
Submitted:
06 June 2023
Posted:
07 June 2023
You are already at the latest version
Abstract
Background: Liquid biopsies are revolutionary tools to detect tumor-specific genetic alterations in body fluids, and cell-free DNA (cfDNA) can be used for molecular diagnosis in cancer patients. In brain tumors cerebrospinal fluid (CSF) cfDNA might be more informative than plasma cfDNA. Here, we assess the use of CSF cfDNA in pediatric embryonal brain tumors (EBT) for molecular diagnosis.
Methods: CSF cfDNA of pediatric patients with medulloblastoma (n=18), ATRT (n=3), ETMR (n=1), CNS NB FOXR2 (n=2) and pediatric EBT NOS (n=1) (mean cfDNA concentration 48 ng/mL; range 4-442 ng/mL) and matched tumor genomic DNA were sequenced by WES, and/or a targeted sequencing approach, to determine single nucleotide variations (SNVs)/copy number alterations (CNA). A specific capture covering transcription start sites (TSS) of genes of interest was used for nucleosome footprinting in CSF cfDNA.
Results: 15/25 CSF cfDNA samples yielded informative results, with informative CNA and SNVs in 11 and 15 cases, respectively. For cases with paired tumor and CSF cfDNA WES (n=15), a mean of 83 (range 1-160) shared SNVs was observed, including SNVs in classical medulloblastoma genes such as SMO and KMT2D. Interestingly, tumor-specific SNVs (mean 18; range 1-62) or CSF-specific SNVs (mean 5; range 0-25) were observed suggesting clonal heterogeneity. The TSS panel resulted in differential coverage profiles across all 112 studied genes in 7 cases, indicating distinct promoter accessibility.
Conclusion: CSF cfDNA sequencing yielded informative results in 60% of cases (15/25), with WES feasible in 83% (15/18). These results pave the way for implementation of these novel approaches for molecular diagnosis and minimal residual disease monitoring.
Keywords:
Pediatric embryonal brain tumors
; liquid biopsy
; cell-free DNA
; molecular diagnosis
; nucleosome footprinting
1. Introduction
Pediatric central nervous system (CNS) tumors are the most common solid tumors in children and comprise 15% to 20% of all malignancies in children. Among these, embryonal brain tumors (EBT) represent a complex group, characterized by high aggressivity, including entities such as medulloblastoma (MB), atypical teratoid rhabdoid tumor (ATRT), and embryonal tumors with multilayered rosettes (ETMR), as well as recently described embryonal tumors with FOXR2 activation or BCOR alteration [1,2,3].
MB, the most prevalent EBT, constitutes a heterogeneous disease with four main groups associated with variable outcomes [4]. The combination of histological and molecular features is now systematically used in the clinical management of patients with MB. WNT MB are mostly characterized by CTNNB1 or APC mutations and monosomy of chromosome 6; Sonic hedgehog (SHH) MB can harbor genomic alterations in several genes including PTCH1, SUFU, MYCN, ELP1 and TP53; group 3 and group 4 MBs have few specific genomic alterations, but MYC amplification in group 3 MB is an important prognostic marker associated with a poor outcome [5].
ATRT is the second most frequent EBT; this aggressive disease occurs in early childhood either in infra- or supra-tentorial regions. The genomic hallmark of ATRT is the biallelic inactivation of SMARCB1 with an otherwise extremely stable genome, whereas three epigenetically defined subtypes have been identified [6]. The SHH ATRT subgroup is characterized by an overexpression of GLI2, TYR ATRT are characterized by an overexpression of several melanosomal markers and OTX2, and MYC ATRT by an overexpression of the MYC oncogene.
The very aggressive ETMR are supra- or infra-tentorial tumors of early childhood, features shared with the also aggressive CNS BCOR-ITD; while the former are characterized by the amplification of a miRNA cluster on chromosome 19 (C19MC amplification), the latter harbor an internal tandem duplication of the BCOR gene. CNS Neuroblastoma FOXR2 activated tumors show complex rearrangements of the FOXR2 locus.
Finally, other rare EBT, defined by their specific histopathological features together with their genomic, expression-based or methylation-based profiling, have also been described. Other rare EBT, NEC (not elsewere classified) are defined by the absence of diagnostic criteria qualifying histologically and molecularly defined CNS EBT [4,7].
Tumor characterization based on pathological and molecular analyses of primary tumor tissue obtained at diagnosis by frontline surgery, or biopsy, is of high importance in determining the exact diagnosis and risk group for affected patients. Indeed, several of these genetic alterations such as SMARCB1 inactivation, BCOR ITD, or amplification of C19MC are considered to be specific enough to allow a definitive diagnosis [3]. Multi-omic profiling might also contribute to the identification of predictive biomarkers and to the introduction of much-needed novel targeted therapies in relapse and, importantly, upfront novel treatment approaches [8,9].
However, lack of a sufficient tumor sample, low quality nucleic acid content following formalin fixation of a tumor sample, or the presence of non-tumor tissue can create inaccuracies in tumor classification and hamper detailed molecular explorations; rarely, the neurosurgical procedures (resection or biopsy) are deemed too risky for the patient, and for some patients, treatment may start without a definitive documented diagnosis based on tissue sampling. There is an ongoing need to improve the approaches for further characterization of EBT at diagnosis and to document molecular alterations in case of relapse.
Liquid biopsies represent recent, noninvasive methods to characterize tumors. The study of cell-free DNA (cfDNA), small DNA fragments released into either the blood stream or other body fluids from cells undergoing apoptosis, necroptosis or other cellular degradation processes, has led to the recent development of revolutionary tools for molecular diagnosis. The study of circulating tumor DNA (ctDNA), a variable fraction of the overall cfDNA, extracted from blood plasma is rapidly moving into standard clinical care for cancer patients. For patients with brain tumors, varying amounts of ctDNA have been demonstrated in cfDNA extracted from plasma, and it is thought that the blood-brain barrier might limit the detection of tumor-specific DNA fragments in the blood stream [10]. Importantly recent reports have indicated that cfDNA can be extracted from cerebrospinal fluid (CSF), and that significant amounts of ctDNA can be detected in patients with different types of brain tumors, including pediatric EBT [2,11,12,13,14]. However, to date only limited data has been reported on the use of cfDNA extracted from CSF for the complete molecular characterization of pediatric EBT [12,13,15].
In addition to genetic analysis, cfDNA can also enable inference of epigenetic features. Methylation data has been generated from CSF cfDNA of brain tumor patients [16]. Recent approaches have explored the feasibility of the study of nucleosome footprints in cfDNA, based on the coverage in whole genome sequencing (WGS) data around nucleosome positions. It is hypothesized that a dense occupation by nucleosomes, corresponding to silencing of expression at transcription start sites (TSS), might protect DNA from degradation by nucleases upon release from cells into body fluids, and that upon WGS across TSS the resulting coverage patterns might identify the tissue of origin and prediction of expression patterns [17,18,19,20].
Altogether, liquid biopsies are revolutionary tools to detect tumor-specific genetic and epigenetic alterations in body fluids. Here, we assess whether the cfDNA in CSF could be used for molecular diagnosis of pediatric EBT.
2. Patients and Methods
2.1. Patients
Patients with embryonal brain tumors treated at Institut Curie or Centre Leon Berard (CLB) were included in this study if CSF was available for extraction of cfDNA (Supplementary Table S1). Twenty-five cases were identified. The CSF samples were obtained at diagnosis for all patients but one for whom the sample was obtained at relapse. Treatment was given according to national or international protocols according to the disease type. For all patients, written informed consent was obtained from parents/guardians according to national law. In addition to clinical molecular analyses, comprehensive molecular characterization was performed following inclusion in the national MICCHADO (NCT03496402) or MAPPYACTS studies (NCT02613962) [8] in 2 and 3 cases, respectively. This study was approved by the Institutional Review Board of Institut Curie (Reference DATA210043).
2.2. Sample Collection and Processing
For each patient, genomic DNA was extracted from a tumor sample obtained during surgery, or biopsy, at diagnosis or disease recurrence according to standard procedures. CSF was obtained during a clinically indicated lumbar puncture, with a mean delay between surgery and lumbar puncture of 17.9 days (range: 3 days before to 67 days after surgery). In 1 case (patient 8), CSF was obtained after the first two cycles of chemotherapy. Cytological analysis revealed the presence of tumor cells in the CSF in 5/25 samples. For cfDNA studies, 5-6 droplets (>300 µl) of CSF were prepared by centrifugation at 2,000 rpm for 10 minutes followed by careful aliquoting of the supernatant and freezing at −80°C within 1 to 24 hours after collection. Germline DNA extracted from blood leucocytes was available in 6 cases [21].
