
Submitted:
12 June 2023
Posted:
12 June 2023
You are already at the latest version
Abstract
Background: Cord blood represents a link between intrauterine and early extrauterine development. Cord blood cells map an important time frame in human immune imprinting processes. It is unknown whether sex of the newborn affects the lymphocyte subpopulations in cord blood. Methods: 9 B and 21 T cell subpopulations were characterized by flow cytometry in human cord blood from 16 male and 21 female newborns, respectively. Results: Except for marginal zone B cells and transitional B cells, B cell count in all subsets was higher in cord blood of male newborns than in female newborns. Frequency of naive thymus negative Th cells was significantly higher in male cord blood whereas the remaining T cell subpopulations showed a higher count in cord blood of female newborns. Conclusion: Our study is the first revealing sex differences in B and T cell subpopulations of human cord blood. These results indicate that sex might have a higher impact for the developing immune system urging the need to expand research in this area.
Keywords:
B cells
; T cells
; B1 cells
; human
; white blood cells
; cord blood
; development
; ontogeny
; sex differences
; gender medicine
1. Introduction
The development of the human immune system begins intrauterine and is not completed at birth.[1] After birth, many processes are initiated, also affecting immune cells in particular due to changed environmental antigen exposure.[2] In the neonatal period, the immune system is immature and certain components of the adaptive immune response are not fully developed yet. Moreover, there are notable differences in the antibody response compared to adults. Although neonates benefit from maternal antibodies providing maternal passive immunity, antibody titers are lower than in adult blood.[3,4] During infancy, the immune system matures. As a continuous learning process, immunity is built up during puberty, also by intercurrent infections and vaccinations. The immune system achieves the similar functional capacities compared to adult mature immune system. [5]
The maturation of immune cells is a complex multi-layered process. Griffin et al. [6] described human B1 cells as CD20+CD27+CD43+ B cell subpopulation based on typical B1 functions in murine B1 cells: spontaneous IgM secretion, efficient T cell stimulation and continuous intracellular signal[6]. B1 cells have a huge percentage in cord blood (CB) (50%) but decrease over the years.[6],[7]
Marginal zone B (MZ B cells) cells are noncirculating mature B cells, forming an interface between blood and lymph nodes.[8],[2] Splenic MZ B cells located in the marginal zone of the spleen are part of the innate immune system responding to blood-borne antigens. Although accounting only for 5% of the innate and adaptive immune system, marginal zone B cells are of utmost importance for early immune response by interacting effectively with rapid antibody reactions on viral and bacterial pathogens.[8]
Thymus negative cells are a premature stage of T helper (Th-) lymphocytes before positive selection in thymus. In the positive selection process, Th cells can bind major histocompatibility complexes (MHC) in order to receive positive growing signals and enter cell cycle. After positive selection process, cells that bind self-antigens are induced to apoptosis.[9] These selection processes take place in the thymus postnatally assuming that maturation of Th cells does not finish by the time of birth.[10]
In this context, viral and microbial colonization by perinatal transmission are important steps in the maturation process of the innate and adaptive immune system. Microbial dysbiosis within the first 100 days of life seems to play an important role, for the susceptibility to develop allergies later in life.[11]
One important and long neglected fact are differences between females and males. Investigating biological (sex) and sociocultural (gender) differences has gained more importance over the past few years. Within the field of medical research, most of the performed researches and published data refers to male individuals. Sex differences have been found in certain diseases like asthma, diabetes and cardiovascular disease. Women are at higher risk of developing diseases like Takotsubo cardiomyopathy or show different manifestations of common diseases like heart failure or myocardial infarction. More over autoimmune diseases as systemic lupus erythematosus (SLE), multiple sclerosis (MS), rheumatoid arthritis and Hashimoto´s thyroiditis are more common in women than in men.[12],[13]
There is a constant increasing testosterone serum level in male newborns over the first three months after birth, with a decrease at 7-12 months, followed by maintaining pre – puberty low levels. In female newborns, there is a decrease of testosterone serum levels after birth. These gender differences are seen within the first year of life, as there is no difference in testosterone levels pre-puberty between male and female until onset of puberty.[14] Overall, Oestrogen stimulates the proliferation of B cells, while testosterone supresses.[15] Regarding T cell subpopulations, testosterone also supresses lymphopoiesis, in early stages. Passing several developmental steps, T cells abandon to express androgen receptors.
Another key role beside sexual hormones plays the X chromosome. On X chromosome, there are coding regions for genes involved in immune responses, as toll-like receptors, cytokine receptors and transcription factors.[17] During embryogenesis, there is a random silencing of one X chromosome, and at the same time, there is an escape out of this silencing process by 15 % of the respective immune related genes, with 10 % of the genes underlaying different levels of inactivation.[18] Thus, an overexpression of immune related genes is associated to X disomy. Genes associated to autoimmunity are also found on X chromosomes, as expression of TLR-7 described in lupus-associated autoimmunity.[19] Although female neutrophils and macrophages showed a better phagocyte activity, [20] testosterone reduces the production of TNF, expression of TLR4 and increases cytokines. Therefore, autoimmune mediated diseases are more frequent in women.[14]
A recent example for sex disparity and associated different clinical courses and outcomes are infections with severe acute respiratory syndrome coronavirus 2. Underlying mechanism remain elusive, the influence of oestrogens and androgens, as well as androgen-sensitive genes coding for angiotensin-converting enzyme 2 (ACE2) and cell surface transmembrane protease serine 2 (TMPRSS2) are under discussion. Besides, sociocultural influences are probable.[21]
There is only limited evidence for sex differences in leucocytes in cord blood. Hence, our study aims to gain more information about the composition of B and T cell subpopulations in cord blood depending on sex.
2. Results
2.1. B cells
We analysed cord blood samples of 16 male newborns and 21 female newborns by flow cytometry. The proportion of B1 cells was significantly higher in cord blood of male neonates than in cord blood of female neonates (male 3.7%±2.9 vs. female 1.82%±1.5, p=0,023, (Figure 1). As shown in table 5 B1 cells are marked CD20+CD27+CD43+ and are given as a percentage of the number of total CD20+ cells. Marginal zone memory B cells, marked as CD19+CD27+IgD+IgM+ and given as percentage of total CD19+ cells, were also significantly higher in male cord blood (male 1.67%± 2,1 vs. female 0.79%± 1,2, p=0.01, Figure 2). Except for the naive B cells and transitional B cells, B cell count in all other subsets (innate B cells, class switched memory B cells, late memory B cells, plasmablasts, transitional B cells, B2 cells) was higher in cord blood of male newborns (Table 1).
2.2. T cells
We analysed T cell subpopulations in two panels (table 5). In T cell panel 1 we analysed 16 male and 21 female and in T cell panel 2 we analysed 11 male and 16 female newborns, by flow cytometry.
Cytotoxic T cells, naive effector Th cells, naive effector cytotoxic T cells and naive thymus negative Th cells showed a trend to be higher in cord blood of male newborns. Naive thymus negative Th cells were significantly more abundant in male cord blood (male 24.4% ± 9.8 vs. female 15.9± 5.7, p=0.05, Figure 3). 12 other T cell subpopulations (T helper cells, T helper cells with αβ-TCR, activated cytotoxic T helper cells with αβ-TCR, memory effector T helper cells, naive central T helper cells, naive central T helper cells, nemory effector cytotoxic T cells, memory central cytotoxic T cells, naive central cytotoxic T cells and naive thymus positive T helper cells) showed a non-significant higher count in cord blood of female newborns than in male newborns (Table 2). The remaining subpopulations (T helper cells 1, Naive T helper cells 1, Memory T helper cells 1, T helper cells 2, Naive T helper cells 2, Memory T helper cells 2, Regulatory T cells) showed similar cell counts in both sexes.
3. Discussion
In our study, we found significant differences in B and T cell populations between male and female newborns. We were able to generate reference values for nine B cell subpopulations and 21 T cell subpopulations, respectively.
