
Submitted:
10 June 2023
Posted:
12 June 2023
You are already at the latest version
Abstract
Modern medical needs call for the efficient remedies against bacterial diseases including severe pulmonary infections reported as a secondary SARS-Cov-2 infection. Here, we propose imprinted drug ciprofloxacin (CF) into the polymer based on methyl-β-cyclodextrin (MCD) via template synthesis. The obtained polycarbamide nanoparticles possess CF's sustained drug release. The interaction of human serum albumin (HSA) with CF and CF-MCD carriers was conducted by FRET, FTIR, fluorescence, and circular dichroism spectroscopy. These studies uncovered that MCD decreases CF's binding efficiency by 2 times, whereas CF’s encapsulation in polymer matrix doubles the Ka value. The changes in HSA’s secondary structure indicate no alterations in the main mechanism of complex formation between CF and HSA in the presence of MCD-based carries. The drug delivery system demonstrates prolonged CF release at pH 7.4 in presence of HSA (model conditions of blood plasma). CF-MCD carriers inhibit E. coli and B. subtilis growth, but for MAX systems we observed a MIC’s increase (~2 times). We believe that our findings are important for further development of new efficient drug forms.
Keywords:
ciprofloxacin
; cyclodextrin
; template synthesis
; human serum albumin
; FRET
1. Introduction
According to the World Health Organization, bacterial infections remain one of the leading causes of death in the world [1,2]. The infections are especially threatening among with reduced human immunity; likewise secondary bacterial infections might follow surgery or serious illnesses. For example, COVID-19 pandemic demonstrated the occurrence of severe pneumonia, requiring complex and lengthy treatment regimen at high doses of antibiotics, and challenged scientists to design new drug forms [3]. Currently, the development of novel drugs and their clinical trials take a long time and require expensive research. Therefore, the most urgent task of pharmacology is the design new efficient drug delivery systems to improve the properties of existed bioactive molecules. Such systems can contribute to the increase of therapy effectiveness, controlled release rate, targeted delivery, and reduction of the side effects probability [1,2,4].
Ciprofloxacin (СF) demonstrates numberless properties of an «ideal» antibacterial drug: a wide spectrum of activity in vitro against gram-negative and gram-positive, aerobic and anaerobic bacteria and even mycobacteria; chemical and biological stability. However, long-term CF therapy and high drug dosage might lead to a number of side effects. Most of the undesirable effects occur in the digestive system: nausea, abdominal discomfort, vomiting and diarrhea. In addition, CF has a negative effect on the central nervous system: dizziness, headaches, nervousness, sleep. Skin irritations have also been reported: a rash or itching may appear, probably of allergic origin [5,6,7]. Moreover, often patients have weakness, fever and heart palpitations. The CF’s dosage decrease helps to reduce the side effects probability [8,9].
An increase in solubility and, as a result, bioavailability of CF can be achieved using drug delivery systems. The promising approach is to obtain non-covalent CF complexes with oligosaccharides cyclodextrins. We propose the use of cyclodextrins (CDs) and their derivatives as they approved by FDA and proved to be effective drug carriers. CDs find a wide spectrum of practical applications in various fields, for example, CDs are used in food and textile industry. CDs are valuable in pharmaceutics as they increase drugs’ bioavailability, solubility and stability and reduce the toxicity due to the formation of guest-host complexes [10]. CF’s complexes with different CD derivatives are well described in the literature [11]. The complexes form by loading the CF’s aromatic fragment into CD’s hydrophobic cavity and stabilized by the additional interactions between CD’s substitutes and other CF’s functional groups.
Recently, the particular attention is drawn to CD polymers synthesized by «template» synthesis, in which the polymerization process takes place in the presence of another organic molecule (such as a medicinal substance). The template «imprinting» might also result in a specific polymer network formation with unique properties [12,13,14]. Encapsulation of the drug by the method might also provide a uniform distribution of the drug in the carrier through the particle volume, as well as high loading efficiency [15]. For example, dihydroxyphenylalanine (L-DOPA) was incorporated with 90% efficiency into a β-CD polymer using N,N'-carbonyldiimidazole as a linker; the obtained sample demonstrated prolonged release [16].
Thus, we are interested in the design of CF’s novel drug delivery systems based on CDs and template synthesis to develop highly effective drug forms with prolonged action. Besides the physical-chemical properties, we also study the interactions of CF’s drug forms with biological macromolecule – human serum albumin (HSA), since protein binding affects the distribution, excretion and therapeutic efficacy of drug molecules. We apply a number of spectroscopic methods to uncover the details of the complex system on molecular level. Besides, we investigate CF’s drug forms antibacterial properties in the presence of HSA to simulate in vitro action.
2. Materials and Methods
2.1. Materials
Methyl-β-cyclodextrin (MCD; 1.5-2.1 -CH3 group per one anhydroglucose), ciprofloxacin (CF), human serum albumin (HSA), 1,6–hexamethylene diisocyanate (HMI), dimethyl sulfoxide (DMSO) from Sigma-Aldrich (St. Louis, MO, USA). Hydrochloric acid is from - Reachim (Moscow, Russia). The tablets for the preparation of Phosphate buffered saline (PBS) (pH 7.4) were purchased from Pan-Eco (Moscow, Russia).
Escherichia coli ATCC 25922 and Bacillus subtitlis ATCC 6633 are from the VKPM «Kurchatov Institute» (Moscow, Russia).
2.2. Preparation of Ciprofloxacin Complexes with Methyl-β-Cyclodextrin:
The MCD powder was dissolved in aqueous solution (0.03 M, HCl pH 4.0) and then CF solution was added (0.03 M, pH 4.0 HCl) to achieve a 1:1 molar ratio. The resulting mixture was incubated at 37°C with gentle shaking for 1 h.
2.3. Synthesis MAX Systems via Crosslinking MCD-CF Complexes
1320 μL of 7.5 mM MCD-CF’s complex (1:1 mol : mol) solution (pH 4.0 HCl) were kept at 37°C for 20-30 min. Then, 1120 μl of dimethyl sulfoxide (DMSO) were added with active stirring. After 2 min, 200 μl of hexamethylene diisocyanate (HDI) solution in DMSO (0.1 M) were added dropwise, so that a molar ratio of HMI : monomer maintained 1. The mixture was incubated for 3 h before being placed in a refrigerator at 4°C for 12 h. The sample purification was performed by 6 h dialysis (the membrane with MWCO 3 kDa (Serva, USA)). The resulting solution was freeze-dried at -70°C for 24 h and then lyophilized at -60°C for 2 days (Edwards 5, BOC Edwards, UK).
The yield of the reaction (by MCD) (the percentage of MCD included in the MAX particles in relation to the added during synthesis) was calculated from the intensity of the absorption band of 1047 cm-1, corresponding to the oscillation of the C-O-C bond of the CD, in the IR spectrum of the MAX. The CF’s encapsulation efficiency into MCD polymer matrix determined by UV spectroscopy. The ratio of CD-torus to CF molecules in MAX was estimated by data obtained by UV- and FTIR spectroscopy.
2.4. Preparation of Binary (CF–HSA) and Ternary (HSA−(CF+CD Carrier)) Systems
A certain amount of CF (binary system) or CF+MCD/MAX (ternary system) solution was transferred to the sodium-phosphate buffer (pH 7.4) and HSA solution was added (pH 7.4). The volume was adjusted to 1 ml. The HSA concentration was maintained 0.06 mM, while the molar excess of free CF or CF in MCD/MAX was ranged from 0.25 to 20. The protein-drug form incubated at 37°C with gentle shaking. For UV-spectroscopy, circular dichroism spectroscopy and fluorescence spectroscopy analysis, the solutions were diluted three times directly before the spectra registration.
2.5. FTIR-Spectroscopy
Tensor 27 spectrometer (Bruker, Ettlingen, Germany) with an attenuated total reflection cell and ZnSe single-flection crystal was used to obtain FTIR-spectra of sample’s solutions. The spectrometer is equipped with an MCT detector cooled with liquid-N2, a thermo-stat (Huber, Offenburg, Germany). FTIR-spectra were recorded in the range of 2500–900 cm−1 with a 1 cm−1 resolution 3 times (70 scans each) at 22°C). Dry nitrogen was used to purge the system with an air compressor (Brezza NiGen HF-1, CLAIND, Italy). The spectra were analyzed by Opus 7.0.
By analyzing the FTIR-spectra in the range of 1700-1600 cm−1 for samples with albumin, the content of secondary structure was determined. The primary bands were detected by the second derivative of the spectrum, and each spectrum was deconvoluted using the Levenberg-Marquardt algorithm.
2.6. Circular Dichroism
The J-815 spectrometer from «Jasco» (Tokyo, Japan) was applied to record circular dichroism spectra. The measurements were performed at 25°C using a quartz cuvette (l = 1 mm) within a wavelength range of 200−260 nm. Spectra were scanned five times at 1 nm step with HSA's concentration 0.02 mM. To determine the secondary structure content the spectra were analyzed with CDNN program Version 2. The experiment was conducted 3 times, the results were performed with SD=3 (n = 3).
2.7. UV-Spectroscopy
The Ultrospec 2100 pro equipment from Amersham Biosciences (UK) was used to record UV-spectra. The CF/CF-MCD/MAX absorption spectra were measured in a quartz cell with an optical path length of 1 cm from Hellma Analytics (Jena, Germany) within a wavelength range of 200-450 nm.
2.8. Fluorescense Spectroscopy
The Varian Cary Eclipse spectrophotometer (Agilent Technologies, United States) was applied to conduct fluorescence measurements. The HSA’s emission spectra were recorded at 37 ± 0.1°C in a 10 mm quartz cuvette, with an excitation wavelength of 280 nm and a 1 nm step in the range of 290−550 nm. The protein concentration in all samples was maintained 0.02 mM (phosphate buffer solution with a pH 7.4).
The Stern-Volmer equation (1) was employed to analyze the quenching of protein fluorescence by small molecules, considering both statistical and dynamic quenching effects [17].
The equation includes and , which represent the fluorescence intensities in the absence and presence of a drug form (quencher), is a Stern-Volmer constant, [Q] - the molar concentration of the quencher, and - the bimolecular constants of quenching rate and the lifetime of HSA's fluorescence in the absence of the quencher respectively. The binding constant of HSA to the drug form () and the number of binding sites (n) were determined using equation (2) at 37 ± 0.1°C:
2.9. Fluorescence Anisotropy
For studying steady-state fluorescence anisotropy () another series of solutions was prepared. To the solution of 50 μM CF/CF+MCD (PBS, pH 7.4) the required volume of 5 mM HSA's (PBS, pH 7.4) solution was added and the volume was adjusted to 1 ml. The concentration of CF/CF+MCD was kept constant at 0.01 mM, while the molar excess of HSA ranged from 0 to 20. The complexes were then incubated at 37°C with gentle shaking.
