
Submitted:
07 June 2023
Posted:
07 June 2023
You are already at the latest version
Abstract
Polyglycolic acid (PGA) is a promising polymer in the packaging field owing to excellent hydrolysis, heat resistance and gas barrier properties, but there are limited in application due to poor toughness. For this reason, a covalently bonded chain extender is introduced to increase compatibility with flexible polymers. However, covalent bonds are unfavorable for application to degradable plastics because of require a lot of energy for reverse reactions. Therefore, it is intended to effectively control the ductility of blending plastics by using a novel ionic chain extender having a relatively weaker non-covalent bond than the existing covalent bond. Polycaprolactone (PCL), which has biodegradability and flexibility, was selected as a blending polymer. For comparison, a covalently reactive chain extender (G-CE) and a non-covalently ionic chain extender (D-CE) were synthesized and compounded to blending plastics. Each chain extenders were improved the compatibility between PGA and PCL, and the ductility of the PGA/PCL blending plastics were greatly enhanced with non-covalently bonding D-CE than with covalently bonding G-CE. At this time, the ductility of PGA/PCL blending plastic without CE is 7.2%, the ductility of 10D with D-CE is 26.6%, and the ductility of 10G with G-CE is 18.45%. Therefore, it was confirmed that the novel ionic chain extender inducing non-covalent bonds improves the compatibility between PGA and PCL and is more advantageous in enhancing ductility through a reversible reaction.
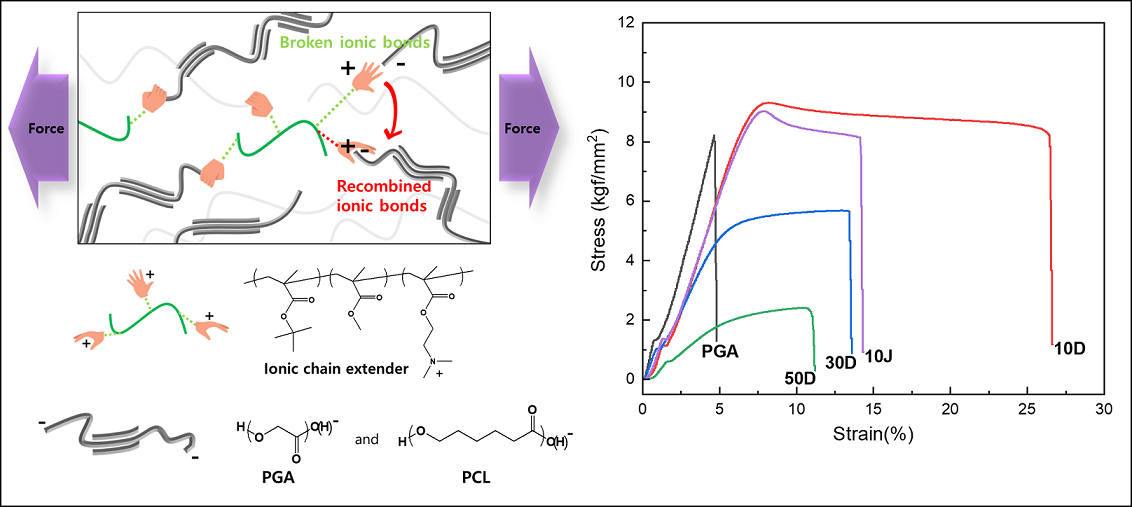
Keywords:
Ionic chain extender
; Non-covalent bonds
; Ductility
; Polyglycolic acid
; Blending plastics
1. Introduction
Polyglycolic acid (PGA) is composed of relatively simple aliphatic polyester, and is one of the representative biodegradable plastics. The PGA are decomposed into low molecules by hydrolysis of ester groups in the main chain and then finally decomposed into carbon dioxide through a biodegradation pathway in which they are metabolized [1,2,3]. In addition, the chemical structure of PGA is simple aliphatic polyester without side chains. Therefore, the molecular structure has high density, high crystallinity, and small free volume. For this reason, it shows a high level of mechanical strength compared to other biodegradable plastics [1,4,5]. Recently, PGA make an attempt to applicate in the packaging fields, because it became possible production from synthesis gas and industrial tail gas [6,7].
However, it is limited in application to the packaging field due to its brittleness and poor ductility, and some papers have reported that PGA has a very low elongation at break of less than about 5% [8,9]. In addition, PGA has difficulty selected processing temperature because the pyrolysis temperature and melting temperature are adjacent to about 250-280 ℃ and about 220-230 ℃ [10,11]. Therefore, plans have been attempted to improve the processability of PGA through compounding of additives for increasing the thermal decomposition temperature, or blending with a flexible polymer to improve ductility. Recently, biodegradable polymers have been blended as flexible polymers which poly(butylene adipate-co-terephthalate) (PBAT) [17], poly(propylene carbonate) (PPC) [18], polyhydroxyalkanoates (PHAs) [19] and polycaprolactone (PCL) [20]. However, most of the flexible polymers showed a limited effect on improving the ductility due to low compatibility with a PGA matrix which strong inter-intermolecular interactions through hydrogen bonds [21,22]. Accordingly, chain extender (CE) as a compatibilizer is added to increase the interface interaction between the flexible polymer and PGA, and the most representative chain extender is Joncryl® CE (BASF Inc., USA). The Joncryl® CEs have functional groups of epoxy or isocyanate, and exhibit the characteristics of increasing viscoelasticity and reducing melt flow index through covalently bonding with the blending polymer chains [23,24,25].
In addition, these chain extenders improve the stretchability when used in an ap-propriate amount, but there is a problem that the hardness increases due to a cross-linking reaction depending on the amount introduced and the stretchability is inhibited. Many studies were reported in order to control to ductility of PGA by using CEs, but all of the CEs were chemical reactive compatibilizers by covalent bonding [23,26]. These reactive CEs are required a lot of energy for the reverse reaction, and they are hard to separate in the hydrolytic degradation process. Usually, the reactive CEs could inhibit the degradation rate due to capping the end group of PGA after covalent bonding. This behavior becomes a factor that hinders the use of hydrolyzable plastics [26,27].
