
Submitted:
06 June 2023
Posted:
07 June 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The recent global emergence of the SARS-CoV-2 pandemic has accelerated research in several areas of science whose valuable outputs and findings can help to address future health challenges in the event of emerging infectious agents. We performed a multifocus shotgun analysis to compare differences in bacterial spectrum and viral presence through culture-independent RNA sequencing. We compared the microbiome of healthy people versus those with varying degrees of COVID-19 severity. We observed a significant increase in the diversity of microbial species in patients with COVID-19 regardless of disease severity. Some bacterial phyla, such as Actinobacteria, are significantly more abundant in healthy people than in infected people, whereas Bacteroides are less abundant in the latter. Infected people, regardless of severity and symptoms, have the same proportional representation of Firmicutes, Proteobacteria, Actinobacteria, Bacteroidetes, and Fusobacteriales the same. In addition to SARS-CoV-2 and numerous phage groups, we identified sequences of clinically significant viruses such as Human Herpes Virus 1, Human Mastadenovirus D, Molluscum Contagiosum Virus, and Rhinovirus A in several samples. Analyses were performed retrospectively, therefore, in the case of SARS-CoV-2 various WHO variants such as Alpha (B.1.1.7), Delta (B.1.617.2), Omicron (B.1.1.529), and 20C strains are represented. Additionally, the presence of specific virus strains has a certain effect on the distribution of individual microbial taxa.
Keywords:
metatranscriptome
; microbiome
; SARS-CoV-2
; virome
1. Introduction
At every breath, we inhale an airborne cocktail containing thousands of microbial particles, perhaps as many as a million a day. The upper respiratory tract is a major gateway for a large number of viruses during infection, not just respiratory ones. At the same time, the mucosa of the nasal cavity is colonized by a wide spectrum of microorganisms whose specific importance is not well elucidated. Although this massive burden may sound frightening, it is undoubtedly essential in building the immunity of individual hosts. A healthy upper respiratory tract microbiota works in tandem with its host, colonizing mainly the anterior nares and nasopharynx to provide an innate barrier that defends against pathogens and modulates the immune responses that occur when exposed to external triggers such as smoke, dust, allergens, chemicals, temperature changes, and microorganisms (Burbank et al., 2017). Under the influence of the recent COVID-19 pandemic, interest in how viral infection affects/modifies the overall microbiome of the airways, particularly the well-accessed upper respiratory tract, has gained prominence. Currently, many scientific reports explore microbial diversity in the context of COVID-19 disease severity (Bai et al., 2022; Candel et al., 2023; Zhu et al., 2022) unfortunately often with controversial outcomes. Besides COVID-19, many studies have demonstrated that unique microbial communities have complex interactions with the host to maintain balance with the host immune system (di Simone et al., 2023; Kumpitsch et al., 2019).
The nasal microbiome of healthy humans is composed primarily of Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria phyla, with a predominance of representatives of the genera Bifidobacterium, Corynebacterium, Staphylococcus, Streptococcus, Dolosigranulum, and Moraxella (Bassis et al., 2014a; Stearns et al., 2015). Archaea are not typically found in the upper respiratory tract of humans, as they are not known to be part of the normal microbiota. However, there have been some studies that have identified the presence of archaea (Methanoarchaea) in the nasal cavity and other parts of body (Baehren et al., 2022).
Among the bacteria, Bacteroidetes and Firmicutes predominate in most of the anatomical regions studied, including upper respiratory tract (URT), and are the most investigated components of the human microbiota (Nemergut et al., 2013).
A somewhat less well-known fact is that humans are colonized by a remarkable number of DNA and RNA viruses that are referred to as the virome capable of causing acute, persistent, or latent infection, and retroviral elements integrated to host chromosomes. The human virome consists of bacteriophages (phages) that infect bacteria, archaeal viruses, eukaryotic viruses that infect human cells, and viruses transient in food (Minot et al., 2013; Rajagopala et al., 2021). Among the viral genomes identified, Picornaviruses, Anelloviruses, and bacteriophages of the family Siphoviridae were the most prevalent in the upper respiratory tract (Rovira Rubió et al., 2023). The human virome comprehends commensals and opportunistic pathogens. The balance between being a commensal or becoming a pathogen is determined by different factors of the viral community itself and the host, such as genetic factors and immune status (Jankauskaitė et al., 2018). It has been well documented that resident viruses can cause exacerbation of chronic pulmonary diseases, cystic fibrosis, and asthma. Moreover, they may contribute to the pathogenesis of community-acquired respiratory virus infections (Porto, 2022).
Generally, there are two major approaches to detecting microbial spectra: 16S rRNA amplicon sequencing and metagenomic shotgun sequencing. The former is attractive at lower cost, is preferred in environments with lower microbial diversity, and identifies mainly dominant microorganisms (Tessler et al., 2017). The latter is characterized by a more robust resolution and covers the metagenome community including the human genome, viruses, and fungi, which cannot be captured by 16S amplicon sequencing (Brumfield et al., 2020). In our approach, we replace the analysis of metagenome with the metatranscriptome to cover also RNA viruses which include SARS-CoV-2. However, we actually did not sequence total RNA but depleted the dominating human 18S rRNA in the initial steps of library preparation to increase the relative abundance of other RNA belonging to humans and other microbial agents. In general, 18S rRNA is estimated to make up around 80-90% of the total RNA in a typical mammalian cell (Mortazavi et al., 2008) .
2. Material and methods
2.1. Patients and samples
The study was conducted in accordance with the approval of the Ethics Committee of the Bratislava Self-Governing Region under the identification number 03228/2021/HF dated 12 January 2021. All patients signed informed consent and received questionnaires with relevant information regarding their medical history and health status in relation to COVID-19. All 151 patients were from Slovakia and were divided into four groups: asymptomatic (n=24), patients with mild/moderate (n=25), severe COVID-19 (n=30), and a negative control group (n=72). The group of patients with severe COVID-19 was recruited from two university hospitals in Bratislava: Ruzinov and Kramare (Slovakia).