The Varian Cary Eclipse spectrophotometer (Agilent Technologies, United States) equipped with polarizer was used for spectra registration. The emission spectra were recorded in four positions of polarizers: HH, VH, HV, VV, where first letter means horizontal (H) or vertical (V) orientation of the excitation polarizer, and the second - horizontal (H) or vertical (V) orientation of the emission polarizer. The recording was conducted at a temperature of 37 ± 0.1°C in a 10 mm thick cuvette, with an excitation wavelength of 340 nm and a step of 1 nm in the range of 340−550 nm. To calculate anisotropy () of fluorophore the equation (3) was used:
where – intensity of emission of fluorescence in different positions, and represents correction factor for the instrument's detector sensitivity [18,19,20].
The calculation the binding constants (Ka) CF or CF+MCD with HSA considering the equilibrium AM+nHSA↔AM×nHSA was conducted by equation (4) [17,21]:
We varied the concentration of HSA and analyzed the changes in the anisotropy by Hill’s linearization in n-binding site model (5) [22]:
where θ is a fraction of the bounded CF’s molecules calculated as (6)
where ξ0 and ξ are the peaks intensities of fluorescence spectra of CF in the absence and the presence of HSA, ξ∞ is the intensity of horizontal asymptote.
2.9. Fluorescence Resonance Energy Transfer (FRET)
In order to determine the average distance (r) between the ligand (CF in drug form) and HSA (Trp-214) equations (7−9) were applied. Formula (7)
was employed to calculate energy transfer efficiency (E). In equation (7) R0 is critical radius for 50% energy transfer efficiency that was determined by equation (8):
where is a factor describing the mutual orientation in the space of the dipole moments of the transitions of the donor and acceptor (2/3 for randomly orientation as in fluid solution); is the refractive index of the medium (1.33 in present case); is the quantum yield of donor in the absence of acceptor (0.118 for Trp in HSA); is the overlap integral of donor fluorescence emission and the acceptor absorption spectra [23]. was calculated according to the equation (9)
where is the fluorescence intensity of the donor at wavelength λ, is the molar coefficient of light absorption of the donor at wavelength λ.
2.11. Release Kinetic Studies
1 mL of the sample was placed in a dialysis bag with a MWCO of 3.5 kDa (Orange Scientific, Belgium) that was immersed in 1 mL phosphate buffer (pH 7.4). The system was incubated at 37°C 150 rpm, and UV-spectra of external solution were recorded regularly.
2.12. In Vitro Studies
The antimicrobial properties of CF drug forms were studied by agar well diffusion test using gram-negative Escherichia coli ATCC 25922 and gram-positive Bacillus subtitlis ATCC 6633 strains. The overnight culture (Luria Bertuni medium, pH 7.4) was diluted to match 0.5 McFarland standard. After the distribution of bacteria on Petri dishes 4 wells were cut using 1 mL sterile plastic tip. 50 μL of each sample was put into wells: sterile buffer as negative control, CF, CF+MCD, MAX, HSA−CF, HSA−(MCD+CF) or HSA−MAX (CCF = 0.2, 0.3, 0.5 μg/mL for E. coli ATCC 25922 and CCF = 0.4, 0.6, 0.8 μg/mL for B. subtitles ATCC 6633). The Petri dishes were incubated at 37℃ for 22-24 h. The diameters of appeared inhibition zones of bacterial growth were analyzed. The minimum inhibition concentration (MIC) was estimated as the sample’s concentration at which the inhibition zone area equals the incised agar area:
where c is CF’s concentration (μg/mL), Sinh is the inhibition zone area (mm2). Each sample was tested 3 times, and the data is presented with standard deviations.
3. Results and Discussion
Here, we design the CF’s drug delivery system via template synthesis (MAX) and investigate the main patterns of its interaction with HSA. CD’s oligomers and polymers are of particular interest, since they can adsorb «guest» molecules not only in the pores of the branched polymer, but also in the cavities of the CDs [24]. The use of such carriers for the drug’s encapsulation seems to be advantageous comparing to common drug-CD complexes. For example, formation of these systems might lead to prolonged release [25], affect the lipid membrane permeability and increase drug’s antibacterial activity [26,27]. Thus, in this study we consider three drug forms: CF, CF+MCD (common non-covalent complex) and MAX (imprinted CF into polymer matrix) to compare how different kinds of CD carriers affect CF’s properties.
First, we obtained MAX system with encapsulated CF molecules by crosslinking CF-MCD complexes with 1.6-hexamethelene diisocyanate. Figure 1 presents the chemical structures of different CF's forms under consideration and the scheme of MAX synthesis.
Synthesized MAX represents white powder. The MAX’s structure was investigated by FTIR. In the MAX’s spectra we found the band 1047 cm−1 attributed to stretching vibrations of C−O−C band as in free MCD, but the band’s shoulder appears. That might be explained due to the changes in the microenvironment of C–O–C bands between glucopyranose residues in β-CD derivatives after crosslinking. Also, we observed the appearance of new low-intensive bands: 951 cm−1 and 1540 − 1570 cm−1 and 1610 – 1680 cm−1 corresponded to C−N, amide I, and amide II of disubstituted carbamide. The CN groups might appear due to hydrolysis of isocyanate groups. The insignificant intensity of these bands might be explained by the low degree of crosslinking. Comparing to MCD FTIR-spectra we also observed the bands in range 1150 − 1400 cm−1 in MAX’s FTIR spectra that indicate the presence of CF. However, their analysis is complicated due to the low CF’s content.
The yield of the reaction (by MCD) (the percentage of MCD included in the MAX particles in relation to the one added during synthesis) was ca. 40%. The CF’s encapsulation efficiency into MCD polymer matrix determined by UV-spectroscopy was approximately 0.6 μg per 1 mg of MAX. The low degree of CF’s encapsulation in the MAX structure might be due to the low binding constant of CF+MCD (Ka = (1.3 ± 0.3) × 103 M−1 [11]) as well as drug’s limited solubility (CF prefers the organic phase instead of MCD tori). The estimated ratio of CD-torus to CF molecules in MAX is 100 : 1. The yield of the reaction and proportion of encapsulated CF was lower than for resemble system based on sulfobutyl-β-cyclodextrin and moxifloxacin [14]. We suppose, that the main differences are caused by higher degree of MCD substitution (1.5-2.1 per one glucose residue), so less -OH groups are available for reaction with HMI and therefore less CF molecules are fixed in polymer network; furthermore, negatively charged −SO3- groups in sulfobutyl-β-cyclodextrin contributed to the drug’s retention, while CF apparently passes into the organic phase.
3.1. Fluorescense Spectroscopy
HSA is the major protein of human’s blood plasma. The albumin binds and transports biologically active molecules, so the drug-HSA complex formation might significantly affect bioactivity and bioavailability of small drugs [28]. With the rise of attention to CD polymers, we are interested in how CD carries would change the CF’s binding with HSA. Fluorescence is a valuable approach for studying the interaction between small molecules and proteins. When tryptophan (Trp), tyrosine (Tyr) are present in the protein, their aromatic fragments can emit intrinsic fluorescence upon absorption of UV light, that indicate changes in the protein's structure and microenvironment of these residues. The changes in the protein’s fluorescence via interaction with other molecules uncover the binding mechanism, mode, constants, intermolecular distances, etc. Commonly, the molecular interactions cause fluorescence quenching including reactions in the excited state, energy transfer, and static and dynamic quenching. Intrinsic fluorescence quenching measurement of proteins is widely used to elucidate the mechanism of their interaction with ligands or drug molecules [29,30,31].
The interaction between CF and HSA is well studied [23,29,32,33] in literature, while ternary systems such as HSA-CF+MCD or HSA-MAX are not reported. With an excitation wavelength of 280 nm the individual CF and HSA demonstrate a fluorescence emission peaks at 415 and 340 nm, correspondingly. The spectra of both components are resolved, so it is possible to investigate the state of each component in binary and ternary systems simultaneously (Figure 2). The complex formation of CF with MCD as well as CF’s encapsulation in MAX does not pronouncedly influence the position of drug’s emission maximum.
With the increase of CF’s concentration, the HSA’s fluorescence intensity decreased significantly, and the band shifts on 7 nm towards the higher wavelengths (Figure 2). The effect suggests that Trp-214 microenvironment changes to more hydrophilic via CF’s binding. The data is confirmed by [34,35]. In our previous research we showed that free MCD doesn’t affect the intensity and maximum position of the emission spectrum of HSA [36,37]. For HSA−(CF+MCD) we observed the less pronounced quenching and more significant red shift (11 nm) compared to HSA−CF. We suggest that more hydrophilic CF+MCD complex (than free CF) has difficulty binding to hydrophobic pocket of HSA in subdomain IIA where CF binds [36,38,39], or the competition for CF’s binding between MCD and HSA takes place.
Unexpectedly, no spectrum emission shift (for both HSA and CF) in case of MAX was observed. The effect might be explained that encapsulated CF is less available for interactions with HSA. Moreover, MAX demonstrates more significant quenching of HSA's emission that free CF. That fact could indicate the different mechanism of interaction between HSA-CF and HSA-MAX systems. We suppose the interaction between the MAX and HSA is the key parameter of the observed effect. The MAX itself might induce the quenching HSA's fluorescence. As MAX may interact only with the surface of HSA no significant changes of Trp’s microenvironment occur and maximum of HSA's emission doesn’t change.
The decrease in the relative fluorescence intensities was analyzed using the Stern-Volmer equation (1) and the results are presented in Figure 3 and Table 1. KSV values for both binary and ternary systems were ca. 105 M−1, alluding that a significant interaction between HSA and CF was responsible for the quenching mechanism. The linear dependence of quenching suggested that only one type of quenching mechanism (either static or dynamic) dominated. To elucidate the most likely mechanism that causes the quenching of protein fluorescence, the values of the quenching rate constants kq were determined. Equation (1) was used to calculate kq. KSV was defined as the slope of the Stern-Volmer dependence, and τ0 = 5.71 × 10−9 s is a constant [20]. The observed value of kq is significantly higher than the bimolecular quenching rate constant of 2 × 1010 M-1×s-1 indicating the prevailing contribution of statistical quenching [17,40], i.e. the quenching was not initiated from dynamic collision, but via complex formation.