Accordingly, we would like to propose that the possibility of a new chain extender can control the ductility using an ionic bond with relatively weak bonding strength than a covalent bond. The ionic bonds are being applied in the field of self-healing becuase can repeat decomposition and recombination under conditions such as temperature or presence of ions [28,29,30]. Some literatures are mentioned that composites with quaternary ammonium (QA) can increase the fluidity of the polymer matrix and reduce frictional resistance under shear stress by causing a decrease in pour viscosity [31,32].
Therefore, a novel ionic chain extender was studied to control the ductility of brittle PGA, and applied as blending plastics to confirm the physical property behavior. Additionally, PCL was selected as a blending polymer owing to enable ionic bonding like PGA which a carboxyl group at the end group.
2. Materials and Methods
2.1. Materials
The methyl methacrylate (MMA), tert-butyl methacrylate (t-BMA), glycidyl methacrylate (GMA), and 2-(dimethylamino)ethyl methacrylate (DMAEMA), which monomers were all purchased from Sigma-Aldrich for chain extenders synthesis. Furthermore, 2,2'-azobisisobutyronitrile (AIBN) as an initiator, toluene as a solvent, and hexane as a precipitation solvent after synthesizing the chain extender, were also supplied from Sigma-Aldrich. The PGA (inherent viscosity 1.3-1.7 dL/g) was supplied by META BIOMED Co. Ltd., Republic of Korea, and the PCL (Mn 80,000 g/mol, Mw/Mn <2) was obtained from Sigma-Aldrich. The Joncryl® (ADR-4368) was purchased from BASF Co. Ltd for compare to mechanical properties of blending plastics.
2.2. Preparation of Chain Extenders
The chain extenders were synthesized using different monomers according to the bonding method of functional groups. The covalently bonded chain extender (G-CE) was synthesized by radical polymerization. The monomers (GMA 3 g, MMA 6g, and t-BMA 6g) were mixed with solvent (toluene 15 g) in round bottom flask with magnetic stirring at 200 rpm. And the above mixture was thermostated by oil bath at 70 ℃ in an argon atmosphere. Then, the initiator (AIBN 0.15 g, dissolved in 10 ml of solvent in advance) was slowly dropwise for 1 hour using a syringe pump, and then synthesized for 4 hours. After the reaction, the reactant was cooling to room temperature, and washed with repeated 4 or more times by precipitation and re-dissolution using hexane. Finally, the product was obtained by drying overnight in a vacuum oven. The ionic chain extender (D-CE) was synthesized in the same method above, and was prepared by changing the monomers to DMAEMA 1.5 g, MMA 7.5 g, and t-BMA 6 g.
2.3. Compounding of PGA/PCL Blending Plastics with/without Chain Extenders
In order to confirm the effect of the synthesized chain extenders on the blending plastic, PGA and PCL were compounded according to their respective compositions as shown in Table 1. The PGA and PCL were dried overnight in vacuum oven prior to compounding. The PGA/PCL blending plastics were prepared by internal mixer (QM310S, Republic of Korea) with screw speed of 60 rpm at 230 ℃. Then, the samples prepared through each compounding were formed into specimens at a temperature of 230 ℃ using a Hydraulic Laboratory Press (Carver, USA).
2.4. Characterization
The chemical structure of synthesized chain extenders were determined by proton nuclear magnetic resonance spectroscopy (1H-NMR, Bruker 500 MHz, Germany) with deuterated chloroform (CDCl3) as the solvent. Melt flow index (MFI) of PGA/PCL blending plastics with/without chain extender were measured using QM280A (QMESYS Inc., Republic of Korea) according to ASTM D1238. To confirm the crystallinity behavior of the blending plastic according to the introduction of the chain extenders were analyzed by differential scanning calorimetry (DSC 8500, PerkinElmer Inc., USA). Mechanical properties of blending plastics were measured using a universal testing machine (UTM, QMESYS Inc., Republic of Korea) at the crosshead speed of 10 mm/min. The morphologies of PGA/PCL blending plastics were obtained using a JSM-6701F field emission-scanning electron microscope (FE-SEM, JEOL Inc., Japan). Each PGA/PCL blending plastic samples were immersed in liquid nitrogen and then crushed, before being coated with gold for the cross-section measurements.
3. Results
3.1. Chemical Structure of the Chain Extenders which Functionalized to Different Groups
To confirm the chemical structures of the two synthesized chain extenders were conducted to H-NMR analysis (Figure S1). The chain extenders were synthesized using similar monomers, whereas functional monomer is different. The G-CE has epoxy functional groups capable of chemical bonding and the D-CE has tertiary amine functional groups capable of ionic bonding. Therefore, these chain extenders were shown similar backbone of the methyl groups, in addition to indicate the peaks corresponding to polyacrylates derived from MMA and t-BMA. The common peaks of methyl protons were measured that the signals of MMA, t-BMA, and backbone were observed at 3.5-3.7, 1.4-1.5, and 0.7-1.3 ppm, respectively (Figure S1.a and b) [33].
On the other hand, these were confirmed to different peaks of functional groups. The G-CE was shown that it has epoxy groups as a functional group through the peaks at 2.6 and 2.8 ppm corresponding to -CH2 in the epoxy group (Figure S1.a). And, the D-CE was confirmed to the peaks of tertiary amine groups at 2.3 ppm of -CH3 and 2.6 ppm of -CH2, which indicates that the tertiary amine functional groups capable of ionic bonding have been well introduced to new chain extender (Figure S1.b) [33,34].
3.2. Evaluation of Changes in Melt Flow Index Characteristics according to Heat Exposure Times and Chain Extender Contents
Melt flow index (MFI) was measured to determine the melt processability of PGA/PCL according to the blending ratio and the introduction of the chain extender (Table S1). In the first, the melt index of PGA over time were analyzed to explore the thermal decomposition time upon melting of PGA before the introduction of the chain extenders (Figure 1.a. inside graph). The initial MFI value of PGA was 24.5 g/10min, and the MFI increased as the heat treatment time increased. In particular, the MFI are gradually increased to 26.94 g/10min until the heat treatment time of 6 minutes, but rapidly increased to 30 g/10min or more after 8 minutes, which indicating that the thermal decomposition of PGA proceeded rapidly. Accordingly, the experiment was conducted by setting the PGA compounding time to within 6 minutes.
Furthermore, the MFI of the PGA/PCL blending plastics were analyzed which confirmed to effect of introducing each synthesized chain extenders (Figure 1.a). When 0.2 wt% of the G-CE or D-CE was added, these were confirmed that the MFI significantly decreased compared to without addition of any chain extenders. At this time, the MFI were measured at 18.39 g/10min at 10G_0.2 and 22.66 g/10min at 10D_0.2.
