (Figure 13a). The corrosion of Mg alloy substrate at the substrate/coating interface causes the formation of corrosion products (e.g., Mg(OH)2 and MgO) and evolution of H2, leading to the accumulation of corrosion products and gas blister under the coating, and gradually resulting in the damage of coating integrity (Figure 13b). Besides, Cl- can migrate through the coating and oxide, playing a crucial role in the dissolution of passivating hydroxide or oxide of magnesium and causing pitting corrosion. Due to the continuous erosion of Cl-, local corrosion such as pitting and blistering under the coating will be aggravated, and finally the epoxy coating will collapse as shown in Figure 13c.
According to the aforementioned corrosion mechanism and anti-corrosion mechanism of 2D layered materials in coatings [14,16,17], the anti-corrosion mechanism of sericite nanosheets in uniform orientation sericite/epoxy coating is discussed, as conceptually illustrated in Figure 13d. Under the influence of electric field, an appropriate amount of sericite will be aligned in the coating, which can form a tight barrier network against diffusion of O2, H2O, and Cl-. For the substrate protected by a coating containing sericite, the low availability of O2 and H2O suppresses the cathodic reactions, which in turn suppresses the anodic reaction of Mg dissolution. In addition, the corrosion rate of the substrate is also reduced because fewer Cl- ions pass through the coating. Generally, the addition of more layered fillers can increase the density of mico-/nano-plates in the coating, leaving fewer penetration paths for corrosive species, thereby further improving the impermeability of the coating [45]. Consequently, our study reveals that the corrosion resistance of E-S4 and E-S6 is superior to that of samples containing a lower concentration of sericite. When the concentration of sericite reaches a certain threshold, however, the dispersion and orderliness of nanosheets in epoxy begin to diminish. The aggregation of nanosheets within a coating can result in defects in the surrounding area, allowing corrosive media to penetrate. This may help explain why the anticorrosion performance of E-S6 coatings is inferior to that of E-S4 coatings.
5. Conclusions
In summary, the CTAB-intercalated and PEI-modified sericite nanosheets were used to prepare a highly-orientated sericite/epoxy coating on AZ31 Mg alloy via electrophoretic deposition. The anticorrosion performance of sericite/epoxy composite coatings was evaluated, and the results indicated that the highly-orientated sericite nanosheets significantly improved the corrosion resistance of epoxy coatings. The potentiodynamic polarization test reveals that the E-S4 coating has the smallest icorr, which is approximately two orders of magnitude lower than the pure epoxy coating and six orders of magnitude lower than the AZ31B Mg substrate. The long-term immersion test demonstrates that that the E-S4 coating can still effectively protect the substrate from severe corrosion after 14 days of immersion in 3.5 wt.% NaCl solution. The increased corrosion resistance is attributed to the barrier effects of the highly-orientated sericite nanosheets, which significantly delay the penetration of corrosive media. The enhanced corrosion resistance stems from the barrier effects of the sericite nanosheets arranged in parallel, which significantly retards the intrusion of corrosive media. This work provides insights into the design of high-performance and low-cost electrophoretic anticorrosion coatings, which have great application potential in the corrosion protection of metals.
Author Contributions
Conceptualization, Hao Wu; Data curation, Ke Xi; Formal analysis, Hao Wu; Funding acquisition, Hao Wu and Chilou Zhou; Investigation, Hao Wu, Ke Xi, Yan Huang, Zena Zheng, Zhenghua Wu and Ruolin Liu; Methodology, Hao Wu; Project administration, Hao Wu, Chilou Zhou and Yansheng Yin; Resources, Yao Xu; Supervision, Yansheng Yin; Validation, Chilou Zhou; Visualization, Hao Wu; Writing – original draft, Hao Wu; Writing – review & editing, Hao Wu. All authors have read and agreed to the published version of the manuscript.
Funding
The work was financially supported by the National Natural Science Foundation of China (No. 51905177 and No. 52071091), China Postdoctoral Science Foundation (No. 2019M662899), Fundamental Research Funds for the Central Universities (No. 2019MS061), as well as Guangdong Basic and Applied Basic Research Foundation (No. 2019A1515011157).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef]
- Esmaily, M.; Svensson, J.E.; Fajardo, S.; Birbilis, N.; Frankel, G.S.; Virtanen, S.; Arrabal, R.; Thomas, S.; Johansson, L.G. Fundamentals and advances in magnesium alloy corrosion. Progress in Materials Science 2017, 89, 92–193. [Google Scholar] [CrossRef]
- Yu, D.; Qiu, H.; Mou, X.; Dou, Z.; Zhou, N.; Guo, Q.; Lyu, N.; Lu, L.; Yang, Z.; Huang, N. One-Pot but Two-Step Vapor-Based Amine- and Fluorine-Bearing Dual-Layer Coating for Improving Anticorrosion and Biocompatibility of Magnesium Alloy. ACS Biomater Sci Eng 2019, 5, 4331–4340. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Sun, J.; Han, J.; Yang, Z.; Zhou, H.; Xiao, L.; Xu, S.; Han, Y.; Ma, A.; Wu, G. Effect of hierarchical precipitates on corrosion behavior of fine-grain magnesium-gadolinium-silver alloy. Corrosion Science 2022, 194, 109924. [Google Scholar] [CrossRef]