To determine the binding constants () and stoichiometric coefficient (n), equation (1) was converted into equation (2) due to the statistical nature of fluorescence quenching at low CF’s concentrations. Figure 3b demonstrates the plots of versus . The binding constant for HSA−CF complex was (1.0 ± 0.3) × 105 M−1 at 37℃ and there was only one binding site of CF on HSA. The formation of ternary systems leaded to the changes of Ka values, while the stoichiometry remains 1. For HSA-(CF+MCD) binding constant was almost in 2 times lower compared to the binary system. The finding might be due to the competition between albumin and MCD for drug binding. Since the binding efficiency of HSA−CF and CF+MCD are close ( M−1 [11]) the drug distribute between two ligands.
Interesting, that for MAX we observed the opposite effect of CD based carrier: the increase of Ka value in 1.5 times. We suppose the MAX particles interact with HSA, bringing together CF and Trp's residue and hiding the solvent interactions with HSA's hydrophobic pocket.
3.2. Steady-State Fluorescence Anisotropy
For monitoring the modulation of the fluorescence characteristics of CF in the presence of HSA involves excitation of the samples at λex ~ 340 nm. At these wavelength n-π* transitions in absorption spectrum of CF occurred [41], and hence assert no light absorption by HSA. Figure 4 illustrates the impact of added HSA on the steady-state fluorescence anisotropy of CF, providing further evidence of the CF-HSA interaction. The steep increase in anisotropy within the protein indicates significant motional constraints on the drug molecules upon binding to the protein, followed by saturation at higher protein concentrations. This finding is consistent with previous studies [41,42]. In ternary systems we also observed the increase of anisotropy with the increasing the HSA's concentration. But for the ternary system the highest magnitude of anisotropy was ~1,5 times smaller than for binary system HSA+CF, therefore part of CF is bounded with MCD even when plato was reached. We suggest that MCD and HSA concurred for binding with CF. The calculated Ka for binary and ternary systems ( M−1 and M−1) agree with the data obtained by the analysis of quenching fluorescence emission of HSA.
3.2. Fluorescence Resonance Energy Transfer (FRET)
The use of fluorescence resonance energy transfer (FRET) as a spectroscopic method allows monitoring the fluorophore proximity and angular orientation, making it a valuable tool for measuring molecular distances in complex biological systems. Forster's nonradiative energy transfer theory outlines the conditions necessary for energy transfer to occur, including fluorescent light production by the donor, overlap of the donor's fluorescence emission spectrum and the acceptor's UV absorbance spectrum, and a distance of approach between donor and acceptor of less than 7 nm. Equations (7-9) can be used with FRET to calculate the distance between drugs and HSA.
The results of calculations are presented in Table 2. The parameters are corresponded to the literature data for HSA−CF and BSA−CF [29,43]. We found that ternary systems have higher efficiency of energy transfer than binary system. The probability of energy transfer from HSA to CF is high, as the distance between them was less than 7 nm and between 0.5R0 and 1.5R0.
3.3. The Analysis of HSA’s Secondary Structure
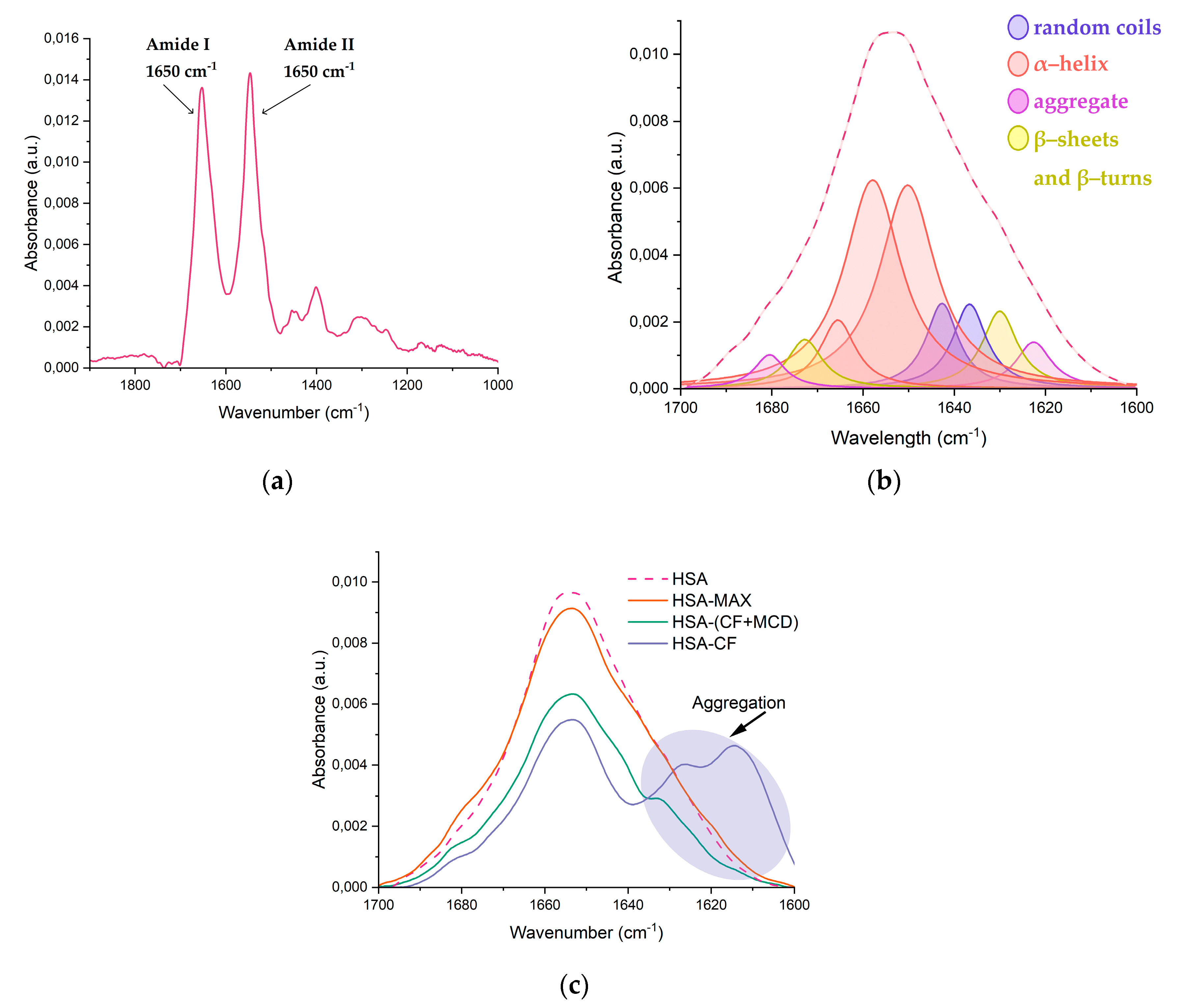
Several studies have reported that the albumin's secondary structure undergoes changes upon the formation of the drug-HSA complex [19,35,42,44,45]. Our research involved the use of FTIR [46,47,48] and circular dichroism spectroscopy [35,49,50,51] to investigate the changes in the albumin’s secondary structure content upon the formation of drug form-HSA complexes. We focused on the Amide I and Amide II bands in the FTIR spectra, which are sensitive to changes in the protein's secondary structure. The Amide I band (1600-1700 cm-1) represents the oscillation of ν(С=О)~80% and ν(C-N)~15%, while the Amide II band (1500-1600 cm-1) corresponds to the δ(N-H)~60%, ν(С-N)~20%, and (C-C)~10% (Figure 6a). By the deconvolution of Amide I band, we determined the content of α-helix, β-structures, and random coils, providing important insights into changes in HSA's secondary structure (Figure 6b).
Our findings confirmed that native HSA contains approximately 65% α-helix [18,34,38,47,52]. The presence of MCD does not affect HSA's secondary structure content, while CF causes a noticeable decrease in α-helix by 6-10%, primarily due to an increase in β-structures and random coils that was reported for other drug-HSA systems [47,52,53]. We observed similar changes in HSA's structure in case of CF+MCD, although the effect was less pronounced for the ternary system. MCD slightly altered CF-HSA binding, possibly by preventing CF from binding to HSA. The alterations in HSA's secondary structure for CF+MCD was similar to the binary system, but less pronounced, so indeed MCD and HSA concur for CF binding. For HSA-MAX system the secondary structure of protein changes insignificantly. Our previous works [37,54] demonstrated that the complex formation between fluoroquinolone drugs and CD’s might be accompanied by the increasing role of hydrogen bond formation and decreasing the force of hydrophobic associations [17,35,38]. So, we might suggest that CF+MCD and MAX interact mainly with the surface of the protein due to the formation of hydrogen interactions, therefore, in these systems, changes in the secondary structure are minimal (Table 3).
Protein aggregation plays a key role in the pathophysiology of many diseases and may have serious consequences [55,56]. In FTIR spectra of proteins characteristic peaks could be observed in region 1614−1618, 1622−1626, and 1685−1696 cm−1 which correspond to anti-parallel β-layers and which are sufficiently distant from the peaks of parallel β-layers and from other elements of the secondary structure [57]. These peaks were not considered in calculating the percentages in Table 3 as aggregation refers to intermolecular contacts. We consider how CF and CF incapsulated into CD-carries would affect the process of aggregation (Figure 6c). When CF was added to HSA the intensity of 1626 and 1616 cm−1 increased. So, it might be concluded that high CF’s concentrations leads to aggregation of HSA. Surprisingly, we observe much less expressed aggregation of protein in HSA−(CF+MCD) (Figure 6c). We suppose MCD makes the surface of protein more hydrophilic, that prevent the albumin’s aggregation. In HSA−MAX system protein aggregation almost does not occur even with the large CF’s molar excesses. As MAX could form more hydrophilic surface of the protein than MCD the effect of preventing aggregation is more significant. These observations let us to suggest that CD’s carries might significantly decrease adverse events associated with CF.
Circular dichroism (CD) was also used to study the changes in the secondary structure of HSA that occur during the interaction of albumin with CF and their complexes with CD. In the range 200-260 nm neither CF nor MCD have a CD spectrum, which makes it possible to study complex multicomponent system. Since α-helices are mainly present in the secondary structure of albumin, as expected, the minimum ellipticity values in the CD spectrum are observed at 208 nm and 220 nm, corresponding to n−π* transitions of the peptide bond in α-helices [39] (Figure 7). The proportion of α helices in free HSA at pH = 7.4 is about 64%, which is consistent with the literature data and data obtained from FTIR (Table 3). The interaction of protein with CF’s forms leads to changes in the secondary structure of the protein, and the carrier for CF has a significant impact on these changes. Circular dichroism spectroscopy confirmed the structural changes observed in HSA obtained by FTIR-spectroscopy.