Although sex related differences have been observed in children and adults, little is known about sex differences at birth. Research data about the latter are sparse and pathophysiologic pathways and mechanism remain elusive. The production of androgens in male fetus begins within the first trimenon at ten weeks of gestation marking an early initiation of sex differentiation also affecting the developing immune system. Sex differences in immune responses are traced back to two main factors: The influence of sex hormones as testosterone and oestrogen, and differences in the number of immune related genes on the X chromosome.[14]
The influence of immune related genes on X chromosome, leading to sex differences in the development of immune related diseases starts early in life. Here, umbilical cord blood allows a special insight into intrauterine lymphocyte profiles, as it represents the interphase between pre – and postnatal physiology. In our analysis, B cell populations were higher in male newborns compared to females, except for naive B cells and transitional B cells. These results are contrary to the relation of B cells in adult blood with higher B cell count in females than in male.[22] Possible reasons are changes of sex hormone levels in utero and in cord blood: Oestrogen stimulates the proliferation of B cells, while testosterone supresses.[15] After birth, there is a drastic increase of testosterone level in male newborns over the first three months, before dropping to a much lower pre-adolescence level at the age of 7-12 months. Females show a decrease of testosterone immediately after birth. Beyond the first year of life until adolescence, there are no sex specific differences in testosterone levels.[23],[14] This fact might lead to the assumption that sex hormones are less relevant during this period of life. Here, partially unknown factors, apart from sex chromosomes, might play an important role for the developing immune system.
Nevertheless, men are more prone to infection-induced inflammation[24], like respiratory tract infections (RTI). Being more susceptible to RTI than women are, severe courses of RTI infections are more often seen in male. This might be due to innate immune responses and its sex differences. A possible explanation is an observed imbalance of TLR-2 and TLR-4: there is a higher expression of TLR-4 in macrophages of male mice following endotoxic shock, leading to extensive production of pro-inflammatory cytokines.[25] A protective factor seems to be the expression of TLR-2, higher in female mice augmenting resistance against viral infections (especially coxsackie-virus).[26] Oestrogen on the other hand suppresses lung inflammation in animal models.[26] X-chromosome and its linked immune related genes as TLR 7/8 encoding genes might also be responsible for sex differences in infection-induced inflammation.[27] X disomy leads to a higher genetic variety in immune related genes. TLR 7/8 is able to detect viral single-stranded RNA (ss-RNA) and to induce protective cytokine responses, in particular IFN responses. Although male and female innate immune cells do not differ significantly on their overall TLR 7/8 expression, a humanised mouse model showed a positive influences on TLR7 ligation on the plasmacytoid dendritic cells and its IFN-α and TNF response.[28] Besides, micro-RNAs (mi-RNA) as non-coding RNAs are associated with inflammatory diseases. Loci of mi-RNA are to be found in a higher amount on X chromosomes than in Y-chromosomes and in autosomes.[28] X-linked miR223 seems to play a crucial role: studies suggest that miR223-negative mice showed more inflammatory symptoms compared to non-deficient mice following Candida albicans infection, miR223 is supposed to affect granulocyte generation and maturation negatively.[29,30]
Former research showed a postnatal increase of CD19+ B cells and CD3+ T cells, which remains stable until the age of 2 years, followed by a gradual decrease until adulthood. While CD3+CD4+ T lymphocytes comply this trend, the subpopulation of CD3+CD8+ T lymphocytes remains stable for the first two years of life followed by a constant decrease until reaching levels similar to adults.[31] Overall, no sex differences were considered in the aforementioned studies. Various influences on lymphocyte populations within this period, such as X chromosomal genes must be considered when analysing data on the developing immune system. There is less evidence on sex related changes in lymphocyte subpopulation and more research is necessary to illuminate all factors leading to the observed changes.
In vitro, estradiol increases the accumulation of B cells, but in healthy doses, it has no effect on proliferation response. Moreover, it displays the same effect on male and female lymphocytes. Non-toxic concentrations of testosterone do not influence the maturation of in vitro B cells.[1]
Sexual hormones also influence T cell subpopulations. Testosterone supresses the lymphopoesis only in early stages. After further development, T cells do not express androgen receptors. However, oestrogen receptors are expressed at any stage of the T cell development: low doses of oestrogen increase CD4+ cells, high doses of oestrogen reduce CD4 and CD8 cell counts.[16] In our analysis, four out of 14 T cell subpopulations (Cytotoxic T cells, naive effector Th cells, naive effector cytotoxic T cells, naive thymus negative Th cells) showed a trend to be higher in cord blood of male newborns. The remaining T cell subpopulations were expected to be higher in female newborns.
Even though there is much data about CD4+ and CD8+ T cell sex differences in general, there is still few evidence about sex differences in smaller lymphocyte subpopulations. We provide reverence values for 14 different T cell subpopulations. There is a need of further studies to gain more data about sex related differences in these subgroups. This could be important for identifying risk factors for diseases.
Regardless of sparse data on sex differences in immunity (or immune development) of neonates, published research indicates that male newborns are more prone to develop a robust innate immunity, characterised by higher counts of NK cells, monocytes and basophils. Male newborns additionally show a better pro inflammatory response compared to females.[17] However, females do have higher CD4+ T cells and CD4+:CD8+ ratios, lower CD8+ T cells and lower Treg cell frequencies.[17] Numbers of B cells, IgG and IgM are similar in both sexes, whilst male newborns show higher IgA and IgE than females.[17] Our data suggest that there are no changes in the total population of B or T cells, but there are changes in their subpopulations. Subpopulations of B and T lymphocytes are very inhomogeneous groups that underlay different influences and react differently to certain stimulations. More studies are needed to identify factors leading to changes in lymphocyte subpopulations.[17]
Sex differences in immune response can be found throughout childhood. Male show higher inflammation and higher NK cell counts; CD4/CD8 ratios, CD8+ cells and CD4+ cells and B cell numbers are similar.[17],[33] Puberty marks a shift in sex differential immunity, which is potentially caused by sex steroids. Adult females show inflammation, CD4/CD8 ratios, CD4+ cells. While male adults show higher counts of CD8+ cells. B cells and immunoglobulin are higher in females. Treg cells are higher in males.[17] After menopause, pro-inflammatory response in males is higher, while females show a higher T cell activation. Other sex differences as CD4+CD8+ cell count an CD4/CD8 ratio remain distributed equally.[17]
We found a significant difference in marginal zone B cells (p=0,025). Even though they only count 5% of all B cells, they play an important role in early immune response. MZ B cells are located at a strategic interface between blood and lymph nodes, so they are able to react promptly with antibody response and bacterial pathogens.[1] They close the gap between early immune response and late adaptive antibody reactions of follicular B cells.[8] A very interesting finding within our data marks the higher count of marginal zone B cells in male newborns. This finding is contradictory to published data, as females of all ages were found to show greater B cell numbers in total. The latter might be an example for changes in composition of lymphocyte populations after birth, which might lead to sex related differences in diseases in neonatal period and later in life.
B1 cells, defined as CD20+CD27+CD43+, also showed a significant difference (p=0.01). B1 cells are the main part of B lymphocytes in newborns, but reduce towards 10% until adulthood.[6] This illustrates that B1 cells are a dynamic part of the immune system, underlying constant changes during the development towards a “mature” immune response. B1 cells are an important part of innate immune system and have been investigated thoroughly. Yet some underlying pathomechanisms remain elusive.[33] Establishing a consistent definition of immune markers leads studies on B1 cell (sub)-populations to be more comparable which is of utmost importance. As a major finding of our study, higher count of B1 cells in male newborns were found. B1 cells are associated with the pathogenesis of asthma and other allergies, in particular with production of IgE antibodies, the regulation of inflammatory processes[34],[35] and the development of autoimmune diseases, mostly affecting women.[36] Our findings suggest changes in the composition of B1 cells after birth, which might be partially due to environmental influences, antigen exposure, or the development of the adaptive immune response.