The timeframe of sampling in hospitalized patients was different in the two hospitals. At Ruzinov Hospital, patients were enrolled in the study at approximately day 7-8 (median 7) after admission to the hospital, at Kramare Hospital it was day 3-4 (median 3.5). The treatment regimen was also different in the two hospitals. In Ruzinov hospital 100% of patients received antibiotic treatment and corticotherapy (corticosteroid treatment) (18/18), more than 60% of patients received immunomodulatory therapy (20/28), and no patient received antiviral treatment. In the second hospital, on the other hand, only less than 30% received antibiotic therapy (3/11) in combination with corticosteroids (9/11), immunomodulatory and antiviral therapy (6/11). None of the above treatments had been administered in one severe patient at study inclusion and material collection.
Age and sex characteristics are summarized in Table 1. Samples were analyzed in the order in which they arrived, in proportional representation, and no preference was given to age, sex, or disease severity when selecting for sequencing. Nasopharyngeal swabs were collected from March 2021 to October 2022 in viRNAtrap transport medium (GeneSpector, Czech Republic) and stored at 4 °C in a refrigerator until processed.
2.2. RNA isolation, RT-PCR and library preparation
RNA was isolated retrospectively from a nasopharyngeal swab-originated collection medium representing samples from patients with positive findings for COVID-19, regardless of disease severity, without selection based on viral load (high or low Ct values) and from covid-negative controls using the Sera-XtractaTM virus/Pathogen Kit (Cytiva, UK) on a KingFisher Flex benchtop automated machine (ThermoFisher Scientific, UK). RNA quantity was measured fluorometrically using QubitTM RNA High sensitivity (Invitrogen, USA). RNA isolates were stored at -80°C until processed. Genomic libraries were prepared using Kappa HyperPrep with RiboErase Kit (Roche, USA) with depletion of eukaryotic RNA for 18S rRNA according to the manufacturer´s recommendations. Illumina's TruSeq CD dual adapters were used for sample indexing. The resulting libraries were quantified by fluorometric analysis using the QubitTM dsDNA HS Assay kit (Invitrogen, USA) and fragment analysis using the High Sensitivity DNA reagent kit (Agilent Technologies, Lithuania).
All RNAs underwent RT-PCR to confirm or exclude SARS-CoV-2 positivity using the COVID-19 Real-Time Multiplex RT-PCR Kit (Labsystems Diagnostics, Finland) and the ABI QuantStudio 6 Real-Time PCR System RT-qPCR platform (ThermoFisher, USA) using the original manufacturer's protocols. A Ct value < 40 was required to evaluate a sample as positive.
2.3. Sequencing and bioinformatic analysis
Paired-end sequencing - 2 × 75 and 2 × 100) was performed on NextSeq500/550 and NextSeq2000 (Illumina, USA) platforms, respectively. Paired-end sequencing data were subjected to quality control by FastQC v0.11.9 (Andrews, 2010) and reads were processed by Trimmomatic v0.39 (CROP:96 HEADCROP:10 LEADING:22 TRAILING:22 SLIDINGWINDOW:4:22 MINLEN:25 and our own set of adapter sequences were used in ILLUMINACLIP step) (Bolger et al., 2014). First, reads were mapped to the human reference hg38 (GRCh38) by the BWA-MEM algorithm (from bwa v0.7.17) (Heng Li, 2013). Unmapped reads, extracted using samtools view from samtools v1.6 (Danecek et al., 2021) and Picard SamToFastq from Picard v2.27.4 (picard toolkit, 2019), longer than 50 bp were further processed for microbiome identification. To identify and quantify bacterial species, we used Kraken 2 v2.1.2 (Wood & Salzberg, 2014), as we previously observed to be well-functional for this purpose (Hadzega et al., 2021). Kraken 2 was run in paired-end mode with --minimum-base-quality set on 20, while -minimum-hit-groups parameter was set on 2 and standard database was used (built with –standard flag, installed on August 2, 2022). Flag -use-mpa-style was used to produce output, which can be useful in downstream analysis and is also more easily readable.
For the SARS-CoV-2 virus, we applied de novo assembly algorithm coronaspades.py from Spades v3.15.5 (Meleshko et al., 2021). SARS-COV-2 lineage/clade assignment was done as proposed in Galaxy tutorial for a pipeline constructed by Maier and Batut, 2023 (Maier & Batut, 2023).
2.4. Statistical analysis
To statistically analyze the results of Kraken 2, mpa-style outputs were first brought together by combine_mpa.py from KrakenTools v1.2 (Lu et al., 2022). Then, counts were normalized by division by the total read pairs number in the given sample (after Trimmomatic step) and subsequently multiplied by the average read pairs number. The difference in the number of bacterial transcripts detected between groups was statistically analyzed by LefSE tool (Galaxy Version 1.0) (Segata et al., 2011).
The normalized data were further analyzed using the R programming language. Firstly, Bray-Curtis dissimilarity distance matrices were computed for all pairwise patient group comparisons. PERMANOVA was then performed to compare the distribution of bacterial genera and species between each pair of groups. Mann-Whitney U test (Wilcoxon test) was used to compare the counts of reads matching sequences from Kraken2 database between each pair of groups. This test was also applied to compare Ct values between each pair of positive groups, thus excluding negative controls. To account for multiple comparisons, Bonferroni correction was used to set a threshold of significance for p-values.
3. Results
3.1. Prokaryotic microbiome
By metatranscriptome shotgun sequencing, we analyzed 151 samples categorized into 4 main groups: asymptomatic (24, A), mild (25, M), severely affected (30, S) and control group (72, NC). The mean number of read pairs per sample after trimming was 45.3 M (27.7-133 M). Eukaryotic 18S rRNA from nasopharyngeal swabs was previously removed during DNA library preparation (detailed in Methods).