3.4. Study of the Kinetics of Ciprofloxacin Release
The method of equilibrium dialysis was used as a model system for studying the effect of complexation on the drug’s kinetic release, since the parameter is crucial for discovering the trend to prolonged drug release. Drug’s complexation with CD might slightly affect its release. For instance, in [11] the formation of CF complexes with β-CD derivatives leads to a slowdown of drug’s release that correlates with the dissociation constants of the complexes. In this work, we studied not only the CD carrier’s effect on CF’s release, but also the role of HSA’s presence (Figure 8). The dialysis membrane’s pore diameter was chosen 3.5 kDa, so only CF can be released.
As was expected the formation of the CF+MCD complex leads to a slowdown in the release of CF by ~15% (according to the tangent of the initial section) compared to free CF (Figure 8a). It is important to note that in [11] the drug’s release from the complex at pH 4.0 (when complex is more stable) leads to more significant decrease of the CF’s release rate.
Surprisingly, there was no significant change in the CF’s release profile from MAX within the first 20 minutes compared to free CF. But after 40 minutes we observed the noticeable decrease of release rate from MAX. The effect might be explained by the fast release of CF molecules located near MAX surface and then slower release of CFs encapsulated in the inner MAX volume. Thus, MAX demonstrates the prolonged drug release.
Let us consider how HSA affects CF’s release profile. We used tenfold excess of HSA to modelling in vitro the physiological conditions where the drug surrounded by many molecules of HSA. For HSA−CF system there is a noticeable decrease in the release rate by ~ 56% in 60 min. 100% of CF releases at 3 h that is in 2.5 times longer than in case of free CF. A significant decrease in the release rate might be due to the binding of CF in a complex with HSA.
In case of CF+MCD complex (ratio 1 : 1) preincubated with HSA we observed the dramatic decrease in the CF’s release rate (by ~ 90% in 1 h), whereas the plato is reached at 220 minutes (2.75 times longer than free CF) (Figure 8b). The additional decrease in the release rate of ~20% in the presence of MCD is probably due to competition of MCD and HSA for CF’s binding.
Figure 8.
(a) Free CF, CF complex with MCD, MAX release curves at pH 7.4; (b) Free CF (black), HSA with a tenfold molar excess of free CF (purple); HSA with a tenfold molar excess of CF complex with MCD (in a ratio of 1:1) (green); HSA with a tenfold molar excess of MAX (orange), 37°C, pH 7.4.
Figure 8.
(a) Free CF, CF complex with MCD, MAX release curves at pH 7.4; (b) Free CF (black), HSA with a tenfold molar excess of free CF (purple); HSA with a tenfold molar excess of CF complex with MCD (in a ratio of 1:1) (green); HSA with a tenfold molar excess of MAX (orange), 37°C, pH 7.4.
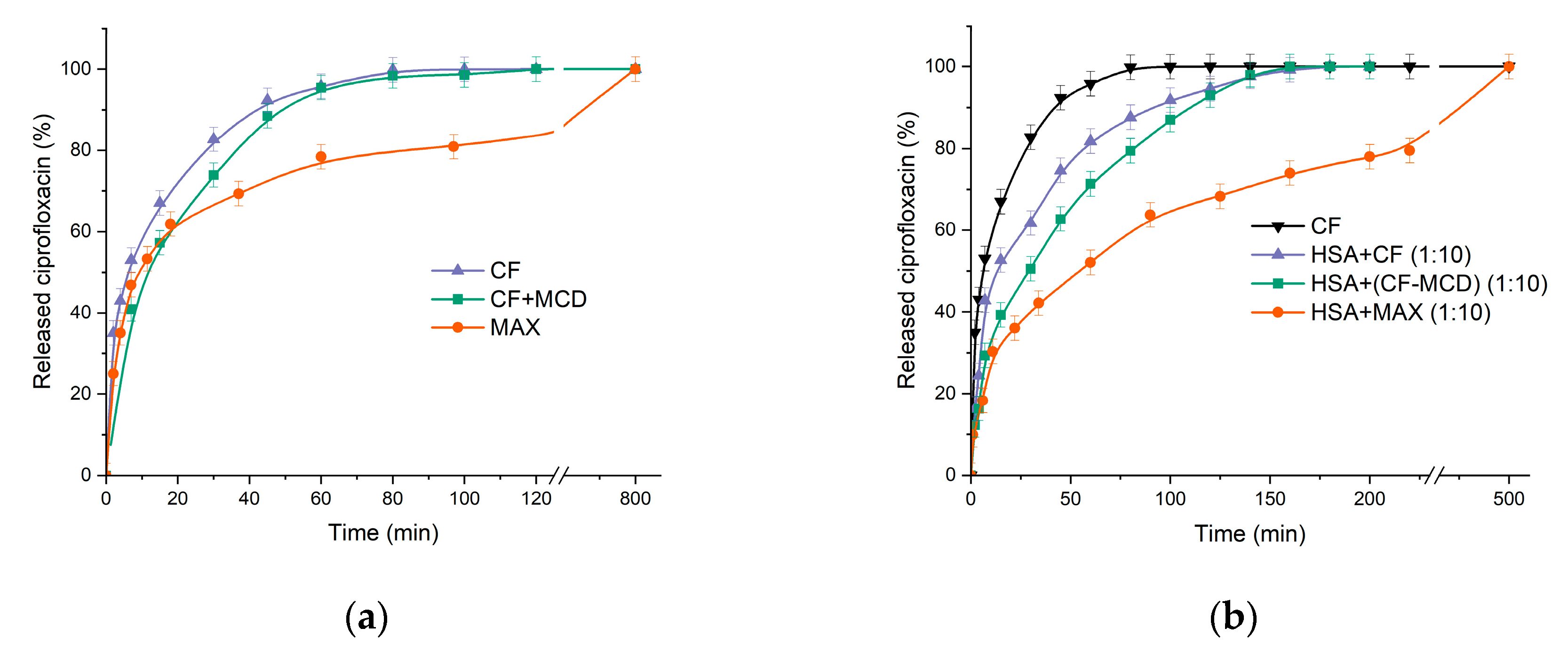
The lowest release rate occurs in the HSA−MAX system. In 3 h only 70% of CF is released. The data obtained allow us to suggest that CF in MCD carries would a long while circulate in the blood flow and transported by protein for a longer time. Slow-release CF from MAX and formation the protein crown may change the antibacterial properties of CF in MAX.
3.5. Antibacterial Activity of CF’s Drug forms in the Presence of HSA
We used gram-positive Bacillus subtilis and gram-negative Escherichia coli to investigate the effect of complex formation between HSA and CF in different forms on minimum inhibition concentration (MIC) by the rapid and reliable agar–well diffusion test. First, we compared the action of free CF, CF+MCD, and MAX: MICCF = MICCF+MCD = 0.02 μg/mL (E. coli) and 0.14 μg/mL (B. subtilis), whereas MICMAX = 0.06 and 0.2 μg/mL for E. coli and B. subtilis respectively. We suggested that the increase of CF’s MIC value for MAX is due to the limited agar diffusion. In addition, the lower CF’s release rate from MAX might contribute to the effect. We also found that incubation CF’s forms with HSA does not influence on MIC (Table 5). The same results were obtained earlier in our research group [54].
4. Conclusions
For the first time the MCD based polymer with encapsulated CF was obtained via template synthesis. We compared MAX to the common CF+MCD non–covalent complex in their binding to HSA, release kinetics and antibacterial activity. MCD prevents CF’s binding to albumin probably due to the competition with HSA for drug binding, whereas for MAX the increase of Ka (~2.5 times) is observed. This effect might be explained by the formation of hydrogen bonds between MAX and HSA that limits CF’s dissociation from protein’s binding site. This effect might contribute to the pronounced decrease of CF’s release rate from HSA−MAX (100% is achieved at 500 min) that might consequently enhance the drug’s lifetime in vivo and prolong the therapeutic effect. Also, preventing the HSA's aggregation during formation complex with CF was achieved in ternary systems. MCD does not change CF’s in vitro activity, whereas MAX’s MIC values for E. coli and B. subtilis are 0.06 and 0.20 μg/mL, correspondingly, which is ~2.5−3 times higher than for free СF.
Although only 0.6 mg of encapsulated CF was per 1 mg of MAX, we suppose the proposed system is still more promising compared to MCD due to prolonged drug release, the increase of CF’s binding efficiency to HSA and satisfied in vitro activity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization: A.A.S. and E.V.K.; experimental work: T.Yu.K.; methodology of microbiological experiments, N.G.B; data analysis and interpretation: T.Yu.K.; writing—original draft preparation: T.Yu.K.; writing—review and editing: A.A.S. and E.V.K..; supervision: A.A.S. and E.V.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Science Foundation grant number 22-24-00604.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its Supplementary Information.
