Submitted:
13 June 2023
Posted:
14 June 2023
You are already at the latest version
Abstract
This study aims to investigate the spectroscopic and structural properties of the compound Lumefantrine which is important in pharmacology because of its anti-malarial effect. The structural and spectroscopic properties of this molecule such as bond lengths, bond angles, FT-IR and NMR spectra were handled computationally using a computational chemistry suite: Spar-tan’14. Both HF and DFT methods were used with different basis sets for the calculations. The re-sults calculated by the software were compared to experimental results from the literature. Both Computational and experimental results were exhibited as tables. Also some calculated results such as HOMO-LUMO boundary orbitals and electrostatic potential map, were given as graphics and pictures.
Keywords:
Lumefantrine
; Anti-malarial activity
; Spectral analysis
; HOMO-LUMO
; Molecular structure
1. Introduction
Malaria is a parasitic disease in humans that causes thousands of deaths every year [1]. According to World Malaria Report 2020 by WHO, every year, 409,000 people died because of this disease. As a promising development from the same report, the malaria deaths have reduced from 736,000 to 409,000 between the years 2000 and 2019. Report also underlines, globally an estimated 1.5 billion malaria cases and 7.6 million of ma-lar-ian deaths have been prevented in the same period [2].
Lumefantrine is an amino alcohol developed by Chinese scientists at around the same time as mefloquine. It has been the mainstay of the most widely used combination therapy with artemether. So far a significant resistance has not been seen because Lu-mefantrine has never been used as monotherapy [3]. Lumefantrine has been used with artemether since 1994. Bombarding by two different drugs with different destructive mechanisms the parasite cannot develop a resistance against the both [4].
This study has been inspired by a former study by Friedrich Research group. In their study which has been published in an elite scientific journal, they investigated the structural and spectral properties of Lumefantrine [1].
SPARTAN'XX is a package of computational chemistry that was published by Wavefunction INC. [5,6]., Some researchers has used this software for investigating different kinds of organic molecules such as pyrazoles [7], boronic acid derivatives [8], semicarbazides [9], dimethoxycoumarin [10], triphenylphosphorusanilideneacetaldehyde [11] and ethoxycoumarin [12]. The package has improved itself on predicting structural and spectral properties very near to experimental results.
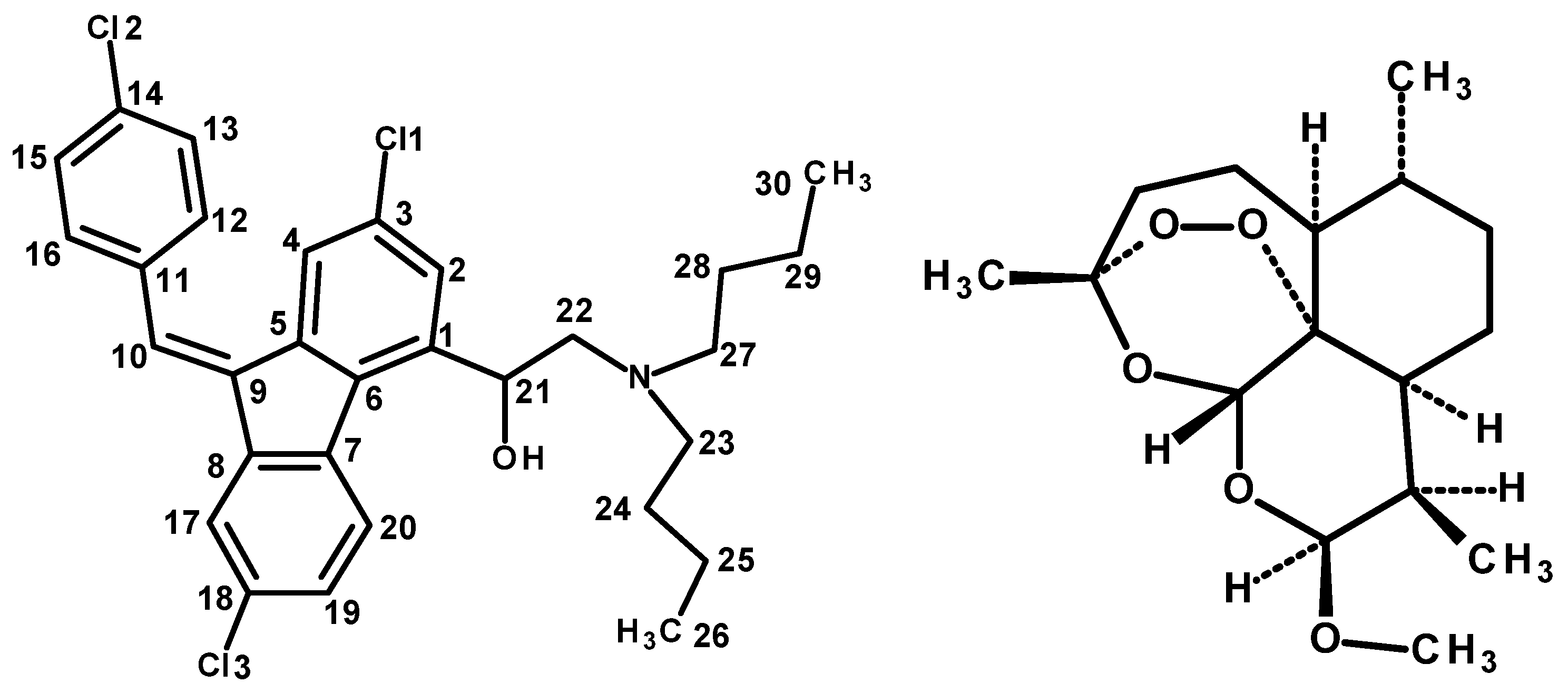
Figure 1.
a) Basic molecular structure and atom numbering of Lumefantrine b) Molecular formula of artemether.
Figure 1.
a) Basic molecular structure and atom numbering of Lumefantrine b) Molecular formula of artemether.
2. Materials and Methods
2.1. Computational Details
For computational analysis of the molecule, two main methods were used with two basis sets for each, in the SPARTAN'14 quantum chemistry suit. HF method was used with 321G and 6-31G* basis sets while DFT was used with EDF2 and B3LYP in 6-31G*[13,14] and, the obtained results have been tabulated and discussed under corresponding divisions. Calculated spectral graphics and MO surfaces have been depicted in corresponding figures. For vibrational analysis, the calculated results which were produced by the software have been corrected by a scaling factor of 0.962.
In the last decade, the DFT method has been given increasing importance among computational chemists. Many researchers investigating especially d-block metal complexes prefer to use this method with or instead of HF. Some small molecules, such as boronic acid derivatives or benzoic acid derivatives, have been investigated using both DFT and HF. The results of the DFT method were found to be more consistent with the experimental results compared to the HF method in most of the studies [15]. The DFT method has also been used in some studies to investigate other types of small molecules such as pyrazole derivatives and promising results have been obtained [16].
3. Results and Discussion
3.1. Structure of the Compound
The structure of any compound is determined by its bond lengths, bond angles and dihedral angles. For this reason, in this study, the structural properties were calculated and tabulated in the following pages. In addition, the potential energy, which shows the stability of the molecule, has been calculated and tabulated for the molecule.
First thing first, it can come to mind, while there is no experimental data on the molecular structure, how can we trust these computational values. For getting rid of this hesitation, we can apply the spectroscopic data for the correlation between the experimental and calculated data. This correlation can be used for the prediction of the molecular structures too. Since the number of atoms of the molecule is very large, the bonds and angles are also numerous. Therefore, the tables showing the bond lengths and angles are also quite large. For this reason, to keep the study short, these tables were included in the Supplementary Information File (Figure S1).
3.2. Electronic and Spectral Properties
Electronic transitions and spectral properties were calculated in each method, HOMO and LUMO molecular orbital surfaces were calculated and depicted, also their energy values are tabulated and compared to each other. ESPMap (Electrostatic potential Map) calculated and presented as figures (Figure 2)
Lumefantrine has 30 C atoms, 32 H atoms, 3 Cl, 1 O and 1 N atoms, 67 atoms in total. So it is better to handle the molecule in two graphics for Mulliken charge distribution. The first graphic shows the charge distribution of H atoms and the second one depicts C and heteroatoms.
Like every molecule, atoms of lumefantrine have different electronegativities, so the electron distribution on the molecule is heterogeneous. More electronegative atoms attract and gather up more electrons than the electropositive ones. So electronic density around these atoms rises and these parts of the molecule gain a partially negative charge. This heterogeneity causes atoms to have different partial charges on the molecule. The more electron-rich a particular part of a molecule is, the more susceptible it is to electrophilic attacks, and of course, the reverse is also true, the more electron-poor it is, the more susceptible it will be to nucleophilic attacks. For this reason, The Mulliken charge distribution is an important data for predicting the possible reaction mechanisms in which the molecule participates [17] The ESPMap surface determines the distance at which a given positive charge can interact with the molecule enough that it can produce an attraction or repulsion so that they can have a bonding probability. Molecular ESP (Vr) is calculated by Equation-1 [18].