Thymus negative T cells were significantly higher in male newborns (p=0,005). Innate T cells migrate from bone marrow to thymus, where they undergo a positive and negative selection. During positive selection process, Th cells able to bind MHC complexes receive positive growing signal and entrance cell cycle. This positive selection takes place in the postnatal thymus, indicating that maturation of Th cells is an ongoing process by the time of birth.[1]
Thymus negative T cells are innate cells, without completed positive and negative selection. After negative selection, mature T cells leaving thymus usually carry CD31 marker. Negative selection plays an important part in preventing development of autoimmune reactive cells. There are tissue specific antigens expressed in thymus. Autoimmune regulator genes (AIRE) control the expression of these antigens. Mutations in this gene may lead to an autoimmune disease called autoimmune polyglandular syndrome 1 (APS-1).[9] Therefore, changes in thymus negative T cell count could be involved in development of autoimmune diseases as APS-1. There are studies hinting correlation between a failure in negative thymus selection and rheumatoid arthritis.[37] Changes in thymus negative counts could induce dysregulations in the immune system leading to reduced or incorrect pathogen detection.
In conclusion, we found significant differences in cord blood lymphocytes, related to male and female neonates, respectively. Sex related differences undergo changes throughout the development towards a mature immune response. Multiple factors are responsible for sex differences in immunological responses throughout life. Pro-inflammatory responses follow changes in puberty and post-menopause indicating oestrogen influence. Other factors remain constant from birth to adulthood. By establishing reference values for nine B cell and 21 T cell subpopulations in cord blood, our study provides important data on the influence of biological sex in the developing immune system. Investigation of influencing factors, such as genetic conditions and sexual hormones after birth are important for a deeper comprehension of early immune development and its sex related differences in neonates, infants, children and young adults.
4. Materials and Methods
4.1. Patient samples
We collected blood samples from umbilical cord blood of term neonates (range 37-40 weeks gestation, n=37) without any infectious, immunologic or chronical disease or antibiotic treatment in the previous two weeks, before planned caesarean section. The Ethics Committee of Saarland approved the study protocol (198/20, 06.08.2020). The written informed consent of the parents was obtained.
4.2. Cell isolation, storage and counting
Cord blood mononuclear cells (CBMC) were isolated by density gradient centrifugation using Lymphocyte Separation Medium 1077. (Linaris biological products, Dossenheim, Germany). Subsequently, CBMCs were stored in FCS+10% DSMO in cryoconservation at a temperature of -80 °C. Prior to the cell analysis, the cryopreserved cells were gradually thawed by first acclimating the frozen vial on ice and second thawing the cell pellet in a water bath at room temperature. Pellets were washed using PBS (Sigma Aldrich, Steinheim, Germany) and stained with Acridinorange/Propidiumiodid (AO/PI) (Logos Biosystems, Gyeonggi-do 14055 South Korea) before they were counted by LUNA-FL™ Automated Fluorescence Cell Counter (Logos Biosystems, Gyeonggi-do 14055 South Korea) to determine the number of viable cells from the donor.
4.3. Flow cytometry
To quantify the lymphocyte subsets, cells were labelled according to table 4. B lymphocytes were marked with anti-CD5, anti-CD19, anti-CD20, anti-CD27, anti-CD38, anti-CD43, anti-IgM and anti-IgD, T lymphocytes with anti-CD3, anti-CD4, anti-CD8, anti-TCR αβ+, anti-CD62L, anti-CD69, anti-CD45RO anti-CD31, anti-CCR4, anti-CCR6, anti-CRTH2, anti-CD25, anti-CD127 and anti-CD183, respectively.
BD Horizon Fixable Viability Stain 780 (Becton, Dickinson & Company, Heidelberg, Germany) was used for was subsequent discrimination of dead cells. After incubation protected from light, followed by washing steps, cell suspension was partitioned and prepared for cell staining. Cell pellets were resuspended in panel specific antibody mix and incubated protected from light at room temperature for 30 minutes. Remaining Erythrocytes were lysed with BD FACS™ Lysing Solution (Becton, Dickinson & Company, Heidelberg, Germany) according to the manufacturers protocol. After centrifugation, the supernatant was discarded, and pellets were resuspended in PBS and stored in a light-protected manner on ice until measurement.
Flow cytometry was performed on a three laser FACS Celesta flow cytometer (Becton, Dickinson & Company, Heidelberg, Germany). By forward scatter-high (FSC-H) plotted against forward scatter-area (FSC-A) doublets and cell aggregates were excluded (1.2). Gates were preset and the measurements were performed blinded for sample identity. Dead cells were excluded from the analysis by staining with BD Horizon Fixable Viability Stain 780.

Table 3.
Surface markers matched with Fluorophores and clones.
| marker | fluorophore | clone | |
|---|---|---|---|
| CD127 | Alexa-Fluor 647 | HIL-7R-M21 | |
| CD183 | BV480 | CXCR3 | |
| CD19 | APC-R700 | HIB19 | |
| CD194 (CCR4) | PE | 1G1 | |
| CD196 (CCR6) | APC-R700 | CCR6 | |
| CD20 | BB700 | 2H7 | |
| CD21 | BV421 | B-Ly4 | |
| CD25 | BV421 | M-A251 | |
| CD27 | APC | L128 | |
| CD294 (CRTH2) | BV650 | BM16 | |
| CD3 | BV786 | SK7 | |
| CD31 | BV421 | L133.1 | |
| CD38 | PE | HB-7 | |
| CD4 | PE-CF594 | SK3 | |
| CD43 | BV605 | 1G10 | |
| CD45RO | BV605 | UCHL1 | |
| CD5 | BV650 | L17F12 | |
| CD62L | BB700 | SK11 | |
| CD69 | BV480 | FN50 | |
| CD8 | APC-R700 | SK1 | |
| IgD | PE-CF594 | IA6-2 | |
| IgM | BV480 | G20-127 | |
| TCRαβ | FITC | WT31 | |
| TCRγδ | PE | 11F2 |
Figure 4.
Gating Strategy of Marginal zone B Cells: 1.1 FSC-H x FSC-A. 1.2. Histogramm of Comp-Cy7-A x FVS780. 1.3 Histogramm of Comp-PerCP-Cy5-5-A x CD20. 1.4. Histogramm of Comp-APC-R700-A x CD27. 1.5.Comp-PE-CF594-A: IgD x Comp-BV510-A: IgM.
Figure 4.
Gating Strategy of Marginal zone B Cells: 1.1 FSC-H x FSC-A. 1.2. Histogramm of Comp-Cy7-A x FVS780. 1.3 Histogramm of Comp-PerCP-Cy5-5-A x CD20. 1.4. Histogramm of Comp-APC-R700-A x CD27. 1.5.Comp-PE-CF594-A: IgD x Comp-BV510-A: IgM.

Figure 5.
Gating Strategy of B1 Cells: 1.1 FSC-H x FSC-A 1.2. Histogramm of Comp-Cy7-A x FVS780. 1.3. Histogramm of Comp-PerCP-Cy5-5-A x CD20. 1.4. Histogramm of Comp-APC-A x CD27. 1.5.SSC-A x Comp-BV605-A:CD43.
Figure 5.
Gating Strategy of B1 Cells: 1.1 FSC-H x FSC-A 1.2. Histogramm of Comp-Cy7-A x FVS780. 1.3. Histogramm of Comp-PerCP-Cy5-5-A x CD20. 1.4. Histogramm of Comp-APC-A x CD27. 1.5.SSC-A x Comp-BV605-A:CD43.