- Lu, X.; Li, Y.; Ju, P.; Chen, Y.; Yang, J.; Qian, K.; Zhang, T.; Wang, F. Unveiling the inhibition mechanism of an effective inhibitor for AZ91 Mg alloy. Corrosion Science 2019, 148, 264–271. [Google Scholar] [CrossRef]
- Wang, C.; Wu, L.; Xue, F.; Ma, R.; Etim, I.-I.N.; Hao, X.; Dong, J.; Ke, W. Electrochemical noise analysis on the pit corrosion susceptibility of biodegradable AZ31 magnesium alloy in four types of simulated body solutions. Journal of Materials Science & Technology 2018, 34, 1876–1884. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, X.; Blawert, C.; Zheludkevich, M.L.; Zhang, T.; Wang, F. Formation of self-lubricating PEO coating via in-situ incorporation of PTFE particles. Surface and Coatings Technology 2018, 337, 379–388. [Google Scholar] [CrossRef]
- Song, Z.; Xie, Z.; Ding, L.; Zhang, Y.; Hu, X. Preparation of corrosion-resistant MgAl-LDH/Ni composite coating on Mg alloy AZ31B. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2022, 632. [Google Scholar] [CrossRef]
- Yilmaz, O. A hybrid polyacrylate/OMMT nanocomposite latex: Synthesis, characterization and its application as a coating binder. Progress in Organic Coatings 2014, 77, 110–117. [Google Scholar] [CrossRef]
- González-García, Y.; González, S.; Souto, R.M. Electrochemical and structural properties of a polyurethane coating on steel substrates for corrosion protection. Corrosion Science 2007, 49, 3514–3526. [Google Scholar] [CrossRef]
- Luo, X.; Zhong, J.; Zhou, Q.; Du, S.; Yuan, S.; Liu, Y. Cationic Reduced Graphene Oxide as Self-Aligned Nanofiller in the Epoxy Nanocomposite Coating with Excellent Anticorrosive Performance and Its High Antibacterial Activity. ACS Appl Mater Interfaces 2018, 10, 18400–18415. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, R.; Liu, Q.; Liu, J.; Yu, J.; Wang, C.; Zhang, M.; Liu, P.; Wang, J. Fabrication of ZnO/epoxy resin superhydrophobic coating on AZ31 magnesium alloy. Chemical Engineering Journal 2019, 368, 261–272. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, J.; Zhong, J.; Shi, X. Thin Nacre-Biomimetic Coating with Super-Anticorrosion Performance. ACS Nano 2018, 12, 10189–10200. [Google Scholar] [CrossRef]
- Yan, H.; Cai, M.; Li, W.; Fan, X.; Zhu, M. Amino-functionalized Ti3C2T with anti-corrosive/wear function for waterborne epoxy coating. Journal of Materials Science & Technology 2020, 54, 144–159. [Google Scholar] [CrossRef]
- He, L.-J.; Shao, Y.; Li, S.-Q.; Cui, L.-Y.; Ji, X.-J.; Zhao, Y.-B.; Zeng, R.-C. Advances in layer-by-layer self-assembled coatings upon biodegradable magnesium alloys. Science China Materials 2021, 64, 2093–2106. [Google Scholar] [CrossRef]
- Zhu, X.; Yan, Q.; Cheng, L.; Wu, H.; Zhao, H.; Wang, L. Self-alignment of cationic graphene oxide nanosheets for anticorrosive reinforcement of epoxy coatings. Chemical Engineering Journal 2020, 389. [Google Scholar] [CrossRef]
- Hosseini, M.G.; Jafari, M.; Najjar, R. Effect of polyaniline–montmorillonite nanocomposite powders addition on corrosion performance of epoxy coatings on Al 5000. Surface and Coatings Technology 2011, 206, 280–286. [Google Scholar] [CrossRef]
- Guo, L.; Wu, W.; Zhou, Y.; Zhang, F.; Zeng, R.; Zeng, J. Layered double hydroxide coatings on magnesium alloys: A review. Journal of Materials Science & Technology 2018, 34, 1455–1466. [Google Scholar] [CrossRef]
- Schriver, M.; Regan, W.; Gannett, W.J.; Zaniewski, A.M.; Crommie, M.F.; Zettl, A. Graphene as a long-term metal oxidation barrier: worse than nothing. ACS nano 2013, 7, 5763–5768. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Lim, A.T.O.; Huang, J. A cautionary note on graphene anti-corrosion coatings. Nat Nanotechnol 2017, 12, 834–835. [Google Scholar] [CrossRef] [PubMed]
- Xi, K.; Wu, H.; Zhou, C.; Qi, Z.; Yang, K.; Fu, R.K.Y.; Xiao, S.; Wu, G.; Ding, K.; Chen, G.; et al. Improved corrosion and wear resistance of micro-arc oxidation coatings on the 2024 aluminum alloy by incorporation of quasi-two-dimensional sericite microplates. Applied Surface Science 2022, 585, 152693. [Google Scholar] [CrossRef]
- Abdullah, N.H.; Shameli, K.; Abdullah, E.C.; Abdullah, L.C. Low cost and efficient synthesis of magnetic iron oxide/activated sericite nanocomposites for rapid removal of methylene blue and crystal violet dyes. Materials Characterization 2020, 163. [Google Scholar] [CrossRef]
- Pan, X.F.; Gao, H.L.; Lu, Y.; Wu, C.Y.; Wu, Y.D.; Wang, X.Y.; Pan, Z.Q.; Dong, L.; Song, Y.H.; Cong, H.P.; et al. Transforming ground mica into high-performance biomimetic polymeric mica film. Nat Commun 2018, 9, 2974. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.; Shen, Y. Swelling of sericite by LiNO3-hydrothermal treatment. Applied Clay Science 2009, 43, 282–288. [Google Scholar] [CrossRef]
- Ding, H.; Wang, Y.; Liang, Y.; Qin, F. Preparation and Characterization of Cetyl Trimethylammonium Intercalated Sericite. Advances in Materials Science and Engineering 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Lalhmunsiama; Tiwari, D.; Lee, S.-M. Surface-functionalized activated sericite for the simultaneous removal of cadmium and phenol from aqueous solutions: Mechanistic insights. Chemical Engineering Journal 2016, 283, 1414–1423. [Google Scholar] [CrossRef]