The Mulliken charges of Lumefantrine are exhibited in Figure 3 and Table 1. These results are also given in Figure 2 as a colour graphic. At a glance the most important points are briefly:
1- All H atoms have positive charges. At first sight H12 dramatically huge charge almost two times bigger than most of the others. H12 also draws attention to the fact that while the HF321 method calculates the highest value for all H atoms, it takes the highest value from the HF 631G* method.
2- C atoms of the molecule are generally negatively charged except for C21 which carries the only –OH group of the molecule and C9 which is neighbor to the olefinic bridge.
3- For most carbon atoms all methods agree with slightly different positive charges, but for C1, C5, C6, C7, C8 and C11 the first two methods predict small negative charges.
4- The O atom of the –OH group has the highest negative charge as expected because of its electronegativity.
5- The only N atom of the molecule has the second highest negative charge depending on its electronegativity. In addition, this part represents one of the most vulnerable parts of the molecule to electrophilic attacks and forms a basic site on the molecule.
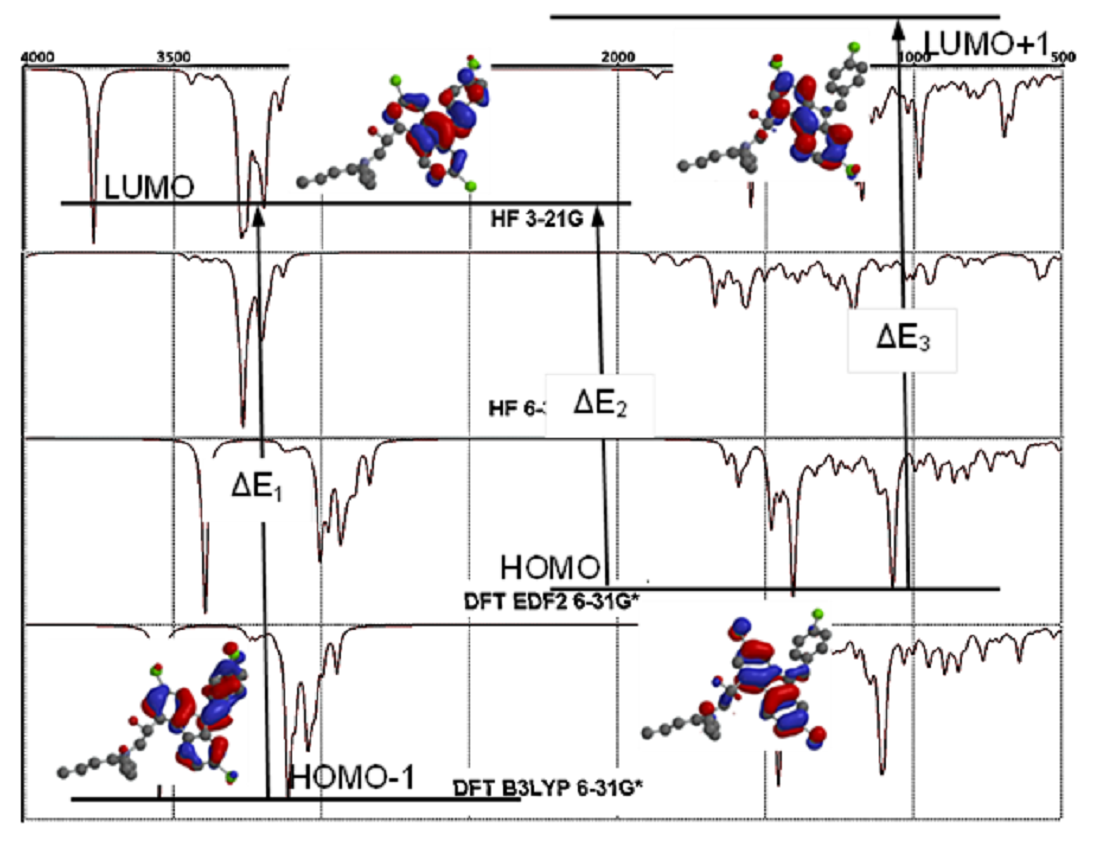
3.3. Molecular Orbitals Surfaces (HOMO LUMO) Analysis and UV-Vis Spectra
Highest Occupied Molecular Orbital (HOMO) represents the highest energy level in which at least one electron exists around the molecule. As just opposite Lowest Unoccupied Molecular Orbital (LUMO) gives the lowest energy level around the molecule in which there is no electron. The gap between these two values is very significant in the biological and chemical reactivities of the molecules. The molecules with small energy gaps are accepted as soft molecules and expected to involve in chemical reactions easily. These energy levels also determine the acidity and basicity of the molecules.
In Lumefantrine the energy gap between HOMO-LUMO was calculated to be 9.9 eV, 9.6 eV, 3.5 eV, and 3.7 eV according to the methods respectively. As can be seen easily, DFT methods give dramatically smaller values than the HF calculations (Table 2 and Table 3). According to these values Lumefantrine can be supposed to be a semi-rigid molecule (Figure 4). The larger HOMO-LUMO gap gives the molecule greater kinetic stability and lower chemical reactivity. Molecule’s hardness can be predicted via Equation 2a and softness (S) can be derived from hardness via Equation 2b.
η=(ԐLUMO-ԐHOMO)
S=1/η
As can be easily guessed, molecules with large energy gaps, defined as hard mole-cules, do not change their electron density very easily. On the other hand, molecules with small energy gaps called soft molecules, change their electron densities relatively easily [19, 20].
3.4. FT-IR Vibrational Analysis
The molecule of Lumefantrine has 67 atoms and that means 195 vibrational modes. These 195 modes can be handled in three parts. The groups in which the aromatic rings are located are the calm parts that make slow movements and oscillations without disturbing their planarity as can be expected. And again, as can be easily predicted, the most mobile and agile part of the molecule is the aliphatic part, which consists of two branches. This second part which consists of C21 and beyond, with O and N atoms exhibits every kind of mechanical motion such as swinging, rocking, etc.
Figure 6.
Calculated FT-IR spectra for Lumefantrine.
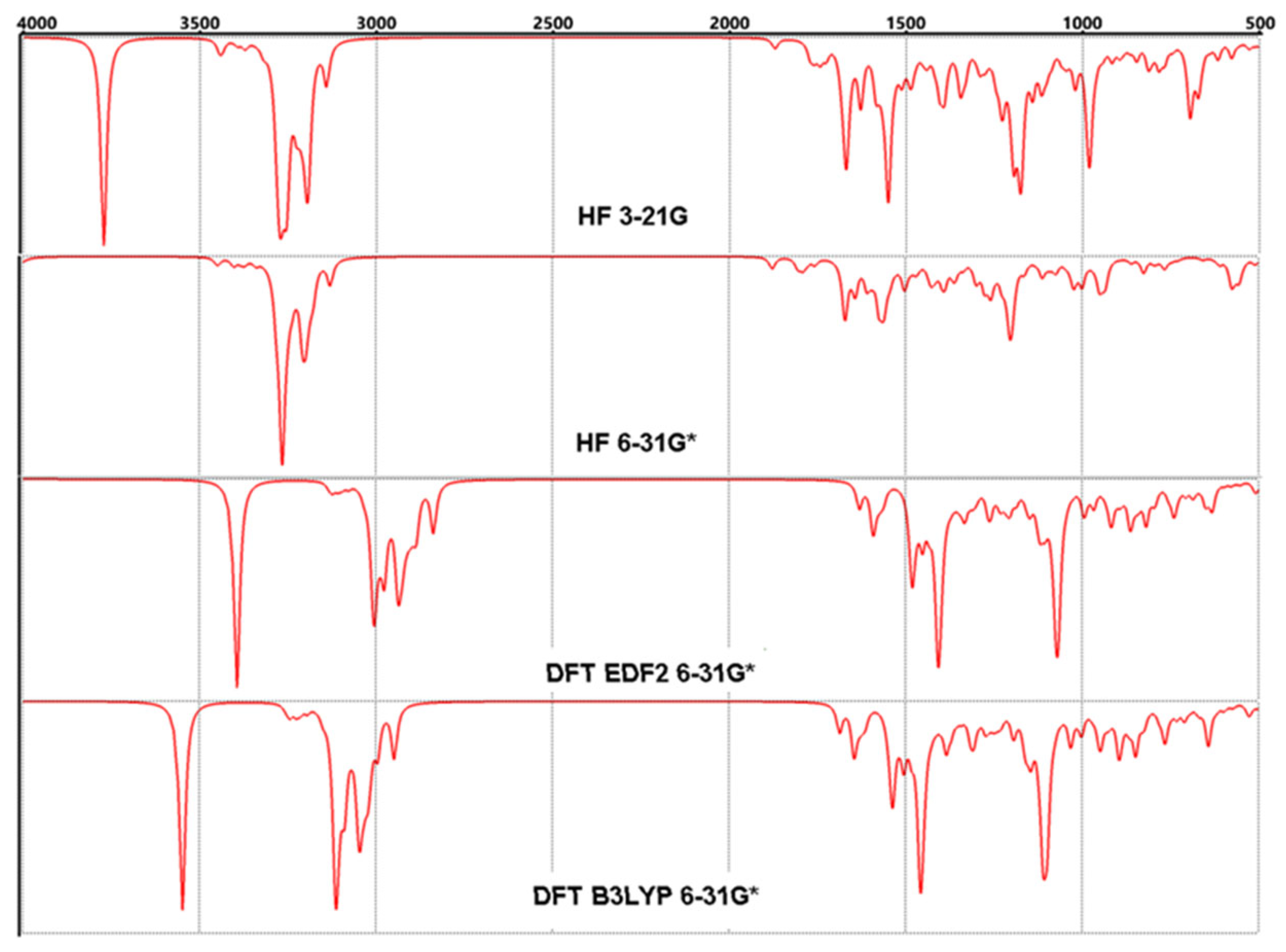
As a brief analysis, the frequencies have been tabulated in Table 4 and depicted in Figure 7. In the table, the calculated values according to four methods were involved comparatively. Although there are no experimental data for FTIR, the computational values available can be considered significant due to the compatibility between the experimental and computational NMR data, which will be seen in the following sections. Even so, some important points should be underlined [21,22].