In the case of G-CE, with a covalent bond, were not largely changed their MFI according to the introduced amount of the chain extender. In detail, the lowest MFI was 15.56 g/10min at 10G_0.5, and the MFI tended to increase slightly to 16.28 g/10min at 10G_1.0. Since PGA and PCL have one carboxyl group at the end group, they can exhibit the highest molecular weight when fully reacted with the epoxy groups of the G-CE. However, when the chain extender is added in excess, the molecular weight is relatively reduced because the content of the epoxy groups is greater than the content of the adjacent carboxyl groups. As a result, G-CE was tended to relatively increased of MFI, when it is added in amounts of 1 wt% or more.
On the other hand, novel chain extender of D-CE, with a ionic bond, were showed high MFI compared to the control except for content of 2 wt% (10D_0.2). The MFI measured of PGA/PCL blending plastics according to the introduced amount of D-CE were 22.66, 44.66, and 52.82 g/10min, corresponding to 10D_0.2, 10D_0.5, and 10D_1.0 respectively. Those ionic chain extender is forming the ions from the quaternary ammonium groups for ionic bonding with the carboxylic groups of PGA. Moreover, the composite with quaternary ammonium (QA) can increase the fluidity of the matrix after mixing with polymer as PGA and PCL, and reduce the frictional resistance during shear stress by causing a decrease in pour viscosity [31,32]. For this reason, the MFI of the PGA/PCL blending plastics were greatly increased to use of the D-CE with a ionic bond compared with introducing G-CE with a covalent bond. Here, we can propose that the introduction of reversible ionic chain extenders can dramatically change the melt viscosity of blending plastics than covalent bonds.
Next, the effects of the two chain extenders on the melt viscosity behavior of the PGA/PCL blended plastic were confirmed (Figure 1.b). In general, as the content of PCL with a low melting point increases, the MFI of the PGA/PCL blending plastic increases. As shown in Figure 1.b, the MFI of the sample according to the PCL ratio was 37.02, 63.42, and 44.07 g/10min at 10, 30, and 50, respectively. Exceptionally, the '50' sample with a PCL content of 50% showed a lower MFI than the '30' sample. can be said to be the result. In fact, it can be inferred that the compatibility between the polymers is significantly lower than that of other samples based on the largest MFI error width for the sample. On the other hand, it was found that the samples to which the two chain extenders were added had significantly low MFI, and it was confirmed that the change in MFI according to the increase in PCL content was small. As a result, it was confirmed that the two chain extenders improved the compatibility of PGA and PCL and induced low MFI through interaction with the polymer.
3.3. Crystallinity of PGA/PCL Blending Plastics according with Chain Extenders
DSC was analyzed for PGA/PCL blended plastics to confirm the effect of the introduction of covalent and non-covalent chain extenders on the crystallinity of polymers (Figure 2 and Table S2). In general, pure PGA has a chemical structure of high chain regularity without side chains, so it has a melting point (Tm) of about 200-225 ° C and a crystallization temperature (Tc) of about 190-195 ° C, which higher than those of pseudolinear aliphatic polyesters [10,35]. These Tm and Tc were almost unchanged when blended with PCL without a chain extender, suggesting that PCL had little effect on the thermal properties of PGA.
On the other hand, the crystallization temperature of PGA was shifted lower, and the interaction between PGA molecules was limited due to chain entanglement and cross-linking structure formation by covalent bonds.
Furthermore, the low Tc equivalent to Tc of PGA shifted to lower temperatures with the reaction with G-CE, suggesting that there are more nucleation centers in the compatible system (Figure 2.b). We reasoned that this is the effect of the nucleation of the domains of PGA or PCL themselves and the nucleation impurities of their matrix polymers [35,36]. Inhibited the crystal growth process of entangled chains. Therefore, crystallinity, crystallization enthalpy and melting temperature decreased. The PGA/PCL blend exhibits two melting endothermic peaks, suggesting that the crystal was not perfect due to the complexity of the macromolecular structure. G-CE shifted the crystallization temperature of PCL to a higher level and slightly increased the crystallinity of PCL segments due to covalent bonds.
3.4. Ductile properties of PGA/PCL Blending Plastics with Novel Chain Extenders
To confirm the effect of the chain extender using covalent or ionic bonds on the blending plastic matrix, mechanical properties were measured and ductility change behavior was evaluated. Covalent G-type and ionic D-type chain extenders were analyzed by applying them to PGA/PCL blended plastics each containing different PCL contents (Figure 3). In addition, the 10J in graph is a PGA/PCL10 blending sample with containing the Joncryl® (ADR-4368), which a typical chain extender used in the packaging industry, and it was prepared to compare the mechanical properties of the synthesized chain extenders. The elongation tended to decrease as the amount of PCL introduced increased with or without a chain extender, and it was confirmed that the same results of morphological behavior was showed in FE-SEM (Figure 3.a and 4). The compatibility is inhibited with increased content of PCL, and it can be inferred that these are become due to the particle size of the dispersed PCL in the PGA matrix increases and more non-uniform.
In common, the elongation rate increased and the tensile strength decreased according to the introduction of the two chain extenders, showing similar behavior (Figure 3.b and c). However, increased ductility which the D-CE of ionic bonding were shown compared to the G-CE of the covalent bonding (Figure 3.c). As the PCL content increased, the elongation rates when G-CE was introduced were 18.45% at 10G, 11.15% at 30G, and 7.20% at 50G, respectively. And, the samples introduced with D-CE were 26.60% at 10D and 13.60% at 30D, respectively. and 11.20% at 50G. Compared to the case of PCL 10 wt% blending, which showed the greatest change in ductility, the ductility of pure blending plastic without CE increased by about 3.69 times compared to 7.20% of blending plastic with ionic bond D-CE. In addition, all samples introduced with ionic bonded D-CE showed relatively higher tensile strength than covalently bonded G-CE. The tensile strength of the blended plastic according to the type of chain extender was 8.990 kgf/mm2 at 10G and 9.314 kgf/mm2 at 10D, 4.645 kgf/mm2 at 30G and 5.686 kgf/mm2 at 30D, 2.146 kgf/mm2 at 50G and 2.411 kgf/mm2 at 50D, respectively (Table S3).
