Submitted:
29 May 2023
Posted:
13 June 2023
You are already at the latest version
Abstract
Charcot–Marie–Tooth disease (CMT) and associated neuropathies constitute the most predominant genetically transmitted neuromuscular conditions; however, effective pharmacological treatments remain elusive. The extensive genetic heterogeneity of CMT, which impacts the peripheral nerves and results in a lifelong disability, presents a significant barrier to the development of comprehensive treatments. An estimated 100 loci within the human genome are linked to various forms of CMT and its related inherited neuropathies. This review delves into prospective therapeutic strategies used for the most frequently encountered CMT variants, namely CMT1A, CMT1B, CMTX1, and CMT2A. Compounds such as PXT3003, which are currently under clinical and preclinical investigation, and a broad array of therapeutic agents and their corresponding mechanisms have been discussed in this review. Furthermore, the progress in established gene therapy techniques, including gene replacement via viral vectors, exon skipping using antisense oligonucleotides, splicing modification, and gene knockdown, has been appraised. Each of these gene therapies has the potential for substantial advancements in future research.
Keywords:
Charcot–Marie–Tooth disease
; PXT3003
; gene therapy
1. Introduction
One of the major known genetically and heterogeneously transmitted peripheral neuropathies is the Charcot–Marie–Tooth (CMT) disorder which affects individuals at any age. Patients typically exhibit distal muscle weakness and atrophy, weakened ankle dorsiflexion, lessened tendon reflexes, and noticeable foot arches (Pes cavus deformities). A slight to moderate loss of the distal sensory, mainly observed in a symmetric pattern known as the stocking-glove distribution, frequently coincides with muscle weakness [1]. CMT is the major hereditary neurological disorder, with an estimated incidence of 10 to 40 per 100,000 individuals. Based on this pattern, the neuropathies can be classified into three main types: autosomal dominant (demyelinating [CMT1] and axonal [CMT2]), X-linked (CMTX1), and autosomal recessive [2,3]. Based on nerve conduction velocity (NCV), it is imperative and enormously helpful to establish a historical clinical segregation. A consistent slow NCV of < 38 m/s in the arms represents the demyelinating form of CMT1, whereas value > 38 m/s is distinctive of the axonal form of CMT2. Transitional NCVs (25–45 m/s) are often observed in males with CMTX1 and in patients with other transitional patterns of CMT [4,5].
The genetic diagnosis of CMT has been made by implementing the targeted next-generation sequencing and whole-exome sequencing approaches. Over 120 gene mutations have been related to the pathogenesis of CMT and associated neuropathies [6,7]. The frequency of the various types of CMT varies by region and the age at which they were published. In the study by Saporta et al., 787 out of 1,024 patients were diagnosed with CMT; a genetic subtype was identified in 527 (67%) of these patients, while the remaining 260 patients did not present with any identifiable mutation. Among the genetically defined cases, CMT1A (PMP22) emerged as the most prevalent subtype, comprising 55% of the cases, followed by CMTX1 (GJB1; 15.2%), hereditary neuropathy with liability to pressure palsy (HNPP; 9.2%), CMT1B (myelin protein zero (MPZ); 8.5%), and CMT2A (MFN2; 4%) [8]. DiVincenzo et al. described the frequency, detection rate, and mutation types in 14 representative genes (PMP22, GJB1, MPZ, MFN2, SH3TC2, GDAP1, NEFL, LITAF, GARS, HSPB1, FIG4, EGR2, PRX, and RAB7A) related to CMT in a cohort study of 17,880 patients tested in a commercial genetic laboratory. Genetic anomalies were detected in 18.5% (n = 3312) of the total population. Sanger sequencing and multiplex ligation-dependent probe amplification revealed that duplications (56.7%) or deletions (21.9%) in PMP22 reported for most of the positive results; next, mutations were detected in GJB1 (6.7%), MPZ (5.3%), and MFN2 (4.3%). GJB1 deletions and mutations in the residual genes counted 5.3% of the anomalies. Of the individuals presenting a positive genetic outcome in a CMT-related gene, 94.9% had a mutation in one of the following four genes (PMP22, GJB1, MPZ, or MFN2) [9].
Effective pharmacological treatments for CMT are currently lacking. This review discusses the treatment of CMT with the four main causative genes. Numerous factors, such as myelin development and conservation, transcription factors for myelin genes, gap junctions and channels, axonal transport (both retrograde and anterograde, involving kinesins, dynein, and dynactin), mitochondrial dynamics, and vesicle trafficking, contribute to the pathomechanisms of CMT and associated neuropathies. Therapeutic strategies may target specific defects in select CMT subtypes or focus on addressing shared pathomechanisms via a broader approach applicable to various categories of CMT and other neuropathies [10]. The challenges in developing an effective treatment for CMT can be attributed to three main factors: (1) extensive genetic heterogeneity, with more than 1500 identified underlying point alterations, counting the 1.4 Mb CMT1A duplication, coupled with overlapping disease phenotypes; (2) the relative scarcity of individuals per genotype, which reduces interest from both research communities and pharmaceutical companies; and (3) the complexity involved in transitioning preclinical researches from rodent and cellular models to human clinical investigations [6]. This study explores the current landscape of potential therapeutic interventions for CMT, encompassing agents under clinical evaluation that show promise for future clinical use and notable compounds in the preclinical pipeline expected to catalyze the continued research efforts. Thus, building on the momentum of the past two decades, significant strides have been made toward developing gene therapies for CMT. Preclinical studies have established a proof of concept for specific therapeutic approaches, underpinning their potential for clinical translation. This has been further bolstered by recent successes in gene therapy for other neuromuscular disorders, as evidenced by clinical trials for TTR-related hereditary amyloidosis and spinal muscular atrophy (SMA). These advancements have invariably invigorated the pursuit of gene therapy for CMT neuropathies.
2. The different forms of Major CMTs
2.1. Demyelinated CMT
2.1.1. CMT1A (PMP22)
Peripheral myelin protein-22 (PMP22), a 22-kDa intrinsic tetraspan glycoprotein, is mainly observed in the compact myelin of the peripheral nervous system. PMP22 is principally produced for myelin creation and preservation by myelinating Schwann cells (SCs) during growth and accounts for about 2%–5% of the compact myelin in the peripheral nervous system. This protein is essential for myelogenesis, myelin thickness, the growth and differentiation of SCs, and maintaining the axons and myelin of the PNS [1,11,12,13,14]. It is particularly relevant in the context of inherited peripheral neuropathies, counting > 50% of the total cases, comprising CMT1A, hereditary neuropathy with liability to pressure palsy (HNPP), and CMT1E. Alterations in PMP22 levels due to gene mutations, such as trisomy in CMT1A, heterozygous deletion in HNPP, and point mutations in CMT1E, lead to varying phenotypes; overexpression and point mutations result in gain-of-function effects and deletion produces loss-of-function. CMT1A, the major known form of CMT, is the result of a 1.4 Mb PMP22 duplication on chromosome 17p11.2, leading to disrupted myelin formation and compromised nerve function. Patients typically present a "classical CMT phenotype" within the first 20 years of life, characterized by progressive muscle feebleness, atrophy, lack of sensory, hyporeflexia, and skeletal malformations. Although the disorder progresses slowly and patients may require ankle-foot orthotics, they generally maintain ambulation and experience no significant impact during their life span. Extensive research on PMP22 and its role in CMT1A has advanced our knowledge of the illness and improved the potential therapeutic techniques [3,6].
2.1.2. CMT1B (MPZ)
CMT1B accounts for 10% of the total CMT1 cases and arises from MPZ mutations, located on chromosome 1q22-q23; the majority of the peripheral nerve myelin protein is encoded by CMT1B. The role of Po protein as a homophilic adhesion molecule is to enable myelin compaction [15]. MPZ variants lead to the etiology of demyelinating neuropathy CMT1B (OMIM 118200). Some mutations result in axonal neuropathy CMT2I/J (OMIM 607677/607736) and the additionally critical juvenile-onset Dejerine–Sottas syndrome (OMIM 145900) and congenital hypomyelination neuropathy 2 (OMIM 618184). Furthermore, MPZ variants are related to dominant intermediate CMT disease D (CMTDID; OMIM 607791). The phenotype of CMT resulting by MPZ variants varies from major pediatric-onset to minor adult onset. Approximately 250 variants of this gene have been identified as causes of inherited peripheral neuropathy [16].
2.1.3. CMTX1 (GJB)
The most known X-linked form of CMT disease and the second most common form of CMT is the X-linked CMT disease type 1 (CMTX1), caused by mutations in GJB1, which encode gap junction protein beta 1, also recognized as connexin 32 (Cx32). The latter is expressed by SCs and oligodendrocytes, while the gap junction formed by Cx32 is critical in the homeostasis of myelinated axons [17]. While men present moderate to severe symptoms, heterozygous women are generally less affected. CMTX1 is assumed as an X-linked dominant trait since it affects female carriers. In general, females are affected with a minor version of the phenotype, and the onset is delayed compared to males. Clinically, CMTX1 may be largely represented as demyelinating or axonal neuropathy, although earlier electrophysiological and pathological researches propose a more prominent axonal endorsement. Further to peripheral neuropathy, severe episodic central nervous system dysfunctions are distinctive of CMTX1 and usually manifest with numerous combinations of paralysis, dysarthria, dysphagia, ataxia, dyspnea, somnolence, and behavior anomalies [3,18,19,20,21].
2.2. Axonal CMT
2.2.1. CMT2A (MFN2)
The location of the mitofusin 2 gene (MFN2) mutations are on chromosome 1p36 and are accountable for CMT2A; the most dominant form of CMT2 (approximately 20% of the total cases of CMT) [3]. A previous study showed that CMT2A constituted the majority (91%) of strictly affected CMT2 patients and showed a smaller proportion (11%) of the mild to moderate cases [22]. The inherited form is mainly autosomal dominant (AD) [23], with sporadic autosomal recessive or semi-dominant cases caused by both homozygous and compound heterozygous MFN2 mutations [24]. MFN2, which is a GTPase protein anchored in the outer mitochondrial membrane, has a mediated function by the two transmembrane domains located near the C-terminus [25]. Mutations in MFN2 result in irregular mitochondrial aggregation and occupation accompanied by dysfunctional subcellular mitochondrial trafficking. In CMT2A patients, the most affected nerves are the peripherals comprising longer axon projections, probably because of advanced energy demands compared to other cell types.
