
Submitted:
09 June 2023
Posted:
12 June 2023
You are already at the latest version
Abstract
(1) Background: The HIV subtype D is generally associated with a faster decline in CD4+ T cell counts, a higher viral load, and a faster progression to AIDS. However, it is still poorly characterized in Brazil. In this study, we used genomics and epidemiological data to investigate the transmission dynamics of HIV subtype D in the state of Bahia, Northeast Brazil. (2) Methods: to achieve this goal, we obtained four novel HIV-1 subtype D partial pol genome sequences using the Sanger method. To understand the emergence of this novel subtype in the state of Bahia, we used phylodynamic analysis on a dataset comprising 3,704 pol genome sequences downloaded from the Los Alamos database. (3) Results: Our analysis revealed three branching patterns, indicating multiple introductions of the HIV-1 subtype D in Brazil from the late 1980s to the late 2000s and a single introduction event in the state of Bahia. Our data further suggest that these introductions most likely originated from European, Eastern African, Western African and Southern African countries. (4) Conclusion: Understanding the distribution of HIV-1 viral strains and their temporal dynamics is crucial for monitoring the real-time evolution of circulating subtypes and recombinant forms, as well as for designing novel diagnostic and vaccination strategies. We advocate for a shift to active surveillance, to ensure adequate preparedness for future epidemics mediated by emerging viral strains.
Keywords:
HIV-1 subtype D
; Phylodynamics
; Genomic surveillance
1. Introduction
The Human Immunodeficiency Virus type 1 (HIV-1), the etiological agent of acquired immunodeficiency syndrome (AIDS), infects around 38,4 million people worldwide. It presents a highly diverse genome of approximately 9.5 kb in length, formed by two single RNA strands. Its genetic diversity can be classified in a wide variety of groups (M, N, O and P). The HIV-1 group M viruses can be further subdivided into subtypes (A1, A2, A3, A4, A6, A7, A8, B, C, D, F1, F2, G, H, J and K), unique recombinant (URF) and circulating (CRF) forms [1].
Since the 1990s, molecular epidemiological studies of HIV-1 in Brazil have aimed to identify and understand the distribution of different subtypes and recombinant forms throughout the country [2,3,4]. In the past decade, other studies have shed light on the dissemination, incidence of primary resistance mutation, and origin of several HIV-1 subtypes, particularly B and C, in Brazil [5,6,7,8,9,10]. Such studies play a vital role in conducting HIV-1 Genomic Surveillance in Brazil and have the potential to uncover the emergence of more pathogenic variants within the country, similar to previous findings of the CRF19 recombinant in Cuba and the recently described“VB” variant in the Netherlands [11,12].
The prevalence of different viral subtypes in Brazil varies by region, with the most common subtypes being B, F, C, and the recombinant form BF [13]. In Northeast Brazil, the prevalence of subtypes B, F, C, and BF recombinants is 76%, 8%, 2%, and 7%, respectively [14]. A similar distribution of subtypes is observed in Bahia, with subtype B being the first to be identified in the state. The estimated prevalence for subtypes B, F, C, and BF recombinants ranges from 67.2% to 91.8%, 1.8% to 14.4%, 1.7% to 4.1%, and 3.3% to 24.1%, respectively [15,16,17,18,19].
Despite the high frequency of those subtypes, other viral strains have already been described in Brazil, including subtype D, which has been suggested to be more pathogenic than other forms [20,21,22,23,24,25]. However, despite the importance of the HIV-1 subtype D surveillance, there is still a paucity of studies in Brazil that describe its phylogenetic relationship with sequences from other countries and its transmission dynamics within Brazilian regions.
In this study, we provide insights into the spread of subtype D in the state of Bahia by sequencing the pol region of the first four HIV-1 positive patients belonging to this subtype. To gain a comprehensive understanding of subtype D's dispersion in Brazil, we conducted a phylodynamic analysis using referene sequences from the Los Alamos database, including all available Brazilian strains. Our analysis revealed multiple introduction events of subtype D in Brazil from Europe and Africa and highlighted South Africa as the primary source driving its nationwide spread.
2. Materials and Methods
HIV-1 Samples from Bahia
A total of four subjects were diagnosed as HIV-1 subtype D positive between 2014-2015 and received follow-up at the Specialized Center for Diagnosis, Care, and Research (CEDAP), a state government public health reference service located in the city of Salvador, Bahia, Northeast Brazil. This study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of the Instituto Gonçalo Moniz (IGM-FIOCRUZ) (protocol number 1.764.505).