As shown in the schematic diagram of Figure 3.d, the ionic bond reacted in the amorphous region is pulled in both directions, and the bond is broken and recombination with a new adjacent carboxyl group is repeated. The quaternary ammonium can increase the fluidity of the polymer matrix and reduce frictional resistance under shear stress by causing a decrease in pour viscosity. It was argued that this cause was due to repeated cleavage and recombination of ionic bonds. In this study, this tendency was clearly shown, and it indicated a higher level of ductility than the existing covalent chain extenders [37].
3.5. Morphology Behavior of PGA/PCL Blending Polymers Matrix according to Type of Chain Extenders
The morphology behavior was analyzed to confirm the miscibility change between PGA and PCL by the ionic chain extender (Figure 4). Because mechanical properties are closely related to the shape and compatibility of the complex [38].
The cross section of the PGA/PCL blending plastic, which blending 10 wt% of PCL without CE, was shown that the size is about 0.2-1 μm dispersed in the PGA matrix (Figure 4.a). And, the blending plastic with D-CE was indicated that the size is uniformly distributed about 0.5-0.7 μm (Figure 4.b). This suggests that the condensation was relieved by the interaction with the PGA interface by the chain extender without the PCL particles independently aggregating. These properties are similar to existing covalent chain extenders, and it is show that ionic chain extenders with relatively low bond strength can also play a sufficient role in improving the compatibility of PGA and PCL. In addition, this compatibility improvement is responsible for the increase in ductility described above.
In particular, PCL particles are form non-uniform 'islands' of about 100 μm in size when PGA and PCL are blended at a ratio of 50 wt% without CE. Furthermore, the phase is partially reversed to form PGA in the PCL area, and it was also dispersed in the form of particles (Figure 4.c and d).
On the other hand, phase inversion of some polymers was observed even when D-CE was added, but was relatively uniform the size of the dispersed particles in the matrix. The uniform dispersion of PGA and PCL can provide balanced strength. Moreover, it can be confirmed by increasing ductility due to improved compatibility, as shown in the previous mechanical property analysis results [39].
4. Discussion
A novel ionic chain extender for controlling the ductility of PGA was studied. Among the amorphous blending polymers, PCL capable of ionic bonding was selected and blended to confirm the effect of the ionic chain extender. Through the MFI behavior, it was possible to establish the processing conditions of PGA within 6 minutes, and it was confirmed that the covalent (G-CE) and non-covalent (D-CE) bond chain extenders can lower the melt viscosity of the blending plastics. Exceptionally, the non-covalent ionic chain extender (D-CE) showed a sharp change in MFI as the introduced content increased, unlike the covalent chain extenders. This suggests that it may be more advantageous to control the melt viscosity.
Significantly increased ductility was confirmed in all cases that G-CE and D-CE were applied to PGA/PCL blending plastics, respectively. Compared to the elongation of 7.20% which PGA 90 wt% and PCL 10 wt% blending plastic without chain extender, the blending plastic with D-CE (10D) showed the largest change in ductility with the elongation of 26.60%. Moreover, these ionic bonded blending plastics with D-CE exhibited relatively higher tensile strength than covalent bonded blending plastics with G-CE. As the amount of PCL increases, the elongation increases but the tensile strength decreases, which is similar to the trade-off of elongation and tensile strength of general polymers.
This trend was also confirmed in the cross-sectional morphology of the blending plastics, and it were showed that the PCL were distributed in the form of 'islands' as the content increased. The 'islands' dispersion hinders interactions between PGA matrix with PCL and lowers dispersibility, resulting in reduced tensile strength and elongation.
Despite this tendency, it was confirmed that the elongation and tensile strength increased with the ionic bonded chain extender (D-CE) than without chain extenders at high content of PCL. Each chain extenders were improved the compatibility between PGA and PCL, and the ductility of the PGA/PCL blending plastics were greatly enhanced with non-covalently bonding D-CE than with covalently bonding G-CE.
Therefore, it was confirmed that the ionic bond proposed in this study can exert a sufficient effect as a chain extender, like to the covalent bond.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Chemical structure analysis of synthesized covalent a) and non-covalent b) chain extenders.; Table S1: Melt flow index (MFI) results of PGA/PCL blending plastics according to blending ratio and introducing with covalent or non-covalent chain extenders.; Table S2: Differential scanning calorimetry (DSC) of PGA and PGA/PCL blending plastics with or without chain extenders.; Table S3: Mechanical properties of PGA/PCL blending plastics according to blending ratio and introducing with or without chain extenders.
Author Contributions
Conceptualization, H.J.K. and K.H.; methodology, H.J.K., J.Y. L. and K.H.; validation, J.Y.L. and K.H.; formal analysis, H.J.K.; investigation, H.J.K., J.J. and K.H.; resources, H.J.K., J.J. and K.H.; data curation, H.J.K. and K.H.; writing—original draft preparation, H.J.K.; writing—review and editing, H.J.K., W.G.K, J.Y.L. and K.H.; visualization, K.H.; supervision, W.G.K, J.Y.L. and K.H.; project administration, K.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) through (High value-added Food Technology Development Program), funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (NA-23-0005), and has been conducted with the support of the Korea Institute of Industrial Technology as " Development of high barrier PGA polymerization technology with processability (kitech IR-21-0036)".