- Lu, C.; Mai, Y.-W. Influence of Aspect Ratio on Barrier Properties of Polymer-Clay Nanocomposites. Physical Review Letters 2005, 95, 088303. [Google Scholar] [CrossRef]
- Sinha Ray, S.; Okamoto, M. Polymer/layered silicate nanocomposites: a review from preparation to processing. Progress in Polymer Science 2003, 28, 1539–1641. [Google Scholar] [CrossRef]
- Paul, B.; Martens, W.N.; Frost, R.L. Organosilane grafted acid-activated beidellite clay for the removal of non-ionic alachlor and anionic imazaquin. Applied Surface Science 2011, 257, 5552–5558. [Google Scholar] [CrossRef]
- Musso, T.B.; Parolo, M.E.; Pettinari, G.; Francisca, F.M. Cu(II) and Zn(II) adsorption capacity of three different clay liner materials. J Environ Manage 2014, 146, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Zhang, Z.; Luo, H.; Hu, B.; Dang, Z.; Yang, C.; Li, L. Adsorption of arsenic on modified montmorillonite. Applied Clay Science 2014, 97-98, 17–23. [Google Scholar] [CrossRef]
- Zhao, R.; Kong, W.; Sun, M.; Yang, Y.; Liu, W.; Lv, M.; Song, S.; Wang, L.; Song, H.; Hao, R. Highly Stable Graphene-Based Nanocomposite (GO-PEI-Ag) with Broad-Spectrum, Long-Term Antimicrobial Activity and Antibiofilm Effects. ACS Appl Mater Interfaces 2018, 10, 17617–17629. [Google Scholar] [CrossRef]
- Huang, X.; Yu, L.; Dong, Y. Corrosion resistance of a novel ceria doped aluminum phosphate ceramic coating on cast Al-Si alloy by steam-assisted curing. Corrosion Science 2021, 182, 109256. [Google Scholar] [CrossRef]
- Cui, X.-j.; Lin, X.-z.; Liu, C.-h.; Yang, R.-s.; Zheng, X.-w.; Gong, M. Fabrication and corrosion resistance of a hydrophobic micro-arc oxidation coating on AZ31 Mg alloy. Corrosion Science 2015, 90, 402–412. [Google Scholar] [CrossRef]
- Zoltowski, P. On the electrical capacitance of interfaces exhibiting constant phase element behaviour. Journal of Electroanalytical Chemistry 1998, 443, 149–154. [Google Scholar] [CrossRef]
- Wu, H.; Qasim, A.M.; Xiao, S.; Huang, Q.; Zhang, F.; Wu, Z.; Fu, R.K.Y.; Wu, G.; Chu, P.K. Magnetron-sputtered fluorocarbon polymeric film on magnesium for corrosion protection. Surface and Coatings Technology 2018, 352, 437–444. [Google Scholar] [CrossRef]
- Wu, H.; Xi, K.; Xiao, S.; Qasim, A.M.; Fu, R.K.Y.; Shi, K.; Ding, K.; Chen, G.; Wu, G.; Chu, P.K. Formation of self-layered hydrothermal coating on magnesium aided by titanium ion implantation: Synergistic control of corrosion resistance and cytocompatibility. Surface and Coatings Technology 2020, 401, 126251. [Google Scholar] [CrossRef]
- Wu, G.; Zhang, X.; Zhao, Y.; Ibrahim, J.M.; Yuan, G.; Chu, P.K. Plasma modified Mg–Nd–Zn–Zr alloy with enhanced surface corrosion resistance. Corrosion science 2014, 78, 121–129. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, C.; Wang, F.; Li, W. Electrochemical behavior of anodized Mg alloy AZ91D in chloride containing aqueous solution. Corrosion Science 2005, 47, 2816–2831. [Google Scholar] [CrossRef]
- Xin, Y.; Hu, T.; Chu, P.K. Degradation behaviour of pure magnesium in simulated body fluids with different concentrations of HCO3. Corrosion Science 2011, 53, 1522–1528. [Google Scholar] [CrossRef]
- Wan, H.; Song, D.; Li, X.; Zhang, D.; Gao, J.; Du, C. A new understanding of the failure of waterborne acrylic coatings. RSC advances 2017, 7, 38135–38148. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, B.; Liu, M.; Ge, Y. Corrosion characterization of micro-arc oxidization composite electrophoretic coating on AZ31B magnesium alloy. Journal of Alloys and Compounds 2015, 621, 53–61. [Google Scholar] [CrossRef]
- Wang, H.; Sun, T.; Chang, L.; Liu, F.; Liu, B.; Zhao, C.; Xue, X.; Xiong, X. Preparation of Ca doping ZrO 2 coating on NiTi shape memory alloy by cathodic plasma electrolytic deposition and its structure, in-vitro bioactivity and biocompatibility analysis. Surface and Coatings Technology 2017, 325, 136–144. [Google Scholar] [CrossRef]
- Wu, H.; Shi, Z.; Zhang, X.; Qasim, A.M.; Xiao, S.; Zhang, F.; Wu, Z.; Wu, G.; Ding, K.; Chu, P.K. Achieving an acid resistant surface on magnesium alloy via bio-inspired design. Applied Surface Science 2019, 478, 150–161. [Google Scholar] [CrossRef]
- Fan, X.; Yan, H.; Cai, M.; Song, S.; Huang, Y.; Zhu, M. Achieving parallelly-arranged Ti3C2Tx in epoxy coating for anti-corrosive/wear high-efficiency protection. Composites Part B: Engineering 2022, 231, 109581. [Google Scholar] [CrossRef]
Figure 2.
(a and b) XRD spectra of as-recieved and modified sericite; (d and e) SEM micrographs of SER (d) and PEI-SER (e), insets depict the magnified images of selected area; (f) SER and PEI-SER solution with concentration of 0.5 mg/mL after standing for 1 h; (g and h) Zeta potential distribution (g) and particle size distribution (h) of SER, CTAB-SER, and PEI-SER.
Figure 2.
(a and b) XRD spectra of as-recieved and modified sericite; (d and e) SEM micrographs of SER (d) and PEI-SER (e), insets depict the magnified images of selected area; (f) SER and PEI-SER solution with concentration of 0.5 mg/mL after standing for 1 h; (g and h) Zeta potential distribution (g) and particle size distribution (h) of SER, CTAB-SER, and PEI-SER.
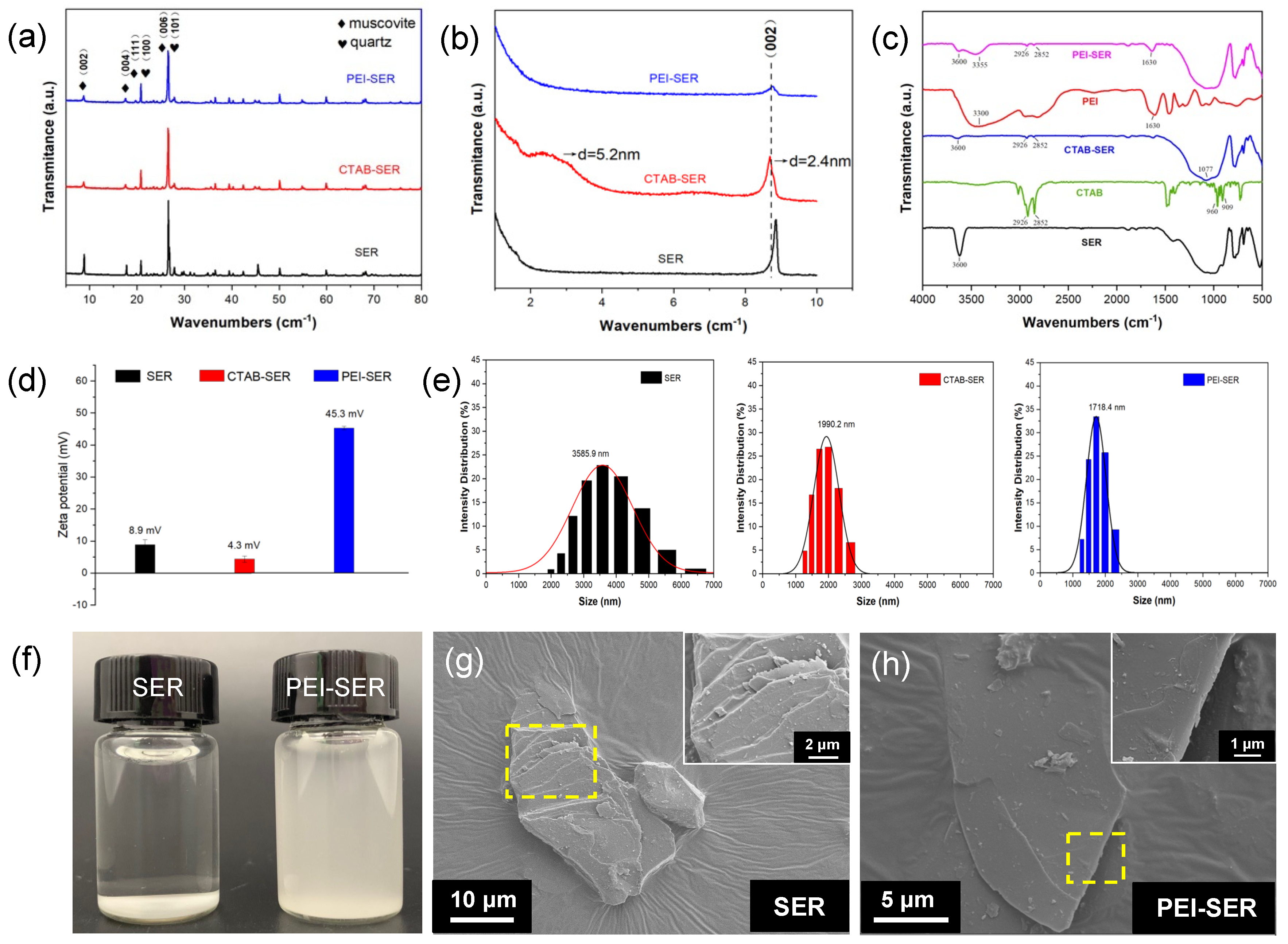
Figure 4.
Potentiodynamic polarization curves of the AZ31B alloy and coating samples after immersion in 3.5 wt.% NaCl solution for 1h; (b) Comparison of the corrosion potentials (Ecorr) and corrosion current densities (Icorr) calculated from the polarization curves.