3. CMT treatment with compounds and drugs (Table 1)
3.1. Clinical research (previous and current)
3.1.1. Ascorbic acid
Ascorbic acid (AA; Vitamin C) was among the initial therapies explored for CMT1A. AA therapy has been used on a large scale in several countries. Although the effectiveness of this therapy has not been verified yet, it has contributed valuable insights into the treatment strategies for CMT. It is crucial to note that safety and acceptability also played a role in the widespread adoption of AA therapy. Many intractable neurological disorders require a longer duration to exhibit the efficacy of a treatment. Preclinical researches have emphasized the significance of PMP22 measure in the CMT1A pathogenesis; therefore, therapeutic efforts have focused on lessening this gene expression and endorsing effective myelination [25]. AA is recognized for its antioxidant properties, neuromodulatory actions, and critical function in myelination. Increased concentrations of AA block adenylate cyclase activity, the enzyme responsible for making cyclic adenosine monophosphate [26]. In vivo studies in C22 mice verified that the expression of PMP22 is suppressed by AA and confirmed the motor function improvement in treated animals [27]. These encouraging findings, coupled with the well-established safety profile of AA, led to multiple clinical trials. Several human studies have been conducted in CMT1A patients, testing various doses of vitamin C (1 to 4 g/day) for up to two years in both adults and children; however, these trials did not show the clinical efficiency of AA in patients with CMT1A [28,29].

Table 1.
Outline of the treatment methods for Charcot–Marie–Tooth disease and associated neuropathies.
Table 1.
Outline of the treatment methods for Charcot–Marie–Tooth disease and associated neuropathies.
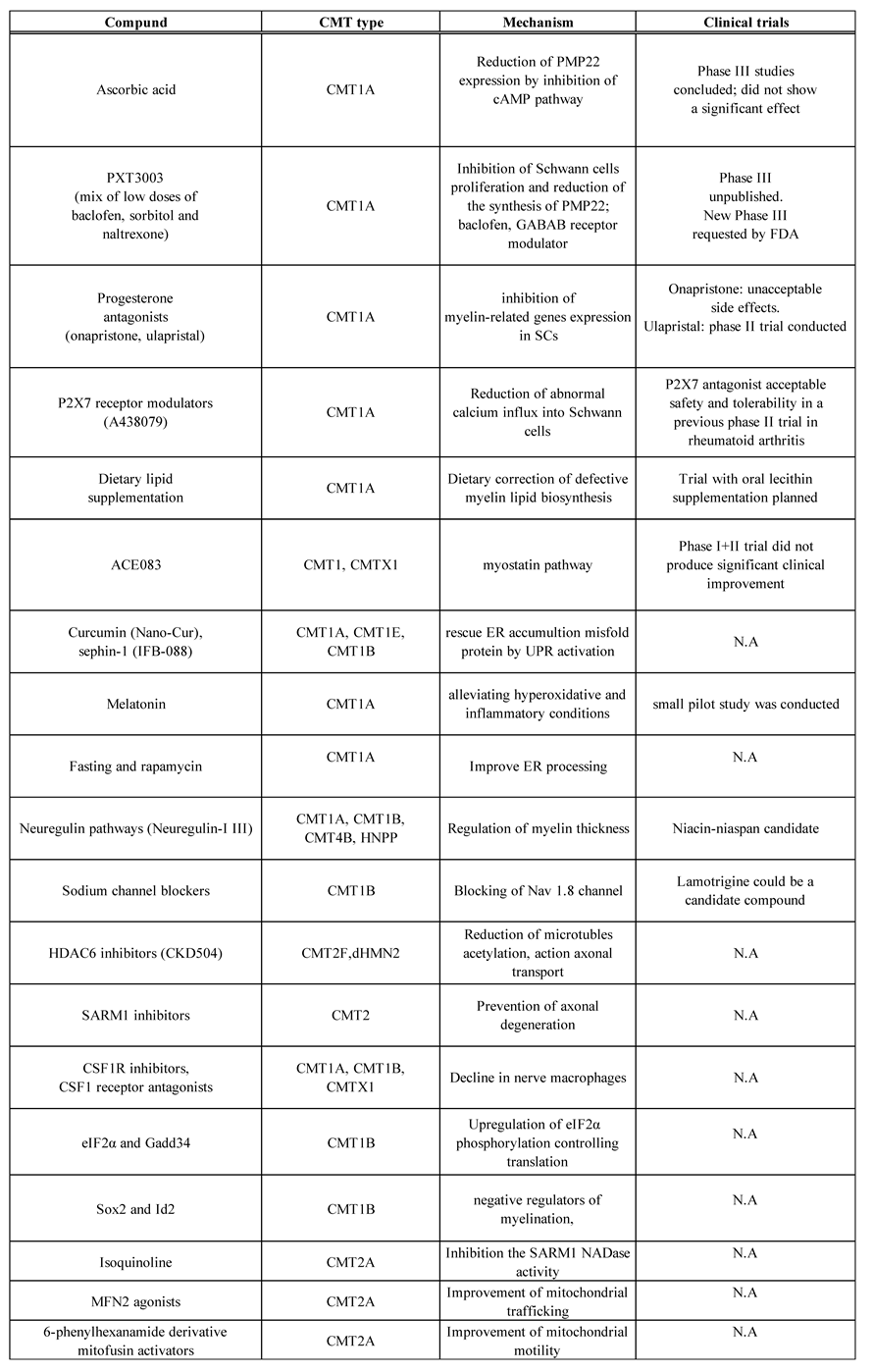
cAMP, cyclic adenosine monophosphate; CMT, Charcot-Marie-Tooth disease, CSF1R, colony-stimulating factor 1 receptor; eIF2α, eukaryotic translation initiation factor 2α kinase; ER, endoplasmic reticulum; FDA, Food and Drug Administration; GABAB, gammaaminobutyric acid B receptor; GADD34, growth arrest and DNA damage gene; HDAC6, histone deacetylase 6; dHMN, distal hereditary motor neuropathy; HNPP, hereditary neuropathy with liability to pressure palsies; Id2, Inhibitor of DNA binding 2; MFN2, mitofusin 2; N.A. , not applicable; NAD, nicotinamide adenine dinucleotide; SARM1, sterile alpha and TIR motif containing 1; SCs, Schwann cells; UPR, unfolded protein response. This table was adapted with some changes from C. Pisciotta et al., Expert Rev Neurother. 2021 Jun;21(6):701-713 and M.Stavrou et al., Int J Mol Sci. 2021 Jun 3;22(11):6048.
3.1.2. PXT3000
PXT3003 is a new fixed dose of synergistic mixture of baclofen, naltrexone, and sorbitol, expressed as an oral solution administered two times daily. The selection of drugs for CMT1A polytherapy was made from a group of approved medications for unconnected diseases by means of a systems biology method followed by pharmacological safety considerations. PXT3003 has been demonstrated to reduce Pmp22 mRNA levels, promote myelination, improve the balance of the phosphatidylinositol 4,5-bisphosphate 3-kinase (PI3K)- AKT murine thymoma viral oncogene homolog 1 (AKT)/ mitogen-activated protein kinase (MEK)- extracellular signal-regulated kinases (ERK) signaling pathways, intensify the number of functional neuromuscular junctions, and improve SC differentiation. A phase III trial expressed worries regarding the steadiness of a high concentration of PXT3003, leading to its termination (NCT02579759). Subsequently, a novel clinical test was initiated in 2021 (NCT04762758), in which a determined concentration of PXT3003 was orally given two times daily. High-dose PXT3003 significantly improved the Overall Neuropathy Limitations Scale total score compared to placebos [1,30,31,32].
3.1.3. Progesterone receptor antagonist
P0 promoter and promoter 1 of the PMP22 gene in SCs upregulates the expression of SOX-10 and KROX-20, further driving PMP22 expression. Furthermore, progesterone derivatives induce myelin gene expression by gamma-aminobutyric acid A receptor (GABAA) receptors activation in SCs [33,34,35]. Onapristone, a progesterone receptor antagonist, decreased the levels of PMP22 and enhanced axonal profiles, thereby significantly rescuing behavioral abnormalities in a rat model of CMT1A [36]. Clinical experiments of Onapristone have not been applied in CMT1A patients because of the critical disadvantages detected in cancer patients treated with this medicine. A Phase II experiment of Ulapristal was performed in France but failed to employ the target number of patients, and the outcomes have not been issued (NCT02600286) [1,30].
3.1.4. ACE-083
Acceleron Pharma spearheaded an innovative therapeutic approach for focal or asymmetric myopathies by developing ACE-083, a locally acting, follistatin-based fusion protein that adopts muscle growth and functionality by sequestering specific ligands extracellularly. Preclinical studies have shown that ACE-083 triggered localized, dose-responsive hypertrophy in the injected muscle, devoid of any systemic muscular effects or endocrine interference; this was observable in both wild-type mice and mouse models of CMT. Additionally, ACE-083 amplified the force of isometric contractions in the anterior tibialis muscle and augmented ankle dorsiflexion torque in CMT mice, suggesting its potential efficacy in treating CMT patients. A Phase I dose escalation trial was subsequently conducted, followed by a Phase II study, where up to 250 mg of ACE-083 was administered bilaterally into the anterior tibialis muscles every three weeks for a maximum of nine doses. The study comprised 62 patients diagnosed with CMT1 and CMTX1. Unfortunately, the program was prematurely terminated before advancing to phase III due to disappointing results; despite an observed increase in muscle volume, this physiological change failed to yield significant enhancements in practical or quality-of-life procedures when related with the placebo (NCT03124459) [37].
3.1.5. P2X7 purinoreceptors
Intracellular Ca (2+) concentration within SCs from CMT1A rats demonstrated abnormal elevation, an effect attributed to Pmp22-induced overexpression of the P2X7 purinoceptor. A pharmacological P2X7 receptor antagonist, A438079, fostered improved myelination in dorsal root ganglion cultures from CMT1A rats in vitro. The administration of A438079 resulted in a notable enhancement of muscle strength in CMT1A-affected rats, in significant contrast to placebo-controlled subjects. Noteworthily, a marked upsurge in the total count of myelinated axons in the tibial nerves was unveiled through histological examination. A438079 appeared to remedy the defect in SC differentiation observed in CMT1A rats. The findings of this study suggest that the pharmacological suppression of the P2X7 receptor is tolerable in CMT1A rats, thus offering an evidence-based suggestion that inhibiting this pathway could rectify the molecular abnormalities and enhance the clinical presentation of CMT1A neuropathy [38]. The transition to clinical experiments for CMT1A is made smoother by prior Phase II clinical trials involving rheumatoid arthritis, where a P2X7 antagonist demonstrated satisfactory safety and tolerability [39].
