
Submitted:
08 June 2023
Posted:
09 June 2023
You are already at the latest version
Abstract
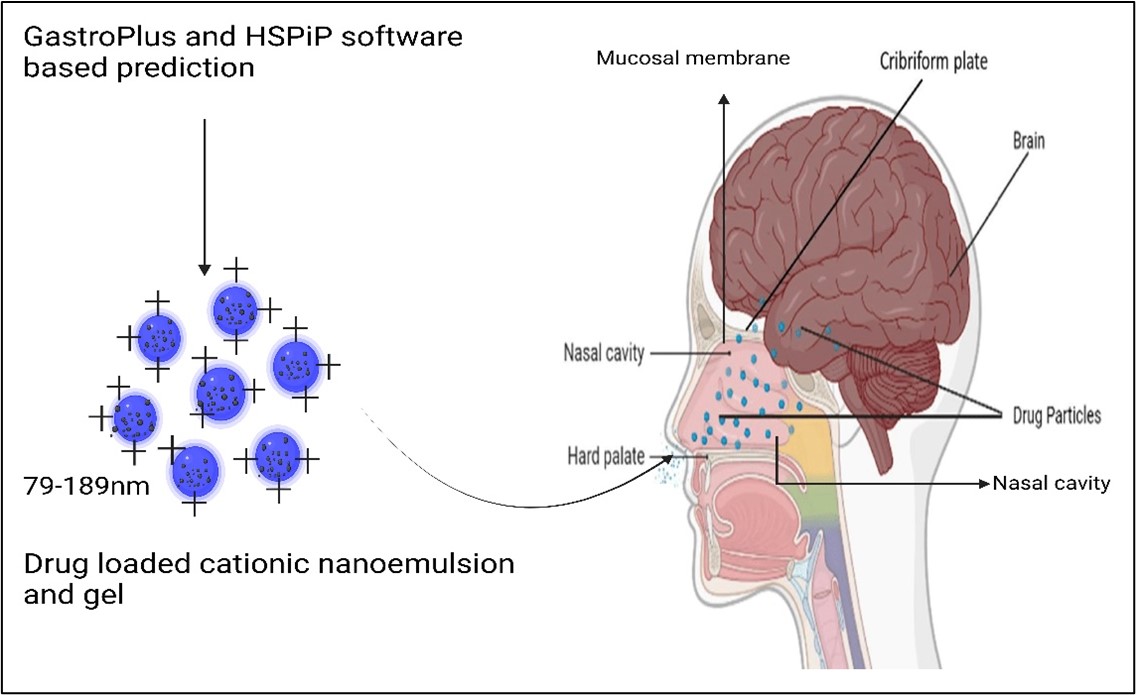
Oral and parenteral delivery of first-line anticonvulsant Valproic cid (VA) are associated with serious adverse effects, high hepatic metabolism, high clearance, and low bioavailability in brain. GastroPlus program was used to predict in vivo performance of immediate (IR) and sustained release (SR) products in human. HSPiP software predicted excipients with maximum possible miscibility of the drug. Based on GastroPlus and HSPiP program, various excipients were screened for experimental solubility study and prepared cationic nanoemulsions and respective gels for nasal to brain delivery. These were characterised for size, size distribution, polydispersity index, zeta potential, morphology, pH, % transmittance, drug content, and viscosity. In vitro drug release, ex vivo permeation profile (goat nasal mucosa), and penetration studies were conducted. Result showed that in-vivo oral drug dissolution and absorption were predicted as 98.6 mg and 18.8 mg, respectively from both tablets (IR and SR) at 8 h using GastroPlus. The drug access to the portal vein was relatively predicted high in IR (115 mg) as compared to SR (82.6 mg). Plasma drug concentration-time profile predicted was in good agreement with published reports. The program predicted duodenum and jejunum as the prime site of drug absorption and no effect of nanonization on Tmax for sustained release formulation. Hansen parameters suggested suitable selection of excipients. The program recommended for nasal to brain delivery of the drug using cationic mucoadhesive product. The optimized stable CVE6 was associated with optimal size (113 nm), low PDI (0.26), high zeta potential (+34.7 mV), high transmittance (97.8%), and high strength (0.7 %w/w). In vitro release and ex vivo permeation of CVE6 were found to be substantially high as compared to anionic AVE6 and respective gels. Penetration study executed high fluorescence intensity with CVE6 and CVE6-gel as compared to suspension and ANE6 which may be attributed to electrostatic interaction between mucosal membrane and nanoglobules. Thus, cationic nanoemulsion and respective mucoadhesive gel are promising strategy for delivery of VA to brain through intransal administration for the treatment of seizure and convulsion.
Keywords:
Valproic acid
; GastroPlus based prediction
; Cationic nanoemulsion
; Gels
; In vitro– ex vivo permeation profile
; CLSM study
1. Introduction
Epilepsy is defined as a group of neurological issues of central nervous system and characterized as a predisposition to epileptic seizures due to complexity of its characteristics. World Health Organization estimated that 50 million people are affected with the disease annually around the world [1]. In the USA, 2.3 million adults and 500,000 children are affected with varied form of epilepsy due to unknown and known possible reasons (genetics, trauma, stroke, brain tumor, and any factors responsible to disturb the normal pattern of brain circuit) [2]. In Saudi Arabia, the reported prevalence cases of epilepsy is 6.45 per 1000 people which is responsible to affect children mental health, behavior and academic performance [3].
Valproic acid (VA) is the most effective and first line anticonvulsant to control grand mal epilepsy and tonic – clonic fits (seizure), all types of seizure, and idiopathic generalized seizure. Low molecular weight (144 g/mol), hydrophobic nature (log p =2.54), high oral dose (not more than 600 mg/kg/day), high first pass metabolism (methylation, sulfation, and glucuronidation), and poor brain bioavailability after oral administration are possible reasons for nasal delivery of the drug to control seizure [4]. Commercial products (oral and parenteral) showed high plasma level of active metabolites (90% such as 4-ene-VA and undergoes to beta oxidation of fatty acid) due to hepatic metabolism (causing hepatotoxicity) and rapid clearance due to efflux (P-gp pump of microvessel endothelial cell in blood brain barrier). Parenteral delivery causes serious side effect possibly due to reticuloendothelial system (Kuffer cells) based metabolism and low bioavailability to brain. Hammond et al. investigated pharmacokinetics profiles of the drug in cat model (six adult cats) after rapid intravenous infusion (60 mg/kg within 3 min of infusion using saline) wherein the maximum level of the drug was obtained at 1 min (brain distribution half-life as 6 min estimated from α-phase) followed by rapid clearance (mean elimination half-life of 41 min) and volume of distribution (Vd) as 0.125 L/kg. Low in vivo uptake (low brain: plasma ratio), low volume of distribution, and rapid clearance (brain elimination half-life as 41 min estimated from β-phase) from brain indicated poor binding of the drug to the cerebral cortex [5]. As per US FDA label of DEPAKENE, oral absorption varies subject to subject (age dependent) and dosage form to dosage form (tablet versus capsule). In adult patient, absorption rate on monotherapy (250 mg of oral delivery) is nonlinear and the kinetics of unbound drug is linear. Notably, the drug is primarily metabolized through liver (30-50% as glucuronide conjugate), mitochondrial β-oxidation (˃ 40%), and urine (3% as unchanged). In human mean plasma clearance and Vd values were reported as 0.56 L/Kg and 11 L, respectively following 250 mg of oral administration in adult (70 kg or 1.73 m2 as body surface area) [6].
Several drugs (35-40 molecules) have been exploited for brain delivery using nasal route of administration. The route is the most preferred one to circumvent the aforementioned issues on oral and parenteral delivery in conventional dosage form. Various nanocarriers have been reported for the drug delivery using nasal route. These are lipidic nanocarriers (lipid nanoparticles, nanoemulsion, liposomes), nanotubes, and dendrimer [7,8,9,10,11]. Tan et al. tailored stable nanoemulsion comprised of safflower (70-80% linolenic acid rich natural oil capable to delivery drug across blood brain barrier and cerebrospinal fluid barrier) for delivery of the drug to the brain and brain bioavailability was improved [10,12].
Nanoemulsion is well explored nanocarrier for drug delivery due to desired innate features such as nanoscale globular size, suitability to load small molecule, thermodynamically stable isotropic mixture. Imposed cationic charge on nanoglobule further improve its pharmaceutical utility for facilitated permeation across biological membrane and extended residence time. Nasal drug delivery is usually challenged with short residence time, and washout after nasal administration. Cationic charged nanocarriers interact with the biological membrane for maximized internalization for increased passive permeation and drug deposition (enhanced drug access across the biological membrane) [13,14]. Nasal route of administered offered several advantages over oral and parenteral routes such as (a) high patient compliance, (b) avoids hepatic metabolisms and related drug degradation, (c) direct drug access from olfactory region to cranial cavity of brain, (d) avoids unnecessary administration of excipients, (e) dose mitigation and reduction in dose related side effects, (f) low therapy cost, (g) ease in regulatory constraints for approval, (h) safety and biocompatibility [14].
In this study, we predicted in vivo performance of the drug using GastroPlus predictive and simulation program using literature, program default value, and experimental data. The program assisted to predict dose dependent pharmacokinetic parameters (time required to reach Cmax as Tmax, area under the curve as AUC, and maximum drug concentration reached in blood as Cmax) considering oral commercial dose (250 mg) and dose form (tablet) in healthy human adult. Moreover, cationic nanoemulsions were prepared, optimized, and characterized for in-vitro (size, size distribution, zeta potential, morphology, thermodynamic stability, release profile at pH 5.5 and 6.8), and ex-vivo performance (permeation flux, drug deposition, and enhancement ratio) (goat nasal tissue).
2. Materials and methods
2.1. Materials
Valproic acid sodium salt (VA, 98.0 % pure) and Polysorbate-80 were procured from Sigma Aldrich (Merck), Mumbai, Maharashtra, India). Soya lecithin powder (97%) was purchased from Otto Chemie Pvt. Ltd., Mumbai, Maharastra, India. HPLC (high performance liquid chromatography) grade solvents (methanol, ethanol, acetonitrile, and buffering reagents were obtained from Merck, Mumbai, Maharashtra, India. Edible safflower, flaxseed and grape seed oils were purchased from local medical shop. Buffer reagents (potassium dihydrogen phosphate, sodium chloride, and sodium hydroxide) were procured from S.D. Fine S.D. In house installed distilled water was used as aqueous solvent. For HPLC mobile phase preparation, Milli-Q water was used (Millipore, USA).
2.2. Methods
2.2.1. Prediction and simulation study using GastroPlus for oral tablet
The program was used to predict pharmacokinetic parameters (PK) of orally delivered VA tablet to an adult patient with dose of 250 mg. In literature and DEPAKENE tablet label, varied bioavailability, absorption rate and PK parameters have been described depending upon patient body weight. To avoid preclinical and clinical study due to expensive and tedious work, the programme assisted to predict various PK parameters in targeted patient for desired dose, dosage form, dosage volume, and frequency of dosing frequency. For this, the program used three basic tabs such as (a) compound tab, (b) formulation tabs and (c) pharmacokinetic tabs. We used literature data, experimental value, and by-default program suggested values to run the simulation and prediction (table 1). Moreover, parameter sensitivity assessment (PSA) was used to know the impact of various factors (physicochemical properties of the drug, physiological conditions such as intestinal lumen and related factors) affecting PK parameters of the drug. Physicochemical properties of the drug include reference solubulity, particle size, volume, density, logP, pKa, and molecular weight. Physiological factors include gastrointestinal pH, stomach volume, residence time, and radius. Formulation factors are nanosize, shape, and solubility. Regional compartmental model predicts regional absorption of the drug through nine different GIT sections (stomach, duodenum, ileum-1,2, jejunum-1,2, ascending colon, colon, and caecum). Total absorption indicates the sum of absorption from the GIT of patient. Prediction and simulation were carried out considering fast subject to avoid food interaction in prediction model. Simulation time was 24 h for each run of prediction and simulation [15,16].
Hansen solubility parameters for VA and excipients
Hansen solubility parameters have been exploited for various solvents, co-solvents, drug and human skin (normal and abnormal). The parameters were estimated using HSPiP program. The fundamental of the software is based on the physicochemical interactions (in term of cohesive energy) of a solute into a particular solvent. These parameters are dispersion energy (δd), polarity (δp), and hydrogen bonding energy (δh) [17,18]. Therefore, a solute interacts into a solvent through these cohesive forces working together. Thus, the difference of any parameter between solute and solvent close to zero is considered as miscible or soluble. Thus, the program predicted relevant excipients based on these HSP values of each excipient close to the HSP values of the model drug. The program estimated these values as shown in table 1. The HSP values of oils, lecithin, and PC were obtained from literature and calculated manually based on percent composition of linoleic acid or phosphatidylcholine (PC) present [19].
Solubility of valproate sodium in various excipients
Solubility of VA was carried out in various lipids, surfactants, and co-surfactants to identify the most suitable and biocompatible excipients for nanoemulsion for brain delivery. Stearylamine was added in organic phase to impose cationic charge on globular surface for adhesive purpose [13]. Flaxseed (50-70%), safflower (70-78%), and grape seed oil (70%) are prime source of linoleic acid. Tween 80, span-80, transcutol, propylene glycol, and lecithin were used as surfactants and co-surfactants. In brief, a fixed amount of each excipient was transferred to a clean glass vial. A weighed amount of the drug was added to each vial containing individual excipient. The glass vial was closely closed and sealed for solubility study. The vial was placed inside water shaker bath (Remi Shaker, Mumbai, India) set at fixed temperature (40 °C) and shaking rate (75 rpm). The study was continued for 72 h to achieve equilibrium. Then, the mixture was centrifuged to get supernatant liquid. The amount of the drug dissolved was assayed using UV Vis spectrophotometer (U 1800, Japan) at 210 nm [10]. The study was repeated to get mean and standard deviation (n = 3, st).