Figure 4.
Potentiodynamic polarization curves of the AZ31B alloy and coating samples after immersion in 3.5 wt.% NaCl solution for 1h; (b) Comparison of the corrosion potentials (Ecorr) and corrosion current densities (Icorr) calculated from the polarization curves.
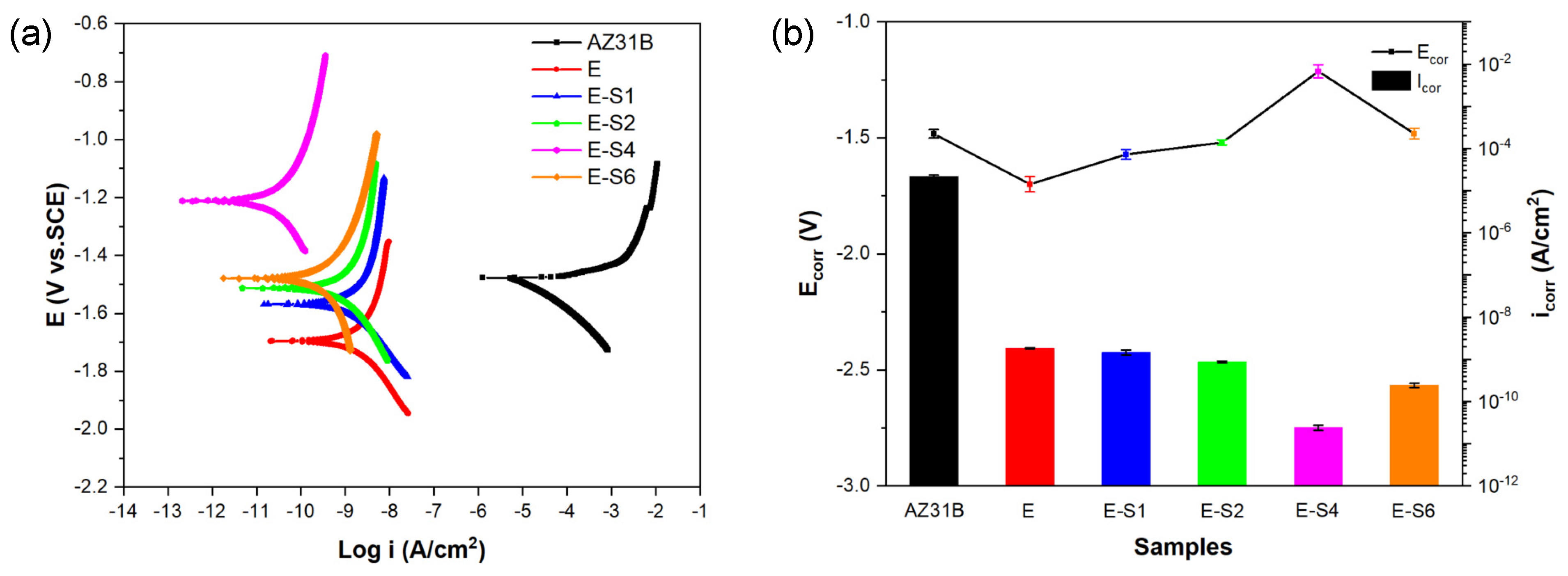
Figure 5.
(a-c) Nyquist, Bode-impedance and Bode-Phase plots of the AZ31B Mg substrate, E, E-S1, E-S2, E-S4 and E-S6 after immersion in 3.5 wt.% NaCl for 1 h.
Figure 5.
(a-c) Nyquist, Bode-impedance and Bode-Phase plots of the AZ31B Mg substrate, E, E-S1, E-S2, E-S4 and E-S6 after immersion in 3.5 wt.% NaCl for 1 h.
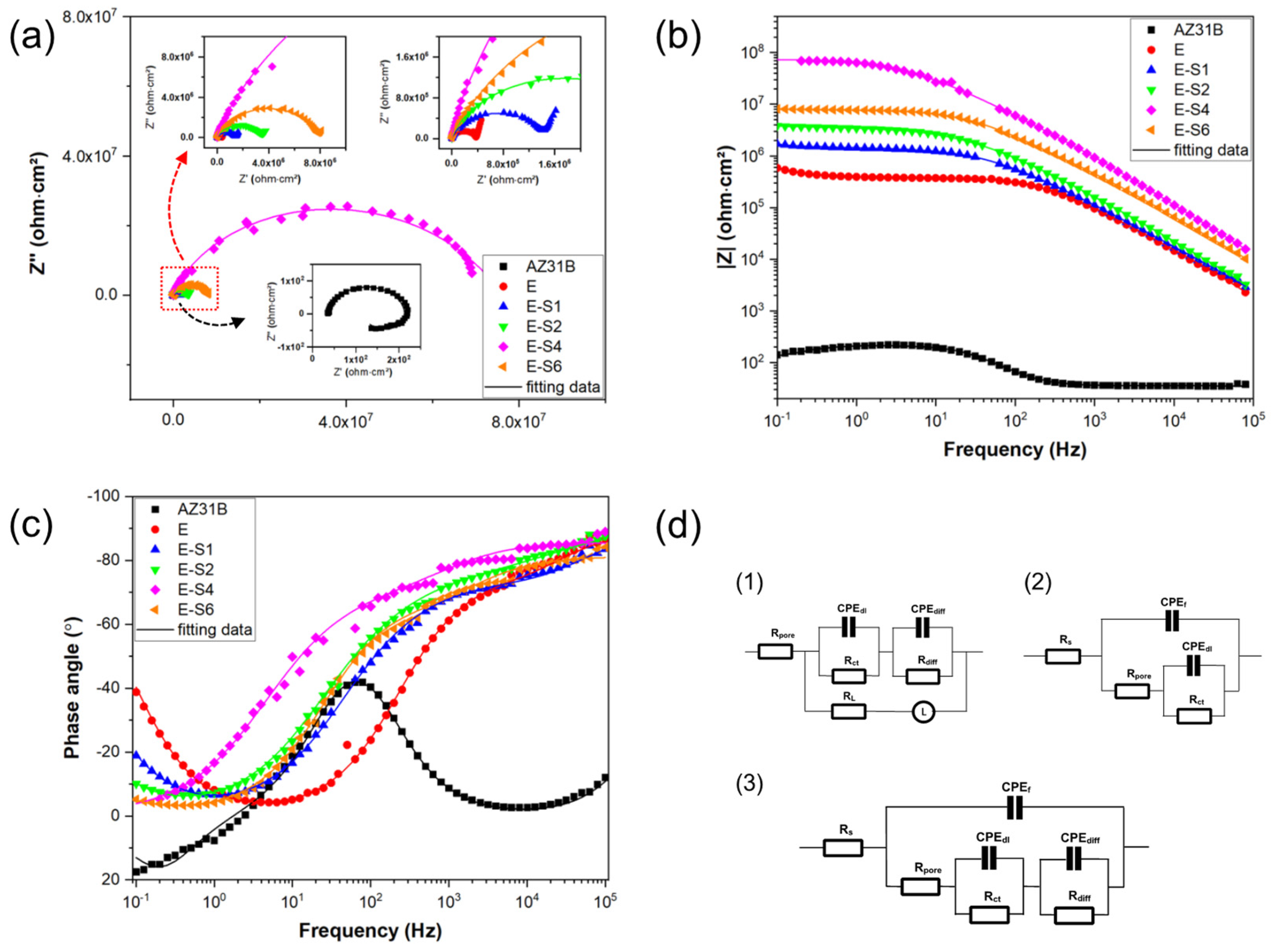
Figure 6.