3.1.6. Lipid supplementation
In a rat model of CMT1A, myelinating SCs display a developmental deficiency characterized by lessened genes transcription necessary for myelin lipid biosynthesis. Dietary supplementation with phosphatidylcholine and phosphatidylethanolamine enhanced myelin biosynthesis and nerve function in this type. While clinical experiments have shown no disadvantages from this therapy, it is still uncertain whether high amounts of dietary phospholipids can help CMT1A patients [40,41]. Clinical translation is feasible due to the absence of any significant side effects with dietary phospholipids; therefore, a clinical experiment with oral lecithin supplementation has been set [30].
3.2. Preclinical research
3.2.1. Neuregulin-1 type I (NRG1)
The peripheral myelination of the nerves is endorsed by axonal neuregulin 1 type III (Nrg1-III) through downstream signaling pathways activation, such as PI3K/Akt and mitogen-activated protein kinase (MAPK)/ERK, which ultimately regulate the master transcriptional regulators of myelin genes, such as Krox20 [42]. The thickness of the myelin is controlled by the amount of Nrg1-III , whom activity is moderated by the secretases beta-site amyloid precursor protein cleaving enzyme 1 (BACE1) and tumor necrosis factor-alpha converting enzyme (TACE ) through distinct extracellular cleavage mechanisms [10]. Niacin/Niaspan (nicotinic acid) may improve focal hypermyelination, avoid myelin deterioration, and reserve axonal physiology by increasing TACE activity and downregulating Nrg1 type III. Niacin/Niaspan has been shown to reduce hypermyelination in the vimentin-null model and is related to improved Nrg1 type III expression. Additionally, Niaspan was reported to reduce myelin out folding in the nerves of Mtmr2-/- mice, a model of CMT4B1 neuropathy, and diminish tomacula in the nerves of Pmp22+/- mice, a model of HNPP neuropathy [43]. In a mouse model of demyelinating CMT1B, the neurophysiological and morphological parameters were promoted by the genetic overexpression of Nrg1-III without aggravating the toxic gain-of-function underlying the neuropathy. Moreover, stimulation of Nrg1-III signaling through pharmacological repression of the Nrg1-III inhibitor TACE enhanced neuropathy [42]. Ameliorated PI3K-Akt signaling by axonally overexpressed Nrg1 leads to contaminated SCs to differentiate and conserves peripheral nerve axons. Notably, in a preclinical treatment trial using a CMT1A rat model, soluble NRG1 successfully overcame impaired peripheral nerve development and restored axon survival into adulthood when treatment was limited to early postnatal development [14]. Thus, modulating Nrg1-III levels is a potential method for treating both hypomyelinating and hypermyelinating neuropathies.
3.2.2. Curcumin
Mutant protein retention in the endoplasmic reticulum (ER) triggers the unfolded protein response (UPR) activation, an adaptive and protective mechanism to alleviate stress caused by misfolded proteins. Curcumin, a polyphenolic compound found in the dietary spice turmeric, exhibits a range of pharmacological effects, including anti-inflammatory, antioxidant, antiproliferative, and antiangiogenic properties. Despite its low bioavailability, the effective treatment of curcumin against several human disorders such as cancer, cardiovascular diseases, and diabetes has been documented [44]. Many myelin gene mutations causing severe diseases, including those in MPZ and PMP22, produce aberrant proteins that predominantly accumulate in the ER, leading to SC apoptosis and, subsequently, peripheral neuropathy. In an in vitro study, Khajavi et al. demonstrated that supplementation with curcumin could counteract ER retention and aggregation-induced apoptosis related to MPZ and PMP22 mutants. Moreover, they showed that oral curcumin administration partially alleviated the severe neuropathy phenotype of the Trembler-J (Tr-J) mouse model (with the L16P mutation, a model of CMT1E) in a dose-dependent manner, featuring dominantly transmitted Pmp22 missense mutations [45,46]. Okamoto et al. observed the activation of different UPR branches in Tr-J mice and found that curcumin therapy leading to a reduction in the expression of the UPR marker, suggesting that it mitigated ER stress in the sciatic nerves of the mice [47]. Additionally, curcumin melted in sesame oil or phosphatidylcholine ameliorated peripheral neuropathy in R98C mice, an accurate model of CMT1B, by lessening ER stress and UPR activation, while promoting SC differentiation [48]. Curcumin-cyclodextrin/cellulose nanoparticles (Nano-Cur) were developed to overcome the limited pharmacokinetics of curcumin [49]. In vitro and in vivo testing of Nano-Cur in a rat model of CMT1A showed a decrease in reactive oxygen species with improved mitochondrial membrane potential and integrity, leading to improved myelination and nerve function [50].
3.2.3. Sephin-1
Sephin-1 (IFB-088), which inhibitseIF2a dephosphorylation (a kinase in the PERK arm of the UPR) and lengthens protein translation reduction in response to stress, showed beneficial in both S63del and R98C Mpz mouse models by sustaining the response and avoiding molecular, morphological, and motor flaws of neuropathy [51].
At present, Sephin-1 is being experienced in CMT1A rodent models, making it a potential candidate for clinical experiments in both CMT1A and CMT1B [30]. Theoretically, this technique may also be appreciated for other mutants retained in the ER.
3.2.4. Eukaryotic initiation factor 2-phosphorylation (eIF2α) and Gadd34
Human CMT1B neuropathy and similar demyelinating in transgenic mice are caused by the mutant P0S63del, which is reserved in the ER, activating a UPR. Under ER stress situations, protein kinase R-like endoplasmic reticulum kinase (PERK) phosphorylates eIF2α to decrease the global translation, thus lessening the misfolded protein overload in the ER. Genetic and pharmacological inactivation of Gadd34, a subunit of the PP1 phosphatase complex that enables the dephosphorylation of eIF2α, was reported to extend eIF2α phosphorylation and lessen motor, neurophysiological, and morphological deficits in S63del mice [52]. Consequently, silencing Gadd34 or directly increasing eIF2 phosphorylation may be useful on CMT1B.
3.2.5. Melatonin
Melatonin has antioxidant and anti-inflammatory properties. Melatonin supplements are Food and Drug Administration (FDA) approved and have been used extensively in clinical trials. In a trivial pilot study including three CMT1A patients treated with melatonin, plasma levels of lipid peroxidation (LPO), nitrites (NOx), and IL-1β, IL-2, IL-6, TNF-α, INF-γ, the ratio of oxidized to reduced glutathione (GSSG/GSH), and the activities of superoxide dismutase (SOD), glutathione-S-transferase (GST), glutathione peroxidase (GPx), and reductase (GRd) in erythrocytes were evaluated. The outcomes presented amplified SOD, GST, GPx, and GRd activities in CMT1A patients, which subsequently decreased after 3 and 6 months of treatment. The GSSG/GSH ratio increased significantly in the patients, but returned to normal levels after melatonin treatment. The presence of inflammation was verified by the elevated levels of all the pro-inflammatory cytokines measured, which were regulated by melatonin; in addition, elevated LPO and NOx levels in the patients were also regulated by melatonin. These findings indicate that melatonin may have therapeutic effects on CMT1A patients by alleviating hyperoxidative and inflammatory conditions and reducing the degenerative process [1,53].
3.2.6. HDAC6 inhibitor
The microtubules acetylation is imperative for axonal transport. Defective axonal transport is observed in numerous neuropathy models, comprising mice with mutations in the HSPB1 gene. HSPB1 encodes heat shock protein (HSP27), and mutations in this gene have been implicated in axonal CMT disease type 2F and distal hereditary motor neuropathy (dHMN). HDAC6 inhibitor has been displayed to correct axonal transport defects and save the phenotype of HSPB1 mutant mice [54]. Furthermore, the HDAC6 inhibitor controls the acetylation of nuclear and cytosolic proteins [55], comprising HSP90 and HSP70, which are involved in the folding/refolding of proteins such as PMP22 [56,57]. The deletion of Hdac6 prohibited motor and sensory dysfunctions in an MFN2 mouse model of CMT2A. These outcomes collectively propose that reduced acetylated alpha-tubulin could characterize a shared pathomechanism among various axonal neuropathies, with HDAC6 inhibitors potentially offering therapeutic benefits for these conditions [58]. HDAC6 is implicated in various conditions, including tumors, neurological disorders, and inflammatory diseases; consequently, targeting HDAC6 has become a potential therapeutic approach recently. ACY-1215 (ricolinostat) is the first orally existing extremely selective HDAC6 inhibitor, and its efficiency and beneficial effects are being continuously confirmed [59].
Recessive CMT phenotypes are mainly caused by a loss or extreme decrease in gene function. Gene replacement treatment may enhance effectiveness once the timing and cell delivery issues are determined. However, dominant CMT phenotypes often include a mutant allele that is toxic to the peripheral nerve; therefore, gene knockdown may be mandatory to improve the phenotypes [60].
3.2.7. Fasting and rapamycin
Tr-J mouse model of CMT1A was put on an intermittent fasting regimen, which improved their locomotor performance compared to the control group [61]. The functional benefits of dietary restriction include augmented the myelin proteins expression, a thicker myelin sheath, less redundant basal lamina, and lessened aberrant SC proliferation. These findings show that dietary limitation is helpful for peripheral nerve function in Tr-J neuropathic mice since it encourages the conservation of locomotor performance [61]. However, intermittent fasting is not a clinically available treatment; therefore, studies using rapamycin, an FDA-approved calorie restriction mimetic, were conducted. The drug was administered to explant cultures of C22 mice, which improved PMP22 processing and myelin internodal profile and increased the production of other myelin-related proteins; nevertheless, despite these potential in vitro outcomes, rapamycin did not recover neuromuscular performance in the Tr-J mouse model in vivo [62,63].
