
Submitted:
07 June 2023
Posted:
08 June 2023
You are already at the latest version
Abstract
The use of hydrogen fuel cells as a mobile source of electricity might prove to be a keystone in future decarbonization of heavy duty road and marine transport. Due to complex interplay of various physicochemical processes in the fuel cells, further development of these devices will rely on concerted effort of researchers from various fields, e.g. chemistry, physics, electric and mechanical engineering. This results in knowledge gaps which are filled by the information, dispersed in wide range of literature, but rarely covered in a short and condensed form. To address this issue and with the aim of providing a concise and simple explanation of fuel cell operation, we propose a simple educational model of the most relevant processes in the fuel cell, aimed at proper description of causal relations. The derivation of model equations provides an intuitive understanding of electric potentials acting on protons on the microscopic level and relates this knowledge to the common terminology in fuel cell research community, such as catalyst electric overpotential and internal membrane resistance. The results of the model fit well with experimental data, indicating that a simple description, provided in the paper, is sufficient for intuitive understanding of fuel cell operation.
Keywords:
Fuel Cell
; PEMFC
; Modelling
1. Introduction
Electrochemical devices such as fuel cells, electrolysers and batteries are key building blocks that enable achieving future environmental and climate goals. They are indispensable components of future energy systems and of electrified powertrains of vehicles. Therefore, a wide spectrum of stakeholders is involved in development and use of these devices, covering the entire chain from the core components over system integration to integration of the fuel cell systems into the energy of vehicle systems, which crucially influence design, functioning and use of these devices. Due to widely different academic backgrounds, the stakeholder often lack in-depth knowledge and understanding of electrochemical and transport processes in the fuel cells, which can lead to unnecessary frustration in hinder the progress in understanding and development of named systems. These facts provide the main motivation of the present paper, where we intend to systematically elucidate physical background of basic electrochemical phenomena in fuel cells.
In this systematic journey that starts with basic thermodynamics, continues with its application to real materials encountered in fuel cells and electrolysers and ends with elucidating processes in non-equilibrium, i.e. finite current, conditions, following key questions will be answered using insightful phenomenological explanations and their interrelations to main governing equations:
- Why do two different reactants fed to two electrodes produce a finite voltage or potential difference?
- Why and how does a selection of reactants influences fuel cell voltage?
- Why hydrogen molecules at the anode of the fuel cell favour decomposition to two protons and two electrons despite the fact that products feature much higher chemical energy?
- What is overpotential? Or a more intriguing version of this question: Is overpotential a cause or a consequence of electrochemical process in the fuel cell?
- Why does a fuel cell voltage decreases with increasing current density?
- What forces the protons to travel through the membrane from anode to cathode side of the fuel cell?
The answers to these questions are generally known, but are mostly dispersed across large volume of books and papers, ranging from basic electrochemistry [1] to more focused works on fuel cell operation principles [2,3]. Basic electrochemistry books [1] do well in explaining microscopic principles of electrochemical processes, but do not apply them to specific selection of chemical species, materials and "experimental" conditions, found in a realistic fuel cell device. Literature, concerned with fuel cell operation principles, on the other hand, takes the results of electrochemical studies as a given fact and applies them to realistic systems without detailed explanation of their origin, often avoiding a proper explanation of causal relations between processes. Most notably, the electric current through the fuel cell is often posed as a driving force of the processes in the device, from which overpotential on interfaces are determined. In literature there are roughly four distinct ways how this modelling approach is being handled. The first is the use of a data-driven approach [4,5,6,7,8], which is widely used in system-level analyses, but has limited applicability in studies aiming at simultaneous optimisation of performance and lifetime. This is because the accuracy of data-driven models does not usually extend beyond the parameter variation learning space and training data-driven models on a variation space with very high dimensionality proves difficult and time consuming. The second approach is based on electrochemical models based on the Tafel equation derived from the BV equation, where the forward reaction is superimposed on the backward reaction. This is a reasonable approximation for operating points with high current densities and consequently high activation overpotentials. However, it has a major drawback in the low current density region where the error of the approximation increases exponentially as the current density approaches zero, which inevitably means that the activation losses cannot be well characterised. The procedure described is used in the publications [9,10,11,12,13,14,15,16,17,18,19]. The third approach is based on electrochemical models based on the Tafel equation, which is extended with different corrections for the low current density range in order to reduce the calibration error of the terms in the activation loss expression. In references [20,21], a linearisation of the BV equation for the anode side and the use of the Tafel equation for the cathode side is proposed to solve this problem. In the references [22,23,24], the subsequent replacement of the natural logarithm in the Tafel equation by a sine hyperbolic fitting is proposed. The proposed substitution successfully reduces the total error in the low current density range, but leads to incorrectly placed individual terms in the activation loss equation, which affects the calibration procedure and the performance of the model. The fourth approach is based on a thermodynamically consistent derivation of the [25,26] model and provides a consistent analytically derived expression for the polarisation curve in the low current density regions where the parameters describing the activation losses are typically prone to calibration errors. While these approaches are in general computationally less demanding and can often work well in stationary conditions yielding plausible results, they can lead to incomplete or misguided understanding of causal relations between processes. On the other hand, mean-field elementary kinetics models [27,28], relying on binding energies of reactants and intermediates on catalyst surfaces in electrochemical environments intuitively and in detail cover causal relation between the processes on the micro-scale and serve as direct scale bridging platform to results of density functional theory calculations [29,30,31]. However, owing to high computational expense they cannot be directly utilized in the fuel cell operation simulations.
Compared to these models, which are aimed at precise and efficient description of fuel cell operation, the proposed model focuses on improving the intuitive understanding of the physical and chemical processes underlying the fuel cell operation. The paper at hand thus enriches the existing literature by systematically addressing the causal chain of phenomena from basics electrochemistry, over microscopic principles to fuel cell operation principles. Proper understanding of physical processes in the fuel cell is crucial for efficient modelling of the devices, which can in turn give additional insight of fuel cell operation. We, therefore, in parallel with explanation of processes also propose a simple model fuel cell, focused primarily on proper understanding of causal relations between species concentrations and electric potentials in the system.
2. Model description
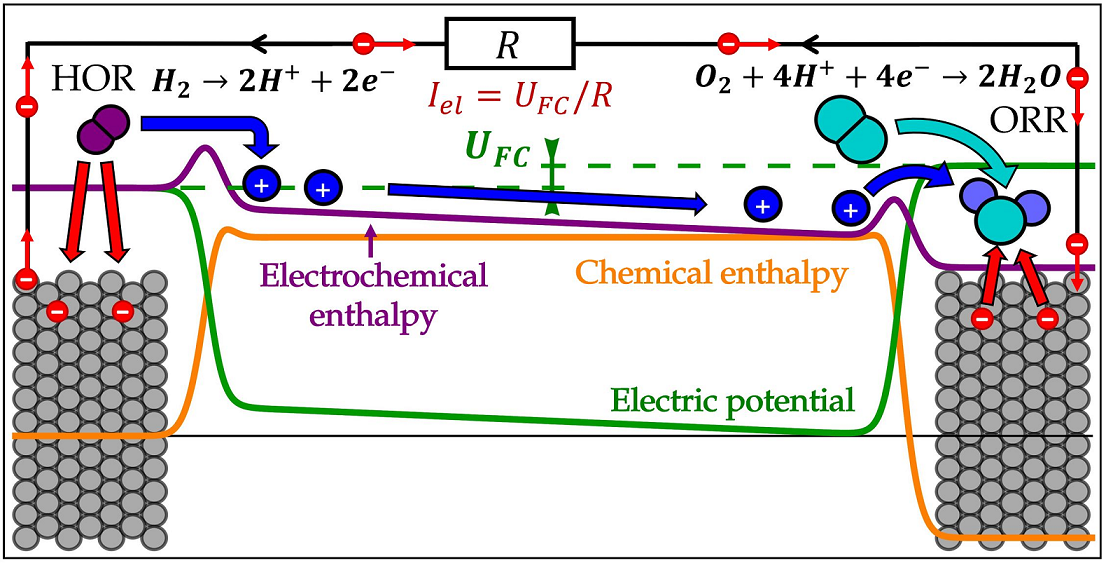
The fuel cell produces electric energy from hydrogen and oxygen by feeding each of reactants to the gas feed channels each side of the cell, schematically presented in Figure 1. Hydrogen diffuses through the anode gas diffusion layer (GDL) to the anode catalyst layer, composed of carbon support structure on which catalyst (typically platinum) nanoparticles are deposited. Catalyst surface serves as an active area on which electrochemical hydrogen oxidation reaction (HOR) takes place, splitting hydrogen molecule into two electrons and two protons. Catalyst carbon support and GDL are both electrically conductive, so the electrons, produced in HOR can travel through them and through the external electric load R towards the the cathode side of the fuel cell. Protons produced in HOR enter the ionomer, a solid proton conducting material, typically made out of Nafion or similar polymer. Ionomer coveres the catalyst particles and connects them with the membrane, made out of the same material, which allows the protons to travel from anode to cathode side, while (nearly fully) blocking the diffusion of gaseous reactants. In the cathode catalyst layer, the protons are recombined in electrochemical oxygen reduction reaction (ORR) with electrons and oxygen, which diffuses from the cathode feed channels through cathode GDL to the cathode catalyst layer. This results in formation of water, which diffuses back through the GDL to the gas channels and through the membrane to the anode side making possible proton transport via the hydronium ion and enhancing the Grotthuss mechanism [32]. The aim of the model is to describe these processes with simple mathematical relation, focused on explaining what are the driving forces behind the processes and how they can be used to extracted useful electric work from the fuel cell.
The state of the fuel cell will be described by 7 time dependent quantities (marked with red in Figure 1):
- - hydrogen concentration in anode catalyst layer,
- - oxygen concentration in cathode catalyst layer,
- - water concentration in cathode catalyst layer,
- - proton concentration in anode catalyst layer,
- - proton concentration in cathode catalyst layer,
- - anode catalyst electric potential,
- - cathode catalyst electric potential.
Operating conditions of the fuel cell are determined by the species concentrations in the channels, influenced by the feed rate and pressure, temperature controlled by cooling system, and external electric load. These quantities are used as the input parameters of the model and are marked in blue in Figure 1:
- - hydrogen concentration in anode gas feed channel,
- - oxygen concentration in cathode gas feed channel,
- - water concentration in cathode gas feed channel,
- - fuel cell temperature,
- - electric resistivity of external load.
The dynamics of model variables are determined by a simple set of ordinary differential equations, which describe the following processes:
- electrochemical (EC) processes in the anode and cathode catalyst layer
- diffusion (dif) of gaseous reactants and products through the GDL
- proton transport through the membrane (mem),
- electron (el) transport through the external electric load.
In Eq. 1 we list the contributions of specific processes to the time dynamics of each of the model variables.
Note that all the modelled variables are affected by electrochemical processes (EC), describing the production/consumption of species, while the species transport is described by either diffusion (gaseous species), membrane transport (protons) or electric conduction (electrons).
In the rest of the paper, we will explain in detail the underlying physics of each of the processes and how it relates to different model parameters, deriving mathematical expression for each of the terms in Eq. (1). We start in Section 3 with general consideration of electrochemical processes to explain the dynamics in the anode electrode, and extend the explanation to the cathode electrode in Section 4. Gas diffusion is explained in Section 5, proton transport in the membrane in Section 6 and electron transport in Section 7.
3. Electrochemical processes on hydrogen electrode
3.1. Microscopic picture of electrochemical reaction
We start our discussion by explaining the processes that occur on the anode side of the fuel cell. The reason for this is that the anode reaction is much simpler compared to cathode reaction, comprising only a reaction between hydrogen, protons and electrons:
This reaction is the same as the one used in standard hydrogen electrode, which is defined as a porous platinum electrode, dipped in 1 M acid solution and surrounded by hydrogen bubbling around it at pressure of 1 atm [1]. These idealized conditions are very similar to the ones in fuel cell anode and will be used for explaining the basic principles of electrochemical processes, with detailed differences between both systems discussed later in Section 3.6.
The first step in understanding the electrochemical reaction (2) is to identify how do species, involved in the reaction, relate to the definition of the standard hydrogen electrode. The protons on the left-hand side of the reaction are present in the acid solution. Strong acids completely dissociate in water, resulting in concentration of protons being equal to the molarity of the acid. To form gaseous hydrogen (right-hand side of Eq. (2)), the protons are combined with the electrons. These are provided from the metal surface, in our case platinum catalyst, in which the electrons are not strongly bound, but form an electron cloud and can, therefore, be easily detached from the metal in certain conditions.
The two-sided arrows in Eq. (2) indicate that the reaction can proceed in both ways, depending on several conditions, which we will discuss in detail later in the paper. In general, the direction of reaction is determined by which configuration is energetically favourable. Comparing the energy of isolated protons and electrons to the hydrogen molecule shows that state of particles, bound into a molecule, is energetically favourable and the preferred direction of reaction will be from right to the left [33]. We, therefore, first examine the situation where no hydrogen is present and only protons from the acid interact with electrons from the platinum and where initially the acid and the catalyst are at the same electric potential. When we dip the electrode in acid, the electrons on its surface will come into contact with protons in acid and proton reduction reaction
will take place, consuming protons from the acid and electrons from the electrode. This will slightly lower the proton concentration in the acid, decreasing its acidity (increasing pH value). Even more importantly, if the electrode is electrically insulated, the deficit of negatively charged electrodes will result in the increase in its electric potential. As a result, the negatively charged ions from the acid will be attracted to its surface, forming the so-called electric double layer [1]. While the detailed picture of its formation and properties are important for the reaction dynamics on the electrode surface, a simple picture of finite increase in electric potential over a small surface layer, as shown in Figure 2, is sufficient for basic understanding of its effect on reaction dynamics. Since electric potential difference between electrolyte and electrode is now positive, double layer will represent an energy barrier for protons that want to react with electrons, therefore slowing down the reaction. This intuitive explanation neatly introduces two important factors, affecting the rate of the reaction: 1. the concentration of reactants needed for the reaction to occur, and 2. the energy difference between initial and final state, which is closely related to the electric phenomena on the electrode surface since interacting particles are electrically charged.
The hydrogen oxidation reaction,
does not occur spontaneously since it is energetically unfavourable for hydrogen to decompose into protons. However, as presented in the next section, proper management of species concentration and electric potential drop across the double layer can result in reaction also proceeding in this direction.