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Samantaray, P.K.; Little, A.; Haddleton, D.M.; McNally, T.; Tan, B.; Sun, Z.; Huang, W.; Ji, Y.; Wan, C. Poly(glycolic Acid) (PGA): A Versatile Building Block Expanding High Performance and Sustainable Bioplastic Applications. Green Chem. 2020, 22, 4055–4081. [Google Scholar] [CrossRef]
- Swider, E.; Koshkina, O.; Tel, J.; Cruz, L. J.; de Vries, I. J. M.; Srinivas, M. Customizing poly (lactic-co-glycolic acid) particles for biomedical applications. Acta biomaterialia. 2018, 73, 38–51. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, B.; Xi, Z.; Zhao, L.; Cen, L.; Yang, Y. A Comparable Study of Polyglycolic acid’s Degradation on macrophages’ Activation. Mater. Sci. Eng. C. 2020, 109, 110574. [Google Scholar] [CrossRef] [PubMed]
- Jem, K.J.; Tan, B. The Development and Challenges of Poly (lactic Acid) and Poly (glycolic Acid). Adv. Ind. Eng. Polym. Res. 2020, 3, 60–70. [Google Scholar] [CrossRef]
- Yamane, K.; Sato, H.; Ichikawa, Y.; Sunagawa, K.; Shigaki, Y. Development of an industrial production technology for high-molecular-weight polyglycolic acid. Polym. J, 2014; 46, 769–775. [Google Scholar]
- Schmidt, C.; Behl, M.; Lendlein, A.; Beuermann, S. Synthesis of High Molecular Weight Polyglycolide in Supercritical Carbon Dioxide. RSC Adv. 2014, 4, 35099–35105. [Google Scholar] [CrossRef]
- Göktürk, E.; Pemba, A.G.; Miller, S.A. Polyglycolic Acid from the Direct Polymerization of Renewable C1 Feedstocks. Polym. Chem. 2015, 6, 3918–3925. [Google Scholar] [CrossRef]
- Samantaray, P. K.; Ellingford, C.; Farris, S.; O’Sullivan, C.; Tan, B.; Sun, Z.; McNally, T.; Wan, C. Electron beam-mediated cross-linking of blown film-extruded biodegradable PGA/PBAT blends toward high toughness and low oxygen permeation. ACS Sustain. Chem. Eng, 2022; 10, 1267–1276. [Google Scholar]
- Rigotti, D.; Soccio, M.; Dorigato, A.; Gazzano, M.; Siracusa, V.; Fredi, G.; Lotti, N. Novel biobased polylactic acid/poly (pentamethylene 2, 5-furanoate) blends for sustainable food packaging. ACS Sustain. Chem. Eng, 2021; 9, 13742–13750. [Google Scholar]
- Nakafuku, C.; Yoshimura, H. Melting parameters of poly (glycolic acid). Polymer. 2014, 45, 3583–3585. [Google Scholar] [CrossRef]
- Gautier, E.; Fuertes, P.; Cassagnau, P.; Pascault, J. P.; Fleury, E. Synthesis and rheology of biodegradable poly (glycolic acid) prepared by melt ring-opening polymerization of glycolide. J. Polym. Sci., Part A1, 2009; 47, 1440–1449. [Google Scholar]
- Wang, K.; Shen, J.; Ma, Z.; Zhang, Y.; Xu, N.; Pang, S. Preparation and Properties of Poly (ethylene glycol-co-cyclohexane-1, 4-dimethanol terephthalate)/Polyglycolic Acid (PETG/PGA) Blends. Polymers. 2021, 13, 452. [Google Scholar] [CrossRef] [PubMed]
- Liu, G. C.; He, Y. S.; Zeng, J. B.; Xu, Y.; Wang, Y. Z. In situ formed crosslinked polyurethane toughened polylactide. Polym. Chem. 2014, 5, 2530–2539. [Google Scholar] [CrossRef]
- Yang, X.; Zhong, S. Properties of maleic anhydride-modified lignin nanoparticles/polybutylene adipate-co-terephthalate composites. J. Appl. Polym. Sci. 2020, 137, 49025. [Google Scholar] [CrossRef]
- Pinheiro, I. F.; Ferreira, F. V.; Alves, G. F.; Rodolfo, A.; Morales, A. R.; Mei, L. H. I. Biodegradable PBAT-based nanocomposites reinforced with functionalized cellulose nanocrystals from Pseudobombax munguba: rheological, thermal, mechanical and biodegradability properties. J. Polym. Environ. 2019, 27, 757–766. [Google Scholar] [CrossRef]
- Lai, L.; Li, J.; Liu, P.; Wu, L.; Severtson, S. J.; Wang, W. J. Mechanically reinforced biodegradable Poly (butylene adipate-co-terephthalate) with interactive nanoinclusions. Polymer. 2020, 197, 122518. [Google Scholar] [CrossRef]
- Andrzejewski, J.; Cheng, J.; Anstey, A.; Mohanty, A. K.; Misra, M. Development of toughened blends of poly (lactic acid) and poly (butylene adipate-co-terephthalate) for 3d printing applications: Compatibilization methods and material performance evaluation. ACS Sustainable Chem. Eng, 2020; 8, 6576–6589. [Google Scholar]
- Hedrick, M. M.; Wu, F.; Mohanty, A. K.; Misra, M. Morphology and performance relationship studies on biodegradable ternary blends of poly (3-hydroxybutyrate-co-3-hydroxyvalerate), polylactic acid, and polypropylene carbonate. Rsc Advances. 2020, 10, 44624–44632. [Google Scholar] [CrossRef]
- Kumar, V.; Sehgal, R.; Gupta, R. Blends and composites of polyhydroxyalkanoates (PHAs) and their applications. Eur. Polym. J. 2021, 161, 110824. [Google Scholar] [CrossRef]
- Przybysz-Romatowska, M.; Haponiuk, J.; Formela, K. Poly (ε-caprolactone)/poly (lactic acid) blends compatibilized by peroxide initiators: Comparison of two strategies. Polymers. 2020, 12, 228. [Google Scholar] [CrossRef]
- Nishimura, F.; Hoshina, H.; Ozaki, Y.; Sato, H. Isothermal crystallization of poly (glycolic acid) studied by terahertz and infrared spectroscopy and SAXS/WAXD simultaneous measurements. Polym. J. 2019, 51, 237–245. [Google Scholar] [CrossRef]