2.3. Molecular Analysis of Primary Tumors
Clinical molecular diagnosis of primary pediatric EBT involved array-CGH, targeted sequencing and/or Nanostring profiling, according to the tumor entity, as reported [22,23,24]. Whole-exome sequencing (WES) of genomic DNA extracted from primary tumors, and paired germline genomic DNA, was performed in 18 and 6 patients, respectively (Supplementary methods) [8].
2.4. cfDNA Extraction, Library Construction and Whole Exome Sequencing of cfDNA
cfDNA was extracted from approximately 500 µl (5-6 droplets) of CSF using QIAamp Circulating Nucleic Acid Kit (Qiagen) [25]. The total cfDNA concentration per mL of CSF was calculated. cfDNA sequencing libraries were constructed without fragmentation. The Medexome Enrichment Kit (Nimblegen Roche Sequencing) was used for whole exome capture. Paired-end (100bp) Illumina™ WES was performed (expected coverage of 100×; Supplementary methods).
2.5. Targeted Sequencing Panel Design
For custom deep coverage targeted capture sequencing to infer expression based on the coverage around the TSS, a panel was designed to cover EBT-relevant genes and their TSS. One hundred and twelve genes which contribute to the genetic classification of embryonic tumors such as MYC (in MB and ATRT MYC subtype), MYCN (in MB and ATRT SHH subtype), TYR (in ATRT TYR subtype) or JAK3 are used for a diagnostic Nanostring panel design at Institut Curie [7,26,27]. The regions (+/- 1 kb) surrounding the TSS of these genes were included in the design. In addition, the coding sequences of six genes (CTNNB1, PTCH1, SMARCB1, SMARCA4, SMO, and SUFU) were added, as well as five glioma hotspots mutations (H3F3A_K27M, HIST1H3B_K27M, BRAF_V600E, IDH1_R132C and IDH2_R172K) [28,29]. For the prediction of expression, eight TSS were selected as controls : 1) 4 ubiquitously expressed genes with strong or moderate expression (ACTB, B2M, GAPDH or SDHA431) 2) 2 genes not expressed in the central nervous system (BAGE4 and ACPT), and 3) 2 genes expressed in the CNS (SLC32A1 and CNTNAP2) were selected from the Protein Atlas database [30,31] This panel design, referred to as the targeted sequencing TSS panel, was used on CSF samples, sequencing to a theoretical coverage of 500x.
2.6. Bioinformatics Analysis:
2.6.1. Copy Number and SNV Analysis
The WES and target sequencing raw reads were mapped to the reference human genome assembly GRCh37/hg19 using BWA v0.7.15, with default parameters, and coverage scores were computed [32]. Variants were called with GATK 4.1.7.0 / Mutect2, annotated, and further filters applied prior to IGV visual inspection (see supplementary methods for details). Copy number analyses were performed with snp-pileup_0.5.14 and FACETS v0.5.11.
2.6.2. Sequencing across the TSS to Infer Expression Profiles
The bigwig files containing the coverage scores representing the number of reads per bin size of 1 were generated with the option BAM coverage using BAM files [32].
Matrices with mean score per genomic region were computed. The window capture of the TSSs had a size of 2 kb around the TSS (-1 kb downstream and +1 kb upstream of the TSS) to include the nucleosome depleted region (NDR). K-means clustering using deeptools, with the number of clusters set to 2, was used to classify genes based on the coverage across the TSS [18,19].
2.6.3. Sequencing Coverage, Quality Statistics and Data Availability
For WES and targeted sequencing panel analysis, the sequencing coverage and quality statistics for each sample are summarized in Supplementary Tables S2 and S3, respectively. The reference genome assembly GRCh37 was used to map the reads. Sequencing data (.vcf files) will be made available upon reasonable request.
3. Results
Tumor samples and CSF from 25 pediatric EBT patients were analyzed in this study: medulloblastoma (n=18), ATRT (n=3), ETMR (n=1), CNS NB FOXR2 (n=2) and pediatric EBT NOS (n=1) (supplementary Table S1).
3.1. cfDNA Concentrations
Following cfDNA extraction from CSF, variable cfDNA concentrations were obtained (mean 48 ng/ml of CSF; range 4 – 442 ng/ml). cfDNA concentrations were independent of the tumor type, and no correlation between cfDNA concentration and disease status (localized versus metastatic disease), the presence/absence of a postsurgical residue, or the delay between surgery and CSF sampling was observed (supplementary Figure S1). The extracted cfDNA was then used for molecular characterization by different sequencing approaches.
3.2. Determining Informative Sequencing Approaches and ctDNA Content in CSF cfDNA
Primary tumor DNA was sequenced by targeted sequencing using an in-house diagnostic panel in seven cases (combined with aCGH), WES in 17 cases, and both approaches in one case, with paired germline available for WES in six cases. These approaches identified large-scale CNA in tumor DNA in all but one case, and SNVs in all cases (Figure 1, supplementary Table S1). Thus, primary tumor analysis was informative in all cases. For cfDNA analysis, sequencing results were considered informative in case of identification of at least one CNA or SNV. cfDNA extracted from CSF was sequenced by WES in 10 cases, using the TSS target panel in seven cases, or both approaches in eight cases. These approaches identified CNAs and SNVs in 11 cases, and SNVs only in 4 additional cases (Figure 1, supplementary Table S1). In 10 CSF cfDNA samples (the 7 samples analyzed by targeted sequencing only, and 3 samples by WES, with very low coverage), no CNA nor SNV could be detected.
The fraction of ctDNA in the CSF cfDNA was determined in 6 patients for whom germline DNA was available, with a mean tumor content of 60% in the tumor samples (range 29 – 93%), and a mean ctDNA content in the CSF cfDNA of 30% (range < 15% - 86%). The highest ctDNA content was observed in the ETMR case.
3.3. Diagnostic Genetic Alterations Can Be Identified in CSF cfDNA
Next we sought to determine whether molecular genetic alterations routinely used as typical diagnostic or subgrouping markers could also be detected in the CSF cfDNA. In 3/3 WNT MBs we identified the typical subgroup driver mutations, i.e. two CTNNB1 heterozygous mutations (c.100G>A/p.(Gly34Arg) and c.98C>G/p.(Ser33Cys)) and one APC homozygous mutation (c.3758del/p.(Ser1253Leufs*12)). In 2/3 ATRT the SMARCB1 mutation and/or deletion was found in CSF cfDNA. However, in 1/3 ATRT case, CSF cfDNA analysis did not detect the previously identified SMARCB1 mutation, and in 1 MB, 1 NB-FOXR2 and 1 EBT NOS, CSF cfDNA analysis did not enable detection of typical PTCH1, PTPRK and TP53/KRAS mutations (Table 1). The typically diagnostic C19MC amplification was identified from CSF cfDNA in 1/1 ETMR, and the suggestive 1q gain in 1/2 NB-FOXR2. Altogether, in 11/25 cases diagnostic SNV or CNA could be identified in CSF cfDNA.
3.4. Further Molecular Characterization: Copy Number Profiling
In the 11 cases with informative copy number profiles in both the primary tumor and CSF cfDNA samples, comparison of CNA between the primary tumor and the cfDNA showed an excellent concordance (Figure 2), including amplifications of MYCN, a region on chromosome 20 and a region on chromosome 19 (n=4), homozygous deletions, or heterozygous copy number losses or gains. CNA detected only in the primary tumor were seen in four cases and involved heterozygous copy number changes (loss of chromosome 6, deletions of chromosome 1p, 10q, 16q, gain of chromosome 7, 8q, 10p).
3.5. Further Molecular Characterization: SNVs
In 15 cases with informative SNV analysis in the primary tumor and CSF cfDNA, comparison of the variant allele frequency (VAF) of SNVs showed a good correlation (r=0.5, p<0.0001 ; Figure 3). However, in some cases, SNVs were only seen in the tumor but not in the corresponding CSF cfDNA, for instance ROBO1, APC, PIK3CA or ARID1A. On the other hand, in other cases SNVs in genes of interest were detected only in the CSF cfDNA but not in the primary tumor, including SNVs in the genes BCORL1 or NCOR1.
3.6. Intratumor Heterogeneity
Taking into account all SNVs observed among the 15 cases with informative SNV analysis in the primary tumor and CSF cfDNA by WES, a mean of 83 (range 1-60) SNVs were observed in common between the tumor and CSF cfDNA, with 5 (range 0-25) and 18 (range 1-62) seen only in the CSF cfDNA and tumor, respectively, indicating clonal heterogeneity in all EBT types (Figure 4). Importantly, copy number analyses also indicated heterogeneity, with a MYC amplification seen in the CSF cfDNA but not the primary tumor, and later emerging at the time of relapse (patient 17; supplementary Figure S2).