HIV-1 Sequencing, Assembly, and Subtyping
The viral RNA isolation was performed using QIAamp Viral RNA Mini Kit (Qiagen, Germany) according to the manufacturer’s instructions. The protease/reverse transcriptase (PR/RT) region was amplified and sequenced as previously described [26]. The outer polymerase chain reaction (PCR) was performed using a SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Thermo Fisher Scientific, United States of America) and the following primers: K1 (CAGAGCCAACAGCCCCACC) and K2 (TTTCCCCACTAACTTCTGTATGTCATTGACA) [27]. Inner PCR was performed using Platinum Taq DNA Polymerase High Fidelity (Thermo Fisher Scientific, United States of America) and with the following primers: DP16 (CCTCAAATCACTCTTTGGCAAC) and RT4 (AGTTCATAACCCATCCAAAG) [28]. The generated inner PCR products were then sequenced using ABI 3500xL Genetic Analyzer (Applied Biosystems, United States of America) with the following primers: F1 (GTTGACTCAGATTGGTTGCAC), F2 (GTATGTCATTGACAGTCCAGC) [29], DP10 (CAACTCCCTCTCAGAAGCAGGAGCCG), DP11 (CCATTCCTGGCTTTAATTTTACTGGTA) [30], RT4 (AGTTCATAACCCATCCAAAG), GABO1 (CTCARGACTTYTGGGAAGTTC) and GABO2 (GCATCHCCCACATCYAGTACTG) [26].
Sequence visualization, editing and assembly were performed using Geneious v.10.0.8 software [31]. Subtyping was determined using the REGA HIV-1 Subtyping Tool v.3.46 available at Genome Detective (https://www.genomedetective.com) and the jpHMM (jumping profile Hidden Markov Model), which is a probabilistic generalization of the jumping-alignment approach [32].
HIV-1 POL Reference sequences from the Los Alamos Database
To perform phylodynamic and phylogeographic analyses, HIV-1 subtype D pol genome sequences over 700 base pairs covering the protease and reverse transcriptase (starting from genomic position 2085 and ending at position 5086 relative to the HXB2 reference), along with the sample collection date and location, were downloaded from Los Alamos HIV Sequence Database (https://www.hiv.lanl.gov) up to July 15th, 2022. Sequences identified as duplicates or with 100% identity, belonging to the same country and year were excluded.
Maximum Likelihood
Sequences were aligned using MAFFT v.7.455 [33,34] and manually edited using Geneious software [29]. Sequences that were too short or did not correspond to the analyzed region were excluded. The phylogenetic signal and the best fitting evolutionary model were evaluated using the software IQ-TREE v.2.0.3 [35]. A maximum likelihood (ML) tree was estimated using IQ-TREE v.2.0.3 [36] under GTR+F+I+G4 nucleotide substitution model [37,38] with 1,000 replicates and an ultrafast bootstrap [39]. Bootstrap was considered significant when >90%. The ML trees were visualized using FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/) and plotted using the ggtree package in RStudio v4.2.2 (https://www.r-project.org) [40].
Molecular Clock Phylogenetic Analysis
To determine the tMRCA (time to Most Recent Common Ancestor) of clades that include Brazilian sequences, a Bayesian analysis was performed [41]. The presence of a temporal signal was evaluated using TempEst v.1.5.3[42]. Time-scaled phylogenetic trees were inferred using the BEAST v.1.10.4 package [41] with BEAGLE v4.0.0 to improve the computational performance [43,44]. We employed a stringent model selection analysis using both path-sampling (PS) and steppingstone (SS) procedures to estimate the most appropriate molecular clock model for the Bayesian phylogenetic analysis [45]. The uncorrelated relaxed molecular clock model was chosen for all datasets as indicated by estimating marginal likelihoods, also employing the codon-based SRD06 model of nucleotide substitution and the nonparametric Bayesian Skyline coalescent model. MCMC analyses were performed in BEAST v.1.10.4, running in duplicate for 300 million interactions and sampling every 30,000 steps in the chain [46,47]. Convergence for each run was assessed in Tracer (effective sample size for all relevant model parameters >200). MCC trees for each run were summarized using TreeAnnotator v.1.10.4 after discarding the initial 10% as burn-in. Posterior probability was considered significant when ≥ 0.9.
3. Results
To understand the introduction and spread of HIV-1 subtype D in Brazil, a worldwide dataset was built. First, 3,808 sequences of pol region were downloaded from Los Alamos database. Among them, 104 were excluded for not belonging to the analyzed region of interest. Of the remaining 3,704 sequences, 1,759 were excluded for being identical, or highly similar with other sequences belonging to the same collection date and location. The 1,945 remaining sequences, which included 27 (1996-2018) previously published sequences from Brazil (11 from Rio de Janeiro, four from Rio Grande do Sul, five from Pará, one from Goiás, two from São Paulo and four not informed) and four newly identified sequences from the state of Bahia (2014-2015) (Table 1), were used to reconstruct the ML tree (Supplementary Figure S1). The clades that included Brazilian sequences were then extracted for the Bayesian analysis, resulting in 203 sequences being analyzed (Figure S1).