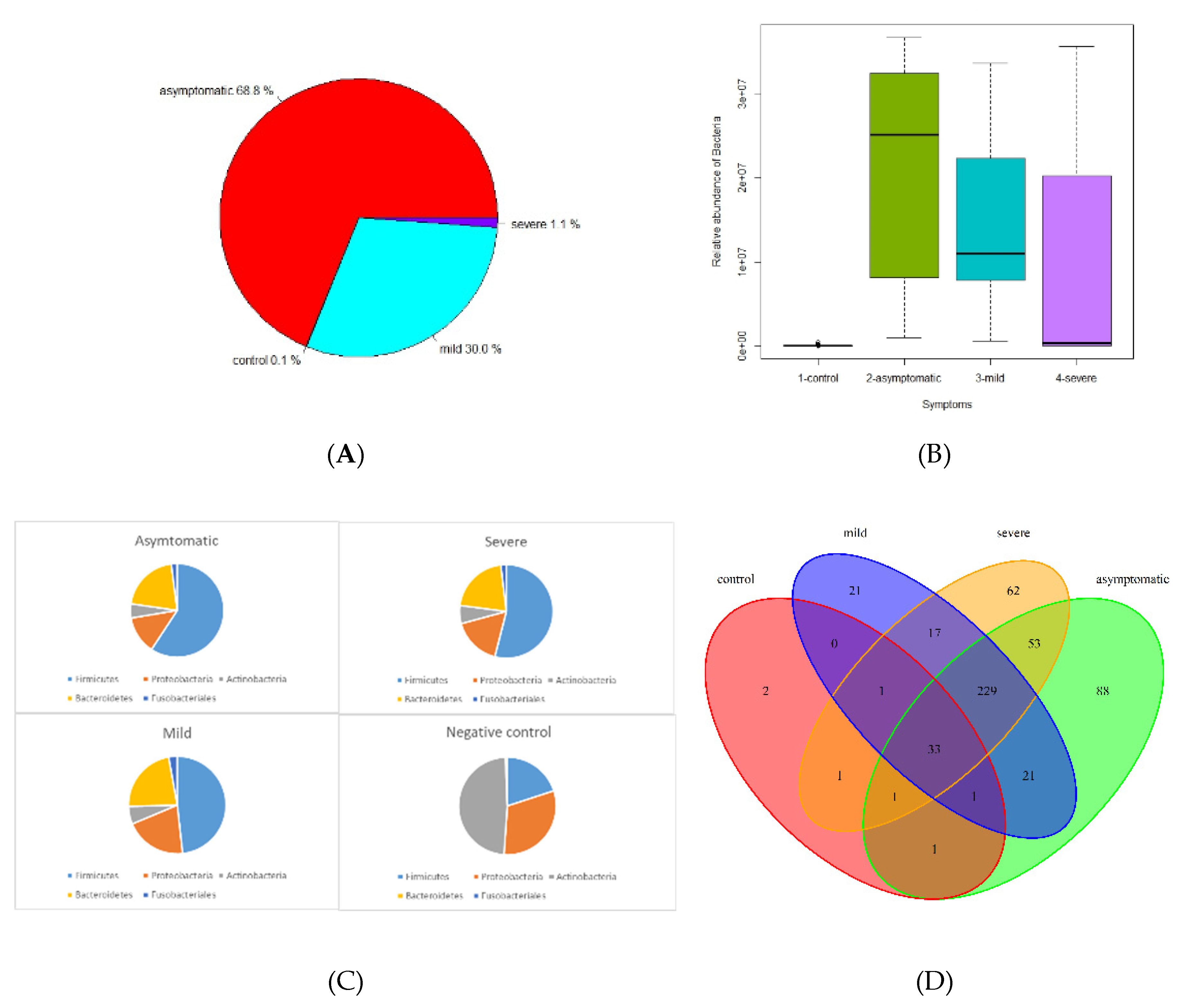
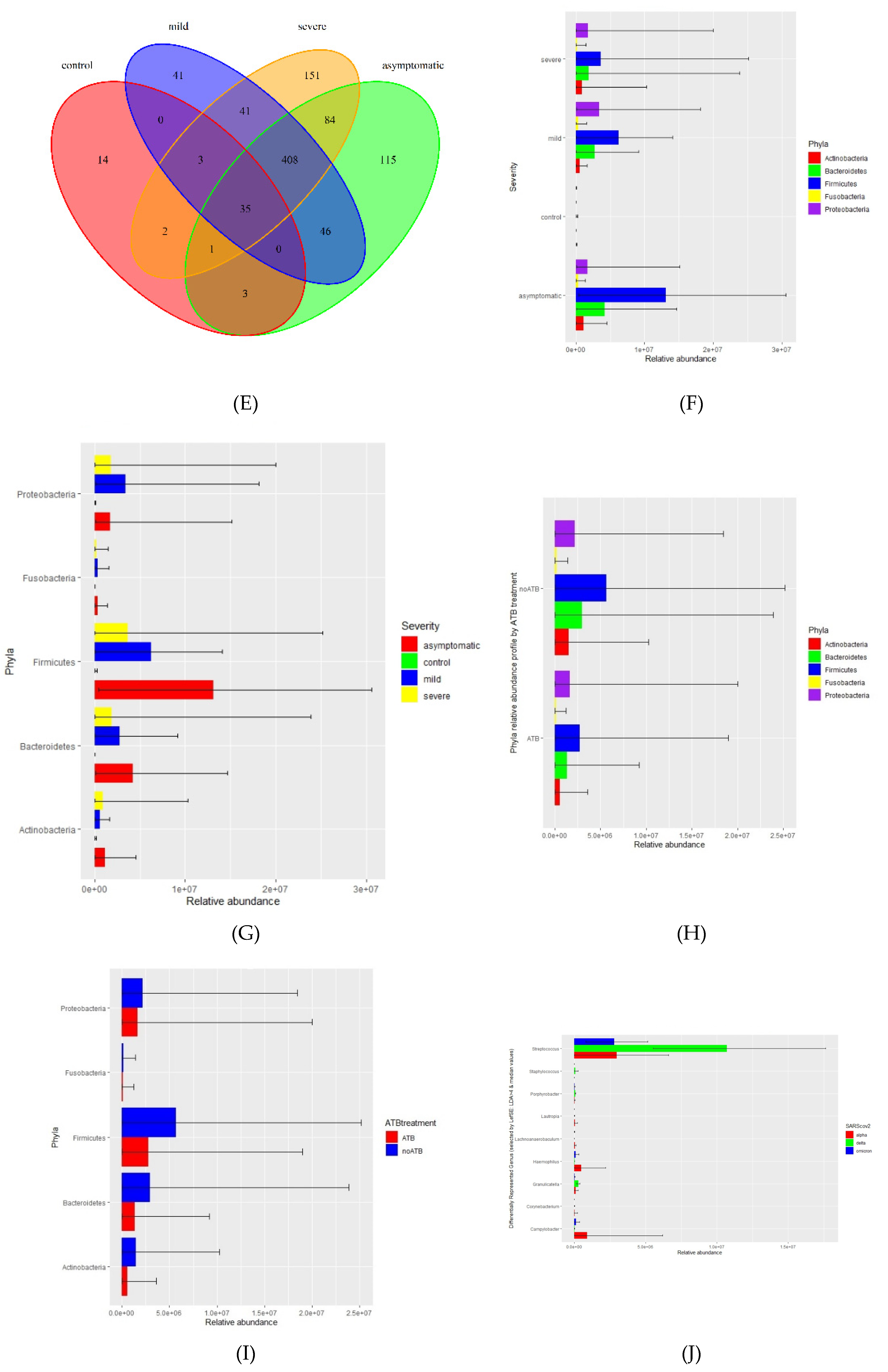
The microbiome fraction or simply the number of reads matching sequences from standard Kraken2 database, distribution, and representation of bacterial taxa were different across all 4 groups. The relative abundance of bacteria according to mapped sequence reads was significantly higher in asymptomatic and mild patients, whereas severe and negative groups were only 1.1% and 0.1%, respectively (Figure 1A, B). Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, and Fusobacteriales were the most abundant phyla among the positives. On the contrary, in the control group Actinobacteria, Proteobacteria, and Firmicutes were the most abundant group while Bacteroides and Fusobacteriales had only a negligible proportion (Figure 1C, F, G). In this context, it is important to mention that a significant proportion of severe patients from hospitals were affected by antibiotic treatment (ATB), which had the effect of reducing the total amount of bacteria, but the ratio of individual bacterial taxa was nevertheless maintained (Figure 1H, I).