1- The peak which refers –O—H group’s stretching, appears in 3629 cm-1, 3884 cm-1, 3393 cm-1, and 3124 cm-1 (mode 195) according to method respectively [23].
2- The peak in mode 171(3088 cm-1, 3083 cm-1, 2929 cm-1, and 2926 cm-1) refers to the C-H stretching motions which belong to the aliphatic branches.
3- Two kinds of aromatic groups of which stretchings are seen as mode 150 (1607 cm-1, 1582 cm-1, 1470 cm-1, and 1474 cm-1) for the single ring and mode 138 (1494 cm-1, 1507 cm-1, 1396 cm-1, and 1392 cm-1)
4-The stretching of the olefinic group (C9=C10) appears as the peak mode 163 (1801 cm-1, 1806 cm-1, 1631 cm-1, and 1622 cm-1).
3.5. NMR Analysis
Let the last thing to be said first, the experimental values from the literature [1] and the calculated ones in this study are in great agreement. This harmony between the data also increases our confidence in the data presented in the previous sections, which have no empirical counterparts. The calculated and experimental data are tabulated in Table 5. The same spectra have been depicted in Figure 7 and Figure 8. Also, COSY, HSQC and HMBC spectra have been presented in Figure 9. The experimental spectra can be seen in the article by Friedrich's research group [1].
Some significant peaks in the spectra can be interpreted as follows
1- H21,22,23 and H30,31,32 the terminal H atoms appeared 0.96 ppm in experimental studies. Their calculated values have been found between 0.93 and 1.19. EDF2 method gave the nearest calculated value to the experimental results.
2- H12 has been found to give a peak near 3 to 4.16 ppm. But its peak isn’t seen in the experimental spectra which is typical for O-H groups.
3- H11 which is on the same C21 with the O atom has given a peak of 5.35 ppm in experimental spectra. Its calculated values are predicted 4.85 ppm to 5.24 ppm.
4- C3, C14 and C18 which bear Cl1, Cl2 and Cl3 respectively were found to give peaks at 136.72 ppm 137.0 ppm and 134.3 ppm experimentally. Their calculated values were found 124 ppm to 136.72 ppm. As with most C atoms, the nearest calculated results came from DFT methods, especially EDF2.
4. Conclusions
Molecular structure and HOMO-LUMO analysis of lumefantrine were performed using the SPARTAN-14 package at EDF2 and B3LYP levels and different base sets by HF and DFT methods. In addition, FT-IR and FT-RAMAN spectra were calculated and compared with the inter-method results. Lumefantrine has been studied by different research groups specifically for its pharmacological activities, but this study is a computational study aimed at investigating the SPARTAN software versus experimental NMR studies.
The calculated values were compared with the experimental values found in the literature and were found to be very close without significant deviation. As a result of all studies, it was seen that the calculated values and the experimental results were very close and consistent with each other.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Data Availability
Supplementary Materials and data can be requested from the author by e-mail. (ahmettkunduracioglu@gmail.com).
Acknowledgments
The software SPARTAN-14 used in this study was bought with the financial support of Pamukkale University Scientific Research Support Unit (Project no: HZL-2014/5).
Conflicts of Interest
The author has no conflict of interests to declare.
References
- Pansuriya P.B.; Maguire G.E.M..; Friedrich H.B. Structural Characterization and Thermal Properties of the Anti-malarial Drug: Lumefantrine. S.Afr. J. Chem 2019, 72: 253-262. https://doi.org/10.17159/0379-4350/2019/v72a33. [CrossRef]
- World Health Organization (WHO). World Malaria Report. 2020.
- Gaur D.; Chitnis C.; Chauhan V. Advances in Malaria Research. The Wiley-IUBMB Series on Biochemistry and Molecular Biology. Wiley-Blackwell, 1st edition. 2016. https://doi.org/10.1002/9781118493816. [CrossRef]
- Shah S. The Fever, How Malaria has ruled humankind for 500,000 years-Penguin Books, 2018. https://doi.org/10.1590/1413-81232020256.20542019. [CrossRef]
- Hehre W. J. SPARTAN’14 Wavefunction Inc. Irvine CA, USA 2014.
- Hehre W. J. SPARTAN’14: Tutorial and User’s Guide. Irvine CA USA, Wavefunction, Inc. 2014.
- Kunduracioglu A. A Novel Pyrazolium Salt with Phthalimide Functional Groups Synthesis Spectroscopic (NMR&FT-IR) and Computational Analysis, Fresenius Environmental Bulletin., 2021 30(068): pages 7551-7560.
- Kunduracioglu A. 2-thienylboronic Acid: A DFT Study For the Spectral, Structural and Molecular Orbital Analysis. El-Cezerî Journal of Science and Engineering., 2021 8(1): 397-409. https://doi.org/10.31202/ecjse.825888. [CrossRef]
- Dhandapani A.; Manivarman S.; Subashchandrabose S.; Saleem H. Molecular structure and vibrational analysis on (E)‒1‒(3‒methyl‒2,6‒diphenyl piperidine‒4‒ylidene) semicarbazide. J. Mol. Struct., 2014 1058: 41‒50 DOI: https://doi.org/10.1016/j.molstruc.2013.09.052. [CrossRef]
- Karakaş‒Sarıkaya E.; Dereli Ö. Study on Molecular Structure and Vibrational Spectra of 5,7‒Dimethoxycoumarin Using DFT: A Combined Experimental and Quantum Chemical Approach. Opt. Spectrosc., 2014 117(2): 240‒249. https://doi.org/10.1134/s0030400x14070108. [CrossRef]
- Dereli Ö.; Erdoğdu Y.; Güllüoğlu M.T., Study on molecular structure and vibrational spectra of (triphenylphosphoranylidene) acetaldehyde using DFT: A combined experimental and quantum chemical approach. J. Mol. Struct., 2012 1012: 105‒112. https://doi.org/10.1016/j.molstruc.2011.12.040. [CrossRef]
- Sarıkaya E.K.; Dereli Ö.; Erdoğdu Y.; Güllüoğlu M. T. Molecular structure and vibrational spectra of 7‒Ethoxycoumarin by density functional method. J. Mol. Struct. 2013, 1049: 220‒226. https://doi.org/10.1016/j.molstruc.2013.06.026. [CrossRef]
- Becke A.D. Density-functional thermochemistry. III.The role of exact exchange, J. Chem. Phys., 1993 98: 5648-5652. https://doi.org/10.1063/1.464913. [CrossRef]
- Lee C.; Yang W.; Parr R.G., Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B Condens. Matter., 1998 37: 785-799. https://doi.org/10.1103/physrevb.37.785. [CrossRef]
- Kemer-Kotan G.; Yuksek H. Experimental (FT-IR, NMR) and Theoretical (B3PW91, B3LYP, HF) Analyses of 2-(3-Ethyl-4,5-Dihydro-1H-1,2,4-Triazole-5-on-4-yl)-azomethine)-Benzoic Acid, Caucasian Journal of Science, 2019 6 (1): 64-75.
- Turhan-Irak Z.; Beytur M. 4-Benzilidenamino-4,5-dihidro-1H-1,2,4-triazol-5-on Türevlerinin Antioksidan Aktivitelerinin Teorik Olarak İncelenmesi [Theoretical Study on The Investigation of Antioxidant Properties of Some 4-Benzylidenamino-4,5-dihydro-1H-1,2,4-triazol-5-one Derivatives] Iğdır Üniversitesi Fen Bilimleri Enstitüsü Dergisi, 2019 9(1): 512-521.