Acknowledgments
The work was performed using equipment (FTIR spectrometer Bruker Tensor 27 and Jasco J-815 CD Spectrometer AFM microscope NTEGRA II) of the program for the development of Moscow State University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Talebi Bezmin Abadi, A.; Rizvanov, A.A.; Haertlé, T.; Blatt, N.L. World Health Organization Report: Current Crisis of Antibiotic Resistance. Bionanoscience BioNanoScience, 2019, 9, 778–788. [Google Scholar] [CrossRef]
- Mahady, G. Medicinal Plants for the Prevention and Treatment of Bacterial Infections. Curr. Pharm. Des. 2005, 11, 2405–2427. [Google Scholar] [CrossRef]
- Chong, W.H.; Saha, B.K.; Ramani, A.; Chopra, A. State-of-the-art review of secondary pulmonary infections in patients with COVID-19 pneumonia. Infection 2021, 49, 591–605. [Google Scholar] [CrossRef]
- Tiwari, G. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef]
- Bennett, A.C.; Bennett, C.L.; Witherspoon, B.J.; Knopf, K.B. An evaluation of reports of ciprofloxacin, levofloxacin, and moxifloxacin-association neuropsychiatric toxicities, long-term disability, and aortic aneurysms/dissections disseminated by the Food and Drug Administration and the European Medicines Agency. Expert Opin. Drug Saf. 2019, 18, 1055–1063. [Google Scholar] [CrossRef]
- Alhajj, N.; O’Reilly, N.J.; Cathcart, H. Developing ciprofloxacin dry powder for inhalation: A story of challenges and rational design in the treatment of cystic fibrosis lung infection. Int. J. Pharm. 2022, 613, 121388. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, J.S.; Hooper, D.C. Fluoroquinolone antimicrobial agents. Clin. Microbiol. Rev. / ed. Oates J.A., Wood A.J.J. 1989, 2, 378–424. [Google Scholar] [CrossRef] [PubMed]
- Yayehrad, A.T.; Wondie, G.B.; Marew, T. Different Nanotechnology Approaches for Ciprofloxacin Delivery Against Multidrug-Resistant Microbes. Infect. Drug Resist. 2022, 15, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Schacht, P.; Arcieri, G.; Hullmann, R. Safety of oral ciprofloxacin. Am. J. Med. 1989, 87, S98–S102. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Skuredina, A.A.; Kopnova, T.Y.; Le-deygen, I.M.; Kudryashova, E. V. Physical and Chemical Properties of the Guest – Host Inclusion Complexes of Cyprofloxacin with β -Cyclodextrin Derivatives. Moscow Univ. Chem. Bull. 2020, 75, 218–224. [Google Scholar] [CrossRef]
- Chen, G.; Jiang, M. Cyclodextrin-based inclusion complexation bridging supramolecular chemistry and macromolecular self-assembly. Chem. Soc. Rev. 2011, 40, 2254. [Google Scholar] [CrossRef] [PubMed]
- Hishiya, T.; Asanuma, H.; Komiyama, M. Spectroscopic Anatomy of Molecular-Imprinting of Cyclodextrin. Evidence for Preferential Formation of Ordered Cyclodextrin Assemblies. J. Am. Chem. Soc. 2002, 124, 570–575. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Tychinina, A.S.; Le-Deygen, I.M.; Golyshev, S.A.; Belogurova, N.G.; Kudryashova, E. V. The formation of quasi-regular polymeric network of cross-linked sulfobutyl ether derivative of β-cyclodextrin synthesized with moxifloxacin as a template. React. Funct. Polym. Elsevier B.V., 2021, 159, 104811. [Google Scholar] [CrossRef]
- Egawa, Y.; Shimura, Y.; Nowatari, Y.; Aiba, D.; Juni, K. Preparation of molecularly imprinted cyclodextrin microspheres. Int. J. Pharm. 2005, 293, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Trotta, F. Molecularly imprinted cyclodextrin nanosponges for the controlled delivery of L-DOPA: perspectives for the treatment of Parkinson’s disease. Expert Opin. Drug Deliv. 2016, 13, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-W.; Wang, K.; Zhang, X.-X. Study of the interactions between fluoroquinolones and human serum albumin by affinity capillary electrophoresis and fluorescence method. Anal. Chim. Acta 2007, 603, 101–110. [Google Scholar] [CrossRef]
- McCarty, T.A.; Page, P.M.; Baker, G.A.; Bright, F. V. Behavior of Acrylodan-Labeled Human Serum Albumin Dissolved in Ionic Liquids. Ind. Eng. Chem. Res. 2008, 47, 560–569. [Google Scholar] [CrossRef]
- Vlasova, I.M.; Bukharova, E.M.; Kuleshova, A.A.; Saletsky, A.M. Spectroscopic investigations of interaction of fluorescent nanomarkers of fluorescein family with human serum albumin at different values of pH. Curr. Appl. Phys. Elsevier B.V, 2011, 11, 1126–1132. [Google Scholar] [CrossRef]
- Rehman, M.T.; Shamsi, H.; Khan, A.U. Insight into the binding mechanism of imipenem to human serum albumin by spectroscopic and computational approaches. Mol. Pharm. 2014, 11, 1785–1797. [Google Scholar] [CrossRef]
- Astray, G.; Mejuto, J.C.; Morales, J.; Rial-Otero, R.; Simal-Gándara, J. Factors controlling flavors binding constants to cyclodextrins and their applications in foods. Food Res. Int. 2010, 43, 1212–1218. [Google Scholar] [CrossRef]
- Zlotnikov, I.D.; Belogurova, N.G.; Krylov, S.S.; Semenova, M.N.; Semenov, V. V.; Kudryashova, E. V. Plant Alkylbenzenes and Terpenoids in the Form of Cyclodextrin Inclusion Complexes as Antibacterial Agents and Levofloxacin Synergists. Pharmaceuticals 2022, 15, 861. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Wang, Q.; Shi, Z.-H.; Xia, X.-H.; Sun, H.-W. Interaction Characteristic Studies of Ciprofloxacin and/or Sulphadiazine with Bovine Serum Albumin by Spectroscopic Technique. Asian J. Chem. 2015, 27, 818–826. [Google Scholar] [CrossRef]
- Sainz-Rozas, P.R.; Isasi, J.R.; González-Gaitano, G. Binding of dibenzofuran and its derivatives to water-soluble β-cyclodextrin polymers. J. Photochem. Photobiol. A Chem. 2005, 173, 248–257. [Google Scholar] [CrossRef]
- Argenziano, M. In Vitro Enhanced Skin Permeation and Retention of Imiquimod Loaded in β-Cyclodextrin Nanosponge Hydrogel. Pharmaceutics 2019, 11, 138. [Google Scholar] [CrossRef]
- Skuredina, A.A. Cyclodextrins and Their Polymers Affect the Lipid Membrane Permeability and Increase Levofloxacin’s Antibacterial Activity In Vitro. Polymers (Basel). 2022, 14, 4476. [Google Scholar] [CrossRef]
- Skuredina, A.A. The New Strategy for Studying Drug-Delivery Systems with Prolonged Release: Seven-Day In Vitro Antibacterial Action. Molecules 2022, 27, 8026. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. Elsevier Ltd, 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Iranfar, H.; Rajabi, O.; Salari, R.; Chamani, J. Probing the Interaction of Human Serum Albumin with Ciprofloxacin in the Presence of Silver Nanoparticles of Three Sizes: Multispectroscopic and ζ Potential Investigation. J. Phys. Chem. B 2012, 116, 1951–1964. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Wang, Y.-Q.; Jiang, M.-L. A fluorimetric study of the interaction of C.I. Solvent Red 24 with haemoglobin. Dye. Pigment. 2009, 82, 156–163. [Google Scholar] [CrossRef]
- Wei, Y.; Li, J.; Dong, C.; Shuang, S.; Liu, D.; Huie, C.W. Investigation of the association behaviors between biliverdin and bovine serum albumin by fluorescence spectroscopy. Talanta 2006, 70, 377–382. [Google Scholar] [CrossRef]
- Kumar, P.V.; Jain, N.K. Suppression of agglomeration of ciprofloxacin-loaded human serum albumin nanoparticles. AAPS PharmSciTech 2007, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Fick, A.C.; Reinscheid, U.M. Characterization of the binding epitope of ciprofloxacin bound to human serum albumin. J. Pharm. Biomed. Anal. 2006, 41, 1025–1028. [Google Scholar] [CrossRef]
- Ahmad, B.; Parveen, S.; Khan, R.H. Effect of Albumin Conformation on the Binding of Ciprofloxacin to Human Serum Albumin: A Novel Approach Directly Assigning Binding Site. Biomacromolecules 2006, 7, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Seedher, N.; Agarwal, P. Complexation of fluoroquinolone antibiotics with human serum albumin: A fluorescence quenching study. J. Lumin. North-Holland, 2010, 130, 1841–1848. [Google Scholar] [CrossRef]
- Yakupova, L.R.; Kopnova, T.Y.; Skuredina, A.A.; Kudryashova, E. V. Effect of Methyl-β-Cyclodextrin on the Interaction of Fluoroquinolones with Human Serum Albumin. Russ. J. Bioorganic Chem. 2022, 48, 163–172. [Google Scholar] [CrossRef]
- Yakupova, L.R. The Formation of β-Cyclodextrin Complexes with Levofloxacin and Ceftriaxone as an Approach to the Regulation of Drugs’ Pharmacokinetic. Colloid J. 2023, 85, 114–127. [Google Scholar] [CrossRef]
- Seedher, N.; Agarwal, P. Competitive binding of fluoroquinolone antibiotics and some other drugs to human serum albumin: a luminescence spectroscopic study. Luminescence 2013, 28, 562–568. [Google Scholar] [CrossRef]
- Varshney, A. Analysis of Binding Interaction Between Antibacterial Ciprofloxacin and Human Serum Albumin by Spectroscopic Techniques. Cell Biochem. Biophys. 2014, 70, 93–101. [Google Scholar] [CrossRef]
- Kaur, A.; Khan, I.A.; Banipal, P.K.; Banipal, T.S. Deciphering the complexation process of a fluoroquinolone antibiotic, levofloxacin, with bovine serum albumin in the presence of additives. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. Elsevier B.V., 2018, 191, 259–270. [Google Scholar] [CrossRef]
- Paul, B.K.; Guchhait, N.; Bhattacharya, S.C. Binding of ciprofloxacin to bovine serum albumin: Photophysical and thermodynamic aspects. J. Photochem. Photobiol. B Biol. 2017, 172, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.K.; Ghosh, N.; Mukherjee, S. Interplay of Multiple Interaction Forces: Binding of Norfloxacin to Human Serum Albumin. J. Phys. Chem. B 2015, 119, 13093–13102. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.J.; Ou-Yang, Y.; Zhang, Y.; Liu, Y. Affinity and specificity of ciprofloxacin-bovine serum albumin interactions: Spectroscopic approach. Protein J. 2010, 29, 234–241. [Google Scholar] [CrossRef]
- Abu, T.M.M.; Ghithan, J.; Abu-Taha, M.I.; Darwish, S.M.; Abu-hadid, M.M. Spectroscopic approach of the interaction study of ceftriaxone and human serum albumin. J. Biophys. Struct. Biol. 2014, 6, 1–12. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Hirano, A.; Arakawa, T.; Shiraki, K. Effects of alcohol on the solubility and structure of native and disulfide-modified bovine serum albumin. Int. J. Biol. Macromol. Elsevier B.V., 2012, 50, 1286–1291. [Google Scholar] [CrossRef]
- Tatulian, S.A. Structural Characterization of Membrane Proteins and Peptides by FTIR and ATR-FTIR Spectroscopy 2013, 177–218.