3.7. Inferring Expression Patterns for Tumor Classification Based on the Coverage around the TSS
To study nucleosome footprints of TSS in the cfDNA samples, the targeted sequencing TSS panel was used for sequencing CSF cfDNA in 15 cases. The observed mean coverage across the targeted sites was 194x (range 4–628). Interestingly, different profiles of coverage across the TSS were observed, ranging from a uniform coverage across the entire captured region, to a dip in coverage over the nucleosome depleted region (NDR).
For seven samples, the coverage profile clearly distinguished two different patterns, suggesting 2 distinct states of nucleosome occupancy (Figure 5A). In these seven samples, where the concentration of cfDNA was > 10 ng/mL (Figure 5C), TSS clustering was deemed informative for downstream analysis.
In eight other samples, these two distinct patterns could not be clearly distinguished (Figure 5B). Seven of these samples had a library size below 1 million reads. For all eight samples, the concentration of cfDNA was < 10 ng/mL suggesting that the concentration of cfDNA may highly impact the TSS footprint analysis and clustering (Figure 5C). These eight cases were deemed not informative for TSS clustering.
We then performed a global heatmap on the NDR regions of the 112 genes for which we captured the TSS. This enabled hierarchical clustering on the genes. The heatmap shows differences on coverage around the TSSs across the seven samples (Figure 5D), reflecting differences in nucleosome occupancy and identifying genes that might be "expressed" as opposed to others that might be "silent". Among all seven informative samples nearly all genes had variations in the TSS coverage profiles, with an identical coverage only observed for one gene.
TSS clustering of the different samples, taking into account the disease type, showed that two out of three MB samples clustered together, whereas other disease types were more dispersed (supplementary Figure S2).
For downstream analysis, we focused on the set of genes for which the coverage scores were consistently close between ATRT cases and different from MB cases, and vice versa, to determine a subset of genes for which TSS nucleosome occupancy might be "ATRT specific" and "MB specific". To achieve this, the coverage normalized scores (row z-score), identifying a threshold that allows to classify the genes into "expressed" and "silent", was determined. With scores ranging from -1.83 to 2.24, values under 0 are considered expressed values, and over 0 are considered silent. This definition of TSS coverage specific to ATRT or MB identified 46 genes with a clear difference between the disease types ATRT and MB (Figure 5E). Six of these genes show a NDR coverage profile suggesting gene expression in MB but not in ATRT (PDLIM3, RELL1, EMX2, UNC5D, FOXN3, BAGE3), with 40 others showing a NDR coverage suggesting gene expression in ATRT but not MB. However, no correlation with expression data determined by Nanostring was observed.
Altogether, somatic genetic alterations (SNVs and/or CNA) could be observed in 15/25 CSF cfDNA samples, and specific nucleosome footprints were observed in 7/15 cases.
4. Discussion
Molecular characterization is an integral part of diagnostic procedures of CNS EBT [3,4]. Nevertheless, in many instances the quantity of available tumor tissue is limited. Furthermore, modifications in genetic and epigenetic profiles might occur under therapy, with only limited possibilities of exploration.
Liquid biopsies are revolutionary tools to detect tumor-specific genetic alterations in body fluids, and cell-free DNA (cfDNA) can be used for molecular diagnosis, the follow-up of disease burden, or study of clonal evolution in patients with different cancers, including pediatric solid tumors such as neuroblastoma, rhabdomyosarcoma or other solid malignancies [8,25]. In EBT, to date only few studies address the feasibility and clinical utility CSF cfDNA [15,33,34,35]. In particular, low coverage WGS has been used for calling of tumor-specific CNA at diagnosis and as a surrogate of minimal residual disease in MB [15,33].
We confirm in our series that CSF can be a reliable source of cfDNA in pediatric EBT. Furthermore, we show that this cfDNA, although obtained in small quantities, can lead to informative results for CNA and SNV calling in 11/18 and 15/25 cases, respectively. Non-informative cases might be linked to the small amounts of CSF, low ctDNA content, or also to the fact that CSF was obtained with a mean of 17.9 days after surgery (range 3 days prior to 67 days after surgery), with no tumor any longer in place in cases with complete excision.
In our series, CNA could be detected in 11/18 CSF cfDNA samples for which the performed techniques would permit calling of copy number changes [34]. All cases with a ctDNA content > 20% showed copy number changes in the CSF cfDNA. Furthermore SNVs could be detected in 15/18 cases for which WES could be performed, confirming the higher sensitivity for detection of SNVs as compared to CNA.
Our results also demonstrate genetic clonal heterogeneity, with tumor-and CSF-specific SNVs observed in all EBT, underlining the usefulness of CSF cfDNA to further evaluate this phenomenon [5,36].
Given the feasibility of molecular characterization of EBT using CSF cfDNA, these approaches might be of clinical usefulness to address multiple questions in the future. First, it may help making a diagnosis by analyzing cfDNA without any tumor biopsy when the biopsy is impossible or deemed too risky; it may in particular be of use to distinguish between a tumor relapse and a secondary malignancy. Second, it will be feasible to use these approaches for the assessment of tumor burden, and the evaluation of minimal residual disease [33,37]. Furthermore, the study of CSF cfDNA paves the way towards serial evaluation of genetic alterations, as temporal heterogeneity has been described in EBT [36]. In addition, this will enable the study of emergence of resistance mutations or other genetic alterations under (targeted) therapies [25] .
In addition to molecular diagnosis based on CNA or SNVs, classification of EBT strongly depends on methylation and expression profiles [38,39,40]. Due to the limited amount of cfDNA available for each sample, methylation profiling on CSF cfDNA could not be performed. We sought to determine whether in CSF cfDNA expression profiles could be inferred from nucleosome footprints based on the average coverage, upon sequencing, across the TSS of genes of interest. Nucleosome occupancy at transcription start sites (TSS) can be reflected by cutting patterns of DNA by nucleases upon release from the nucleus, leading to distinct profiles of coverage with expressed genes having a dip in the coverage pattern over the TSS due to lower nucleosome occupancy, as opposed to silent genes, which present a relatively flat coverage profile due to densely packed nucleosomes [17,18,19,20].
We applied a targeted sequencing approach, aiming for a higher depth of coverage than that of low coverage WGS, targeting the TSS of 112 EBT-relevant genes. Importantly, distinct coverage profiles suggesting different nucleosome occupancy across the studied regions were observed in 7/15 studied samples. The samples non-contributive for TSS analysis were those with lower CSF cfDNA concentrations.
Distinct patterns of nucleosome occupancy across the studied TSSs predicted genes to be over- or under-expressed in different tumor entities. However, clustering by disease type did not enable to cluster all specific disease types together, with one MB clustering with the ATRT. This might be due to technical bias or poor cfDNA concentration compared to other samples, with a lower cfDNA concentration compared to the other MB (15 ng/mL versus at least 40 ng/mL for the other cases) observed in this case. In addition to nucleosome occupancy, many other molecular steps are involved in gene expression, and it has been suggested that altered promotor nucleosome positioning is an early event in gene silencing, with more permanent gene silencing linked to hypermethylation [41]. We show distinct TSS nucleosome footprints in different tumors; however the absence of robust tumor clustering as opposed to that seen by expression profiles could be linked an absence of a direct correlation between TSS nucleosome footprints and expression levels in the studied genes. However, no direct correlation with expression data from paired samples could be studied. Analyzing TSS nucleosome footprints across the whole genome, and correlating this with expression profiles, could lead to further comprehension of mechanisms implicated in gene regulation in particular in EBT.
Altogether, our study provides further insight into the clinical usefulness of cfDNA extracted from CSF obtained during clinical routine procedure, with informative results in 60% (15/25) of all cases. Further studies, including correlation with methylation studies, will enable to explore the important observation of specific nucleosome occupancy around the TSS of EBT-relevant genes.
Future decision algorithms could propose CSF cfDNA-based molecular diagnosis as a first step either if molecular diagnosis is compatible with neo-adjuvant chemotherapy prior to surgery, or in unresectable pediatric brain tumors, with a biopsy then proposed secondarily if necessary, and CSF cfDNA analyses could be integrated into disease surveillance during treatment and follow-up.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Experimental design : MC, YI, JMP, VC, VA, OD, JW, FB, GS; Study implementation (including collection of biological samples) : MC, YI, JMP, VC, VA, ASC, DF, CFC, KB, PV, BG, SB, GP, YB, FD, OD, JW, FB, GS; Analysis and interpretation of the data : MC, YI, JMP, OD, JW, FB, GS; Writing of the manuscript at draft : MC, YI, JMP, VC, VA, ASC, DF, CFC, KB, PV, BG, SB, GP, YB, FD, OD, JW, FB, GS; Approval of the final manuscript version : MC, YI, JMP, VC, VA, ASC, DF, CFC, KB, PV, BG, SB, GP, YB, FD, OD, JW, FB, GS.