3.2. Simple kinetic model of electro-chemical reaction rates
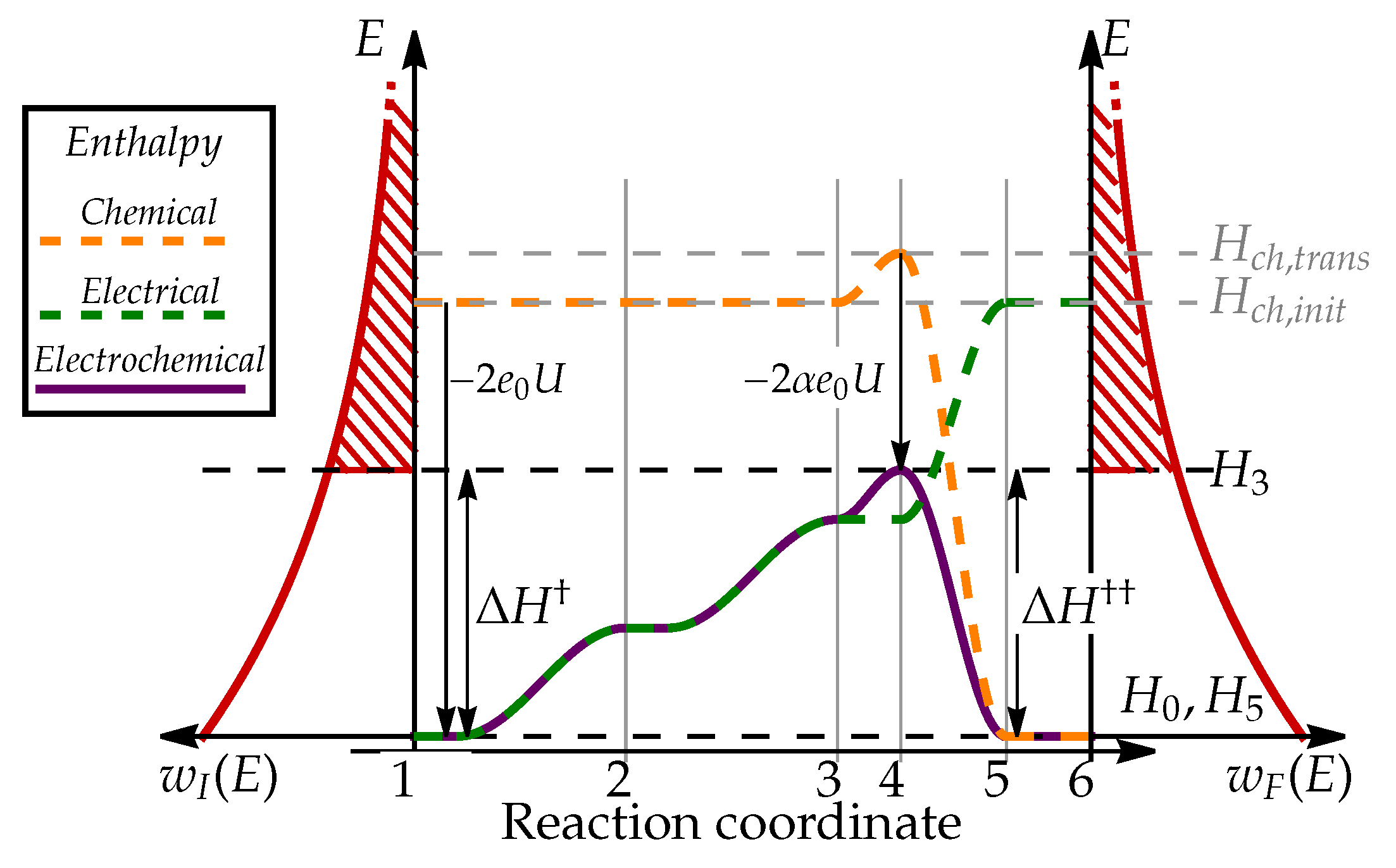
To present the process in mathematical terms, we will use a simple kinetic model for describing the rates of electrochemical reactions [1]. To describe the reaction kinetics, we identify 6 distinct states, schematically presented in Figure 2 through which the system transits from the initial state (state 1 in Figure 2) to the final state (state 6). Chemical enthalpy of i-th state is denoted , electric energy as , and total electrochemical enthalpy as .
When electrode is not electrically charged, the transition of protons from bulk electrolyte to electrode surface () does result in any change of system enthalpy (), as schematically presented in Figure 3 a). In the next step (), the protons need to arrange themselves in a configuration where they can bond into hydrogen molecule using two electrons. This so-called transition state is energetically unfavorable due to electrostatic repulsion between two positively charged particles, resulting in chemical enthalpy of transition state being higher than enthalpy of other configurations (as schematically presented in Figure 3). When protons and electrons form hydrogen molecule (), the enthalpy decreases significantly due to formation of chemically stable and electrically neutral molecule with enthalpy .
When electrode is electrically charged, electric double layer, formed on its surface, acts as a electric potential step of magnitude
which affects the energy of electrically charged species, e.g. protons, when they approach the surface, as shown in Figure 2. Since the hydrogen molecule is electrically neutral, electrochemical enthalpy of states 5 and 6 will remain unchanged
The enthalpy change of the initial states 1 can be explained in two ways. If we assume that electrode potential was increased compared to fixed potential in the electrolyte, the energy of each electron will decrease by . If, on the other hand, we assume that electrolyte potential was lowered compared to fixed electrode potential, the energy of each proton will decrease by . Both descriptions are aligned and indicate that the energy decrease is proportional to the number of charged particles in each state, resulting in enthalpy
To describe the electric energy of transition states, we consider the energy needed for transition of proton from bulk electrolyte to the electrode surface, which requires approximately . However, if we consider that proton adsorbed to the electrode surface still feels EDL electric field and that it charge is partially neutralized by surface electrons, the resulting in energy difference is a bit lower (see Appendix A). The difference in proton electrostatic energy between electrolyte and electrode surface is parameterized by so-called charge transfer coefficient , () [1], which describes how the energy of transition states relates to the electrode potential U. Enthalpy of transition states 2 and 3 is therefore
We further assume that the charge configuration of transition state 4 is similar to state 3:
This relation communicates an intuitive explanation of charge transfer coefficient as a description of energy change of transition state on charged electrode. As we will further explore in Section 3.5, charge transfer coefficient plays significant role in how the rates of oxidation and reduction are affected by the changes in electric potential U.
This intuitive description of energy landscape of reaction coordinate will allow us to calculate the rate of the reaction (2). We start with assumption that the rate of reaction is proportional to the concentration of particles, involved in the reaction: the higher the concentration, the more likely the reaction is to occur. Secondly, only the particles with sufficiently high energy to overcome the enthalpy barrier can react. This energy is provided by thermal fluctuations. We know from thermodynamics that in any system at finite temperature, the energy of individual constituents is not a fixed value, but distributed among them according to the Boltzmann distribution [34]. As a result (see Appendix B for derivation), the probability of a proton to have sufficient energy to overcome the potential barrier is determined as
Using this rule, we can determine the rate of proton adsorption to the electrode surface
and probability of adsorbed protons forming a transition state
Transformation from transition state to the hydrogen molecule () and its desorption from the surface () decreases the enthalpy, so the process can be characterised by a constant probability , which can be absorbed into a rate factor . To calculate the actual rate of reaction, we need to further multiply the probabilities with the surface of electrode S on which the reaction takes place:
While the value of factor can nowadays be determined ab-initio by rather complex calculations (e. g. [35]), the detailed value is not crucial for basic understanding we are trying to communicate. Note that in terms of enthalpy levels the reaction rate depends only on the enthalpy difference between transition and initial state
denotes the difference in chemical enthalpy between initial and transition state, while the exact enthalpies of intermediate states are irrelevant at this level of detail.
For easier tracking of units, the expression (16) is conventionally rewritten as a molar flux density
where we defined species activity coefficients by multiplying the probability factor with some reference concentration of protons [1] This results in both reaction rate and reaction rate constant being expressed as a molar flux density in units .
Similar procedure for constructing the reaction rate can be carried for oxidation direction of Eq. (4), describing the decay of hydrogen into protons. The reaction rate will now be proportional to the concentration of hydrogen and exponentially dependant on the difference in enthalpy between transition state and state of molecular hydrogen
denotes the difference in chemical enthalpy between transition and final state, and term describes how the enthalpy difference is affected by electrode charging. Oxidation reaction rate is then written similarly to (18)
We again defined reference concentration to preserve units of reaction rate constant .
By combining both terms we can calculate the overall rate of reaction (2) as a difference between reduction and oxidation reaction rate. To simplify the expression, we furthermore define species activities as a ratio between actual and reference concentration and . This results in
In this expression we already see some terms, familiar from Butler-Volmer equation [1], namely terms and in exponents. Most importantly, the rate and direction of reaction is affected by the electric potential difference U between bulk electrolyte and electrode, caused by the EDL. The increase in potential U will decrease the exponential term in the reduction direction and increase the exponential term in the oxidation direction, thus decreasing the consumption of protons, which is consistent with our previous explanation of protons being repulsed by the electric field in EDL. The decrease in U will have the opposite effect: increase in reduction exponential term will promote the reaction of protons to hydrogen and suppress the hydrogen decay into protons.
3.3. Equilibrium electric potential
This raises an obvious question: does the reaction ever reach equilibrium and does this happen spontaneously? To answer these questions, we also need to consider the changes in the electrode potential due reaction taking place on its surface. The reduction direction of reaction from protons to hydrogen also consumes electrons and will, therefore, increase the potential of electrically isolated electrode. This will in turn decrease the rate of reduction reaction and increase the rate of oxidation reaction, decreasing also the rate at which the potential on the electrode is changing. In simple mathematical terms, the rate of change of electrode electric potential can be described as
where S is electrode surface, its capacitance, F Faraday constant and factor 2 describes the number of electrons exchanged in the reaction.
Figure 4.
Electrochemical enthalpy landscape of reaction (2) with electric potential close to equilibrium. Electric charging results in electrochemical enthalpy of the initial and the final state being similar, further resulting in similar transition state barriers in both directions. Small deviations between enthalpy levels can be a result of different activities of reactants and products, which can promote the reaction rate in reduction or oxidation direction.
Figure 4.
Electrochemical enthalpy landscape of reaction (2) with electric potential close to equilibrium. Electric charging results in electrochemical enthalpy of the initial and the final state being similar, further resulting in similar transition state barriers in both directions. Small deviations between enthalpy levels can be a result of different activities of reactants and products, which can promote the reaction rate in reduction or oxidation direction.
The electric potential of the isolated electrode will, therefore, reach equilibrium once the reduction and oxidation rate of reaction will be equal, , resulting in net zero reaction rate . The equilibrium value of electric potential, calculated from Eq. (21), is
As we see, the equilibrium electric potential depends on activities of reactants and products in the system. At some specified standard conditions, at which concentrations of protons and hydrogen equal their reference concentrations and , respectively, the activities equal unity. This conditions are used to define standard equilibrium potential
which is nearly proportional to enthalpy difference between initial state of two protons in electrolyte and final state of hydrogen molecule , corrected with the logarithm of ratio of species activity coefficients and . The potential difference can in fact be calculated by rather complex microscopic models, taking into account the detailed properties of EDL and interaction between proton and surrounding electrolyte, yielding a value [36]. The potential difference is indeed rather large and certainly needs to be taken into account if we try to properly understand the microscopic conditions on the electrode surface.
But how does this result compare to the value of standard hydrogen electrode potential, found in every chemistry textbook, where by definition? This question would best be answered by direct measurement: put one measurement probe in electrolyte, the other on electrode, and measure the voltage. Unfortunately, such measurement would not give us the answer we are looking for. When the voltage measurement probe, inevitably made out of electric conducting material, is dipped into electrolyte, the electrochemical processes very similar to the ones we just explained, would take place on its surface, forming EDL and thus increasing its electric potential compared to the bulk electrolyte. We are therefore unable to measure the potential of bulk electrolyte potential by the very nature of the system that we want to study. All we can do is measure the potential differences between different electrodes, which are all shifted from electrolyte potential by certain EDL voltage shift. In this perspective, it is reasonable to define standardized hydrogen electrode as reference point to which other electrode types are compared.
3.4. Nernst equation
Now we will answer the question: how does the equilibrium electrode potential change if the concentration of reactants are shifted from their reference values? Intuitive picture, developed thus far, can easily support answering this question. If the EDL serves as a barrier preventing protons from reacting with electrons in the metal, higher proton concentration will require higher EDL potential difference for the equilibrium state to be achieved. Higher hydrogen concentration, on the other hand, will cause more hydrogen molecules to dissociate into protons, donating the deficit electrons to the electrode, thus lowering equilibrium electrode potential.
This equilibrium can mathematically be described by Eq. (23), which we simplify by expressing enthalpy difference in terms of . This results in a well known Nernst equation [1]:
where we multiplied numerator and denominator of logarithm prefactor by Avogadro constant and applied definitions and .
From this derivation it is clear that the equilibrium potential will change with species concentration due to their effect on reduction and oxidation reaction rate, which is proportional to their concentration. Note, however, that this potential is only achieved on electrically isolated electrode in equilibrium and in absence of any side reactions, e.g. reduction of potential in reactions with the cross over gasses, and that it does not hold any real physical significance in conditions where either net reduction or oxidation takes place.
3.5. Butler-Volmer equation
As explained phenomenologically in Section 3.1 and mathematically in Eq. (22), the electric potential on electrode tend to stabilize at some equilibrium potential. Every imbalance in reduction or oxidation reaction rate will lead to either accumulation or deficit of electrons in the electrode, resulting in the shift in electric potential, which will quickly lead to equilibration of reaction. If we want a steady rate of reaction, we therefore need to provide some external potential that forces the system out of equilibrium. This can be achieved by draining or supplying the electrode with additional electrons via external voltage or current source.
It is convenient to express the reaction rate in terms of overpotential which denotes the difference between actual electrode potential U and standard equilibrium potential . Note that constant value (Eq. (24)), chosen as a reference, is unaffected by Nernst potential shift (Eq. (25)), which will make the final result easier to understand in terms of concentration effects on reaction rates.
Substituting in Eq. (21) and using from Eq. (24), we arrive after some simplification to the following expression
We make a further simplification by defining exchange current density as
which is completely defined by properties and energy levels of the reaction and independent of electric potential U. This leads to a standard form of the Butler-Volmer reaction
known from chemistry textbooks. From this form we can clearly see how the increase or decrease in electric potential U will affect the reaction rate. If U is increased above (), the exponent in second term will increase, promoting the reaction from hydrogen to protons. On the microscopic level, the hydrogen more willingly donates its electrons to more positively charged electrode and therefore more easily decomposes to protons. The protons are on the other hand repulsed by higher potential and their reaction with electrons in the electrode is therefore suppressed. If the potential is lowered (), the situation is reversed. Lower EDL potential barrier is less effective at repulsing protons, which will more easily react with electrons and form hydrogen. The hydrogen, on the other hand, is less willing to donate its electrons to negatively charged electrode and will therefore more likely stay in its molecular form.