- Jensen F. Introduction to Computational Chemistry. 2016 Chichester: Wiley.
- Peter K., Vollhardt C., Schore N. E., Organic chemistry: Structure and function. 6th ed. 2011 Freeman&Comp. NY-US... https://doi.org/10.1007/978-1-319-19197-9. [CrossRef]
- Şahin Z.S.; Kaya-Kantar G.; Şaşmaz S.; Büyükgüngör O. Synthesis, molecular structure, spectroscopic analysis, thermodynamic parameters and molecular modeling studies of (2-methoxyphenyl)oxalate, J. Mol. Struct., 2015 1087:104-112. https://doi.org/10.1016/j.molstruc.2015.01.039. [CrossRef]
- Pearson R.G. Chemical hardness and density functional theory, Chem. Sci. J., 2005 117(5): 369-377. https://doi.org/10.1007/bf02708340. [CrossRef]
- Silverstein R.M.; Webster F.X.; Kiemle D.J. Spectrometric Identification of Organic Compounds. 7th Ed. 2005 John Wiley Sons INC.. https://doi.org/10.1002/ange.19650771675. [CrossRef]
- Ramachandran K.I.; Deepa G.; Namboori K. Computational Chemistry and Molecular Modeling: Principles and Applications. 2008 Springer-Verlag Heidelberg. Berlin:.. https://doi.org/10.1007/978-3-540-77304-7. [CrossRef]
- Lu G.P.; Voigtritter K.R.; Cai C.; Lipshutz B.H., Ligand effects on the stereochemical outcome of Suzuki-Miyaura couplings. J. Org. Chem., 2012 77(8): 3700-3703.. https://doi.org/10.1021/jo300437t. [CrossRef]
Figure 2.
Calculated ESPMaps for Lumefantrine.
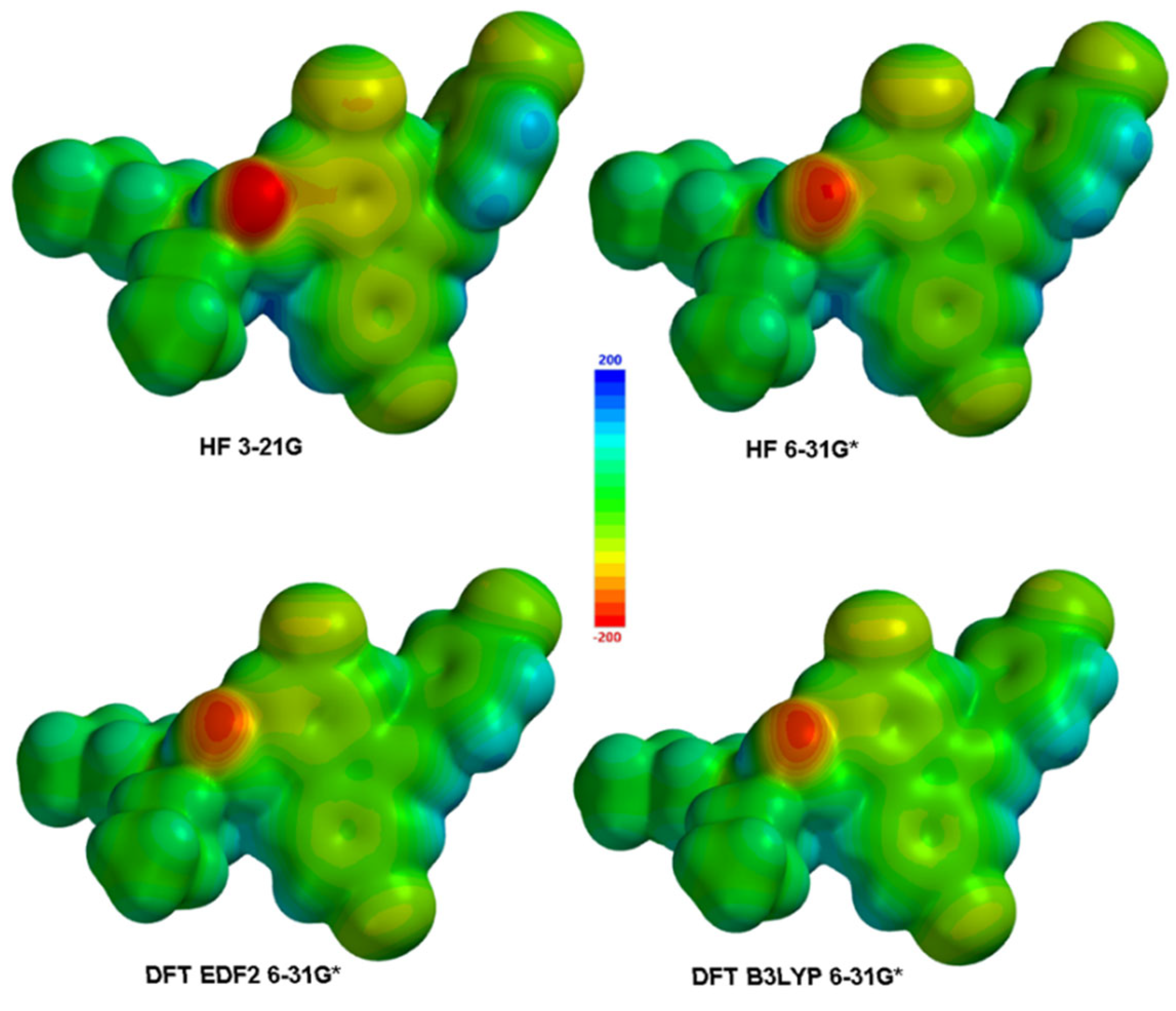
Figure 3.
Mulliken charges of the atoms in Lumefantrine molecule (a) H atoms, (b) C&heteroatoms).
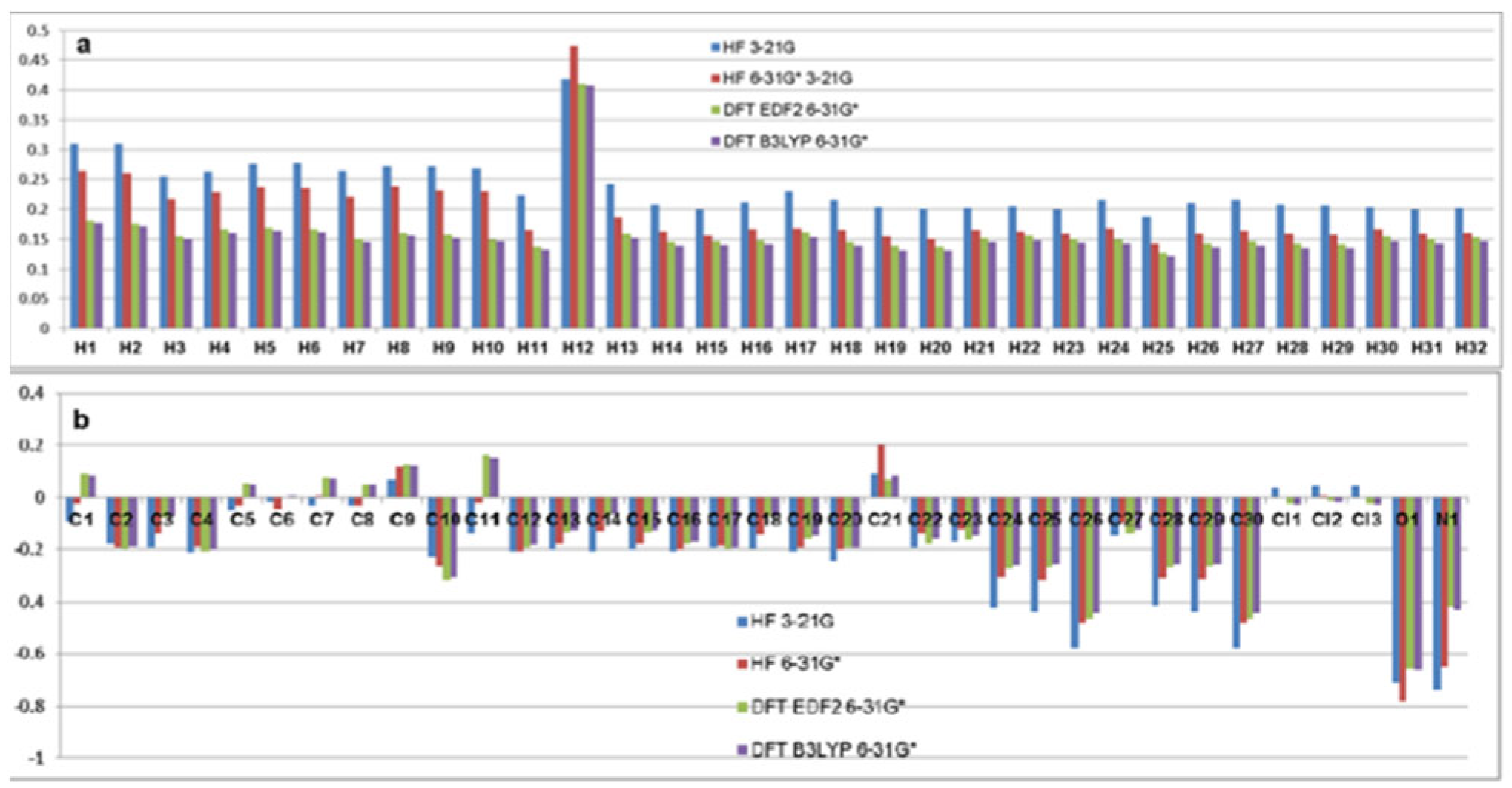
Figure 4.