Funding
This work was supported by grants from the following associations and funding organisations: the French Society of Pediatric Hematology and Oncology (Société Française de lutte contre les Cancers et Les Leucémies de l’Enfant et de l’adolescent; SFCE), Association Enfants, Cancers et Santé, Association Hubert Gouin – Enfance et Cancer, the Annenberg Foundation,the Nelia et Amadeo Barletta Foundation, and Fondation ARC pour la Recherche sur le Cancer. High-throughput sequencing was performed by the ICGex NGS platform of the Institut Curie supported by the grants ANR-10-EQPX-03 (Equipex) and ANR-10-INBS-09-08 (France Génomique Consortium) from the Agence Nationale de la Recherche ("Investissements d’Avenir" program), by the ITMO-Cancer Aviesan (Plan Cancer III) and by the SiRIC-Curie program (SiRIC Grant INCa-DGOS- 4654). The MAPPYACTS protocol is funded by the French National Cancer Institute, Imagine for Margo and Fondation ARC pour la Recherche sur le Cancer. MICCHADO is supported by the French National Cancer Institute, Imagine for Margo, Association Hubert Gouin Enfance et Cancer, KickCancer as well as research grants from BMS, MSDavenir, and Roche.
References
- Grobner SN: Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, Johann PD, Balasubramanian GP, Segura-Wang M, Brabetz S et al: The landscape of genomic alterations across childhood cancers. Nature 2018, 555(7696):321-327. [CrossRef]
- Li BK, Al-Karmi S, Huang A, Bouffet E: Pediatric embryonal brain tumors in the molecular era. Expert Rev Mol Diagn 2020, 20(3):293-303. 3. [CrossRef]
- Pfister SM, Reyes-Mugica M, Chan JKC, Hasle H, Lazar AJ, Rossi S, Ferrari A, Jarzembowski JA, Pritchard-Jones K, Hill DA et al: A Summary of the Inaugural WHO Classification of Pediatric Tumors: Transitioning from the Optical into the Molecular Era. Cancer Discov 2022, 12(2):331-355. [CrossRef]
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G et al: The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 2021, 23(8):1231-1251. [CrossRef]
- Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, Garzia L, Torchia J, Nor C, Morrissy AS et al: Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31(6):737-754 e736. [CrossRef]
- Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Fruhwald M, Hasselblatt M, Bourdeaut F, Williamson D, Huang A et al: Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol 2020, 22(5):613-624. [CrossRef]
- Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I et al: New Brain Tumor Entities Emerge from Molecular Classification of CNS-PNETs. Cell 2016, 164(5):1060-1072. [CrossRef]
- Berlanga P, Pierron G, Lacroix L, Chicard M, Adam de Beaumais T, Marchais A, Harttrampf AC, Iddir Y, Larive A, Soriano Fernandez A et al: The European MAPPYACTS Trial: Precision Medicine Program in Pediatric and Adolescent Patients with Recurrent Malignancies. Cancer Discov 2022, 12(5):1266-1281. [CrossRef]
- van Tilburg CM, Pfaff E, Pajtler KW, Langenberg KPS, Fiesel P, Jones BC, Balasubramanian GP, Stark S, Johann PD, Blattner-Johnson M et al: The Pediatric Precision Oncology INFORM Registry: Clinical Outcome and Benefit for Patients with Very High-Evidence Targets. Cancer Discov 2021, 11(11):2764-2779. [CrossRef]
- Seoane J, De Mattos-Arruda L, Le Rhun E, Bardelli A, Weller M: Cerebrospinal fluid cell-free tumour DNA as a liquid biopsy for primary brain tumours and central nervous system metastases. Ann Oncol 2019, 30(2):211-218. [CrossRef]
- Bounajem MT, Karsy M, Jensen RL: Liquid biopsies for the diagnosis and surveillance of primary pediatric central nervous system tumors: a review for practicing neurosurgeons. Neurosurg Focus 2020, 48(1):E8. 1. [CrossRef]
- Li J, Zhao S, Lee M, Yin Y, Zhou Y, Ballester LY, Esquenazi Y, Dashwood RH, Davies PJA, Parsons DW et al: Reliable tumor detection by whole-genome methylation sequencing of cell-free DNA in cerebrospinal fluid of pediatric medulloblastoma. Sci Adv 2020, 6(42). [CrossRef]
- Sun Y, Li M, Ren S, Liu Y, Zhang J, Li S, Gao W, Gong X, Liu J, Wang Y et al: Exploring genetic alterations in circulating tumor DNA from cerebrospinal fluid of pediatric medulloblastoma. Sci Rep 2021, 11(1):5638. [CrossRef]
- Stankunaite R, Marshall LV, Carceller F, Chesler L, Hubank M, George SL: Liquid biopsy for children with central nervous system tumours: Clinical integration and technical considerations. Front Pediatr 2022, 10: 957944. [CrossRef]
- Liu APY, Smith KS, Kumar R, Paul L, Bihannic L, Lin T, Maass KK, Pajtler KW, Chintagumpala M, Su JM et al: Serial assessment of measurable residual disease in medulloblastoma liquid biopsies. Cancer Cell 2021. [CrossRef]
- Zuccato JA, Patil V, Mansouri S, Voisin M, Chakravarthy A, Shen SY, Nassiri F, Mikolajewicz N, Trifoi M, Skakodub A et al: Cerebrospinal fluid methylome-based liquid biopsies for accurate malignant brain neoplasm classification. Neuro Oncol 2022. [CrossRef]
- Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J: Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164(1-2):57-68. [CrossRef]
- Ulz P, Perakis S, Zhou Q, Moser T, Belic J, Lazzeri I, Wolfler A, Zebisch A, Gerger A, Pristauz G et al: Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat Commun 2019, 10(1):4666. [CrossRef]
- Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, Abete L, Pristauz G, Petru E, Geigl JB et al: Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet 2016, 48(10):1273-1278. [CrossRef]
- Vanderstichele A, Busschaert P, Landolfo C, Olbrecht S, Coosemans A, Froyman W, Loverix L, Concin N, Braicu EI, Wimberger P et al: Nucleosome footprinting in plasma cell-free DNA for the pre-surgical diagnosis of ovarian cancer. NPJ Genom Med 2022, 7(1):30. [CrossRef]
- Oh S, Geistlinger L, Ramos M, Morgan M, Waldron L, Riester M: Reliable Analysis of Clinical Tumor-Only Whole-Exome Sequencing Data. JCO Clin Cancer Inform 2020, 4:321-335. [CrossRef]
- Euskirchen P, Bielle F, Labreche K, Kloosterman WP, Rosenberg S, Daniau M, Schmitt C, Masliah-Planchon J, Bourdeaut F, Dehais C et al: Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol 2017, 134(5):691-703. [CrossRef]