Charge transfer coefficient determines how strongly the changes in electric potential, described above, affect the reduction and oxidation reaction rate. If is close to 1, the energy of transition state behaves similarly to initial state, resulting in difference between them staying constant, so the rate of reduction reaction will not be much affected. The energy difference between transition and final state will in that case change significantly, so the electric potential will have strong effect on oxidation reaction rate. For close to zero, situation is reversed. The energy of transition state remains constant regardless of electric potential, resulting in unchanged oxidation reaction rate and strongly affected reduction reaction rate.
Butler-Volmer equation can also be expressed in slightly different form by comparing potential U to the Nernst equilibrium potential (25) instead of standard equilibrium potential (24). Reaction rate (28) can in this case be expressed as
where exchange current density
depends on the concentrations of chemical species. While this form is sometimes more convenient to use in calculations, note that in this notation both equilibrium potential and exchange current density are functions of the reactant’s and product’s activities, which makes their effect on reaction rate less intuitive.
3.6. Electrochemical processes in the fuel cell anode catalyst layer
We will now apply the insights, obtained thus far, to the anode catalyst layer of the hydrogen fuel cell. The aim is to propose some simple equations which will describe how the anode processes affects the temporal dynamics of hydrogen concentration , proton concentration and anode EDL potential difference .
In hydrogen fuel cell, the hydrogen oxidation reaction takes place on the anode catalyst layer, consuming the hydrogen and transforming it into protons, so the reaction rate,
should be negative . As explained in previous section, this requires that the electric potential on the anode is larger than the equilibrium potential, . The removal of electrons from the anode, required to achieve such potential, is achieved by the external electric current from the cathode to the anode, which will be explained in detail in Section 7.
If this condition is achieved, electrochemical reaction will consume hydrogen to produce protons and electrons. To describe how the process affects the concentrations of these three species, we model the anode catalyst layer as a porous structure with thickness , volume ratio of ionomer material , and volume ratio of void space . The rest of the volume is filled with catalyst material, in low-temperature fuel cells typically composed of Pt nanoparticles dispersed on the surface of highly porous carbon structure. The structure of catalyst is schematically presented in Figure 5. Electrochemically active catalyst surface area of electrode
can be estimated from macroscopic fuel cell surface area m, Pt loading g/m, describing the total mass of Pt dispersed over specific FC surface, and catalyst electrochemical surface area m/g], describing the catalyst surface per mass of the dispersed catalyst. This quantity mostly depends on the size of catalyst particles and can be estimated as [37], where is the bulk density of platinum.
To describe how the concentration of hydrogen in the catalyst layer changes with time, we multiply the rate of electrochemical reaction (28) with catalyst surface area to calculate the molar rate of consumption, and divide it by the void volume inside the catalyst layer, accessible to the hydrogen , where is the total catalyst layer volume:
Please note that anode reaction rate , so the concentration of hydrogen is decreasing.
Concentration of protons is governed by similar equation
but with different accessible volume since protons reside in ionomer.
The concentration of electrons is reflected in electric charging of the electrode. Since the thickness of EDL - over which the potential drop takes place is relatively small compared to the electrochemically active surface area of electrode , the anode can be effectively described as a plane capacitor with capacitance
where is vacuum permittivity and is dielectric constant of ionomer. The rate of change of the electric potential of the anode due to electrochemical reaction is then obtained by dividing the electric current by capacitance
Electrochemical reaction that consumes hydrogen to produce protons and electrons, therefore, tends to decrease the electric potential on the anode, which would in turn decrease the rate of electrochemical reaction (31). If no external electric current is provided to the anode, as for example in open circuit fuel cell operation, the potential decreases to the equilibrium potential, defined by Nernst equation (25), and electrochemical reaction stops.
Equations (33), (34) and (36) describe the effects of electrochemical reactions in the anode on the concentrations of hydrogen and protons, and electric charging of the anode electrode. Note that these quantities are also affected by other physical processes in the fuel cell, such as diffusion and conduction, which will be discussed in more detail in Section 5, Section 6 and Section 7.
4. Cathode electrochemical processes
4.1. Kinetic model of cathode reaction rates
In this section we will describe what happens with electrons and protons, produced in anode electrochemical reaction, when they approach the cathode electrode, where additional species, oxygen, is introduced. We will use the understanding of electrochemical processes, obtained on a simpler example of hydrogen electrode to describe a more complex electrochemical reaction taking place on the cathode electrode, to explain the reaction between protons and electrons in the presence of oxygen. The details on how protons and electrons are transported from the anode to the cathode will be discussed in detail in Section 6 and Section 7.
Since more species are involved in cathode reaction, protons , electrons and oxygen , the cathode chemistry is richer and more than one reaction can take place. Most prominent is 4 electron oxygen reduction, resulting in formation of water
As described on the example of anode reaction, the rate of electrochemical reaction is determined by concentrations of reactants and xxxxx ta del do vejice se mi ne bere najbolj tekoce: the probability they have of forming a transition state, which is in turn determined by the enthalpy differences between initial , transition and final state . Note that the enthalpies for cathode reaction are labeled with to distinguish them from anode label H.
The initial and the final state are well defined and thus also their enthalpy, which, similarly to the anode reaction, change with electrode potential U proportionally to the charge transferred during the reaction, which equals
The transition between the initial and the final state is now much more complex compared to anode reaction, comprising various transition states of molecular and atomic oxygen adsorbed to the electrode surface with different number of protons bound to them. The reaction can thus proceed along different reaction pathways and even more importantly, the oxygen reduction and water oxidation pathways can be significantly different, which is demonstrated by DFT calculations, for example in Anderson et al. [38]. Exact description of reaction rates, equivalent to anode reaction, would therefore require inclusion of various reaction steps and calculation of concentration of all intermediate species. This procedure is complicated and does not bring much intuition. We, therefore, try to simplify the expression to a more simple and intuitive form. We can argue that since the probability of transition between two consequent intermediate states in reaction depends exponentially on the difference in enthalpy values, we can assume that the rate of reaction will be limited by largest increase in encountered along the chain of states. This transition is labeled as the rate-limiting step as it determines the rate of reaction in specified direction. Reduction and oxidation reaction can feature different rate-limiting transition states with different enthalpies, as shown schematically in Figure 6.
Regardless of exact details of relevant reaction pathway, we label the enthalpy of reduction transition state with and assume it is changing with electrode potential U as
where charge transfer coefficient describes how the enthalpy of relevant transition state changes with electrode potential U.
The enthalpy of transition state, relevant for oxidation direction, is described similarly
with charge transfer coefficient .
The reaction rates of cathode reactions in reduction and oxidation direction can then be constructed similarly as in the anode case,
The reaction rate proportional to the activities of relevant species (, , ) and exponential of enthalpy differences between relevant states.
The differences in enthalpy again depends on the electrode potential U,
schematically presented in Figure 6. As in anode case, and denote the enthalpy differences on electrically neutral electrode.
As for the case of anode reaction, the rates of reduction
and oxidation
are proportional to the activities of the species involved in the reaction and to the exponent of the relevant enthalpy increase ( for reduction and for oxidation, see Figure 6). Total rate of cathode electrochemical reaction on charged electrode is calculated as a difference between reduction and oxidation reaction rate
The expression can be significantly simplified by introducing the equilibrium potential at which the net reaction rate is zero. As on the anode, the reduction reaction consumes electrons and thus charges the electrode positively. This in turn increases the enthalpy difference and suppresses the reduction reaction rate, while simultaneously decreases , thus increasing the oxidation reaction rate. In standard conditions, i.e. at , the reactions balance out at potential
dominated mostly by enthalpy difference between initial and final state , but also affected by difference in transition states . If both reduction and oxidation reaction feature the same rate-limiting transition state, as the hydrogen reaction, the term simply vanishes. Standard equilibrium potential is further affected by difference in charge transfer coefficients .
The value of standard equilibrium potential of reaction (37) is V compared to standard hydrogen electrode reaction (2), since the energy released in reaction of protons with oxygen, resulting in water, is higher than in protons reacting to hydrogen molecule. The electric potential change across the EDL on cathode electrode is therefore larger than on the anode electrode. The absolute value is obtained as a sum of EDL potential of standard hydrogen electrode and relative cathode potential compared to SHE: 1.23 V + 4.44 V = 5.67 V.
The Nernst potential for 4-electron oxygen reaction is obtained in a similar manner as for the standard equilibrium potential, but with concentrations xxxx prosim preveri, ce je izraz OK: featuring non-standard values, resulting in
The equilibrium potential increases with oxygen and proton activity, since larger electric repulsion is needed to suppress the reaction when more protons are present, similarly as in case of anode reaction.
4.2. Butler-Volmer reaction aaaa - morda raje equation
The Butler-Volmer equation for cathode reaction rate is obtained by expressing the transition state enthalpies in terms of standard equilibrium potential, which simplifies Eq. (48) to
where overpotential is expressed as a difference between actual potential U and standard equilibrium potential for cathode reaction . Note that exchange current densities are now different for reduction and oxidation part of the equation:
with
The reaction rate constants and are defined by the detailed structure of transition states of the reaction, but independent of either reactant and product concentration or electric potential of the electrode. The effects of concentrations and the potential are expressed in a very similar way as for the anode Butler-Volmer equation (31): the reaction rate in each direction is proportional to the concentration of relevant species with appropriate power law and exponentially dependent on the overpotential . The reaction will proceed in reduction direction, consuming oxygen and hydrogen and producing water, when the potential is lower than equilibrium potential (), as indicated by negative exponential in first term. If potential U is higher than (), the direction can be reversed, promoting water dissociation into protons and oxygen.
4.3. Electrochemical processes in fuel cell cathode catalyst layer
Electrochemical reactions on the cathode electrode of fuel cell take place in a similar environment as on the anode side, i. e. in catalyst layer consisting of porous carbon supported catalyst particles mixed with ionomer. For simplicity, we assume the same catalyst (), ionomer () and void volume () fractions, and catalyst thickness in both anode and cathode catalyst layer.
Cathode electrochemical reaction
depends on and affects the concentrations of all three species involved, oxygen (), water () and protons (), and on potential difference between cathode catalyst and ionomer .
The rates of electrochemical consumption or production are expressed as time derivatives of concentrations
In addition to electrochemical production and consumption, the concentrations are also affected by diffusion of gases from GDL and conduction of protons through the membrane, which will be discussed in more detail in Section 5 and Section 6.
The consumption or production of electrons is expressed similarly as on anode side, by treating the EDL layer as a capacitor, resulting in electric potential change
During fuel cell operation, oxygen, protons and electrons are consumed in cathode catalyst, resulting in positive value of , increasing the cathode electric potential . If no external electric current flows through the cell, the potential will increase, until it reaches the value, determined by species concentration via Nernst equation (50), when reaction stops. In actual operating conditions, however, the external electric current flows from cathode to anode (see Section 7), ensuring sufficiently low electric potential to maintain positive sign of the cathode reaction . The dynamics of catalyst ionomer electric potential will be further discussed in Section 6.
5. Gaseous species transport
As we discussed in the previous section, the oxygen and hydrogen are consumed during the fuel cell operation and the water is produced. Where do these species come from and how are they transported to the catalyst layer where they react?
The reactants and products involved in the electrochemical processes in the fuel cell appear in gaseous form at temperatures at which the fuel cells typically operate. Reactants are fed to the fuel cell via channels, from which they travel to the catalyst layer through the gas diffusion layer (GDL). The transport is driven by differences in concentration and pressure. At the anode, the hydrogen is consumed in the reaction, resulting in reduced pressure in the catalyst layer compared to the pressure in the channel. Detailed modelling of the gas diffusion can be rather complicated due to porous structure of GDL [39], but the main causal relation can still be described by modelling a molar flux as being proportional to concentration difference
where and denote effective GDL diffusivity and thickness, respectively, and is the hydrogen concentration in the gas feed channel.
The transport processes on cathode side are similar, with oxygen being transported from gas feed channel to the catalyst
The water transport is more complex, since phase change from gaseous to liquid state can result in wide range of additional phenomena inside the GDL [26]. However, from simple perspective of causal relations, we will model this transport also by a simple effective diffusion
where effective diffusion coefficients for water is different from the oxygen coefficient .
Diffusion fluxes replenish the reactants, consumed in the electrochemical reaction, and dispose of the products from the catalyst layer. The following equations describe their effects on species concentrations in the catalyst layer
where denotes the catalyst layer thickness and we assume that gasses only reside in the void fraction of the catalyst layer. For simplicity, we assume the same catalyst properties on the anode and the cathode side.
Transport of reactant plays a crucial role in fuel cell performance at high current densities when their consumption in electrochemical reactions is high. Since the lowest possible value of oxygen and hydrogen concentrations in the catalyst is zero, the transport fluxes (60) and (61) are theoretically limited by values and . When this limit is approached, the reactants concentrations in catalyst is reduced due to imbalance between electrochemical consumption and diffusion transport, causing the reactant depletion and consequent reduction in electrochemical reaction rates.
6. Proton transport in the fuel cell membrane
We have already explained in Section 3.6 how the protons are produced in electrochemical reaction in the anode catalyst layer at sufficient hydrogen concentration and sufficiently high electric potential, and in Section 4.3 how protons are consumed in cathode catalyst layer. To maintain a steady reaction, the protons, therefore, need to be transferred from anode to cathode side of the fuel cell.
In the hydrogen fuel cell, the protons are transported via proton conducting membrane, which prevents (or minimizes) the transport of gaseous species, i. e. hydrogen and oxygen, but allows the transport of protons. In principle, the proton transport can occur via two mechanisms: by diffusion, caused by the gradient of proton concentration, or by electric conduction, where proton movement is forced by internal electric field in the membrane. In real systems, the diffusivity of protons is relatively low compared to the conductivity, therefore the main mechanism of proton transport is the electric conduction [3].
This raises the question where does the electric field, forcing the transport of protons, originate from? To answer this question, we need to understand the basic properties of proton conducting polymers from which the membrane is produced. The polymer consists of long perfluorinated chains of carbon atoms in which side chains are attached [40]. This side chains end with sulphonic groups (), from which the proton can easily detach when membrane is sufficiently hydrated. This effectively results in a fixed volume concentration of immobile negatively charged groups, overlapped by concentration of positively charged protons , , which can move through the membrane material. In equilibrium, the concentrations of both species is equal , resulting in zero net electric charge concentration
where Faraday constants F transforms molar concentration to electric charge concentration.