3.2.8. Sox2 and Id2
The myelination status of SCs appears to be detected by the stability between opposing signaling systems. Positive regulators such as Krox-20, Oct-6, Sox-10, Brn2, and NF-κB are predominant in normal nerves, but the balance shifts to negative regulators such as c-Jun, Notch, Pax-3, Sox2, and Id2 in the injured and pathological nerves. In addition, negative regulators may be important in the onset and myelination rate during growth (59). The continued expression of Sox2 and Id2 seems to perform a protective function in neuropathy since their elimination in P0S63del mice—a CMT1B mouse model—led to an exacerbation of the dysmyelinating phenotype [64]. Therefore, the overexpression of these genes may have a therapeutic effect on CMT1B.
3.2.9. Colony-stimulating factor 1 receptor inhibitor
Neuropathic phenotype is promoted by low-grade inflammation from phagocytizing macrophages in mice models for CMT1A, CMT1B, and CMTX1. Colony-stimulating factor (CSF1) is a macrophage activator expressed by endoneurial fibroblasts. It mediates macrophage-related neural impairment in mice and humans with CMT. Consequently, oral administration of the CSF1 receptor (CSF1R) inhibitor PLX5622 followed by a vigorous decrease in the number of macrophages in the peripheral nerves of Cx32def mice; moreover, long-term CSF1R inhibition was reported to improve axonal integrity in these mice. However, although long-term CSF1R inhibition reduced the neuropathic patterns in P0het mutants, it did not produce the same effect in PMP22tg mutants [65].
3.2.10. Sodium channel blockers
Dysmyelination in Mpz knockout mice associates with the ectopic expression of the sensory neuron-specific sodium channel isoform NaV1.8 on motor axons. Progressive impairment of motor performance in MPZ-deficient mice can be inverted by NaV1.8 blocker treatment [66]. Oral subtype-selective NaV1.8 blockade can treat severe demyelinating motor dysfunction, including CMT1B and probably other demyelinating CMT types [67].
3.2.11. SARM1 Pathway
Axonal degeneration, a shared endpoint across all CMT types, occurs irrespective of the primary pathology (i.e., myelinopathy with secondary axonal damage or primary axonopathy). Axons are hardwired to self-destruct under specific circumstances, such as stress, disease, or during the developmental phases. Gerdts et al. proposed a biochemical mechanism that regulates axonal degeneration by manipulating Sterile Alpha and TIR Motif Containing 1 (SARM1) variants, which could be alternately activated or inhibited within cells [68]. Axonal degeneration, a defining feature of numerous neurological disorders, including CMT2A, is conceptualized as a hereditarily encoded program of subcellular self-destruction in which the SARM1 protein have an essential function. Activation of SARM1 instigates axonal degeneration, even without any discernible damage [69]. A reduction in mitochondrial membrane likely precipitates a lower survival factor in the axon nicotinamide mononucleotide adenylyl-transferase 2, which triggers SARM1 and leads to axonal degeneration [70,71]. Inhibitors of SARM1, currently under development, hold significant potential for treating all CMT types and related neuropathies. A genetic treatment using dominant negative SARM1 mutants packed synergistically into an AAV8 capsid was recently introduced [72]. Postmitochondrial injury, axonal rescue, and recovery are feasible with rotenone via SARM1 inhibition, even when axons have already transitioned into a metastable state [73]. Consequently, SARM1 inhibition continues to be a primary therapeutic target not only for CMT2A but also for other axonal CMT types and demyelinating forms that exhibit secondary axonal degeneration [1].
3.2.12. MFN2 agonist and MFN1
Crucially, MFN2 shares significant homology with MFN1; research across diverse experimental systems has revealed their direct interaction capabilities and cooperative role in mitochondrial fusion, in addition to the potential of MFN1 to reimburse for an MFN2 shortage. The low expression of MFN1 in axons elucidated the heightened susceptibility of motor and sensory neurons to MFN2 mutations [74,75]. The dimerization of mitofusin molecules on neighboring mitochondria is enabled by the transition of the HR2 domain from a "closed" state, where it binds to HR1 of the same MFN2 molecule, to an "open" state, where it binds to HR2 of a different MFN2 molecule. In a recent study, molecules that target HR1/HR2 of MFN2, dubbed "mitofusin agonists," could shift the mitofusin equilibrium toward the active/open state and improve mitochondrial axon trafficking deficits in cultured motor neurons from Mfn2 T105M mutant mice [76]. MiM111, an orally administered mitofusin agonist, alleviated the disease symptoms in Mfn2 T105M mutant mice [77,78]. Considering the dominant negative impact of mutant MFN2 when the MFN1 isoform is present in reduced quantities (such as in neurons), the increase in the levels of MFN1 or WT MFN2 might serve as a promising treatment against CMT2A [79]. Notably, overexpression of MFN1 in the nervous system of Mfn2 R94Q mutant mice led to enhanced body weight, improved behavioral performance and visual acuity, extended survival, and diminished mitochondrial aggregation and axon degeneration. Hence, it is evident that the MFN1/MFN2 ratio is a critical determinant of neuronal vulnerability to the detrimental effects of mutant MFN2. Augmenting the levels of MFN1 or WT MFN2 could be a feasible beneficial approach for CMT2A[79].
3.3. CMT treatment with gene-mediated therapy (Table 2)
3.3.1. Viral vector-based therapy and the growth factors neurotrophin 3
Gene therapy involves the genetic material delivery to an individual, predominantly via viral vectors. One significant advantage of viral vector genetic treatment is its ability to cross both the blood-brain-barrier and the blood-nerve-barrier, in addition to being a one-time therapy that offers long-term effects. Lentiviral vectors were among the first to enable the efficient delivery of therapeutic genes for CMT models treatment. Clinical experiments have shown the local use of integrative lentiviral vectors for other illnesses; however, their applicability for CMT-related neurological conditions is still incomplete due to their integration into the host genome [80,81,82,83,84,85]. Given that both neurons and SCs targeted by CMT therapies are extremely differentiated and non-proliferating, the episomal persistence of adeno-associated viruses (AAVs) without integration into the host genome does not compromise the treatment stability. AAV vectors have been used in promising preclinical study to treat CMT neuropathies, either by bringing the expression of trophic factors or by targeting the culpable genes in neurons or SCs. AAV1 has been employed for a CMT1A clinical experiment (NCT03520751), while other serotypes such as AAV9 have been clinically applied to treat other neuromuscular diseases, particularly SMA (NCT03306277) [86,87,88,89,90,91].
The growth factor neurotrophin 3 (NT-3) is most frequently used in therapeutic studies with viral vectors; it promotes nerve regeneration following injury and SC survival. Consequently, the injection of recombinant NT-3 has been found to enhance regeneration and remyelination in animal models[86,92,93]. SCs expressing endogenous NT-3 contribute the autocrine loop, permitting the cells to develop and endure without the axon and motivating neurite outgrowth and myelination [94,95,96,97]. A pilot study involving eight patients with CMT1A demonstrated increased myelinated fiber density, reduced neurological disability, and improved sensory scores compared to a placebo [92]. Nevertheless, long-term therapy was not possible due to its highly short serum half-life. Additional investigations have furnished preclinical data indicating the effectiveness of AAV-mediated NT-3 genetic treatment (AAV1-NT-3) in a mouse model of CMT1A and more lately in Cx32 knockout mice [86,90,93]. Therefore, a study has been devised to intramuscularly administer rising dosages of the AAV1-NT-3 gene in both legs of CMT1A subjects (NCT03520751). Gene therapy aimed at replacing the defective gene is under investigation for various types of CMT characterized by loss-of-function mutations, such as CMTX1 and the recessive forms of CMT [30]. Intrathecal delivery of GJB1 was successfully carried out by Kleopa et al. using either lentivirus or AAV9 vectors along with the myelin-specific Mpz promoter in GJB1-knockout mice lacking Cx32 expression. This gene delivery yielded a stable Cx32 expression in SCs and peripheral nerves, accompanied by clinical improvement. However, owing to the possibility of damaging interactions between the delivered wild-type Cx32 and mutant Cx32 forms, the benefits of this approach in GJB1 mutants, which could result in protein construction, need to be established [80,81,91].
3.3.2. Gene silencing therapy
RNA interference is a well-established technique that involves small interfering RNA (siRNA), short hairpin RNA (shRNA), and microRNA (miRNA). RNA interference (RNAi) has been one of the fundamental marks in CMT1A genetic treatment. In vivo, the effectiveness of allele-specific siRNA, which precisely and selectively lessened the level of expression of the mutant allele, was assessed through intraperitoneal administration in Tr-J mice [98]. This treatment significantly improved motor function and muscle volume in the Tr-J mice, as evidenced by results from the rotarod test and magnetic resonance imaging analysis, respectively. Intravenous administration of Pmp22-targeting siRNA coupled to squalenoyl nanoparticles in JP18 and JP18/JY13 mice models of CMT1A resulted in decreased PMP22 levels and improved locomotor function; nevertheless, this therapeutic method has short-lasting effects and needs recurrent doses [99]. A long-lasting RNAi therapy was achieved by administering an intraneural injection of an AAV2/9 vector expressing murine Pmp22-targeting shRNA in a CMT1A rat model; this model normalized MPZ and PMP22 protein levels and enhanced myelination and function [100]. The transfection of regular and humanized transgenic neuropathic mouse SCs with a microRNA 29a (miR-29a) expression plasmid lessened the levels of endogenous mouse and transgenic human PMP22 transcripts compared to those in the control vector [87]. Additionally, ectopic expression of miR-29a resulted in a considerable (approximately 50%) decrease in Pmp22 mRNA, corresponding to an approximate 20% decrease in PMP22 protein levels [87]. Antisense oligonucleotides (ASOs) are single-stranded synthetic nucleic acids that specifically bind to mRNA sequences, promoting their degradation in an RNaseH-dependent manner. ASOs have been demonstrated to successfully conquer Pmp22 mRNA in affected nerves in two murine CMT1A models; notably, starting ASO therapy following the onset of the disease reestablished myelination, motor nerve conduction verocity, and compound muscle action potential nearly to the levels observed in wild-type animals [101].
3.3.3. CRISPR/Cas9
The CRISPR/Cas9 approach targets controlling elements in the PMP22 gene to lower transcription. This approach was applied in a rat SC line to remove an upstream region of the Pmp22 gene, a region foreseen to comprise an enhancer or promoter for the gene; the removal of this region led to a decrease in Pmp22 mRNA levels. CRISPR/Cas9-mediated removal of the TATA-box promoter of Pmp22 gene in C22 mice downregulated Pmp22 mRNA and enhanced nerve pathology [102,103].