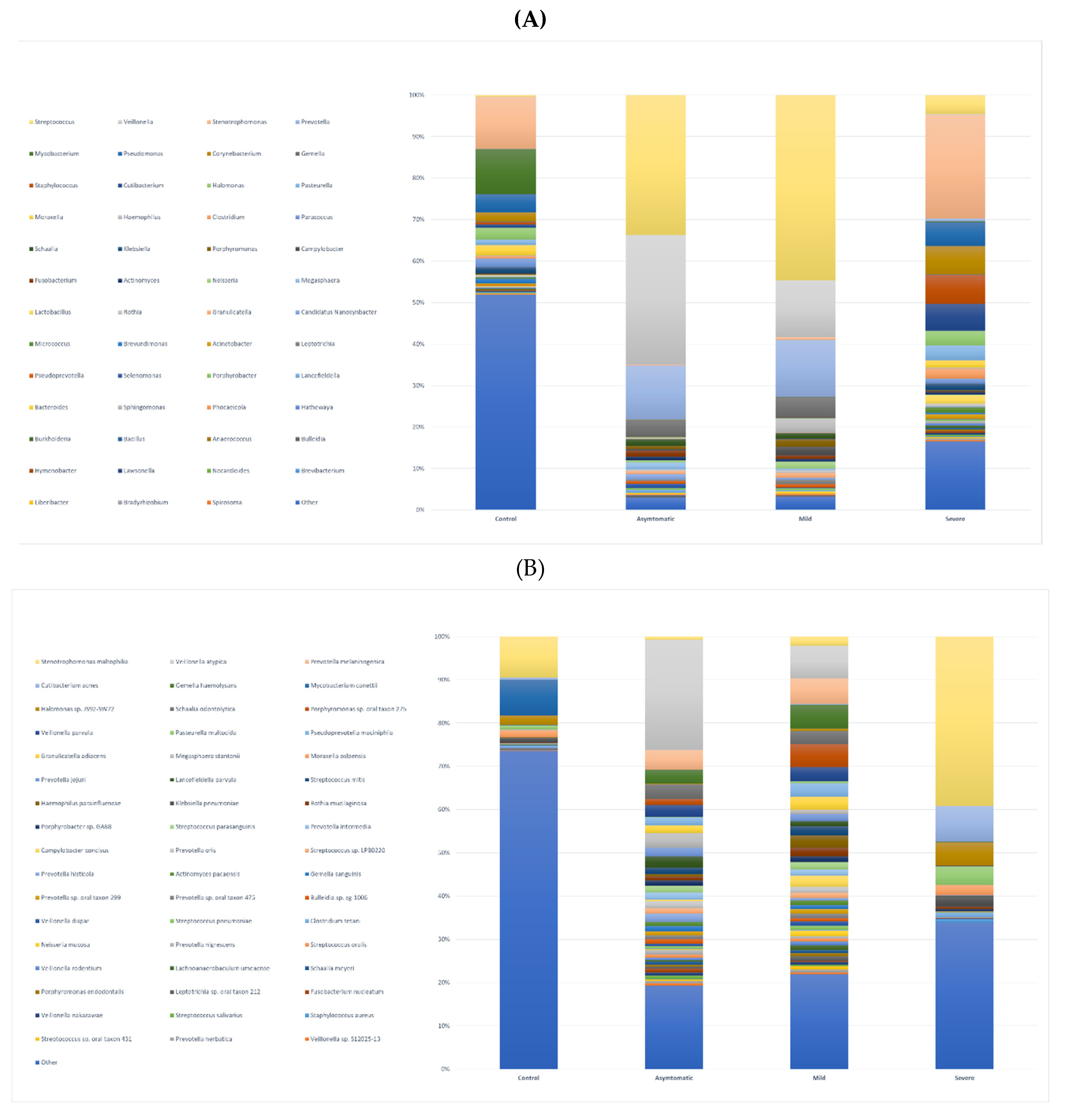
Using a threshold of 1000 reads in at least one sample, we identified a total of 944 species, 531 genera, 218 families, 110 orders, 28 classes, and 25 phyla. The highest number of species was in the severe (725), asymptomatic (692), mild (574), and less than tenfold lower numbers in the negative control group (58) (Figure 1D, E). We further profiled the microbial composition at the genus and species level in healthy controls and various COVID-19 groups. In the nasopharyngeal swab samples, there is a striking enrichment of Streptococcus, Prevotella, and Veillonella, the most abundant genera in the mild and asymptomatic group (Figure 2A, Supplementary table). Stenotrophomonas, Staphylococcus, and Corynebacterium are dominating genera in the severe group. Surprisingly again, Stenotrophomonas, Mycobacterium, and Pseudomonas are the most abundant in healthy participants. At the species level, Stenotrophomonas maltophilia, Cutibacterium acnes, and Halomonas sp. JS92-SW72 was enriched in severe patients. Veillonella atypica and Prevotella melaninogenica were most abundant in mild and asymptomatic group, and the total numbers of other species of the genus Prevotella (P. jejuni, P. histicola, P. intermedia, P. oris) and Streptococcus (S. parasanguinis, S. mitis, S. sp. LPB0220) were very similar (Figure 2B, Supplementary Table). The most frequently present species in the healthy control group as well as in the severe group were Stenotrophomonas maltophilia, Halomonas sp. JS92-SW72 and Mycobacterium canettii. Of the clinically relevant opportunistic bacterial species, e.g. Klebsiella pneumoniae, Staphylococcus aureus, Streptococcus pneumoniae and Haemophilus parainfluenzae were present in all four, although they did not significantly dominate in any of the groups and the differences between the groups were not statistically significant; rather, their prevalence was similar in the pairs of severe and negative control versus mild and asymptomatic. Of particular interest was the presence of bacterial species in all groups that are not entirely typical of the human upper respiratory tract microbiome. Both gram-negative coccobacilli, Moraxella osloensis is rarely isolated from clinical specimens and Pasteurella multocida is part of the normal flora in the nasopharynx of many wild and domestic animals, including cats and dogs.