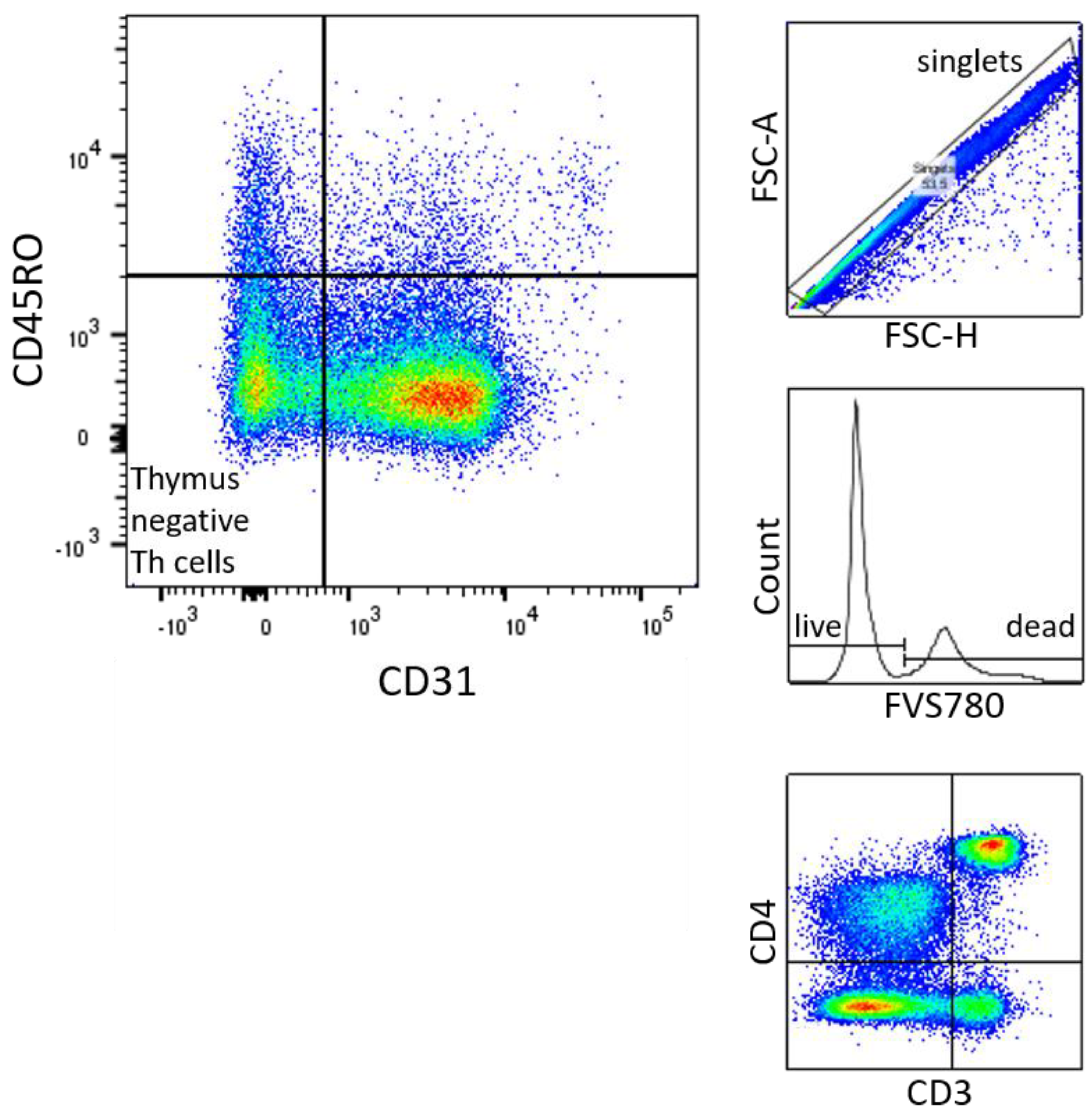
Figure 6.
Gating Strategy of naive thymus negative T Cells: 1.1 FSC-A x FSC-H. 1.2. Histogramm of Count x FVS780. 1.3. CD4 x CD3 1.4. CD45RO x CD31.
Figure 6.
Gating Strategy of naive thymus negative T Cells: 1.1 FSC-A x FSC-H. 1.2. Histogramm of Count x FVS780. 1.3. CD4 x CD3 1.4. CD45RO x CD31.
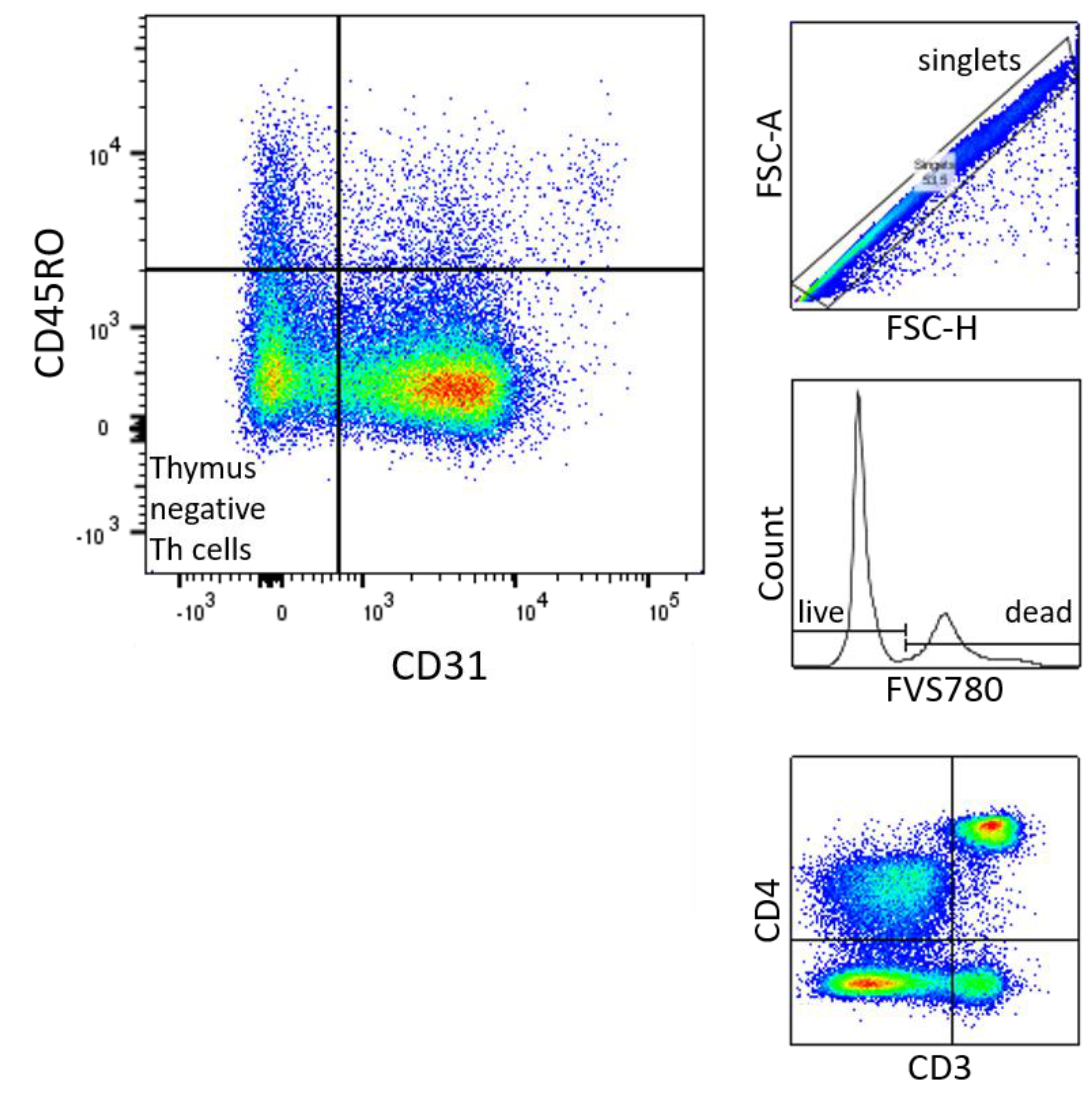
Table 4.
Definition of B and T lymphocyte subpopulations. Surface markers for 11 B and T lymphocytes.
Table 4.