- Poureshghi, F.; Ghandforoushan, P.; Safarnejad, A.; Soltani, S. Interaction of an antiepileptic drug, lamotrigine with human serum albumin (HSA): Application of spectroscopic techniques and molecular modeling methods. J. Photochem. Photobiol. B Biol. Elsevier B.V., 2017, 166, 187–192. [Google Scholar] [CrossRef]
- Tretiakova, D.; Le-Deigen, I.; Onishchenko, N.; Kuntsche, J.; Kudryashova, E.; Vodovozova, E. Phosphatidylinositol Stabilizes Fluid-Phase Liposomes Loaded with a Melphalan Lipophilic Prodrug. Pharmaceutics 2021, 13, 473. [Google Scholar] [CrossRef]
- Yan, J.; Wu, D.; Ma, X.; Wang, L.; Xu, K.; Li, H. Spectral and molecular modeling studies on the influence of β-cyclodextrin and its derivatives on aripiprazole-human serum albumin binding. Carbohydr. Polym. 2015, 131, 65–74. [Google Scholar] [CrossRef]
- Paul, B.K.; Guchhait, N. A spectral deciphering of the binding interaction of an intramolecular charge transfer fluorescence probe with a cationic protein: Thermodynamic analysis of the binding phenomenon combined with blind docking study. Photochem. Photobiol. Sci. 2011, 10, 980–991. [Google Scholar] [CrossRef]
- Dockal, M.; Carter, D.C.; Rüker, F. Conformational Transitions of the Three Recombinant Domains of Human Serum Albumin Depending on pH. J. Biol. Chem. 2000, 275, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Abu, T.M.M.; Ghithan, J.; Abu-Taha, M.I.; Darwish, S.M.; Abu-hadid, M.M. Spectroscopic approach of the interaction study of ceftriaxone and human serum albumin. J. Biophys. Struct. Biol. 2014, 6, 1–12. [Google Scholar] [CrossRef]
- Trynda-Lemiesz, L. Paclitaxel-HSA interaction. Binding sites on HSA molecule. Bioorganic Med. Chem. 2004, 12, 3269–3275. [Google Scholar] [CrossRef]
- Skuredina, A.A.; Yakupova, L.R.; Kopnova, T.Y.; Le-Deygen, I.M.; Belogurova, N.G.; Kudryashova, E. V. Cyclodextrins and Their Polymers Affect Human Serum Albumin’s Interaction with Drugs Used in the Treatment of Pulmonary Infections. Pharmaceutics 2023, 15, 1598. [Google Scholar] [CrossRef]
- Rostagno, A.; Ghiso, J.A. Misfolding, Aggregation, and Amyloid Formation: The Dark Side of Proteins. Protein Fold. Disord. Cent. Nerv. Syst. WORLD SCIENTIFIC, 2017, 1–31.
- Akter, R. Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res. 2016, 2016, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, E. V. Reversible self-association of ovalbumin at air-water interfaces and the consequences for the exerted surface pressure. Protein Sci. 2005, 14, 483–493. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
CF’s and MCD chemical structures and the schematic representation of MAX synthesis.
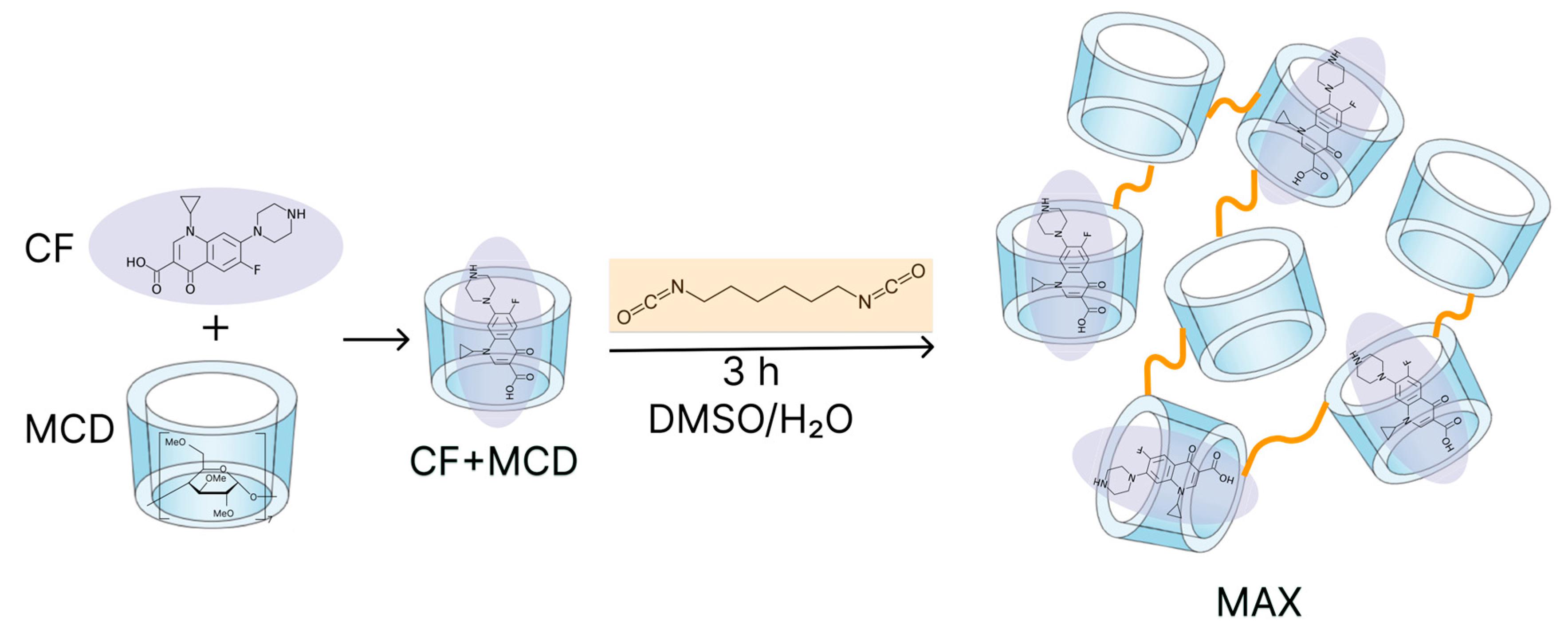
Figure 2.
(a) Normalized emission spectra of HSA-(CF+MCD), λex = 280 nm, CHSA = 0.02 mM, CCF+MCD = 0 – 0.02 mM, 37℃, PBS, pH 7.4; (b) The intensity of HSA’s peak via the increase of drug’s concentration, λex = 280 nm, CHSA = 0.02 mM, pH 7.4, 37 °C.
Figure 2.
(a) Normalized emission spectra of HSA-(CF+MCD), λex = 280 nm, CHSA = 0.02 mM, CCF+MCD = 0 – 0.02 mM, 37℃, PBS, pH 7.4; (b) The intensity of HSA’s peak via the increase of drug’s concentration, λex = 280 nm, CHSA = 0.02 mM, pH 7.4, 37 °C.
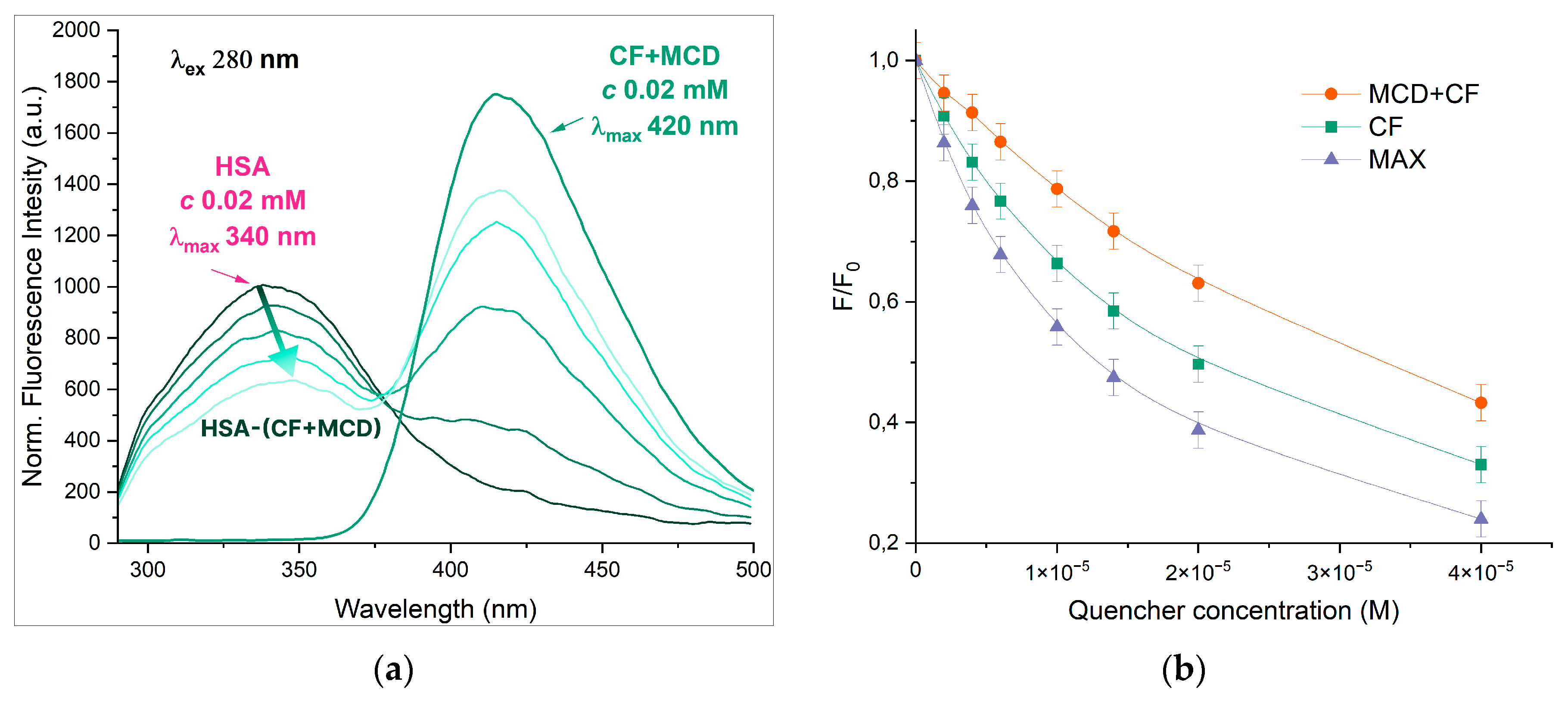
Figure 3.