3.3.4. Hepatocyte Growth factor: Engenesis®️ VM202
VM202 denotes a nonviral vector that encodes a unique genomic cDNA hybrid of human hepatocyte growth factor (HGF) [104]. HGF facilitates peripheral nerve regeneration through stimulation of SC repair. Specifically, HGF enhances the migration and proliferation of cultured SCs while concurrently upregulating the expression of multiple genes, including glial cell line-derived neurotrophic factor and leukemia inhibitory factor, likely via the activation of the ERK pathways [105]. The FDA has designated VM202 for expedited development and review, otherwise known as the 'fast track' status; several clinical trials evaluating this agent for other neurological disorders have been conducted [106,107]. While the outcomes of the CMT1A study (NCT05361031) are yet to be reported, a previous study involving recurrent intramuscular doses of the vector in ischemic heart illness patients observed a reduction in the beneficial effects of VM202 after several months [108], indicating that it may serve as a short-term symptomatic treatment.
4. Conclusions
Currently, the primary challenge faced by CMT research lies in identifying disease-modifying treatments. To date, no definitive pharmacological treatment for any CMT variant has been established; nonetheless, recent years have witnessed substantial progress. For example, AA therapy has made its way into clinical trials, marking a critical milestone in the field. Numerous other trials are underway or in the pipeline, and various therapeutic strategies are currently being evaluated in experimental models. Notably, these were the inaugural large scale, multicenter clinical experiments in the realm of CMT, although they yielded negative results for the primary outcome measure. Despite the significant time and cost invested in these trials, the potential benefits of AA remain nebulous due to the inability of these trials to detect a meaningful response. Nevertheless, these trials have underscored the importance of developing more effective clinical research outcome measures and validated biomarkers [109]. The emerging biomarkers of disease activity and burden, such as muscle MRI and plasma neurofilament light chain, along with biomarkers of target engagement for the most prevalent CMT subtype, offer promise [109]. The ongoing investigation of PXT3003, currently the most studied candidate, could yield significant results if it modifies the typically progressive course of the disease and demonstrates a sustained therapeutic effect over an extended treatment period. However, this achievement alone will not suffice. Further advancements from the multitude of ongoing preclinical studies are required to produce symptomatic improvements. Although gene therapy has revolutionized treatment for other neuromuscular diseases, its potential benefits have not been effectively translated to CMT therapy. Hence, future research is anticipated to usher in the much-needed advancement in this area.
Author Contributions
Y.O. (Yuji Okamoto) and H.T. (Hiroshi Takashima) were responsible for conception and design for this study. Y.O. was responsible for writing the manuscript.
Acknowledgments
The authors are assisted by Enago (www.enago.jp) for reviewing the English in this review.
Conflicts of Interest
None.
References
- Stavrou, M.; Sargiannidou, I.; Georgiou, E.; Kagiava, A.; Kleopa, K.A. Emerging therapies for Charcot-Marie-Tooth inherited neuropathies. Int J Mol Sci 2021, 22. [CrossRef]
- Pareyson, D.; Saveri, P.; Pisciotta, C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr Opin Neurol 2017, 30, 471–480. [CrossRef]
- Patzkó, A.; Shy, M.E. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep 2011, 11, 78–88. [CrossRef]
- Dyck, P.J.; Lambert, E.H. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch Neurol 1968, 18, 619–625. [CrossRef]
- Dyck, P.J.; Lambert, E.H. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol 1968, 18, 603–618. [CrossRef]
- Juneja, M.; Burns, J.; Saporta, M.A.; Timmerman, V. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J Neurol Neurosurg Psychiatry 2019, 90, 58–67. [CrossRef]
- Higuchi, Y.; Takashima, H. Clinical genetics of Charcot-Marie-Tooth disease. J Hum Genet 2023, 68, 199–214. [CrossRef]
- Saporta, A.S.; Sottile, S.L.; Miller, L.J.; Feely, S.M.; Siskind, C.E.; Shy, M.E. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol 2011, 69, 22–33. [CrossRef]
- DiVincenzo, C.; Elzinga, C.D.; Medeiros, A.C.; Karbassi, I.; Jones, J.R.; Evans, M.C.; Braastad, C.D.; Bishop, C.M.; Jaremko, M.; Wang, Z.; Liaquat, K.; Hoffman, C.A.; York, M.D.; Batish, S.D.; Lupski, J.R.; Higgins, J.J. The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med 2014, 2, 522–529. [CrossRef]
- Pisciotta, C.; Saveri, P.; Pareyson, D. Challenges in treating Charcot-Marie-Tooth disease and related neuropathies: Current management and future perspectives. Brain Sci 2021, 11. [CrossRef]
- Stavrou, M.; Sargiannidou, I.; Christofi, T.; Kleopa, K.A. Genetic mechanisms of peripheral nerve disease. Neurosci Lett 2021, 742, 135357. [CrossRef]
- Snipes, G.J.; Suter, U.; Welcher, A.A.; Shooter, E.M. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J Cell Biol 1992, 117, 225–238. [CrossRef]
- Adlkofer, K.; Martini, R.; Aguzzi, A.; Zielasek, J.; Toyka, K.V.; Suter, U. Hypermyelination and demyelinating peripheral neuropathy in Pmp22-deficient mice. Nat Genet 1995, 11, 274–280. [CrossRef]
- Fledrich, R.; Stassart, R.M.; Klink, A.; Rasch, L.M.; Prukop, T.; Haag, L.; Czesnik, D.; Kungl, T.; Abdelaal, T.A.; Keric, N.; Stadelmann, C.; Brück, W.; Nave, K.A.; Sereda, M.W. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat Med 2014, 20, 1055–1061. [CrossRef]
- Shy, M.E.; Jáni, A.; Krajewski, K.; Grandis, M.; Lewis, R.A.; Li, J.; Shy, R.R.; Balsamo, J.; Lilien, J.; Garbern, J.Y.; Kamholz, J. Phenotypic clustering in MPZ mutations. Brain 2004, 127, 371–384. [CrossRef]
- Taniguchi, T.; Ando, M.; Okamoto, Y.; Yoshimura, A.; Higuchi, Y.; Hashiguchi, A.; Shiga, K.; Hayashida, A.; Hatano, T.; Ishiura, H.; Mitsui, J.; Hattori, N.; Mizuno, T.; Nakagawa, M.; Tsuji, S.; Takashima, H. Genetic spectrum of Charcot-Marie-Tooth disease associated with myelin protein zero gene variants in Japan. Clin Genet 2021, 99, 359–375. [CrossRef]
- Scherer, S.S.; Kleopa, K.A. X-linked Charcot-Marie-Tooth disease. J Peripher Nerv Syst 2012, 17(suppl 3), 9–13. [CrossRef]
- Yuan, J.H.; Sakiyama, Y.; Hashiguchi, A.; Ando, M.; Okamoto, Y.; Yoshimura, A.; Higuchi, Y.; Takashima, H. Genetic and phenotypic profile of 112 patients with X-linked Charcot-Marie-Tooth disease type 1. Eur J Neurol 2018, 25, 1454–1461. [CrossRef]
- Abrams, C.K.; Freidin, M.; Bukauskas, F.; Dobrenis, K.; Bargiello, T.A.; Verselis, V.K.; Bennett, M.V.; Chen, L.; Sahenk, Z. Pathogenesis of X-linked Charcot-Marie-Tooth disease: Differential effects of two mutations in connexin 32. J Neurosci 2003, 23, 10548–10558. [CrossRef]
- Yiu, E.M.; Geevasinga, N.; Nicholson, G.A.; Fagan, E.R.; Ryan, M.M.; Ouvrier, R.A. A retrospective review of X-linked Charcot-Marie-Tooth disease in childhood. Neurology 2011, 76, 461–466. [CrossRef]
- Siskind, C.; Feely, S.M.; Bernes, S.; Shy, M.E.; Garbern, J.Y. Persistent CNS dysfunction in a boy with CMT1X. J Neurol Sci 2009, 279, 109–113. [CrossRef]
- Feely, S.M.; Laura, M.; Siskind, C.E.; Sottile, S.; Davis, M.; Gibbons, V.S.; Reilly, M.M.; Shy, M.E. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 2011, 76, 1690–1696. [CrossRef]