Electron transitions and corresponding E and ΔE values for Lumefantrine.
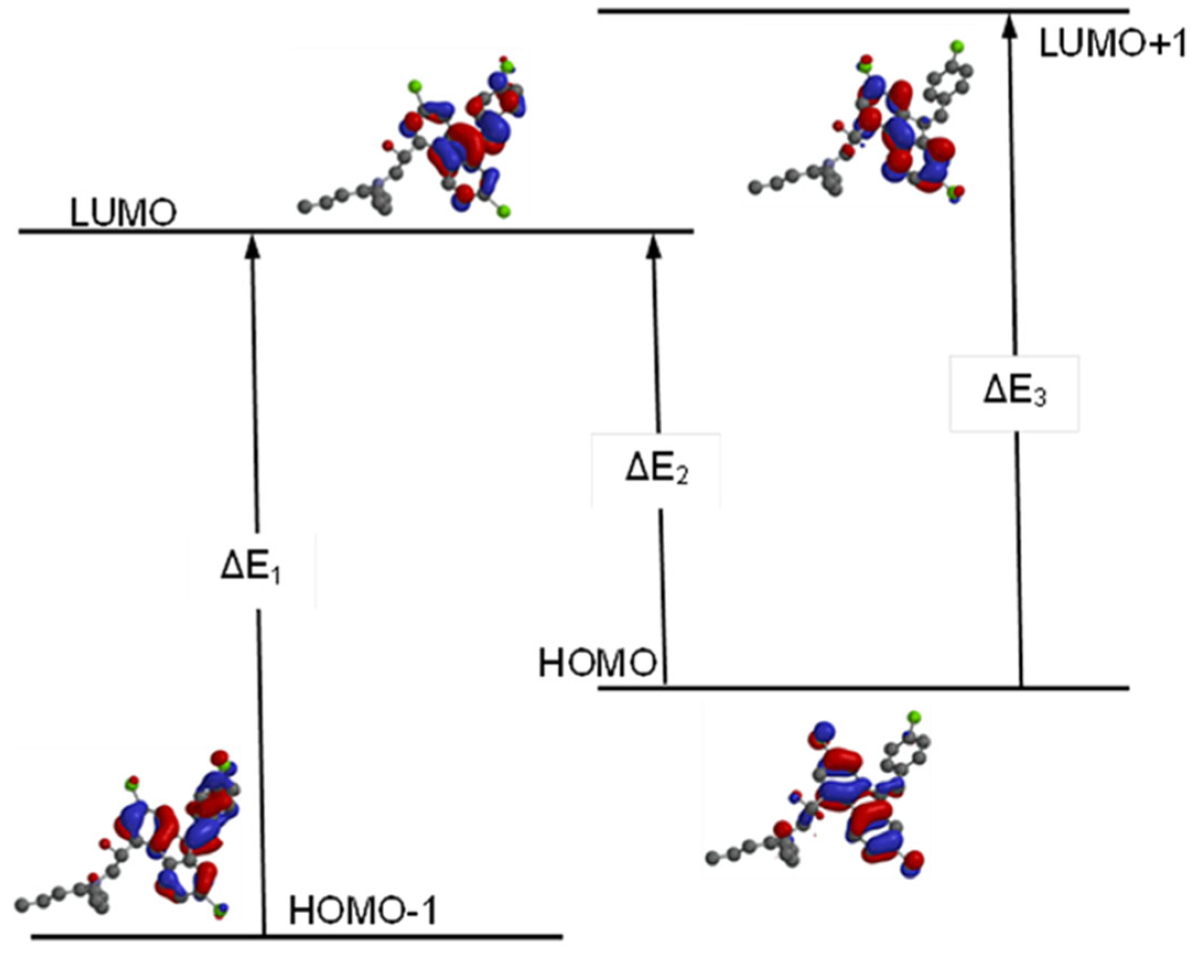
Figure 5.
Calculated UV-Vis spectra for Lumefantrine.
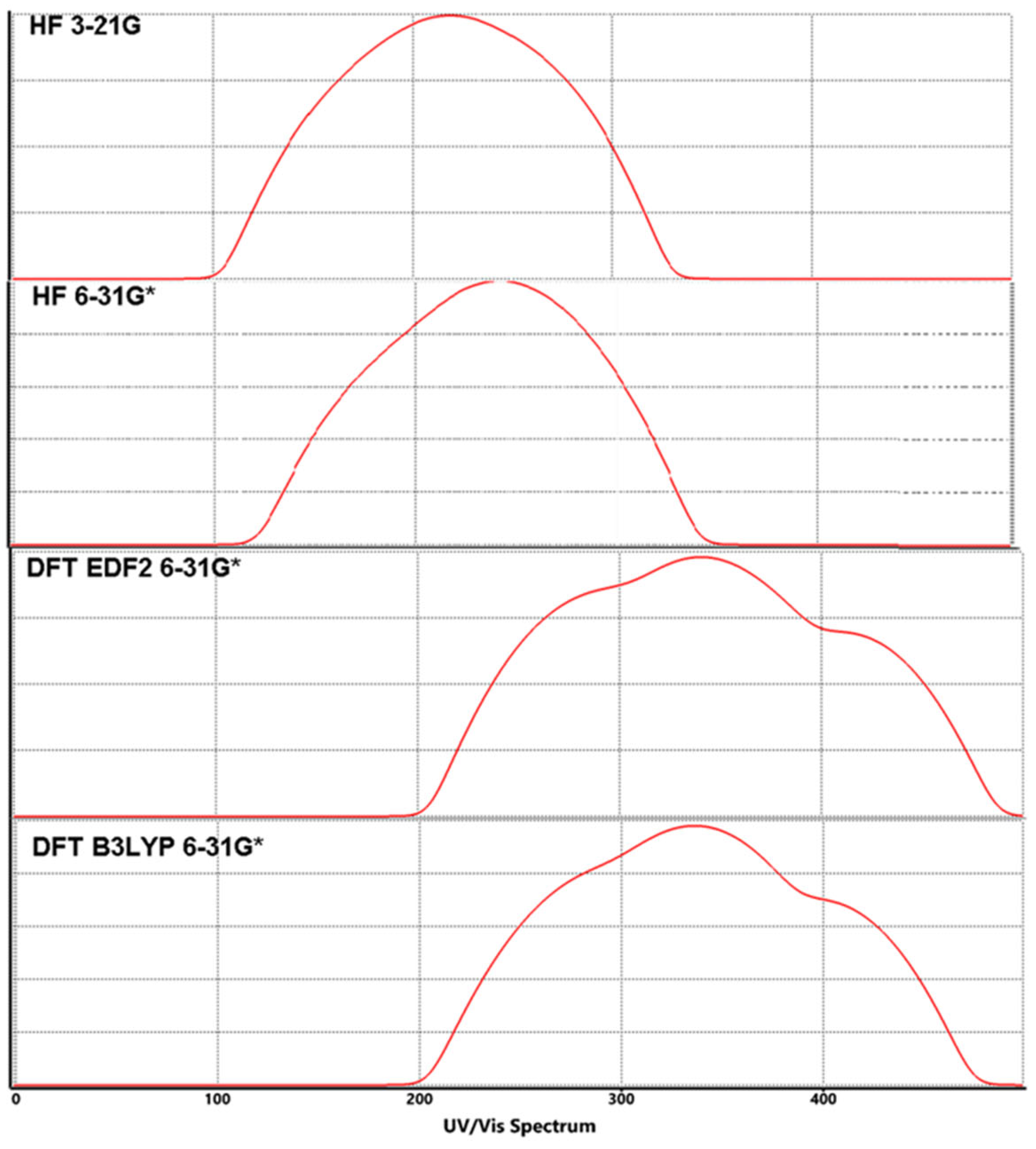
Figure 7.
Calculated 1H NMR spectra for Lumefantrine.