When electrochemical reaction takes place in the fuel cell, the local concentration of protons on the electrode surface changes: concentration increases on the anode surface and decreases on the cathode surface. This results in a local change of net electric charge concentration
This electric charge is a source of electric field according to Gauss law
where is the value of dielectric constant inside the electrolyte.
If we assume that catalyst layer of the fuel cell is relatively thin compared the fuel cell surface area, the charge density due to the proton concentration changes can effectively be described as a surface charge density , where is the catalyst thickness and is the ionomer volumetric ratio. The anode and cathode catalyst layer can then be seen as two parallel plates of a plate capacitor, with charges on plates producing combined electric field of magnitude
pointing from the anode to the cathode. The difference between charge densities can be expressed in terms of proton concentrations and using Eq. (67)-(68) as
The electric potential drop across the membrane is calculated as a product between electric field and the membrane thickness
Note that this electric field is fully contained in the electrolyte and shielded from external electric field by EDL. The field is, therefore, distinct from the electric field between the anode and the cathode electrode outside of the fuel cell, which points from the cathode towards the anode. The profile of electric field and electric potential through the MEA is schematically presented in Figure 7.
The electric potential in the membrane affects the fuel cell dynamics in two ways. As explained in Section 3.6 and Section 4.3, the electrochemical reaction rates depend on the electric potential difference between the electrode and ionomer, . The membrane potential creates the difference in ionomer potential between anode and cathode side . Since the zero value of electric potential can be chosen arbitrarily, we can set ionomer potential on cathode surface to zero and calculate ionomer potential on anode surface as
Ionomer electric potentials and thus depend directly on concentrations of protons and in anode and cathode catalyst layer.
In addition to changing the ionomer potential, the electric field in the electrolyte is also a cause of electric force on charged particles, in this case protons, and forces them to move in the direction of the field, that is from the anode to the cathode, resulting in electric current density , where is electrolyte conductivity. The molar flux of protons is proportional to electric current , which can, therefore, be expressed by membrane voltage drop (72) as
This equation shows that the transport of protons is a direct consequence of imbalance between concentration of protons in the anode and the cathode, causing the electric field inside the electrolyte.
The protons flux results in the balancing of proton concentrations:
Characteristic time of this relaxation can be estimated from the electrolyte conductivity and the dielectric constant. For Nafion with S/m [2] and [41], relaxation is fast, s, indicating that any inhomogeneity in proton concentration inside the membrane will be balanced out almost instantly. The electric charge imbalance, generated by the difference in proton and sulphonic group concentrations, can, therefore, only appear very close to the electrode surface, where the protons are generated or consumed. This justifies our assumption that the net electric charge density inside the membrane is zero and that the system can, therefore, effectively be described as a plane capacitor. The magnitude of proton concentration deviations can be estimated from typical electric current density in the fuel cell A/m using Eq. (74), resulting in mol/m, which is very small compared to concentration of protons and sulphonic groups in Nafion mol/m [40].
Short relaxation times and small concentration differences show that the concentration of protons in the membrane can in most cases be considered homogeneous and the modelling of its dynamics can usually be avoided, unless the model aims at describing the dynamics in frequency range . The explanation of these processes nevertheless serves an important educational role in properly understanding the causal chain between proton production and consumption in the catalyst layer and mechanism of their transport inside the membrane.
7. Electron transport
As explained in Section 3 and Section 4, the electrochemical reactions on anode and cathode in the fuel cell are a source and sink of electrons, which cause the change in the electric potential of electrodes. The equilibrium electric potential for electrochemical reaction on cathode is higher than on the anode, which results in electric potential difference between cathode and anode which can be exploited to extract useful electric work from the fuel cell. Fuel cell electric voltage equals the electric potential difference between cathode and anode . Electric current through the external load with resistance R, attached to the fuel cell, is than
This current results in potentially useful electric work done on the load . From the perspective of electrochemical processes in the fuel cell, the electric current I represents additional sink and source of electrons in the anode and cathode electrode, which affects their electric potential as
with and introduced previously in Section 3.6 and Section 4.3. Combining these two equations with electrochemically-induced changes in electrode potential allows us to calculate the time evolution of potential on each of the electrodes.
It is instructive to write the fuel cell voltage also in terms of voltage drops across the anode and the cathode EDL and across the membrane (5), resulting in expression
which clearly communicates that the fuel cell voltage is affected by both the electrochemical reaction rates and proton transport in the membrane. In terms of anode and cathode overpotentials and , fuel cell voltage can be written as
where we defined ideal fuel cell voltage . The actual voltage is smaller than ideal value because of anode overpotential , cathode overpotential and membrane potential drop as well as the gas crossover, which is present in real systems.
8. Modelling results and discussion
The model was tested by simulating 4 polarization curves, measured at different inlet gas pressures of hydrogen and oxygen: bar. Gas flows were set by constant stoichiometry on anode side and cathode side . The temperature was assumed to be constant and determined by the cooling medium with temperature C. Calibration of model parameters is explained in detail in Appendix C.
Measured polarization curves are compared in Figure 8 with the modelled results, calculated using the calibrated model parameters. The values are in good agreement, indicating that a simple proposed model is capable of sufficient explanation of the main physical processes taking place in the fuel cell. Please note that this model is introduced with a pedagogical aim and, therefore, it does not include sub-models of specific detailed phenomena, as for example formation and transport of liquid water [26], which enhances accuracy of results at high current densities.
As explained with Eq. (81), the drop of fuel cell voltage from its maximal theoretical value of 1.23 V can be attributed to three contributions: anode overpotential , cathode overpotential and membrane voltage drop . These contributions are plotted for different polarization curved in Figure 9.
The contribution of cathode overpotential is significantly larger than of the anode overpotential, which is a result of cathode electrochemical reaction rate being much smaller than anode reaction rate. Both overpotentials depend significantly on the gas inlet pressure: higher pressure results in higher reactant concentrations, requiring lower overpotential for reaction to proceed at a rate, determined by external current. In the stationary conditions in which the polarization curve is measured, the electric current density through the fuel cell is linearly proportional to the proton flux through the membrane. The membrane voltage drop (72) therefore increases linearly with current density and is independent of gas inlet pressure.
The model also provides insight into the concentration profiles of other chemical species, i. e. oxygen, hydrogen and water, plotted in Figure 10. As expected, the concentrations of reactants decrease linearly with current density, which causes a linear increase in reactants flux through the GDL at constant species concentrations in gas channels. Note that at sufficiently large current densities, the oxygen concentration on cathode surface is so low that it results in significant increase in cathode overpotential (see black line in Figure 9, required to maintain sufficient reaction rate at decreased reactant concentration. The concentration of water (dotted line) is steadily increasing with increased current density as a result of faster production.
The explanation of electrochemical reactions and transport processes, provided in Section 3, Section 4, and Section 6, and the results, calculated using the model, make possible to schematically present the contributions to proton energy as a function of its position in the membrane-electrode assembly in Figure 11. The total electro-chemical energy of the proton (purple) can be split into two contributions: chemical enthalpy, resulting from reaction between species (orange) and electric energy (green) as a result of proton interaction with electric field in the fuel cell.
In the anode (), the protons are bound in hydrogen molecules with low chemical enthalpy. When hydrogen molecule is split into protons (which enter the ionomer membrane), their chemical enthalpy is increased, but the effect is compensated by the electric energy due to proton transition through the EDL. The overall change in enthalpy is, therefore, quite small. When no current flows through the cell (case a) in Figure 11), small differences in enthalpy levels are a result of concentration contributions to Nernst potential. In case of electric current through the cell (case b) in Figure 11), the enthalpy of hydrogen in the anode is higher than the enthalpy of protons in the membrane, forcing the hydrogen to split into protons.
The chemical part of enthalpy of protons in the membrane is constant, while the electric part is affected by the internal electric field (70). The field is only present when electric current flows through the cell and can be seen in Figure 11 b) as a linear change in electric energy of protons through the membrane.
Transition between the membrane and the cathode is characterised by significant chemical enthalpy decrease when protons from the ionomer are bound with oxygen to form water. The chemical contribution is again balanced by the increase in electric energy, resulting from EDL, resulting in nearly equal electrochemical enthalpy in membrane and cathode when electric current is zero (Figure 11 a)). In case of non-zero electric current, the electric potential on the cathode is decreased, resulting in lower electrochemical enthalpy on the cathode, which forces increased reaction rate.
9. Conclusion
The paper presents a simple educational model of electrochemical reactions and species transport in the hydrogen fuel cell. The aim of the paper is to provide an intuitive explanation of physico-chemical processes in the fuel cell, such as electrochemical reactions, species diffusion and conduction, and describe them in form of simple set of differential equations which enable easy understanding of causal relationships between the relevant quantities.
The description of electrochemical reactions is based on intuitive description of molecular dynamics on charged electrode. As explained in the paper, the electrode charging causes a formation of electric double layer in electrolyte at the electrode surface, which affects the movement of protons involved in electrochemical reactions. The rate of reactions is, therefore, determined by the species concentration and the interplay between energy distribution of species due to thermal excitations, chemical enthalpy of species, and the magnitude of EDL energy barrier. We show that even for hydrogen oxidation reaction with standard potential , the actual electric potential difference across the EDL needs to be large () to provide a balance between chemical and electrical contribution to the enthalpy of the reaction. Oxygen reduction reaction on the cathode results in even larger EDL potential of (. This understanding was used to provide a sound and intuitive mathematical derivation of Butler-Volmer equation and apply it to the case of hydrogen oxidation reaction in the anode and oxygen reduction reaction in the cathode catalyst layer of hydrogen fuel cell.
Furthermore, we use a simple model of plane capacitor to explain how electrochemical production and consumption of protons in the catalyst layers affects the electric field inside the electrolyte and how it affects the electric potential difference across the membrane. This allows a simple description of proton transport in the membrane. When combined with understanding of EDL dynamics on electrode surface, a clear picture of electric potential profile across the membrane-electrode assembly is established: electric potential drops significantly on the junction between anode electrode and electrolyte (), changes only slightly across the membrane and again rises significantly across the cathode surface EDL ().
Finally, we explain how the electric potential of electrodes is affected by the production and consumption of electrons in electrochemical reactions and by the transport through the external load. The time dynamics of electric potential is mathematically described with a simple picture of electrode surface as a plane capacitor with EDL potential difference being linearly related to the excess charge on the electrode. This allows complete understanding of what happens in the fuel cell when external load is connected between the anode and the cathode. The current of electrons travels from low electric potential on the anode to the high potential on the cathode, resulting in the increase of the anode electric potential. This shifts the reaction balance on the anode, promoting the oxidation of hydrogen into two protons, deposited in electrolyte, and two electrons, which try to balance the increased anode potential by its negative electric charge. The electrons traveling to the cathode are decreasing its electric potential and thus shifting the reaction balance in the direction of protons bonding with oxygen and excess electrons, which acts towards the increase of electric potential on the cathode. The imbalance between proton concentrations in anode and cathode catalyst layers results in establishment of the electric field in the membrane which forces the protons to travel between the anode and cathode, balancing the electrochemical production and consumption.
Mathematical descriptions of electrochemical reactions and proton, gaseous species and electrons’ transport were coupled into a model of fuel cell operation, described as a set of 7 first order differential equations for electric potentials and species concentrations in anode and cathode catalyst layer. Electric potentials on anode and cathode, calculated by the proposed model, fit well with experimental data, indicating that the model describes all the relevant processes in the fuel cell with sufficient level of detail, while at the same provides a high degree of intuitive understanding of the underlying processes. Most importantly, the structure of the model provides proper understanding of causal relations between physical quantities describing the fuel cell operation. Due to its intuitiveness, we believe that the proposed model will be a great asset in training and explaining the details of fuel cell operation to the new generations of researchers.
Acknowledgments
The authors acknowledge financial support from the Slovenian Research Agency (research core funding No. P2-0401 and project J2-2494). This work has been partly conducted within the MORELife project and has received funding from the Fuel Cells and Hydrogen 2 Joint Undertaking (JU) under grant agreement No 101007170. The work was also partially funded by project ARRS-CEA NC-0025.
Conflicts of Interest
There are no conflicts to declare.
Appendix A. Electrostatic energy of transition states
Electrostatic contribution to the energy of the transition state, composed of protons adsorbed to the electrode surface, can be estimated using a simple schematic picture of electric charge distribution, shown in Figure A1.
Figure A1.
Schematic representation of energy distribution function and number of particles with sufficient energy for reaction.
Figure A1.
Schematic representation of energy distribution function and number of particles with sufficient energy for reaction.
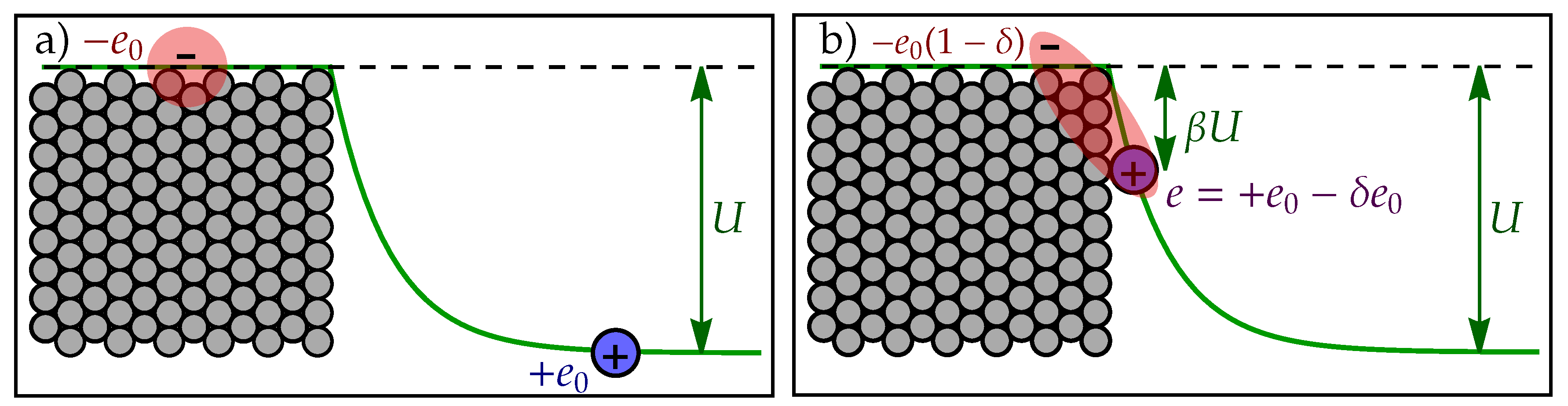
In initial state (Figure A1 a)), the proton with charge (blue circle) lies at negative potential due to EDL and electron (red cloud) at potential 0, resulting in electrostatic energy . In transition state (Figure A1 b)), the proton is adsorbed to the electrode surface, residing at negative potential , where is defined by steepness of EDL potential (i.e. surface electric field) and proton distance from electrode surface. Additionally, the proton charge can be partially screened by electron cloud of effective charge , , resulting in effective charge of adsorbed proton . Electrostatic energy of asorbed proton can thus be estimated as
where we reduced the number of parameters by defining charge transfer coefficient , .