Pseudo ternary phase diagram, cationic nanoemulsions, and nanoemulsion gel
To construct cationic nanoemulsion, a constant amount of stearylamine (0.1%) was used in the formulation. Based on HSP values and experimental solubility of AV, excipients were selected. Excipient possessing HSP values close to the HSP values of AV and excipients with the highest solubility of AV were selected for cationic nanoemulsion. Thus, safflower seed oil, tween 80+lecithin (1:1), and PG were selected as oil, surfactant, and co-surfactant, respectively. To impose cationic charge, a constant value (0.1%) of SA (stearylamine as cationic lipid) was incorporated into the organic phase of each formulation [20]. Various pseudoternary phase diagrams were constructed to identify a right ratio of surfactant to co-surfactant (Smix). Slow and spontaneous titration method was adopted to prepare nanoemulsion by varying lipid to Smix ratio [15]. Transparent and isotropic cationic nanoemulsion was selected for further characterizations and studies. To prepare a nanoemulsion gel, cationic nanoemulsion was incorporated into carbopol gel (1%). The final strength of gel was 0.5%. Each nanoemulsion and respective gel contained a constant amount of VA. For this, a weighed amount of carbopol 934 was dispersed in a warm distilled water to get final strength of 1%w/w. The dispersed gel was vigorously stirred with a mixer at high speed (10000 rpm). The obtained gel was treated with few drops (3-5 drops) of triethanolamine (base) as cross linking agent. The acidic solution of carbopol dispersion was triggered for cross linking under triethanolamine and become transparent viscous gel. Equal weight of gel and lyophilized formulation was mixed together using homogenizer to get a gel of 0.5% gel strength. Final concentration of AV in gel product was approximately 5%w/w. The final pH of each formulation was adjusted to 6.8 to get good consistency and compatibility with nasal mucosa.
Thermodynamic stability of cationic nanoemulsion: Freeze–thaw cycle and ultracentrifugation
Each developed nanoemulsion was subjected to extreme physical (ultracentrifugation) and thermal stress (extreme low and extreme high temperature). For this, each cationic nanoemulsion was stored in a clear glass vial, labelled and sealed. Each formulation was separately stored in stability chamber with set temperature. A cycle of exposure to low as freeze (-21 °C) and high as thaw (40 °C) temperature was repeated thrice followed by room temperature. Each sample was withdrawn from both temperature and kept at room temperature (25 °C) to resume its original stable form (isotropic liquid). In second phase, each stable formulation was subjected to ultracentrifugation step (22000 rpm for 5 min). Any sign of physical instability (drug precipitation, colour, creaming, and phase separation) was considered as unstable product and dropped out from further study. This freeze-thaw cycle was mandatory to identify the most stable product.
2.2.2. Evaluation of cationic nanoemulsions and gels
Nanoemulsions were evaluated for globular size, size distribution and zeta potential. These parameters were determined using Zetasizer (zetasizer, ZP, USA). Formulations were diluted with distilled water before scanning for size analysis. In case of zeta potential, the formulations were analysed without dilution to get tangible zeta values. This value was expected as positive for cationic nanoemulsion whereas nanoemulsion without stearylamine was anticipated to be negative. The analysis was carried out at room temperature. Viscosity of each formulation was determined using viscometer (Bohlin viscometer, NJ, USA). The sample was processed under room temperature (25 °C). The study was replicated for the mean and standard deviation (n =3). The values of pH were estimated using calibrated digital pH meter.
Morphological evaluation of the optimized cationic nanoemulsion and respective gel
The optimized cationic nanoemulsion and respective gel were observed under transmission electron microscopy. The tool assisted to visualize globular size, size distribution, and morphology (shape). For this, the sample was placed on a glass cover slip previously coated with poly-L-lysin (a fixative) [21]. The sample was air dried before scanning. Then, the sample was transferred to copper grid using double adhesive tape such as that the sample faced air. The sample was stained with phosphatungstic acid (0.1%) before coating. Finally, the dried sample was scanned at various magnification and resolution. A fixed location was located and scanned for the sample. The process was conducted at room temperature. The wet sample was avoided to scan due to poor scanning, and resolution of images as observed by interfered electronic beam.
Drug content estimation
The drug content was estimated from the optimized anionic nanoemulsion, cationic nanoemulsion, and respective gel formulations. In brief, a weighed amount of the formulation was dissolved in methanol-chloroform mixture (1:10). The mixture was stirred for 10 min to extract the drug. The mixture centrifuged for 15 min at 12000 rpm. The supernatant was used to estimate the total drug content. The drug was assayed using validated HPLC method at 210 nm. The experiment was repeated to get mean and standard deviation. In case of gel formulation, a weighed amount of gel was dispersed into water-ethanol mixture (1:2) to get the extracted drug. Then, the mixture was stirred for 15 min followed by centrifugation. The supernatant was used to estimate the drug content.
2.2.3. In vitro drug release profile
In vitro drug release profile for each nanoemulsions and respective gel were performed using a dialysis membrane with molecular weight cut-off of 12-14K Dalton (HiMedia, Mumbai, India). For this, a fixed dimension of the dialysis membrane was cut from ribbon and soaked in saline for 12 h before use. The activated dialysis bag was filled with the test sample and both ends were clipped with a plastic clipper. This helped to maintain constant effective surface area for the drug release. The release medium (500 mL) was phosphate buffer at pH 6.8 and pH 7.4. A glass beaker containing the release medium was used for the drug release. The test sample bag was suspended in the release medium already placed on a heating magnetic stirrer. A teflon coated magnetic bead was used to maintain the temperature and uniform drug distribution within the bulk volume released from the bag. Sample was collated at different time points (0.5, 1, 2, 3, and 6 h). The withdrawn volume was replaced with the fresh release medium. The withdrawn sample was filtered and used for the drug content released after each time point. The drug was analysed using HPLC method. The release medium chamber maintained at temperature of 32±1°C throughout the study. The effective surface area for passive diffusion of the drug was 1.34 cm2 functional at 32±1°C [22,23].
2.2.4. Ex vivo drug permeation and drug deposition using goat nasal mucosal tissue
Drug permeation and deposition studies were performed using an excised goat nasal mucosa obtained from a local slaughter house. The excised tissue was used immediately after 20 min of sacrifice time to avoid tissue damage and death. The intact nose was obtained and the skin was removed. Then, the nose was stored in a cold phosphate buffer solution (pH 7.4) [24]. The nasal mucosa was removed using surgical scissor and forceps without making any surgical cut in effective desired area of mucosal membrane. The obtained mucosal tissue was immersed into a freshly prepared Ringer’s solution with proper aeration [25]. The excised tissue has dimension of 0.2 mm × 10 mm with effective surface area of diffusion of 1.78 cm2. The tissue was mounted between the receptor and donor chamber. The receptor chamber was filled with SNF (simulated nasal fluid) at pH 6.8 [26]. The release medium was maintained at 37 ± 1 °C by circulating hot water in jacketed system around the chamber. A rice bead was placed inside receptor chamber rotating at 300 rpm on magnetic stirrer. A constant amount of the sample was placed on the mucosal adhesive side for drug permeation. Four groups were categorized as (a) CVE6-1, AVE6, CVE6-gel, and AVE6-gel. For comparison, drug solution was used as control in gel formulation. In each case, equivalent amount of the drug was loaded on the effective permeation area. The sampling (1 mL using syringe) was conducted at different time points as 0.5, 1, 1.5, 2, 2.5, and 3 h. The withdrawn sample volume was replaced with equal volume of the fresh medium. The sample was filtered using membrane filter (0.2 µm) and the content of the drug was estimated using HPLC method. The study was replicated to get mean and standard values. The result was expressed as the percent of the drug permeated for brain delivery or percent diffusion for brain access or availability of the drug to the brain ex vivo. The permeation parameters (steady state flux, targeted flux, permeability coefficient, and enhancement ratio) were estimated using equation below.
Jss = (dM/dt) × (1/A) = PC
Wherein A, P, and C represent as the effective surface area for diffusion, permeability diffusion coefficient, and the initial loaded content of the drug, respectively. Jss indicates the steady state flux of the solution as per Fick diffusion equation (1). The value of dm indicates the amount of the drug diffused across the mucosal membrane in given time point (dt). The study was conducted up to 360 min to avoid loss of natural integrity of tissue and tissue viability [27].
Drug deposition study was conducted after completion of ex vivo permeation study. The mounted tissue was removed from the diffusion cell. Each tissue was separately sliced into small pieces. The sliced pieces were transferred into a vial containing methanol and chloroform (1:2). The mixture was stirred for 12 h under closed condition using magnetic bead. The drug was extracted from the tissue and subjected for centrifugation. Fatty debris and tissues were settled at bottom as pellet and the supernatant clear solution was removed for the drug analysis. The supernatant was filtered using a membrane filter and analysed using HPLC method [10,28].
2.2.5. Confocal laser scanning microscopy (CLSM)
To visualize the degree of the drug penetration, the same formulations and control were reformulated using rhodamine 123 as probe in the formulation. The composition and experimental conditions were kept constant as in ex vivo permeation and drug deposition study section. The dye was present as 0.01% w/v in each formulation. The Franz diffusion cell, tissue mounting, release SNF, volume, pH, and loaded dose were constant as in ex vivo study for 6 h. After 6 h of permeation study, the tissue were removed for each group (five groups) and the adhered material was washed with running water. The tissue was sliced as per CLSM requirement. The treated and the untreated skin were sliced into small pieces using microtome. The tissue specimen was placed on the glass coverslip and air dried for 12 h. Each tissue was visualized under CLSM and evaluated for globular penetration across mucosal membrane (Fluorescence Correlation Microscope-Olympus FluoView FV1000, Olympus, Melville, NY) with an argon laser beam with excitation at 488 nm and emission at 590 nm [29,30].
3. Results and discussion
3.1. Prediction and simulation study using GastroPlus for oral tablets
VA is orally administered as different dosage forms such as immediate release table, oral solution, oral capsule, and sustained release tablet. Considering 200 mg as an adult dose in IR tablet and SR tablet, the program was run for simulation of 24 h. The program was used to predict PK parameters (Cmax, AUC, and Tmax) for both of them in adult human. Limited data are available for comparative PK studies of VA using IR and SR tablet in human. No data are available for predicting PK parameters using GastroPlus program and comparison of IR tablet and SR tablet at fixed dose and dosing frequency. Teixeira-da-Silva et al. predicted population pharmacokinetics of VA monotherapy considering different doses, body weight, and age group. The regimen depicted were designed to achieve VA concentration within the acceptable therapeutic range. The steady state plasma concentrations were predicted as ˃ 120 mg/l for age group of 15 (dose of 1000 mg in tablet) and 35 years (dose of 1200 mg in tablet) whereas this value was predicted as ˂ 100 mg/l for 1 (dose of 100 mg in solution) and 6 years (dose of 200 mg in solution) children [31]. Thus, authors found that there was no significant difference in plasma drug concentration from tablet of 1000 mg or 1200 mg in adult of different age (15 versus 35 year), and body weight (56 versus 70 kg) [31]. In the present study, we used 200 mg dose for an adult of 70 kg body to predict in vivo dissolution and in vivo absorption of IR tablet and SR tablet of VA. The result is illustrated in Figure 1A-B. It is clear that the predicted pattern of in vivo dissolution of IR VA tablet and SR VA tablet are closely related without substantial difference in fast state adult. Interestingly, the amount of the drug absorbed to the portal vein (AmtPV-1) is quite higher in IR VA tablet as compared to SR VA tablet (Figure 1A) as predicted in the program. This may be prudent to correlate the difference in dissolution rate between IR and SR tablet. IR tablet exhibits rapidly drug dissolution in gastric content for profound availability of the drug for absorption at intestinal mucosa of lumen. On the hand, SR tablet follows a different dissolution process due to rate limiting membrane of polymer coated on the tablet. Slow and sustained release of the drug caused slow and extended absorption as predicted in Figure 1B. Total amount of the drug absorbed form both types of tablet is approximately same as predicted in the program (green bold colour) suggesting no significant difference on modified form of tablet over period of 24 h. This may be due to slightly acidic nature of VA (pKa = 5.14) suitable for absorption from intestinal area as the prime site of the drug absorption. Therapeutic window of the drug is 30 – 100 mg/L after oral administration in human [32]. The drug is rapidly absorbed from oral dosage form and the drug access to the brain is limited due to high protein binding capacity (90%). Low volume of distribution (0.125 L/Kg) is very similar to that found in human suggesting no significant bounding of the drug to the brain. Therefore, this needs high blood plasma level by administering high oral doses. Limited free drug in the plasma is available for brain access. In a study, it was observed that VA transport to brain is through the monocarboxylic acid transport system. The plasma level of VA ˂ 60 µg/ml results in low level of the drug in brain. For clinical effectiveness in human, it is only realized with relatively high plasma concentration above 55 µg/mL [33].