Our analysis revealed that the Brazilian sequences are distributed across nine distinct clades, suggesting multiple introductions events of this subtype in the country (Supplementary Figure S1A). The Bayesian analysis shows that the multiple introductions of HIV-1 subtype D in Brazil occurred at different times from the late 1980s to the late 2000s (Figure 1) and likely originated from Europe (Portugal, France, and Spain), Eastern Africa (Kenya, Congo and, the Democratic Republic of the Congo) and Western Africa (Senegal), and Southern Africa (South Africa).
One sequence collected in 2017 from São Paulo was found to be closely related to sequences from Portugal with a bootstrap value of 100% (Supplementary Figure S1E). This clade was estimated to have originated around 1999, with 95% Bayesian high posterior density (HPD) between 1993-10-01 and 2003-02-17 (pp=0.99) (Figure 1). Two sequences isolated from Rio de Janeiro in 2016 and 2017 were grouped with two sequences from France and one from Spain with a statistical support of 90% (Supplementary Figure S1G). However, this clade did not have statistical support on the Bayesian tree (pp=0.53). Nonetheless, the clade with the sequence isolated in 2016 sharing a common ancestor with sequences from France has a tMRCA of 1995 (95% HPD: 1991-07-31:2000-02-17, pp=0.98) (Figure 1). These findings are similar to those of HIV-1 subtype C and F1, which also suggest that Europe was the source of the introduction of these viruses in Brazil [48,49,50].
A group of four sequences from Rio Grande do Sul collected between 2010 and 2017 were grouped with a sequence from Kenya (bootstrap value=98%) (Supplementary Figure S1B). This group of sequences from Rio Grande do Sul and the sequence from Kenya shared a common ancestor around 1992 (95% HPD: 1988-01-11:1996-11-10, pp=0.98) (Figure 1). One sequence isolated from Pará, a state located in the north region of Brazil, in 2018 clustered together with sequences from Uganda (bootstrap value=100%). The tMRCA for this clade is 2008 (95 % HPD: 2005-03-07:2009-12-24, pp=1) (Figure 1). The sequence from Goiás (Midwest region), collected in 2017 (bootstrap value=100%), was clustered with two sequences from Uganda (Supplementary Figure S1D). Another sequence from Pará, collected in 2015, was clustered with sequences from the Democratic Republic of the Congo and Senegal (bootstrap value=97%) (Supplementary Figure S1F). A Brazilian sequence from Rio de Janeiro, collected in 2006, also grouped with a sequence from the Democratic Republic of the Congo (bootstrap value=100%) (Supplementary Figure S1H). These clades from Goiás, Pará, and Rio de Janeiro did not have statistical support for tMRCA inference, with pp values of 0.49, 0.02, and 0.11 respectively. Another sequence isolated in Pará in 2017 clustered together with sequences from Uganda but without statistical significance and was excluded from Bayesian inference (bootstrap value=69%) (Supplementary Figure S1J). These findings are in accordance with studies that show the relationship between Brazilian and African sequences, especially in HIV-1 subtypes C and HIV-1 CRF02_AG [6,51].
The nineteen remaining Brazilian sequences, which correspond to 61% of total subtype D Brazilian sequences, including those sequenced in this study, were clustered in a monophyletic group with 100% bootstrap statistical support (Supplementary Figure S1I). This clade contains sequences from samples collected between 1996 to 2017 from different regions (Northeast, North, and Southeast) and shares a common ancestor around 1987 with statistical support (95% HPD: 1983-05-27:1990-07-31, pp=0.99) (Figure 1). The four new sequences from Bahia were also grouped into a monophyletic cluster inside this Brazilian cluster, suggesting a single introduction of this virus in the state, sharing a common ancestor with a sequence from Pará with tMRCA of 1997 (95% HPD: 1990-03-04:2007-05-05, pp=1) (Figure 1). No epidemiological relationship was observed among these sequences. Of note, this Brazilian clade, which contains 19 sequences, including the four new sequences from Bahia, was clustered with sequences isolated from South Africa between 1984 to 1990, sharing a common ancestor with an introduction date of 1983 with statistical support (95% HPD: 1983-08-15:1984-01-22, pp=0.99).
Multiple introductions of HIV-1 subtype D in Brazil from different world regions, including Africa and Europe, have occurred at different times over the last few decades. These findings are consistent with other viral subtypes, where Africa is the epicenter of the HIV epidemic and the place of origin of the virus, and Europe serves as a transitory source for the passage of these viruses [6,48,52]. Although HIV-1 subtype D is more pathogenic and, therefore, should be considered a public health concern, this is the first study in Brazil that demonstrates the origin and dispersion dynamic of this subtype in the country, reporting the first pol sequences of this subtype from the Brazilian Northeast region. However, a complete genome analysis with a larger number of sequences needs to be performed to clarify the subtype D dispersion dynamics and increase the power of genomic surveillance in Brazil.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Phylogenetic tree of HIV-1 subtype D, branches with Brazilian sequences are highlighted: (a) Maximum likelihood tree of 1945 HIV-1 subtype D sequences; (b) Highlighted branch with Brazilian sequences from Rio Grande do Sul; (c) Highlighted branch with a Brazilian sequence from Pará; (d) Highlighted branch with a Brazilian sequence from Goiás; (e) Highlighted branch with a Brazilian sequence from São Paulo; (f) Highlighted branch with another Brazilian sequence from Pará; (g) Highlighted branch with Brazilian sequences from Rio de Janeiro; (h) Highlighted branch with a Brazilian sequence from Rio de Janeiro; (i) Highlighted branch of the major Brazilian clade; (j) Highlighted branch with another Brazilian sequence from Pará.