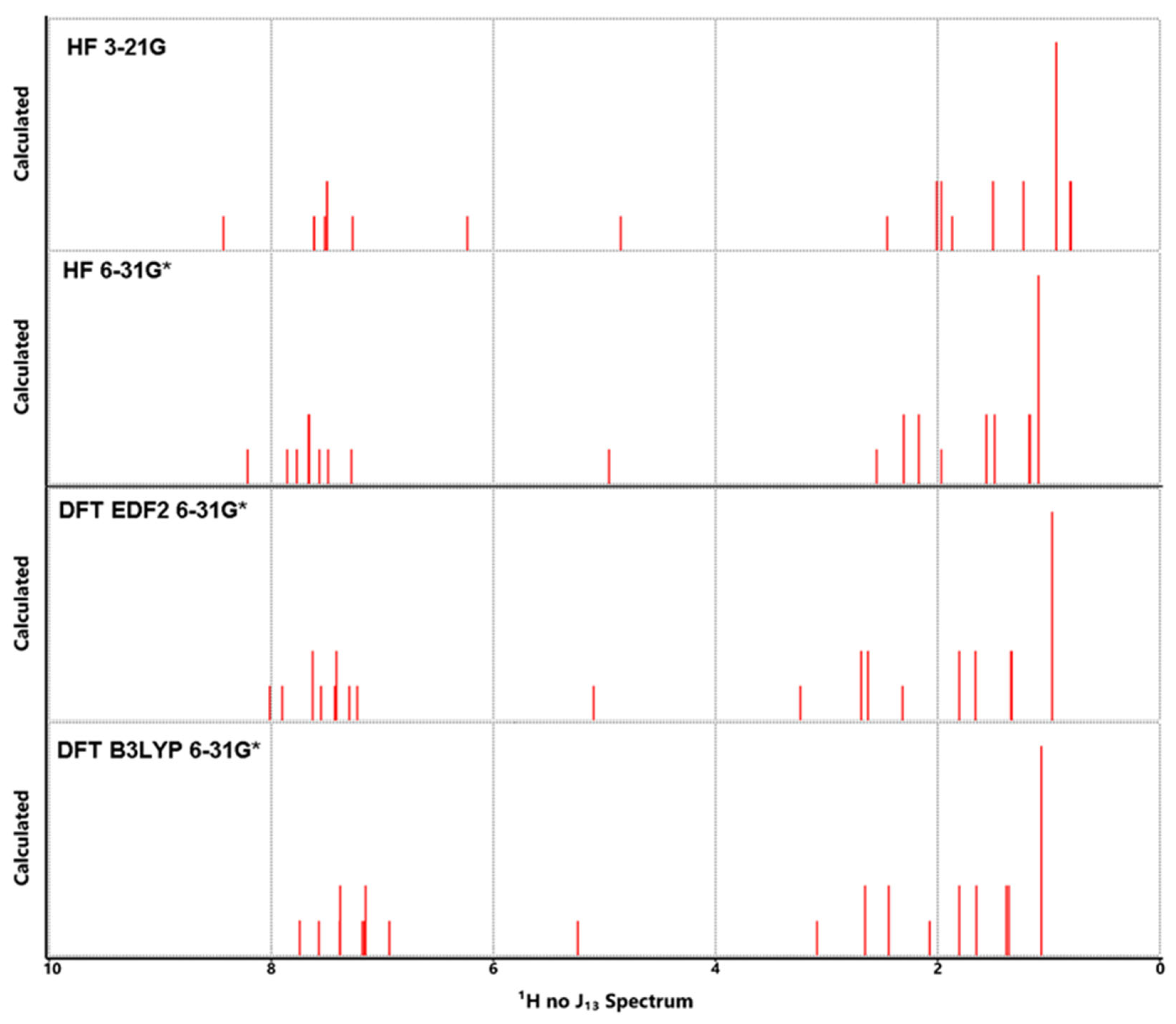
Figure 8.
Calculated 13C NMR spectra for Lumefantrine.
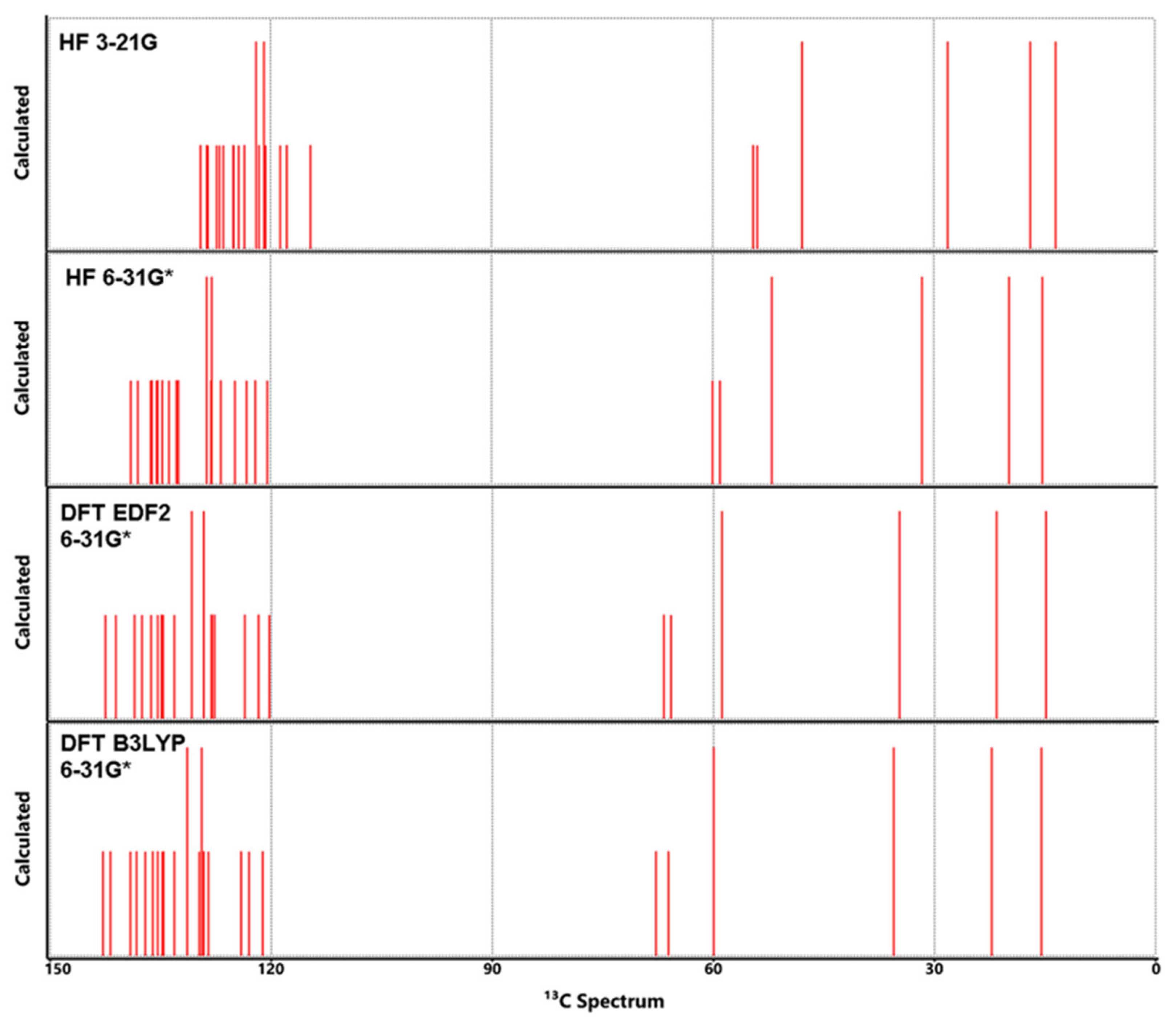
Figure 9.
Calculated COSY, HSQC and HMBC spectra for Lumefantrine.