- Passeri T, Dahmani A, Masliah-Planchon J, Naguez A, Michou M, El Botty R, Vacher S, Bouarich R, Nicolas A, Polivka M et al: Dramatic In Vivo Efficacy of the EZH2-Inhibitor Tazemetostat in PBRM1-Mutated Human Chordoma Xenograft. Cancers (Basel) 2022, 14(6). [CrossRef]
- Richer W, Masliah-Planchon J, Clement N, Jimenez I, Maillot L, Gentien D, Albaud B, Chemlali W, Galant C, Larousserie F et al: Embryonic signature distinguishes pediatric and adult rhabdoid tumors from other SMARCB1-deficient cancers. Oncotarget 2017, 8(21):34245-34257. [CrossRef]
- Chicard M, Colmet-Daage L, Clement N, Danzon A, Bohec M, Bernard V, Baulande S, Bellini A, Deveau P, Pierron G et al: Whole-Exome Sequencing of Cell-Free DNA Reveals Temporo-spatial Heterogeneity and Identifies Treatment-Resistant Clones in Neuroblastoma. Clin Cancer Res 2018, 24(4):939-949. [CrossRef]
- Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, Jones DTW, Sturm D, Hermann C, Segura Wang M et al: Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 2016, 29(3):379-393. [CrossRef]
- Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, Shih DJ, Luu B, Dubuc AM, Northcott PA et al: Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 2013, 14(12):1200-1207. [CrossRef]
- Behling F, Schittenhelm J: Oncogenic BRAF Alterations and Their Role in Brain Tumors. Cancers (Basel) 2019, 11(6). [CrossRef]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J et al: The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet 2014, 46(5):444-450. [CrossRef]
- Consortium GT: The Genotype-Tissue Expression (GTEx) project. Nat Genet 2013, 45(6):580-585. [CrossRef]
- Lizio M, Harshbarger J, Shimoji H, Severin J, Kasukawa T, Sahin S, Abugessaisa I, Fukuda S, Hori F, Ishikawa-Kato S et al: Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol 2015, 16(1):22 . [CrossRef]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, Manke T: deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 2016, 44(W1):W160-165. [CrossRef]
- Liu APY, Smith KS, Kumar R, Robinson GW, Northcott PA: Low-coverage whole-genome sequencing of cerebrospinal-fluid-derived cell-free DNA in brain tumor patients. STAR Protoc 2022, 3(2):101292. [CrossRef]
- Jones DTW, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, Cho Y-J, Pugh TJ, Hovestadt V, Stütz AM et al: Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488(7409):100-105. [CrossRef]
- Escudero L, Llort A, Arias A, Diaz-Navarro A, Martinez-Ricarte F, Rubio-Perez C, Mayor R, Caratu G, Martinez-Saez E, Vazquez-Mendez E et al: Circulating tumour DNA from the cerebrospinal fluid allows the characterisation and monitoring of medulloblastoma. Nat Commun 2020, 11(1):5376. [CrossRef]
- Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, Boult JK, Williamson D, Ahmad Z, Hallsworth A et al: Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015, 27(1):72-84. [CrossRef]
- Izquierdo E, Proszek P, Pericoli G, Temelso S, Clarke M, Carvalho DM, Mackay A, Marshall LV, Carceller F, Hargrave D et al: Droplet digital PCR-based detection of circulating tumor DNA from pediatric high grade and diffuse midline glioma patients. Neurooncol Adv 2021, 3(1):vdab013. [CrossRef]
- Pan-cancer analysis of whole genomes. Nature 2020, 578(7793):82-93. [CrossRef]
- Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE et al: DNA methylation-based classification of central nervous system tumours. Nature 2018, 555(7697):469-474. [CrossRef]
- Lambo S, Grobner SN, Rausch T, Waszak SM, Schmidt C, Gorthi A, Romero JC, Mauermann M, Brabetz S, Krausert S et al: The molecular landscape of ETMR at diagnosis and relapse. Nature 2019, 576(7786):274-280. 2019; 576, 7786, 274–280. [CrossRef]
- Hesson LB, Sloane MA, Wong JW, Nunez AC, Srivastava S, Ng B, Hawkins NJ, Bourke MJ, Ward RL: Altered promoter nucleosome positioning is an early event in gene silencing. Epigenetics 2014, 9(10):1422-1430. [CrossRef]
Figure 1.
Genetic alterations detected in tumor tissue and cfDNA extracted from CSF. A. Copy number profile in an EMTR showing amplification of C19MC in both samples (patient 23). B. Copy number profile of an EBT NOS with an amplification on chr 20 in both samples, as well as gains of chromosomes 2,7,12 and 18 (patient 25). C. SNVs detected in WES of the primary tumor, and WES and targeted sequencing of cfDNA, with labels indicating the number of reads supporting the SNVs, coverage at the given position and percentage of variant allele fraction : a SMARCB1 mutation in ATRT (patient 1), a CTNNB1 mutation seen by WES of the primary and targeted sequencing of cfDNA (patient 5). Heterogeneity of SNVS observed in a MB with a SMARCA4 alteration seen in the primary tumor but not in CSF, and DNMT3A seen in CSF but not in the primary (patient 14). Heterogeneity of SNVs with a KMT2D alterations seen in CSF but not in the primary tumor, and a FANCI SNV seen in both (patient 17).
Figure 1.
Genetic alterations detected in tumor tissue and cfDNA extracted from CSF. A. Copy number profile in an EMTR showing amplification of C19MC in both samples (patient 23). B. Copy number profile of an EBT NOS with an amplification on chr 20 in both samples, as well as gains of chromosomes 2,7,12 and 18 (patient 25). C. SNVs detected in WES of the primary tumor, and WES and targeted sequencing of cfDNA, with labels indicating the number of reads supporting the SNVs, coverage at the given position and percentage of variant allele fraction : a SMARCB1 mutation in ATRT (patient 1), a CTNNB1 mutation seen by WES of the primary and targeted sequencing of cfDNA (patient 5). Heterogeneity of SNVS observed in a MB with a SMARCA4 alteration seen in the primary tumor but not in CSF, and DNMT3A seen in CSF but not in the primary (patient 14). Heterogeneity of SNVs with a KMT2D alterations seen in CSF but not in the primary tumor, and a FANCI SNV seen in both (patient 17).
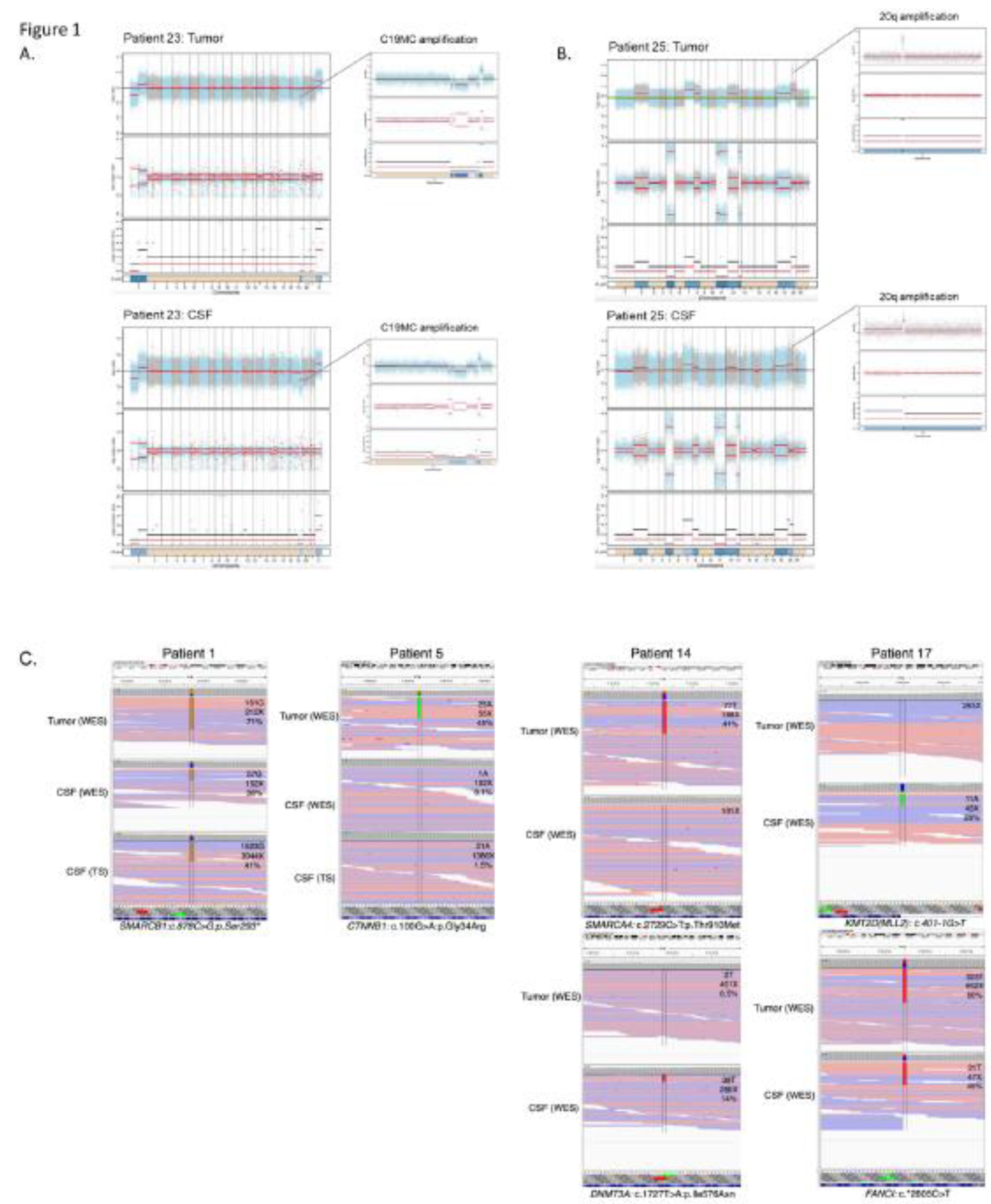
Figure 2.