Author Contributions
Conceptualization, L.A.S., J.V.W., M.G., RK.; Data curation, F.F.A.R., F.G.T., L.dM.; Formal analysis, F.F.A.R., M.G.; Investigation, J.A.G.S., F.G.T., M.O.S., M.P.P.S.; Methodology, R.K.; Resources, R.K..; Writing – original draft, F.F.A.R.; Writing – review & editing, F.F.A.R., L.A.S., M.G., L.dM., R.K. All the authors discussed the structure of the manuscript and contributed to the final manuscript.
Funding
This research was funded by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) [Finance Code 001, R.K.], Fundação de Amparo à Pesquisa do Estado da Bahia (FAPESB, grant APP0032/2016, R.K.), Brazilian National Council for Scientific and Technological Development (CNPq, grant 65083/2015-8, L.A.S.); Fonds voor Wetenschappelijk Onderzoek Vlaanderen (grant G0A0621N, J.W.). MG is funded by PON “Ricerca e Innovazione” 2014-2020. None of the funding organizations had any role in the study design, data collection, data interpretation, or writing of this report.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the Gonçalo Moniz Institute (IGM-FIOCRUZ) (CEP/CAAE: 51733115.1.0000.0040; approval number: 1.764.505).
Informed Consent Statement
Patient consent was waived due to the retrospective nature of the study and the use of serum samples used in this research were collected for diagnostic purposes. These samples were accompanied by their respective epidemiological sheets, with patient identification already encoded.
Data Availability Statement
The new sequences have been deposited in NCBI GenBank under accession numbers MW596909, MW596999, MW597006, and MW597008.
Acknowledgments
The sequencing was performed at the RPT01B Genomic Platform from Gonçalo Moniz Institute (IGM-FIOCRUZ).
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D.HIV subtype diversity worldwide. Curr Opin HIV AIDS. 2019 May;14(3):153-160. [CrossRef]
- Couto-Fernandez, J. C.; Morgado, M. G.; Bongertz, V.; Tanuri, A.; Andrade, T.; Brites, C.; Galvão-Castro, B. HIV-1 subtyping in Salvador, Bahia, Brazil: A city with African Sociodemographic characteritics. J Acquir Immune Defic Syndr . 1999 Nov 1;22(3):288-93. [CrossRef]
- Tanuri, A.; Swanson, P.; Devare, S.; Berro, O. J.; Savedra, A.; Costa, L. J.; Telles, J. G.; Brindeiro, R.; Schable, C.; Pieniazek, D.; Rayfield, M.HIV-1 subtypes among blood donors from Rio de Janeiro, Brazil. J Acquir Immune Defic Syndr . 1999 Jan 1;20(1):60-6. [CrossRef]
- Bongertz, V.; Bou-Habib, D. C.; Brígido, L. F.; Caseiro, M.; Chequer, P. J.; Couto-Fernandez, J. C.; Ferreira, P. C.; Galvão-Castro, B.; Greco, D.; Guimarães, M. L.; Linhares de Carvalho, M. I.; Morgado, M. G.; Oliveira, C. A.; Osmanov, S.; Ramos, C. A.; Rossini, M.; Sabino, E.; Tanuri, A.; Ueda, M.HIV-1 diversity in Brazil: genetic, biologic, and immunologic characterization of HIV-1 strains in three potential HIV vaccine evaluation sites. Brazilian Network for HIV Isolation and Characterization. J Acquir Immune Defic Syndr. 2000 Feb 1;23(2):184-93. [CrossRef]
- Bello, G.; Zanotto, P. M.; Iamarino, A.; Gräf, T.; Pinto, A. R.; Couto-Fernandez, J. C.; Morgado, M. G. Phylogeographic analysis of HIV-1 subtype C dissemination in Southern Brazil. PLoS One. 2012;7(4):e35649. [CrossRef]
- Delatorre, E.; Couto-Fernandez, J. C.; Guimarães, M. L.; Vaz Cardoso, L. P.; de Alcantara, K. C.; Stefani, M. M.; Romero, H.; Freire, C. C.; Iamarino, A.; de A Zanotto, P. M.; Morgado, M. G.; Bello, G.Tracing the origin and northward dissemination dynamics of HIV-1 subtype C in Brazil. PLoS One. 2013 Sep 12;8(9):e74072. [CrossRef]
- Gräf, T.; Vrancken, B.; Maletich Junqueira, D.; de Medeiros, R. M.; Suchard, M. A.; Lemey, P.; Esteves de Matos Almeida, S.; Pinto, A. R. Contribution of Epidemiological Predictors in Unraveling the Phylogeographic History of HIV-1 Subtype C in Brazil. J Virol. 2015 Dec;89(24):12341-8. [CrossRef]