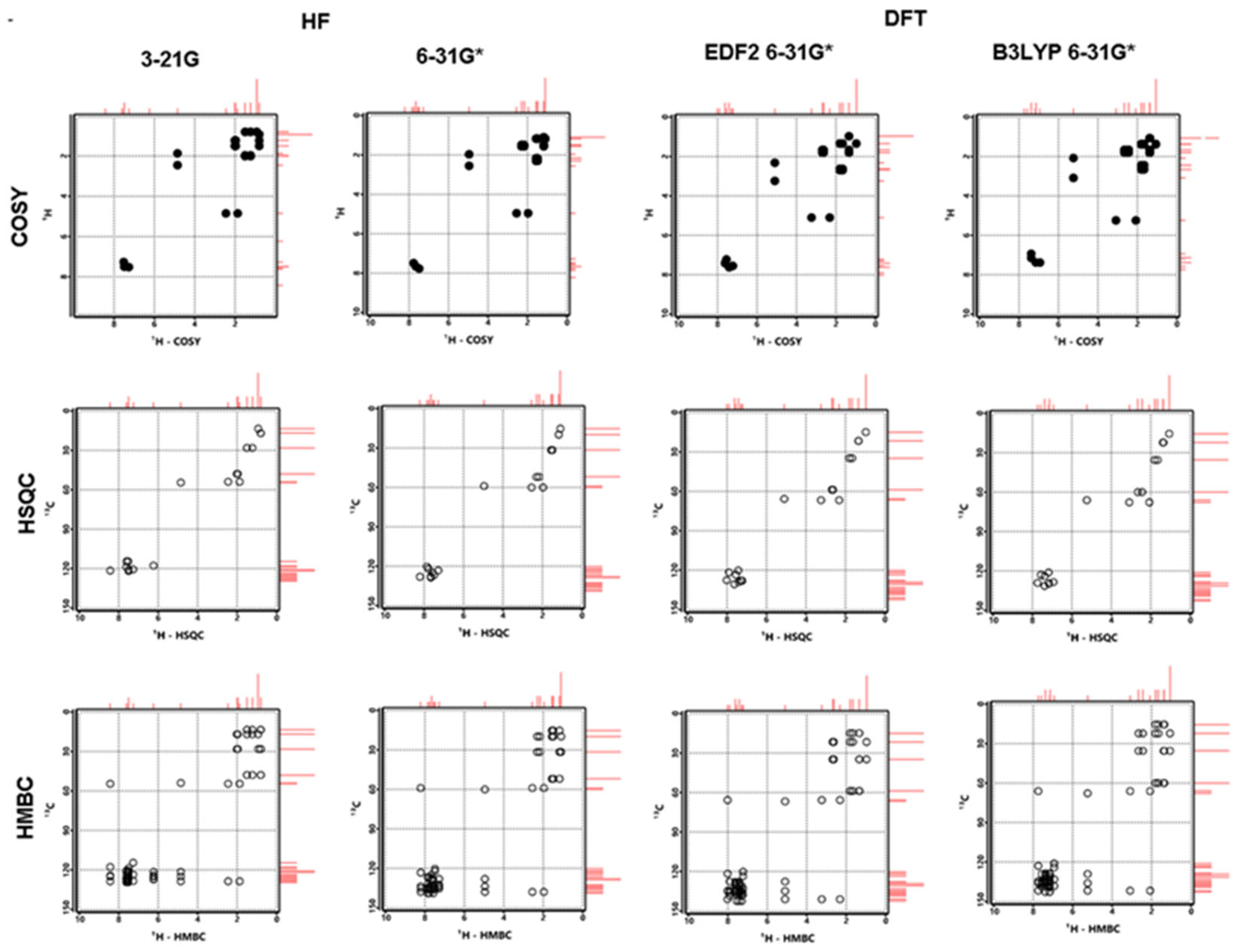

Table 1.
Mulliken Charge distribution of the atoms of Lumefantrine.
| Atom | HF | DFT EDF2 | DFT B3LYP | Atom | HF | DFT EDF2 | DFT B3LYP | ||
|---|---|---|---|---|---|---|---|---|---|
| 3-21G | 6-31G* | 6-31G* | 6-31G* | 3-21G | 6-31G* | 6-31G* | 6-31G* | ||
| C1 | -0.087 | -0.019 | 0.093 | 0.083 | H1 | 0.310 | 0.265 | 0.181 | 0.177 |
| C2 | -0.178 | -0.195 | -0.199 | -0.190 | H2 | 0.310 | 0.261 | 0.176 | 0.172 |
| C3 | -0.194 | -0.142 | -0.075 | -0.071 | H3 | 0.255 | 0.217 | 0.154 | 0.151 |
| C4 | -0.209 | -0.190 | -0.208 | -0.202 | H4 | 0.263 | 0.229 | 0.167 | 0.160 |
| C5 | -0.047 | -0.029 | 0.054 | 0.051 | H5 | 0.276 | 0.236 | 0.169 | 0.164 |
| C6 | -0.012 | -0.044 | -0.001 | 0.001 | H6 | 0.277 | 0.235 | 0.166 | 0.161 |
| C7 | -0.030 | 0.006 | 0.075 | 0.072 | H7 | 0.264 | 0.221 | 0.150 | 0.145 |
| C8 | -0.031 | -0.031 | 0.051 | 0.051 | H8 | 0.273 | 0.238 | 0.160 | 0.156 |
| C9 | 0.067 | 0.121 | 0.126 | 0.124 | H9 | 0.273 | 0.231 | 0.157 | 0.152 |
| C10 | -0.226 | -0.266 | -0.319 | -0.307 | H10 | 0.268 | 0.230 | 0.149 | 0.147 |
| C11 | -0.142 | -0.014 | 0.166 | 0.154 | H11 | 0.223 | 0.165 | 0.137 | 0.132 |
| C12 | -0.206 | -0.208 | -0.192 | -0.182 | H12 | 0.418 | 0.474 | 0.410 | 0.408 |
| C13 | -0.203 | -0.177 | -0.136 | -0.130 | H13 | 0.242 | 0.186 | 0.159 | 0.152 |
| C14 | -0.207 | -0.135 | -0.066 | -0.064 | H14 | 0.208 | 0.162 | 0.145 | 0.138 |
| C15 | -0.203 | -0.179 | -0.138 | -0.131 | H15 | 0.200 | 0.156 | 0.146 | 0.140 |
| C16 | -0.207 | -0.202 | -0.180 | -0.172 | H16 | 0.211 | 0.167 | 0.148 | 0.141 |
| C17 | -0.197 | -0.184 | -0.201 | -0.194 | H17 | 0.230 | 0.168 | 0.161 | 0.153 |
| C18 | -0.199 | -0.145 | -0.072 | -0.070 | H18 | 0.215 | 0.165 | 0.145 | 0.139 |
| C19 | -0.206 | -0.196 | -0.158 | -0.149 | H19 | 0.203 | 0.155 | 0.138 | 0.131 |
| C20 | -0.245 | -0.203 | -0.195 | -0.193 | H20 | 0.201 | 0.151 | 0.137 | 0.130 |
| C21 | 0.092 | 0.200 | 0.068 | 0.085 | H21 | 0.202 | 0.165 | 0.152 | 0.145 |
| C22 | -0.194 | -0.140 | -0.180 | -0.161 | H22 | 0.205 | 0.162 | 0.156 | 0.148 |
| C23 | -0.173 | -0.124 | -0.166 | -0.148 | H23 | 0.200 | 0.159 | 0.151 | 0.144 |
| C24 | -0.423 | -0.307 | -0.273 | -0.260 | H24 | 0.216 | 0.168 | 0.149 | 0.142 |
| C25 | -0.436 | -0.316 | -0.268 | -0.255 | H25 | 0.188 | 0.142 | 0.127 | 0.121 |
| C26 | -0.580 | -0.479 | -0.463 | -0.442 | H26 | 0.210 | 0.158 | 0.142 | 0.136 |
| C27 | -0.148 | -0.096 | -0.140 | -0.123 | H27 | 0.215 | 0.164 | 0.146 | 0.139 |
| C28 | -0.416 | -0.311 | -0.270 | -0.256 | H28 | 0.208 | 0.158 | 0.143 | 0.135 |
| C29 | -0.434 | -0.314 | -0.267 | -0.254 | H29 | 0.206 | 0.157 | 0.141 | 0.134 |
| C30 | -0.581 | -0.479 | -0.464 | -0.442 | H30 | 0.204 | 0.166 | 0.154 | 0.146 |
| Cl1 | 0.038 | -0.002 | -0.021 | -0.026 | H31 | 0.199 | 0.158 | 0.150 | 0.143 |
| Cl2 | 0.044 | 0.003 | -0.008 | -0.013 | H32 | 0.202 | 0.160 | 0.153 | 0.146 |
| Cl3 | 0.042 | 0.000 | -0.020 | -0.025 | |||||
| O1 | -0.708 | -0.783 | -0.656 | -0.659 | |||||
| N1 | -0.737 | -0.650 | -0.417 | -0.430 | |||||
Table 2.
MO energies for Lumefantrine molecule.
| MOs | HF | DFT | Average | ||
|---|---|---|---|---|---|
| 321G | 6-31G* | EDF2 6-31G* |
B3LYP 6-31G* |
||
| LUMO{+1} | 2.5 | 2.4 | -1.2 | -1.1 | 0.7 |
| LUMO | 1.6 | 1.5 | -2.3 | -2.2 | -0.4 |
| HOMO | -8.3 | -8.1 | -5.8 | -5.9 | -7.0 |
| HOMO{-1} | -8.7 | -8.3 | -5.9 | -6.0 | -7.2 |
| HOMO{-2} | -9.4 | -9.4 | -6.1 | -6.3 | -7.8 |
| HOMO{-3} | -9.6 | -9.5 | -6.9 | -7.0 | -8.3 |
| HOMO{-4} | -9.9 | -9.7 | -7.2 | -7.3 | -8.5 |
| HOMO{-5} | -10.0 | -10.0 | -7.2 | -7.4 | -8.7 |
| HOMO{-6} | -10.4 | -10.2 | -7.3 | -7.4 | -8.8 |
| HOMO{-7} | -11.6 | -11.6 | -7.3 | -7.5 | -9.5 |
| HOMO{-8} | -12.1 | -12.1 | -8.2 | -8.4 | -10.2 |
| HOMO{-9} | -12.2 | -12.2 | -8.4 | -8.5 | -10.3 |
Table 3.
