Submitted:
13 June 2023
Posted:
13 June 2023
You are already at the latest version
Abstract
CD8 T cells and Natural Killer (NK) cells are cytotoxic lymphocytes important in the response to intracellular pathogens and cancer. Their activity depends on the integration of a large set of intracellular and environmental cues, including antigenic signal, cytokine stimulation and nutrients availability. This integration is achieved by signaling hubs such as the mechanistic target of Rapamycin (mTOR). mTOR is a conserved protein kinase, controlling cellular growth and metabolism in eukaryotic cells and therefore is essential for lymphocyte development and maturation. However, our current understanding of mTOR signaling comes mostly from studies performed in transformed cell lines, which constitute a poor model to comprehend metabolic pathway regulation. Therefore, it is only quite recently that the regulation of mTOR in primary cells has been assessed. Here we review the signaling pathways leading to mTOR activation in CD8 T and NK cells, focusing on activation by cytokines. We also discussed how this knowledge can contribute to immunotherapy development for intracellular pathogen and cancer treatment.
Keywords:
mTOR
; CD8 T cell
; NK cell
; cytokine signaling
1. Introduction
The Target of Rapamycin (TOR) pathway orchestrates metabolic regulation from yeast to plant and human. More specifically, activation of the TOR kinase, the effector of the TOR pathway, promotes anabolic pathways such as translation or synthesis of nucleotides and lipids while it represses catabolic activities such as autophagy. TOR kinase activity is regulated by intracellular (eg ATP) and extracellular cues (eg amino acids), so that anabolic pathways are only activated when the building blocks they require are present while catabolic activities and a more economic behavior are switched-on in less prosperous conditions. In multicellular organisms however, the organism homeostasis maintains nutrients availability [1], thus calling for a second control mechanism to prevent unrestricted growth, a hallmark of cancer [2]. During evolution, extracellular signals controlling cell activation and growth have thus been co-opted as inputs regulating TOR signaling network. This explains why mechanistic TOR (mTOR), behaves as a coincidence detector being active only when both nutrients and growth signals are present. Importantly, the surface receptors that detect such growth signals are different from one cell type to another to ensure independent regulation of different cell types. As a consequence, the classes of receptors involved in growth signal detection are varied, ranging from tyrosine kinase receptors to G-protein coupled receptors. This diversity suggests that the signaling pathways leading to mTOR activation can differ from one cell type to another and even inside a given cell type if two distinct receptors are triggered. Historically, the study of mTOR signaling has been conducted in vitro in transformed cell lines using the pathway triggered by insulin as a model. This pathway and its components are conserved whichn most cell types making it a good model system. However, it does not reveal the whole breadth of the signaling network regulating mTOR activity. In addition, the complete rewiring of cellular metabolism in cultured transformed cell lines makes them a poor model for specific regulatory steps taking place in primary resting cells. Here we will review the signaling pathways leading to mTOR activation in lymphocytes taking the example of CD8 T cells and Natural Killer (NK) cells. Indeed, various steps of these cells’ biology are regulated by cytokines, a subgroup of growth factors, that stimulate the mTOR pathway. We also focused this review on mTORC1 activation, the complex primarily involved in metabolic regulation, since we and others have demonstrated the importance of this complex in the regulation of lymphocyte functions and activity.
2. CD8 T and NK cell development and functions: common themes and divergences
CD8 T lymphocytes and Natural Killer (NK) cells are cytotoxic lymphocytes specialized in the response to intracellular pathogens and cancer. They are able to kill infected or tumor cells through directed exocytosis of granules containing perforin and granzyme and produce large quantities of pro-inflammatory cytokines such as IFN-γ [3]. These capacities place them both at the forefront of anti-tumoral and antiviral responses. They however diverge in the conditions that lead to the acquisition of these effector functions: while naïve CD8 T cells require clonal expansion and subsequent differentiation into cytotoxic T lymphocytes (CTL) to acquire these capacities, NK cells acquire them during their development. This translates into very different response kinetics during immune responses: while NK cells display their effector functions in a matter of hours, CD8 T cells require a differentiation step of several days. Moreover, the signals leading to this acquisition also differ. Indeed, naïve T cells differentiation into CTL is triggered upon engagement of the T cell receptor (TCR) and this first signal is complemented by the help of Interleukin (IL)-2, secreted by helper CD4+ T cells or to a lesser extent by CD8+ T cells themselves. In contrast, NK cell development and acquisition of effector potential is principally driven by the cytokine IL-15. IL-2 and IL-15 bind to partially overlapping receptors comprised of IL2Rβ and the γ chain for the common part while the alpha chains: IL2Rα and IL15Rα, bring the specificity. Since the α chains are unable to signal, the signal arising from IL-2 or IL-15 receptors is almost undistinguishable [4]. Other cytokines such as cytokines of the IL-1 family also take part in the activation of certain functions of CD8 and NK cells. Indeed, cytokines from the IL-1 family are important for terminal differentiation and acquisition of effector functions, such as IL-1β which increases CD8+ T cell cytotoxic capacity [5] or IL-18 which, in association with IL-12, is the main driver of IFN-γ production by both CD8 T cells and NK cells [6]. In addition, it becomes clear that additional factors such as the presence of metabolites in the extracellular milieu or the bioenergetic state of the cell strongly influence lymphocyte fate decisions. Therefore, lymphocyte activation depends on the integration of a large set of intracellular and environmental cues, including antigenic signal, cytokine stimulation and nutrients availability. This integration is achieved by signaling hubs such”as m’ORC1.
3. A brief overview of the mTOR pathway
3.1. mTOR complexes and targets
mTOR was identified thanks to its sensitivity to Rapamycin, a macrolide compound isolated from the bacteria Streptomyces hygroscopicus found in a soil sample of Easter Island (Rapa Nui being the native name of the island). It is the core kinase of two complexes, the mTOR complexes 1 (mTORC1) and 2 (mTORC2). mTORC1 is composed of the kinase mTOR associated with the protein mammalian lethal with SEC13 protein 8 (mLST8) and the scaffold protein Regulatory-associated protein of mTOR (RAPTOR). Two endogenous inhibitors also belong to the complex, the proline-rich AKT substrate of 40 kDa (PRAS40) and the DEP domain-containing mTOR-interacting protein (DEPTOR). mTORC2 is organized around mTOR, the protein mLST8 and the scaffold protein Rapamycin-insensitive companion of mammalian target of rapamycin (RICTOR) in lieu of RAPTOR, along with DEPTOR (as in mTORC1), mammalian stress-activated protein kinase interacting protein 1 (mSIN1) and protein observed with RICTOR 1 and 2 (PROTOR ½). The two complexes differ by their sensitivity to Rapamycin, mTORC1 being sensitive and mTORC2 not directly sensitive to it [7], but also by their substrates and functions. mTORC1 plays a well-known role in the control of protein synthesis through the phosphorylation of 4E-BP and S6K, which are classical hallmarks of mTORC1 activation. In addition, mTORC1 also activates glycolysis, lipids and nucleotides synthesis, and represses autophagy therefore supporting anabolism and energy synthesis. Altogether, this explains that mTORC1 activation is usually associated with cell growth and proliferation. The role of mTORC2 is less understood; it phosphorylates AKT on Ser473, thereby potentiating its kinase activity which represses transcription factors FOXO1/3 in lymphocytes [8]. Such factors regulate the expression of multiple genes associated with lymphocyte quiescence or trafficking. Moreover, in some systems mTORC2 regulates cytoskeleton dynamics. As stated above, this review will mainly discuss mTORC1 signaling and control.
3.2. The importance of mTOR in lymphocytes
The notion that mTORC1 activity is essential for lymphocyte activation and functions stems from early studies that discovered the immunosuppressive activity of Rapamycin [9]. mTORC1 inhibitors have thus been used in clinic, notably in context of lymphoproliferative diseases [10,11,12] or organ transplantation [13,14]. Therefore, the essential role of mTOR for immune cell development and function is now widely recognized, especially in lymphocytes [15]. Immune signals delivered by the TCR and costimulatory molecules in T cells, NK activating receptors in NK cells, or cytokine receptors in both T and NK cells, trigger mTORC1 activation which switches-on the metabolic pathways required to support cellular activation. mTOR also has a crucial role during NK cell development by controlling their maturation and maintaining their reactivity [16,17]. Furthermore, memory CD8 T cell differentiation also relies on a tight regulation of mTOR activity [18,19,20].
3.3. mTOR upstream regulation
In eukaryotic cells, mTORC1 senses two broad categories of signals: growth factors and nutrients. The concomitance of these two signals is required for mTORC1 activation as it regulates both mTORC1 kinase activity and localisation (Figure 1).