Copy number changes detected in cases with informative copy number analysis both in tumor and CSF cfDNA. For patients with an informative copy number analysis in CSF cfDNA (x-axis), copy number alterations are indicated, according to their detection in tumor (blue triangle) or CSF cfDNA (red triangle).
Figure 2.
Copy number changes detected in cases with informative copy number analysis both in tumor and CSF cfDNA. For patients with an informative copy number analysis in CSF cfDNA (x-axis), copy number alterations are indicated, according to their detection in tumor (blue triangle) or CSF cfDNA (red triangle).
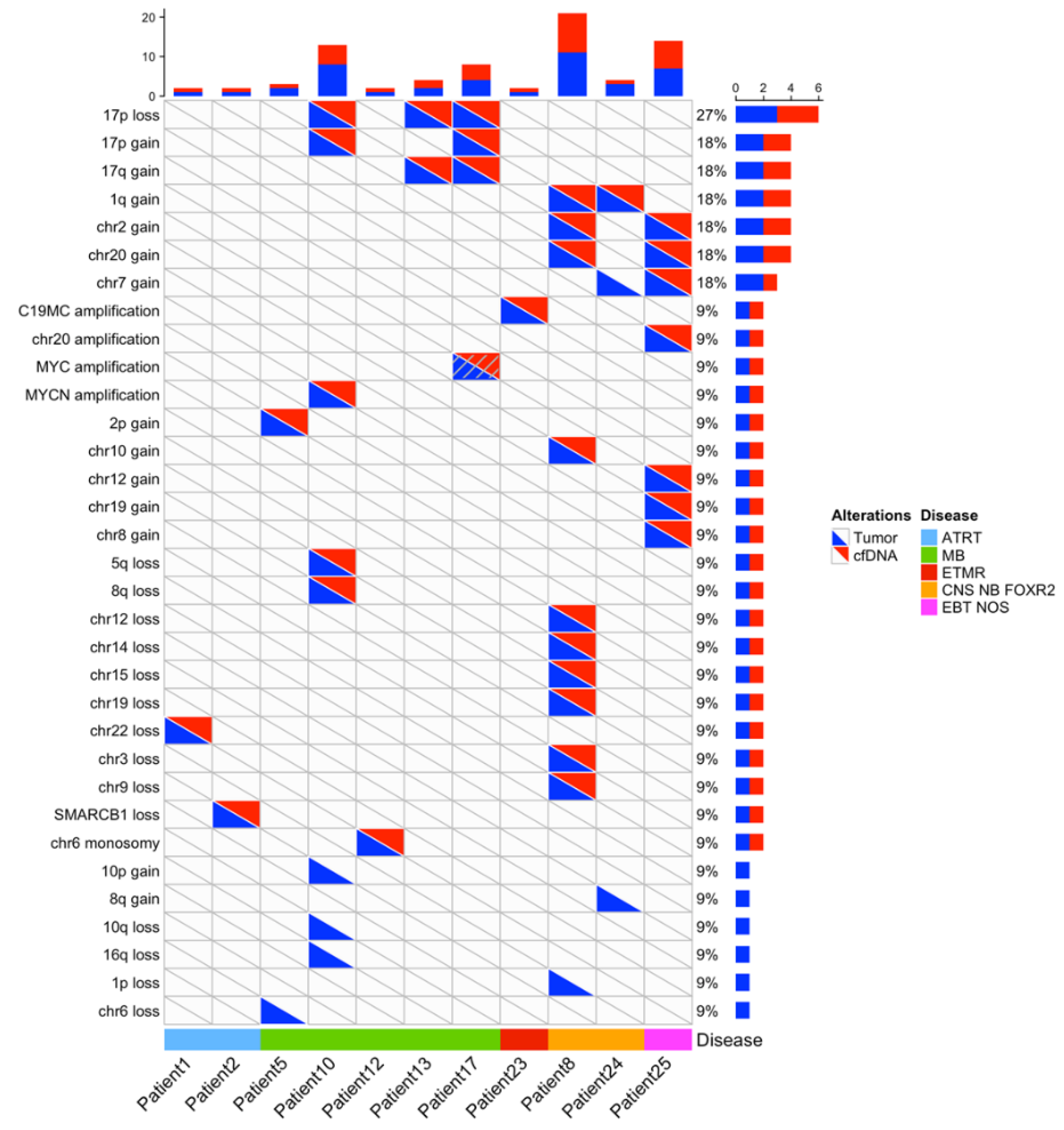
Figure 3.
Variant allele fractions of SNVs of genes included in the targeted sequencing panel. In 15 cases with informative SNV analysis in the primary tumor and CSF cfDNA, the mutated allele fractions of SNVs in the tumor sample (x-axis) and in the CSF cfDNA (y-axis) is indicated.
Figure 3.
Variant allele fractions of SNVs of genes included in the targeted sequencing panel. In 15 cases with informative SNV analysis in the primary tumor and CSF cfDNA, the mutated allele fractions of SNVs in the tumor sample (x-axis) and in the CSF cfDNA (y-axis) is indicated.
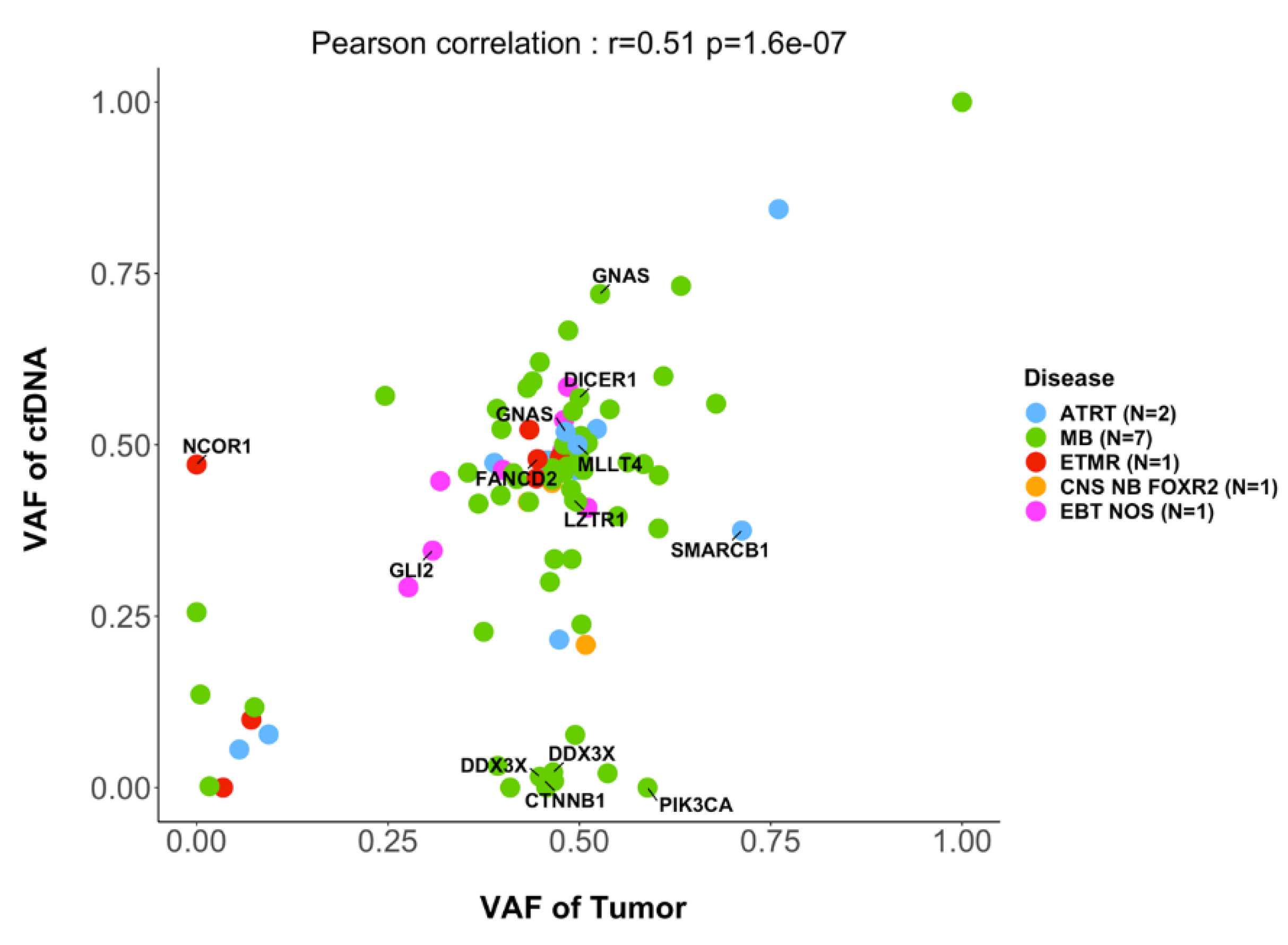
Figure 4.