(a) Stern-Vollmer dependences expressing the effect of HSA quenching via binding CF’s forms F0/F, C(HSA) = 0.02 mM, 37°C, pH 7.4. (b) Plot versus for HSA-drugs, C(HSA) = 0.02 mM, 37°C, pH 7.4.
Figure 3.
(a) Stern-Vollmer dependences expressing the effect of HSA quenching via binding CF’s forms F0/F, C(HSA) = 0.02 mM, 37°C, pH 7.4. (b) Plot versus for HSA-drugs, C(HSA) = 0.02 mM, 37°C, pH 7.4.
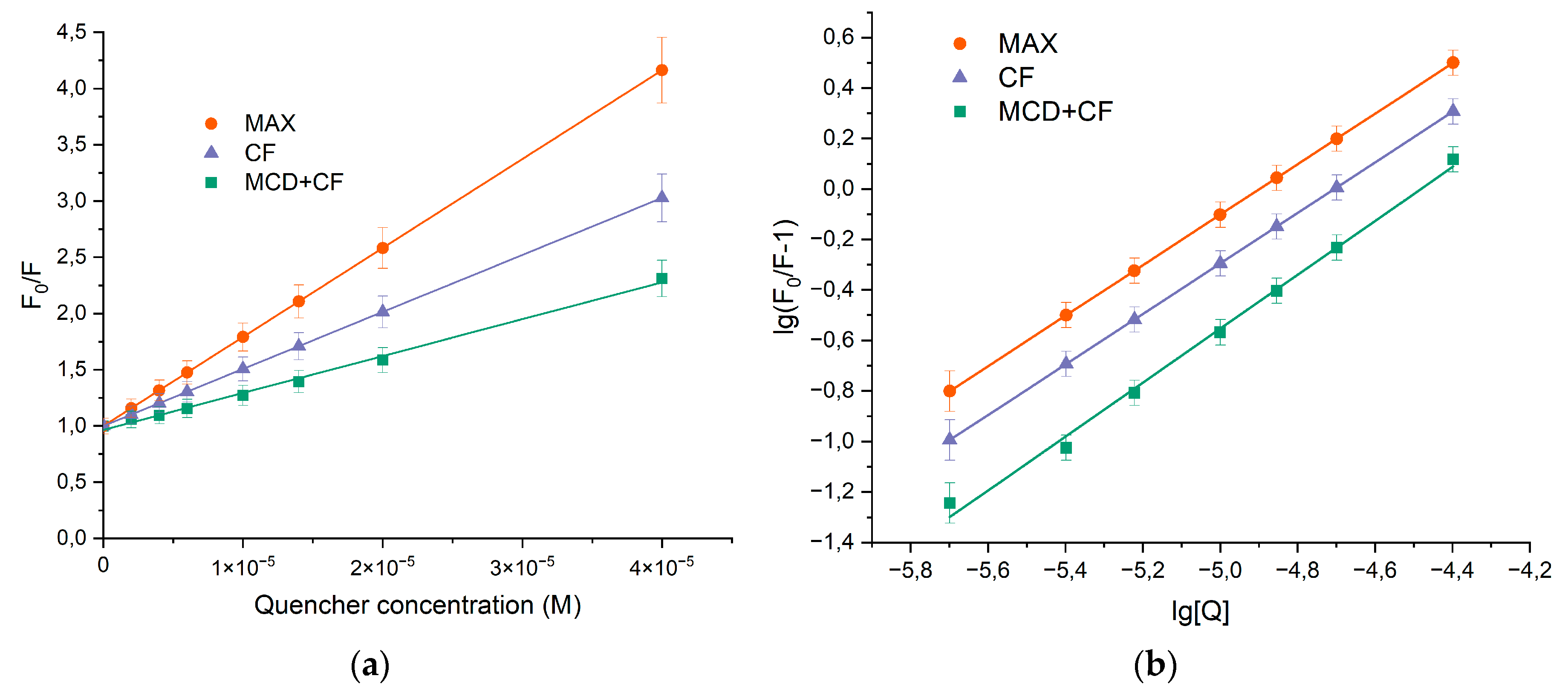
Figure 4.
Anisotropy changes as HSA was added to CF’s (purple) and (CF+MCD)’s (green) solution, PBS, 37℃, CCF = C(CF+MCD) = 0.01 mM.
Figure 4.
Anisotropy changes as HSA was added to CF’s (purple) and (CF+MCD)’s (green) solution, PBS, 37℃, CCF = C(CF+MCD) = 0.01 mM.
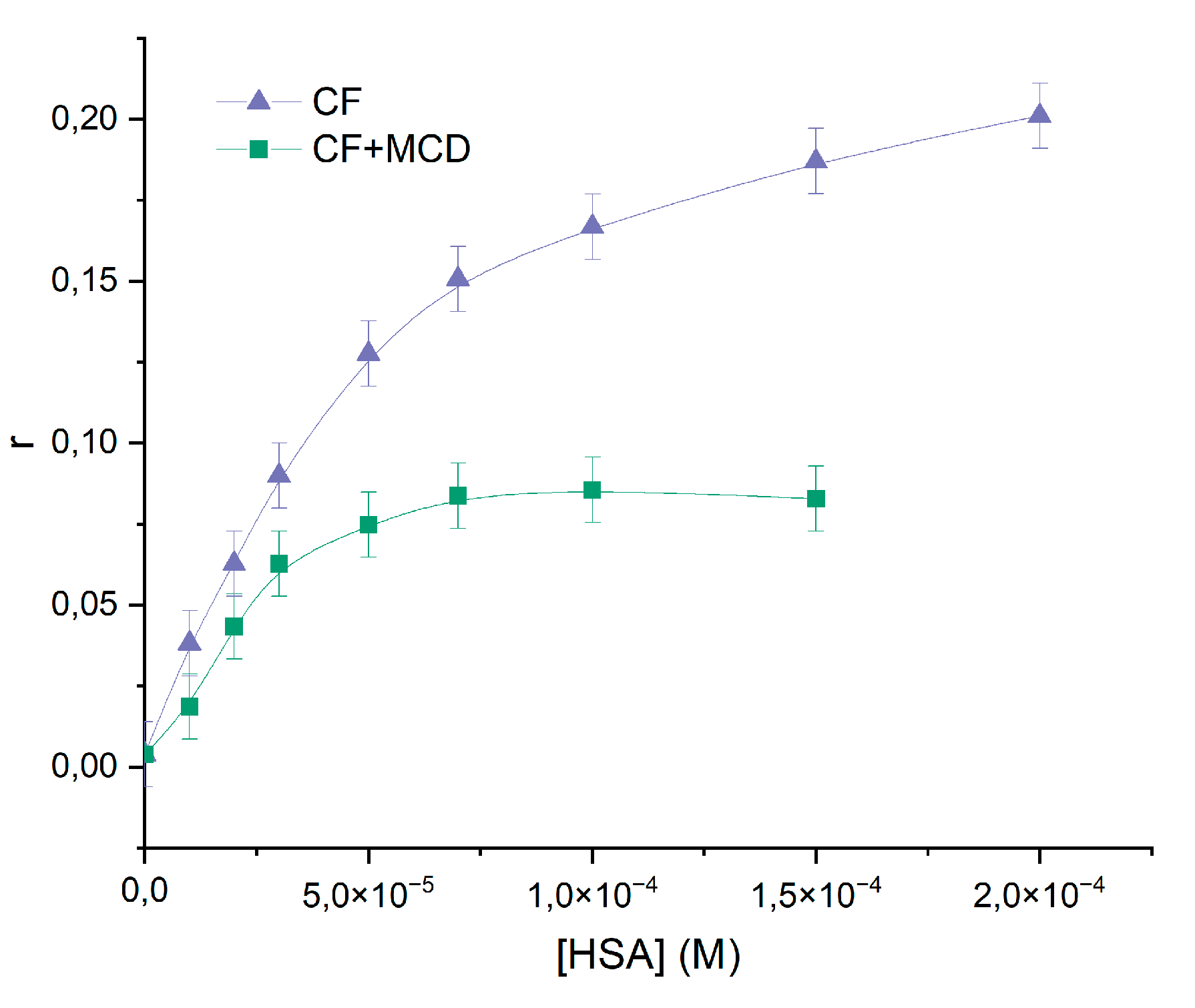
Figure 5.
Normalized overlap of the CF absorption spectra with the fluorescence spectra of HSA at 25℃, both in PBS pH 7.4.
Figure 5.
Normalized overlap of the CF absorption spectra with the fluorescence spectra of HSA at 25℃, both in PBS pH 7.4.
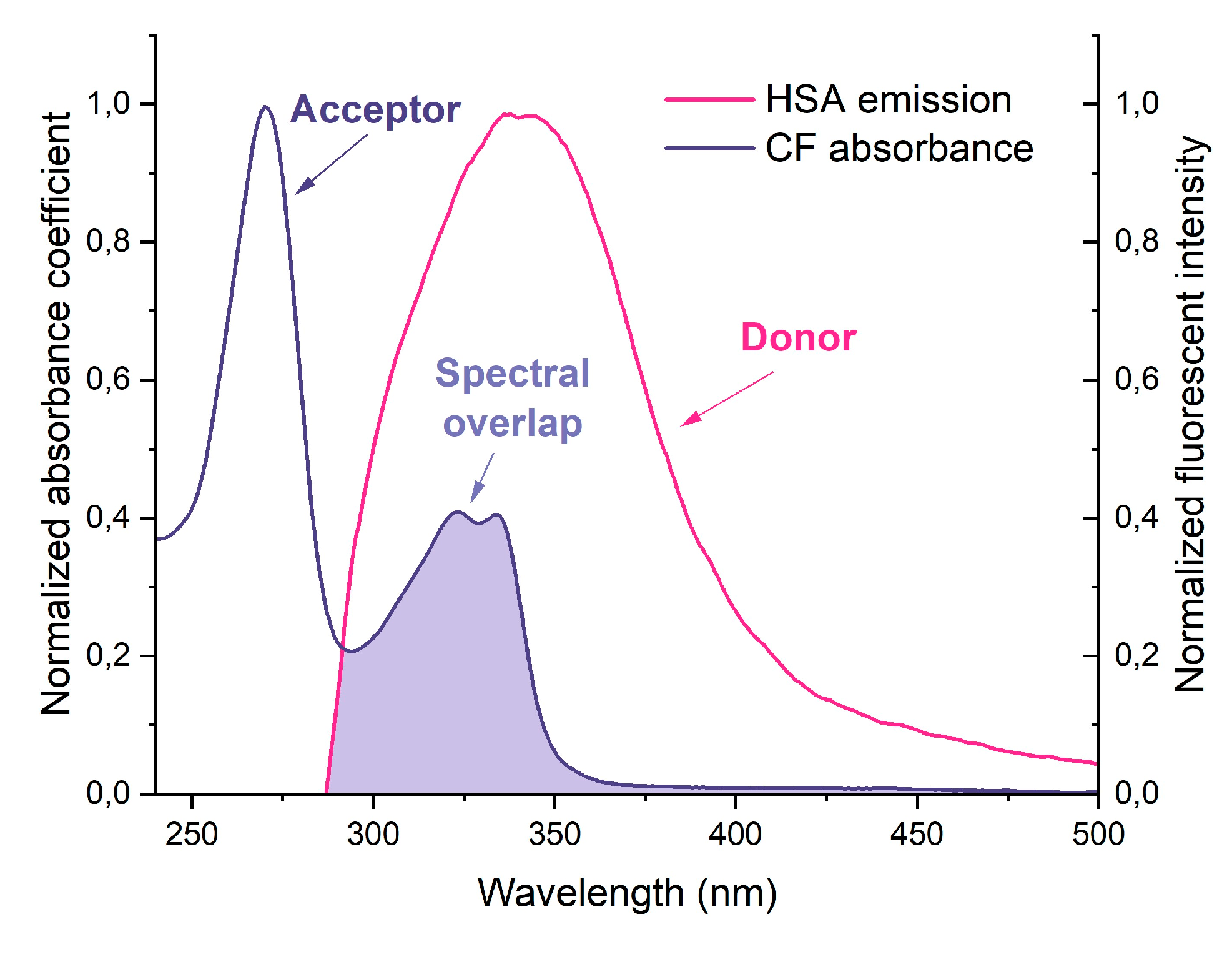
Figure 6.