Definition of B and T lymphocyte subpopulations. Surface markers for 11 B and T lymphocytes.
| Lymphocyte subsets | Definition | |
|---|---|---|
| B lymphocytes | B cells | CD19+ |
| Innate B cells | CD19+CD27-IgD-IgM- | |
| Naive B cells | CD19+CD27-IgD+IgM+ | |
| Memory B1 cells | CD19+CD27+ | |
| Marginal zone memory B cells | CD19+CD27+IgD+IgM+ | |
| Class switched memory B cells | CD19+CD27+IgD-IgM- | |
| Late memory B cells | CD19+CD27+CD38+IgM+ | |
| Plasmablasts | CD19+CD27+CD38++IgM- | |
| Transitional B cells | CD19+CD20+CD27-CD38+ | |
| B1 cells | CD20+CD27+CD43+ | |
| B2 cells | CD20+CD27+CD43- | |
| T lymphocytes | T cells | CD3+ |
| T helper cells | CD3+CD4+ | |
| Cytotoxic T cells | CD3+CD8+ | |
| T helper cells with αβ-TCR | TCRαβ+CD4+ | |
| Cytotoxic T helper cells with αβ-TCR | TCRαβ+CD8+ | |
| Activated T helper cells αβ-TCR | TCRαβ+CD4+CD69+ | |
| Activated cytotoxic T helper cells with αβ-TCR | TZR αβ+ CD8+CD69+ | |
| Memory effector T helper cells | CD3+CD4+CD62L-CD45RO+ | |
| Naive central T helper cells | CD3+CD4+CD62L+CD45RO+ | |
| Naive effector T helper cells | CD3+CD4+CD62L-CD45RO- | |
| Naive central T helper cells | CD3+CD4+CD62L+CD45RO- | |
| Memory effector cytotoxic T cells | CD3+CD8+CD62L-CD45RO+ | |
| Memory central cytotoxic T cells | CD3+CD8+CD62L+CD45RO+ | |
| Naive effector cytotoxic T cells | CD3+CD8+CD62L-CD45RO- | |
| Naive central cytotoxic T cells | CD3+CD8+CD62L+CD45RO- | |
| Naive thymus negative T helper cells | CD3+CD4+CD31-CD45RO- | |
| Naive thymus positive T helper cells | CD3+CD4+CD31+CD45RO- | |
| T helper cells 1 | CD3+CD4+CD183+CCR6+ | |
| Naive T helper cells 1 | CD3+CD4+CD183+CD45RO- | |
| Memory T helper cells 1 | CD3+CD4 CD183+CD45RO+ | |
| T helper cells 2 | CD3+CD4+CCR4+CRTH2+ | |
| Naive T helper cells 2 | CD3+CD4+CD45RO-CRTH2+ | |
| Memory T helper cells 2 | CD3+CD4+CD45RO+CRTH2+ | |
| Regulatory T cells | CD3+CD4+CD25+CD127- | |
4.4. Statistical Analysis
Statistical analysis was performed using SPSS 28.0.0. (IBM, Chicago, USA). Group differences were tested using Mann-Whitney U Test assuming non-normally distributed data. Differences with p-values of p < 0.05 were deemed significant. Means are given with standard deviation (SD). Boxplots in the figures show 10 to 90 percentile (whiskers).
Author Contributions
MB: conception and design, coordination of sample collection, data analysis and interpretation, manuscript writing, final approval of manuscript. CS: collection and assembly of data, manuscript writing, final approval of manuscript. ST: coordination of sample collection, manuscript writing, final approval of manuscript. BH: manuscript writing: final approval of manuscript. RW: MH: collection and assembly of data, data analysis, manuscript writing, final approval of manuscript. EK: collection and assembly of data, manuscript writing. GW: data analysis and interpretation, manuscript writing, final approval of manuscript. NNT: manuscript writing: final approval of manuscript. MZ: conception and design, data analysis and interpretation, financial support, manuscript writing, final approval of manuscript. SGF: conception and design, coordination of sample collection, data analysis and interpretation, manuscript writing, final approval of manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Funded by a grant from the HOMFOR Foundation of Saarland University Medical School, by the Else-Kröner-Fresenius Stiftung, by the Staatskanzlei Saarbrücken, by Centre of Digital Neurotechnologies Saar, by Werner-Zeh-Stiftung and by the BMBF (PRIMAL Clinical Study FKZ: 01GL1746D).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Ethics Committee of Saarland University (198/20, 06.08.2020).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank Ellen Maurer for her excellent technical help.
Conflicts of Interest
The authors declare no conflict of interest .
References
- LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. 2013, 13, 118–132. [Google Scholar] [CrossRef]
- Kumar SKM, Bhat BV. Distinct mechanisms of the newborn innate immunity. Immunology Letters. 2016, 173, 42–54. [Google Scholar] [CrossRef]
- Basha S, Surendran N, Pichichero M. Immune Responses in Neonates. Expert Rev Clin Immunol. 2014, 10, 1171–1184. [Google Scholar] [CrossRef]
- Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70-. J Exp Med. 2011, 208, 67–80. [CrossRef]
- Prabhu SB, Rathore DK, Nair D, Chaudhary A, Raza S, Kanodia P, et al. Comparison of Human Neonatal and Adult Blood Leukocyte Subset Composition Phenotypes. PLOS ONE. 2016, 11, e0162242. [Google Scholar] [CrossRef]
- Lopes-Carvalho T, Kearney JF. Development and selection of marginal zone B cells. Immunological Reviews. 2004, 197, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Murphy K, Weaver C. Die Entwicklung der B- und T-Lymphocyten. Janeway Immunologie. 2018; 377–440. [CrossRef]
- Lind EF, Prockop SE, Porritt HE, Petrie HT. Mapping Precursor Movement through the Postnatal Thymus Reveals Specific Microenvironments Supporting Defined Stages of Early Lymphoid Development. Journal of Experimental Medicine. 2001, 194, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Arrieta M-C, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Science Translational Medicine. 2015, 7, 307ra152. [Google Scholar] [CrossRef]
- Cooper GS, Stroehla BC. The epidemiology of autoimmune diseases. Autoimmunity Reviews. 2003, 2, 119–125. [Google Scholar] [CrossRef]