3.3.1. Linking growth factors to mTOR activation: the exemple of the insulin signaling
Growth factors, which we will refer here to molecules capable of stimulating cell proliferation and survival, are a first set of signals that trigger mTORC1 activation. In 1998, insulin was found to activate mTOR [21] and became since the best documented example of mTOR activation by a growth factor. The insulin receptor is a receptor tyrosine kinase expressed by virtually all cells. Insulin binding to its receptor triggers receptor autophosphorylation on several cytoplasmic tyrosines, driving the recruitment and phosphorylation of the adaptor protein Insulin Receptor Substrate (IRS). Mammals express 4 IRS isoforms, the main ones being IRS1 and IRS2 [22]. Phosphorylated IRS then serve as binding site to recruit proteins with SH2 domains such as phosphatidylinositol 3-kinases (PI3K). Class I PI3K are formed by a 110-kDa catalytic subunit (p110) among the isoforms α, β, γ, δ, associated with a regulatory subunit among p85α, p85β or p55γ. The 4 p110 catalytic subunits can act upstream of mTOR, however, they are recruited downstream different receptors, p110γ being only recruited downstream G-protein coupled receptors. In mature lymphocytes, the most expressed subunit is p110δ; it associates with p85α or p85β [23]. Class I PI3K catalyze the phosphorylation of phosphatidylinositol (4,5) biphosphate (PI(4,5)P2) into PI(3,4,5)P3, which acts as a docking site for proteins containing pleckstrin homology (PH) domains such as the Protein kinase B (PKB also named AKT) and 3-phosphoinositide-dependent protein kinase-1 (PDK1). This allows PDK1 to phosphorylate the activation loop of AKT at Thr-308, an essential event for AKT activation. In addition, AKT is also phosphorylated on Ser-473 primarily by mTORC2 even though this modification can be added by other kinases such as TBK1 [24,25,26]. This second phosphorylation event stabilizes the Thr308 phosphorylation and increases AKT kinase activity. IRS also recruits the adaptor GRB2, which binds SOS, a guanine nucleotide exchange factor for RAS. Stimulation of RAS constitute the starting point of the activation of the ERK Mitogen-activated protein kinases (MAPK) cascade.
Regarding the mTOR pathway, a major convergence point of AKT and ERK pathways is the inhibition of the tuberous sclerosis complex (TSC), a key inhibitor of mTORC1. TSC is composed of three proteins, TSC1, TSC2 and TBC1D7 [27] and acts as a GTPase-activating protein (GAP) for the small GTPase RHEB [28,29], an essential mTORC1 activator. By inhibiting TSC, growth factors such as insulin allow thus mTOR to be fully activated by RHEB. Structural studies show that RHEB loaded with GTP binds to mTOR distally from the kinase site and causes a global conformational change of the complex [30]. This molecular mechanism explains that GTP-loaded RHEB behaves as an allosteric activator increasing mTORC1 activity by several orders of magnitude. Of note, RHEB has unusually slow intrinsic GTPase activity. As a results, TSC, that stimulates RHEB GTPase activity and therefore inhibits this small GTPase, behaves as a gatekeeper of mTORC1 activity. In addition to this well described canonical pathway, some studies also suggest that the mTORC1 complex itself can be post-translationally modified following growth factor stimulation [31,32]. The relevance of such modifications is unclear.
The activity of TSC Is tightly regulated by multiple proteins. AKT phosphorylates TSC2 at different sites, thus suppressing its control of RHEB and mTOR [33]. TSC is also inhibited by phosphorylation from ERK [34] and the p90 ribosomal S6 kinase (RSK) [35], another downstream kinase of the Ras-ERK pathway. Other studies described regulation of TSC by growth factors through the control of its recruitment to the lysosome [36,37,38,39]. Regulation of TSC through the disruption of the complex itself or through its degradation are still debated (reviewed in[40]). Overall, these results define TSC as a major hub collecting inputs negatively regulating mTORC1.
3.3.2. mTOR control by nutrients and cellular stress
In addition to growth factors, nutrients are essential for mTORC1 activation. In particular, the role of amino acids has long been recognized and involves a set of 4 GTPases, the RAG GTPase A,B,C and D that act as heterodimers combining RAG A or B and RAG C or D [41,42]. In conditions of amino acids sufficiency, the RAG GTPases are locked in an “on-state”, with RAG A/B bound to GTP and RAG C/D bound to GDP, and vice-versa in case of amino acid scarcity. In their “on-state”, RAG GTPases mediate mTORC1 attachment to the lysosome surface via a multi-proteic complex called the Ragulator [43,44], bringing mTORC1 in close vicinity to its activator RHEB that also lies on the external membrane of the lysosome. Thus, when nutrients are abundant, mTORC1 is tethered to the lysosome surface where it can be fully activated by mitogen signals such as insulin or other growth factors. In the contrary, when nutrients are limiting, mTORC1 is released from the lysosome surface and thus inactivated. This two-step mechanism explains the importance of the coincidence of both nutrients and growth signals for mTORC1 activation and support its role as a signal integrator. Several proteins are involved in amino-acid sensing both inside the lysosome and the cytoplasm, and constitute today a topic of active research (review in [45]). On note, not all amino acids seem to have the same potency. Arginine, Leucine and Methionine have been described as direct regulators of mTORC1, each one possessing its dedicated sensor [46]. Other nutrients such as glucose [47] or cholesterol [48,49,50] have also been shown to impinge directly onto mTORC1 pathway in a manner partially overlapping the amino acid pathway.
In addition to this direct control, the lack of nutrients can also be sensed via the decrease of ATP content resulting from it. Indeed, energy drop or cellular stress also negatively regulate mTOR activity. Glucose depletion activates the AMP-activated protein kinase (AMPK), which senses the decrease in ATP/ADP ratio and inhibits mTORC1 either directly through phosphorylation of Raptor on inhibitory sites [51] or indirectly through activating phosphorylation of TSC2 [52]. Independently of AMPK, hypoxia also negatively regulates mTORC1 through TSC activation [53,54]. Several mTORC1 negative regulators, including TSC or AMPK, are targets of p53 which plays a central role in DNA damage sensing. [55]. Finally, endoplasmic reticulum stress or oxidative stress also negatively regulate mTORC1 function (review in [56]).
4. mTOR activation downstream γc cytokines receptor in lymphocytes
Cytokines are soluble proteins with signaling functions in the immune system and for some of them, a growth factor activity. Physiologically, γ chain cytokines, such as IL-2 and IL-15 are potent mitogens for T and NK cells. IL-1 family cytokines increase cytotoxic functions and cytokine production of lymphocytes, two processes that require energy. Therefore, it is no surprise that these cytokines activate the mTORC1 pathway. The next two sections discuss our current knowledge on mTORC1 regulation in T and NK cells, focusing on γ chain and IL-1 type cytokines.
4.1. Overview of the γc cytokines family
The γ chain (γc) family regroups cytokines that signal through the common receptor γ chain namely IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21. Γc was first discovered as a component of the IL-2 receptor, making IL-2 the archetypal member of this family [57,58]. IL-2 receptor is composed of three proteins; CD25: the α chain of the IL2R (IL-2Rα), CD122: the β chain of the IL2R (IL-2Rβ), and the γ chain CD132. As mentioned before, IL-15 signals through two of the three IL-2 receptor chains, the γc and IL-2Rβ while its unique IL-15Rα chain serves to present IL-15 in trans from neighbouring cells [59,60]. In terms of signalling, most of our knowledge comes from studies on IL-2 receptor. Contrary to insulin receptor, IL-2 receptor lacks intrinsic kinase activity. IL-2Rβ and γc are associated with the kinases JAK1 and JAK3 respectively, and the binding of IL-2 results in JAK1/3 activation and phosphorylation of tyrosine residues on IL-2Rβ [61,62,63]. These residues serve as docking sites for proteins containing SH2 domain. Phosphorylated IL-2Rβ binds the p85 subunit of PI3K [64], which triggers the activation of the PI3K-AKT pathway. Phosphorylated IL-2Rβ also serves as a docking site for the GRB2-SOS complex through the adaptor SHC, whichh constitutes the starting point of the Ras-ERK MAPK cascade. The transcription factors STAT5 is also recruited to the activated IL-2R and is of particularly importance for IL-2 signaling [65].
4.2. mTOR regulation by γc cytokines in T cells
The first evidences of mTOR activation by γc cytokines comes from studies using IL-2 [66,67]. In T cells, IL-2 treatment triggers ribosomal protein S6 phosphorylation, a marker of S6K activity downstream mTORC1. IL-2 treatment is also associated with an increase in cell size and metabolism, other indirect hallmarks of mTORC1 activation [68,69,70,71]. Several studies show an increase in AKT phosphorylation few minutes after IL-2 treatment, confirming that IL-2 activates this pathway in T cells [69,70,72]. However, the importance of PI3K-AKT in mTORC1 activation in T cells is debated. Studies from the Cantrell’s group show that the inhibition of AKT or PI3Kδ does not decrease mTORC1 activity following IL-2 treatment in CTL [69]. The same group however showed that PDK1 is required for mTORC1 activity, since CTL deleted for this protein display decreased S6 phosphorylation, glucose uptake and cell proliferation [68]. Conversely, deletion of PTEN, a negative regulator of PI3K signaling that can drive the activation of AKT, is not sufficient to activate mTORC1 and mTORC1-dependent glucose metabolism [73]. These evidences suggest that PI3K-AKT pathway is activated by IL-2 but dispensable for mTORC1 activation. How PDK1 controls mTORC1 independently of the PI3K-AKT pathway remains to be deciphered. One hypothesis would be that its activity is not required extemporaneously but more upstream to phosphorylate and license the neosynthetized S6K [74].