- Sato, H.; Miyada, M.; Yamamoto, S.; Reddy, K. R.; Ozaki, Y. C–H⋯ O (ether) hydrogen bonding along the (110) direction in polyglycolic acid studied by infrared spectroscopy, wide-angle X-ray diffraction, quantum chemical calculations and natural bond orbital calculations. RSC advances. 2016, 6, 16817–16823. [Google Scholar] [CrossRef]
- Rasselet, D.; Caro-Bretelle, A. S.; Taguet, A.; Lopez-Cuesta, J. M. Reactive compatibilization of PLA/PA11 blends and their application in additive manufacturing. Materials. 2019, 12, 485. [Google Scholar] [CrossRef]
- Kahraman, Y.; Özdemir, B.; Gümüş, B. E.; Nofar, M. Morphological, rheological, and mechanical properties of PLA/TPU/nanoclay blends compatibilized with epoxy-based Joncryl chain extender. Colloid and Polymer Science. 2023, 301, 51–62. [Google Scholar] [CrossRef]
- Matumba, K. I.; Motloung, M. P.; Ojijo, V.; Ray, S. S.; Sadiku, E. R. Investigation of the Effects of Chain Extender on Material Properties of PLA/PCL and PLA/PEG Blends: Comparative Study between Polycaprolactone and Polyethylene Glycol. Polymers. 2023, 15, 2230. [Google Scholar] [CrossRef]
- Palsikowski, P. A.; Kuchnier, C. N.; Pinheiro, I. F.; Morales, A. R. Biodegradation in soil of PLA/PBAT blends compatibilized with chain extender. Journal of Polymers and the Environment. 2018, 26, 330–341. [Google Scholar] [CrossRef]
- Tatai, L.; Moore, T. G.; Adhikari, R.; Malherbe, F.; Jayasekara, R.; Griffiths, I.; Gunatillake, P. A. Thermoplastic biodegradable polyurethanes: the effect of chain extender structure on properties and in-vitro degradation. Biomaterials. 2007, 28, 5407–5417. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Liu, R.; Liang, Z.; Yang, J.; Zhang, R.; Zhou, Z.; Nie, Y. Design of Self-Healing Rubber by Introducing Ionic Interaction to Construct a Network Composed of Ionic and Covalent Cross-Linking. Ind. Eng. Chem. Res, 2019; 58, 14848–14858. [Google Scholar]
- Yang, Y.; Urban, M. Self-Healing of Polymers via Supramolecular Chemistry. Adv. Mater. Interfaces. 2018, 5, 1800384. [Google Scholar] [CrossRef]
- Mai, D.; Mo, J.; Shan, S.; Lin, Y.; Zhang, A. Self-Healing, Self-Adhesive Strain Sensors Made with Carbon Nanotubes/Polysiloxanes Based on Unsaturated Carboxyl–Amine Ionic Interactions. ACS Applied Materials & Interfaces, 2021; 13, 49266–49278. [Google Scholar]
- Wah, C. A.; Choong, L. Y.; Neon, G. S. Effects of titanate coupling agent on rheological behaviour, dispersion characteristics and mechanical properties of talc filled polypropylene. European Polymer Journal. 2000, 36, 789–801. [Google Scholar] [CrossRef]
- Liu, Z.; Gilbert, M. Structure and properties of talc-filled polypropylene: Effect of phosphate coating. J. Appl. Polym, Sci, 1996; 59, 1087–1098. [Google Scholar]
- Liu, L.; Zhang, S.; Shi, B.; Mai, D.; Du, X.; Lin, B. Synthesis and thermal analysis of methacrylate ester-based linear triblock copolymers-grafted multiwalled carbon nanotubes. Journal of Thermal Analysis and Calorimetry. 2015, 119, 2029–2037. [Google Scholar] [CrossRef]
- Comí, M.; Lligadas, G.; Ronda, J. C.; Galià, M.; Cádiz, V. Adaptive bio-based polyurethane elastomers engineered by ionic hydrogen bonding interactions. European Polymer Journal. 2017, 91, 408–419. [Google Scholar] [CrossRef]
- Magazzini, L.; Grilli, S.; Fenni, S. E.; Donetti, A.; Cavallo, D.; Monticelli, O. The Blending of Poly (glycolic acid) with Polycaprolactone and Poly (l-lactide): Promising Combinations. Polymers. 2021, 13, 2780. [Google Scholar] [CrossRef] [PubMed]
- Righetti, M.C.; Marchese, P.; Vannini, M.; Celli, A.; Lorenzetti, C.; Cavallo, D.; Ocando, C.; Müller, A.J.; Androsch, R. Polymorphism and Multiple Melting Behavior of Bio-Based Poly(propylene 2,5-furandicarboxylate). Biomacromolecules. 2020, 21, 2622–2634. [Google Scholar] [CrossRef]
- Comí, M.; Lligadas, G.; Ronda, J. C.; Galià, M.; Cádiz, V. Adaptive bio-based polyurethane elastomers engineered by ionic hydrogen bonding interactions. European Polymer Journal. 2017, 91, 408–419. [Google Scholar] [CrossRef]
- Wang, X.; Peng, S. X.; Chen, H.; Yu, X. L.; Zhao, X. P. Mechanical properties, rheological behaviors, and phase morphologies of high-toughness PLA/PBAT blends by in-situ reactive compatibilization. Compos. B Eng. 2019, 173, 107028. [Google Scholar] [CrossRef]
- Sangroniz, L.; Wang, B.; Su, Y.; Liu, G.; Cavallo, D.; Wang, D.; Müller, A.J. Fractionated crystallization in semicrystalline polymers. Prog. Polym. Sci. 2021, 115, 101376. [Google Scholar] [CrossRef]
Figure 1.
Melt flow index (MFI) of PGA/PCL/CE blending plastics according to ratio of chain extenders a) and PGA/PCL blending with/without CEs according to blending ratio b).
Figure 1.
Melt flow index (MFI) of PGA/PCL/CE blending plastics according to ratio of chain extenders a) and PGA/PCL blending with/without CEs according to blending ratio b).