Prediction of plasma drug concentration time profile
The program predicted the plasma drug concentration time profile of IR tablet and SR tablet of VA considering 200 mg of dose for 70 Kg adult. The result of predicted PK profile is executed in Figure 2A-B wherein Cmax values of VA IR tablet and VA SR tablet were predicted as 159.3 µg/ml and 82.5 µg/ml, respectively. The predicted values are quite interesting and convincing as explained before for therapeutic effectiveness. Both values are enough to produce substantial level of the drug in plasma for brain access (˃ 55 µg/ml) [33]. The acidic form of the drug is suitable for solubility in water and acidic medium (pKa 5.4). Therefore, IR tablet showed rapid drug dissolution for immediate drug absorption. Therefore, IR tablet (2.1 h) showed relatively low Tmax as compared to SR tablet (5.2 h) in prediction. These predicted Tmax values are in good agreement with the published report for oral solution and SR tablet [34]. This indicated that the model is in good fit (as observed in high Akaike value) for simulation and prediction. The result can be correlated to the difference of oral bioavailability of VA in drug solution and SR formulation. In literature, drug solution and SR formulation resulted in 100 % and 80-90% bioavailability for VA [35]. Thus, the predicted pattern of VA SR tablet suggested slow and sustained delivery of VA for long term effect within therapeutic window (200 mg). However, the drug is limited to brain access due to various possible reasons. These may be high protein binding capacity, high hepatic drug metabolism, low solubility of the drug, and extra hepatic drug metabolism. Sustained release tablet slightly decreased the drug absorption to the portal vein (Figure 1B).
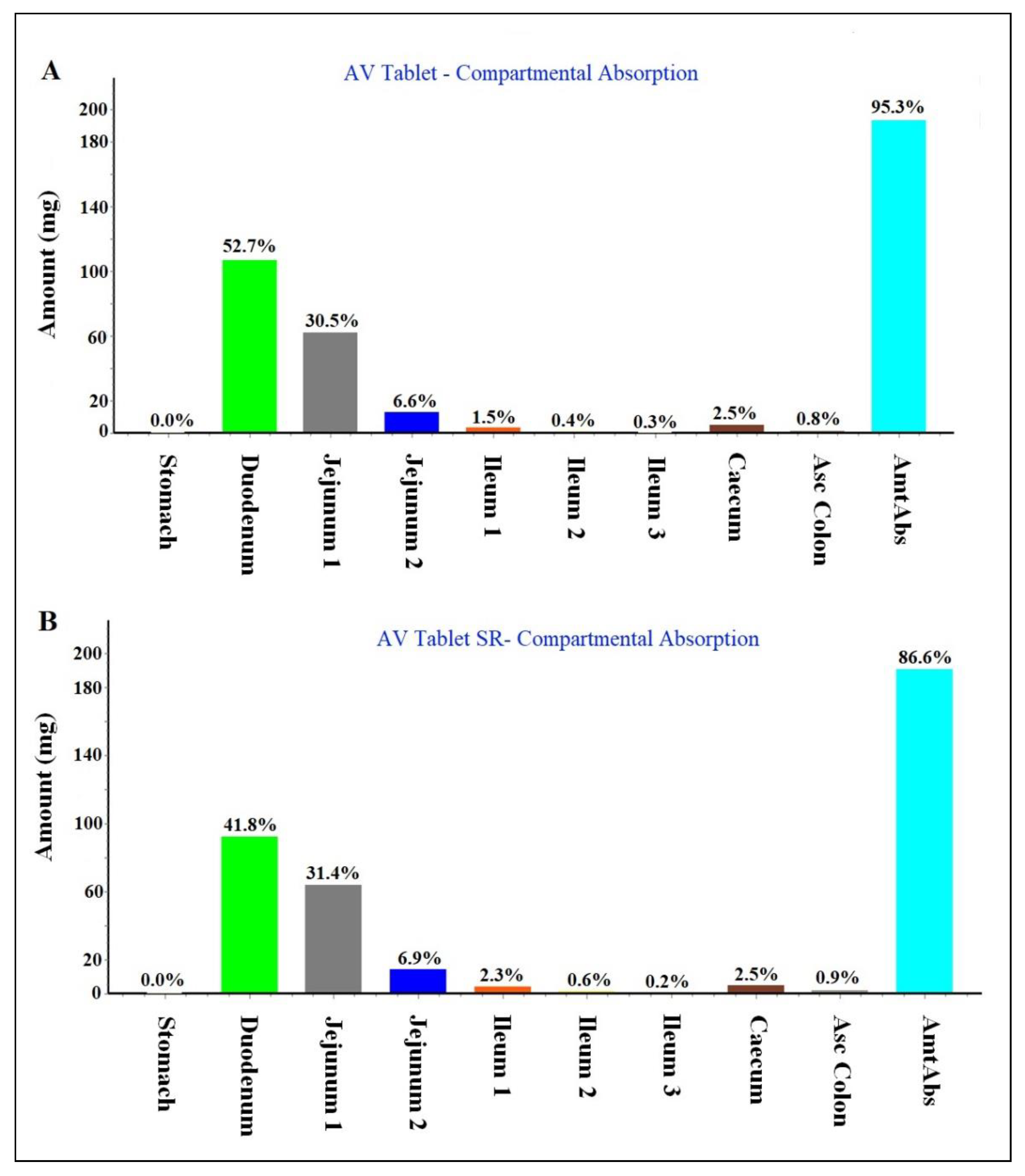
Regional compartmental absorption of both tablets
The program predicted nine compartmental absorption sites in GIT. Both IR and SR tablets were processed in the system to estimate percent regional absorption of the drug. The result is elicited in Figure 3A-B wherein IR tablet and SR tablet predicted overall total absorption as 95.3 % and 86.6%, respectively. VA is a slightly acidic drug with a pka value of 5.6. Therefore, the drug was predicted to be absorbed primarily from proximal portion of GIT. Thus, duodenum and jejunum are the main sites of oral absorption. The drug is considered poorly absorbed from the distal GIT region as shown in Figure 3A-B. The predicted values are in agreement with the published report of oral bioavailability for drug solution (100%) and SR tablet (80 – 90%) [35].
PSA assessment (parameter sensitivity analysis)
Parameter sensitivity analysis (PSA) assessment was performed to identify relevant factors responsible to affect PK parameters of VA tablet on oral administration. The analysis was carried out GastroPlus program considering fast state condition of subject. This avoided any interaction of food. In the study, we attempted to predict the impact of nanocarrier system for oral drug delivery and their impact on PK parameters such as Cmax, Tmax, and AUC (area under the curve). It is clear in the prediction study and literature based findings that conventional dosage forms of VA (tablet, SR tablet, and solution) do not have much difference in term of bioavailability in human. Therefore, oral bioavailability values of predicted one is almost similar to reported values (as described before). However, the program predicted that nano effect has no impact on PK parameters on oral administration. This can be correlated to the lipophilic nature of drug being absorbed but poor dissolution (BCS class II) [15,16]. Conclusively, GastroPlus simulation and prediction program assisted me to understand that nanonized product of VA for oral delivery could be of no benefit for brain delivery. Therefore, it is better to formulate a nanocarrier based drug delivery for brain delivery using nasal route of administration. The basal route contains olfactory chamber directly linked to brain for the drug access. Thus, the purpose of GastroPlus based prediction was to understand the feasibility of oral nanocarrier for brain delivery using human data (obtained from literature). The program provided various predicted in vivo values for human trial.
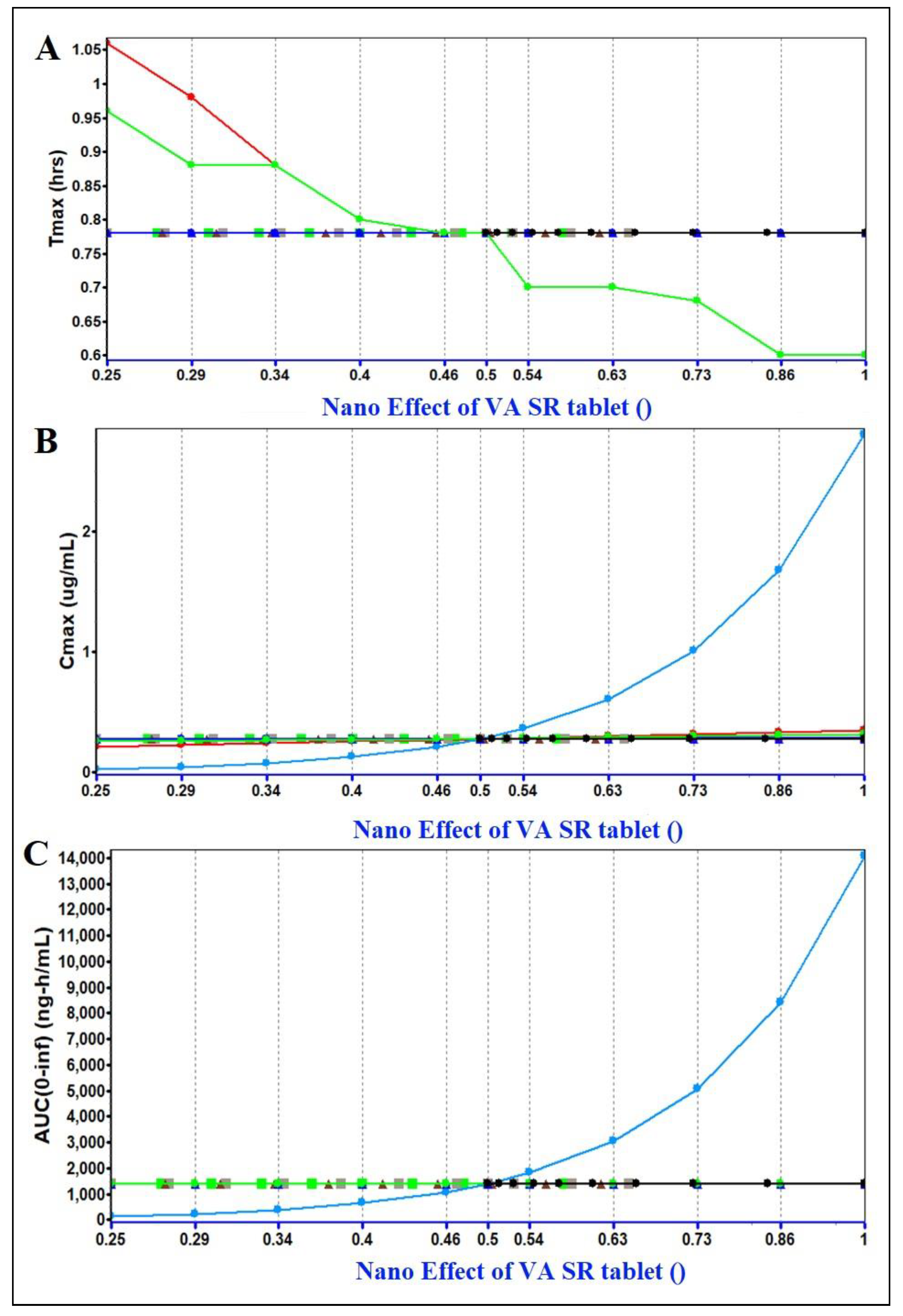
Figure 4.
Parameter sensitivity analysis assessment using GastroPlus simulation and prediction program. (A) Impact of nanonized VA product (oral delivery) on Tmax, (B) impact of nanonized product on Cmax, and (C) impact of nanonized product on AUC values. Blue bold line indicates the impact of Nano Effect. Red bold line indicated “Peff” permeability coefficient across mucosal membrane, and effect of duodenum ASF (absorption scale factor).
Figure 4.
Parameter sensitivity analysis assessment using GastroPlus simulation and prediction program. (A) Impact of nanonized VA product (oral delivery) on Tmax, (B) impact of nanonized product on Cmax, and (C) impact of nanonized product on AUC values. Blue bold line indicates the impact of Nano Effect. Red bold line indicated “Peff” permeability coefficient across mucosal membrane, and effect of duodenum ASF (absorption scale factor).

Table 1.
Summary of input data for GastroPlus simulation and prediction of VA sodium.
| Parameter | Values |
|---|---|
| Molecular formula** | C8H15NaO2 |
| Molecular weight (g/mol)** | 166.19 |
| Melting point (°C) | 300 |
| Aqueous solubility (mg/ml) at 25 °C | ˂ 1 |
| Density (g/mL) | 0.9 |
| Pka | 5.14 |
| Log p | 3.08 |
| ***Apparent permeability coefficient (cm/h) across hCMEC/D3 and CC-2565 of in vitro blood brain barrier | 0.625 |
| Dose (mg) | 200 |
| Body weight (Kg) | 70 |
| Dosing volume (mL) | 1 |
| Mean precipitation time (s) | 30 |
| AUC (µg. h /mL)ψ | 10 – 160 |
| Cmax (mg/L)## | ~ 120 |
| Tmax (h) (mean) | 5 |
| Elimination half-life (h)* | 8-15 |
| Clearance (L/h)# | 0.206 – 1.154 |
| Plasma protein binding (%)# | 90 - 95 |
| Vd (L)# | 8.4-23.3 |
| pH for reference solubility** | 7.0 |
| Simulation time (h)** | 24 |
Hansen solubility parameters for VA and excipients
HSP values helped to select excipients possibly exhibiting maximum drug solubility via cohesive interaction (cohesive forces) [41]. The program is well exploited for solute miscibility/solubility in a particular solvent. The HSP values of the drug and each excipient have been summarized in table 2. It is easy to understand that the lipid, the surfactant, and the co-surfactant possessing HSP close to the values of the drug could be the most appropriate and suitable for the drug solubility. The values of δd, δp, and δh of the drug are 16.1, 4.3, and 9.0 MPa1/2, respectively. The δh value of tween 80 is quite close to the HSP values of VA as compared to span 80 (δh of span 80 is 12.4 as compared to 9 of tween 80). Therefore, a solute interacts into a solvent through these cohesive forces working together. Thus, the difference of any parameter between solute and solvent close to zero is considered as miscible or soluble. Thus, the program predicted relevant excipients based on these HSP values of each excipient close to the HSP values of the model drug. The program estimated these values as shown in table 2. The HSP values of oils (safflower, Flaxseed oil, and grape seed oil), lecithin, and PC were obtained from literature and calculated manually based on percent composition of linoleic acid or phosphatidylcholine (PC) present [19]. Among oils, safflower seed oil might be the most suitable for tailoring cationic nanoemulsion due to predicted miscibility of the drug in term of HSP. The oil has been reported with high content of linoleic acid (78%) and linoleic acid is considered as a promoter for diffusion of lipophilic drug across blood brain barrier [42].