How can IL-2 activate mTORC1 if not through the well described PI3K-AKT axis? One possibility could be through the JAK-STAT pathway. CD4+ T cells deficient for STAT5 treated with IL-2 show decreased mTORC1 activity, as measured by S6 phosphorylation, associated with a reduction in cell size, proliferation and metabolism compared to their WT counterpart. However, STAT5 deficient T cells have also decreased levels of AKT phosphorylation at Thr308, suggesting that STAT5 may act upstream AKT. To directly test the relationship between STAT5 and AKT, T cells deleted for STAT5 were complemented with either constitutively activated STAT5 or constitutively activated AKT. Only STAT5 transduction rescued mTORC1 activation whereas transduction of AKT did not, suggesting a mechanism whereby mTOR activation relies on STAT5 while it is independent of AKT [70]. To explain the activation of AKT, Ross and Cantrell proposed a mechanism involving pathways depending on the SRC family kinases LCK and FYN. This signaling, independent of STAT5, would participate in the context of “preorganized” signal transduction pathways which integrates with IL-2-STAT5 signaling [72]. Of note, STAT5 is a transcription factor and activates the transcription of several key proteins relevant for mTORC1 signaling such as IL-2R chains, AKT, RHEB or amino acid transporters and sensors, which participate in longer term activation of mTORC1 [70,71].
In T cells, the γc cytokine IL-7 is also known to activate mTORC1, although the molecular details are less documented than for IL-2. Indeed, IL-7 treatment enhances S6 phosphorylation [75] as well as cell size and survival [76] and glucose uptake [77]. This last metabolic change seems to require AKT and STAT5 activation [77], although no clear evidence has demonstrated a molecular link between these pathways and mTORC1 in IL-7 treated lymphocytes.
4.3. mTOR regulation by γc cytokines in NK cells
NK cell maturation and survival is governed by the γc cytokine IL-15. IL-15 treatment activates mTORC1 which increases cellular metabolism [16]. IL-15 also activates STAT5, AKT [16], ERK [78] and its downstream target RSK [79]. Several studies have investigated the role of these different proteins in the activation of mTORC1. Using pharmacological inhibitors, Nandagopal and col. Showed that PI3K or STAT5 inhibition following IL-15 treatment decreased NK cell production of IFN-γ and granzyme B as well as their proliferative capacity to the same extent that rapamycin. However, the status of mTORC1 activation itself was not measured in this study [80]. NK cells deficient for PTEN, an antagonist of PI3K, display increased AKT and S6 phosphorylation [81] while NK cells deficient for the catalytic subunits of PI3K p110γ and p110δ display a defective maturation and function [82]. Interestingly, the defect observed included a strong decrease in circulating NK cells and a near complete disappearance of the most mature CD11b+ subset, phenocopies of mTOR or Raptor deficiencies [16,83,84]. In addition, IL-15 activates the ERK pathway. The use of a MEK inhibitor suggests that ERK could participate in mTORC1 activation in NK cells as ERK inhibition leads to decreased S6 phosphorylation following IL-15 treatment [78]. Overall, these data are indicative of a role of both the PI3K-AKT, and ERK pathways in the control of mTORC1 in NK cells. On the contrary, a study conducted in IL-2/-12 in vitro activated NK cells reached the opposite conclusion that the PI3K-AKT pathway is not involved in mTORC1 activation. Indeed, in this last study, NK cells treated with AKT inhibitors show no decrease in cell size, IFN-γ production, granzyme expression or S6 phosphorylation [85]. This suggest that mTORC1 regulation differs when comparing CD8 T cells and resting or activated NK cells and that its regulation can be adapted to the cellular activation status. The reason for these differences is unknown at this point (Figure 2).
4.4. The role of the TSC axis in lymphocytes
As described above, TSC constitutes a hub controlling mTORC1 activation thanks to its capacity to integrate both negative and positive inputs. As a result, TSC null cell lines show maximal mTORC1 activity, to the same extent than insulin treated ones [86,87] whereas TSC overexpression results in drastic decreases of mTORC1 activity [33,52,88,89]. A similar derepression of mTORC1 is observed in vivo in primary muscle cells [90] or in hepatocytes [91] upon TSC inactivation. For these reasons, different groups looked at the importance of the TSC hub in T and NK cells.
Four distinct studies have investigated the role of TSC in T cells and concluded that TSC is required to maintain cell quiescence in the absence of antigen stimulation [20,92,93,94]. TSC1 specific deletion in immature T cells, using the T cell specific CD4-Cre deleter, leads to a decrease in CD4+ and CD8+ T cell number [92,93,94]. The remaining T cells show an increase in cell proliferation and display activation of the intrinsic apoptotic pathway [93,94], characterized by a decrease in the anti-apoptotic protein Bcl-2 [93]. They also display elevated cellular ROS which could be reversed by antioxidant treatments which restore T cell numbers [93,94]. This phenotype is associated with an increase in mTORC1 activity and a decreased activity of mTORC2 [93,94]. Such a decrease in mTORC2 activity is supposed to arise from the negative feedback loop leading from S6K to targets lying upstream of mTORC2 [45]. Focusing on CD8+ T cell, Pollizzi et al. show that TSC2 deficient T cells display characteristics of terminally differentiated effector T cell and are unable to transition to the memory state. Confirming these results, T cells deficient for the GTPase RHEB fail to differentiate into effector cells but retained memory characteristics [20].
In NK cell, TSC1 deletion at an early stage produces a similar phenotype than TSC1 deletion in immature T cells, with a decrease in NK cell number, associated with an increase in cell proliferation and apoptosis and a constitutive mTORC1 activation [95]. However, deletion of TSC1 in mature cells does not have any effect on NK cell phenotype, function or response to cytokines [95]. In the same line, inducible inhibition of TSC1 at latter stage in T cell using ER-Tamoxifen construct has only a mild effect on T cell population [93]. Of note, TSC is mainly express by progenitors and its expression decreases during the maturation process, both in NK and T cells [95], which could account for the more important phenotype of TSC deletion in immature cells compared to mature ones. These results also echo with those from the Cantrell’s group, suggesting that PI3K-AKT signaling is not mandatory for mTOR regulation by γc chain cytokines. In fact, T and NK cell homeostasis mainly depends on the γc cytokine IL-2 and IL-15 respectively, which as discussed earlier, may not depend on the classical pathway involving AKT and therefore TSC, one of the main target of AKT, for their signaling in lymphocyte. Of note, even if these studies show the importance of TSC in the maintenance of cell quiescence, TSC1 mutations in lymphocytes results in mild phenotype compared to other cell types. For instance, mice bearing liver specific TSC1 ablation developed hepatocellular carcinoma [91]. This suggests other mechanisms of regulation involving TSC-independent control of mTOR in mature lymphocytes.
5. mTOR activation downstream IL-1β family receptor in lymphocytes
5.1. Overview of the IL-1β cytokines family
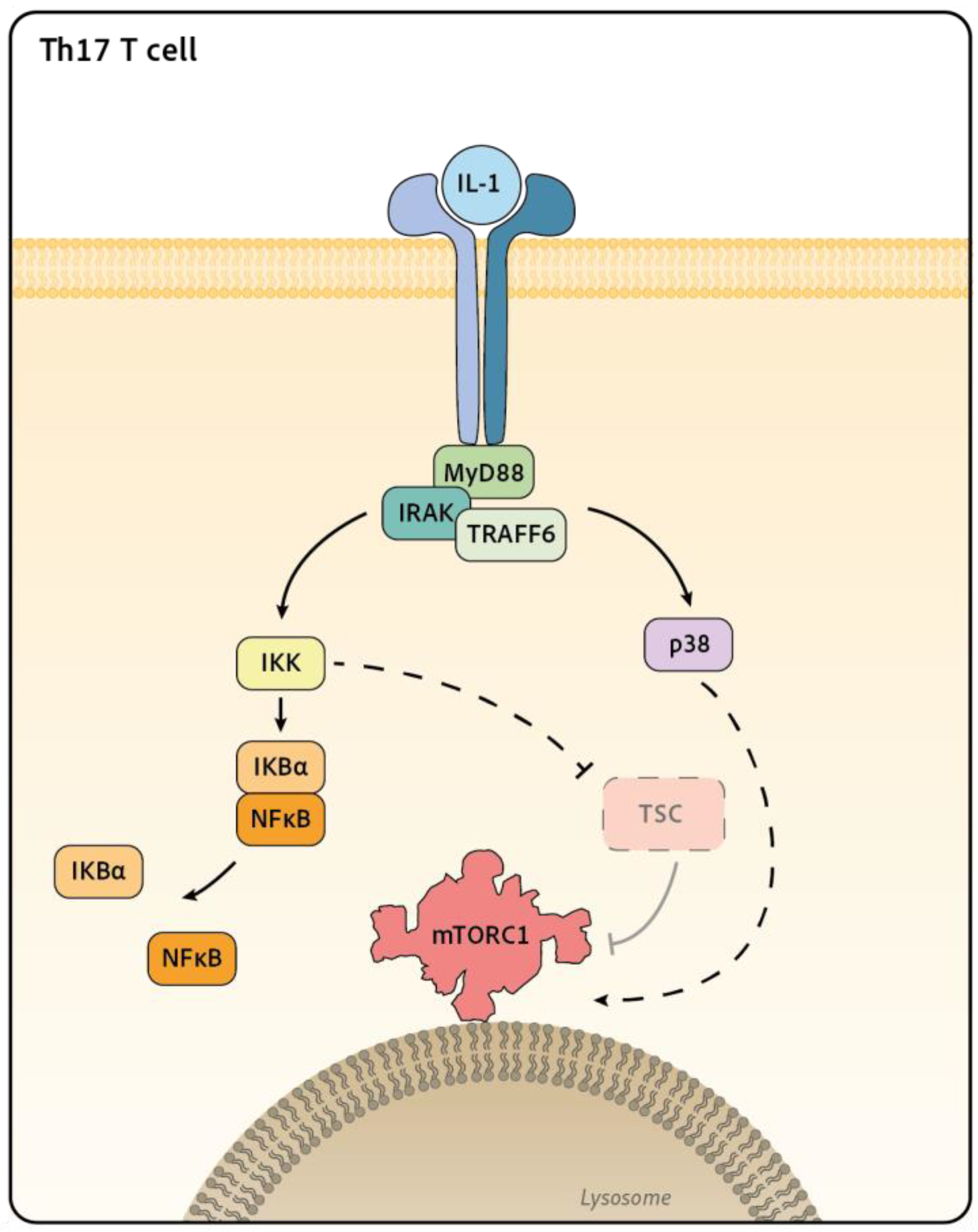
The IL-1 family is a large group of cytokines composed of agonist molecules such as IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, IL-36γ, IL-37 and antagonist ones like IL-1Rα. They all signal through receptors composed of two different chains, which are all characterized by the presence of a conserved cytosolic region containing a signalling domain called the Toll/IL-1 Receptor (TIR) domain. The binding of the cytokine to the first receptor chain triggers the recruitment of the second one and the juxtaposition of their TIR domains. This induces the recruitment of the protein MyD88 and the kinase IRAK4. The latter is activated by autophosphorylation leading to the subsequent recruitment and phosphorylation of IRAK1 and 2, two adaptors and kinase proteins. MyD88 and its IRAK partners then form a protein platform that recruits the E3 ubiquitin ligase TRAF6, which triggers the activation of different pathways such as the MAPK and NF-κB pathways [96].