- Piscosquito, G.; Saveri, P.; Magri, S.; Ciano, C.; Di Bella, D.; Milani, M.; Taroni, F.; Pareyson, D. Mutational mechanisms in MFN2-related neuropathy: Compound heterozygosity for recessive and semidominant mutations. J Peripher Nerv Syst 2015, 20, 380–386. [CrossRef]
- Barbullushi, K.; Abati, E.; Rizzo, F.; Bresolin, N.; Comi, G.P.; Corti, S. Disease modeling and therapeutic strategies in CMT2A: State of the art. Mol Neurobiol 2019, 56, 6460–6471. [CrossRef]
- Kiepura, A.J.; Kochański, A. Charcot-Marie-Tooth type 1A drug therapies: Role of adenylyl cyclase activity and G-protein coupled receptors in disease pathomechanism. Acta Neurobiol Exp (Wars) 2018, 78, 198–209. [CrossRef]
- Kaya, F.; Belin, S.; Diamantidis, G.; Fontes, M. Ascorbic acid is a regulator of the intracellular cAMP concentration: Old molecule, new functions? FEBS Lett 2008, 582, 3614–3618. [CrossRef]
- Passage, E.; Norreel, J.C.; Noack-Fraissignes, P.; Sanguedolce, V.; Pizant, J.; Thirion, X.; Robaglia-Schlupp, A.; Pellissier, J.F.; Fontés, M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med 2004, 10, 396–401. [CrossRef]
- Pareyson, D.; Reilly, M.M.; Schenone, A.; Fabrizi, G.M.; Cavallaro, T.; Santoro, L.; Vita, G.; Quattrone, A.; Padua, L.; Gemignani, F.; Visioli, F.; Laurà, M.; Radice, D.; Calabrese, D.; Hughes, R.A.; Solari, A.; CMT-TRIAAL; CMT-TRAUK groups Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): A double-blind randomised trial. Lancet Neurol 2011, 10, 320–328. [CrossRef]
- Gess, B.; Baets, J.; De Jonghe, P.; Reilly, M.M.; Pareyson, D.; Young, P. Ascorbic acid for the treatment of Charcot-Marie-Tooth disease. Cochrane Database Syst Rev 2015, 2015, CD011952. [CrossRef]
- Pisciotta, C.; Saveri, P.; Pareyson, D. Updated review of therapeutic strategies for Charcot-Marie-Tooth disease and related neuropathies. Expert Rev Neurother 2021, 21, 701–713. [CrossRef]
- Chumakov, I.; Milet, A.; Cholet, N.; Primas, G.; Boucard, A.; Pereira, Y.; Graudens, E.; Mandel, J.; Laffaire, J.; Foucquier, J.; Glibert, F.; Bertrand, V.; Nave, K.A.; Sereda, M.W.; Vial, E.; Guedj, M.; Hajj, R.; Nabirotchkin, S.; Cohen, D. Polytherapy with a combination of three repurposed drugs (PXT3003) down-regulates Pmp22 over-expression and improves myelination, axonal and functional parameters in models of CMT1A neuropathy. Orphanet J Rare Dis 2014, 9, 201. [CrossRef]
- Attarian, S.; Young, P.; Brannagan, T.H.; Adams, D.; Van Damme, P.; Thomas, F.P.; Casanovas, C.; Tard, C.; Walter, M.C.; Péréon, Y.; Walk, D.; Stino, A.; de Visser, M.; Verhamme, C.; Amato, A.; Carter, G.; Magy, L.; Statland, J.M.; Felice, K. A double-blind, placebo-controlled, randomized trial of PXT3003 for the treatment of Charcot-Marie-Tooth type 1A. Orphanet J Rare Dis 2021, 16, 433. [CrossRef]
- Désarnaud, F.; Do Thi, A.N.; Brown, A.M.; Lemke, G.; Suter, U.; Baulieu, E.E.; Schumacher, M. Progesterone stimulates the activity of the promoters of peripheral myelin protein-22 and protein zero genes in Schwann cells. J Neurochem 1998, 71, 1765–1768. [CrossRef]
- Magnaghi, V.; Ballabio, M.; Roglio, I.; Melcangi, R.C. Progesterone derivatives increase expression of Krox-20 and Sox-10 in rat Schwann cells. J Mol Neurosci 2007, 31, 149–157. [CrossRef]
- Melcangi, R.C.; Magnaghi, V.; Cavarretta, I.; Zucchi, I.; Bovolin, P.; D'Urso, D.; Martini, L. Progesterone derivatives are able to influence peripheral myelin protein 22 and P0 gene expression: Possible mechanisms of action. J Neurosci Res 1999, 56, 349–357. [CrossRef]
- Sereda, M.W.; Meyer zu Hörste, G.; Suter, U.; Uzma, N.; Nave, K.A. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med 2003, 9, 1533–1537. [CrossRef]
- Thomas, F.P.; Brannagan, T.H., 3rd; Butterfield, R.J.; Desai, U.; Habib, A.A.; Herrmann, D.N.; Eichinger, K.J.; Johnson-Cl, N.E.; Karam, C.; Pestronk, A.; et al. Randomized Phase 2 Study of ACE-083 in Patients With Charcot-Marie-Tooth Disease. Neurology 2022, 98, e2356–2367. [CrossRef]
- Sociali, G.; Visigalli, D.; Prukop, T.; Cervellini, I.; Mannino, E.; Venturi, C.; Bruzzone, S.; Sereda, M.W.; Schenone, A. Tolerability and efficacy study of P2X7 inhibition in experimental Charcot-Marie-Tooth type 1A (CMT1A) neuropathy. Neurobiol Dis 2016, 95, 145–157. [CrossRef]
- Keystone, E.C.; Wang, M.M.; Layton, M.; Hollis, S.; McInnes, I.B.; D1520C00001 Study Team Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann Rheum Dis 2012, 71, 1630–1635. [CrossRef]
- Küllenberg, D.; Taylor, L.A.; Schneider, M.; Massing, U. Health effects of dietary phospholipids. Lipids Health Dis 2012, 11, 3. [CrossRef]
- Fledrich, R.; Abdelaal, T.; Rasch, L.; Bansal, V.; Schütza, V.; Brügger, B.; Lüchtenborg, C.; Prukop, T.; Stenzel, J.; Rahman, R.U.; Hermes, D.; Ewers, D.; Möbius, W.; Ruhwedel, T.; Katona, I.; Weis, J.; Klein, D.; Martini, R.; Brück, W.; Müller, W.C.; Bonn, S.; Bechmann, I.; Nave, K.A.; Stassart, R.M.; Sereda, M.W. Targeting myelin lipid metabolism as a potential therapeutic strategy in a model of CMT1A neuropathy. Nat Commun 2018, 9, 3025. [CrossRef]
- Scapin, C.; Ferri, C.; Pettinato, E.; Zambroni, D.; Bianchi, F.; Del Carro, U.; Belin, S.; Caruso, D.; Mitro, N.; Pellegatta, M.; Taveggia, C.; Schwab, M.H.; Nave, K.A.; Feltri, M.L.; Wrabetz, L.; D’Antonio, M. Enhanced axonal neuregulin-1 type-III signaling ameliorates neurophysiology and hypomyelination in a Charcot-Marie-Tooth type 1B mouse model. Hum Mol Genet 2019, 28, 992–1006. [CrossRef]
- Bolino, A.; Piguet, F.; Alberizzi, V.; Pellegatta, M.; Rivellini, C.; Guerrero-Valero, M.; Noseda, R.; Brombin, C.; Nonis, A.; D'Adamo, P.; Taveggia, C.; Previtali, S.C. Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol Med 2016, 8, 1438–1454. [CrossRef]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol Pharm 2007, 4, 807–818. [CrossRef]
- Khajavi, M.; Inoue, K.; Wiszniewski, W.; Ohyama, T.; Snipes, G.J.; Lupski, J.R. Curcumin treatment abrogates endoplasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am J Hum Genet 2005, 77, 841–850. [CrossRef]
- Khajavi, M.; Shiga, K.; Wiszniewski, W.; He, F.; Shaw, C.A.; Yan, J.; Wensel, T.G.; Snipes, G.J.; Lupski, J.R. Oral curcumin mitigates the clinical and neuropathologic phenotype of the trembler-J mouse: A potential therapy for inherited neuropathy. Am J Hum Genet 2007, 81, 438–453. [CrossRef]
- Okamoto, Y.; Pehlivan, D.; Wiszniewski, W.; Beck, C.R.; Snipes, G.J.; Lupski, J.R.; Khajavi, M. Curcumin facilitates a transitory cellular stress response in trembler-J mice. Hum Mol Genet 2013, 22, 4698–4705. [CrossRef]
- Patzkó, A.; Bai, Y.; Saporta, M.A.; Katona, I.; Wu, X.; Vizzuso, D.; Feltri, M.L.; Wang, S.; Dillon, L.M.; Kamholz, J.; Kirschner, D.; Sarkar, F.H.; Wrabetz, L.; Shy, M.E. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain 2012, 135, 3551–3566. [CrossRef]
- Ndong Ntoutoume, G.M.A.; Granet, R.; Mbakidi, J.P.; Brégier, F.; Léger, D.Y.; Fidanzi-Dugas, C.; Lequart, V.; Joly, N.; Liagre, B.; Chaleix, V.; Sol, V. Development of curcumin-cyclodextrin/cellulose nanocrystals complexes: New anticancer drug delivery systems. Bioorg Med Chem Lett 2016, 26, 941–945. [CrossRef]
- Caillaud, M.; Msheik, Z.; Ndong-Ntoutoume, G.M.; Vignaud, L.; Richard, L.; Favreau, F.; Faye, P.A.; Sturtz, F.; Granet, R.; Vallat, J.M.; Sol, V.; Desmoulière, A.; Billet, F. Curcumin-cyclodextrin/cellulose nanocrystals improve the phenotype of Charcot-Marie-Tooth-1A transgenic rats through the reduction of oxidative stress. Free Radic Biol Med 2020, 161, 246–262. [CrossRef]
- Chen, Y.; Podojil, J.R.; Kunjamma, R.B.; Jones, J.; Weiner, M.; Lin, W.; Miller, S.D.; Popko, B. Sephin1, which prolongs the integrated stress response, is a promising therapeutic for multiple sclerosis. Brain 2019, 142, 344–361. [CrossRef]