By reducing the threshold from 1000 reads to 100 we were able to identify sequences of microorganisms that are not typical of the nares and nasopharynx microbiome e.g. Archaea, (originally Archebacteria) a distinct group separate from bacteria that commonly inhabit extreme biotypes. We identified two phyla Candidatus Micrarchaeota and Euryarchaeota. The most abundant species identified was Natrinema salinisoli.
Apart from researching bacterial content, we compared relative counts of taxa transcripts between different disease symptoms and different SARS-CoV-2 variants (alpha, delta, omicron) by LefSE tool. Here we are showing differentially represented genera under LDA treshold, further filtered by median values (Figure 1J).
Figure 1.
Summary performance of metatranscriptome analysis from nasopharyngeal swabs in 4 groups (from normalized Kraken2 counts). A Pie chart illustrating proportion of bacterial relative abundance between the COVID-19 severity groups (shown by median value) B Box plot graph illustrating relative transcript counts for each of the COVID-19 severity groups. C Pie chart distribution of 5 most frequent taxa in asymptomatic, mild, severe and negative contols. D Genus distribution and richness across each group. E. Species distribution and richness across each group F Phyla relative abundance profile. G Phyla relative abundance by degree of severity H Relative abundance of differentially represented genera by WHO SARS CoV2 variants, shown genera were chosen under conditions: LDA from LefSE > 4 and median of relative abundance in either control group or COVID-positive group > 100 ( graphs F,G, H are showing min, mean and max statistics).
Figure 1.
Summary performance of metatranscriptome analysis from nasopharyngeal swabs in 4 groups (from normalized Kraken2 counts). A Pie chart illustrating proportion of bacterial relative abundance between the COVID-19 severity groups (shown by median value) B Box plot graph illustrating relative transcript counts for each of the COVID-19 severity groups. C Pie chart distribution of 5 most frequent taxa in asymptomatic, mild, severe and negative contols. D Genus distribution and richness across each group. E. Species distribution and richness across each group F Phyla relative abundance profile. G Phyla relative abundance by degree of severity H Relative abundance of differentially represented genera by WHO SARS CoV2 variants, shown genera were chosen under conditions: LDA from LefSE > 4 and median of relative abundance in either control group or COVID-positive group > 100 ( graphs F,G, H are showing min, mean and max statistics).
3.2. Virome analyses
Among the 79 RT-PCR-positive samples, 41 complete SARS-CoV-2 genomes and 19 partially complete SARS-CoV-2 genomes (multiple scaffolds) were assembled by metatranscriptome shotgun sequencing, accounting for 62% (51/79). Additionally, we did not identify SARS-CoV-2 sequences in any of the control group samples (SARS-CoV-2 RT-PCR negative). In the asymptomatic, mild, and severe group, we identified SARS-CoV-2 clades: Alpha (B.1.1.7) – clade 20I, Delta (AY.4; AY.43; AY.43.9; AY.122; AY.9.2;) -clade 21I and in one case 21J, Omicron (BA.1.1; BA.2.9; BA.2; BA.2.67; BA.5) - clade 21L and in one case 22B, and one sample clade 20C. Alpha and Delta variants were predominant in the severe COVID group, however, 30% of analyses failed to assemble the genome due to the insufficient read counts aligned to the SARS-CoV-2 reference. Alpha was dominant in the mild group, while Omicron and Delta in an asymptomatic group with undetermined SARS-CoV-2 clade in 24% and 33% with median Ct values 33.3 and 33.8, respectively.
In addition to SARS-CoV-2, we identified sequences of other human RNA viruses, e.g. from the family Picornaviridae (Rhinovirus A). Expectedly, among the DNA viruses, these were mainly phages from Pedoviridae, Rountreeviridae, and Siphoviridae. In a few individual samples, we identified sequences of Human Herpes Virus 1, HSV-1 (Alphaherpesviridae), Human Mastadenovirus D (Adenoviridae), and Molluscum Contagiosum Virus (Poxviridae).
In terms of age, the differences were mainly between severe COVID patients from the hospital (median 68) and the other groups (M-37, A-42, N-37) (Table 1). But the distribution of taxa in age-similar patients was similar in microbiome representation in all positive participants.
Figure 2.
stacked bar graphs illustrating relative abundance in all groups of A. bacterial genera B. bacterial species.
Figure 2.
stacked bar graphs illustrating relative abundance in all groups of A. bacterial genera B. bacterial species.