IL-1 family cytokines typically engage the MAPK p38. There are 4 isoforms of p38 (p38α, β, γ, δ), the most expressed in mammalians being p38α. As for other MAPKs, p38 activation results from phosphorylation cascades involving different MAP3Ks, such as TAK1 which is classically engaged by IL-1. Furthermore, as described for other MAPK, p38 has the potential to phosphorylate a multitude of targets including other kinases such as the MAPK activated protein kinase (MAPKAPK) MK2, MK3 and MK5/PRAK [97]. Several studies report a control of mTORC1 by p38 in different cell lines, mentioning both activator [98,99,100,101] or inhibitor [102] role for p38 depending on the stimulus involved. In addition to the p38 MAPK pathway, IL-1 family cytokines also engage the NF-κB pathway. NF-κB activation requires activation of the IKK complex through its binding to polyubiquitin chains on several molecules downstream IL-1 receptor such as IRAK1 or TAK1. Activated IKK phosphorylates IκBα which promotes its degradation and the release and nuclear translocation of the NF-κB, promoting the pathway activation. IKK has also been involved in mTORC1 activation. Indeed, in one study, cancer cell lines treated with TNFα activate IKKβ, a component of the IKK complex, that interacts and phosphorylate TSC1, resulting in TSC1 inhibition and mTORC1 activation [103].
5.2. mTOR regulation by IL-1β cytokines family in T and NK cells
In T and NK cells, different cytokines of the IL-1 family have been shown to activate mTORC1, although the molecular pathway involved are not fully understood yet. mTORC1 has been shown to be activated by IL-18 [16,104] or by IL-33 [105] in NK cells, by IL-1 in Th17 [106,107] and IL-36b in CD8+ T cells [108,109]. The most complete mechanistic study comes from Gulen et al. [107]. Working on Th17, the authors showed that IL-1 induces the activation of mTORC1, measured by S6 and 4EBP1 phosphorylation. The authors also demonstrate that IL-1 signals through the IRAK1 and IRAK4 proteins to induce TSC disruption. The precise mechanism of TSC inactivation still awaits to be described. The authors propose that inactivation of this complex is the event leading to mTORC1 activation. In the same study, IL-1 treatment is also shown to induce p38, JNK and IκBα phosphorylation, although the implication of these pathways in TSC disruption was not clearly addressed [107] (Figure 3). Another study demonstrates that T cells deficient for p38α/β or MK2/3 show decreased mTORC1 activity as measured by S6 phosphorylation upon TCR stimulation [110]. This defect correlates with increased regulatory T cell (Treg) numbers in vivo, which is reminiscent of the phenotype of mTOR deficient T cells [111]. This suggests that p38 could control mTORC1 activation in lymphocytes however the significance of this pathway for cytokine signaling remains to be determined. As mentioned earlier, mTORC1 activity is also controlled by nutrients availability. In this context, Almutairi et al. showed that IL-18 treatment leads to enhanced surface expression of the leucine transporter CD98/LAT-1 on NK cells. This resulted in increased leucine import thus probably participating in mTORC1 activation [104]. It should however be noted that CD98 induction by IL-18 takes place after an overnight stimulation, it is thus unlikely that this mechanism is at play in short-term (<4h) activation of mTORC1 upon IL-18 stimulation.
5.3. Signal Integration toward mTOR control
Multiple signaling pathways modulate mTORC1 activity, as reviewed here. The outcome of costimulation with multiple cytokines will have to be deciphered. Indeed, we mostly focused our review of the literature on data reporting the effect of cytokines taken in isolation. However, cells generally encounter cytokines simultaneously. For instance, generation of the so-called CIML (cytokine induced memory like cells) for therapeutic purposes implies the stimulation of NK cells with IL-15 and IL-18, both having the potential to activate mTORC1 with parallel pathways. Moreover, coincident stimulation with different cytokines can arguably lead to contradictory effects. For instance, T cell and NK cell proliferation and cytoxicity induced in vitro with IL-2 can be inhibited by TGF-β treatment [112,113]. Interestingly, TGF-β antagonizes IL-15 activation of mTOR in NK cells, resulting in a reduction in NK cell bioenergetic metabolism. This inhibition is detectable in the minutes following TGF- β treatment and is specific to mTOR since it does not impact STAT5 activation [114]. More recently, TGF-β has been shown to decrease mTOR activity in T cells [115]. In this study, early inhibition of mTOR in precursors of exhausted CD8+ T cells preserved their metabolic capacity and sustains the T cell response during chronic infection. In both studies, the molecular mechanism at play remains elusive but could well involve some of the molecular players described above. Indeed, TGF-β has been shown to potentially activate a large set of pathways in a variety of cell types. This includes TAK1 [116], ERK [117], p38 [118] and PI3K [119]. Another possible mechanism to explain the inhibition of mTORC1 by TGF-β is the phosphatases PP2A. PP2A has been shown to inhibits mTORC1 in Tregs and the ablation of PP2A in Treg leads to severe autoimmunity [120]. The direct link between PP2A and the TGF-β signaling still remains to be determined. Whether activation of these pathways enters in competition with their involvement in mTOR signalling remains to be tested.
Some studies also highlight that several transduction pathways can be engaged for a given stimulus. For instance, in NK cells a low dose of IL-15 only trigger STAT5 activation and cell survival whereas higher doses engage mTORC1 and cell proliferation [16]. Similarly, in another model studying stress granule formation upon arsenite treatment, Heberle et al. addressed the respective importance of PI3K-AKT and p38 pathways in the control of mTORC1 activation in multiple cell lines. Using both in silico and in vitro approaches, the authors showed that PI3K-AKT and p38 pathways act in a hierarchical manner toward mTORC1 activation. Inhibition of PI3K results in residual mTORC1 activation, measured by p70-S6K phosphorylation, which is inhibited by an additional treatment with a p38 inhibitor. Based on these findings, the authors further show that the effect of p38 inhibition on mTORC1 activity are significant when PI3K activity is decreased to 40% or lower. From these results, it appears that a moderate stress activates mTORC1 through p38 whereas increasing the stress signal also engages the PI3K-AKT pathway leading to stronger mTORC1 activation [121]. Even though this study has not been performed on lymphocytes, we can hypothesize that similar mechanisms may take place in T and NK cells in response to different cytokines concentrations.
6. Targeting mTOR activation in lymphocytes for immunotherapy development
Controlling mTOR signaling constitute a promising strategy to modulate lymphocytes activity. The mTOR inhibitor rapamycin and its analogs were first approved as immunosuppressants in the context of transplantation and then autoimmunity. In parallel, mTOR is hyperactivated in up to 80% of cancers [45], founding the use of mTOR inhibitors as antiproliferative drugs in chemotherapies. This is not without side effects. Indeed, studies from our group have shown that patients with metastatic breast cancer treated with the mTOR inhibitor everolimus, display lower level of mTORC1 activation in NK cells as well as a decrease in NK cell number and maturation [122]. As NK cells have crucial antimetastatic activity [123], this side effect of mTOR inhibitors should be considered for the development of next generation therapies. At the same time, mTOR inhibition may also benefit certain types of responses and promote certain differentiation pathways. Indeed, as discussed above, mTORC1 inhibition favors memory CD8+ T cell generation [18,20] and have protective effect against T CD8+ exhaustion in chronic infection [115]. For these reasons, mTORC1 activation level in immune cell has to be well balanced to insure a favorable anti-tumoral effect.
In this context, a large set of therapies are now aiming at boosting lymphocyte functions in particular for adoptive cell transfer therapies in cancer. Chimeric Antigen Receptor (CAR) T and NK cell-based immunotherapies have revolutionized the treatment of certain cancers [124]. The use of such therapies to treat viral infections (EBV and others) is also under assessment [125]. A better understanding of cytokines signaling toward mTOR could lead to improved T and NK cell expansion in vitro. Study of cytokine signaling has also the potential to benefit to tailoring better cytokine cocktails in order to prime lymphocyte in vitro. In the same line of idea, a better understanding of the signal transduction pathways involved downstream cytokine receptors could inform the design of engineered receptors to improve CAR cells effector functions (review in [126]). Therefore, mTORC1 tight regulation is required to ensures a proper response to pathogen and cancer, which will benefit from a better understanding of its specific regulation in immune cell.