(a, d, g, j, m) Nyquist plots, (b, e, h, k, n) Bode impedance plots, and (c, f, i, l, o) Bode phase angle plots of (a, b, c) E, (d, e, f) E-S1, (g, h, i) E-S2, (j, k, l) E-S4, and (m, n, o) E-S6 after immersion for different times.
Figure 6.
(a, d, g, j, m) Nyquist plots, (b, e, h, k, n) Bode impedance plots, and (c, f, i, l, o) Bode phase angle plots of (a, b, c) E, (d, e, f) E-S1, (g, h, i) E-S2, (j, k, l) E-S4, and (m, n, o) E-S6 after immersion for different times.
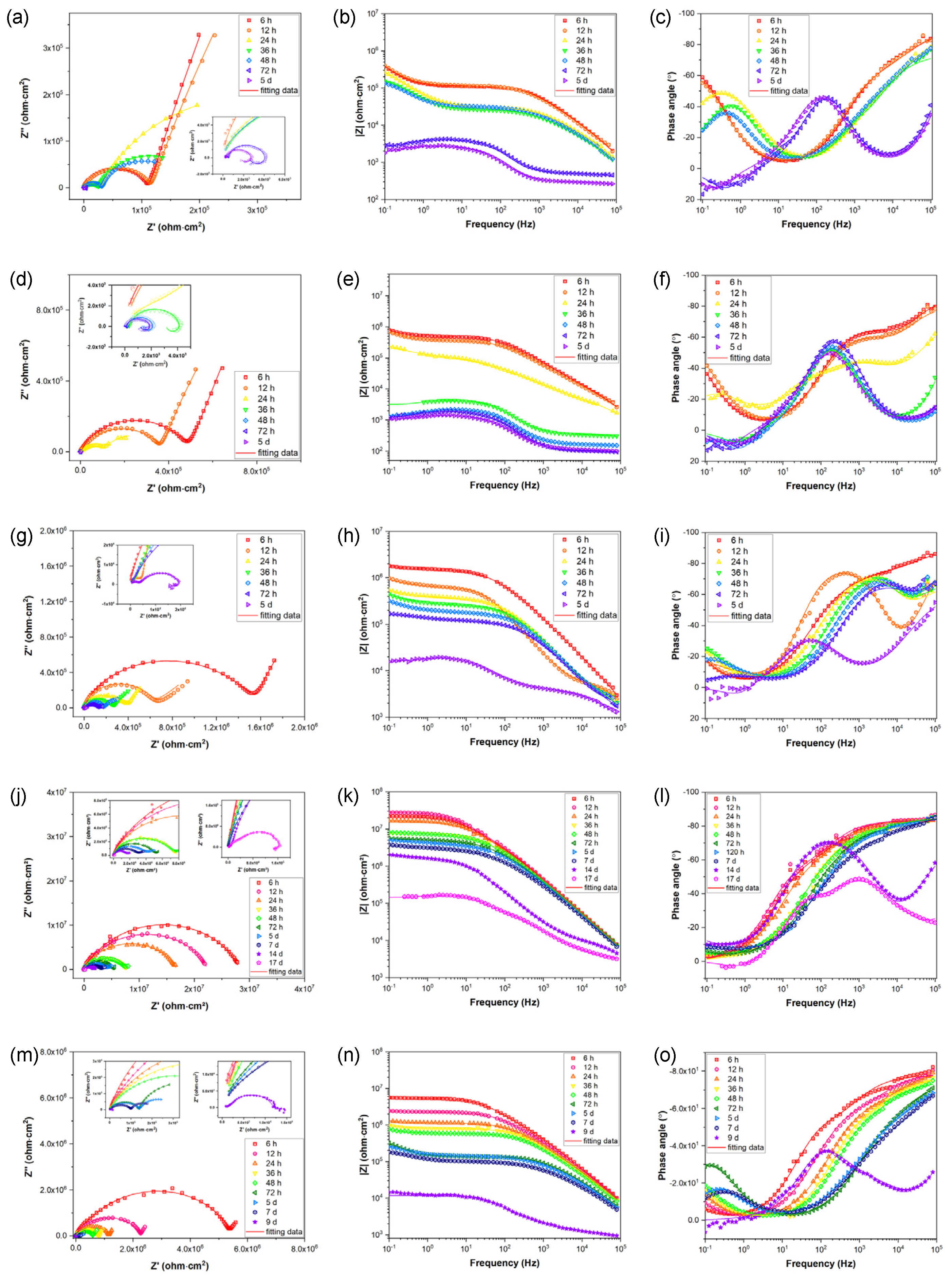
Figure 7.
The change of Rpore and |Z|0.1 Hz against different immersion time from 1h to 5 d.
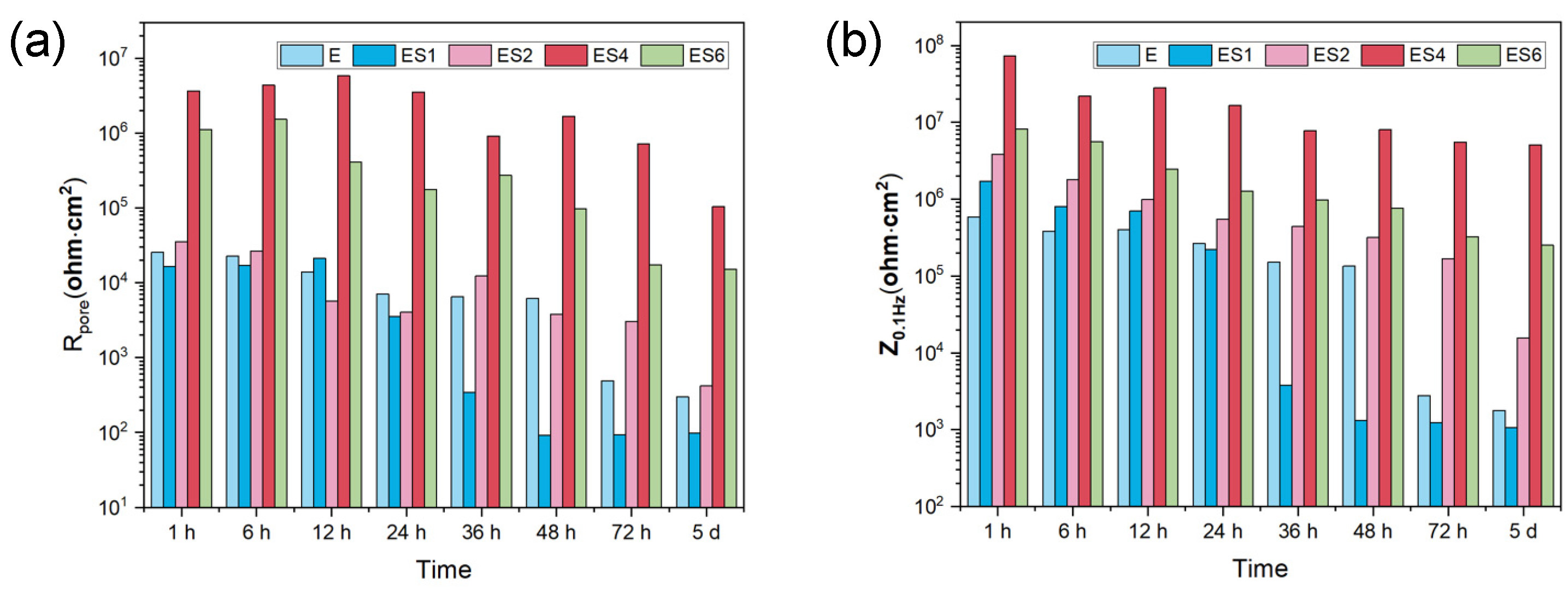
Figure 8.