- Scapin, C.; Ferri, C.; Pettinato, E.; Bianchi, F.; Del Carro, U.; Feltri, M.L.; Kaufman, R.J.; Wrabetz, L.; D'Antonio, M. Phosphorylation of eIF2alpha promotes Schwann cell differentiation and myelination in CMT1B mice with activated UPR. J Neurosci 2020, 40, 8174–8187. [CrossRef]
- Chahbouni, M.; López, M.D.S.; Molina-Carballo, A.; de Haro, T.; Muñoz-Hoyos, A.; Fernández-Ortiz, M.; Guerra-Librero, A.; Acuña-Castroviejo, D. Melatonin treatment reduces oxidative damage and normalizes plasma pro-inflammatory cytokines in patients suffering from Charcot-Marie-Tooth neuropathy: A pilot study in three children. Molecules 2017, 22. [CrossRef]
- d'Ydewalle, C.; Krishnan, J.; Chiheb, D.M.; Van Damme, P.; Irobi, J.; Kozikowski, A.P.; Vanden Berghe, P.; Timmerman, V.; Robberecht, W.; Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 2011, 17, 968–974. [CrossRef]
- Simões-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol Neurodegener 2013, 8, 7. [CrossRef]
- Ha, N.; Choi, Y.I.; Jung, N.; Song, J.Y.; Bae, D.K.; Kim, M.C.; Lee, Y.J.; Song, H.; Kwak, G.; Jeong, S.; Park, S.; Nam, S.H.; Jung, S.C.; Choi, B.O. A novel histone deacetylase 6 inhibitor improves myelination of Schwann cells in a model of Charcot-Marie-Tooth disease type 1A. Br J Pharmacol 2020, 177, 5096–5113. [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; Seto, E.; Bhalla, K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 2005, 280, 26729–26734. [CrossRef]
- Picci, C.; Wong, V.S.C.; Costa, C.J.; McKinnon, M.C.; Goldberg, D.C.; Swift, M.; Alam, N.M.; Prusky, G.T.; Shen, S.; Kozikowski, A.P.; Willis, D.E.; Langley, B. HDAC6 inhibition promotes alpha-tubulin acetylation and ameliorates CMT2A peripheral neuropathy in mice. Exp Neurol 2020, 328, 113281. [CrossRef]
- Li, J.; Yu, M.; Fu, S.; Liu, D.; Tan, Y. Role of selective histone deacetylase 6 inhibitor ACY-1215 in cancer and other human diseases. Front Pharmacol 2022, 13, 907981. [CrossRef]
- Ravi, B.; Antonellis, A.; Sumner, C.J.; Lieberman, A.P. Genetic approaches to the treatment of inherited neuromuscular diseases. Hum Mol Genet 2019, 28, R55–R64. [CrossRef]
- Madorsky, I.; Opalach, K.; Waber, A.; Verrier, J.D.; Solmo, C.; Foster, T.; Dunn, W.A., Jr; Notterpek, L. Intermittent fasting alleviates the neuropathic phenotype in a mouse model of Charcot-Marie-Tooth disease. Neurobiol Dis 2009, 34, 146–154. [CrossRef]
- Rangaraju, S.; Verrier, J.D.; Madorsky, I.; Nicks, J.; Dunn, W.A., Jr; Notterpek, L. Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J Neurosci 2010, 30, 11388–11397. [CrossRef]
- Nicks, J.; Lee, S.; Harris, A.; Falk, D.J.; Todd, A.G.; Arredondo, K.; Dunn, W.A., Jr; Notterpek, L. Rapamycin improves peripheral nerve myelination while it fails to benefit neuromuscular performance in neuropathic mice. Neurobiol Dis 2014, 70, 224–236. [CrossRef]
- Florio, F.; Ferri, C.; Scapin, C.; Feltri, M.L.; Wrabetz, L.; D'Antonio, M. Sustained expression of negative regulators of myelination protects Schwann cells from dysmyelination in a Charcot-Marie-Tooth 1B mouse model. J Neurosci 2018, 38, 4275–4287. [CrossRef]
- Klein, D.; Patzkó, Á.; Schreiber, D.; van Hauwermeiren, A.; Baier, M.; Groh, J.; West, B.L.; Martini, R. Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot-Marie-Tooth disease in mice. Brain 2015, 138, 3193–3205. [CrossRef]
- Rosberg, M.R.; Alvarez, S.; Krarup, C.; Moldovan, M. An oral NaV1.8 blocker improves motor function in mice completely deficient of myelin protein P0. Neurosci Lett 2016, 632, 33–38. [CrossRef]
- Moldovan, M.; Pisciotta, C.; Pareyson, D.; Krarup, C. Myelin protein zero gene dose dependent axonal ion-channel dysfunction in a family with Charcot-Marie-Tooth disease. Clin Neurophysiol 2020, 131, 2440–2451. [CrossRef]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD⁺ destruction. Science 2015, 348, 453–457. [CrossRef]
- Gerdts, J.; Summers, D.W.; Milbrandt, J.; DiAntonio, A. Axon self-destruction: New links among SARM1, MAPKs, and NAD+ metabolism. Neuron 2016, 89, 449–460. [CrossRef]
- Summers, D.W.; Frey, E.; Walker, L.J.; Milbrandt, J.; DiAntonio, A. DLK activation synergizes with mitochondrial dysfunction to downregulate axon survival factors and promote SARM1-dependent axon degeneration. Mol Neurobiol 2020, 57, 1146–1158. [CrossRef]
- Loreto, A.; Hill, C.S.; Hewitt, V.L.; Orsomando, G.; Angeletti, C.; Gilley, J.; Lucci, C.; Sanchez-Martinez, A.; Whitworth, A.J.; Conforti, L.; Dajas-Bailador, F.; Coleman, M.P. Mitochondrial impairment activates the Wallerian pathway through depletion of NMNAT2 leading to SARM1-dependent axon degeneration. Neurobiol Dis 2020, 134, 104678. [CrossRef]
- Geisler, S.; Huang, S.X.; Strickland, A.; Doan, R.A.; Summers, D.W.; Mao, X.; Park, J.; DiAntonio, A.; Milbrandt, J. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J Exp Med 2019, 216, 294-303. [CrossRef]
- Hughes, R.O.; Bosanac, T.; Mao, X.; Engber, T.M.; DiAntonio, A.; Milbrandt, J.; Devraj, R.; Krauss, R. Small molecule SARM1 inhibitors recapitulate the SARM1-/- phenotype and allow recovery of a metastable pool of axons fated to degenerate. Cell Rep 2021, 34, 108588. [CrossRef]
- Detmer, S.A.; Chan, D.C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol 2007, 176, 405–414. [CrossRef]
- Misko, A.L.; Sasaki, Y.; Tuck, E.; Milbrandt, J.; Baloh, R.H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci 2012, 32, 4145–4155. [CrossRef]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; Kitsis, R.N.; Mochly-Rosen, D.; Townsend, R.R.; Gavathiotis, E.; Dorn, G.W. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [CrossRef]
- Dang, X.; Zhang, L.; Franco, A.; Li, J.; Rocha, A.G.; Devanathan, S.; Dolle, R.E.; Bernstein, P.R.; Dorn, G.W., 2nd Discovery of 6-phenylhexanamide derivatives as potent stereoselective mitofusin activators for the treatment of mitochondrial diseases. J Med Chem 2020, 63, 7033–7051. [CrossRef]
- Franco, A.; Dang, X.; Walton, E.K.; Ho, J.N.; Zablocka, B.; Ly, C.; Miller, T.M.; Baloh, R.H.; Shy, M.E.; Yoo, A.S.; Dorn, G.W. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. eLife 2020, 9. [CrossRef]
- Zhou, Y.; Carmona, S.; Muhammad, A.K.M.G.; Bell, S.; Landeros, J.; Vazquez, M.; Ho, R.; Franco, A.; Lu, B.; Dorn, G.W., 2nd; Wang, S.; Lutz, C.M.; Baloh, R.H. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest 2019, 129, 1756–1771. [CrossRef]
- Sargiannidou, I.; Kagiava, A.; Bashiardes, S.; Richter, J.; Christodoulou, C.; Scherer, S.S.; Kleopa, K.A. Intraneural GJB1 gene delivery improves nerve pathology in a model of X-linked Charcot-Marie-Tooth disease. Ann Neurol 2015, 78, 303–316. [CrossRef]
- Kagiava, A.; Karaiskos, C.; Richter, J.; Tryfonos, C.; Lapathitis, G.; Sargiannidou, I.; Christodoulou, C.; Kleopa, K.A. Intrathecal gene therapy in mouse models expressing CMT1X mutations. Hum Mol Genet 2018, 27, 1460–1473. [CrossRef]
- Kagiava, A.; Richter, J.; Tryfonos, C.; Karaiskos, C.; Heslegrave, A.J.; Sargiannidou, I.; Rossor, A.M.; Zetterberg, H.; Reilly, M.M.; Christodoulou, C.; Kleopa, K.A. Gene replacement therapy after neuropathy onset provides therapeutic benefit in a model of CMT1X. Hum Mol Genet 2019, 28, 3528–3542. [CrossRef]
- Kagiava, A.; Sargiannidou, I.; Theophilidis, G.; Karaiskos, C.; Richter, J.; Bashiardes, S.; Schiza, N.; Nearchou, M.; Christodoulou, C.; Scherer, S.S.; Kleopa, K.A. Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc Natl Acad Sci U S A 2016, 113, E2421–E2429. [CrossRef]