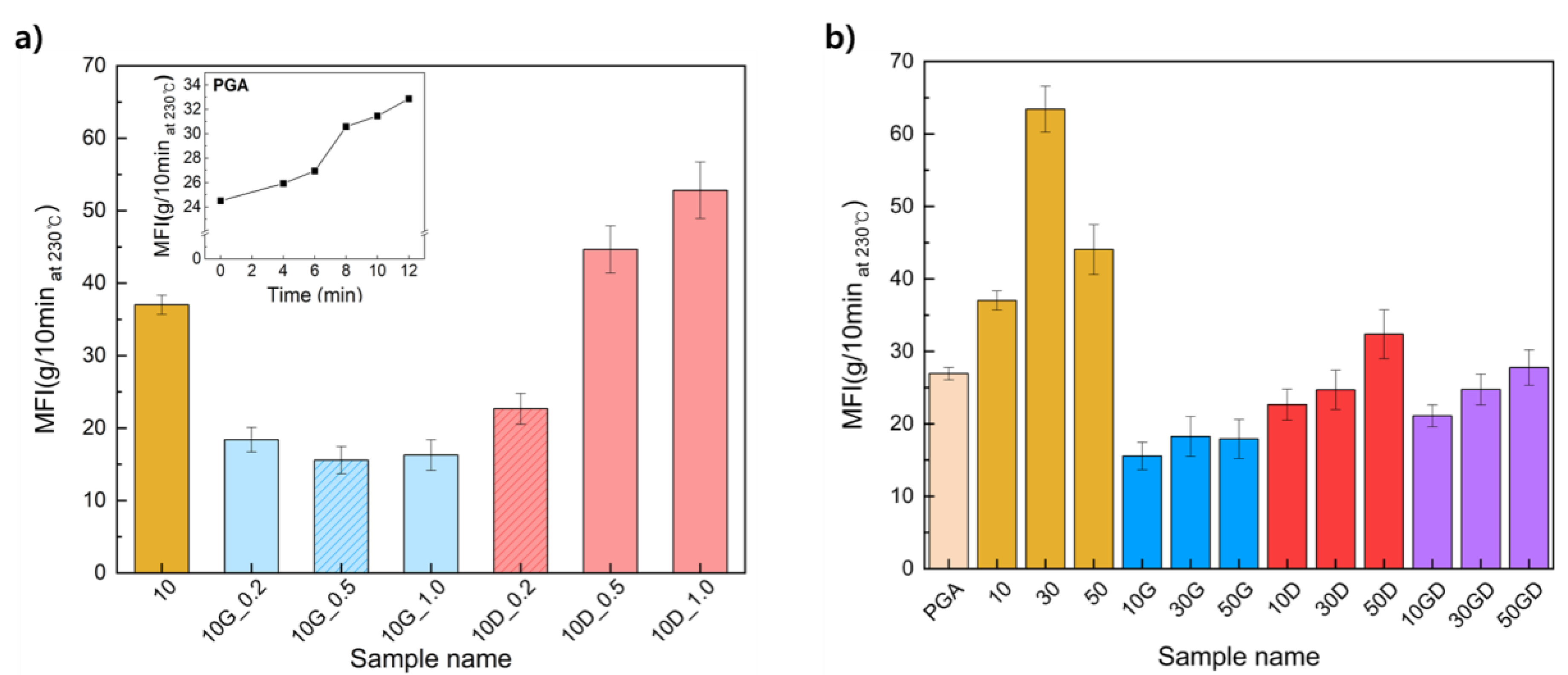
Figure 2.
Differential scanning calorimetry (DSC) thermographs of neat PGA, PGA/PCL, and PGA/PCL/CEs blending plastic samples about Tc1 a), Tc2 b), and Tm c).
Figure 2.
Differential scanning calorimetry (DSC) thermographs of neat PGA, PGA/PCL, and PGA/PCL/CEs blending plastic samples about Tc1 a), Tc2 b), and Tm c).
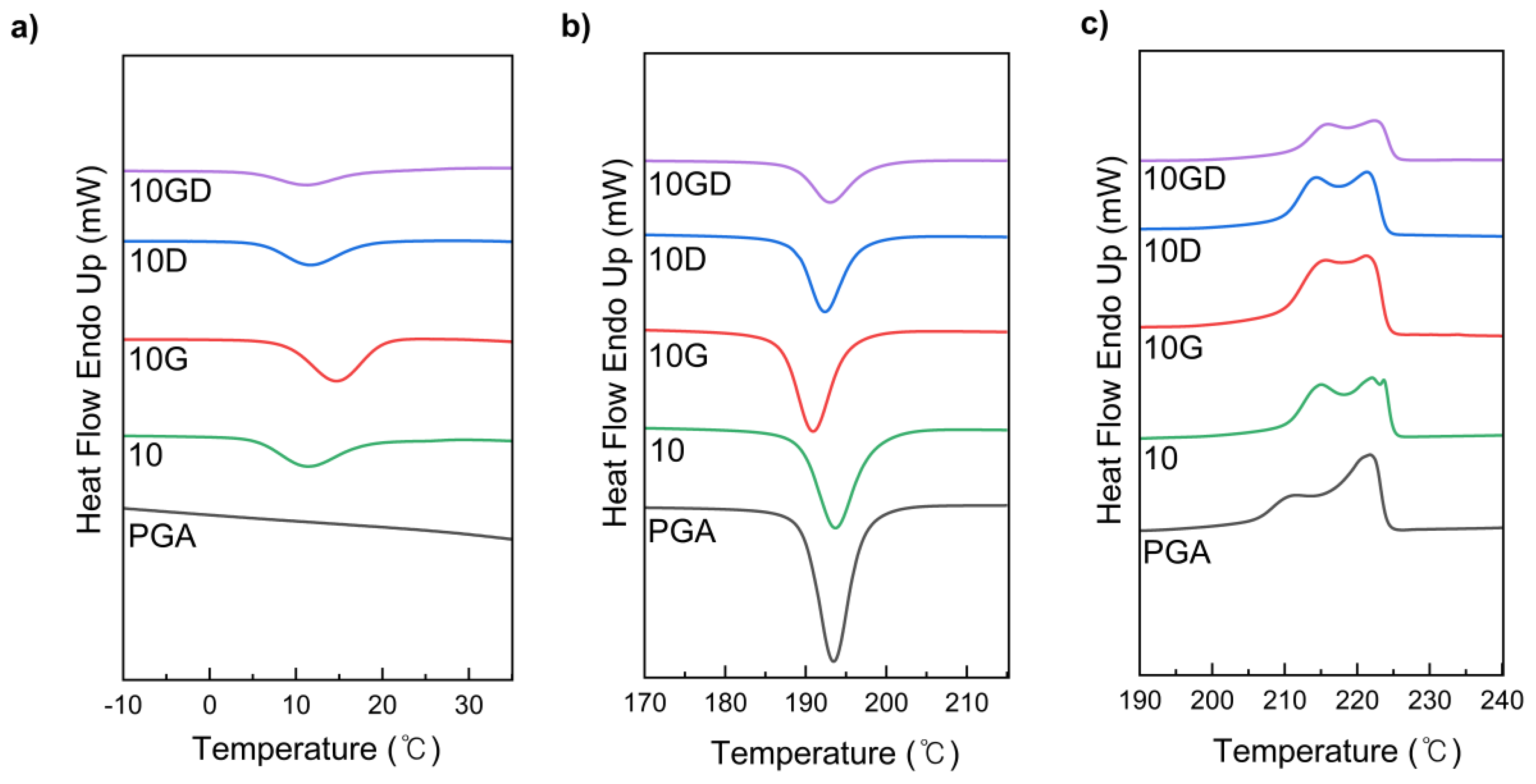
Figure 3.