Comparison of number of SNVs from WES analysis in tumor and CSF cfDNA, among cases with informative analyses, followed by disease type. Among the 15 cases with informative SNV analysis in both the primary tumor and CSF cfDNA by WES, the total number of SNVs seen in the tumor (red circles), in the CSF cfDNA (blue circles), or in both (overlap) is indicated. Among the 15 patients, a mean of 83 SNVs (range 1-160) were observed in common between the tumor and CSF cfDNA, with 5 (range 0-25) and 18 (range 1-62) seen only in the CSF cfDNA and tumor, respectively, indicating clonal heterogeneity.
Figure 4.
Comparison of number of SNVs from WES analysis in tumor and CSF cfDNA, among cases with informative analyses, followed by disease type. Among the 15 cases with informative SNV analysis in both the primary tumor and CSF cfDNA by WES, the total number of SNVs seen in the tumor (red circles), in the CSF cfDNA (blue circles), or in both (overlap) is indicated. Among the 15 patients, a mean of 83 SNVs (range 1-160) were observed in common between the tumor and CSF cfDNA, with 5 (range 0-25) and 18 (range 1-62) seen only in the CSF cfDNA and tumor, respectively, indicating clonal heterogeneity.
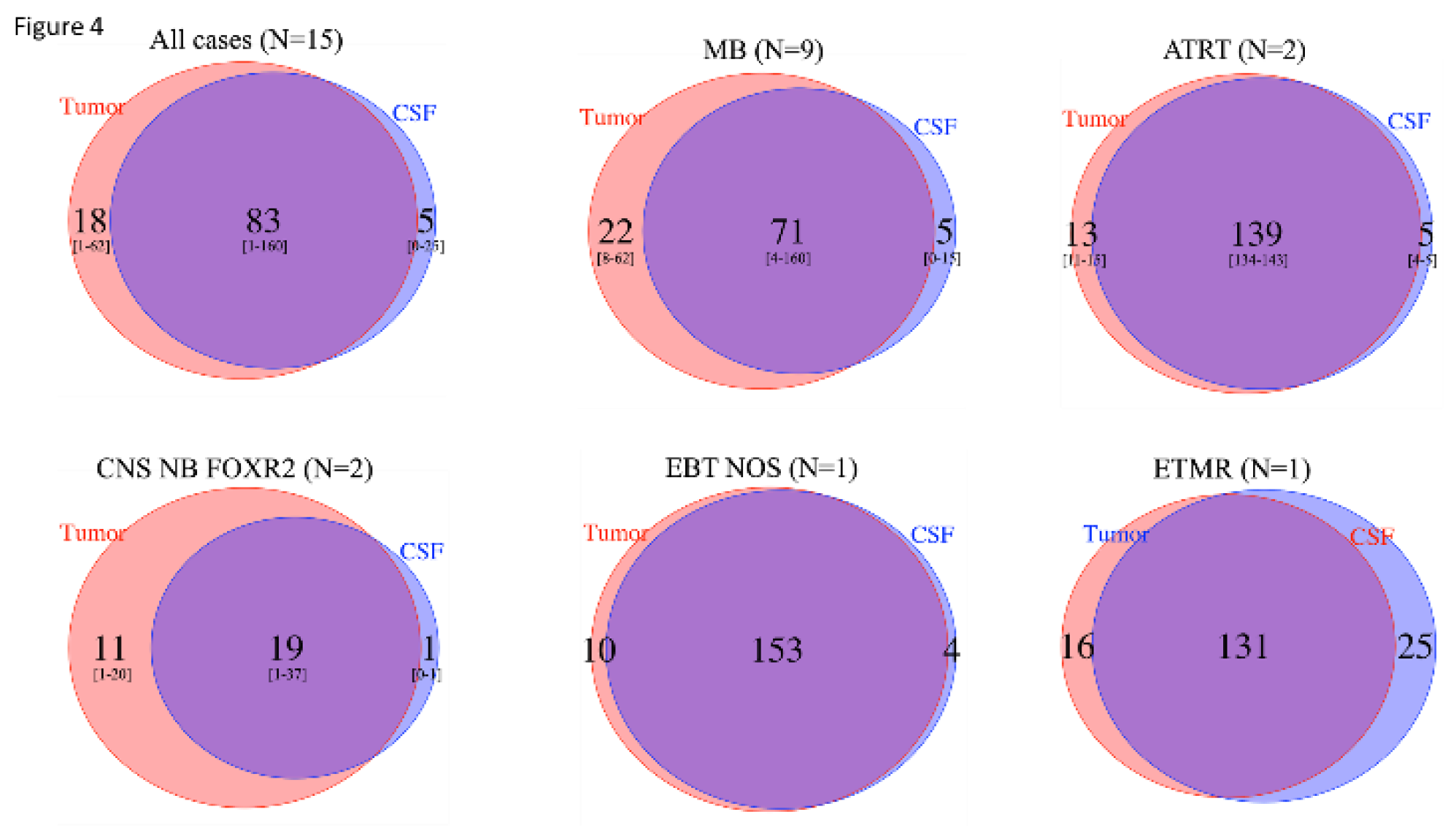
Figure 5.
TSS nucleosome footprinting based on the coverage across the transcription start sites (TSS) of 112 genes in the TSS targeted sequencing panel. A. Coverage across the NDR regions surrounding the TSS enabled to distinguish two different groups with specific patterns in seven cases using k-means clustering. B. Example of a case in which no distinct groups could be distinguished. C. Library size (y-axis: total number of reads after removal of duplicates) depending on the CSF cfDNA concentration (x-axis); samples with un-informative TSS analyses are indicated in red. D. Clustering on the total 112 NDR regions in seven samples for which the k-means clustering of genes into two groups was informative. Hierarchical clustering was performed on NDR regions only. The scores represent the counts per million (CPM) values of coverage sequencing after row scaling (row z-scores). The results of hierarchical clustering on the genes are represented by a dendrogram. Samples were ordered by disease type. The heatmap shows differences on coverage around the TSS across the seven samples. E. Clustering on the 46 NDR regions selected after applying thresholds, on four samples (2 ATRT and 2 MB cases). Hierarchical clustering was performed on NDR and on samples.
Figure 5.
TSS nucleosome footprinting based on the coverage across the transcription start sites (TSS) of 112 genes in the TSS targeted sequencing panel. A. Coverage across the NDR regions surrounding the TSS enabled to distinguish two different groups with specific patterns in seven cases using k-means clustering. B. Example of a case in which no distinct groups could be distinguished. C. Library size (y-axis: total number of reads after removal of duplicates) depending on the CSF cfDNA concentration (x-axis); samples with un-informative TSS analyses are indicated in red. D. Clustering on the total 112 NDR regions in seven samples for which the k-means clustering of genes into two groups was informative. Hierarchical clustering was performed on NDR regions only. The scores represent the counts per million (CPM) values of coverage sequencing after row scaling (row z-scores). The results of hierarchical clustering on the genes are represented by a dendrogram. Samples were ordered by disease type. The heatmap shows differences on coverage around the TSS across the seven samples. E. Clustering on the 46 NDR regions selected after applying thresholds, on four samples (2 ATRT and 2 MB cases). Hierarchical clustering was performed on NDR and on samples.