Energy equivalencies for the transitions according to different methods.
| Method & Basis Set |
HOMO-1 | HOMO | LUMO | LUMO+1 | Energy Diff. (ΔE) | λmax | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔE1 | ΔE2 | ΔE3 | Calculated (Vac.) | ||||||||
| HF | 3-21G | -8.7 | -8.3 | 1.6 | 2.5 | 10.3 | 9.9 | 10.8 | 120.47 | 125.34 | 114.89 |
| 6-31G* | -8.3 | -8.1 | 1.5 | 2.4 | 9.8 | 9.6 | 10.5 | 126.61 | 129.25 | 118.17 | |
| DFT | EDF2 6-31G* |
-5.9 | -5.8 | -2.3 | -1.2 | 3.6 | 3.5 | 4.6 | 344.67 | 354.52 | 269.74 |
| B3LYP 6-31G* |
-6.0 | -5.9 | -2.2 | -1.1 | 3.8 | 3.7 | 4.8 | 326.53 | 335.36 | 258.50 | |
Table 4.
Calculated Vibrational spectra (FT-IR) for Lumefantrine*.
| 321G | 6-31G* | EDF2 631G* |
B3LYP 631G* |
321G | 6-31G* | EDF2 631G* |
B3LYP 631G* |
||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 493 | 490 | 456 | 456 | 11 | 1434 | 1439 | 1333 | 1331 |
| 2 | 616 | 588 | 560 | 558 | 12 | 1494 | 1507 | 1396 | 1392 |
| 3 | 663 | 630 | 596 | 595 | 13 | 1607 | 1582 | 1470 | 1474 |
| 4 | 700 | 670 | 607 | 621 | 14 | 1678 | 1710 | 1570 | 1561 |
| 5 | 783 | 767 | 731 | 732 | 15 | 1801 | 1806 | 1631 | 1622 |
| 6 | 983 | 974 | 886 | 882 | 16 | 3023 | 3010 | 2837 | 2836 |
| 7 | 1296 | 1304 | 1230 | 1226 | 17 | 3088 | 3083 | 2929 | 2926 |
| 8 | 1179 | 1180 | 1100 | 1098 | 18 | 3142 | 3141 | 3003 | 2994 |
| 9 | 1296 | 1304 | 1230 | 1226 | 19 | 3313 | 3316 | 3131 | 3130 |
| 10 | 1352 | 1344 | 1259 | 1261 | 20 | 3629 | 3884 | 3393 | 3124 |
* The full table can be found in the supporting information file.
Table 5.
Calculated and experimental NMR spectra for Lumefantrine.
| Atom | HF | DFT | DFT | Exp** | Atom | HF | DFT | DFT | Exp** | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3-21G | 6-31G* | EDF2 6-31G* | B3LYP 6-31G* | 3-21G | 6-31G* | EDF26-31G* | B3LYP6-31G* | ||||
| C1 | 128.66 | 137.99 | 141.12 | 135.18 | 141.7 | H1 | 8.43 | 8.21 | 8.02 | 7.74 | 7.58 |
| C2 | 121.57 | 128.09 | 127.69 | 122.43 | 124.1 | H2 | 6.24 | 7.28 | 7.90 | 7.57 | 7.58 |
| C3 | 124.34 | 135.48 | 134.71 | 136.72 | 133.3 | H3 | 7.62 | 7.57 | 7.30 | 7.16 | 7.31 |
| C4 | 117.79 | 123.27 | 121.72 | 116.39 | 127.8 | H4 | 7.49 | 7.66 | 7.63 | 7.67 | 7.44 |
| C5 | 126.93 | 135.30 | 138.57 | 132.56 | 140.0 | H5 | 7.50 | 7.67 | 7.41 | 7.12 | 7.44 |
| C6 | 125.01 | 133.79 | 134.90 | 129.57 | 133.0 | H6 | 7.50 | 7.67 | 7.41 | 7.18 | 7.44 |
| C7 | 128.51 | 136.27 | 137.52 | 131.71 | 136.6 | H7 | 7.49 | 7.60 | 7.63 | 7.09 | 7.44 |
| C8 | 129.51 | 138.94 | 142.48 | 136.16 | 138.4 | H8 | 7.61 | 7.86 | 7.43 | 7.18 | 7.67 |
| C9 | 127.29 | 132.49 | 136.33 | 131.10 | 135.1 | H9 | 7.27 | 7.49 | 7.23 | 6.94 | 7.44 |
| C10 | 118.67 | 124.84 | 128.04 | 123.58 | 128.5 | H10 | 7.52 | 7.77 | 7.55 | 7.38 | 7.71 |
| C11 | 126.38 | 132.76 | 134.66 | 128.27 | 135.1 | H11 | 4.85 | 4.96 | 5.10 | 5.24 | 5.35 |
| C12 | 120.90 | 128.68 | 130.80 | 124.41 | 130.7 | H12 | 3.83 | 3.06 | 4.16 | 3.95 | -- |
| C13 | 121.93 | 127.96 | 129.17 | 122.37 | 129.2 | H13 | 1.87 | 1.97 | 2.31 | 2.07 | 2.44 |
| C14 | 125.05 | 136.10 | 135.43 | 137.50 | 134.8 | H14 | 2.45 | 2.55 | 3.24 | 3.09 | 2.87 |
| C15 | 121.93 | 127.96 | 129.17 | 122.99 | 129.2 | H15 | 1.97 | 2.30 | 2.69 | 2.58 | 2.59 |
| C16 | 120.90 | 128.68 | 130.80 | 124.88 | 130.7 | H16 | 2.01 | 2.17 | 2.63 | 2.57 | 2.59 |
| C17 | 114.58 | 120.41 | 120.28 | 114.59 | 126.5 | H17 | 1.50 | 1.56 | 1.80 | 2.00 | 1.49 |
| C18 | 123.53 | 134.68 | 133.14 | 135.27 | 134.3 | H18 | 1.23 | 1.49 | 1.66 | 1.64 | 1.49 |
| C19 | 120.67 | 126.74 | 128.14 | 121.79 | 130.7 | H19 | 0.81 | 1.17 | 1.33 | 1.38 | 1.36 |
| C20 | 114.60 | 122.07 | 123.59 | 117.46 | 120.8 | H20 | 0.80 | 1.18 | 1.34 | 1.42 | 1.36 |
| C21 | 54.52 | 58.98 | 65.74 | 65.49 | 65.6 | H21 | 0.93 | 1.09 | 0.97 | 1.19 | 0.96 |
| C22 | 53.92 | 60.05 | 66.73 | 65.96 | 60.1 | H22 | 0.93 | 1.09 | 0.97 | 1.06 | 0.96 |
| C23 | 47.89 | 52.00 | 58.82 | 56.47 | 53.5 | H23 | 0.93 | 1.09 | 0.97 | 0.98 | 0.96 |
| C24 | 28.11 | 31.58 | 34.76 | 36.97 | 29.2 | H24 | 2.01 | 2.17 | 2.63 | 2.31 | 2.59 |
| C25 | 16.90 | 19.79 | 21.57 | 23.31 | 20.7 | H25 | 1.97 | 2.30 | 2.69 | 2.73 | 2.59 |
| C26 | 13.48 | 15.27 | 14.86 | 15.78 | 14.2 | H26 | 1.50 | 1.56 | 1.80 | 1.61 | 1.49 |
| C27 | 47.89 | 52.00 | 58.82 | 60.59 | 53.5 | H27 | 1.23 | 1.49 | 1.66 | 1.66 | 1.49 |
| C28 | 28.11 | 31.58 | 34.76 | 34.67 | 29.2 | H28 | 0.81 | 1.17 | 1.33 | 1.34 | 1.36 |
| C29 | 16.90 | 19.79 | 21.57 | 23.05 | 20.7 | H29 | 0.80 | 1.18 | 1.34 | 1.34 | 1.36 |
| C30 | 13.48 | 15.27 | 14.86 | 15.89 | 14.2 | H30 | 0.93 | 1.09 | 0.97 | 1.17 | 0.96 |
| H31 | 0.93 | 1.09 | 0.97 | 0.97 | 0.96 | ||||||
| H32 | 0.93 | 1.09 | 0.97 | 1.03 | 0.96 | ||||||
** Borrowed from Reference 1.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