4. Discussion
Our study compares the microbiome including the virome of the nasopharynx among four groups of participants based on the severity of the viral respiratory disease: negative (no signs of illness, RT-PCR negative), mild (benign symptoms, not requiring hospitalization), asymptomatic (no signs of illness, RT-PCR positive), and severely ill COVID-19 patients (breathing difficulties, pneumonia, hospitalized) using metatranscriptome shotgun sequencing. Samples were collected over an extended period of time at mobile sampling centers (NC, asymptomatic, mild) and at two hospitals (severe). In the text, for simplification, we use an abundance of bacteria or viruses, although, in reality, we are still referring to transcripts, which, however, express the representation of the active part of the microbiome. The relative abundance of bacteria is significantly higher in the mild and asymptomatic groups while only a small proportion is present in the severe and negative control (Figure 1A, B). The low abundance of bacteria in severe patients is easily explainable by the intake of antibiotics. The use of antibiotics without a clear indication of bacterial superinfection is controversial, however, it appears to have been common practice in some healthcare centers regardless of the risks of antimicrobial resistance. The side effects of corticosteroids and immunomodulatory therapies on the microbiome of the upper respiratory tract are complex and can vary depending on multiple factors such as dosage, duration, and individual factors, however, they can alter the microbial diversity in both ways: increase or decrease(Hartmann et al., 2021). Few publications report that the bacterial load increased as COVID-19 severity increased, (although differences between groups were not statistically significant) abundance of the microbiome increases with the severity of the disease (Li et al., 2023; Shilts et al., 2022). According to the severity of the disease, Firmicutes are most frequent in asymptomatic, mild, and severe patients, whereas they are significantly less abundant and proportionally represented in healthy controls. (Figure 1C, F, G). We observed a significant decrease in the abundance but not in the diversity of bacterial taxa and in the representation of major taxa in severe patients. Although even in a small group of 30 patients, we divided them into antibiotic-treated and non-antibiotic-treated patients, apart from the apparent decrease in abundance, we did not observe a change in the relative abundance of the different bacterial phyla (Figure 1H, I).
The division into two groups of COVID-positive mild and asymptomatic was motivated by the objective to investigate whether there is any difference between the microbiome of people with symptoms of respiratory disease such as rhinitis, fever, cough and people who, although infected (RT-PCR positive), do not show any symptoms. Our results indicate that both the abundance of bacteria and the proportions of representation of each taxon are very similar in both groups. Although the median Ct values in mild (24.89) and asymptomatic (28.56) appeared to be rather different, they were not statistically significant (Wilcoxon test, p=0.017), the size of both groups (25 vs. 24), and age distribution (37 vs. 42) were otherwise similar. We did not observe any significant differences at the genus and species level, as confirmed by the statistical analysis (PERMANOVA; p=0.053 and 0.024, calculated cut-off p-value was 0.0023). The most enriched genera in both groups were Streptococcus, Veillonella (both phyla Firmicutes), and Prevotella (phyla Bacteroidetes) with Veillonella atypica and Prevotella melaninogenica being the most abundant species (bacterial species and genus obrázky Fig.2a.b). This observation is consistent with findings from similar studies of the microbiome of upper respiratory airways (Liu et al., 2021; Rattanaburi et al., 2022; Ventero et al., 2021). P. melaninogenica is a Gram-negative, obligate anaerobic coccobacillus that can act as an opportunistic pathogen, and there are conflicting hypotheses for its effect on the respiratory tract. For example, P. melaninogenica has been found to be a "beneficial" member of the airway microbiome because it enhances protection against bacterial pneumonia caused mainly by Streptococcus pneumoniae (S. pneumoniae). Further, Prevotella melaninogenica was ranked among the most distinguishing bacterial species separating patients with pneumonia caused by S. pneumoniae (with less P. melaninogenica) from healthy controls (with more P. melaninogenica) (Horn et al., 2022). Interestingly, the severe group with acquired pneumonia of unknown bacterial origin had significantly less P. melaninogenica (Figure 2B, Supplementary table).
We analyzed 72 negative healthy controls, all were additionally verified/tested by RT-PCR (genes E, ORF1ab, N) and confirmed negative. Relative abundance of bacteria based on the number of mapped reads and transcripts represented the lowest proportion (0.1%) of all groups (asymptomatic 68.8%; mild 30%, and severe 1.1%) (Figure 1A). This is not that surprising since any viral infection including that of SARS-CoV-2 predisposes to secondary bacterial superinfection. In healthy adults, skin-associated bacteria are usually present in the nasal cavity, predominantly representatives of Actinobacteria (e.g., Corynebacterium spp., Propionibacterium spp.), followed by Firmicutes (e.g., Staphylococcus spp.) and Proteobacteria (Bassis et al., 2014b). In negative controls, we identified 58 species out of more than 900 species, thus contributing only about 6% to the microbial diversity and richness. Overlapping taxa at the genus and species level are illustrated in Figure 1D and E. The prevalent phyla were Actinobacteria but the dominantly invaded species was Stenotrophomonas maltophilia belonging to Proteobacteria (Pseudomonadota). It has relatively low virulence in immunocompetent persons but in patients with chronic respiratory disease, immunocompromization, prolonged antibiotic use (especially carbapenems) and long-term hospitalization or admission to intensive care unit can be a source of life-threatening complications (Looney et al., 2009).
Not surprisingly, the nasopharyngeal virome in asymptomatic, mild, and severe patients was composed predominantly of different SARS-CoV-2 variants, which correlated unambiguously with the period of occurrence of the particular strains in Slovakia. The sensitivity of metatranscriptome sequencing was only about 60%, on the other hand, the set of SARS-CoV-2 positive samples consisted of different Ct values. At the individual level, for all samples that had Ct of 20 or less, corresponding to higher viral loads, we were able to assemble the whole genome of the SARS-CoV-2 virus. Samples from a group of severe patients were collected 3-7 days after inclusion in the study, and these patients had been treated in the interim with a wide range of drugs (antibiotics, corticotherapy, immunomodulatory therapy, antivirals) that may have influenced viral load in the meantime. The mild, asymptomatic, and healthy controls, based on questionnaire information, were not taking antimicrobial therapy or any other treatment with the potential to affect the microbiome. Despite the limited number of samples in the groups with confirmed Delta, Alpha, and Omicron variants, we were interested in whether they differed specifically at the level of bacterial taxa. The relative abundance of differentially repressed genera by SARS-CoV-2 WHO variant (LefSE) figure shows that the genus Streptococcus is clearly more abundant in the Delta-positive group compared to Alpha and Omicron. Campylobacter and Haemophilus are slightly enriched in the Alpha group (Figure 1J). The clear advantage of the transcriptome approach is its ability to capture not only RNA but also DNA viruses (their transcripts). Besides SARS-CoV-2, unsurprisingly, the most frequently identified sequences were those derived from DNA viruses of bacteria i.e phages of the families Pedoviridae, Rountreeviridae, and Siphoviridae.
Sequences mapped to the human transcriptome were a non-negligible part of the analysis, but they are not the subject of this paper and deserve special attention.