Digital pictures taken from the AZ31B substrate, E, E-S1, E-S2, E-S4 and E-S6 after immersion for different time.
Figure 8.
Digital pictures taken from the AZ31B substrate, E, E-S1, E-S2, E-S4 and E-S6 after immersion for different time.
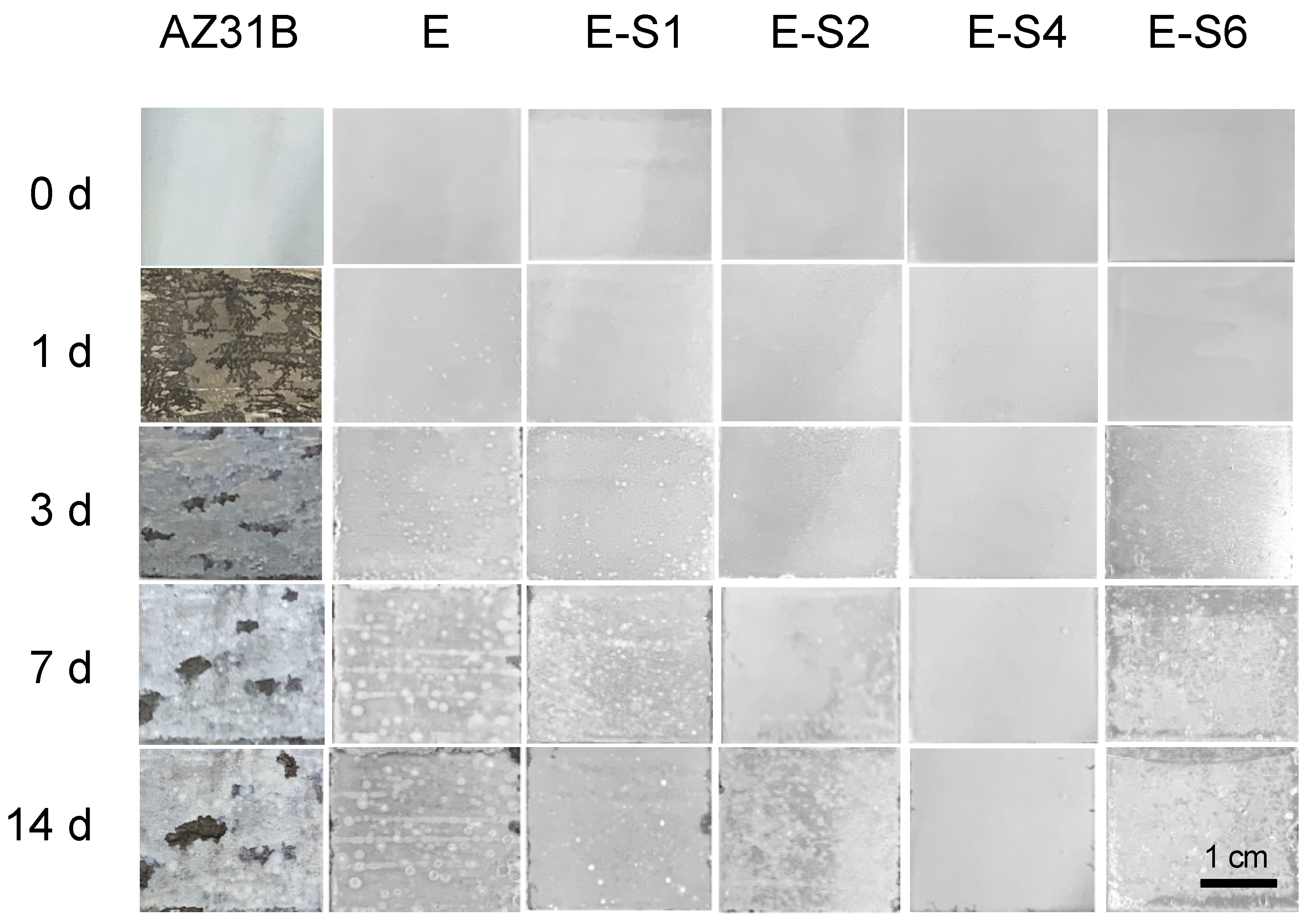
Figure 9.
Confocal laser scanning microscopy (CLSM) and 3D topography images after immersion for 14 days of the untreated and untreated AZ31B Mg samples in 3.5 wt.% NaCl.
Figure 9.
Confocal laser scanning microscopy (CLSM) and 3D topography images after immersion for 14 days of the untreated and untreated AZ31B Mg samples in 3.5 wt.% NaCl.

Figure 10.
GIXRD patterns of samples after immersion in 3.5 wt% NaCl for 14 days.
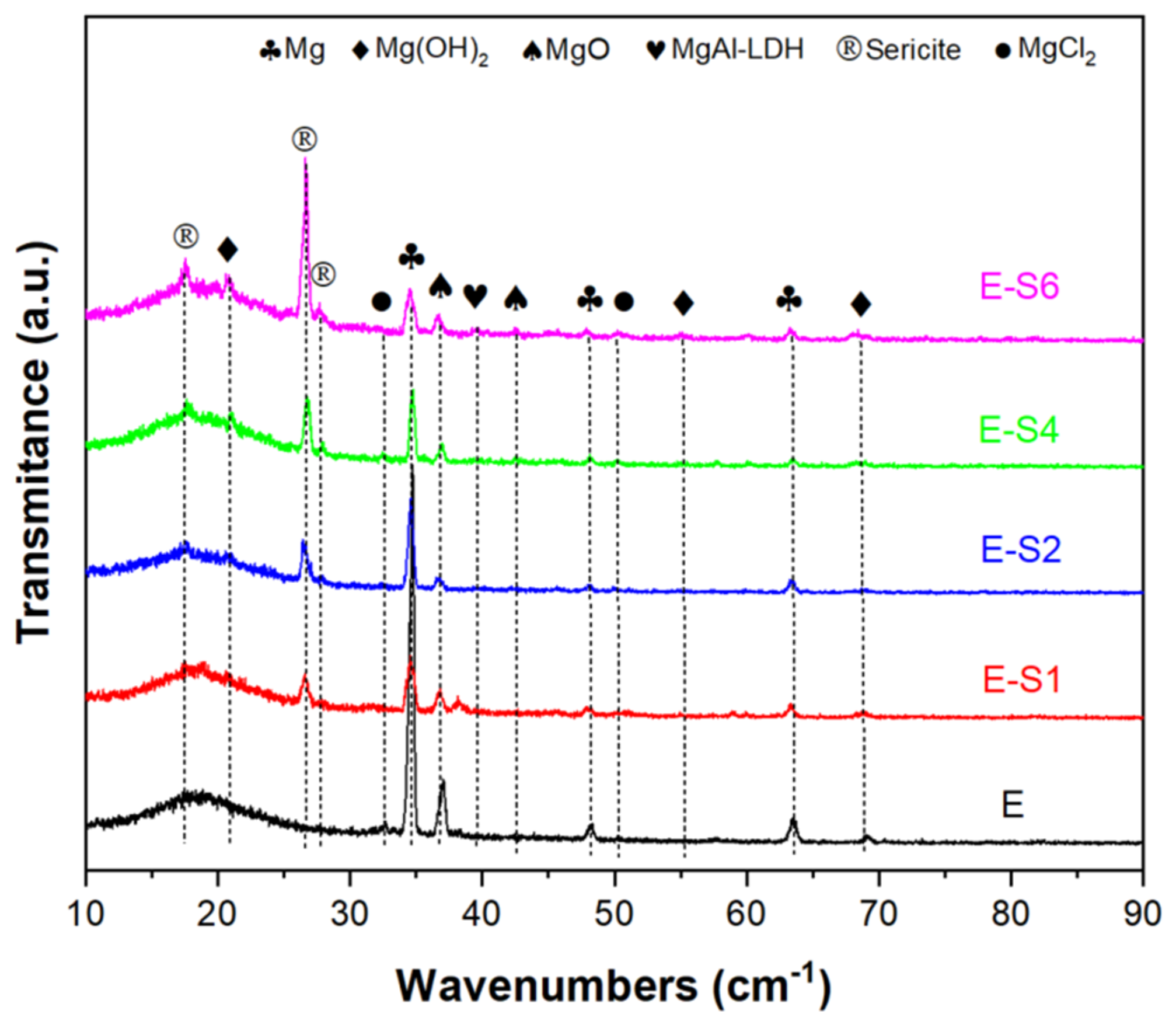
Figure 11.