- Jacobson DL, Gange SJ, Rose NR, Graham NM. Epidemiology and estimated population burden of selected autoimmune diseases in the United States. Clin Immunol Immunopathol. 1997, 84, 223–243. [Google Scholar] [CrossRef]
- Libert C, Dejager L, Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. 2010, 10, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Bereshchenko O, Bruscoli S, Riccardi C. Glucocorticoids, Sex Hormones, and Immunity. Front Immunol. 2018, 9, 1332. [Google Scholar] [CrossRef]
- Lai J-J, Lai K-P, Zeng W, Chuang K-H, Altuwaijri S, Chang C. Androgen Receptor Influences on Body Defense System via Modulation of Innate and Adaptive Immune Systems: Lessons from Conditional AR Knockout Mice. The American Journal of Pathology. 2012, 181, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Lyn-Cook BD, Xie C, Oates J, Treadwell E, Word B, Hammons G, et al. Increased expression of Toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol. 2014, 61, 38–43. [Google Scholar] [CrossRef]
- Spitzer, JA. Gender differences in some host defense mechanisms. Lupus. 1999, 8, 380–383. [Google Scholar] [CrossRef]
- Lott N, Gebhard CE, Bengs S, Haider A, Kuster GM, Regitz-Zagrosek V, et al. Sex hormones in SARS-CoV-2 susceptibility: key players or confounders? Nat Rev Endocrinol. 2023, 19, 217–231. [CrossRef]
- Abdullah M, Chai P-S, Chong M-Y, Tohit ERM, Ramasamy R, Pei CP, et al. Gender effect on in vitro lymphocyte subset levels of healthy individuals. Cellular Immunology. 2012, 272, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Forest MG, Cathiard AM, Bertrand JA. Evidence of testicular activity in early infancy. J Clin Endocrinol Metab. 1973, 37, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Chamekh M, Deny M, Romano M, Lefèvre N, Corazza F, Duchateau J, et al. Differential Susceptibility to Infectious Respiratory Diseases between Males and Females Linked to Sex-Specific Innate Immune Inflammatory Response. Front Immunol. 2017, 8, 1806. [Google Scholar] [CrossRef] [PubMed]
- Lamason R, Zhao P, Rawat R, Davis A, Hall JC, Chae JJ, et al. Sexual dimorphism in immune response genes as a function of puberty. BMC Immunology. 2006, 7, 2. [Google Scholar] [CrossRef]
- Scotland RS, Stables MJ, Madalli S, Watson P, Gilroy DW. Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood. 2011, 118, 5918–5927. [Google Scholar] [CrossRef] [PubMed]
- Kondo Y, Miyazato A, Okamoto K, Tanaka H. Impact of Sex Differences on Mortality in Patients With Sepsis After Trauma: A Nationwide Cohort Study. Front Immunol. 2021, 12, 678156. [Google Scholar] [CrossRef]
- Laffont S, Rouquié N, Azar P, Seillet C, Plumas J, Aspord C, et al. X-Chromosome complement and estrogen receptor signaling independently contribute to the enhanced TLR7-mediated IFN-α production of plasmacytoid dendritic cells from women. J Immunol. 2014, 193, 5444–5452. [Google Scholar] [CrossRef]
- Johnnidis JB, Harris MH, Wheeler RT, Stehling-Sun S, Lam MH, Kirak O, et al. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature. 2008, 451, 1125–1129. [Google Scholar] [CrossRef]
- Haneklaus M, Gerlic M, O’Neill LAJ, Masters SL. miR-223: infection, inflammation and cancer. J Intern Med. 2013, 274, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Comans-Bitter WM, de Groot R, van den Beemd R, Neijens HJ, Hop WC, Groeneveld K, et al. Immunophenotyping of blood lymphocytes in childhood. Reference values for lymphocyte subpopulations. J Pediatr. 1997, 130, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Paavonen T, Andersson LC, Adlercreutz H. Sex hormone regulation of in vitro immune response. Estradiol enhances human B cell maturation via inhibition of suppressor T cells in pokeweed mitogen-stimulated cultures. J Exp Med. 1981, 154, 1935–1945. [Google Scholar] [CrossRef]
- Baumgarth, N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. 2011, 11, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Rothstein TL, Griffin DO, Holodick NE, Quach TD, Kaku H. Human B-1 cells take the stage. Annals of the New York Academy of Sciences. 2013, 1285, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Kawikova I, Paliwal V, Szczepanik M, Itakura A, Fukui M, Campos RA, et al. Airway hyper-reactivity mediated by B-1 cell immunoglobulin M antibody generating complement C5a at 1 day post-immunization in a murine hapten model of non-atopic asthma. Immunology. 2004, 113, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Deng J, Wang X, Chen Q, Sun X, Xiao F, Ko K-H, et al. B1a cells play a pathogenic role in the development of autoimmune arthritis. Oncotarget. 2016, 7, 19299–19311. [Google Scholar] [CrossRef]
- Fournier, C. Where Do T Cells Stand in Rheumatoid Arthritis? Joint Bone Spine. 2005, 72, 527–532. [Google Scholar] [CrossRef]
Figure 1.
B1 lymphocytes (CD20+CD27+CD43+) percentage of CD 20+ [%] were significantly higher in male (n=16) than in female (n=21) newborns, given with median (male = 2.7, female = 1.6), minimum (male = 0.3, female = 0.2) and maximum (male = 10, female = = 5.8); mean ± standard deviation [%] (male = 3.7 ± 2.9, female = 0.8 ± 1.2).
Figure 1.
B1 lymphocytes (CD20+CD27+CD43+) percentage of CD 20+ [%] were significantly higher in male (n=16) than in female (n=21) newborns, given with median (male = 2.7, female = 1.6), minimum (male = 0.3, female = 0.2) and maximum (male = 10, female = = 5.8); mean ± standard deviation [%] (male = 3.7 ± 2.9, female = 0.8 ± 1.2).
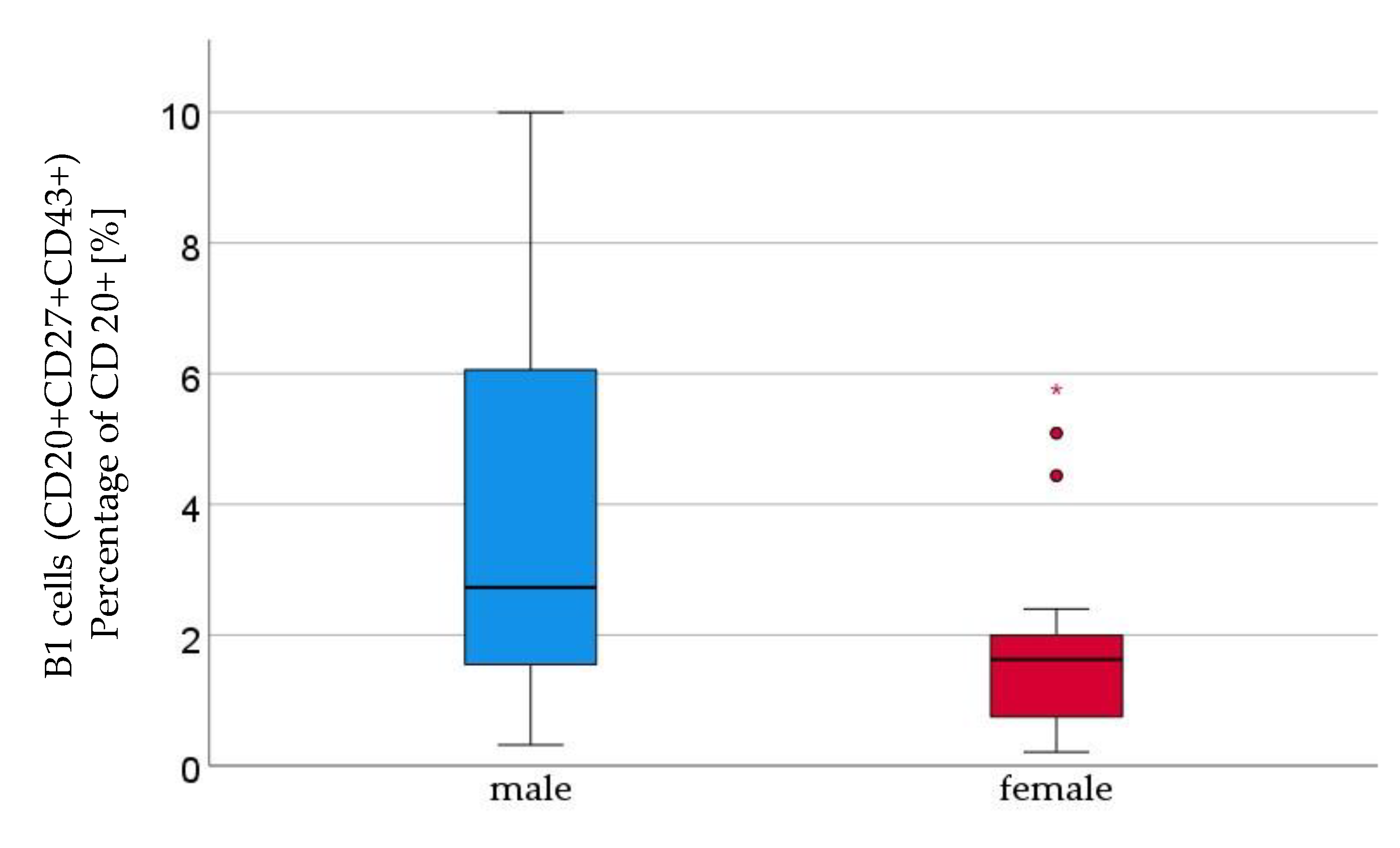
Figure 2.