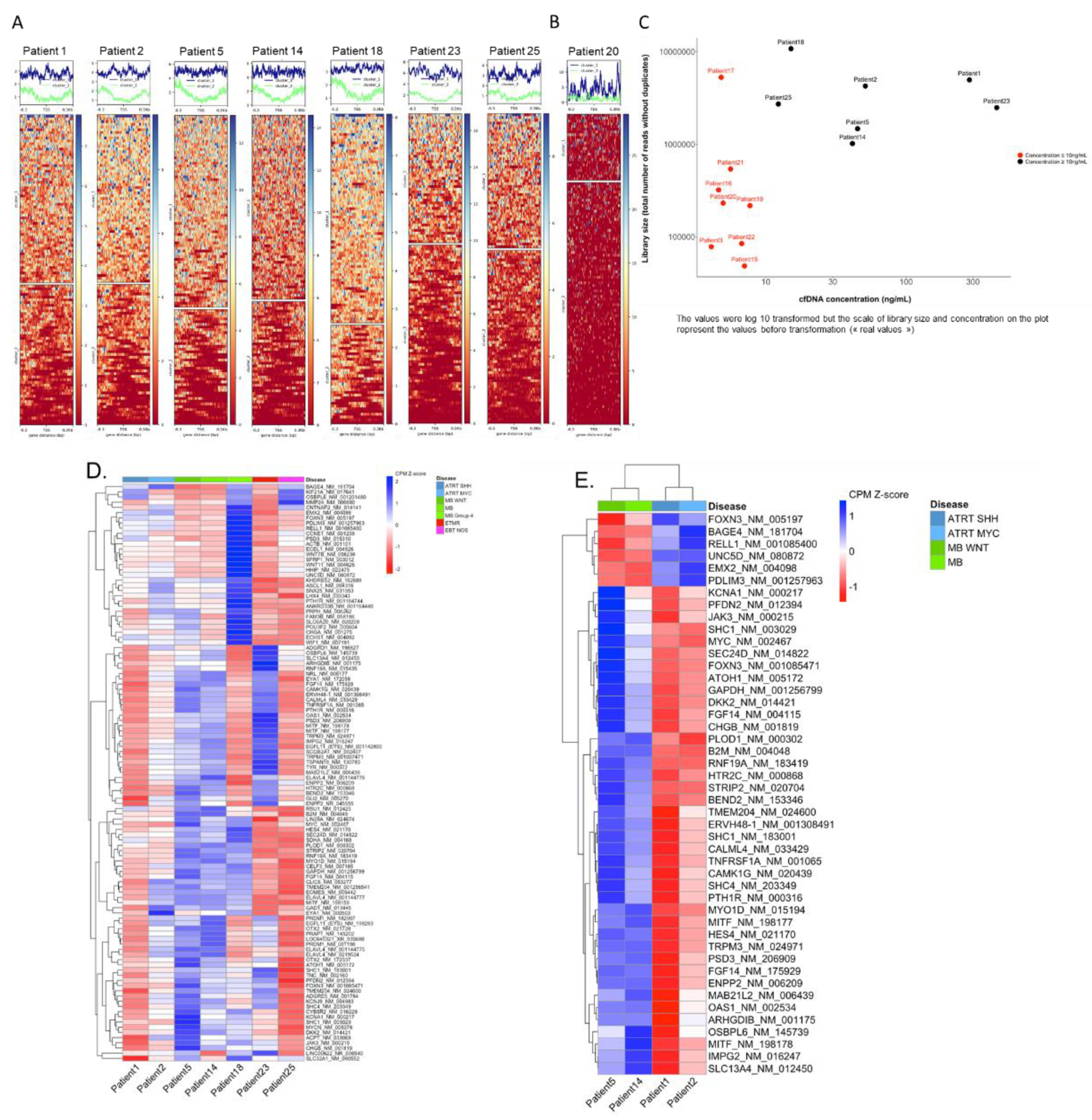

Table 1.
Diagnostic molecular genetic alterations in 25 patients with embryonal brain tumors. Genetic alterations (SNVs and copy number alterations) detected in clinical molecular analysis are listed according to their detection in the primary tumor, and in CSF cfDNA.
Table 1.
Diagnostic molecular genetic alterations in 25 patients with embryonal brain tumors. Genetic alterations (SNVs and copy number alterations) detected in clinical molecular analysis are listed according to their detection in the primary tumor, and in CSF cfDNA.
| Patient number |
Primary tumor molecular diagnosis : SNV | SNV in CSF cfDNA | Primary tumor molecular diagnosis : CNA | CNA in CSF cfDNA |
|---|---|---|---|---|
| 1 | SMARCB1 :c.851C>G / (p.Ser284*) | SMARCB1 :c.851C>G / (p.Ser284*) | SMARCB1 loss | SMARCB1 loss |
| 2 | SMARCB1 loss | SMARCB1 loss | ||
| 3 | SMARCB1 : c.601C>T / p.(Arg201*) | not found/ CSF cfDNA non contributive | ||
| 4 | Gains: 2pter-p22.3 (subclone), 7, 17q12.2-qter Losses: 11pter-p11.12, 17q12.2-qter, |
not found/ CSF cfDNA non contributive | ||
| 5 | CTNNB1:Gly34Arg (c. 100G>A) | CTNNB1:Gly34Arg (c. 100G>A) | chr6 loss chr2pter-p13,2 gain |
chr2pter-p13,2 gain |
| 6 | 17q gain, 17p loss | not found/ CSF cfDNA non contributive | ||
| 7 | Losses: chr10, 2, 11, 13, 16, et 20 | not found/ CSF cfDNA non contributive | ||
| 8 | Gains: 1q, 2, 10, 20 Losses: 1p, 3, 9, 12, 14, 15, 19, X |
Gains: 1q, 2, 10, 20 Losses: 3, 9, 12, 14, 15, 19 |
||
| 9 | 17q loss , 17q gain | not found/ CSF cfDNA non contributive | ||
| 10 | Amplicon 2p24.3-2 containing MYCN. Losses: 5q31,2-ter 8q12,3-ter, 10q, 16q, 17pter-p11,2 Gains: 10p, 17p11.2-qter |
Amplicon 2p24.3-2 containing MYCN. Losss: 5q31,2-ter, 8q12,3-ter, 17pter-p11,2 Gains : 17p11.2-qter |
||
| 11 | PTCH1:c.1359_1360insG (p.cys454valfs*43) | not found/ CSF cfDNA non contributive | 9q loss 3q gain |
not found/ CSF cfDNA non contributive |
| 12 | APC : c.3183_3187del/p.Gln1062* germline APC : c.3758del/p.Ser1253Leufs*12 |
APC : c.3183_3187del/p.Gln1062* germline APC : c.3758del/p.Ser1253Leufs*12 |
chr6 monosomy | chr6 monosomy |
| 13 | 17p loss 17pq gain |
17p loss 17pq gain |
||
| 14 | ||||
| 15 | CCND3 c.774_775delCTinsTG p.(Ser259Ala) COL3A1 c.946G>A p.(Ala316Thr) FANCD2 (c.1588C>T p.(Arg530*) NCKIPSD c.1650_1651delGCinsTT p.(Pro551Ser) PTPRC c.3452A>G p.(Lys1151Arg) |
altered genes not inlcuded in targeted sequencing CSF cfDNA panel | ||
| 16 | Segmental gains : 1q(210,34Mb-tel), 3p(tel-5,17Mb) | not found/ CSF cfDNA non contributive | ||
| 17 | Segmental losses : 17p(tel-18,91Mb) harboring TP53 Segmental gains : 17pq(19,14Mb-tel) |
Segmental losses : 17p(tel-18,91Mb) harboring TP53 Segmental gains : 17pq(19,14Mb-tel) |
||
| 18 | Segmental losses : 11p- , 17p(tel-22,22Mb) Segmental gains : 4q(142,07Mb-tel), 13q(90,81-96,80Mb), 15q(54,53Mb-tel) , 17pq(25,28Mb-tel) Amplicons : 7q(92,20-96,80Mb) harboring CDK6. |
not found/ CSF cfDNA non contributive | ||
| 19 | Segmental losses : 2q(213,21Mb-tel), 8p(tel-10,18Mb), 16q(57,83Mb-tel), 17p(tel-18,15Mb), X(78,64-Tel), X(16,68-17,87Mb) Subclonal segmental losses: 4q(170,70Mb-tel), 5q(170,87Mb-tel) , 8q(55,00-70,23Mb), 9p(tel-11,78Mb), 10q(107,71Mb-tel), 12q(40,77-57,69Mb) |
No CN analyis for target seq data | ||
| 20 | CTNNB1 : c.98C>G / p.(Ser33Cys) PIK3CA : c.311C>G / p.(Pro104Arg) (50,3% ; 193X) |
CTNNB1 : c.98C>G / p.(Ser33Cys) | chr6 monosomy | No CN analyis for target seq data |
| 21 | Segmental losses : 17p(tel-18,91Mb) Segmental gains : 17pq(19,14Mb-tel) |
No CN analyis for target seq data | ||
| 22 | Segmental losses: 16q(66,57Mb-tel) Subclonal segmental losses: 8q(52,57Mb-tel), 11q(75,97Mb-tel), 13q(25,30-31,39Mb), 13q(50,05-81,10Mb) Segmental gains: 7p(tel-4,88Mb), 13q(20,28-25,27Mb), 13q(31,45-49,99Mb), 13q(81,14Mb-tel), 17q(46,78Mb-tel) |
No CN analyis for target seq data | ||
| 23 | 19q13,41 amplification | 19q13,41 amplification | ||
| 24 | PTPRK p.(Thr395AspfsTer6) | not found/ CSF cfDNA non contributive | Gains: 1q, 7, 8q | Gains: 1q |
| 25 | KDR : Hist1144Asp TP53 : Arg175His KRAS : Gly12Asp |
not found/ CSF cfDNA non contributive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