- Fritsch, H. M.; Almeida, S. E. M.; Pinto, A. R.; Gräf, T.Spatiotemporal and demographic history of the HIV-1 circulating recombinant form CRF31_BC in Brazil. Infect Genet Evol. 2018 Jul;61:113-118. [CrossRef]
- Arantes, I.; Esashika Crispim, M. A.; Nogueira da Guarda Reis, M.; Martins Araújo Stefani, M.; Bello, G.Reconstructing the Dissemination Dynamics of the Major HIV-1 Subtype B Non-Pandemic Lineage Circulating in Brazil. Viruses. 2019 Oct 1;11(10):909. [CrossRef]
- Gräf, T.; Bello, G.; Andrade, P.; Arantes, I.; Pereira, J. M.; da Silva, A. B. P.; Veiga, R. V.; Mariani, D.; Boullosa, L. T.; Arruda, M. B.; Fernandez, J. C. C.; Dennis, A. M.; Rasmussen, D. A.; Tanuri, A. HIV-1 molecular diversity in Brazil unveiled by 10 years of sampling by the national genotyping network. Sci Rep. 2021 Aug 4;11(1):15842. [CrossRef]
- Kouri, V.; Khouri, R.; Alemán, Y.; Abrahantes, Y.; Vercauteren, J.; Pineda-Peña, A.C.; Theys, K.; Megens, S.; Moutschen, M.; Pfeifer, N.; Van Weyenbergh, J.; Pérez, AB.; Pérez, J.; Pérez, L.; Van Laethem, K.; Vandamme, A.M. CRF19_cpx is an Evolutionary fit HIV-1 Variant Strongly Associated With Rapid Progression to AIDS in Cuba. EBioMedicine. 2015 Jan 28;2(3):244-54. [CrossRef]
- Wymant, C.; Bezemer, D.; Blanquart, F.; Ferretti, L.; Gall, A.; Hall, M.; Golubchik, T.; Bakker, M.; Ong, S.H.; Zhao, L.; Bonsall, D.; de Cesare, M.; MacIntyre-Cockett, G.; Abeler-Dörner, L.; Albert, J.; Bannert, N.; Fellay, J.; Grabowski, M.K.; Gunsenheimer-Bartmeyer, B.; Günthard, H.F.; Kivelä, P.; Kouyos, R.D.; Laeyendecker, O.; Meyer, L.; Porter, K.; Ristola, M.; van Sighem, A.; Berkhout, B.; Kellam, P.; Cornelissen, M.; Reiss, P.; Fraser C.; Netherlands ATHENA HIV Observational Cohort†; BEEHIVE Collaboration†. A highly virulent variant of HIV-1 circulating in the Netherlands. Science. 2022 Feb 4;375(6580):540-545. [CrossRef]
- Alves, B. M.; Siqueira, J. D.; Prellwitz, I. M.; Botelho, O. M.; Da Hora, V. P.; Sanabani, S.; Recordon-Pinson, P.; Fleury, H.; Soares, E. A.; Soares, M. A.Estimating HIV-1 Genetic Diversity in Brazil Through Next-Generation Sequencing. Front Microbiol. 2019 Apr 9;10:749. [CrossRef]
- da Costa, C. P.; Rodrigues, J. K. F.; de Morais, V. M. S.; de Andrade, C. A. D. N.; Neves, P. A. F.; Lima, K.HIV-1 subtype frequency in Northeast Brazil: A systematic review and meta-analysis. J Med Virol. 2020 Apr 8. Online ahead of print. [CrossRef]
- Monteiro, J. P.; Alcantara, L. C.; de Oliveira, T.; Oliveira, A. M.; Melo, M. A.; Brites, C.; Galvão-Castro, B. Genetic variability of human immunodeficiency virus-1 in Bahia state, Northeast, Brazil: high diversity of HIV genotypes. J Med Virol. 2009 Mar;81(3):391-9. [CrossRef]
- Araujo, A. F.; Brites, C.; Monteiro-Cunha, J.; Santos, L. A.; Galvao-Castro, B.; Alcantara, L. C. Lower prevalence of human immunodeficiency virus type 1 Brazilian subtype B found in northeastern Brazil with slower progression to AIDS. AIDS Res Hum Retroviruses. 2010 Nov;26(11):1249-54. [CrossRef]
- Santos, L. A.; Monteiro-Cunha, J. P.; Araujo, A. F.; Brites, C.; Galvao-Castro, B.; Alcantara, L. C.Detection of distinct human immunodeficiency virus type 1 circulating recombinant forms in northeast Brazil. J Med Virol. 2011 Dec;83(12):2066-72. [CrossRef]
- Monteiro-Cunha, J. P.; Araujo, A. F.; Santos, E.; Galvao-Castro, B.; Alcantara, L. CLack of high-level resistance mutations in HIV type 1 BF recombinant strains circulating in northeast Brazil. AIDS Res Hum Retroviruses. 2011 Jun;27(6):623-31. [CrossRef]
- Amaral, A. G.; Oliveira, I. B.; Carneiro, D. C.; Alcantara, L. C.; Monteiro-Cunha, J. P. An overview of the molecular and epidemiological features of HIV-1 infection in two major cities of Bahia state, Brazil. Mem Inst Oswaldo Cruz. 2017 Jun;112(6):411-418. [CrossRef]