5. Conclusion
We identified differences in the microbiome between different groups of COVID positive and negative controls. Despite differences in abundance, the relative abundance of individual taxa in the covid-positive groups was similar regardless of severity and treatment used. The healthy negative control group had the fewest microbes. Specific SARS-CoV-2 viral strains may potentially influence the presence of a particular type of bacteria. Metatranscriptome sequencing is a tool for the broad-spectrum study of the active part of the prokaryotic microbiome, the virome but also the human transcriptome from a variety of available sources. Although it does not accentuate its diagnostic potential in terms of sensitivity, it can add to the jigsaw puzzle of the mosaic of broad species diversity and help in understanding the complex relationships between the microbiome and human immunity under conditions of a particular disease, its severity, and at different sites in the human body. In our study, the model disease was SARS-CoV-2 with varying severity, ranging from asymptomatic to severe manifestations requiring hospitalization and intensive treatment. The nasopharynx, in turn, is a site that is well accessible for sampling, is colonized by a wide range of microorganisms that affect the local immune response, and is also a site of entry and initial multiplication of a wide range of respiratory viruses.
Funding
This article was created with the support of the OP Integrated Infrastructure for the project: Research on COVID-19 progressive diagnostic methods and biomarkers useful in early detection of individuals at increased risk of severe disease, ITMS: 313011ATA2, co-financed by the European Regional Development Fund.
References
- Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data.
- Baehren, C., Buedding, E., Bellm, A., Schult, F., Pembaur, A., Wirth, S., Ehrhardt, A., Paulsen, F., Postberg, J., & Aydin, M. (2022). The Relevance of the Bacterial Microbiome, Archaeome and Mycobiome in Pediatric Asthma and Respiratory Disorders. Cells, 11(8), 1287. [CrossRef]
- Bai, X. Bai, X., Narayanan, A., Skagerberg, M., Ceña-Diez, R., Giske, C. G., Strålin, K., & Sönnerborg, A. (2022). Characterization of the Upper Respiratory Bacterial Microbiome in Critically Ill COVID-19 Patients. Biomedicines, 10(5), 982. [CrossRef]
- Bassis, C. M., Tang, A. L., Young, V. B., & Pynnonen, M. A. (2014a). The nasal cavity microbiota of healthy adults. Microbiome, 2(1), 27. [CrossRef]
- Bassis, C. M., Tang, A. L., Young, V. B., & Pynnonen, M. A. (2014b). The nasal cavity microbiota of healthy adults. Microbiome, 2(1), 27. [CrossRef]
- Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. [CrossRef]
- Brumfield, K. D., Huq, A., Colwell, R. R., Olds, J. L., & Leddy, M. B. (2020). Microbial resolution of whole genome shotgun and 16S amplicon metagenomic sequencing using publicly available NEON data. PLOS ONE, 15(2), e0228899. [CrossRef]
- Burbank, A. J., Sood, A. K., Kesic, M. J., Peden, D. B., & Hernandez, M. L. (2017). Environmental determinants of allergy and asthma in early life. Journal of Allergy and Clinical Immunology, 140(1), 1–12. [CrossRef]
- Candel, S., Tyrkalska, S. D., Álvarez-Santacruz, C., & Mulero, V. (2023). The nasopharyngeal microbiome in COVID-19. Emerging Microbes & Infections, 12(1). [CrossRef]
- Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., Whitwham, A., Keane, T., McCarthy, S. A., Davies, R. M., & Li, H. (2021). Twelve years of SAMtools and BCFtools. GigaScience, 10(2). [CrossRef]
- di Simone, S. K., Rudloff, I., Nold-Petry, C. A., Forster, S. C., & Nold, M. F. (2023). Understanding respiratory microbiome–immune system interactions in health and disease. Science Translational Medicine, 15(678). [CrossRef]
- Hadzega, D., Minarik, G., Karaba, M., Kalavska, K., Benca, J., Ciernikova, S., Sedlackova, T., Nemcova, P., Bohac, M., Pindak, D., Klucar, L., & Mego, M. (2021). Uncovering Microbial Composition in Human Breast Cancer Primary Tumour Tissue Using Transcriptomic RNA-seq. International Journal of Molecular Sciences, 22(16), 9058. [CrossRef]
- Hartmann, J. E., Albrich, W. C., Dmitrijeva, M., & Kahlert, C. R. (2021). The Effects of Corticosteroids on the Respiratory Microbiome: A Systematic Review. Frontiers in Medicine, 8. [CrossRef]
- Heng Li. (n.d.). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. [CrossRef]
- Horn, K. J., Schopper, M. A., Drigot, Z. G., & Clark, S. E. (2022). Airway Prevotella promote TLR2-dependent neutrophil activation and rapid clearance of Streptococcus pneumoniae from the lung. Nature Communications, 13(1), 3321. [CrossRef]
- Jankauskaitė, L., Misevičienė, V., Vaidelienė, L., & Kėvalas, R. (2018). Lower Airway Virology in Health and Disease—From Invaders to Symbionts. Medicina, 54(5), 72. [CrossRef]
- Kumpitsch, C., Koskinen, K., Schöpf, V., & Moissl-Eichinger, C. (2019). The microbiome of the upper respiratory tract in health and disease. BMC Biology, 17(1), 87. [CrossRef]
- Li, J., Jing, Q., Li, J., Hua, M., Di, L., Song, C., Huang, Y., Wang, J., Chen, C., & Wu, A. R. (2023). Assessment of microbiota in the gut and upper respiratory tract associated with SARS-CoV-2 infection. Microbiome, 11(1), 38. [CrossRef]
- Liu, J., Liu, S., Zhang, Z., Lee, X., Wu, W., Huang, Z., Lei, Z., Xu, W., Chen, D., Wu, X., Guo, Y., Peng, L., Lin, B., Chong, Y., Mou, X., Shi, M., Lan, P., Chen, T., Zhao, W., & Gao, Z. (2021). Association between the nasopharyngeal microbiome and metabolome in patients with COVID-19. Synthetic and Systems Biotechnology, 6(3), 135–143. [CrossRef]
- Looney, W. J., Narita, M., & Mühlemann, K. (2009). Stenotrophomonas maltophilia: an emerging opportunist human pathogen. The Lancet Infectious Diseases, 9(5), 312–323. [CrossRef]
- Lu, J., Rincon, N., Wood, D. E., Breitwieser, F. P., Pockrandt, C., Langmead, B., Salzberg, S. L., & Steinegger, M. (2022). Metagenome analysis using the Kraken software suite. Nature Protocols, 17(12), 2815–2839. [CrossRef]
- Maier, W., & Batut, B. (2023). Mutation calling, viral genome reconstruction and lineage/clade assignment from SARS-CoV-2 sequencing data.