Table 2.
Summary of HSP values for the drug and selected excipients.
| Drug and excipient | δd (MPa1/2) | δp (MPa1/2) | δh (MPa1/2) |
|---|---|---|---|
| AV | 16.1 | 4.3 | 9.0 |
| Safflower seed oil (87%)* | 14.5 | 2.7 | 5.3 |
| Grape seed oil (70%)* | 11.69 | 2.17 | 4.27 |
| Flaxseed oil (60%)* | 10.02 | 1.86 | 3.66 |
| Tween 80 | 16.6 | 5.3 | 7.5 |
| Span 80 | 16.7 | 6.1 | 12.4 |
| Lecithin (PC as 20%)* | 3.2 | 0.54 | 0.64 |
| Linoleic acid* | 16.7 | 3.1 | 6.1 |
| Transcutol HP | 16.0 | 2.8 | 6.2 |
| PGϕ | 16.8 | 10.4 | 21.3 |
| PC* | 16 | 2.7 | 3.2 |
Solubility of valproate (VA) in various excipients
The result of experimental solubility of VA is portrayed in Figure 5. The solubility of the drug was found to be maximum in safflower seed oil (8.9 ± 0.11 mg/mL), tween 80 (5.3 ± 0.09 mg/mL), and transcutol (6.3 ± 0.08 mg/mL). These maximized solubility can be rationalized based on the HSP values as predicted in table 2. The difference value of Δδd is 1.6 (16.1 – 14.5) for the solute (VA) and the solvent (safflower oil) which is quite low for high miscibility/solubility. Similarly, the difference values of Δδp and Δδh are 1.6 (4.3 – 2.7) and 3.7 (9 – 5.3), respectively, for the drug in safflower. These difference are quite convincing for maximized drug solubility due to interactive forces (polarization, hydrogen bonding ability, and dispersion nature). Among co-surfactants, transcutol was selected due to highest solubility and suitability for the drug. Flaxseed (50-70%), safflower (70-87%), and grape seed oil (70%) are prime source of linoleic acid. Linoleic acid rich oils are getting popular in pharmaceutical and cosmeceutical industries due to possessing various skin benefits such as (a) anti-inflammatory, (b) acne reductive, (c) skin softening, (d) moisture retentive, (e) facilitates drug diffusion across blood brain barrier (50-87% linoleic acid), and (f) biocompatibility [10,44]. Thus, safflower, tween 80, and transcutol were selected as oil, surfactant, and co-surfactant. However, a blend of tween 80 and lecithin (1:1) was taken for stable and small sized nanoemulsion. Combination was supposed to stabilize nanoemulsion with small size particle as compared to tween 80 as standalone.
VA loaded cationic nanoemulsions prepared
To construct cationic nanoemulsion, a constant amount of stearylamine (5 mg) was used in each formulation. A series of nanoemulsions (CVE as cationic and AVE as anionic nanoemulsion) were prepared as shown in table 3. Formulation CVE5 exhibited unique characteristic feature among them. The globular size, PDI, zeta potential, %T, and product strength (%w/w) were found to be as 79 nm (the lowest value), 0.11 (the lowest value), +27.1 mV (optimal), 95%, and 0.5%, respectively. The lowest value of PDI is due to the lowest content of oil (9.8%) and the sufficient amount of Smix (21.84%) responsible for efficient emulsification and resulted in homogeneous nature of globular distribution. However, %DC was found to be low (~0.5%) for CVE5 which can be related with the low content of oil (9.8%). The formulation CVE6 had optimal content of oil (14.46%) and Smix (17.15%) to render optimal size (113 nm), high zeta potential (+ 34.7 mV) for enhanced stability, and high %DC (67%) as compared to others. Comparing CVE1, CVE3, and CVE4, it is clear that increasing relative concentration of Smix as compared to oil, the size values were regularly decreased from 185 nm to 148 nm. This may be due to efficient emulsification by surfactant mixture. Comparing CVE2 (189 nm) and CVE4 (148 nm), the size of CVE4 was substantially decreased due to high content of surfactant mixture even decreasing relative content of co-surfactant transcutol (from 1:3 to 1:2) within the Smix. AVE6 was anionic (zeta potential = -22 mV) in nature due to the lack of stearylamine in the formulation and served as control group. Negative zeta potential is due to lipid (triglycerides of fatty acids). The study aimed to address the impact of charge on nanocarrier for permeation behaviour across nasal mucosa followed by blood brain barrier. Notably, all of the nanoemulsions showed %T (%transmittance) higher than 96% suggesting isotropic and transparent nature of cationic and anionic nanoemulsion.
Table 3.
Summary of selected cationic/anionic VA loaded nanoemulsions and their evaluated parameters.
Table 3.
Summary of selected cationic/anionic VA loaded nanoemulsions and their evaluated parameters.
| Code | SO (%) | Smix (%) | Water (%) | Smix ratio | ST (%) | Size (nm) | PDI | ZP (mV) | %T | Product strength (%w/w) |
|---|---|---|---|---|---|---|---|---|---|---|
| CVE1 | 16.46 | 30.21 | 48.59 | 1:2 | 0.04 | 162 | 0.27 | + 24.7 | 98.5 | 0.4 |
| CVE2 | 20.75 | 23.5 | 50.22 | 1:3 | 0.05 | 189 | 0.32 | + 26.8 | 96.8 | 0.5 |
| CVE3 | 14.72 | 21.67 | 57.12 | 1:2 | 0.06 | 185 | 0.31 | + 31.6 | 97.2 | 0.6 |
| CVE4 | 19.88 | 43.51 | 32.16 | 1:2 | 0.04 | 148 | 0.18 | + 23.9 | 96.9 | 0.4 |
| CVE5 | 9.8 | 21.84 | 63.04 | 2:1 | 0.05 | 79 | 0.11 | + 27.1 | 95.3 | 0.5 |
| CVE6 | 14.46 | 17.15 | 60.99 | 3:1 | 0.07 | 113 | 0.26 | + 34.7 | 97.8 | 0.7 |
| AVE6 | 14.46 | 17.15 | 60.92 | 3:1 | 0.0 | 126 | 0.29 | – 22.8 | 95.6 | 0.7 |
| Nanoemulsion gel (0.5%w/w) composition (VA strength) | Evaluated parameters | |||||||||
| 0.5% VE gel | NE (g) | Gel-blank (g) | Triethanolamine (g) | CVE6:gel ratio | Size (nm) | PDI | ZP (mV) | Viscosity (cP) | pH | |
| CVE6 gel (0.35%) | 1 | 0.95 | 0.05 g | 1:1 | 129 | 0.24 | + 21.9 | 1837.3 | 6.8 | |
| AVE6 gel (0.35%) | 1 | 0.95 | 0.05 g | 1:1 | 142 | 0.31 | – 26.5 | 1907.1 | 7.1 | |
Note: SO = Safflower, Smix = Tween 80-lecithin: transcutol, ST = stearylamine (cationic charge inducer), PDI = Polydispersity index; ZP = zeta potential, NE = Nanoemulsion , CVE = Cationic NE, ANE = Anionic NE
Freeze –thaw cycle and ultracentrifugation of nanoemulsions
The developed formualtions CVE1-CVE6, and AVE6 were subjected to ensure stability and capability to withstand thermal and physical stress during storage and transportation. Centrifugation step confirmed physical stability to face attrition and friction triggered phase separation usually observed during transportation [45]. On the other hand, extreme temperature (freeze and accelerated temperatures) assured stability against thermal mediated instability in nanoemulsion. The result is presented in table 4. All of the formulations (cationic and anionic) were physically and thermally stable at explored temperature kept for studied time period. A sequential series of thermal exposure from low to high through room temperature indicated that each product resumed original state of transparent isotropic nature of nanoemulsion with good flowability, consistency, and elegancy. There were no signs of any instability over explored period of time. It was imperative to corroborate thermal and physical stability so that the developed nanoemulsion can be stored and transported accordingly.
Table 4.
Freeze-thaw and centrifugation cycles and observation.
| Formulations | Freezing (- 21 °C) | Room temperature | Thaw (40 °C) | Centrifugation | Inference* |
|---|---|---|---|---|---|
| CVE1 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE2 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE3 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE4 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE5 | ✓ | ✓ | ✓ | ✓ | Stable |
| CVE6 | ✓ | ✓ | ✓ | ✓ | Stable |
| AVE6 | ✓ | ✓ | ✓ | ✓ | Stable |
Note: *Recovery of original form/state of nanoemulsion at room temperature after exposure to the extreme temperature was considered as stable in inference. Formulations exhibiting any signs of instability in term of drug precipitation, phase separation, colour development, and creaming was dropped out from further studies.
3.2.2. Evaluation of cationic and anionic nanoemulsions gels
CVE6 and AVE6 were used to incorporate in 1% carbopol gel (1:1 ratio) to gel respective nanoemulsion gel (0.5%) containing 0.35 % w/w of VA in the gels. Thus, the final product strength was 0.35%w/w in each gels. Both gels were evaluated for size, PDI, ZP, viscosity and final pH as shown in table 3. It is apparent that the pH (from 7.4 to 6.8) and zeta potential (from +34.7 to +21.9 mV) values of CVE6-gel were significantly reduced from respective CVE6 nanoemulsion. This is obvious due to acidic carbopol polymeric gel with free carboxylic acid in its structural backbone. However, globular size values were nearly similar to CVE6 nanoemulsion suggesting no globular aggregation in gel carrier. Viscosity values of CVE6 gel and AVE6 gel were obtained as 1837 and 1907 cP, respectively. These findings are in good agreement with the reported 0.5% carbopol 934 gel for topical application [46]. The viscosity indicates good consistency and shear thinning behaviour after topical application due to oil in water based system. In final selected formulations, fixed amount of SA was used CVE6 and CVE6-gel to achieve concerted positivity on globular surface of nanoemulsion which may facilitate mucoadhesive property (as result of electrostatic interaction) after nasal administration to improve residence time and absorption [20]. Final pH of CVE6, AVE6, CVE6-gel, and AVE-6 gel products was found to be in the range of 6.8 – 7.4 which provided agreeable consistency and compatibility with nasal mucosa.
Morphological evaluation of the optimized cationic nanoemulsion and respective gel
CVE6, AVE6, CVE6-gel, and AVE6-gel were considered as the most stable and optimized nanoemulsion and respective gels. Generally, size, shape, and size distribution are expected to be changed after incorporation of nanoemulsion into a hydrogel carrier. Therefore, it was requisite to visualize CVE6, AVE6, CVE6-gel, and AVE6-gel. Thus, morphology of nanoemulsions was compared after incorporation into gel. The result is portrayed in Figure 6 wherein shape, size, and globular size distribution. The shape of globular particle is approximately similar in nanoemulsion and respective gel. However, cationic nanoemulsion is found to be well dispersed in CVE6 as compared AVE6 which may be due to imposed positive charge. AVE6 is slightly dispersed without forming any aggregation. Similar observation was obtained in respective gel. Thus, hydrogel could not change the shape, size and globular distribution of nanoemulsion. Moreover, there was no observed drug precipitation even after amalgamation of CVE6 or AVE6 into carbopol hydrogel matrix. This suggested substantially firm layer of Smix coated around oil globules containing solubilized VA. It is noteworthy that the size obtained from DLS always differ from the size estimated using TEM. This happens due to instrumental error and difference in the working principle. Both techniques are quite different and followed different sample processing during analysis. Therefore, this error is defined as “Fold error” and estimated using the following formula:
Fold error (FE) = 1/n [logsize of DLS/size of TEM]
In general, the error is considered acceptable when it comes below 2 (≤ 2) [47]. The values of FE for CVE6, AVE6, CVE6-gel, and AVE6 gel were found to be as 1.4, 1.7, 1.3, and 1.9, respectively. For gel, sample was first diluted in water to gel consistency similar to respective nanoemulsion before analysis using DLS technique. The same sample was scanned under TEM. In DLS analysis, and TEM based scanning, temperature was kept constant to avoid any further error in result.
Drug content estimation
The percent drug content of CVE1-CVE6, AVE6, AVE6-gel, and CVE6 gel were estimated using HPLC method. The sample was dissolved in acetonitrile-methanol mixture (30:70) to extract the drug. The sample was filtered and analysed. The percent drug content in each formulation was not less than 99.3%. There was slight loss of drug content during preparation and handling process. The percent strength of each nanoemulsion and gel is presented in table 3.