- Kiwanuka, N.; Robb, M.; Laeyendecker, O.; Kigozi, G.; Wabwire-Mangen, F.; Makumbi, F. E.; Nalugoda, F.; Kagaayi, J.; Eller, M.; Eller, L. A.; Serwadda, D.; Sewankambo, N. K.; Reynolds, S. J.; Quinn, T. C.; Gray, R. H.; Wawer, M. J.; Whalen, C. C. HIV-1 viral subtype differences in the rate of CD4+ T-cell decline among HIV seroincident antiretroviral naive persons in Rakai district, Uganda. J Acquir Immune Defic Syndr . 2010 Jun;54(2):180-4. [CrossRef]
- Baeten, J. M.; Chohan, B.; Lavreys, L.; Chohan, V.; McClelland, R. S.; Certain, L.; Mandaliya, K.; Jaoko, W.; Overbaugh, J. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J Infect Dis. 2007 Apr 15;195(8):1177-80. [CrossRef]
- Kaleebu, P.; French, N.; Mahe, C.; Yirrell, D.; Watera, C.; Lyagoba, F.; Nakiyingi, J.; Rutebemberwa, A.; Morgan, D.; Weber, J.; Gilks, C.; Whitworth, J.Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda.J Infect Dis. 2002 May 1;185(9):1244-50. [CrossRef]
- Ssemwanga, D.; Nsubuga, R. N.; Mayanja, B. N.; Lyagoba, F.; Magambo, B.; Yirrell, D.; Van der Paal, L.; Grosskurth, H.; Kaleebu, P. Effect of HIV-1 subtypes on disease progression in rural Uganda: A prospective clinical cohort study. PLoS One. 2013 Aug 12;8(8):e71768. [CrossRef]
- Vasan, A.; Renjifo, B.; Hertzmark, E.; Chaplin, B.; Msamanga, G.; Essex, M.; Fawzi, W.; Hunter, D. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clin. Infect. Dis. 2006 Mar 15;42(6):843-52. [CrossRef]
- Venner, C. M.; Nankya, I.; Kyeyune, F.; Demers, K.; Kwok, C.; Chen, P. L.; Rwambuya, S.; Munjoma, M.; Chipato, T.; Byamugisha, J.; Van Der Pol, B.; Mugyenyi, P.; Salata, R. A.; Morrison, C. S.; Arts, E. J.Infecting HIV-1 Subtype Predicts Disease Progression in Women of Sub-Saharan Africa. EBioMedicine. 2016 Nov;13:305-314. [CrossRef]
- Barreto, C. C.; Nishyia, A.; Araújo, L. V.; Ferreira, J. E.; Busch, M. P.; Sabino, E. C.Trends in antiretroviral drug resistance and clade distributions among HIV-1-infected blood donors in Sao Paulo, Brazil. J Acquir Immune Defic Syndr. 2006 Mar;41(3):338-41. [CrossRef]
- Kozal, M. J.; Shah, N.; Shen, N.; Yang, R.; Fucini, R.; Merigan, T. C.; Richman, D. D.; Morris, D.; Hubbell, E.; Chee, M.; Gingeras, T. R. Extensive polymorphisms observed in HIV–1 clade B protease gene using high–density oligonucleotide arrays. Nat Med. 1996 Jul;2(7):753-9. [CrossRef]
- Pieniazek, D.; Peralta, J. M.; Ferreira, J. A.; Krebs, J. W.; Owen, S. M.; Sion, F. S.; Filho, C. F.; Sereno, A. B.; de Sa, C. A.; Weniger, B. G.Identification of mixed HIV-1/HIV-2 infections in Brazil by polymerase chain reaction. AIDS. 1991 Nov;5(11):1293-9. [CrossRef]
- Frenkel, L. M.; Wagner, L. E.; 2nd, Atwood, S. M.; Cummins, T. J.; Dewhurst, S. Specific, sensitive, and rapid assay for human immunodeficiency virus type 1 pol mutations associated with resistance to zidovudine and didanosine. J Clin Microbiol. 1995 Feb;33(2):342-7. [CrossRef]
- Janini, L. M.; Pieniazek, D.; Peralta, J. M.; Schechter, M.; Tanuri, A.; Vicente, A. C.; dela Torre, N.; Pieniazek, N. J.; Luo, C. C.; Kalish, M. L.; Schochetman, G.; Rayfield, M. A. Identification of single and dual infections with distinct subtypes of human immunodeficiency virus type 1 by using restriction fragment length polymorphism analysis. Virus Genes. 1996;13(1):69-81. [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; Thierer, T.; Ashton, B.; Meintjes, P.; Drummond, A.Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012 Jun 15;28(12):1647-9. [CrossRef]