SEM images and corresponding EDS results of (a-c) AZ31, (d-f) E, (g-i) E-S1, (j-l) E-S2,(m-o) E-S4 and (p-r) E-S6 after immersion in 3.5 wt% NaCl of 14 days.
Figure 11.
SEM images and corresponding EDS results of (a-c) AZ31, (d-f) E, (g-i) E-S1, (j-l) E-S2,(m-o) E-S4 and (p-r) E-S6 after immersion in 3.5 wt% NaCl of 14 days.

Figure 12.
Cross-sectional SEM images and corresponding EDS mapping of (a) E, (b) E-S1, (c) E-S2, (d) E-S4, and (e) E-S6 after immersion in 3.5 wt% NaCl for 14 days.
Figure 12.
Cross-sectional SEM images and corresponding EDS mapping of (a) E, (b) E-S1, (c) E-S2, (d) E-S4, and (e) E-S6 after immersion in 3.5 wt% NaCl for 14 days.
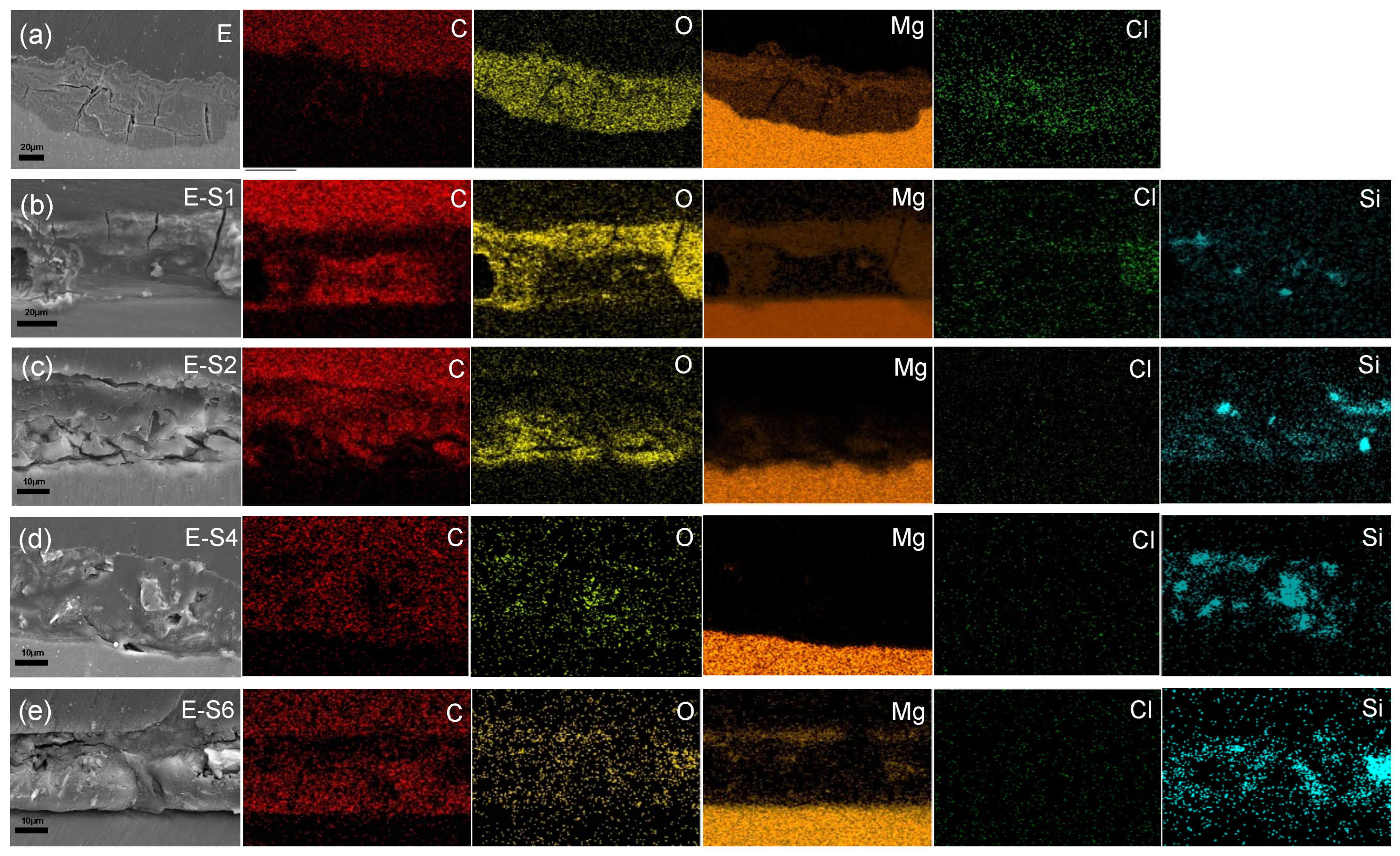
Figure 13.
Schematic diagram of anti-corrosion mechanism: (a-c) different stages of the corrosion process of bare epoxy coating; (d) the anti-corrosion mechanism of the uniform-oriented sericite nanosheets in epoxy coating.
Figure 13.
Schematic diagram of anti-corrosion mechanism: (a-c) different stages of the corrosion process of bare epoxy coating; (d) the anti-corrosion mechanism of the uniform-oriented sericite nanosheets in epoxy coating.