Appendix B. Boltzmann distribution
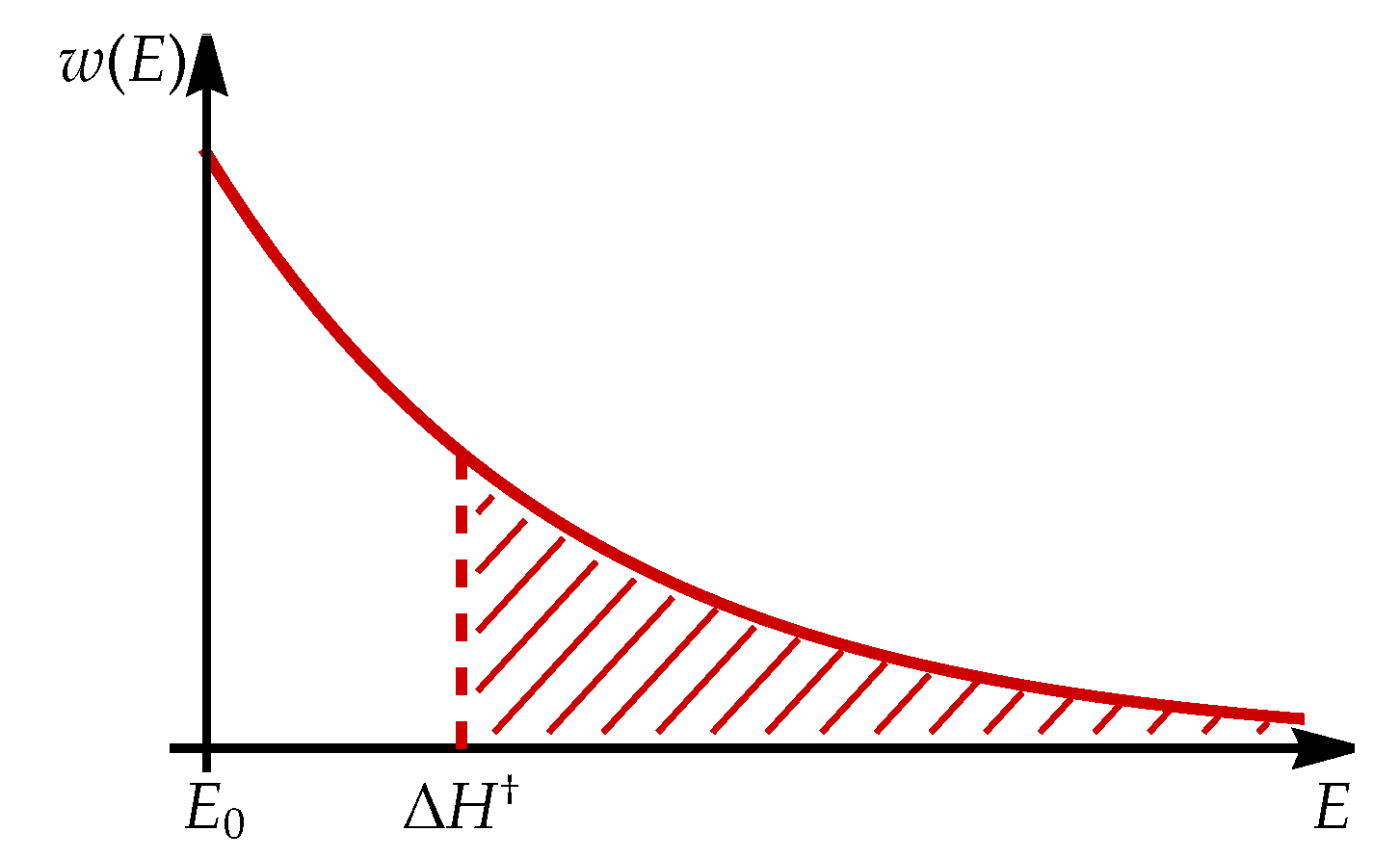
For a macroscopic system at finite temperature, the energy the system is distributed among individual constituents according to Boltzmann distribution [34], describing the probability density of constituent having a specific energy E.
Probability density decays exponentially with energy, with T being temperature, Boltzmann constant. Normalization factor is calculated by integrating from lowest possible energy level to infinity. This means that at any finite temperature, the concentration of reactants with sufficient energy to overcome the potential barrier to the transition state is calculated as a product of concentration and probability for particle having sufficient energy, obtained by integration of probability density:
The procedure is schematically presented in Figure A2.
Figure A2.
Schematic representation of energy distribution function and number of particles with sufficient energy for reaction.
Figure A2.
Schematic representation of energy distribution function and number of particles with sufficient energy for reaction.
Appendix C. Model calibration
The model was calibrated on 4 measured polarization curves (, describing the relation between fuel cell electric current density and voltage , with j describing the measurement points on each curve. Polarization curves differed in inlet pressures in gas feed channels bar, stoichiometry on anode side and cathode side and the temperature, determined by the cooling medium, C.
These data was used to determine the operating conditions of the model Eq. (Section 2). Operating conditions were assumed to be stationary. The average reactants concentrations in the channels were set to constant values, calculated from inlet pressure and stoichiometry
Average water concentration in the cathode channel was calculated from cathode inlet gas humidity using the Antoine equation [42]
with kPa, , K and K. External electric load was determined for each measuring point .
From 19 model parameters, the values for 9 of them were set to characteristic values, found in the literature. The values of parameters are listed in first three columns of Table A1.

Table A1.
List of model parameters.
| Fixed parameter | value | source | Fitted parameter | value | source |
|---|---|---|---|---|---|
| 25 cm | assumed | mol/m s | fitted | ||
| 1 m | assumed | mol/m s | fitted | ||
| 5 m | assumed | 0.65 | fitted | ||
| 30 m | assumed | 0.74 | fitted | ||
| 30% | Ref. [43] | 0.29 | fitted | ||
| 50% | Ref. [43] | m/s | fitted | ||
| 60 m2/g | assumed | S/m | fitted | ||
| 3 g/m | assumed | ||||
| 20 | Ref. [41] | ||||
| 0.122 nm | Calc. from [44] |
9 parameters from the last three columns of Table A1 were obtained by fitting procedure, aimed at minimizing the value of fitness function , calculated by comparing the measured values of fuel cell voltage with fuel cell voltage values , calculated from the model.
Modelled values were obtained by implementing the model (1) in Python programming language. The equations were integrated using solve_ivp routine from Scipy library [45], with constant input values, specified above. Simulation time was set to s to ensure the convergence of all modelled variables. Modelled fuel cell voltage was calculated as for each set of operating conditions . The values of fitted parameters were varied using Nealder-Mead [46] routine from Scipy library, aimed at minimizing the value of . The list of best-fit parameters is listed in Table A1. Since the reverse direction of reaction 51 is strongly suppressed in the fuel cell, the value of oxidation reaction rate constant was assumed to be equal to reduction reaction rate constant .
The use of best-fit parameters in the model result in relatively small value of , which indicate good agreement between model and experiment.
References
- Atkins, P.; de Paula, J. Physical Chemistry, 8 ed.; W. H. Freeman and Company, 2006.
- Zaidi, S.M.J.; Rauf, M.A. Fuel Cell Fundamentals. In Polym. Membr. Fuel Cells; Springer: Boston, MA, USA, 2008; pp. 1–6. [Google Scholar] [CrossRef]
- O’Hayre, R.; Cha, S.W.; Colella, W.; Prinz, F.B. Fuel Cell Fundamentals; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016. [Google Scholar] [CrossRef]
- Lee, T.W.; Tseng, A.A.; Bae, K.S.; Do, Y.H. Simulation of the Proton-Exchange Membrane (PEM) Fuel Cell Life-Cycle Performance with Data-Driven Parameter Estimation. Energy & Fuels 2010, 24, 1882–1888. [Google Scholar] [CrossRef]
- Javed, K.; Gouriveau, R.; Zerhouni, N.; Hissel, D. Prognostics of Proton Exchange Membrane Fuel Cells stack using an ensemble of constraints based connectionist networks. Journal of Power Sources 2016, 324, 745–757. [Google Scholar] [CrossRef]
- Ma, R.; Yang, T.; Breaz, E.; Li, Z.; Briois, P.; Gao, F. Data-driven proton exchange membrane fuel cell degradation predication through deep learning method. Applied Energy 2018, 231, 102–115. [Google Scholar] [CrossRef]
- Silva, R.; Gouriveau, R.; Jemeï, S.; Hissel, D.; Boulon, L.; Agbossou, K.; Yousfi Steiner, N. Proton exchange membrane fuel cell degradation prediction based on Adaptive Neuro-Fuzzy Inference Systems. International Journal of Hydrogen Energy 2014, 39, 11128–11144. [Google Scholar] [CrossRef]
- Liu, H.; Chen, J.; Zhu, C.; Su, H.; Hou, M. Prognostics of Proton Exchange Membrane Fuel Cells Using A Model-based Method. IFAC-PapersOnLine 2017, 50, 4757–4762. [Google Scholar] [CrossRef]
- Pisani, L.; Murgia, G.; Valentini, M.; D’Aguanno, B. A new semi-empirical approach to performance curves of polymer electrolyte fuel cells. Journal of Power Sources 2002, 108, 192–203. [Google Scholar] [CrossRef]
- Haji, S. Analytical modeling of PEM fuel cell i–V curve. Renewable Energy 2011, 36, 451–458. [Google Scholar] [CrossRef]
- Hao, D.; Shen, J.; Hou, Y.; Zhou, Y.; Wang, H. An Improved Empirical Fuel Cell Polarization Curve Model Based on Review Analysis. International Journal of Chemical Engineering 2016, 2016, 10. [Google Scholar] [CrossRef]
- Youssef, M.; Al-Nadi, K.E.; Khalil, M.H. Lumped model for proton exchange membrane fuel cell (PEMFC). International Journal of Electrochemical Science 2010, 5, 267–277. [Google Scholar] [CrossRef]
- Larminie, J. Fuel cell systems explained/James Larminie, Andrew Dicks; J. Wiley: Chichester, West Sussex, 2003. [Google Scholar]
- Amphlett, J.C.; Baumert, R.M.; Mann, R.F.; Peppley, B.A.; Roberge, P.R.; Harris, T.J. Performance Modeling of the Ballard Mark IV Solid Polymer Electrolyte Fuel Cell: II . Empirical Model Development. Journal of The Electrochemical Society 1995, 142, 9–15. [Google Scholar] [CrossRef]
- Mann, R.F.; Amphlett, J.C.; Hooper, M.A.; Jensen, H.M.; Peppley, B.A.; Roberge, P.R. Development and application of a generalised steady-state electrochemical model for a PEM fuel cell. Journal of Power Sources 2000, 86, 173–180. [Google Scholar] [CrossRef]
- J.M. Correa.; F. A. Farret.; L. N. Canha.; M. G. Simoes. An electrochemical-based fuel-cell model suitable for electrical engineering automation approach. IEEE Transactions on Industrial Electronics 2004, 51, 1103–1112. [CrossRef]
- Robin, C.; Gerard, M.; Franco, A.A.; Schott, P. Multi-scale coupling between two dynamical models for PEMFC aging prediction. International Journal of Hydrogen Energy 2013, 38, 4675–4688. [Google Scholar] [CrossRef]
- Chandesris, M.; Vincent, R.; Guetaz, L.; Roch, J.S.; Thoby, D.; Quinaud, M. Membrane degradation in PEM fuel cells: From experimental results to semi-empirical degradation laws. International Journal of Hydrogen Energy 2017, 42, 8139–8149. [Google Scholar] [CrossRef]
- Weydahl, H.; Thomassen, M.S.; Børresen, B.T.; Møller-Holst, S. Response of a proton exchange membrane fuel cell to a sinusoidal current load. Journal of Applied Electrochemistry 2010, 40, 809–819. [Google Scholar] [CrossRef]
- Gu, W.; Baker, D.R.; Liu, Y.; Gasteiger, H.A. Proton exchange membrane fuel cell (PEMFC) down-the-channel performance model. In Handbook of Fuel Cells; American Cancer Society, 2010. [CrossRef]
- Chan, S.; Khor, K.; Xia, Z. Complete polarization model of a solid oxide fuel cell and its sensitivity to the change of cell component thickness. Journal of Power Sources - J POWER SOURCES 2001, 93, 130–140. [Google Scholar] [CrossRef]
- Laoun, B. Simulation of PEMFC performance. Revue des Energies Renouvelables 2011, 14, 441–448. [Google Scholar] [CrossRef]
- Kulikovsky, A.A. A Physically–Based Analytical Polarization Curve of a PEM Fuel Cell. Journal of The Electrochemical Society 2014, 161, F263–F270. [Google Scholar] [CrossRef]
- Kulikovsky, A. Analytical Modelling of Fuel Cells; Elsevier, 2010. [CrossRef]
- Kravos, A.; Ritzberger, D.; Tavčar, G.; Hametner, C.; Jakubek, S.; Katrašnik, T. Thermodynamically consistent reduced dimensionality electrochemical model for proton exchange membrane fuel cell performance modelling and control. Journal of Power Sources 2020, 454, 227930. [Google Scholar] [CrossRef]
- Kravos, A.; Kregar, A.; Penga, Ž.; Barbir, F.; Katrašnik, T. Real-time capable transient model of liquid water dynamics in proton exchange membrane Fuel Cells. J. Power Sources 2022, 541, 231598. [Google Scholar] [CrossRef]
- Eberle, D.; Horstmann, B. Oxygen Reduction on Pt(111) in Aqueous Electrolyte: Elementary Kinetic Modeling. Electrochimica Acta 2014, 137, 714–720. [Google Scholar] [CrossRef]
- Wang, J.X.; Zhang, J.; Adzic, R.R. Double-trap kinetic equation for the oxygen reduction reaction on pt(111) in acidic media. Journal of Physical Chemistry A 2007, 111, 12702–12710. [Google Scholar] [CrossRef]
- Song, C.; Zhang, J. Electrocatalytic Oxygen Reduction Reaction. In PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications; Springer: London, UK, 2008; pp. 89–134. [Google Scholar] [CrossRef]
- Stamenkovic, V.; Mun, B.S.; Mayrhofer, K.J.; Ross, P.N.; Markovic, N.M.; Rossmeisl, J.; Greeley, J.; Nørskov, J.K. Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure. Angewandte Chemie International Edition 2006, 45, 2897–2901. [Google Scholar] [CrossRef] [PubMed]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. The Journal of Physical Chemistry B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Choi, P.; Jalani, N.H.; Datta, R. Thermodynamics and proton transport in Nafion II. Proton diffusion mechanisms and conductivity. Journal of the Electrochemical Society 2005, 152, E123–E130. [Google Scholar] [CrossRef]
- Stoicheff, B. On the dissociation energy of molecular hydrogen. Can. J. Phys. 2001, 79, 165–172. [Google Scholar] [CrossRef]
- L.B. Studien über das Gleichgewicht der lebendigen Kraft zwischen bewegten materiellen Punkten. Wiener Berichte 1868, 58, 517–560. [CrossRef]
- Skúlason, E.; Karlberg, G.S.; Rossmeisl, J.; Bligaard, T.; Greeley, J.; Jónsson, H.; Nørskov, J.K. Density functional theory calculations for the hydrogen evolution reaction in an electrochemical double layer on the Pt(111) electrode. Phys. Chem. Chem. Phys. 2007, 9, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Trasatti, S. The absolute electrode potential: an explanatory note (Recommendations 1986). Pure Appl. Chem. 1986, 58, 955–966. [Google Scholar] [CrossRef]
- Ferreira, P.J.; la O’, G.J.; Shao-Horn, Y.; Morgan, D.; Makharia, R.; Kocha, S.; Gasteiger, H.A. Instability of Pt/C Electrocatalysts in Proton Exchange Membrane Fuel Cells. J. Electrochem. Soc. 2005, 152, A2256. [Google Scholar] [CrossRef]
- Anderson, A.B.; Albu, T.V. Catalytic Effect of Platinum on Oxygen Reduction An Ab Initio Model Including Electrode Potential Dependence. J. Electrochem. Soc. 2000, 147, 4229. [Google Scholar] [CrossRef]
- Cussler, E. Diffusion: Mass Transfer in Fluid Systems; Cambridge Series in Chemical Engineering, Cambridge University Press, 2009.