- Schultz, A. K.; Bulla, I.; Abdou-Chekaraou, M.; Gordien, E.; Morgenstern, B.; Zoaulim, F.; Dény, P.; Stanke, M. jpHMM: recombination analysis in viruses with circular genomes such as the hepatitis B virus. Nucleic Acids Research.2012 Jul;40(Web Server issue):W193-8. [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T.MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002 Jul 15;30(14):3059-66. [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2019 Jul 19;20(4):1160-1166. [CrossRef]
- Kalyaanamoorthy, S.; Minh, B. Q.; Wong, T. K. F.; von Haeseler, A.; Jermiin, L. S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat Methods. 2017 Jun;14(6):587-589. [CrossRef]
- Nguyen, L. T.; Schmidt, H. A.; von Haeseler, A.; Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015 Jan;32(1):268-74. [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22(2):160-74. [CrossRef]
- Yang, Z.Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J Mol Evol. 1994 Sep;39(3):306-14. [CrossRef]
- Hoang, D. T.; Chernomor, O.; von Haeseler, A.; Minh, B. Q.; Vinh, L. S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol Biol Evol. 2018 Feb 1;35(2):518-522. [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y.ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution. 2016;8(1):28-36. [CrossRef]
- Suchard, M. A.; Lemey, P.; Baele, G.; Ayres, D. L.; Drummond, A. J.; Rambaut, A.Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evolution. 2018 Jun 8;4(1):vey016. [CrossRef]
- Rambaut, A.; Lam, T. T.; Max Carvalho, L.; Pybus, O. G.Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016 Apr 9;2(1):vew007. [CrossRef]
- Baele, G.; Ayres, D. L.; Rambaut, A.; Suchard, M. A.; Lemey, P. High-Performance Computing in Bayesian Phylogenetics and Phylodynamics Using BEAGLE. Evolutionary Genomics. 2019;1910:691-722. [CrossRef]
- Ayres, D. L.; Cummings, M. P.; Baele, G.; Darling, A. E.; Lewis, P. O.; Swofford, D. L.; Huelsenbeck, J. P.; Lemey, P.; Rambaut, A.; Suchard, M. A. BEAGLE 3: Improved Performance, Scaling, and Usability for a High-Performance Computing Library for Statistical Phylogenetics. Syst Biol. 2019 Nov 1;68(6):1052-1061. [CrossRef]
- Baele, G.; Li, W. L.; Drummond, A. J.; Suchard, M. A.; Lemey, P. Accurate model selection of re-laxed molecular clocks in bayesian phylogenetics. Mol Biol Evol. 2013 Feb;30(2):239-43. [CrossRef]
- Drummond, A. J.; Ho, S. Y.; Phillips, M. J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biology. 2006 May;4(5):e88. [CrossRef]
- Drummond, A. J.; Rambaut, A.; Shapiro, B.; Pybus, O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005 May;22(5):1185-92. [CrossRef]
- Crispim, M. A. E.; Reis, M. N. D. G.; Abrahim, C.; Kiesslich, D.; Fraiji, N.; Bello, G.; Stefani, M. M. A. Homogenous HIV-1 subtype B from the Brazilian Amazon with infrequent diverse BF1 recombinants, subtypes F1 and C among blood donors. PLoS One. 2019 Sep 9;14(9):e0221151. [CrossRef]
- Silva, G. P. S. A.; Oliveira, R. C.; de Souza, J. S. M.; Giovanetti, M.; Guimarães, M. L.; Brites, C.; Monteiro-Cunha, J. P. Tracing the relationship among HIV-1 sub-subtype F1 strains: a phylodynamic perspective. Mem Inst Oswaldo Cruz. 2023 Jan 20;117:e220109. [CrossRef]
- Gräf, T.; Pinto, A. R.The increasing prevalence of HIV-1 subtype C in Southern Brazil and its dispersion through the continent. Virology. 2013 Jan 5;435(1):170-8. [CrossRef]
- Delatorre, E.; Velasco-De-Castro, C. A.; Pilotto, J. H.; Couto-Fernandez, J. C.; Bello, G.; Morgado, M. G. Short Communication: Reassessing the Origin of the HIV-1 CRF02_AG Lineages Circulating in Brazil. AIDS Res Hum Retroviruses. 2015 Dec;31(12):1230-7. [CrossRef]
- Faria, N.R.; Rambaut, A.; Suchard, M.A.; Baele, G.; Bedford, T.; Ward, M.J.; Tatem, A.J.; Sousa, J.D.; Arinaminpathy, N.; Pépin, J.; Posada, D.; Peeters, M.; Pybus, O.G.; Lemey, P. HIV epidemiology. The early spread and epidemic ignition of HIV-1 in human populations. Science. 2014 Oct 3;346(6205):56-61. [CrossRef]
Figure 1.