- Meleshko, D., Hajirasouliham, I., & Korobeynikov, A. (2021). coronaSPAdes: from biosynthetic gene clusters to RNA viral assemblies. BioRxiv.
- Minot, S., Bryson, A., Chehoud, C., Wu, G. D., Lewis, J. D., & Bushman, F. D. (2013). Rapid evolution of the human gut virome. Proceedings of the National Academy of Sciences, 110(30), 12450–12455. [CrossRef]
- Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., & Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods, 5(7), 621–628. [CrossRef]
- Nemergut, D. R., Schmidt, S. K., Fukami, T., O’Neill, S. P., Bilinski, T. M., Stanish, L. F., Knelman, J. E., Darcy, J. L., Lynch, R. C., Wickey, P., & Ferrenberg, S. (2013). Patterns and Processes of Microbial Community Assembly. Microbiology and Molecular Biology Reviews, 77(3), 342–356. [CrossRef]
- picard toolkit. (2019). Broad Institute, GitHub Repository.
- Porto, B. N. (2022). Insights Into the Role of the Lung Virome During Respiratory Viral Infections. Frontiers in Immunology, 13. [CrossRef]
- Rajagopala, S. v., Bakhoum, N. G., Pakala, S. B., Shilts, M. H., Rosas-Salazar, C., Mai, A., Boone, H. H., McHenry, R., Yooseph, S., Halasa, N., & Das, S. R. (2021). Metatranscriptomics to characterize respiratory virome, microbiome, and host response directly from clinical samples. Cell Reports Methods, 1(6), 100091. [CrossRef]
- Rattanaburi, S., Sawaswong, V., Chitcharoen, S., Sivapornnukul, P., Nimsamer, P., Suntronwong, N., Puenpa, J., Poovorawan, Y., & Payungporn, S. (2022). Bacterial microbiota in upper respiratory tract of COVID-19 and influenza patients. Experimental Biology and Medicine, 247(5), 409–415. [CrossRef]
- Rovira Rubió, J., Megremis, S., Pasioti, M., Lakoumentas, J., Constantinides, B., Xepapadaki, P., Bachert, C., Finotto, S., Jartti, T., Andreakos, E., Stanic, B., Akdis, C. A., Akdis, M., & Papadopoulos, N. G. (2023). Respiratory virome profiles reflect antiviral immune responses. Allergy. [CrossRef]
- Segata, N., Izard, J., Walron, L., Gevers, D., Miropolsky, L., Garrett, W., & Huttenhower, C. (2011). Metagenomic Biomarker Discovery and Explanation.
- Shilts, M. H., Rosas-Salazar, C., Strickland, B. A., Kimura, K. S., Asad, M., Sehanobish, E., Freeman, M. H., Wessinger, B. C., Gupta, V., Brown, H. M., Boone, H. H., Patel, V., Barbi, M., Bottalico, D., O’Neill, M., Akbar, N., Rajagopala, S. V., Mallal, S., Phillips, E., … Das, S. R. (2022). Severe COVID-19 Is Associated With an Altered Upper Respiratory Tract Microbiome. Frontiers in Cellular and Infection Microbiology, 11. [CrossRef]
- Stearns, J. C., Davidson, C. J., McKeon, S., Whelan, F. J., Fontes, M. E., Schryvers, A. B., Bowdish, D. M. E., Kellner, J. D., & Surette, M. G. (2015). Culture and molecular-based profiles show shifts in bacterial communities of the upper respiratory tract that occur with age. The ISME Journal, 9(5), 1246–1259. [CrossRef]
- Tessler, M., Neumann, J. S., Afshinnekoo, E., Pineda, M., Hersch, R., Velho, L. F. M., Segovia, B. T., Lansac-Toha, F. A., Lemke, M., DeSalle, R., Mason, C. E., & Brugler, M. R. (2017). Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Scientific Reports, 7(1), 6589. [CrossRef]
- Ventero, M. P., Cuadrat, R. R. C., Vidal, I., Andrade, B. G. N., Molina-Pardines, C., Haro-Moreno, J. M., Coutinho, F. H., Merino, E., Regitano, L. C. A., Silveira, C. B., Afli, H., López-Pérez, M., & Rodríguez, J. C. (2021). Nasopharyngeal Microbial Communities of Patients Infected With SARS-CoV-2 That Developed COVID-19. Frontiers in Microbiology, 12. [CrossRef]
- Wood, D. E., & Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biology, 15(3), R46. [CrossRef]
- Zhu, T., Jin, J., Chen, M., & Chen, Y. (2022). The impact of infection with COVID-19 on the respiratory microbiome: A narrative review. Virulence, 13(1), 1076–1087. [CrossRef]

Table 1.
Study cohort, Ct mean values (only severe, mild, asymptomatic).
| . | Number | M | F | Median age | SD | M | SD | F | SD |
|---|---|---|---|---|---|---|---|---|---|
| Negative | 72 | 26 | 46 | 37 (25–57) | 10.59 | 42 (25–58) | 9.05 | 35 (25-75) | 11.45 |
| Severe | 30 | 16 | 14 | 68 (32–90) | 13 | 69.5 (32–90) | 13.6 | 64 (41-77) | 11.7 |
| Mild | 25 | 13 | 12 | 37 (17–57) | 11.83 | 38 (17–57) | 12.77 | 32.5 (19-57) | 10.54 |
| Asymptomatic | 24 | 14 | 10 | 42 (20–49) | 7.86 | 39 (21–49) | 9.35 | 43 (36-48) | 3.72 |
| 151 | 69 | 82 | 39.5 | 40.5 yrs | 39 yrs | ||||
| 45.70% | 54.30% | ||||||||
| Median Ct value E gene | SD | ||||||||
| Severe | 28.06 (13.9-37.3) | 7.34 | |||||||
| Mild | 24.89 (15-38.77) | 5.32 | |||||||
| asymptomatic | 28.56 (19.24-33.59) | 4.72 |
M-male, F-female, SD-standard deviation, Ct-threshold cycle in RT-PCR.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