3.2.3. In vitro drug release profile
The model drug is acidic in nature (pKa = 5.2) and poor soluble in water (1.3 mg/ml). The drug is reported to be soluble in alkaline medium such as sodium hydroxide and alcohol. The optimized nanoemulsions (CVE6 and AVE6) and their respective gels showed different release behaviour at pH 6.8 and 7.4 (phosphate buffer solution). The result is presented in Figure 6A-B. The nasal fluid and mucosa pH is about 6.8 and systemic delivery across blood brain barrier is exposed to pH 7.4. Therefore, it was mandatory to investigate the impact of mucosal pH and blood pH when formulations expected to be transported across mucosal and BBB for brain delivery. The result showed two important findings. These were (a) the impact of gel and (b) the impact of release medium pH. It is quite clear that the drug was rapidly released from cationic and anionic nanoemulsion through dialysis membrane as compared to respective gel. This can be correlated to viscous nature of gel and two drug release limiting factors. These drug release rate limiting factors are gel matrix and dialysis membrane slowing down nanoglobules diffusion from matrix to the medium. In case of nanoemulsion, there is only dialysis membrane as drug release rate limiting factor. The low viscosity further facilitated drug diffusion from nanoemulsion to the release medium [22]. The release medium chamber maintained at temperature of 32±1°C throughout the study. The drug suspension (7 mg/ml) was rapidly released (˃ 90%) within 30 min due to its salt solubility (1.3 mg/mL) at pH 6.8 (Figure 6A). Similar pattern was observed at pH 7.4 (˃ 78.4% within 30).
3.2.4. Ex vivo drug permeation and drug deposition using goat nasal mucosal tissue
Various reports have been published for drug delivery to brain using nasal route. Nasal mucosa composition, viscosity of nasal formulation, and mucoadhesiveness, residence time, and nasal pH are major critical factors responsible to control drug release and permeation across nasal epithelium [24,25]. The study was conducted up to 6 h to avoid any loss of natural anatomical structural integrity of mucosal tissue and tissue viability [27]. The study was conducted using simulated nasal fluid with pH 6.8 (to mimic nasal pH) to avoid nasal irritation and discomfort after application [26]. Gel products are relatively viscous and more mucoadhesive as compared to CVE6 and AVE6. Cumulative amount of the drug permeation has been revealed in Figure 7C and the drug deposition into the nasal mucosal tissue is presented in Figure 7D. The values of permeation flux for CVE6, AVE6, CVE6-gel, and AVE6-gel were estimated as 67.64, 48. 01, 57.18, 31.74, and 3.15 µg/cm2/h, respectively across nasal mucosa of goat. The steady state permeation flux values of cationic nanoemulsion and its gel exhibited 21.47- and 18.15 folds higher flux rate as compared to control suspension which may be correlated to cationic and mucoadhesive gel carrier providing electrostatic interaction with negatively charged mucosal surface, extended residence time, respectively, and linoleic acid was reported to facilitate drug permeation across blood brain barrier [48]. Moreover, gel is mucoadhesive, biocompatible, slightly acidic comparable to nasal fluid pH and drug pKa value (5.2 – 5.6). The flux value of CVE6 is very comparable to published report of flux (~ 73 µg/cm2/h) for VA loaded niosomal in situ gel across goat mucosal membrane [50]. Slightly high flux value may be attributed to niosomal loading efficiency greater than nanoemulsion. Fortunately, these parameters are suitable for maximized nasal permeation of the drug in the explored carrier for brain delivery. The drug supposed to remain unionized at nasal pH due to comparable pKa value for enhanced permeation and drug deposition. In addition, considering poorly-vascularized (anterior third of each nasal cavity) and highly vascularized anatomical area (respiratory epithelium and two third posterior portion of cavity) of nose, inhaled particles or nanoglobules were thought to be lodged by three prime mechanisms such as (a) gravitational sedimentation, (b) inertial impaction, and (c) Brownian diffusion (if spayed) playing together [50]. To understand the mechanistic perspective of the drug delivery from nose to the brain, it is imperative to consider the interplay of various critical factors such as formulation characteristics, device, and patient related conditions. These factors are directly involved in the drug laden-nanodroplets for maximized permeation and drug deposition within the nasal cavities and subsequently the drug access to brain. Notably, the exact localization of the drug for deposition recognised as one of the key to success or failure of the nasal product [51]. The sites of the drug localization within nose dictate the purpose local, systemic and brain drug delivery. For drug delivery to the brain, the nasal cavities (innervated with olfactory and trigeminal nerves) are the most ideal site for the drug localization and a potential target for nose to brain delivery using cationic nanoemulsion and gel formulation. Moreover, these cavities rapidly absorb the lodged drug through thin membrane to achieve faster onset of action at low dose, high patient compliance, reduced dose and metabolite (4-eve-VPA) based side effects (hepatic toxicity due to reticuloendothelial system), without hepatic metabolism, and maximized drug access to the brain [52,53,54]. Greater uptake by RES indicates greater drug metabolism and incidence of side effects. Considering formulation related factors such as globular size, shape, zeta potential, viscosity, mucoadhesiveness, the drug solubility, polarity, hydrophilicity, and composition (surfactant and oil) are complementary factors. Linoleic acid rich oils are getting popular in pharmaceutical and cosmeceutical industries due to possessing various skin benefits such as (a) anti-inflammatory, (b) acne reductive, (c) skin softening, (d) moisture retentive, (e) facilitating drug diffusion across blood brain barrier (50-70% linoleic acid), and (f) biocompatibility [10,44]. Tween 80 possessed high hydrophilicity due to high HLB value (14.5) and it is anticipated for maximized emulsification in hydrophilic mucosal layer to keep nanoglobules in emulsified form within mucosal matrix for prolonged systemic circulation time (probably due to long fatty acid chain in lipid such as linoleic acid) in brain or reducing RES uptake. The surfactant is reported to have several benefits for nasal nanoemulsion for VA delivery to brain. These are a) protection of the drug from enzymatic degradation, b) improved brain bioavailability, and c) prolonged circulation time in brain due to long fatty acid and polyunsaturated fatty acid (PUFA) nature of the present oil [10].
The result of the drug deposition has been presented in Figure 7D wherein CVE6, AVE6, CVE6-gel, and AVE6-gel, and suspension showed percent drug deposition as 67.64, 48.0, 57.18, 31.74, and 3.15%, respectively. It is quite clear that greater drug deposition means greater permeation flux as observed in CVE-6 as compared to respective gel and other nanoemulsion. Gel matrix slightly delayed permeation and drug deposition which is good for prolonged drug release and extended effect to control epileptic fits and seizure. However, considering types of patient and working or traveling schedule, both formulations are important. For immediate relief, it is better to spray cationic nanoemulsion being aqueous and free flowing due to low viscosity. In case of planned traveling schedule, gel product is good and suitable as prophylactic dose for prolonged relief from seizure attack. Globular size, surface charge, and pH are another factors controlling the drug deposition and subsequently drug flux. Nanoemulsion size depends upon the oil content (oil content inversely proportional to globular size of nanoemulsion), and content and types of surfactant. Tan et al. revealed reduced globular size of nanoemulsion from 142 nm to 80 nm due to reduced content of oil from 6 % to 1.5%, respectively [10]. In literature, it was reported that VA transport and nanoemulsion permeation across blood brain barrier are mediated via the organic anion transporter and the LDL-mediated endocytosis due to the presence of tween 80, respectively [56,57]. This may explain the significant difference in permeation profiles between the drug suspension and formulations.
3.2.6. Confocal laser scanning microscopy (CLSM)
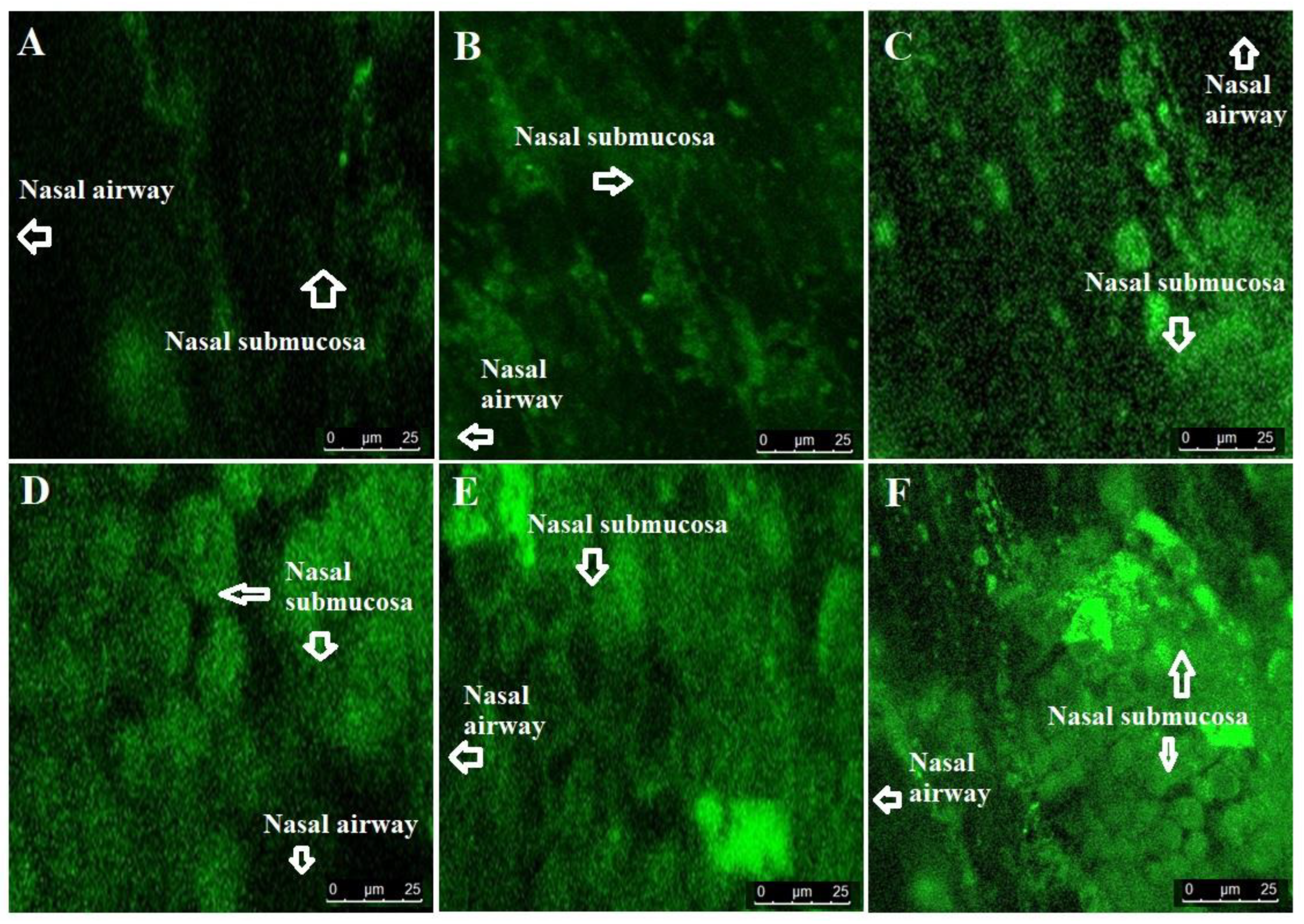
To evaluate the degree of penetration and permeation across the superior nasal concha (nasal membrane), we scanned nasal mucosa membrane treated with formulations under CLSM. For comparison, R123 solution was used as control. The result is provided in Figure 7A-F. It is obvious from the result that the dye solution and suspension were not penetrable across hydrophilic (about 90 – 95% water and glycoprotein giving gel like structure) nasal mucosal membrane as evidenced with poor fluorescence intensity [58]. Drug suspension containing the dye showed approximately similar result due to the drug insolubility and poor permeation behaviour. The fluorescence intensity values of dye solution, suspension, AVE6-R, AVE6-R-gel, CVE6-R, and CVE6-R-gel were obtained as 11.6, 17.3, 65.6, 75.62, 84.7, and 96.11 %, respectively. The lowest fluorescence intensity associated with dye solution and suspension could be attributed to poor dye and drug permeation across hydrophilic nasal mucosa as result of poor drug solubility. However, intense intensity values were observed for both nanoemulsions (AVE6-R and CVE6-R) and gels (AVE6-R-gel and CVE6-R-gel) as shown in Figure 7. High degree of intense fluorescence by the gel and cationic nanoemulsion can be correlated to mucoadhesiveness and prolonged residence time on the nasal mucosa of goat. Carbopol gel is known for good mucoadhesive nature and compatible pH for nasal pH (4.5 – 6.8) without producing any nasal irritation [59]. Nasal pH (4.5 – 6.8) is very suitable for gel consistency maintained after nasal application. Moreover, the drug is slightly acidic to be in stable and non-ionized form if it comes in contact with nasal fluid and mucosal membrane. All these drug and formulation related properties provides suitability for the drug permeation, penetration and compatibility for intransal delivery of the drug to control convulsion in patients. Moreover, imposed positive charge on cationic nanoemulsion facilitated nanoemulsion penetration as compared anionic counterpart as evidenced with remarkably high fluorescence intensity. This can be correlated to electrostatic interaction mediated improved permeation and subsequently drug deposition within the submucosal region of nasal tissues. In addition, intransal delivery of nanocarrier based drug offers several advantages over oral administration of the drug.