Author Contributions
Writing—original draft preparation, L.F. and A.M.; writing—review T.W.
Funding
This work was funded by grants from Ecole Normale supérieure de Lyon and Fondation pour la Recherche Médicale (FDT202304016764).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We like to thank all the member of the Lyacts team and Pr Olivier Thaunat for fruitful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the MTORC1 Pathway. Cell Metab. 2017, 26, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Lanier, L.L. NK Cell Development, Homeostasis and Function: Parallels with CD8+ T Cells. Nat. Rev. Immunol. 2011, 11, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Ring, A.M.; Lin, J.-X.; Feng, D.; Mitra, S.; Rickert, M.; Bowman, G.R.; Pande, V.S.; Li, P.; Moraga, I.; Spolski, R.; et al. Mechanistic and Structural Insight into the Functional Dichotomy between IL-2 and IL-15. Nat. Immunol. 2012, 13, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sasson, S.Z.; Hogg, A.; Hu-Li, J.; Wingfield, P.; Chen, X.; Crank, M.; Caucheteux, S.; Ratner-Hurevich, M.; Berzofsky, J.A.; Nir-Paz, R.; et al. IL-1 Enhances Expansion, Effector Function, Tissue Localization, and Memory Response of Antigen-Specific CD8 T Cells. J. Exp. Med. 2013, 210, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Chaix, J.; Tessmer, M.S.; Hoebe, K.; Fuséri, N.; Ryffel, B.; Dalod, M.; Alexopoulou, L.; Beutler, B.; Brossay, L.; Vivier, E.; et al. Cutting Edge: Priming of NK Cells by IL-18. J. Immunol. Baltim. Md 1950 2008, 181, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Gaubitz, C.; Oliveira, T.M.; Prouteau, M.; Leitner, A.; Karuppasamy, M.; Konstantinidou, G.; Rispal, D.; Eltschinger, S.; Robinson, G.C.; Thore, S.; et al. Molecular Basis of the Rapamycin Insensitivity of Target Of Rapamycin Complex 2. Mol. Cell 2015, 58, 977–988. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Martel, R.R.; Klicius, J.; Galet, S. Inhibition of the Immune Response by Rapamycin, a New Antifungal Antibiotic. Can. J. Physiol. Pharmacol. 1977, 55, 48–51. [Google Scholar] [CrossRef]
- Kuehn, H.S.; Niemela, J.E.; Rangel-Santos, A.; Zhang, M.; Pittaluga, S.; Stoddard, J.L.; Hussey, A.A.; Evbuomwan, M.O.; Priel, D.A.L.; Kuhns, D.B.; et al. Loss-of-Function of the Protein Kinase C δ (PKCδ) Causes a B-Cell Lymphoproliferative Syndrome in Humans. Blood 2013, 121, 3117–3125. [Google Scholar] [CrossRef]
- Gu, H.; Chen, Z.; Ma, J.; Wang, J.; Zhang, R.; Wu, R.; Wang, T. Sirolimus Is Effective in Autoimmune Lymphoproliferative Syndrome-Type III: A Pedigree Case Report with Homozygous Variation PRKCD. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211025936. [Google Scholar] [CrossRef]
- Moreews, M.; Mathieu, A.-L.; Pouxvielh, K.; Reuschlé, Q.; Drouillard, A.; Dessay, P.; Meignien, M.; Zhang, J.; Fallone, L.; Rousseaux, N.; et al. MTOR Activation Underlies Enhanced B Cell Proliferation and Autoimmunity in PrkcdG510S/G510S Mice. J. Immunol. Baltim. Md 1950 2023, 210, 1209–1221. [Google Scholar] [CrossRef]
- Berger, S.P.; Sommerer, C.; Witzke, O.; Tedesco, H.; Chadban, S.; Mulgaonkar, S.; Qazi, Y.; de Fijter, J.W.; Oppenheimer, F.; Cruzado, J.M.; et al. Two-Year Outcomes in de Novo Renal Transplant Recipients Receiving Everolimus-Facilitated Calcineurin Inhibitor Reduction Regimen from the TRANSFORM Study. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2019, 19, 3018–3034. [Google Scholar] [CrossRef]
- Koenig, A.; Chen, C.-C.; Marçais, A.; Barba, T.; Mathias, V.; Sicard, A.; Rabeyrin, M.; Racapé, M.; Duong-Van-Huyen, J.-P.; Bruneval, P.; et al. Missing Self Triggers NK Cell-Mediated Chronic Vascular Rejection of Solid Organ Transplants. Nat. Commun. 2019, 10, 5350. [Google Scholar] [CrossRef]
- Chi, H. Regulation and Function of MTOR Signalling in T Cell Fate Decision. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Marçais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The Metabolic Checkpoint Kinase MTOR Is Essential for IL-15 Signaling during the Development and Activation of NK Cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef]
- Marçais, A.; Marotel, M.; Degouve, S.; Koenig, A.; Fauteux-Daniel, S.; Drouillard, A.; Schlums, H.; Viel, S.; Besson, L.; Allatif, O.; et al. High MTOR Activity Is a Hallmark of Reactive Natural Killer Cells and Amplifies Early Signaling through Activating Receptors. eLife 2017, 6, e26423. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. MTOR Regulates Memory CD8 T-Cell Differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.-S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-Cell Memory by Modulating Fatty Acid Metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Pollizzi, K.N.; Patel, C.H.; Sun, I.-H.; Oh, M.-H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. MTORC1 and MTORC2 Selectively Regulate CD8+ T Cell Differentiation. J. Clin. Invest. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.H.; Brunn, G.J.; Kohn, A.D.; Roth, R.A.; Lawrence, J.C. Evidence of Insulin-Stimulated Phosphorylation and Activation of the Mammalian Target of Rapamycin Mediated by a Protein Kinase B Signaling Pathway. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 7772–7777. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and Cellular Properties of Insulin Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- Lucas, C.L.; Chandra, A.; Nejentsev, S.; Condliffe, A.M.; Okkenhaug, K. PI3Kδ and Primary Immunodeficiencies. Nat. Rev. Immunol. 2016, 16, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Joung, S.M.; Park, Z.-Y.; Rani, S.; Takeuchi, O.; Akira, S.; Lee, J.Y. Akt Contributes to Activation of the TRIF-Dependent Signaling Pathways of TLRs by Interacting with TANK-Binding Kinase 1. J. Immunol. Baltim. Md 1950 2011, 186, 499–507. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, D.; Zhao, B.; Lu, M.-K.; You, M.; Condorelli, G.; Wang, C.-Y.; Guan, K.-L. IκB Kinase ε and TANK-Binding Kinase 1 Activate AKT by Direct Phosphorylation. Proc. Natl. Acad. Sci. 2011, 108, 6474–6479. [Google Scholar] [CrossRef]
- Ou, Y.-H.; Torres, M.; Ram, R.; Formstecher, E.; Roland, C.; Cheng, T.; Brekken, R.; Wurz, R.; Tasker, A.; Polverino, T.; et al. TBK1 Directly Engages Akt/PKB Survival Signaling to Support Oncogenic Transformation. Mol. Cell 2011, 41, 458–470. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 Is a Third Subunit of the TSC1-TSC2 Complex Upstream of MTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase Is a Direct Target of TSC2 GAP Activity and Regulates MTOR Signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex Gene Products, Tuberin and Hamartin, Control MTOR Signaling by Acting as a GTPase-Activating Protein Complex toward Rheb. Curr. Biol. CB 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Structural Mechanisms of MTORC1 Activation by RHEB and Inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Vander Haar, E.; Lee, S.-I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin Signalling to MTOR Mediated by the Akt/PKB Substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the MTORC1 Protein Kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the Tuberous Sclerosis Complex-2 Tumor Suppressor Gene Product Tuberin as a Target of the Phosphoinositide 3-Kinase/Akt Pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and Functional Inactivation of TSC2 by Erk Implications for Tuberous Sclerosis and Cancer Pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef]
- Roux, P.P.; Ballif, B.A.; Anjum, R.; Gygi, S.P.; Blenis, J. Tumor-Promoting Phorbol Esters and Activated Ras Inactivate the Tuberous Sclerosis Tumor Suppressor Complex via P90 Ribosomal S6 Kinase. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13489–13494. [Google Scholar] [CrossRef] [PubMed]
- Demetriades, C.; Doumpas, N.; Teleman, A.A. Regulation of TORC1 in Response to Amino Acid Starvation via Lysosomal Recruitment of TSC2. Cell 2014, 156, 786–799. [Google Scholar] [CrossRef]