Marginal zone B lymphocytes (CD19+CD27+IgD+IgM+), percentage of CD 19+ [%], were higher in male (n=16) than in female (n=21) newborns, given with median (male = 0.8, female = 0.4), minimum (male = 0.1, female = 0.1) and maximum (male = 7, female = 4.3); mean ± standard deviation [%] (male = 1.7 ± 2.1, female = 0.8 ± 1.2).
Figure 2.
Marginal zone B lymphocytes (CD19+CD27+IgD+IgM+), percentage of CD 19+ [%], were higher in male (n=16) than in female (n=21) newborns, given with median (male = 0.8, female = 0.4), minimum (male = 0.1, female = 0.1) and maximum (male = 7, female = 4.3); mean ± standard deviation [%] (male = 1.7 ± 2.1, female = 0.8 ± 1.2).
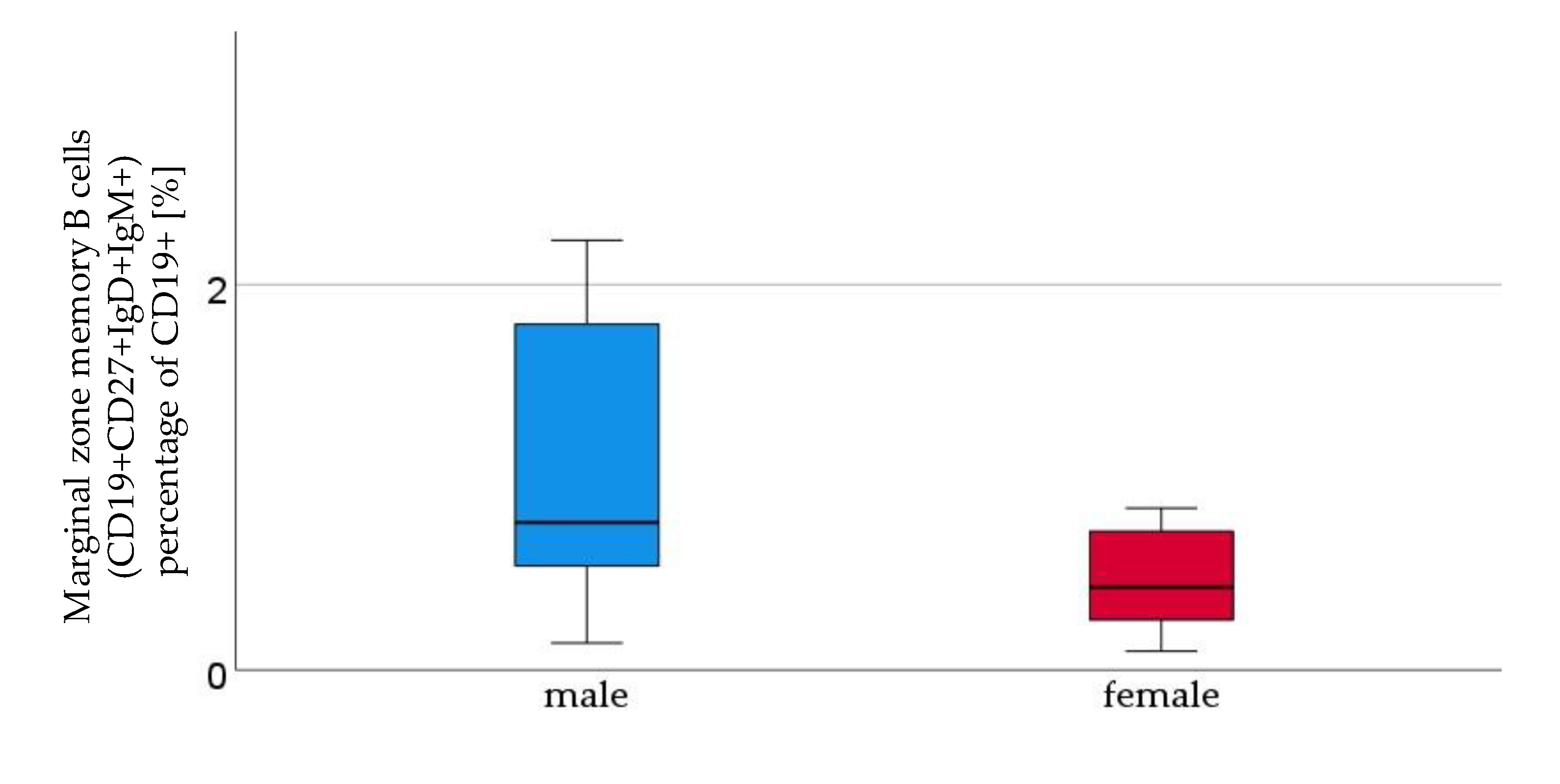
Figure 3.
Thymus negative T lymphocytes (CD3+CD4+CD31-CD45RO-) percentage of CD3+CD4+ [%] were significantly higher in male (n=16) than in female (n=21) newborns (see Figure 1). Given with median (male = 21, female = 16.2), minimum (male 9.4, female 4.2) and maximum (male 42.3, female = 27.1); mean ± standard deviation [%] (male = 24.4 ± 9.8, female = 15.9 ± 5.7).
Figure 3.
Thymus negative T lymphocytes (CD3+CD4+CD31-CD45RO-) percentage of CD3+CD4+ [%] were significantly higher in male (n=16) than in female (n=21) newborns (see Figure 1). Given with median (male = 21, female = 16.2), minimum (male 9.4, female 4.2) and maximum (male 42.3, female = 27.1); mean ± standard deviation [%] (male = 24.4 ± 9.8, female = 15.9 ± 5.7).
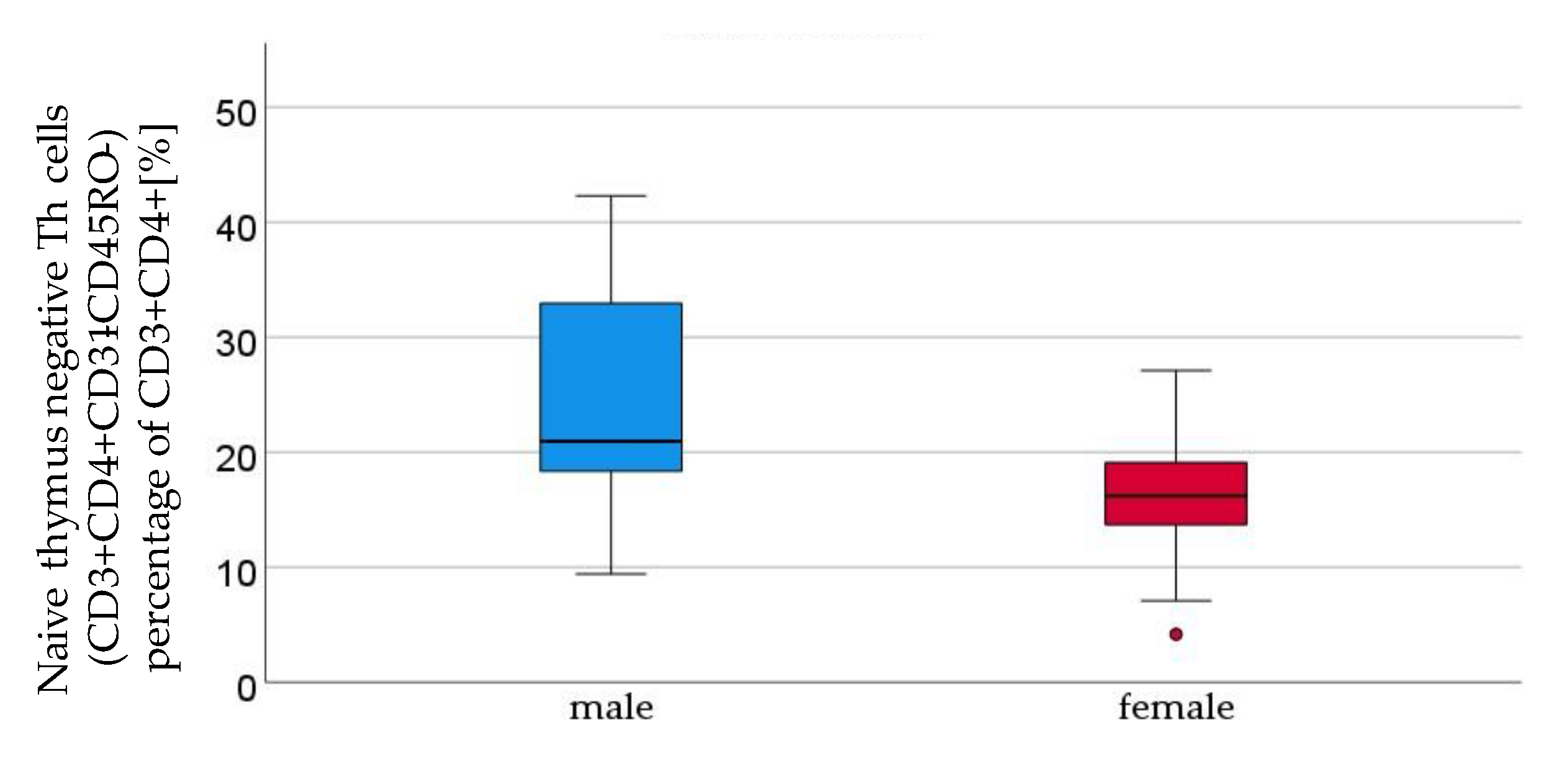
Table 1.