- Schiza, N.; Georgiou, E.; Kagiava, A.; Médard, J.J.; Richter, J.; Tryfonos, C.; Sargiannidou, I.; Heslegrave, A.J.; Rossor, A.M.; Zetterberg, H.; Reilly, M.M.; Christodoulou, C.; Chrast, R.; Kleopa, K.A. Gene replacement therapy in a model of Charcot-Marie-Tooth 4C neuropathy. Brain 2019, 142, 1227–1241. [CrossRef]
- Lee, J.S.; Kwak, G.; Kim, H.J.; Park, H.T.; Choi, B.O.; Hong, Y.B. miR-381 attenuates peripheral neuropathic phenotype caused by overexpression of PMP22. Exp Neurobiol 2019, 28, 279–288. [CrossRef]
- Sahenk, Z.; Galloway, G.; Clark, K.R.; Malik, V.; Rodino-Klapac, L.R.; Kaspar, B.K.; Chen, L.; Braganza, C.; Montgomery, C.; Mendell, J.R. AAV1.NT-3 gene therapy for charcot-marie-tooth neuropathy. Mol Ther 2014, 22, 511–521. [CrossRef]
- Serfecz, J.; Bazick, H.; Al Salihi, M.O.; Turner, P.; Fields, C.; Cruz, P.; Renne, R.; Notterpek, L. Downregulation of the human peripheral myelin protein 22 gene by miR-29a in cellular models of Charcot-Marie-Tooth disease. Gene Ther 2019, 26, 455–464. [CrossRef]
- Morelli, K.H.; Griffin, L.B.; Pyne, N.K.; Wallace, L.M.; Fowler, A.M.; Oprescu, S.N.; Takase, R.; Wei, N.; Meyer-Schuman, R.; Mellacheruvu, D.; Kitzman, J.O.; Kocen, S.G.; Hines, T.J.; Spaulding, E.L.; Lupski, J.R.; Nesvizhskii, A.; Mancias, P.; Butler, I.J.; Yang, X.L.; Hou, Y.M.; Antonellis, A.; Harper, S.Q.; Burgess, R.W. Allele-specific RNA interference prevents neuropathy in Charcot-Marie-Tooth disease type 2D mouse models. J Clin Invest 2019, 129, 5568–5583. [CrossRef]
- Sahenk, Z.; Ozes, B. Gene therapy to promote regeneration in Charcot-Marie-Tooth disease. Brain Res 2020, 1727, 146533. [CrossRef]
- Ozes, B.; Myers, M.; Moss, K.; McKinney, J.; Ridgley, A.; Chen, L.; Bai, S.; Abrams, C.K.; Freidin, M.M.; Mendell, J.R.; Sahenk, Z. AAV1.NT-3 gene therapy for X-linked Charcot-Marie-Tooth neuropathy type 1. Gene Ther 2022, 29, 127–137. [CrossRef]
- Kagiava, A.; Karaiskos, C.; Richter, J.; Tryfonos, C.; Jennings, M.J.; Heslegrave, A.J.; Sargiannidou, I.; Stavrou, M.; Zetterberg, H.; Reilly, M.M.; Christodoulou, C.; Horvath, R.; Kleopa, K.A. AAV9-mediated Schwann cell-targeted gene therapy rescues a model of demyelinating neuropathy. Gene Ther 2021, 28, 659–675. [CrossRef]
- Sahenk, Z.; Nagaraja, H.N.; McCracken, B.S.; King, W.M.; Freimer, M.L.; Cedarbaum, J.M.; Mendell, J.R. NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology 2005, 65, 681–689. [CrossRef]
- Yalvac, M.E.; Amornvit, J.; Chen, L.; Shontz, K.M.; Lewis, S.; Sahenk, Z. AAV1.NT-3 gene therapy increases muscle fiber diameter through activation of mTOR pathway and metabolic remodeling in a CMT mouse model. Gene Ther 2018, 25, 129–138. [CrossRef]
- Jessen, K.R.; Mirsky, R. Schwann cells and their precursors emerge as major regulators of nerve development. Trends Neurosci 1999, 22, 402–410. [CrossRef]
- McTigue, D.M.; Horner, P.J.; Stokes, B.T.; Gage, F.H. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci 1998, 18, 5354–5365. [CrossRef]
- Meier, C.; Parmantier, E.; Brennan, A.; Mirsky, R.; Jessen, K.R. Developing Schwann cells acquire the ability to survive without axons by establishing an autocrine circuit involving insulin-like growth factor, neurotrophin-3, and platelet-derived growth factor-BB. J Neurosci 1999, 19, 3847–3859. [CrossRef]
- Sterne, G.D.; Brown, R.A.; Green, C.J.; Terenghi, G. Neurotrophin-3 delivered locally via fibronectin mats enhances peripheral nerve regeneration. Eur J Neurosci 1997, 9, 1388–1396. [CrossRef]
- Lee, J.S.; Chang, E.H.; Koo, O.J.; Jwa, D.H.; Mo, W.M.; Kwak, G.; Moon, H.W.; Park, H.T.; Hong, Y.B.; Choi, B.O. Pmp22 mutant allele-specific siRNA alleviates demyelinating neuropathic phenotype in vivo. Neurobiol Dis 2017, 100, 99–107. [CrossRef]
- Boutary, S.; Caillaud, M.; El Madani, M.; Vallat, J.M.; Loisel-Duwattez, J.; Rouyer, A.; Richard, L.; Gracia, C.; Urbinati, G.; Desmaële, D.; Echaniz-Laguna, A.; Adams, D.; Couvreur, P.; Schumacher, M.; Massaad, C.; Massaad-Massade, L. Squalenoyl siRNA PMP22 nanoparticles are effective in treating mouse models of Charcot-Marie-Tooth disease type 1 A. Commun Biol 2021, 4, 317. [CrossRef]
- Gautier, B.; Hajjar, H.; Soares, S.; Berthelot, J.; Deck, M.; Abbou, S.; Campbell, G.; Ceprian, M.; Gonzalez, S.; Fovet, C.M.; Schütza, V.; Jouvenel, A.; Rivat, C.; Zerah, M.; François, V.; Le Guiner, C.; Aubourg, P.; Fledrich, R.; Tricaud, N. AAV2/9-mediated silencing of PMP22 prevents the development of pathological features in a rat model of Charcot-Marie-Tooth disease 1 A. Nat Commun 2021, 12, 2356. [CrossRef]
- Zhao, H.T.; Damle, S.; Ikeda-Lee, K.; Kuntz, S.; Li, J.; Mohan, A.; Kim, A.; Hung, G.; Scheideler, M.A.; Scherer, S.S.; Svaren, J.; Swayze, E.E.; Kordasiewicz, H.B. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J Clin Invest 2018, 128, 359–368. [CrossRef]
- Pantera, H.; Moran, J.J.; Hung, H.A.; Pak, E.; Dutra, A.; Svaren, J. Regulation of the neuropathy-associated Pmp22 gene by a distal super-enhancer. Hum Mol Genet 2018, 27, 2830–2839. [CrossRef]
- Lee, J.S.; Lee, J.Y.; Song, D.W.; Bae, H.S.; Doo, H.M.; Yu, H.S.; Lee, K.J.; Kim, H.K.; Hwang, H.; Kwak, G.; Kim, D.; Kim, S.; Hong, Y.B.; Lee, J.M.; Choi, B.O. Targeted PMP22 TATA-box editing by CRISPR/Cas9 reduces demyelinating neuropathy of Charcot-Marie-Tooth disease type 1A in mice. Nucleic Acids Res 2020, 48, 130–140. [CrossRef]
- Henry, T.D.; Hirsch, A.T.; Goldman, J.; Wang, Y.L.; Lips, D.L.; McMillan, W.D.; Duval, S.; Biggs, T.A.; Keo, H.H. Safety of a non-viral plasmid-encoding dual isoforms of hepatocyte growth factor in critical limb ischemia patients: A phase I study. Gene Ther 2011, 18, 788–794. [CrossRef]
- Ko, K.R.; Lee, J.; Lee, D.; Nho, B.; Kim, S. Hepatocyte growth factor (HGF) promotes peripheral nerve regeneration by activating repair Schwann cells. Sci Rep 2018, 8, 8316. [CrossRef]
- Sufit, R.L.; Ajroud-Driss, S.; Casey, P.; Kessler, J.A. Open label study to assess the safety of VM202 in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2017, 18, 269–278. [CrossRef]
- Kessler, J.A.; Shaibani, A.; Sang, C.N.; Christiansen, M.; Kudrow, D.; Vinik, A.; Shin, N.; VM202 study group M.s. Gene therapy for diabetic peripheral neuropathy: A randomized, placebo-controlled phase III study of VM202, a plasmid DNA encoding human hepatocyte growth factor. Clin Transl Sci 2021, 14, 1176–1184. [CrossRef]
- Kim, J.S.; Hwang, H.Y.; Cho, K.R.; Park, E.A.; Lee, W.; Paeng, J.C.; Lee, D.S.; Kim, H.K.; Sohn, D.W.; Kim, K.B. Intramyocardial transfer of hepatocyte growth factor as an adjunct to CABG: Phase I clinical study. Gene Ther 2013, 20, 717–722. [CrossRef]
- Rossor, A.M.; Shy, M.E.; Reilly, M.M. Are we prepared for clinical trials in Charcot-Marie-Tooth disease? Brain Res 2020, 1729, 146625. [CrossRef]
Table 2.
Outline of the gene-mediated treatment methods for Charcot–Marie–Tooth disease and associated neuropathies.
Table 2.
Outline of the gene-mediated treatment methods for Charcot–Marie–Tooth disease and associated neuropathies.
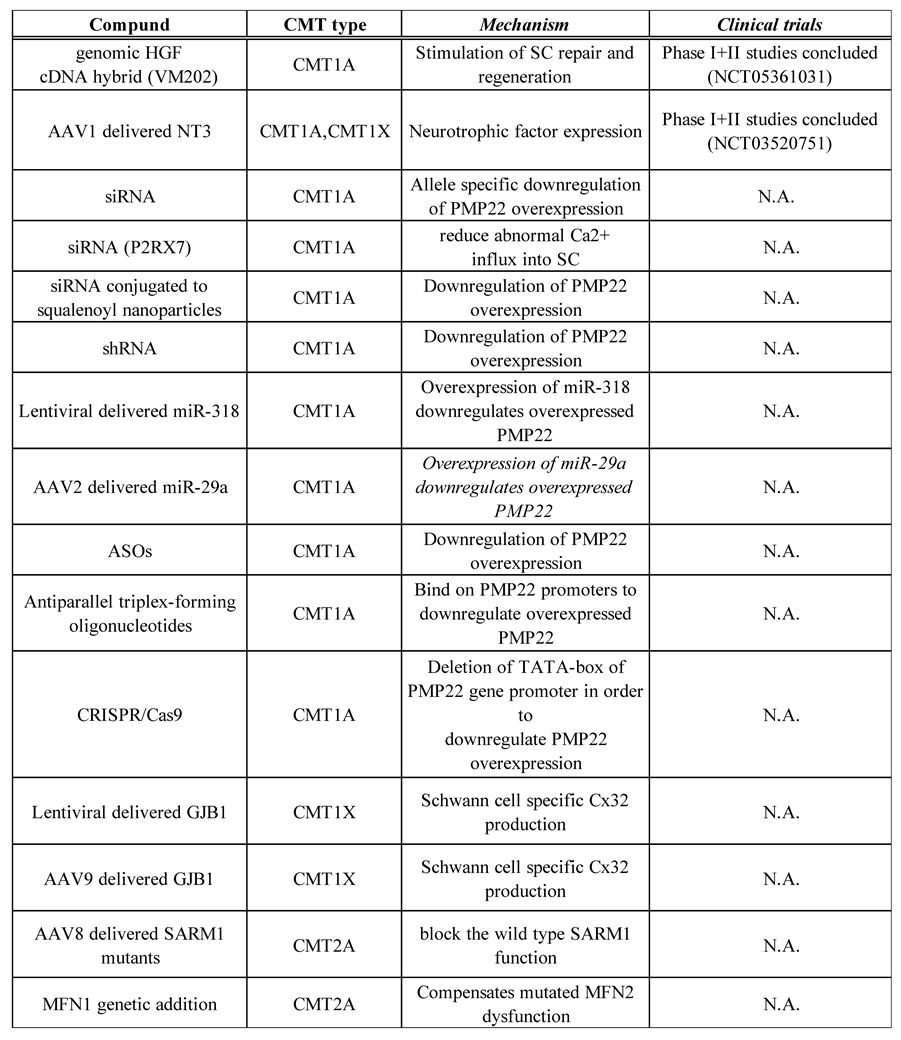
AAV, adeno-associated virus; ASO, antisense oligonucleotide; cDNA, complementary DNA; CRISPR/Cas9, clustered regularly interspaces short palindromic repeats / CRISPR-associated protein 9; HGF, Hepatocyte Growth Factor; MFN1, mitofusin1; miRNA, microRNA; N.A, not applicable; NT-3, neurotrophin-3; SARM1, sterile alpha and TIR motif containing 1; SC, Schwann cell; shRNA, short hairpin RNA; siRNA, small interfering RNA. This table was adapted with some changes from C. Pisciotta et al., Expert Rev Neurother. 2021 Jun;21(6):701-713 and M.Stavrou et al., Int J Mol Sci. 2021 Jun 3;22(11):6048.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