Conclusively, the dye solution and the drug suspension itself are not capable to be penetrated. both nanoemulsions were relatively less viscous as compared to gel formulation. This caused slightly lower residence time in mucosal region. Gel carrier provided hydration and high residence time for nanoemulsion penetration. Finally, cationic globular electrostatic interaction with negatively charged nasal membrane rendered the investigated nanoemulsion suitable for maximized permeation and penetration [60]. Thus, it was hypothesized that optimized viscosity, imposed cationic charge, reduced globular size, and mucoadhesive gel could be working in tandem for the drug delivery to the brain through nasal administration
4. Conclusion
Conventional dosage form of VA is associated with multiple challenges. These challenges are related to the physicochemical properties, pharmacokinetic behavior, and pharmacodynamics properties of the drug. Low bioavailability to brain, high hepatic metabolism, and severe side effects on oral and parenteral delivery gained wide attention to formulation scientists for alternative and high therapeutic benefits. GastroPlus program assisted us to understand the in vivo behavior of the drug in human body at explored dose, dosing frequency, and dosage form. Moreover, the program predicted various factors responsible to affect in vivo pharmacokinetics and drug dissolution. HSPiP software predicted various excipients based on HSP parameters to reduce experimental screening burden. Catonic nanoemulsion could be promising option for maximized drug access to the nasal cavity due to small size (113 nm), high mucoadhesiveness (high positive zeta potential and mucoadhesive carbopol gel), and linoleic acid (as high content in the oil) mediated drug permeation across blood brain barrier. Ex vivo permeation flux, enhancement ratio, drug deposition, and penetration property of CVE6 and CVE6 gel confirmed electrostatic and mucoadhesiveness worked in tandem for extended residence time in nasal mucosa and subsequently augmented drug access to the brain. Conclusively, the strategy is promising and suitable alternative to conventional cream or oral tablet with high therapeutic effectiveness and patient compliance to control seizures.
Author Contributions
Afzal Hussain: Conceptualization, methodology, drafting, and writing, Mohammad A. Altamimi: drafting, review, and analysis, Mohd Aamir Mirza: Review and validation, Mohhammad Ramzan: Analysis, data curation and resources, Tahir Khuroo: Editing and visualization. All authors have read and agreed to the published version of the manuscript.
Ethical statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research at King Saud University for this work.
Conflicts of Interest
Authors report no conflict of interest.
References
- World Health Organization Epilepsy. Available online: https://www.who.int/news-room/fact-sheets/detail/epilepsy (accessed on 20 December 2021).
- Available online: https://www.ninds.nih.gov/current-research/focus-disorders/focus-epilepsy-research/curing-epilepsies-promise-research (accessed on 20 December 2021).
- Al Rajeh S, Awada A, Bademosi O, Gunniyi A. The prevalence of epilepsy and other seizure disorders in an Arab population: a community-based study. Seizure. 2001, 10, 410–414. [CrossRef]
- N. Ishizue, S. Niwano, M. Saito, H. Fukaya, H. Nakamura, T. Igarashi, Fujiishi T., Yoshizawa, T., Oikawa, J., Satoh, A., Kishihara, J., Murakami, M., Niwano, H., Miyaoka, H., Ako, J. Polytherapy with Sodium Channel-Blocking Antiepileptic Drugs Is Associated with Arrhythmogenic ST-T Abnormality in Patients with Epilepsy. Seizure, 2016, 40, 81–87.
- Hammond, E.J., Perchalski, R.J., Villarreal, H.J., Wilder, B. J. (1982). In vivo uptake of valproic acid into brain. Brain Research, 240, 195–198.
- US FDA (US Food and Drug Administration). chrome-extension. Available online: https://efaidnbmnnnibpcajpcglclefindmkaj/https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/018081s065_018082s048lbl.pdf.
- S. Eskandari, J. Varshosaz, M. Minaiyan, M. Tabbakhian, Brain delivery of valproic acid via intranasal administration of nanostructured lipid carriers: in vivo pharmacodynamic studies using rat electroshock model, Int. J. Nanomed. 2011, 6, 363–371.
- H. Wang, Q. Huang, H. Chang, J. Xiao, Y. Cheng, Stimuli-responsive dendrimers in drug delivery, Biomater. Sci. 4 (3) (2016) 375–390.
- Lopez, T. Biocompatible Titania Microtubes Formed by Nanoparticles and its Application in the Drug Delivery of Valproic Acid. Optical Materials, 2006, 29, 70–74. [Google Scholar] [CrossRef]
- an, B.P. Kirby, J. Stanslas, H Bin Basri, Characterization, in-vitro and in-vivo evaluation of valproic acid-loaded nanoemulsion for improved brain bioavailability, J. Pharm. Pharmacol. 2017, 69, 1447–1457.
- Mori, N., Ohta, S. Comparison of anticonvulsant effects of valproic acid entrapped in positively and negatively charged liposomes in amygdaloid-kindled rats, Brain Res. 593 (2), 1992, 329–331.
- Tan, S. F., Masoumi, H. R. F., Karjiban, R. A., Stanslas, J., Kirby, B. P., Basri, M., & Basri, H. B. Ultrasonic emulsification of parenteral valproic acid-loaded nanoemulsion with response surface methodology and evaluation of its stability. Ultrasonics Sonochemistry, 2016, 29, 299–308.
- Yadav, S. , Gandham, S. K., Panicucci, R., Amiji, M. M. Intranasal brain delivery of cationic nanoemulsion-encapsulated TNFα siRNA in prevention of experimental neuroinflammation. Nanomedicine: Nanotechnology, Biology and Medicine, 2016, 12, 987–1002. [Google Scholar]
- Azambuja, J. H. , Schuh, R. S., Michels, L. R., Gelsleichter, N. E., Beckenkamp, L. R., Iser, I. C., Lenz, G.S., de Oliveira, F.H., Venturin, G., Greggio, S., daCosta, J.C., Wink, M.R., Sevigny, J., Stefani, M.A., Battastini, A.M.O., Teixieria H.F., Braganhol, E. Nasal Administration of Cationic Nanoemulsions as CD73-siRNA Delivery System for Glioblastoma Treatment: a New Therapeutical Approach. Molecular Neurobiology. 2020, 57, 635–649. [Google Scholar] [PubMed]
- Hussain, A.; Alshehri, S.; Ramzan, M.; Afzal, O.; Altamimi, A.S.A.; Alossaimi, M.A. Biocompatible solvent selection based on thermodynamic and computational solubility models, in-silico GastroPlus prediction, and cellular studies of ketoconazole for subcutaneous delivery. Journal of Drug Delivery Science and Technology 2021, 65, 102699. [Google Scholar] [CrossRef]
- Alsarra, I. A. , Al-Omar, M., Belal, F. Valproic Acid and Sodium Valproate: Comprehensive Profile. Profiles of Drug Substances, Excipients and Related Methodology, 2005, 32, 209–240. [Google Scholar] [CrossRef]
- Ezati, N.; Roberts, M.S.; Zhang, Q.; Moghimi, H.R. Measurement of Hansen Solubility Parameters of Human Stratum Corneum. Iran J. Pharm. Res. 2020, 19, 572–578. [Google Scholar]
- Hansen, C.M.; Andersen, B. The affinities of organic solvents in biological systems. Am. Ind. Hyg. Assoc. 1988, 49, 301–308. [Google Scholar] [CrossRef]
- De La Peña-Gil, A. , Toro-Vazquez, J. F., Rogers, M.A. Simplifying Hansen Solubility Parameters for Complex Edible Fats and Oils. Food Biophysics, 2016, 11, 283–291. [Google Scholar] [CrossRef]
- Jones, M. N. , Song, Y. -H., Kaszuba, M., Reboiras, M.D. The Interaction of Phospholipid Liposomes with Bacteria and Their Use in the Delivery of Bactericides. Journal of Drug Targeting, 1997, 5, 25–34. [Google Scholar] [CrossRef]
- Hussain, A. , Singh, S.K. Evidences for anti-mycobacterium activities of lipids and surfactants. World Journal of Microbiology and Biotechnology, 2015, 32. [Google Scholar] [CrossRef]
- Christensen, J. M.; Chuong, M. C.; Le, H.; Pham, L.; Bendas, E. Hydrocortisone diffusion through synthetic membrane, mouse skin, and Epiderm™ cultured skin. Archives of Drug Information, 2011, 4, 10–21. [Google Scholar] [CrossRef]
- Hansen, C. M. The significance of the surface condition in solutions to the diffusion equation: explaining “anomalous” sigmoidal, Case II, and Super Case II absorption behavior. European Polymer Journal, 2010, 46, 651–662. [Google Scholar] [CrossRef]
- Yuwanda, A. , Surini, S.; Harahap, Y.; Jufri. M. Study of valproic acid liposomes for delivery into the brain through an intranasal route. Heliyon, 2022, 8, e09030). [Google Scholar] [CrossRef]
- Basu, S. , Maity, S.Preparation and Characterisation of Mucoadhesive Nasal Gel of Venlafaxine Hydrochloride for Treatment of Anxiety Disorders. Indian J. Pharm. Sci., 2012, 74, 428–433. [Google Scholar] [CrossRef]
- Trenkel, M.; Scherlie, R. Nasal Powder Formulations: In-Vitro Characterisation of the Impact of Powders on Nasal Residence Time and Sensory Effects. Pharmaceutics, 2021, 13, 385. [Google Scholar] [CrossRef]
- Khuroo, T. , Khuroo, A., Hussain, A., Mirza, M.A., Panda, A.K., Iqbal, Z. Qbd based and Box-Behnken design assisted oral delivery of stable lactone (active) form of topotecan as polymeric nanoformulation: Cytotoxicity, pharmacokinetic, in vitro, and ex vivo gut permeation studies. Journal of Drug Delivery Science and Technology Available online 6 October 2022, 103850, In press. [CrossRef]
- G。 Nava, E. Pin˜on, L. Mendoza, N. Mendoza, D. Quintanar, A. Ganem, Formulation and in vitro, ex vivo and in vivo evaluation of elastic liposomes for transdermal delivery of ketorolac tromethamine, Pharm. Times 2011, 3, 954–970.
- Hussain, A. , Samad, A.; Singh, S.K.; Ahsan, M.N.; Haque, M.W.; Faruk, A.; Ahmed, F.J. Drug Deliv. 2016, 23, 652–667; [Google Scholar]
- Chen, H.; Chang, X.; Du, D.; Liu, W.; Liu, J.; Weng, T.; Yang, Y.; Xu, H.; Yang, X. Podophyllotoxin-loaded solid lipid nanoparticles for epidermal targeting. Journal of Controlled Release, 2006, 110, 296–306. [Google Scholar] [CrossRef]
- Teixeira-da-Silva, P.; Pérez-Blanco, J.S.; Santos-Buelga, D.; Otero, M.J.; García, M.J. Population pharmacokinetics of valproic acid in pediatric and adult caucasian patients. Pharmaceutics 2022, 14, 811. [Google Scholar] [CrossRef]
- Zaccara, G.; Messori, A.; Moroni, F. Clinical Pharmacokinetics of Valproic Acid - 1988. Clinical Pharmacokinetics, 1988, 15, 367–389. [Google Scholar] [CrossRef]
- Levy, R.H. CSF and plasma pharmacokinetics: relationship to mechanisms of action as exemplified by valproic acid in monkey. In J. S. Lockard and A. A. Ward (Eds.), Epilepsy: A Window to Brain Mechanisms, Raven Press, New York, 1980, 191-200.
- Johannessen, C.U.; Johannessen, S.I. Valproate: past, present, and future. CNS Drug Rev. 2003, 9, 199–216. [Google Scholar] [CrossRef]
- Winter, M.E. Basic Clinical Pharmacokinetics, 5th edn. Philadelphia, PA: Lippincott Williams & Wilkins Health, 2010.