Relative amounts of T lymphocyte subpopulations with mean and standard deviation [%]. Group differences were tested using Mann-Whitney U test. Test groups were divided in male (n=16) and female (n=21) newborns.
Table 1.
Relative amounts of T lymphocyte subpopulations with mean and standard deviation [%]. Group differences were tested using Mann-Whitney U test. Test groups were divided in male (n=16) and female (n=21) newborns.
| sex | Mean ± standard deviation [%] | p-value | |
|---|---|---|---|
| Innate B cells | male | 6.6 ± 4.0 | 0.8 |
| (percent of CD19+ cells ) | female | 6.2 ± 3.7 | |
| Naive B cells | male | 72.0 ± 11.7 | 0.53 |
| (percent of CD19+ cells) | female | 75.3 ± 8.3 | |
| Marginal zone memory B cells | male | 1.7 ± 2.1 | 0.01 |
| (percent of CD19+ cells) | female | 0.8 ± 1.2 | |
| Class switched memory B cells | male | 5.7 ± 4.1 | 0.84 |
| (percent of CD19 + cells) | female | 5.3 ± 2.6 | |
| Late memory B cells | male | 3.2 ± 2.7 | 0.073 |
| (percent of CD19+ cells) | female | 1.7 ± 1.4 | |
| Plasmablasts | male | 5.5 ± 4.0 | 0.84 |
| (percent of CD19+ cells) | female | 5.2 ± 2.6 | |
| Transitonal B cells | male | 82.7 ± 11.0 | 0.89 |
| (percent of CD19+ cells) | female | 85.4 ± 6.7 | |
| B1 cells | male | 3.7 ± 2.9 | 0.023 |
| (percent of CD20+ cells) | female | 1.8 ± 1.5 | |
| B2 cells | male | 0.3 ± 0.3 | 0.13 |
| (percent of CD20+ cells) | female | 0.2 ± 0.3 |
Table 2.
Relative amounts of T lymphocyte subpopulations with mean and standard deviation [%]. Group differences were tested using Mann-Whitney U test.
Table 2.
Relative amounts of T lymphocyte subpopulations with mean and standard deviation [%]. Group differences were tested using Mann-Whitney U test.
| n | sex | Mean ± standard deviation [%] | p-value | |
|---|---|---|---|---|
| Th cells | 16 | male | 74.4 ± 6.6 | 0.22 |
| (percent of CD3+ cells) | 21 | female | 76.8 ± 7.2 | |
| Cytotoxic T cells | 16 | male | 27.9 ± 7.6 | 0.12 |
| (percent of CD3+ cells) | 21 | female | 24.2 ± 7.7 | |
| Th cells with αβ-TCR CD4+CD6+ | 16 | male | 76.0 ± 6.4 | 0.5 |
| (percent of TCRab+ cells) | 21 | female | 77.3 ± 7.1 | |
| Activated cytotoxic Th-cells with αβ-TCR | 16 | male | 0.6 ± 0.6 | 0.27 |
| (percent of TCRab+ cells) | 21 | female | 0.9 ± 0.8 | |
| Memory effector Th cells | 16 | male | 49.5 ± 13.6 | 0.48 |
| (percent of CD3+CD4+ cells) | 21 | female | 52.1 ± 15.8 | |
| Memory central Th cells | 16 | male | 5.7 ± 2.8 | 0.58 |
| (percent of CD3+CD4+ cells) | 21 | female | 6.7 ± 4.1 | |
| Naive effector Th cells | 16 | male | 40.9 ± 15.2 | 0.17 |
| (percent of CD3+CD4+ cells) | 21 | female | 34.9 ± 11.8 | |
| Naive central Th cells | 16 | male | 3.9 ± 1.4 | 0.62 |
| (percent of CD3+CD4+ cells) | 21 | female | 6.4 ± 9.4 | |
| Memory effector cytotoxic T cells | 16 | male | 50.8 ± 13.8 | 0.4 |
| (percent of CD3+CD8+ cells) | 21 | female | 53,.7 ± 14.1 | |
|
Memory central cytotoxic T cells
(percent of CD3+CD8+ cells) |
16 | male | 6.2 ± 4.3 | 0.66 |
| 21 | female | 7.8 ± 6.4 | ||
| Naive effector cytotoxic T cells | 16 | male | 39.4 ± 14.6 | 0.24 |
| (percent of CD3+CD8+ cells) | 21 | female | 34.1 ±1 0.6 | |
| Naive central cytotoxic T cells | 16 | male | 3.6 ± 1.6 | 0.73 |
| (percent of CD3+CD8+ cells) | 21 | female | 4.5 ± 4.1 | |
| Naïve thymus negative Th cells | 16 | male | 24.4 ± 9.8 | 0.005 |
| (percent of CD3+CD4+ cells) | 21 | female | 15.9 ± 5.7 | |
|
Naïve thymus positive Th cells CD31+
(percent of CD3+CD4+ cells) |
16 | male | 65.7 ± 9.4 | 0.6 |
| 21 | female | 70.8 ± 9.6 | ||
|
Th1 cells
(percent of CD3+ cells) |
11 | male | 2.8 ± 1.6 | 0.61 |
| 16 | female | 2.8 ± 1.9 | ||
|
Naive Th1 cells
(percent of CD3+ CD4+ CD183+ cells) |
11 | male | 87.2 ± 7.0 | 0.54 |
| 16 | female | 88.1 ± 6.8 | ||
|
Memory Th1 cells
(percent of CD3+ CD4+ CD183+ cells) |
11 | male | 12.8 ± 7.0 | 0.51 |
| 16 | female | 11.9 ± 6.8 | ||
|
Th2 cells
(percent of number of CD3+ cells) |
11 | male | 0.7 ± 0.5 | 0.54 |
| 16 | female | 0.5 ± 0.3 | ||
|
Naive Th2 cells
(percent of CD3+ CD4+ CRTH2+ cells) |
11 | male | 66.8 ± 8.6 | 0.9 |
| 16 | female | 67.6 ± 11.2 | ||
|
Memory Th2 cells
(percent of CD3+ CD4+ CRTH2+ cells) |
11 | male | 33.2 ± 8.6 | 0.9 |
| 16 | female | 32.4 ± 11.2 | ||
|
Regulatory T cells
(percent of CD3+ cells) |
11 | male | 2.3 ± 0.6 | 0.8 |
| 16 | female | 2.4 ± 0.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