- Demetriades, C.; Plescher, M.; Teleman, A.A. Lysosomal Recruitment of TSC2 Is a Universal Response to Cellular Stress. Nat. Commun. 2016, 7, 10662. [Google Scholar] [CrossRef]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial Control of the TSC Complex Integrates Insulin and Nutrient Regulation of MTORC1 at the Lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef]
- Carroll, B.; Maetzel, D.; Maddocks, O.D.; Otten, G.; Ratcliff, M.; Smith, G.R.; Dunlop, E.A.; Passos, J.F.; Davies, O.R.; Jaenisch, R.; et al. Control of TSC2-Rheb Signaling Axis by Arginine Regulates MTORC1 Activity. eLife 2016, 5, e11058. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 Complex: A Molecular Switchboard Controlling Cell Growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.-L. Regulation of TORC1 by Rag GTPases in Nutrient Response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases Bind Raptor and Mediate Amino Acid Signaling to MTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets MTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator Is a GEF for the Rag GTPases That Signal Amino Acid Levels to MTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. MTOR at the Nexus of Nutrition, Growth, Ageing and Disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Vellai, T. How the Amino Acid Leucine Activates the Key Cell-Growth Regulator MTOR. Nature 2021, 596, 192–194. [Google Scholar] [CrossRef]
- Orozco, J.M.; Krawczyk, P.A.; Scaria, S.M.; Cangelosi, A.L.; Chan, S.H.; Kunchok, T.; Lewis, C.A.; Sabatini, D.M. Dihydroxyacetone Phosphate Signals Glucose Availability to MTORC1. Nat. Metab. 2020, 2, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.N.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal Cholesterol Activates MTORC1 via an SLC38A9-Niemann-Pick C1 Signaling Complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef]
- Lim, C.-Y.; Davis, O.B.; Shin, H.R.; Zhang, J.; Berdan, C.A.; Jiang, X.; Counihan, J.L.; Ory, D.S.; Nomura, D.K.; Zoncu, R. ER-Lysosome Contacts Enable Cholesterol Sensing by MTORC1 and Drive Aberrant Growth Signalling in Niemann-Pick Type C. Nat. Cell Biol. 2019, 21, 1206–1218. [Google Scholar] [CrossRef]
- Shin, H.R.; Citron, Y.R.; Wang, L.; Tribouillard, L.; Goul, C.S.; Stipp, R.; Sugasawa, Y.; Jain, A.; Samson, N.; Lim, C.-Y.; et al. Lysosomal GPCR-like Protein LYCHOS Signals Cholesterol Sufficiency to MTORC1. Science 2022, 377, 1290–1298. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 Is Phosphorylated and Inhibited by Akt and Suppresses MTOR Signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G. Regulation of MTOR Function in Response to Hypoxia by REDD1 and the TSC1/TSC2 Tumor Suppressor Complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia Regulates TSC1/2-MTOR Signaling and Tumor Suppression through REDD1-Mediated 14-3-3 Shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The Regulation of AMPK Beta1, TSC2, and PTEN Expression by P53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-MTOR Pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Heberle, A.M.; Prentzell, M.T.; van Eunen, K.; Bakker, B.M.; Grellscheid, S.N.; Thedieck, K. Molecular Mechanisms of MTOR Regulation by Stress. Mol. Cell. Oncol. 2014, 2, e970489. [Google Scholar] [CrossRef]
- Noguchi, M.; Yi, H.; Rosenblatt, H.M.; Filipovich, A.H.; Adelstein, S.; Modi, W.S.; McBride, O.W.; Leonard, W.J. Interleukin-2 Receptor Gamma Chain Mutation Results in X-Linked Severe Combined Immunodeficiency in Humans. Cell 1993, 73, 147–157. [Google Scholar] [CrossRef]
- DiSanto, J.P.; Müller, W.; Guy-Grand, D.; Fischer, A.; Rajewsky, K. Lymphoid Development in Mice with a Targeted Deletion of the Interleukin 2 Receptor Gamma Chain. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.; Mariner, J.; Waldmann, T.A.; Tagaya, Y. IL-15Ralpha Recycles and Presents IL-15 In Trans to Neighboring Cells. Immunity 2002, 17, 537–547. [Google Scholar] [CrossRef]
- Mortier, E.; Woo, T.; Advincula, R.; Gozalo, S.; Ma, A. IL-15Ralpha Chaperones IL-15 to Stable Dendritic Cell Membrane Complexes That Activate NK Cells via Trans Presentation. J. Exp. Med. 2008, 205, 1213–1225. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Barber, D.L.; Nakarai, T.; Freeman, G.J.; Gribben, J.G.; Bernstein, G.M.; D’Andrea, A.D.; Ritz, J.; Nadler, L.M. Prevention of T Cell Anergy by Signaling through the Gamma c Chain of the IL-2 Receptor. Science 1994, 266, 1039–1042. [Google Scholar] [CrossRef]
- Miyazaki, T.; Kawahara, A.; Fujii, H.; Nakagawa, Y.; Minami, Y.; Liu, Z.J.; Oishi, I.; Silvennoinen, O.; Witthuhn, B.A.; Ihle, J.N. Functional Activation of Jak1 and Jak3 by Selective Association with IL-2 Receptor Subunits. Science 1994, 266, 1045–1047. [Google Scholar] [CrossRef]
- Russell, S.M.; Johnston, J.A.; Noguchi, M.; Kawamura, M.; Bacon, C.M.; Friedmann, M.; Berg, M.; McVicar, D.W.; Witthuhn, B.A.; Silvennoinen, O. Interaction of IL-2R Beta and Gamma c Chains with Jak1 and Jak3: Implications for XSCID and XCID. Science 1994, 266, 1042–1045. [Google Scholar] [CrossRef]
- Truitt, K.E.; Mills, G.B.; Turck, C.W.; Imboden, J.B. SH2-Dependent Association of Phosphatidylinositol 3′-Kinase 85-KDa Regulatory Subunit with the Interleukin-2 Receptor Beta Chain. J. Biol. Chem. 1994, 269, 5937–5943. [Google Scholar] [CrossRef] [PubMed]
- Delespine-Carmagnat, M.; Bouvier, G.; Bertoglio, J. Association of STAT1, STAT3 and STAT5 Proteins with the IL-2 Receptor Involves Different Subdomains of the IL-2 Receptor Beta Chain. Eur. J. Immunol. 2000, 30, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.J.; Chung, J.; Fiorentino, D.F.; Flanagan, W.M.; Blenis, J.; Crabtree, G.R. Rapamycin Selectively Inhibits Interleukin-2 Activation of P70 S6 Kinase. Nature 1992, 358, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Reif, K.; Burgering, B.M.; Cantrell, D.A. Phosphatidylinositol 3-Kinase Links the Interleukin-2 Receptor to Protein Kinase B and P70 S6 Kinase. J. Biol. Chem. 1997, 272, 14426–14433. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, A.N.; Finlay, D.; Preston, G.; Sinclair, L.V.; Waugh, C.M.; Tamas, P.; Feijoo, C.; Okkenhaug, K.; Cantrell, D.A. Protein Kinase B Controls Transcriptional Programs That Direct Cytotoxic T Cell Fate but Is Dispensable for T Cell Metabolism. Immunity 2011, 34, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.K.; Rosenzweig, E.; Sinclair, L.V.; Feijoo-Carnero, C.; Hukelmann, J.L.; Rolf, J.; Panteleyev, A.A.; Okkenhaug, K.; Cantrell, D.A. PDK1 Regulation of MTOR and Hypoxia-Inducible Factor 1 Integrate Metabolism and Migration of CD8+ T Cells. J. Exp. Med. 2012, 209, 2441–2453. [Google Scholar] [CrossRef]
- Villarino, A.V.; Laurence, A.D.; Davis, F.P.; Nivelo, L.; Brooks, S.R.; Sun, H.-W.; Jiang, K.; Afzali, B.; Frasca, D.; Hennighausen, L.; et al. A Central Role for STAT5 in the Transcriptional Programing of T Helper Cell Metabolism. Sci. Immunol. 2022, 7, eabl9467. [Google Scholar] [CrossRef]
- Rollings, C.M.; Sinclair, L.V.; Brady, H.J.M.; Cantrell, D.A.; Ross, S.H. Interleukin-2 Shapes the Cytotoxic T Cell Proteome and Immune Environment-Sensing Programs. Sci. Signal. 2018, 11, eaap8112. [Google Scholar] [CrossRef]
- Ross, S.H.; Rollings, C.; Anderson, K.E.; Hawkins, P.T.; Stephens, L.R.; Cantrell, D.A. Phosphoproteomic Analyses of Interleukin 2 Signaling Reveal Integrated JAK Kinase-Dependent and -Independent Networks in CD8+ T Cells. Immunity 2016, 45, 685–700. [Google Scholar] [CrossRef]
- Grzes, K.M.; Swamy, M.; Hukelmann, J.L.; Emslie, E.; Sinclair, L.V.; Cantrell, D.A. Control of Amino Acid Transport Coordinates Metabolic Reprogramming in T-Cell Malignancy. Leukemia 2017, 31, 2771–2779. [Google Scholar] [CrossRef]
- Newton, A.C. Regulation of the ABC Kinases by Phosphorylation: Protein Kinase C as a Paradigm. Biochem. J. 2003, 370, 361–371. [Google Scholar] [CrossRef]