Bayesian phylogeny containing 203 HIV-1 subtype D sequences from pol region.
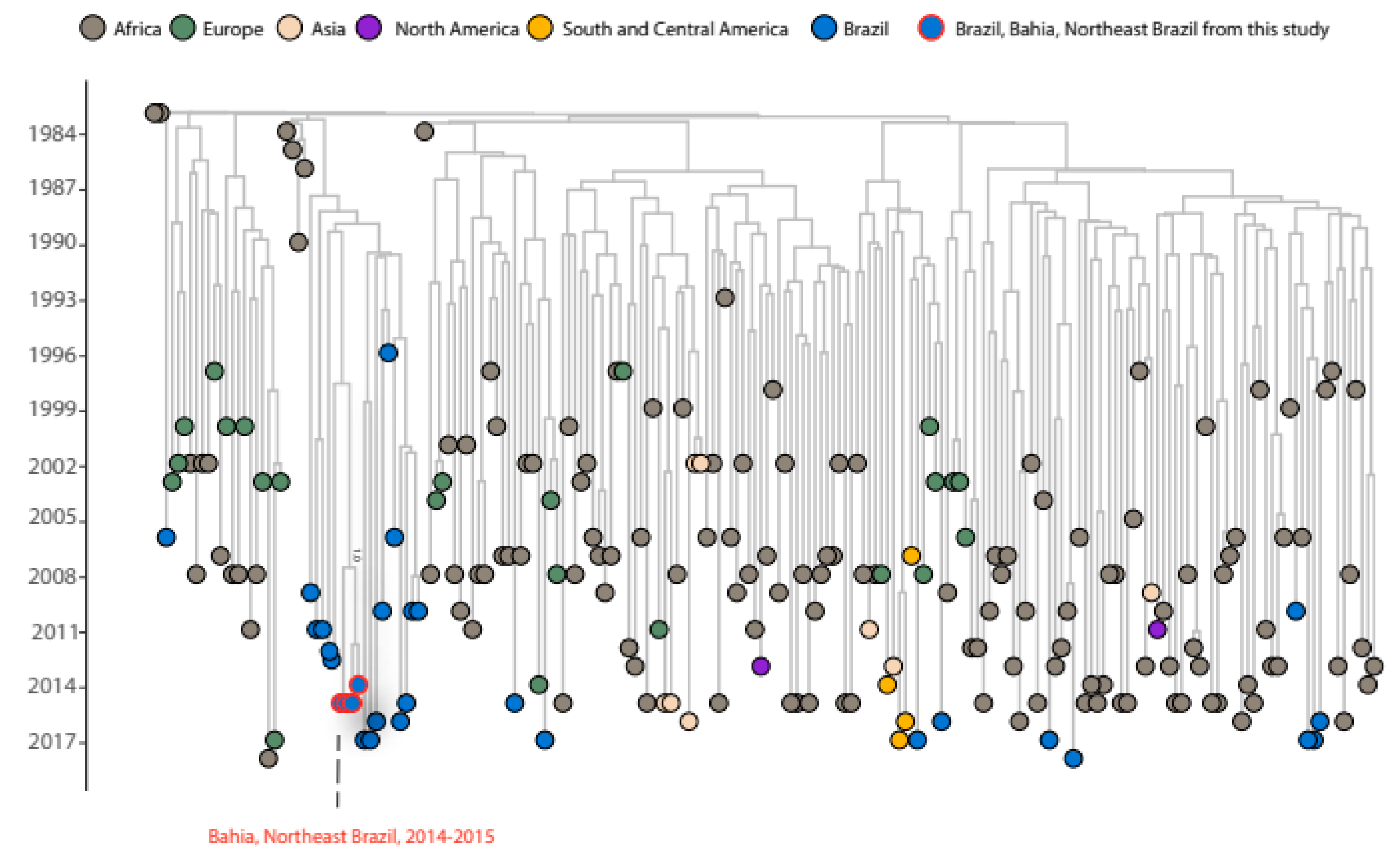

Table 1.
Clinical and demographic characteristics of four patients described in this study.
| Patient ID | Gender | Age | Viral load (copies/ml) | CD4 cell count /m3 | CD8 cell count/m3 | CD4/CD8 ratio | CD45 cell count/m3 |
| HV0018 | Female | 29 | 216 | 1,553 | 1,399 | 1.11 | 4,022 |
| HV0206 | Male | 26 | 50,273 | 704 | 1,231 | 0.57 | 2,315 |
| HV0220 | Male | 31 | 138,217 | 466 | 698 | 0.67 | 2,263 |
| HV0225* | Male | 53 | NI | NI | NI | NI | NI |
NI = not informed * Patient did not return for follow-up after diagnostic.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