Stress–strain curves of PGA/PCL blending plastic sheets without CEs a), with G-CE b), and with D-CE c). The schematic diagram of ionic bonding reaction d).
Figure 3.
Stress–strain curves of PGA/PCL blending plastic sheets without CEs a), with G-CE b), and with D-CE c). The schematic diagram of ionic bonding reaction d).
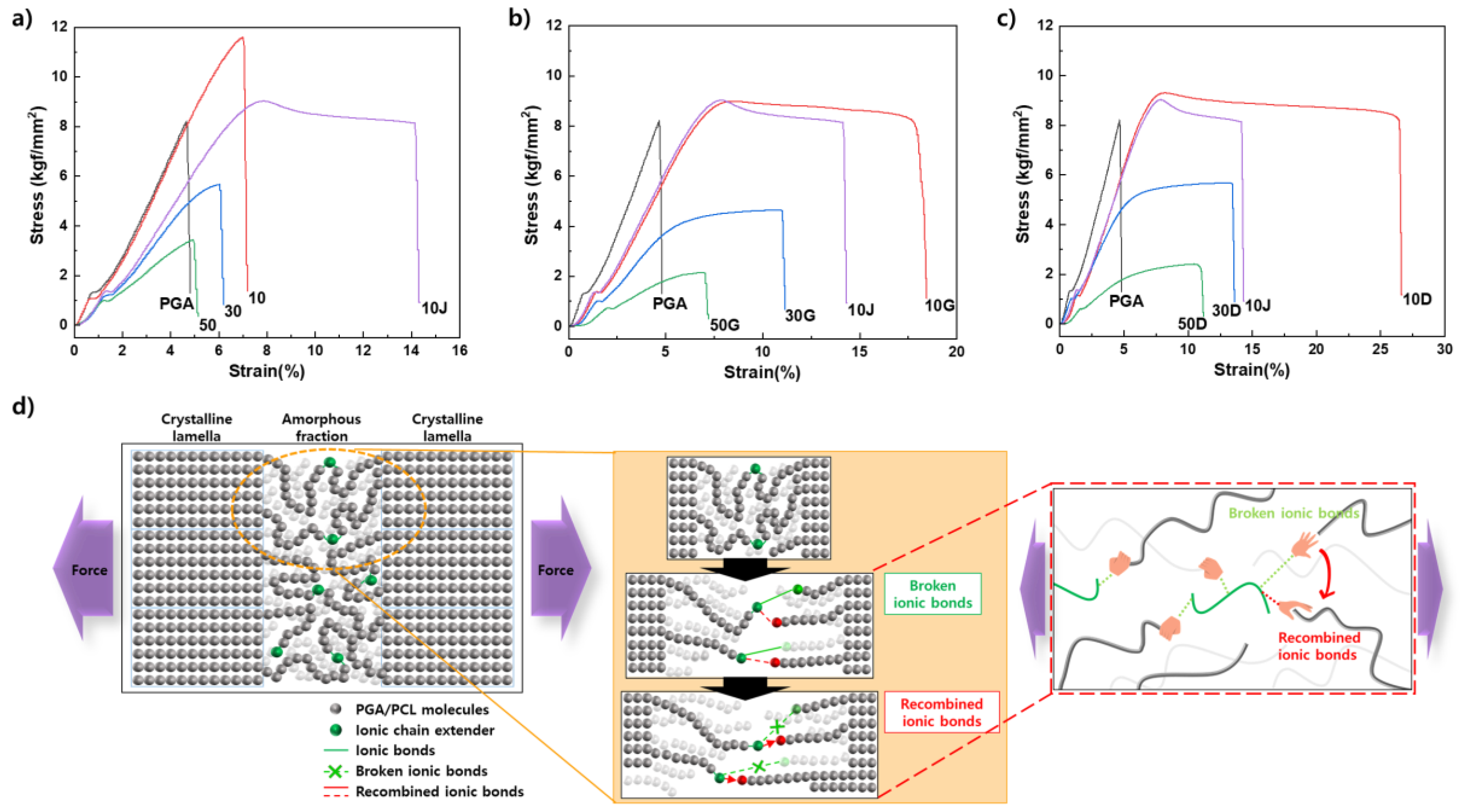
Figure 4.
SEM images for cross-section of PGA/PCL blending plastics with/without D-CE. PGA/PCL(90/10) without CE a), PGA/PCL(90/10) with D-CE b), PGA/PCL(50/50) without CE c), and PGA/PCL(50/50) with D-CE d).
Figure 4.
SEM images for cross-section of PGA/PCL blending plastics with/without D-CE. PGA/PCL(90/10) without CE a), PGA/PCL(90/10) with D-CE b), PGA/PCL(50/50) without CE c), and PGA/PCL(50/50) with D-CE d).


Table 1.
Compositions of PGA/PCL blending plastics and PGA/PGA/CE blending plastic samples.
| Sample name | PGA (g) | PCL (g) | D-CE*a (g) | G-CE*b (g) | ADR-4368*c (g) |
|---|---|---|---|---|---|
| PGA | 150 | - | - | - | - |
| 10 | 135 | 15 | - | - | - |
| 30 | 105 | 45 | - | - | - |
| 50 | 75 | 75 | - | - | - |
| 10D (10D_0.2) | 135 | 15 | 0.30 | - | - |
| 10D_0.5 | 135 | 15 | 0.75 | - | - |
| 10D_1.0 | 135 | 15 | 1.50 | - | - |
| 30D | 105 | 45 | 0.30 | - | - |
| 50D | 75 | 75 | 0.30 | - | - |
| 10G_0.2 | 135 | 15 | - | 0.30 | - |
| 10G (10G_0.5) | 135 | 15 | - | 0.75 | - |
| 10G_1.0 | 135 | 15 | - | 1.50 | - |
| 30G | 105 | 45 | - | 0.30 | - |
| 50G | 75 | 75 | - | 0.30 | - |
| 10J | 135 | 15 | - | - | 1.50 |
*a ; Ionic chain extender (D-CE) *b ; Covalent chain extender (G-CE) *c ; Joncryl® chain extender purchased from BASF Co. Ltd.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