- Frühwirt, P.; Kregar, A.; Törring, J.T.; Katrašnik, T.; Gescheidt, G. Holistic approach to chemical degradation of Nafion membranes in fuel cells: modelling and predictions. Physical Chemistry Chemical Physics 2020, 22, 5647–5666. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Polizos, G.; Macdonald, D.D.; Manias, E. State of Water in Perfluorosulfonic Ionomer (Nafion 117) Proton Exchange Membranes. J. Electrochem. Soc. 2008, 155, B163. [Google Scholar] [CrossRef]
- Antoine, M.C. Nouvelle relation entre les tensions et les temperatures. C. r. held Seanc. Acad. Sci. Paris 1888, 107, 681–684. [Google Scholar]
- Passalacqua, E.; Lufrano, F.; Squadrito, G.; Patti, A.; Giorgi, L. Nafion content in the catalyst layer of polymer electrolyte fuel cells: effects on structure and performance. Electrochim. Acta 2001, 46, 799–805. [Google Scholar] [CrossRef]
- Smith, A.M.; Lee, A.A.; Perkin, S. The Electrostatic Screening Length in Concentrated Electrolytes Increases with Concentration. J. Phys. Chem. Lett. 2016, 7, 2157–2163. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nature Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. The Computer Journal 1965, 7, 308–313. [Google Scholar] [CrossRef]
Figure 1.
Schematic overview of fuel cell cross-section, describing the model variables (red) and external parameters (blue) aaaa niso to robni pogoji za model?aaa. Dimensions of individual fuel cell components are not presented in proper scale.
Figure 1.
Schematic overview of fuel cell cross-section, describing the model variables (red) and external parameters (blue) aaaa niso to robni pogoji za model?aaa. Dimensions of individual fuel cell components are not presented in proper scale.
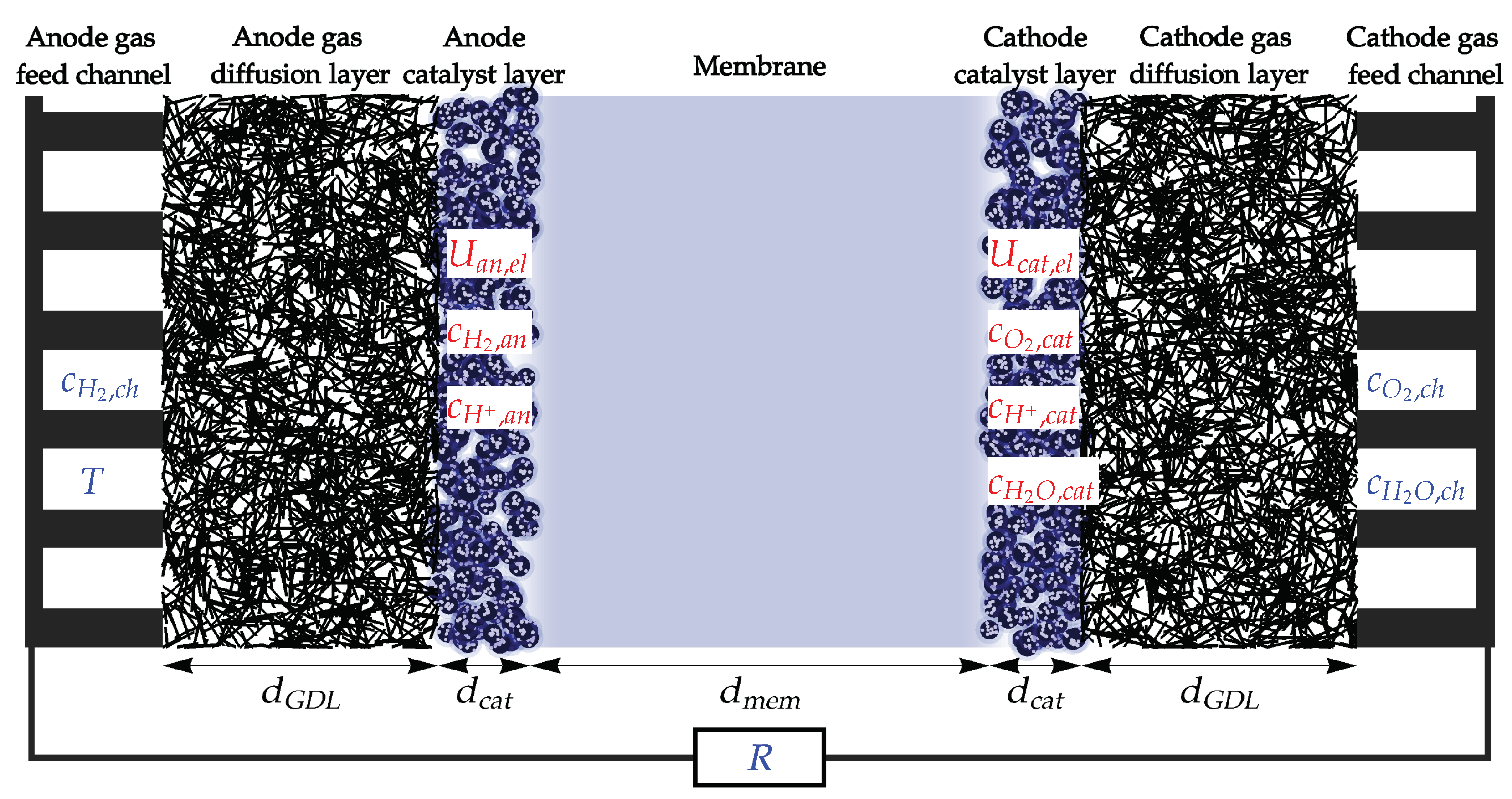
Figure 2.
Schematic representation of configuration of particles, involved in hydrogen reduction and oxidation. In initial state 1, both protons (blue circles) reside in bulk electrolyte, screened from electrode charging by electric double layer (EDL). Proton reduction occurs by protons first adsorbing to the electrode surface (states 2 and 3). With sufficient energy, protons can form activated state (4), which chemically binds into hydrogen molecule (state 5) and finally detaches from the electrode surface (state 6). When hydrogen molecule is oxidized, the reaction proceeds in the opposite direction from state 6 to state 1.
Figure 2.
Schematic representation of configuration of particles, involved in hydrogen reduction and oxidation. In initial state 1, both protons (blue circles) reside in bulk electrolyte, screened from electrode charging by electric double layer (EDL). Proton reduction occurs by protons first adsorbing to the electrode surface (states 2 and 3). With sufficient energy, protons can form activated state (4), which chemically binds into hydrogen molecule (state 5) and finally detaches from the electrode surface (state 6). When hydrogen molecule is oxidized, the reaction proceeds in the opposite direction from state 6 to state 1.
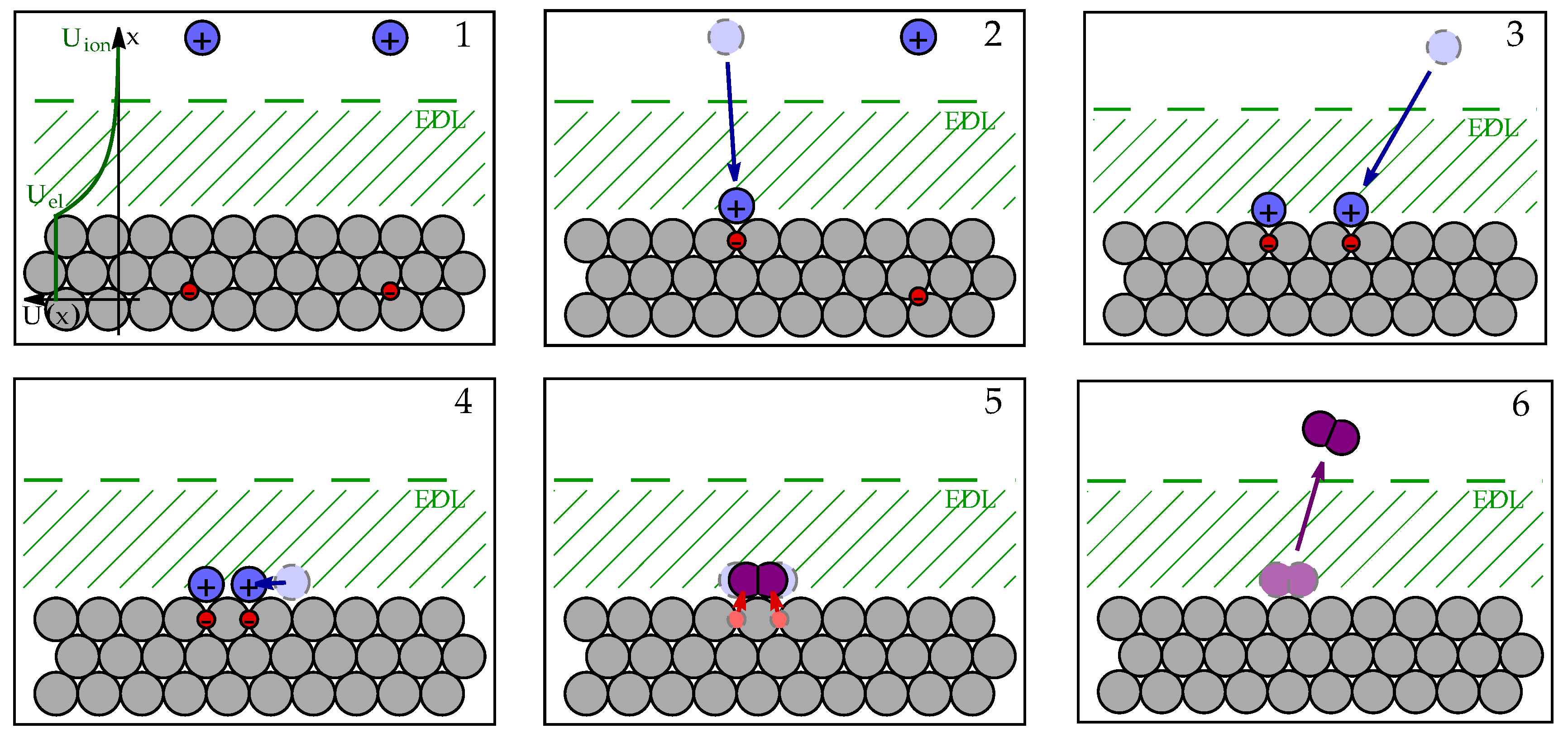
Figure 3.
Schematic presentation of energy levels of states, involved in hydrogen electrochemical reaction on electrically neutral (a) and charged (b) electrode. The numbering of states relates to the Figure 2: 1: , 2: , 3: , 4: , 5: , 6: . On neutral electrode a), electrochemical enthalpy levels (purple) are defined only by chemical enthalpy (purple) with no electric contribution (green). In this case, adsorption of protons to the electrode surface () does not affect the enthalpy, which is increased only when transition state (4) is formed. Formation of hydrogen molecule () results in a significant decrease in chemical enthalpy. When electrode is charged, the proton adsorption requires the transition through EDL, which increases the electric and electrochemical energy of the system. This effectively decreases the electrochemical enthalpy of the initial state 0 by and the transition state 4 by , where is charge transfer coefficient. The change in enthalpy levels affects the share of particles with sufficient thermal energy to overcome the enthalpy barrier to the transition state (red dashed area), thus affecting the rates of electrochemical reaction.
Figure 3.
Schematic presentation of energy levels of states, involved in hydrogen electrochemical reaction on electrically neutral (a) and charged (b) electrode. The numbering of states relates to the Figure 2: 1: , 2: , 3: , 4: , 5: , 6: . On neutral electrode a), electrochemical enthalpy levels (purple) are defined only by chemical enthalpy (purple) with no electric contribution (green). In this case, adsorption of protons to the electrode surface () does not affect the enthalpy, which is increased only when transition state (4) is formed. Formation of hydrogen molecule () results in a significant decrease in chemical enthalpy. When electrode is charged, the proton adsorption requires the transition through EDL, which increases the electric and electrochemical energy of the system. This effectively decreases the electrochemical enthalpy of the initial state 0 by and the transition state 4 by , where is charge transfer coefficient. The change in enthalpy levels affects the share of particles with sufficient thermal energy to overcome the enthalpy barrier to the transition state (red dashed area), thus affecting the rates of electrochemical reaction.