- Xu, A.; Leary, S.C.; Islam, M.F.; Wu, Z.; Bhanumathy, K.K.; Ara, A.; Chibbar, R.; Fleywald, A.; Ahmed, K.A.; Xiang, J. Prosurvival IL-7-Stimulated Weak Strength of MTORC1-S6K Controls T Cell Memory via Transcriptional FOXO1-TCF1-Id3 and Metabolic AMPKα1-ULK1-ATG7 Pathways. J. Immunol. Baltim. Md 1950 2022, 208, 155–168. [Google Scholar] [CrossRef]
- Rathmell, J.C.; Farkash, E.A.; Gao, W.; Thompson, C.B. IL-7 Enhances the Survival and Maintains the Size of Naive T Cells1. J. Immunol. 2001, 167, 6869–6876. [Google Scholar] [CrossRef] [PubMed]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 Promotes Glut1 Trafficking and Glucose Uptake via STAT5-Mediated Activation of Akt to Support T-Cell Survival. Blood 2008, 111, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Niogret, C.; Miah, S.M.S.; Rota, G.; Fonta, N.P.; Wang, H.; Held, W.; Birchmeier, W.; Sexl, V.; Yang, W.; Vivier, E.; et al. Shp-2 Is Critical for ERK and Metabolic Engagement Downstream of IL-15 Receptor in NK Cells. Nat. Commun. 2019, 10, 1444. [Google Scholar] [CrossRef] [PubMed]
- MacMullan, M.A.; Wang, P.; Graham, N.A. Phospho-Proteomics Reveals That RSK Signaling Is Required for Proliferation of Natural Killer Cells Stimulated with IL-2 or IL-15. Cytokine 2022, 157, 155958. [Google Scholar] [CrossRef] [PubMed]
- Nandagopal, N.; Ali, A.K.; Komal, A.K.; Lee, S.-H. The Critical Role of IL-15–PI3K–MTOR Pathway in Natural Killer Cell Effector Functions. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Briercheck, E.L.; Trotta, R.; Chen, L.; Hartlage, A.S.; Cole, J.P.; Cole, T.D.; Mao, C.; Banerjee, P.P.; Hsu, H.-T.; Mace, E.M.; et al. PTEN Is a Negative Regulator of NK Cell Cytolytic Function. J. Immunol. Author Choice 2015, 194, 1832–1840. [Google Scholar] [CrossRef]
- Tassi, I.; Cella, M.; Gilfillan, S.; Turnbull, I.; Diacovo, T.G.; Penninger, J.M.; Colonna, M. P110gamma and P110delta Phosphoinositide 3-Kinase Signaling Pathways Synergize to Control Development and Functions of Murine NK Cells. Immunity 2007, 27, 214–227. [Google Scholar] [CrossRef]
- Yang, C.; Tsaih, S.-W.; Lemke, A.; Flister, M.J.; Thakar, M.S.; Malarkannan, S. MTORC1 and MTORC2 Differentially Promote Natural Killer Cell Development. eLife 2018, 7, e35619. [Google Scholar] [CrossRef]
- Wang, F.; Meng, M.; Mo, B.; Yang, Y.; Ji, Y.; Huang, P.; Lai, W.; Pan, X.; You, T.; Luo, H.; et al. Crosstalks between MTORC1 and MTORC2 Variagate Cytokine Signaling to Control NK Maturation and Effector Function. Nat. Commun. 2018, 9, 4874. [Google Scholar] [CrossRef]
- Loftus, R.M.; Assmann, N.; Kedia-Mehta, N.; O’Brien, K.L.; Garcia, A.; Gillespie, C.; Hukelmann, J.L.; Oefner, P.J.; Lamond, A.I.; Gardiner, C.M.; et al. Amino Acid-Dependent CMyc Expression Is Essential for NK Cell Metabolic and Functional Responses in Mice. Nat. Commun. 2018, 9, 2341. [Google Scholar] [CrossRef]
- Dunkerly-Eyring, B.L.; Pan, S.; Pinilla-Vera, M.; McKoy, D.; Mishra, S.; Martinez, M.I.G.; Oeing, C.U.; Ranek, M.J.; Kass, D.A. Single Serine on TSC2 Exerts Biased Control over MTORC1 Activation Mediated by ERK1/2 but Not Akt. Life Sci. Alliance 2022, 5. [Google Scholar] [CrossRef]
- Zhang, H.; Cicchetti, G.; Onda, H.; Koon, H.B.; Asrican, K.; Bajraszewski, N.; Vazquez, F.; Carpenter, C.L.; Kwiatkowski, D.J. Loss of Tsc1/Tsc2 Activates MTOR and Disrupts PI3K-Akt Signaling through Downregulation of PDGFR. J. Clin. Invest. 2003, 112, 1223–1233. [Google Scholar] [CrossRef]
- Goncharova, E.A.; Goncharov, D.A.; Eszterhas, A.; Hunter, D.S.; Glassberg, M.K.; Yeung, R.S.; Walker, C.L.; Noonan, D.; Kwiatkowski, D.J.; Chou, M.M.; et al. Tuberin Regulates P70 S6 Kinase Activation and Ribosomal Protein S6 Phosphorylation. A Role for the TSC2 Tumor Suppressor Gene in Pulmonary Lymphangioleiomyomatosis (LAM). J. Biol. Chem. 2002, 277, 30958–30967. [Google Scholar] [CrossRef]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex-1 and -2 Gene Products Function Together to Inhibit Mammalian Target of Rapamycin (MTOR)-Mediated Downstream Signaling. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 13571–13576. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Lin, S.; Romanino, K.; Castets, P.; Guridi, M.; Summermatter, S.; Handschin, C.; Tintignac, L.A.; Hall, M.N.; Rüegg, M.A. Differential Response of Skeletal Muscles to MTORC1 Signaling during Atrophy and Hypertrophy. Skelet. Muscle 2013, 3, 6. [Google Scholar] [CrossRef]
- Menon, S.; Yecies, J.L.; Zhang, H.H.; Howell, J.J.; Nicholatos, J.; Harputlugil, E.; Bronson, R.T.; Kwiatkowski, D.J.; Manning, B.D. Chronic Activation of MTOR Complex 1 Is Sufficient to Cause Hepatocellular Carcinoma in Mice. Sci. Signal. 2012, 5, ra24. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, Y.; Chen, C.; Ikenoue, T.; Qiao, Y.; Li, C.-S.; Li, W.; Guan, K.-L.; Liu, Y.; Zheng, P. The Tuberous Sclerosis Complex-Mammalian Target of Rapamycin Pathway Maintains the Quiescence and Survival of Naive T Cells. J. Immunol. Baltim. Md 1950 2011, 187, 1106–1112. [Google Scholar] [CrossRef]
- Yang, K.; Neale, G.; Green, D.R.; He, W.; Chi, H. The Tumor Suppressor Tsc1 Enforces Quiescence of Naive T Cells to Promote Immune Homeostasis and Function. Nat. Immunol. 2011, 12, 888–897. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, T.F.; Gorentla, B.K.; Xie, D.; Srivatsan, S.; McLeod, I.X.; He, Y.-W.; Zhong, X.-P. Regulation of T-Cell Survival and Mitochondrial Homeostasis by TSC1. Eur. J. Immunol. 2011, 41, 3361–3370. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, S.; Du, J.; He, J.; Wang, Y.; Li, Z.; Liu, G.; Peng, W.; Zeng, X.; Li, D.; et al. NK Cell Development Requires Tsc1-Dependent Negative Regulation of IL-15-Triggered MTORC1 Activation. Nat. Commun. 2016, 7, 12730. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.E.; Smith, D.E. The IL-1 Family: Regulators of Immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Nebreda, A.R. Diversity and Versatility of P38 Kinase Signalling in Health and Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef]
- Li, Y.; Inoki, K.; Vacratsis, P.; Guan, K.-L. The P38 and MK2 Kinase Cascade Phosphorylates Tuberin, the Tuberous Sclerosis 2 Gene Product, and Enhances Its Interaction with 14-3-3*. J. Biol. Chem. 2003, 278, 13663–13671. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-N.; Wang, X.-K.; Wu, S.-Q.; Lu, J.; Zheng, M.; Wang, Y.-H.; Zhou, H.; Zhang, H.; Han, J. Phosphorylation of Raptor by P38beta Participates in Arsenite-Induced Mammalian Target of Rapamycin Complex 1 (MTORC1) Activation. J. Biol. Chem. 2011, 286, 31501–31511. [Google Scholar] [CrossRef]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 Polyubiquitination and Activation of MTOR by the P62-TRAF6 Complex in Nutrient-Activated Cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef]
- Linares, J.F.; Duran, A.; Reina-Campos, M.; Aza-Blanc, P.; Campos, A.; Moscat, J.; Diaz-Meco, M.T. Amino Acid Activation of MTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell Rep. 2015, 12, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Wang, Y.-H.; Wu, X.-N.; Wu, S.-Q.; Lu, B.-J.; Dong, M.-Q.; Zhang, H.; Sun, P.; Lin, S.-C.; Guan, K.-L.; et al. Inactivation of Rheb by PRAK-Mediated Phosphorylation Is Essential for Energy-Depletion-Induced Suppression of MTORC1. Nat. Cell Biol. 2011, 13, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-F.; Kuo, H.-P.; Chen, C.-T.; Hsu, J.-M.; Chou, C.-K.; Wei, Y.; Sun, H.-L.; Li, L.-Y.; Ping, B.; Huang, W.-C.; et al. IKK Beta Suppression of TSC1 Links Inflammation and Tumor Angiogenesis via the MTOR Pathway. Cell 2007, 130, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Almutairi, S.M.; Ali, A.K.; He, W.; Yang, D.-S.; Ghorbani, P.; Wang, L.; Fullerton, M.D.; Lee, S.-H. Interleukin-18 up-Regulates Amino Acid Transporters and Facilitates Amino Acid-Induced MTORC1 Activation in Natural Killer Cells. J. Biol. Chem. 2019, 294, 4644–4655. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, A.; Blanc, E.; Picant, V.; Alcazer, V.; Rocca, Y.; Ardin, M.; Voissière, A.; Onodi, F.; Rodriguez, C.; Tonon, L.; et al. IL-33 Drives Polyfunctionality and Antitumor Activity of a Unique ST2+ NK Cell Population 2023, 2023. 02.14.52 8486.