- Loscher, W. Serum protein binding and pharmacokinetics of valproate in man, dog, rat and mouse. J. Pharmacol. Exp. Ther. 1978, 204, 255–261. [Google Scholar] [PubMed]
- Dickinson, R.G.; Harland, R.C.; Ilias, A.M.; Rodgers, R.M.; Kaufman, S.N.; Lynn, R.K.; Gerber, N. Disposition of valproic acid in the rat: dose dependent metabolism, distribution, enterohepatic recirculation and choleretic effect. J. Pharmacol. Exp. Ther. 1979, 211, 583–595. [Google Scholar]
- Hansen, S.; Lehr, C.-M.; Schaefer, U.F. Improved input parameters for diffusion models of skin absorption. Advanced Drug Delivery Reviews, 2013, 65, 251–264. [Google Scholar] [CrossRef]
- Methaneethorn, J. A systematic review of population pharmacokinetics of valproic acid. Br. J. Clin. Pharmacol. 2018, 84, 816–834. [Google Scholar] [CrossRef] [PubMed]
- Gugler, R.; von Unruh, G.E. Clinical Pharmacokinetics of Valproic Acid1. Clinical Pharmacokinetics, 1980, 5, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Vay, K.; Scheler, S.; Frie, W. Application of Hansen solubility parameters for understanding and prediction of drug distribution in microspheres. International Journal of Pharmaceutics, 2011, 416, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A.; Brunaldi, K.A. model for fatty acid transport into the brain. J. Mol. Neurosci. 2007, 33, 12–17. [Google Scholar] [CrossRef]
- Hussain, A.; Altamimi, M.A.; Afzal, O.; Altamimi, A.S.A.; Ali, A.; Ali, A.; Martinez, F.; Siddique, M.U.M.; Acree Jr., W. E.; Jouyban, A. Preferential solvation study of the synthesized aldose reductase inhibitor (SE415) in the {PEG400 (1) + Water (2)} cosolvent mixture and GastroPlus-based prediction. ACS Omega, 2022, 7, 1197–1210. [Google Scholar] [CrossRef]
- Han, X.; Cheng, L.; Zhang, R.; Bi, J. Extraction of safflower seed oil by supercritical CO2. Journal of Food Engineering, 2009, 92, 370–376. [Google Scholar] [CrossRef]
- Afzal, O.; Alshammari, H.A.; Altamimi, M.A.; Hussain, A.; Almohaywi, B. ; Altamimia, A.S. A. Hansen solubility parameters and green nanocarrier based removal of trimethoprim from contaminated aqueous solution. Journal of Molecular Liquids, 2022, 361, 119657 doiorg/101016/jmolliq2022119657. [Google Scholar] [CrossRef]
- Abdullah, G.Z.; Abdulkarim, M.F.; Mallikarjun, C.; Mahdi, E.S.; Basri, M.; Sattar, M.A.; Noor, A.M. Carbopol 934, 940 and Ultrez 10 as viscosity modifiers of palm olein esters based nano-scaled emulsion containing ibuprofen. Pak. J. Pharm. Sci., 2013, 26, 75–83. [Google Scholar] [PubMed]
- Altamimi, M.A.; Hussain, A.; Alshehri, S.; Imam, S.S.; Alnemer, U.A. Development and evaluations of transdermally delivered luteolin loaded cationic nanoemulsion: In vitro and ex vivo evaluations. Pharmaceutics, 2021, 13, 1218. [Google Scholar] [CrossRef]
- Edmond, J. Essential Polyunsaturated Fatty Acids and the Barrier to the Brain: The Components of a Model for Transport. J. Mol. Neurosci. 2001, 16, 181–194. [Google Scholar] [CrossRef]
- Trenkel, M.; Scherlie, R. Nasal Powder Formulations: In-Vitro Characterisation of the Impact of Powders on Nasal Residence Time and Sensory Effects. Pharmaceutics 2021, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- Shilpa, P.; Vibhavari, C.; Chatur, M. Development of Valproic Acid Niosomal in situ Nasal Gel Formulation for Epilepsy. Indian Journal of Pharmaceutical Education and Research, 2013, 47, 31–41. [Google Scholar]
- Ehrick, J.D. , Shah, S.A., Shaw, C., Kulkarni, V.S., Coowanitwong, I., De, S., Suman, J.D. Considerations for the Development of Nasal Dosage Forms. AAPS Advances in the Pharmaceutical Sciences Series, 2013, 6, 99–144. [Google Scholar]
- Vidgren, M.T.; Kublik, H. Nasal delivery systems and their effect on deposition and absorption. Adv. Drug Deliv. Rev. 1998, 29, 157–177. [Google Scholar] [PubMed]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Kumar, S.; Wong, H.; Yeung, S.A.; Riggs, K.W.; Abbott, F.S.; Rurak, D.W. Disposition of valproic acid in maternal, fetal, and newborn sheep II: metabolism and renal elimination. Drug Metab. Dispos. 2000, 28, 845–856. [Google Scholar]
- Tang, W.; Borel, A.G.; Fujimia, T.; Abbott, F.S. Fluorinated analogs as mechanistic probes in valproic acid hepatotoxicity: hepatic microvesicular steatosis and glutathione status. Chem. Res. Toxicol. 1995, 8, 671–682. [Google Scholar] [CrossRef]
- Rossi, MA. Targeting anti-epileptic drug therapy without collateral damage: nanocarrier-based drug delivery. Epilepsy Curr. 2012, 12, 199–200. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E. ; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Andersen I, Proctor DF. Measurement of nasal mucociliary clearance. Eur. J. Respir. Dis. Suppl. 1983, 127, 37–40.
- Patel, R. B. , Patel, M.R., Bhatt, K. K., Patel, B. G. Formulation consideration and characterization of microemulsion drug delivery system for transnasal administration of carbamazepine. Bulletin of Faculty of Pharmacy, Cairo University, 2013, 51, 243–253. [Google Scholar] [CrossRef]
- Yang, Y. , Jing, Y., Yang, J., Yang, Q. Effects of intranasal administration with Bacillus subtilis on immune cells in the nasal mucosa and tonsils of piglets. Experimental and Therapeutic Medicine. 2018, 15, 5189–5198. [Google Scholar]
Figure 1.
Simulation and prediction software (GastroPlus) based analysis of IR tablet and SR tablet for oral administration. (A) Prediction of in vivo dissolution and in vivo absorption of VA IR Tablet (200 mg) for oral delivery (once a day in fast state condition) and (B) Prediction of in vivo dissolution and in vivo absorption of VA SR tablet (200 mg) for oral delivery (once a day in fast state condition).
Figure 1.
Simulation and prediction software (GastroPlus) based analysis of IR tablet and SR tablet for oral administration. (A) Prediction of in vivo dissolution and in vivo absorption of VA IR Tablet (200 mg) for oral delivery (once a day in fast state condition) and (B) Prediction of in vivo dissolution and in vivo absorption of VA SR tablet (200 mg) for oral delivery (once a day in fast state condition).
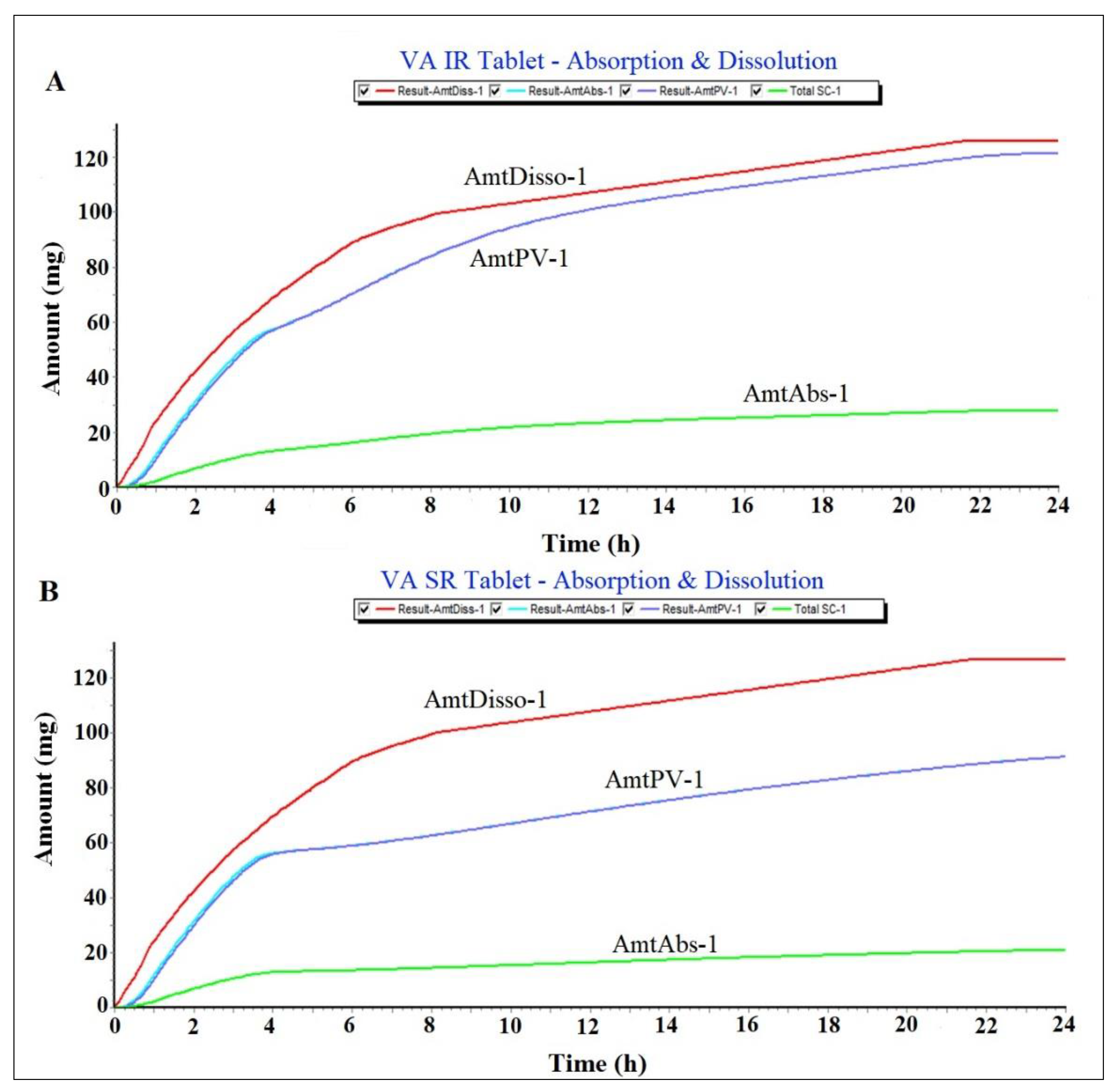
Figure 2.
Plasma drug concentration – time profile predicted for VA IR tablet and VA SR tablet (200 mg). This was predicted profile in human at fast state condition using GastroPlus.
Figure 2.
Plasma drug concentration – time profile predicted for VA IR tablet and VA SR tablet (200 mg). This was predicted profile in human at fast state condition using GastroPlus.
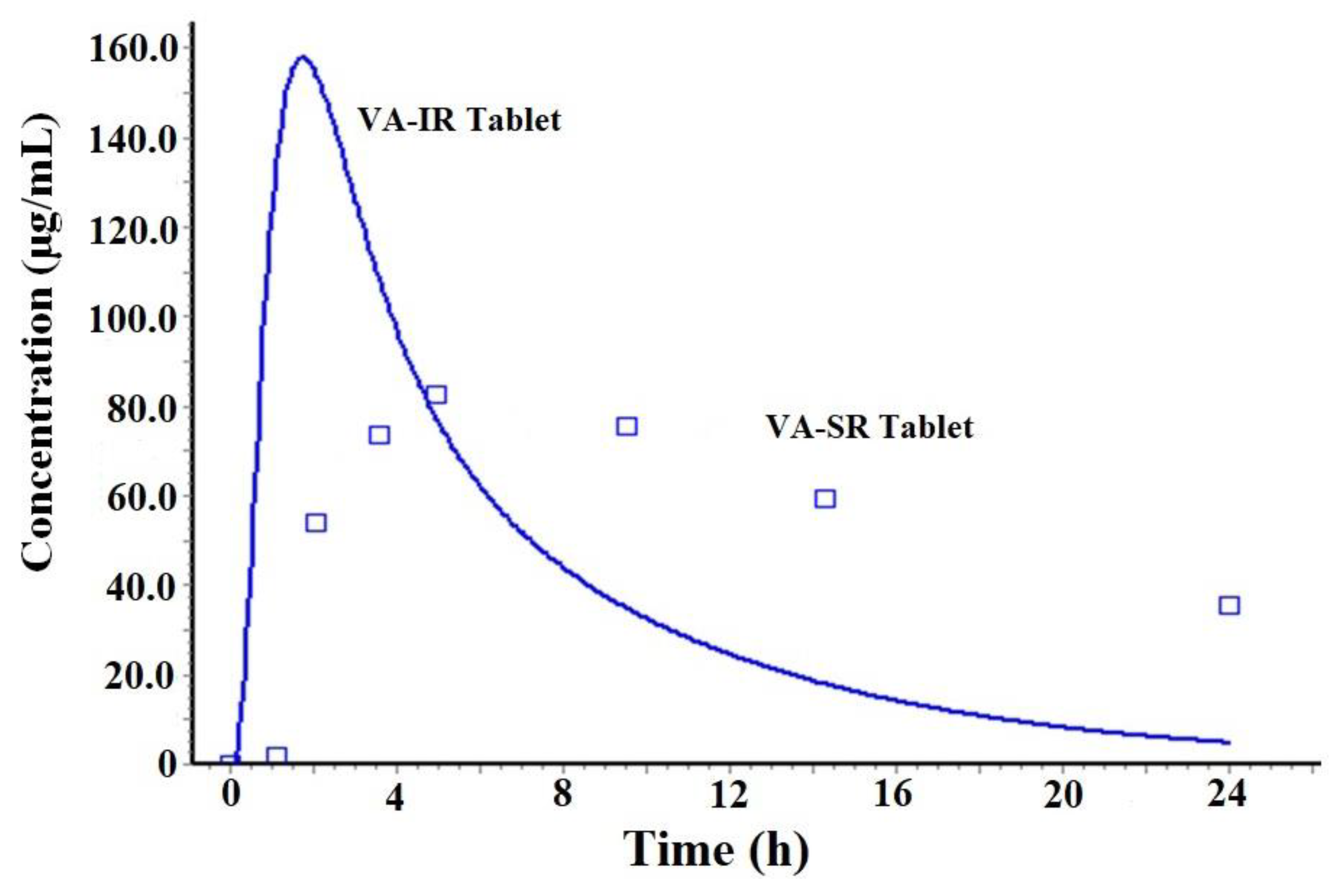
Figure 5.
Experimental solubility of VA in various excipients at 40 °C. Data were expressed as mean ± standard deviation (n = 3).
Figure 5.
Experimental solubility of VA in various excipients at 40 °C. Data were expressed as mean ± standard deviation (n = 3).