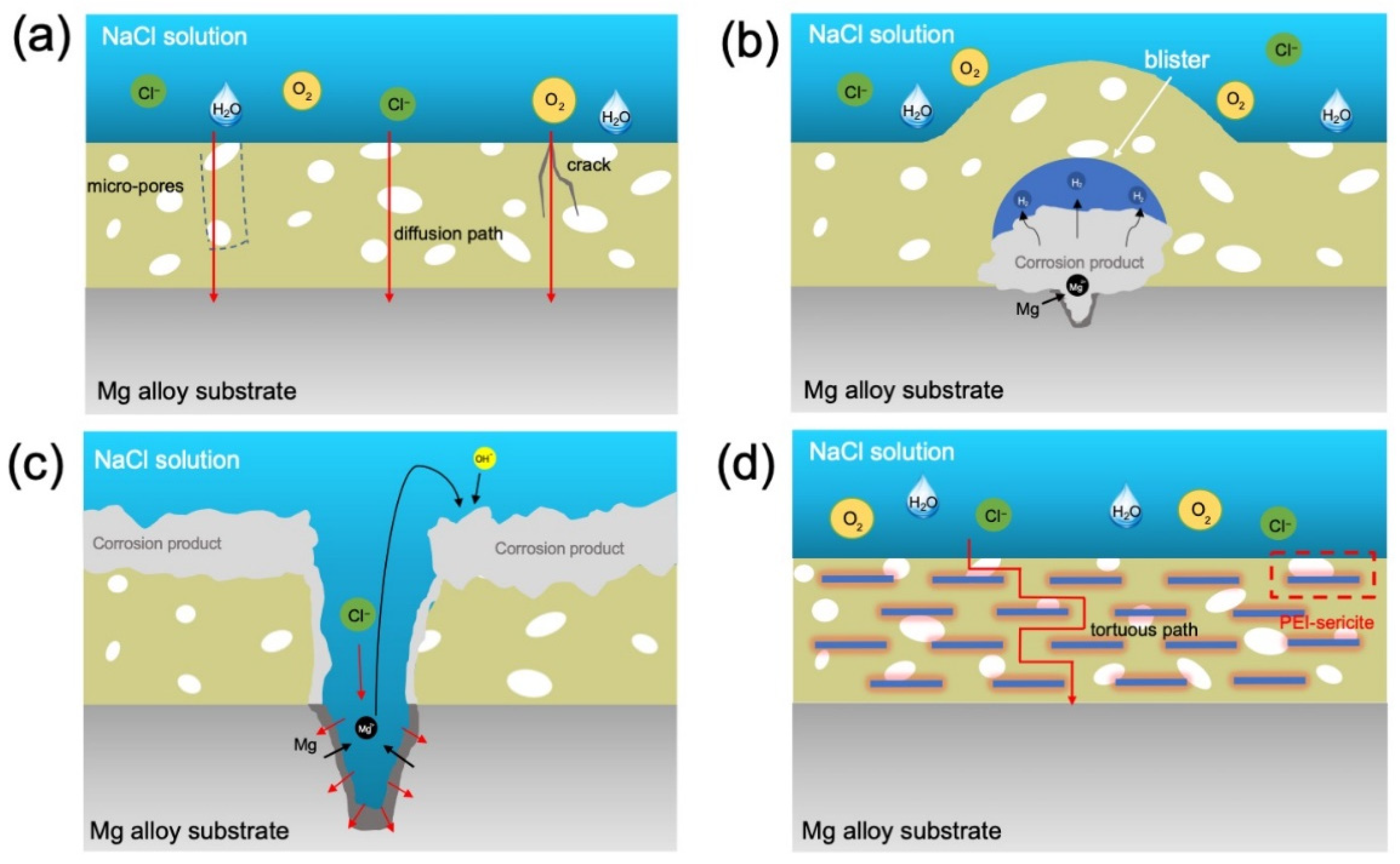

Table 1.
Ecorr, icorr, and βc of different samples in 3.5 wt% NaCl calculated from the polarization curves.
Table 1.
Ecorr, icorr, and βc of different samples in 3.5 wt% NaCl calculated from the polarization curves.
| Sample | Ecorr(V vs.SCE) | icorr(A*cm-2) | βc(V/decade) |
|---|---|---|---|
| AZ31B | -1.483 ± 0.018 | (2.153 ± 0.19) × 10-5 | -0.138 ± 0.006 |
| E | -1.700 ± 0.032 | (1.831 ± 0.05) × 10-9 | -0.216 ± 0.009 |
| E-S1 | -1.571 ± 0.021 | (1.464 ± 0.20) × 10-9 | -0.196 ± 0.008 |
| E-S2 | -1.520 ± 0.011 | (8.723 ± 0.43) × 10-10 | -0.238 ± 0.001 |
| E-S4 | -1.214 ± 0.028 | (2.412 ± 0.35) × 10-11 | -0.260 ± 0.002 |
| E-S6 | -1.482 ± 0.023 | (2.435 ± 0.29) × 10-10 | -0.247 ± 0.002 |
Table 2.
Fitted EIS results of different samples after immersion in 3.5 wt% NaCl solution for 0.5 h based on the corresponding equivalent circuit models.
Table 2.
Fitted EIS results of different samples after immersion in 3.5 wt% NaCl solution for 0.5 h based on the corresponding equivalent circuit models.
| AZ31B | E | E-S1 | E-S2 | E-S4 | E-S6 | |
|---|---|---|---|---|---|---|
| Equivalent Circuit | R(((QR)(QR))RL) | R(Q(R((QR)(QR)))) | R(Q(R((QR)(QR)))) | R(Q(R((QR)(QR)))) | R(Q(R((QR)(QR)))) | R(Q(R((QR)(QR)))) |
| Rs (ohm·cm2) | 15.60 | 11.88 | 12.95 | 10.46 | 14.60 | 4.88 |
| Y0 f (ohm-2·cm-2·S-n) | - | 9.72×10-10 | 6.36×10-10 | 5.94×10-10 | 2.35×10-10 | 6.73×10-10 |
| nf | - | 0.9702 | 1 | 1 | 0.9529 | 0.9040 |
| Rpore (ohm·cm2) | - | 1.57×104 | 1.64×104 | 3.56×104 | 3.64×106 | 1.12×106 |
| Y0 dl (ohm-2·cm-2·S-n) | 2.78×10-8 | 1.29×10-8 | 1.26×10-8 | 1.00×10-8 | 1.35×10-9 | 2.13×10-9 |
| ndl | 1 | 0.6781 | 0.6937 | 0.6506 | 0.6338 | 0.7179 |
| Rct (ohm·cm2) | 20.23 | 1.55×106 | 2.81×106 | 3.61×106 | 7.27×107 | 6.83×106 |
| Y0 diff (ohm-2·cm-2·S-n) | 4.65×10-5 | 2.61×10-6 | 2.654×10-6 | 2.417×10-6 | - | 2.29×10-6 |
| ndiff | 0.9449 | 0.9553 | 0.8420 | 0.8556 | - | 0.8048 |
| Rdiff (ohm·cm2) | 179.6 | 2.48×106 | 9×107 | 2.13×107 | - | 1.32×107 |
| L (H) | 252.2 | - | - | - | - | - |
| RL (ohm·cm2) | 223.1 | - | - | - | - | - |
| χ2 | 1.39×10-3 | 7.86×10-4 | 6.74×10-4 | 7.48×10-4 | 1.67×10-3 | 5.34×10-4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