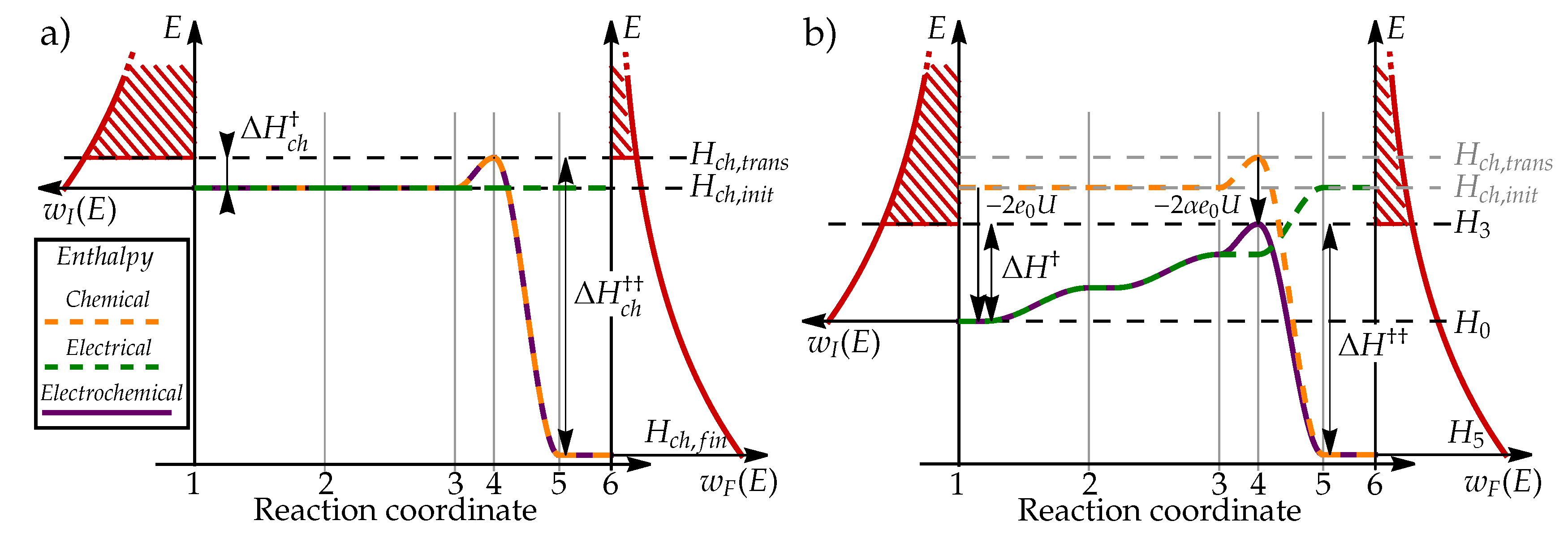
Figure 5.
Schematic presentation of catalyst layer. Catalyst nanoparticles (white) are dispersed on the surface of carbon support structure (black) with volumetric ratio . The catalyst and carbon support are covered with ionomer film (light blue) with volumetric ratio , which allows the protons to travel to the catalyst surface. The gaseous species (hydrogen, oxygen) travel through void volume of the catalyst (white, volumetric ration ).
Figure 5.
Schematic presentation of catalyst layer. Catalyst nanoparticles (white) are dispersed on the surface of carbon support structure (black) with volumetric ratio . The catalyst and carbon support are covered with ionomer film (light blue) with volumetric ratio , which allows the protons to travel to the catalyst surface. The gaseous species (hydrogen, oxygen) travel through void volume of the catalyst (white, volumetric ration ).

Figure 6.
Schematic representation of enthalpy levels of states, involved in electrochemical reaction in fuel cell cathode. Reduction (solid line) and oxidation (dashed line) can proceed between initial (init) and final (fin) state via different transition states (trans,Red and trans,Ox) with different chemical enthalpies (orange), which are differently affected by electric charging (green), resulting in different electrochemical enthalpy barriers (purple) for reduction and oxidation reaction.
Figure 6.
Schematic representation of enthalpy levels of states, involved in electrochemical reaction in fuel cell cathode. Reduction (solid line) and oxidation (dashed line) can proceed between initial (init) and final (fin) state via different transition states (trans,Red and trans,Ox) with different chemical enthalpies (orange), which are differently affected by electric charging (green), resulting in different electrochemical enthalpy barriers (purple) for reduction and oxidation reaction.
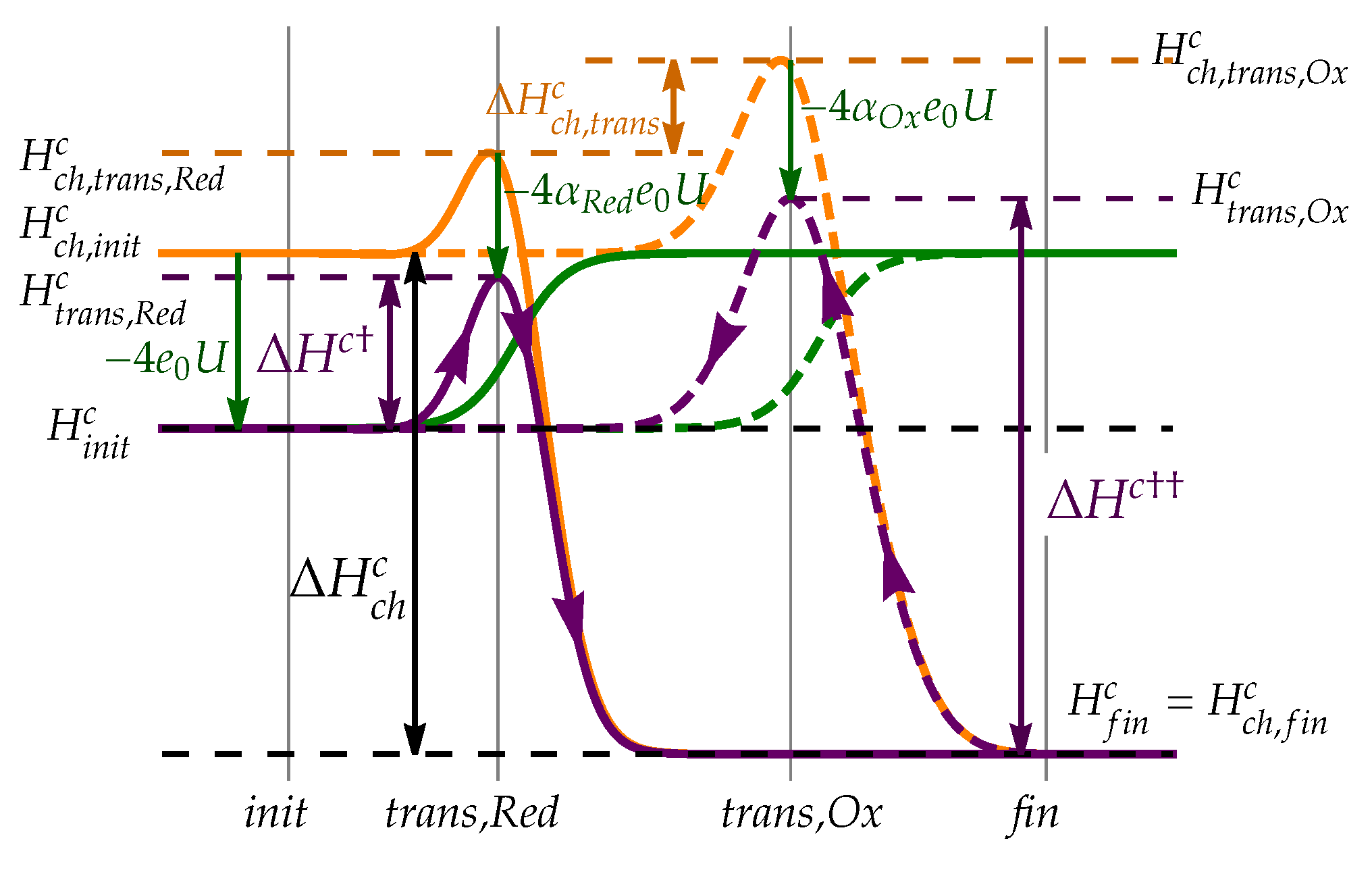
Figure 7.
Schematic presentation of the electric field and the potential in the fuel cell ionomer. Imbalance between proton (red) and sulphonic groups (dashed orange) concentrations in the anode and cathode layer results in positive net charge density on the anode and negative charge density on the cathode (blue). This produces electric field inside the membrane (purple) which forces the protons to move thought the membrane and also results in the difference in ionomer electric potential (green) between the anode and the cathode.
Figure 7.
Schematic presentation of the electric field and the potential in the fuel cell ionomer. Imbalance between proton (red) and sulphonic groups (dashed orange) concentrations in the anode and cathode layer results in positive net charge density on the anode and negative charge density on the cathode (blue). This produces electric field inside the membrane (purple) which forces the protons to move thought the membrane and also results in the difference in ionomer electric potential (green) between the anode and the cathode.
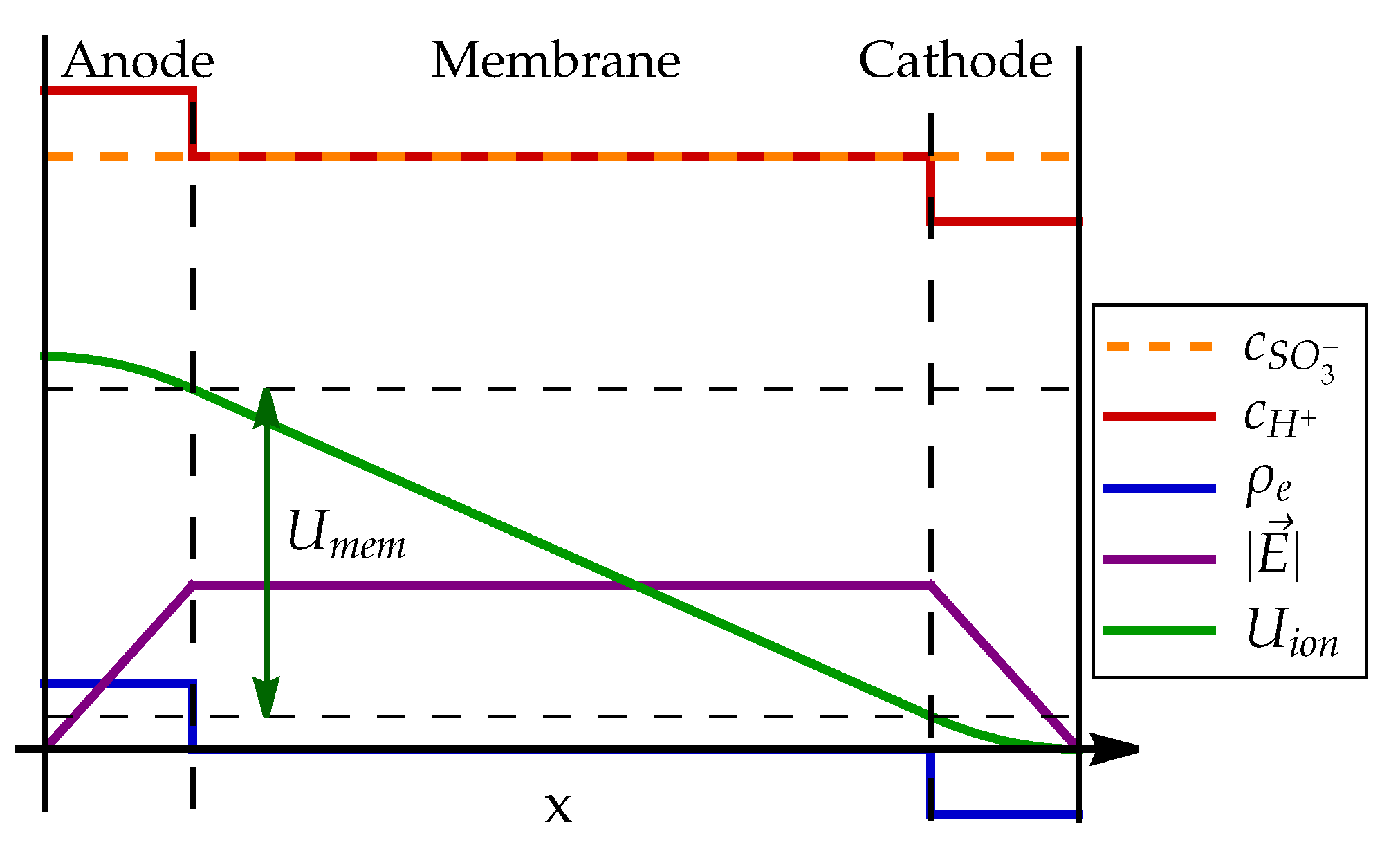
Figure 8.
Comparison between measured polarization curves and the data, produced by the model.
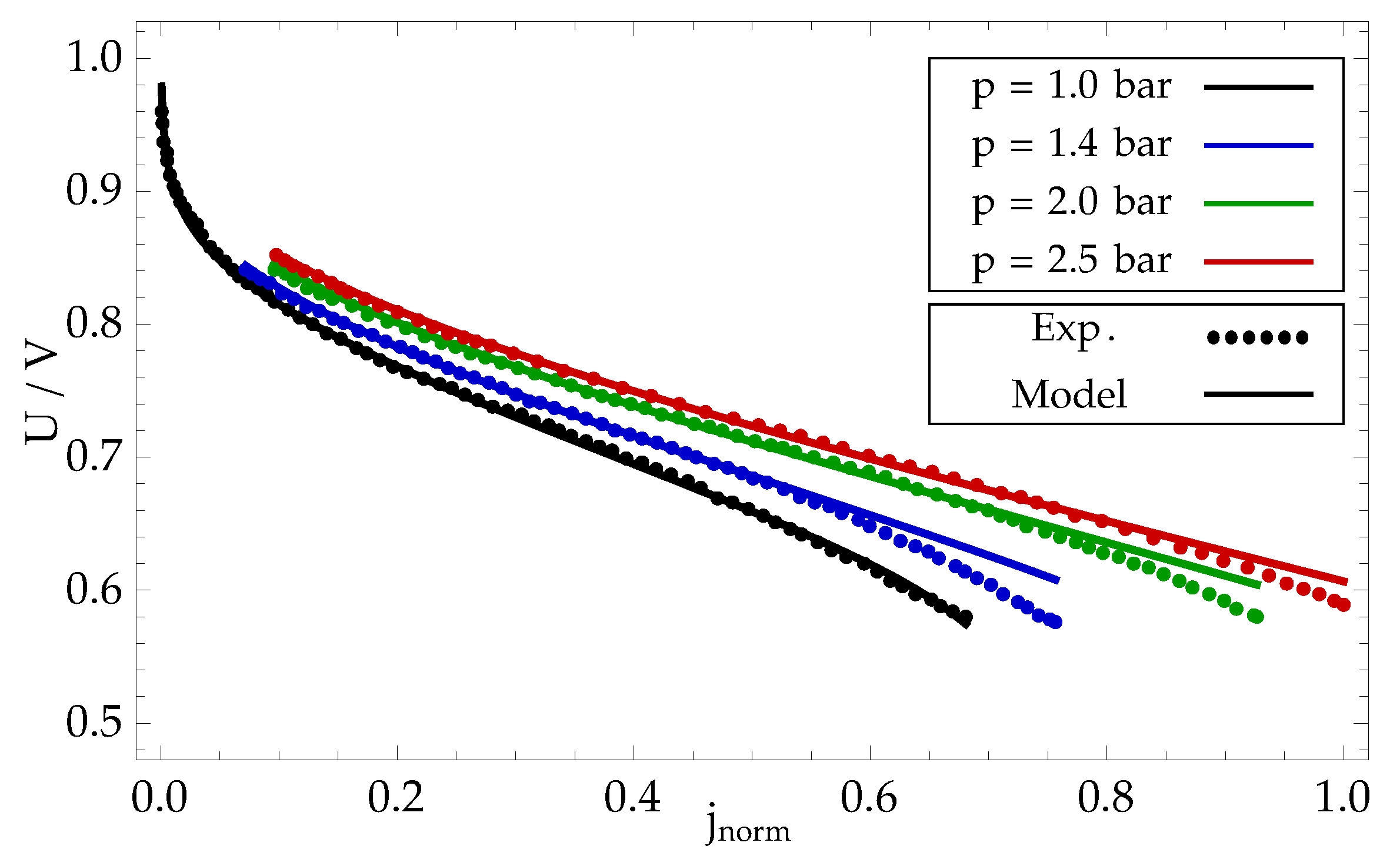
Figure 9.