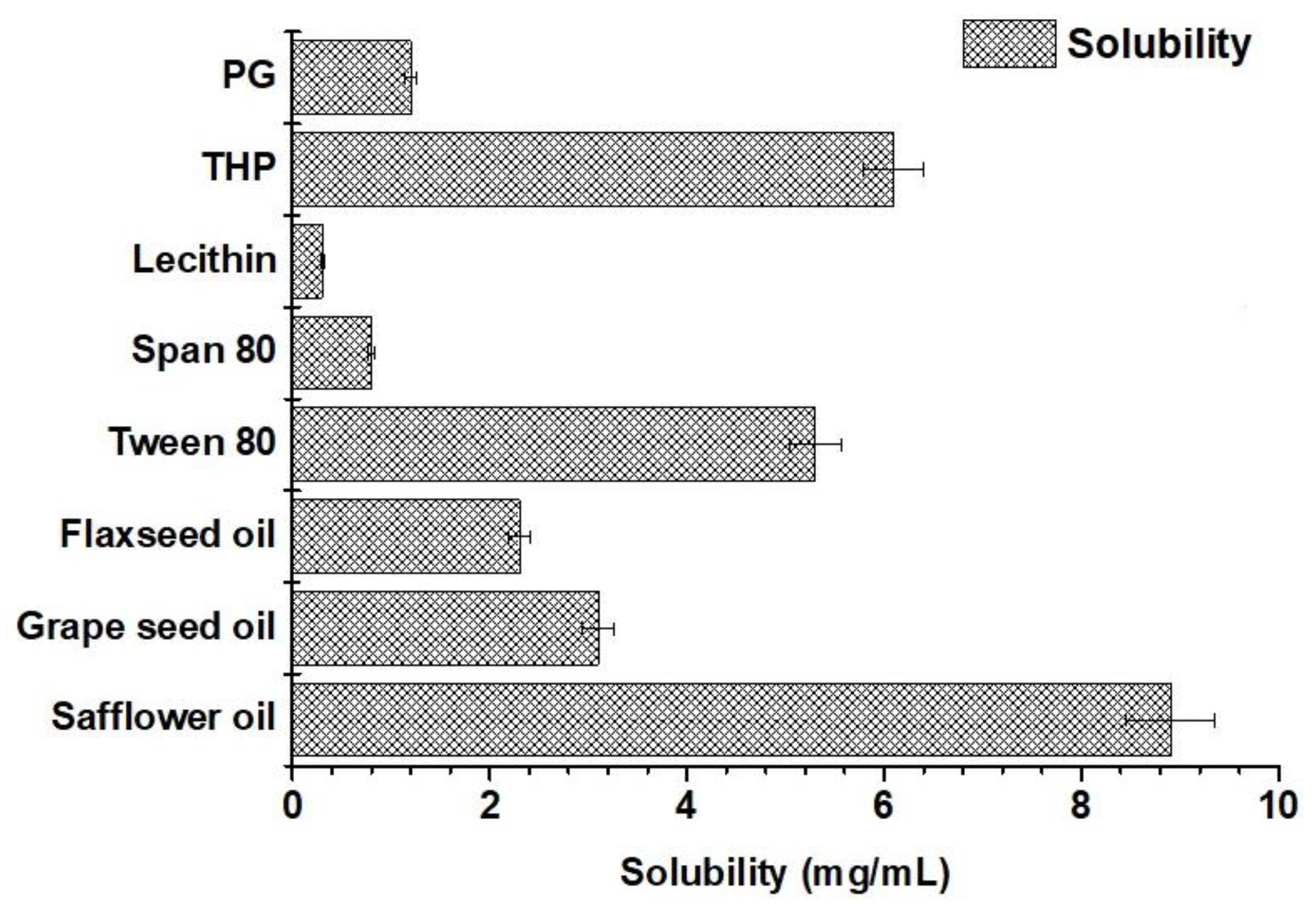
Figure 6.
Representative images of TEM micrographs: (A) CVE6 nanoemulsion, (B) AVE6 nanoemulsion, (C) CVE6 gel, and (D) AVE6 gel. Magnification 49000X.
Figure 6.
Representative images of TEM micrographs: (A) CVE6 nanoemulsion, (B) AVE6 nanoemulsion, (C) CVE6 gel, and (D) AVE6 gel. Magnification 49000X.
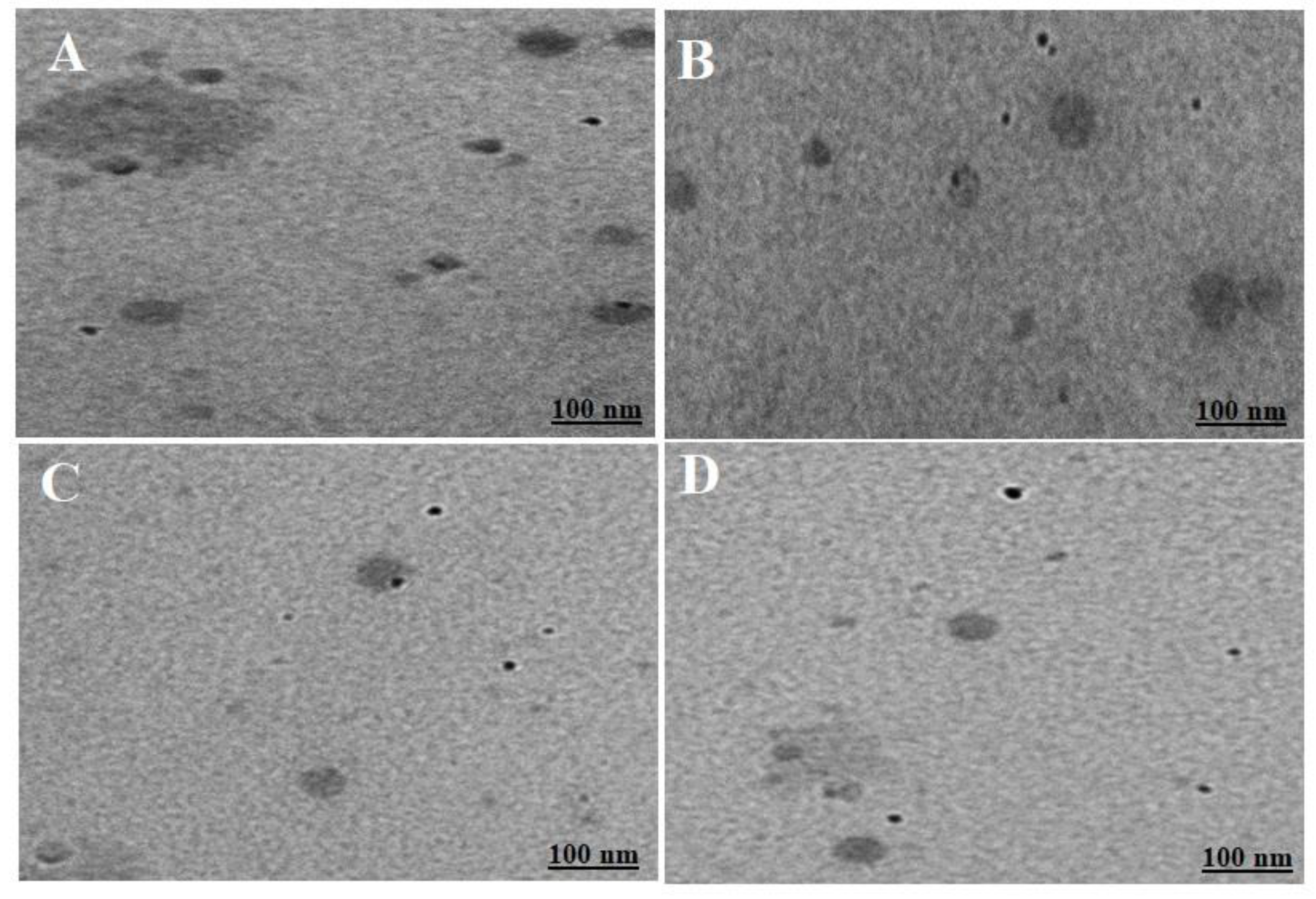
Figure 6.
In vitro drug release using a dialysis membrane: (A) release profile at pH 6.8, (B) release profile at pH 7.4, (C) Ex vivo cumulative drug permeation across nasal mucosa of goat over period of 6 h in simulated nasal fluid, and (D) drug deposition of the drug in nasal mucosa after 6 h of ex vivo permeation at 37±1 °C (data are expressed as mean ± standard deviation, n =3).
Figure 6.
In vitro drug release using a dialysis membrane: (A) release profile at pH 6.8, (B) release profile at pH 7.4, (C) Ex vivo cumulative drug permeation across nasal mucosa of goat over period of 6 h in simulated nasal fluid, and (D) drug deposition of the drug in nasal mucosa after 6 h of ex vivo permeation at 37±1 °C (data are expressed as mean ± standard deviation, n =3).
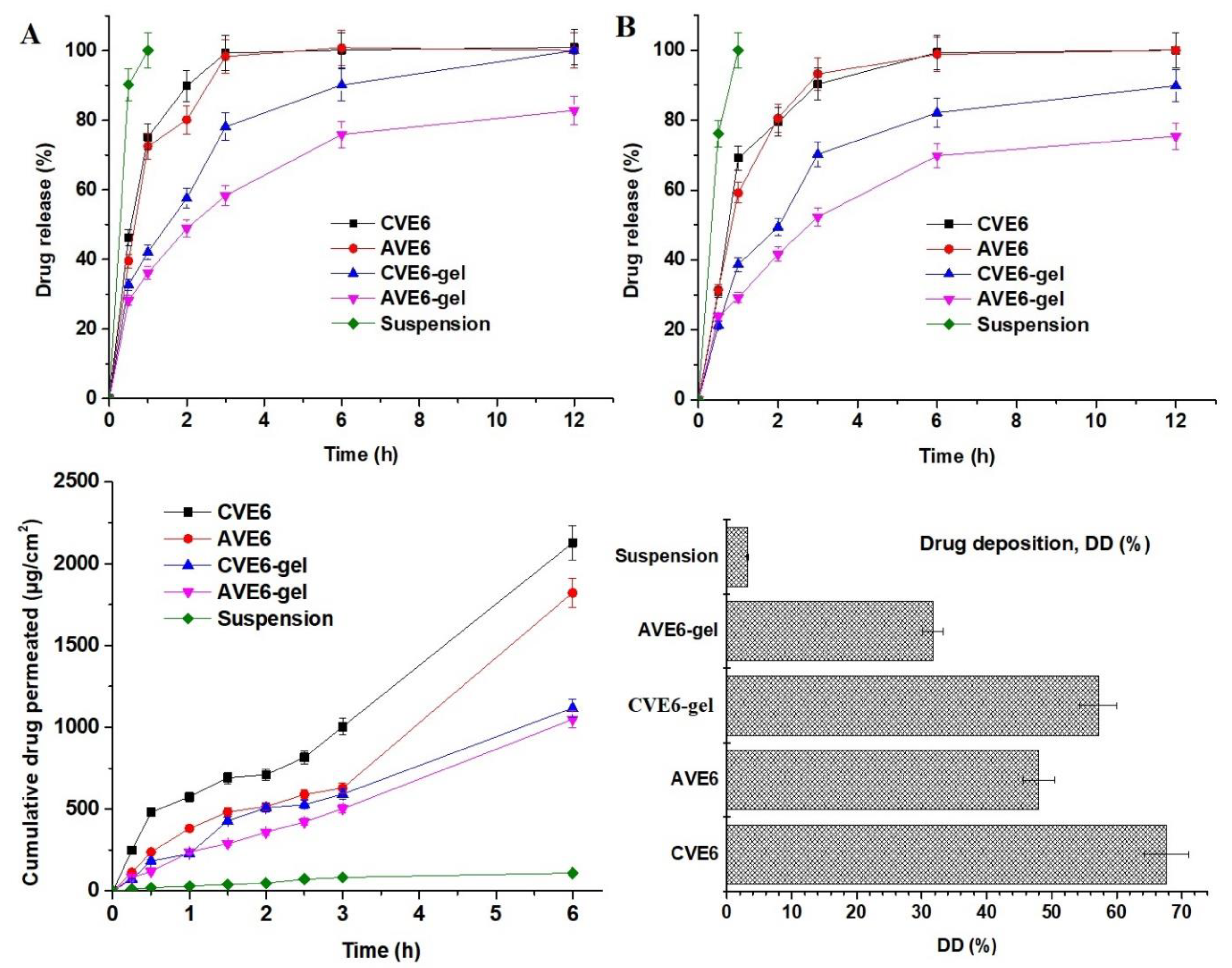
Figure 5.
Penetration and permeation of the optimized nanoemulions and respective gels across nasal epithelium to submucosal and mucosal region using CLSM (confocal laser scanning microscopy). (A) Control using R123 solution, (B) R123-probed drug suspension, (C) AVE6-R nanoemulsion, (D) AVE6-R-gel, (E) CVE6-R nanoemulsion, and (F) CVE6-R-gel. Mean intensity measured using image J software (E).
Figure 5.
Penetration and permeation of the optimized nanoemulions and respective gels across nasal epithelium to submucosal and mucosal region using CLSM (confocal laser scanning microscopy). (A) Control using R123 solution, (B) R123-probed drug suspension, (C) AVE6-R nanoemulsion, (D) AVE6-R-gel, (E) CVE6-R nanoemulsion, and (F) CVE6-R-gel. Mean intensity measured using image J software (E).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