(a) The FTIR spectra of HSA, pH 7.4; (b) The deconvolution of HSA FTIR spectra, CHSA = 0.06 mM, the simulated spectra demonstrated in dashed pink line, pH 7.4; (c) Changes in the structure of Amide I , CHSA = 0.06 mM, CCF = 1.2 mM, pH 7.4.
Figure 6.
(a) The FTIR spectra of HSA, pH 7.4; (b) The deconvolution of HSA FTIR spectra, CHSA = 0.06 mM, the simulated spectra demonstrated in dashed pink line, pH 7.4; (c) Changes in the structure of Amide I , CHSA = 0.06 mM, CCF = 1.2 mM, pH 7.4.
| α-helix | β-structures | Random | |
|---|---|---|---|
| HSA | 64 ± 2% | 19 ± 1% | 17 ± 1% |
| HSA-CF | 58 ± 2% | 22 ± 1% | 20 ± 1% |
| HSA-(CF+MCD) | 61 ± 2% | 21 ± 1% | 18 ± 1% |
| HSA-MAX | 62 ± 2% | 22 ± 1% | 16 ± 1% |
Figure 7.
(a) Circular dichroism spectrum of HSA, C = 0.05 mM, pH 7.4; (b) The content of secondary structures in HSA, CHSA = CCF+MCD = CCF = 0.02 mM, pH 7.4.
Figure 7.
(a) Circular dichroism spectrum of HSA, C = 0.05 mM, pH 7.4; (b) The content of secondary structures in HSA, CHSA = CCF+MCD = CCF = 0.02 mM, pH 7.4.
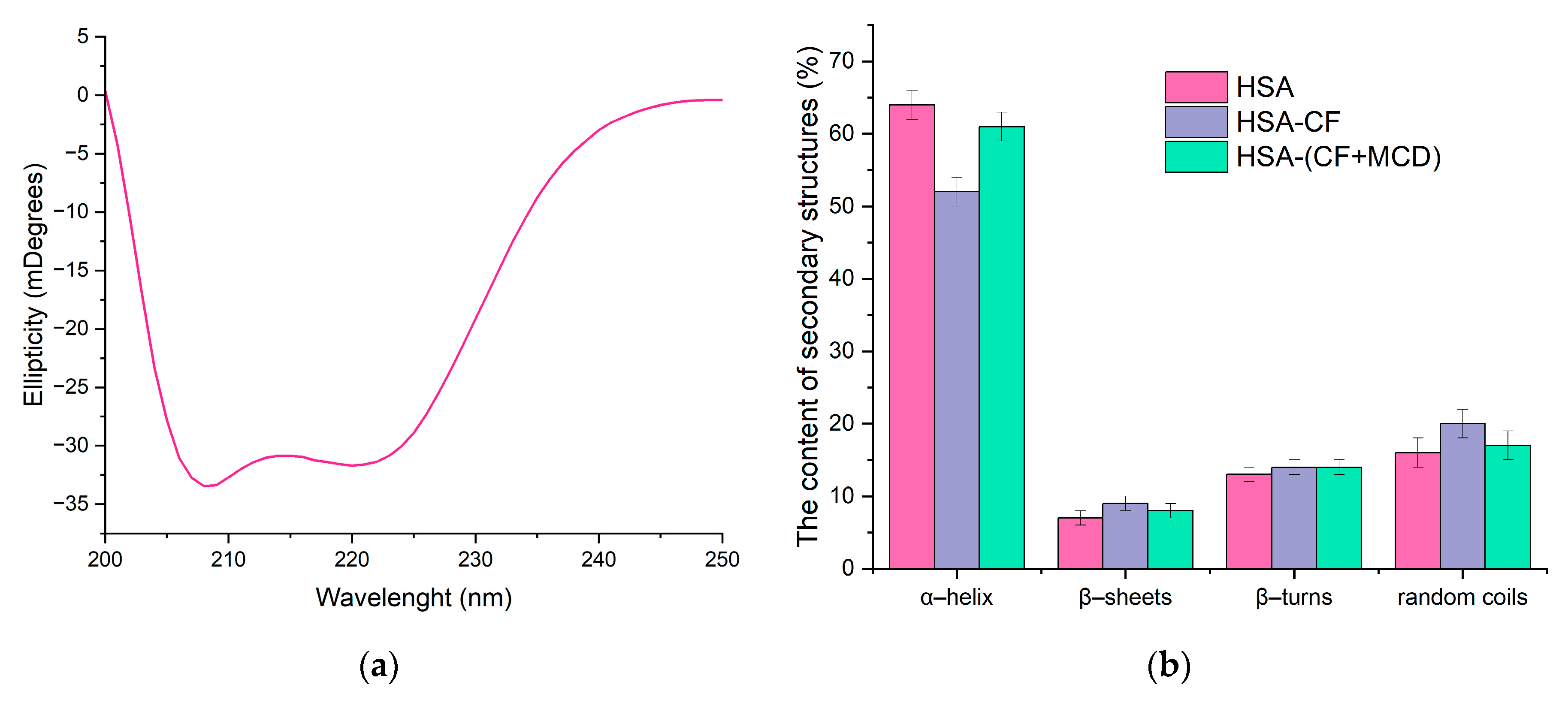

Table 1.
Stern−Volmer Quenching Constants at λex = 280 nm and Binding Parameters for Interaction of drugs with HSA, pH 7.4, CHSA = 0.02 mM.
Table 1.
Stern−Volmer Quenching Constants at λex = 280 nm and Binding Parameters for Interaction of drugs with HSA, pH 7.4, CHSA = 0.02 mM.
| Parameter | HSA-CF | HSA-(CF-MCD) | HSA-MAX |
|---|---|---|---|
| , M−1 | 51 ± 2 | 32.8 ± 0.8 | 79 ± 3 |
| , M-1·s-1 | 8.9 ± 0.4 | 5.75 ± 0.16 | 13.8 ± 0.6 |
| , M−1 | 10 ± 3 | 6 ± 1 | 26 ± 6 |
| n | 1,06 ± 0,06 | 1,07 ± 0,03 | 1,10 ± 0,04 |
Table 2.
Energy transfer parameters for the interactions of the drugs with human serum albumin (25℃, PBS, pH 7.4).
Table 2.
Energy transfer parameters for the interactions of the drugs with human serum albumin (25℃, PBS, pH 7.4).
| Parameter | HSA-CF | HSA-(CF-MCD) | HSA-MAX |
|---|---|---|---|
| , M−1cm3 | 6.54 | 7.29 | 8.08 |
| R0, Å | 22.9 | 23.3 | 23.7 |
| r, Å | 33.5 | 34.5 | 32.2 |
| EFRET | 0.10 | 0.09 | 0.13 |
Table 3.
The content of secondary structures in HSA, CHSA = CCF = 0.06 mM, pH 7.4, SD (n=3).
| α-helix | β-structures | Random | |
|---|---|---|---|
| HSA | 64 ± 2% | 19 ± 1% | 17 ± 1% |
| HSA-CF | 58 ± 2% | 22 ± 1% | 20 ± 1% |
| HSA-(CF+MCD) | 61 ± 2% | 21 ± 1% | 18 ± 1% |
| HSA-MAX | 62 ± 2% | 22 ± 1% | 16 ± 1% |
Table 4.
The release rate of CF or CF+CD carrier in the absence and in the presence of HSA, represented as slopes of the initial sections of the CF release curves (Figure 8), the relative changes in concentration per minute, pH 7.4, 37 °C.
Table 4.
The release rate of CF or CF+CD carrier in the absence and in the presence of HSA, represented as slopes of the initial sections of the CF release curves (Figure 8), the relative changes in concentration per minute, pH 7.4, 37 °C.
| The presence of HSA | CF | CF+MCD | MAX | |
|---|---|---|---|---|
| tg α1 | − | 4.5 ± 0.2 | 3.8 ± 0.2 | 4.0 ± 0.2 |
| + | 3.5 ± 0.2 | 2.6 ± 0.2 | 2.2 ± 0.2 |
1 first 15 minutes were analyzed.
Table 5.
MIC (µg/mL), pH 7.4 (PBS sterile buffer), agar–well diffusion method, 37 °C.
| Strain | The presence of HSA | CF | CF+MCD | MAX |
|---|---|---|---|---|
|
Escherichia coli ATCC 25922 |
− | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.06 ± 0.01 |
| + | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.07 ± 0.01 | |
|
Bacillus subtitlis ATCC 6633 |
− | 0.12 ± 0.03 | 0.13 ± 0.03 | 0.20 ± 0.03 |
| + | 0.14 ± 0.03 | 0.14 ± 0.03 | 0.21 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