- Chang, J.; Burkett, P.R.; Borges, C.M.; Kuchroo, V.K.; Turka, L.A.; Chang, C.-H. MyD88 Is Essential to Sustain MTOR Activation Necessary to Promote T Helper 17 Cell Proliferation by Linking IL-1 and IL-23 Signaling. Proc. Natl. Acad. Sci. 2013, 110, 2270–2275. [Google Scholar] [CrossRef]
- Gulen, M.F.; Kang, Z.; Bulek, K.; Youzhong, W.; Kim, T.W.; Chen, Y.; Altuntas, C.Z.; Sass Bak-Jensen, K.; McGeachy, M.J.; Do, J.-S.; et al. The Receptor SIGIRR Suppresses Th17 Cell Proliferation via Inhibition of the Interleukin-1 Receptor Pathway and MTOR Kinase Activation. Immunity 2010, 32, 54–66. [Google Scholar] [CrossRef]
- Liang, Y.; Wang, X.; Wang, H.; Yang, W.; Yi, P.; Soong, L.; Cong, Y.; Cai, J.; Fan, X.; Sun, J. IL-33 Activates MTORC1 and Modulates Glycolytic Metabolism in CD8+ T Cells. Immunology 2022, 165, 61–73. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, X.; Shen, X.; Tang, P.; Chen, C.; Zhu, Q.; Li, M.; Xia, R.; Yang, X.; Feng, C.; et al. IL-36β Promotes CD8+ T Cell Activation and Antitumor Immune Responses by Activating MTORC1. Front. Immunol. 2019, 10, 1803. [Google Scholar] [CrossRef]
- Hayakawa, M.; Hayakawa, H.; Petrova, T.; Ritprajak, P.; Sutavani, R.V.; Jiménez-Andrade, G.Y.; Sano, Y.; Choo, M.-K.; Seavitt, J.; Venigalla, R.K.C.; et al. Loss of Functionally Redundant P38 Isoforms in T Cells Enhances Regulatory T Cell Induction. J. Biol. Chem. 2017, 292, 1762–1772. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. MTOR Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Ortaldo, J.R.; Mason, A.T.; O’Shea, J.J.; Smyth, M.J.; Falk, L.A.; Kennedy, I.C.; Longo, D.L.; Ruscetti, F.W. Mechanistic Studies of Transforming Growth Factor-Beta Inhibition of IL-2-Dependent Activation of CD3- Large Granular Lymphocyte Functions. Regulation of IL-2R Beta (P75) Signal Transduction. J. Immunol. Baltim. Md 1950 1991, 146, 3791–3798. [Google Scholar] [CrossRef]
- Malygin, A.M.; Meri, S.; Timonen, T. Regulation of Natural Killer Cell Activity by Transforming Growth Factor-Beta and Prostaglandin E2. Scand. J. Immunol. 1993, 37, 71–76. [Google Scholar] [CrossRef]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β Inhibits the Activation and Functions of NK Cells by Repressing the MTOR Pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.S.; Tsui, C.; Chisanga, D.; Weber, F.; Llano-León, M.; Gubser, P.M.; Bartholin, L.; Souza-Fonseca-Guimaraes, F.; Huntington, N.D.; Shi, W.; et al. Transforming Growth Factor-β-Regulated MTOR Activity Preserves Cellular Metabolism to Maintain Long-Term T Cell Responses in Chronic Infection. Immunity 2021, 54, 1698–1714. [Google Scholar] [CrossRef]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.-H.; Landström, M. The Type I TGF-β Receptor Engages TRAF6 to Activate TAK1 in a Receptor Kinase-Independent Manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β Activates Erk MAP Kinase Signalling through Direct Phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 Mediates Smad-Independent Activation of JNK and P38 by TGF-β. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I Transforming Growth Factor Beta Receptor Binds to and Activates Phosphatidylinositol 3-Kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef]
- Apostolidis, S.A.; Rodríguez-Rodríguez, N.; Suárez-Fueyo, A.; Dioufa, N.; Ozcan, E.; Crispín, J.C.; Tsokos, M.G.; Tsokos, G.C. Phosphatase PP2A Is Requisite for the Function of Regulatory T Cells. Nat. Immunol. 2016, 17, 556–564. [Google Scholar] [CrossRef]
- Heberle, A.M.; Navas, P.R.; Langelaar-Makkinje, M.; Kasack, K.; Sadik, A.; Faessler, E.; Hahn, U.; Marx-Stoelting, P.; Opitz, C.A.; Sers, C.; et al. The PI3K and MAPK/P38 Pathways Control Stress Granule Assembly in a Hierarchical Manner. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Besson, L.; Mery, B.; Morelle, M.; Rocca, Y.; Heudel, P.E.; You, B.; Bachelot, T.; Ray-Coquard, I.; Villard, M.; Charrier, E.; et al. Cutting Edge: MTORC1 Inhibition in Metastatic Breast Cancer Patients Negatively Affects Peripheral NK Cell Maturation and Number. J. Immunol. 2021, 206, 2265–2270. [Google Scholar] [CrossRef]
- Chan, I.S.; Ewald, A.J. The Changing Role of Natural Killer Cells in Cancer Metastasis. J. Clin. Invest. 2022, 132, e143762. [Google Scholar] [CrossRef]
- Laskowski, T.J.; Biederstädt, A.; Rezvani, K. Natural Killer Cells in Antitumour Adoptive Cell Immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef]
- Mohammadi, M.; Akhoundi, M.; Malih, S.; Mohammadi, A.; Sheykhhasan, M. Therapeutic Roles of CAR T Cells in Infectious Diseases: Clinical Lessons Learnt from Cancer. Rev. Med. Virol. 2022, 32, e2325. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.; Gottschalk, S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 2021, 12, 684642. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of mTORC1 upstream regulation. mTORC1 activation is control by both growth factor and nutrients. Growth factor such as insulin activates both the PI3K-AKT signaling and the MAPK ERK pathway, which converge on TSC inhibition. TSC acts as a GAP for RHEB, which boost mTORC1 kinase activity. Among nutrients, amino acids are essential for mTORC1 activation as they control mTORC1 translocation to the lysosome through the activation of the RAG GTPase A/B and C/D. At the lysosome, mTORC1 is in close vicinity to its activator RHEB. This two-step mechanism allows mTORC1 to integrate both growth factor and nutrients presence. Other parameters such as energy level or hypoxia further regulates mTORC1.
Figure 1.
Overview of mTORC1 upstream regulation. mTORC1 activation is control by both growth factor and nutrients. Growth factor such as insulin activates both the PI3K-AKT signaling and the MAPK ERK pathway, which converge on TSC inhibition. TSC acts as a GAP for RHEB, which boost mTORC1 kinase activity. Among nutrients, amino acids are essential for mTORC1 activation as they control mTORC1 translocation to the lysosome through the activation of the RAG GTPase A/B and C/D. At the lysosome, mTORC1 is in close vicinity to its activator RHEB. This two-step mechanism allows mTORC1 to integrate both growth factor and nutrients presence. Other parameters such as energy level or hypoxia further regulates mTORC1.

Figure 2.
Signaling downstream IL-2 and IL-15 receptor toward mTORC1 in CD8+ T cell and NK cells. IL-2 and IL-15 receptors share both their IL-2Rβ and γ chain (γc) associated with a unique IL-2Rα or IL-15Rα chain depending on the receptor. IL-2Rβ and γc are associated with the kinase JAK1 and JAK3 respectively. The binding of the cytokine results in JAK1/3 activation and phosphorylation of tyrosine residues on IL-2Rβ, which serves as activating platform for STAT5, PI3K and GRB2-SOS. Even though the PI3K-AKT pathway is engaged by the IL-2 receptor (in orange), its importance in mTORC1 activation remained under debate.
Figure 2.
Signaling downstream IL-2 and IL-15 receptor toward mTORC1 in CD8+ T cell and NK cells. IL-2 and IL-15 receptors share both their IL-2Rβ and γ chain (γc) associated with a unique IL-2Rα or IL-15Rα chain depending on the receptor. IL-2Rβ and γc are associated with the kinase JAK1 and JAK3 respectively. The binding of the cytokine results in JAK1/3 activation and phosphorylation of tyrosine residues on IL-2Rβ, which serves as activating platform for STAT5, PI3K and GRB2-SOS. Even though the PI3K-AKT pathway is engaged by the IL-2 receptor (in orange), its importance in mTORC1 activation remained under debate.

Figure 3.
Signaling downstream IL-1 receptor toward mTORC1 in T cell. IL-1 binding to its receptors induces the recruitment of the adaptors MyD88 and IRAKs. This activates the ubiquitin E3 ligase TRAF6 which triggers the activation of MAPK such as p38 and the NFκB pathways. In Th17 T cell, Gulen et al. show that IL-1 treatment drive the disruption of TSC and the activation of mTORC1. The role of the MAPK p38 or NFκB in this process in lymphocyte still remains to be determine as p38 and IKK have been shown to activates mTOR in other cell types.
Figure 3.
Signaling downstream IL-1 receptor toward mTORC1 in T cell. IL-1 binding to its receptors induces the recruitment of the adaptors MyD88 and IRAKs. This activates the ubiquitin E3 ligase TRAF6 which triggers the activation of MAPK such as p38 and the NFκB pathways. In Th17 T cell, Gulen et al. show that IL-1 treatment drive the disruption of TSC and the activation of mTORC1. The role of the MAPK p38 or NFκB in this process in lymphocyte still remains to be determine as p38 and IKK have been shown to activates mTOR in other cell types.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