Comparison between different contributions to fuel cell voltage drop in Eq. (81) at different gas pressures: anode overpotential (dashed), cathode overpotential (solid) and membrane voltage drop (dotted).
Figure 9.
Comparison between different contributions to fuel cell voltage drop in Eq. (81) at different gas pressures: anode overpotential (dashed), cathode overpotential (solid) and membrane voltage drop (dotted).
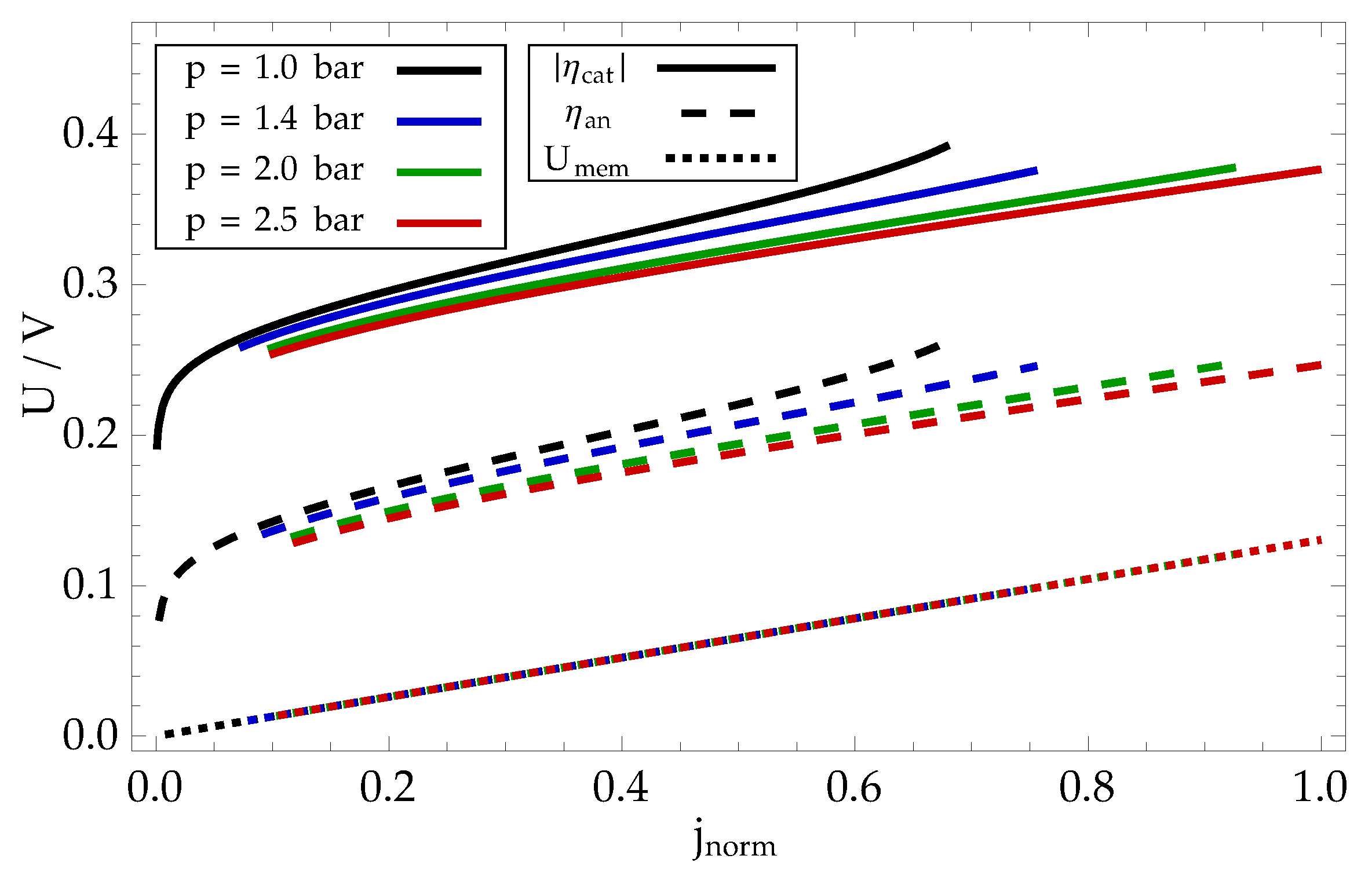
Figure 10.
Changes in the anode catalyst hydrogen concentration (dashed), and the cathode catalyst oxygen (solid) as well as water (dotted linen) concentration with current density at different gas inlet pressures. Increasing current density results in a lower concentration of reactants (hydrogen and oxygen) due to electrochemical consumption and higher concentration of product (water).
Figure 10.
Changes in the anode catalyst hydrogen concentration (dashed), and the cathode catalyst oxygen (solid) as well as water (dotted linen) concentration with current density at different gas inlet pressures. Increasing current density results in a lower concentration of reactants (hydrogen and oxygen) due to electrochemical consumption and higher concentration of product (water).
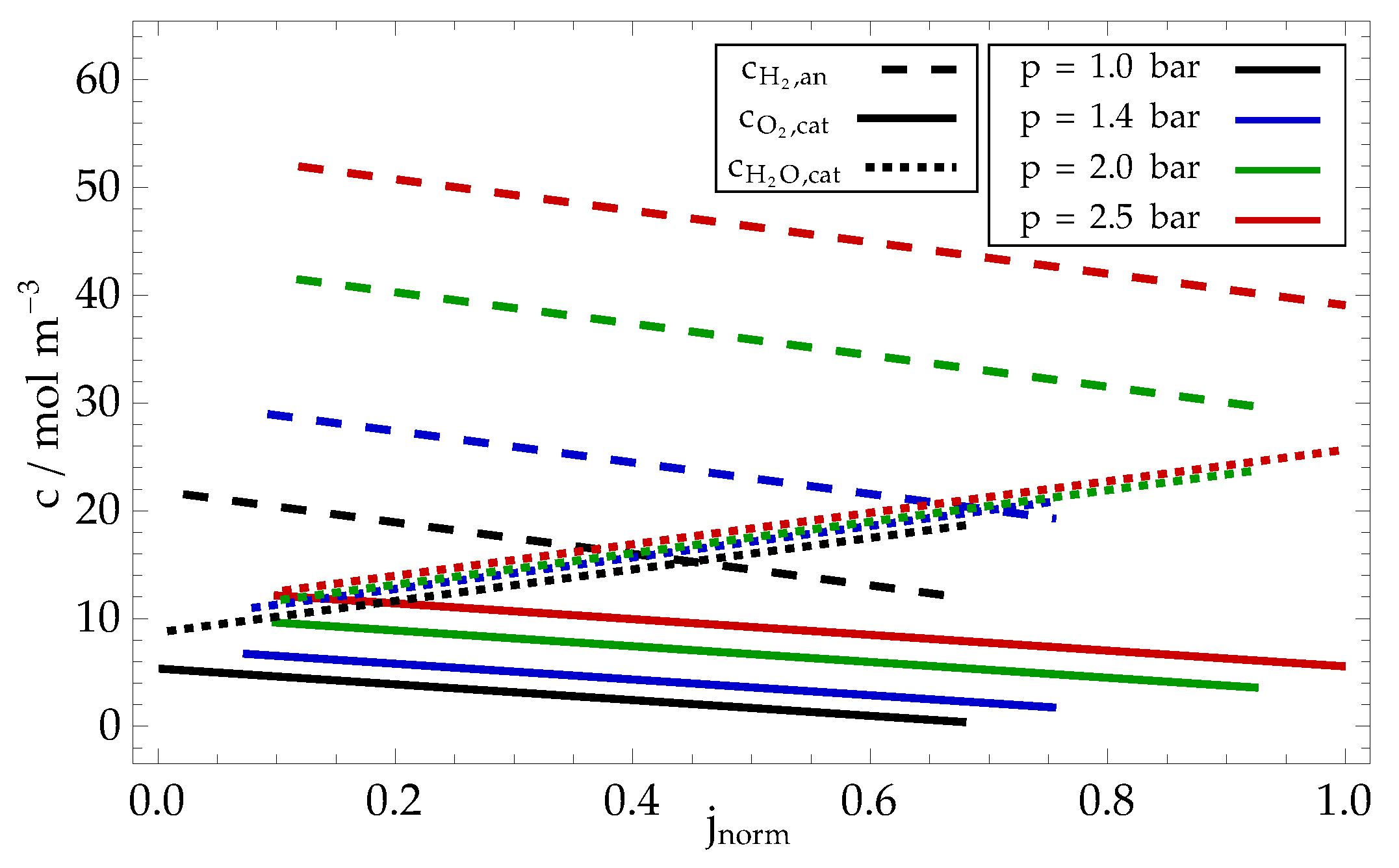
Figure 11.
Schematic presentation of proton enthalpy in the fuel cell electrode-membrane assembly during operation at two different current densities: a) , and b) . Total electrochemical enthalpy (purple) can be separated to chemical (orange) and electrical (green) contributions. Protons in the "Anode" are bound into hydrogen molecule with low chemical enthalpy () and large electric energy (). Transition into "Membrane" requires splitting the molecule into two protons and two electrons, which increases the chemical enthalpy but reduces electric energy due to electrode charging. Reaction with oxygen on cathode results in protons bounding into water molecule in "Cathode" with large decrease in chemical energy () and accompanying increase in electric energy (), resulting in net electric potential difference between cathode and anode (). In OCV conditions with no electric current (Figure a)), electrode overpotential and are determined of Nernst equation (25), while potential drop through the membrane is zero. When current flows (Figure b)) through the cell, larger overpotentials and appear to provide a sufficient rate of electrochemial reactions. Transport through the membrane requires electric potential difference across the membrane.
Figure 11.
Schematic presentation of proton enthalpy in the fuel cell electrode-membrane assembly during operation at two different current densities: a) , and b) . Total electrochemical enthalpy (purple) can be separated to chemical (orange) and electrical (green) contributions. Protons in the "Anode" are bound into hydrogen molecule with low chemical enthalpy () and large electric energy (). Transition into "Membrane" requires splitting the molecule into two protons and two electrons, which increases the chemical enthalpy but reduces electric energy due to electrode charging. Reaction with oxygen on cathode results in protons bounding into water molecule in "Cathode" with large decrease in chemical energy () and accompanying increase in electric energy (), resulting in net electric potential difference between cathode and anode (). In OCV conditions with no electric current (Figure a)), electrode overpotential and are determined of Nernst equation (25), while potential drop through the membrane is zero. When current flows (Figure b)) through the cell, larger overpotentials and appear to provide a sufficient rate of electrochemial reactions. Transport through the membrane requires electric potential difference across the membrane.
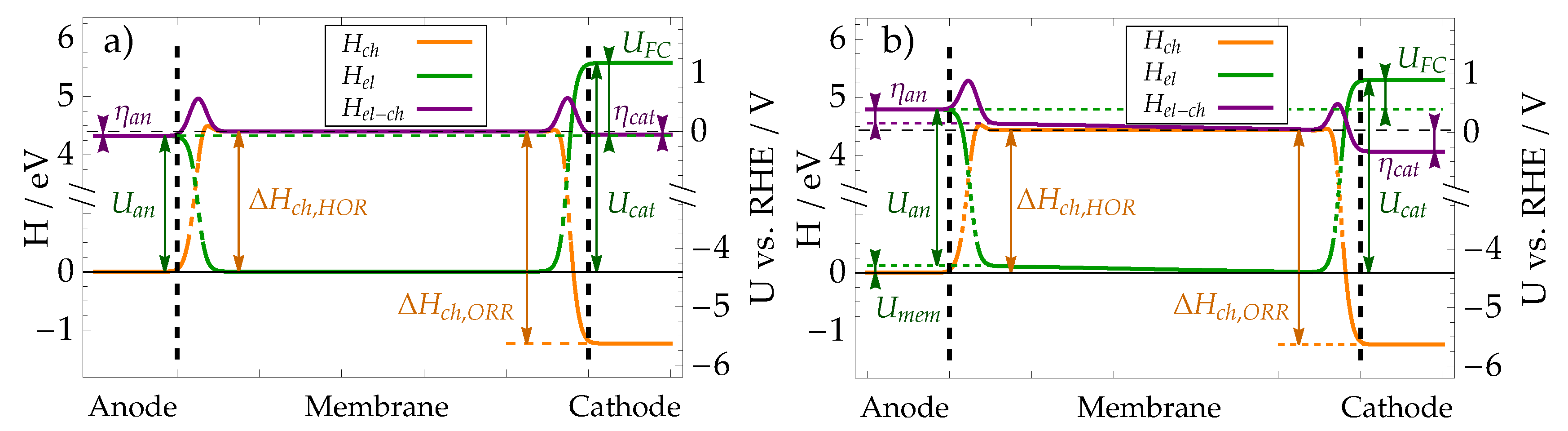
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



