
Submitted:
14 June 2023
Posted:
15 June 2023
Read the latest preprint version here
Abstract
Muscarinic acetylcholine receptors are well-known for their crucial involvement in hippocampus-dependent learning and memory, but the exact roles of the various receptor subtypes (M1-M5) are still not fully understood. Here, we studied how M1 and M3 receptors affect plasticity at the mossy fiber (MF)-CA3 pyramidal cell syn-apse. In hippocampal slices from M1/M3 receptor double knockout (M1/M3-dKO) mice, the signature short-term plasticity of the MF-CA3 synapse was not significantly affected. However, the rather unique, NMDA receptor-independent and presynaptic form of long-term potentiation (LTP) of this synapse was much larger in M1/M3-deficient slices compared to wild type slices, in both field potential and whole-cell recordings. Consistent with its presynaptic origin, induction of MF-LTP strongly en-hanced the excitatory drive onto single CA3 pyramidal cells, with the effect being more pronounced in M1/M3-dKO cells. In an earlier study (Zheng et al., 2012), we found that deletion of M2 receptors in mice disinhibits MF-LTP in a similar fashion, suggesting that endogenous acetylcholine employs both M1/M3 and M2 receptors to constrain MF-LTP. Importantly, such synergism was not observed for MF long-term depression (LTD). Low-frequency stimulation, which reliably induced LTD of MF synapses in control slices, failed to do so in M1/M3-dKO slices and gave rise to LTP instead. In striking contrast, loss of M2 augmented LTD when compared to control slices. Taken together, our data demonstrate convergence of M1/M3 and M2 recep-tors on MF-LTP, but functional divergence on MF-LTD, the net effect being well-balanced bidirectional plasticity of the MF-CA3 pyramidal cell synapse.
Keywords:
muscarinic acetylcholine receptors
; hippocampal CA3 pyramidal cells
; mossy fiber synapses
; frequency facilitation
; long-term depression
; long-term potentiation
1. Introduction
Muscarinic acetylcholine receptors (mAChRs) have been long recognized as essential players in cognitive functions (Levey, 1996; Hasselmo, 2006; Thiele, 2013; Haam and Yakel, 2017; Ananth et al., 2023), yet the particular roles of the five mAChR subtypes (M1-M5) are still not sufficiently resolved. Based on their downstream signaling pathways, mAChRs fall in two groups. The unevenly numbered receptors (M1, M3, M5) couple to Gq/11 (M1-type receptors), whereas the evenly numbered receptors (M2, M4) signal via Gi/o (M2-type receptors) (Felder, 1995; Wess, 1996; Thiele, 2013). In the absence of highly subtype-specific pharmacological tools, the advent of knock-out (KO) mice lacking one or two mAChRs substantially advanced the field, enabling a more detailed analysis of muscarinic effects on cognitive performance (Bymaster et al., 2003; Wess et al., 2007; Thomsen et al., 2018). For example, M2-KO, but not M4-KO mice exhibit deficits in hippocampus-dependent learning tasks (Tzavara et al., 2003; Seeger et al., 2004; Bainbridge et al., 2008; Romberg et al., 2018). Likewise, global and hippocampal-specific deletion of M3 receptors impairslearning and memory (Poulin et al., 2010; Leaderbrand et al., 2016), whereas M1-KO mice show only selective deficits in tasks involving hippocampal-cortical interplay (Miyakawa et al., 2001; Anagnostaras et al., 2003).
As long-term synaptic plasticity is widely accepted as a neurobiological substrate of learning and memory, an obvious question is whether the cognitive deficits of mAChR-KO mice can be attributed to impaired plasticity at the synaptic level. Indeed, for M2-KO mice, we found a significant decline in NMDA receptor-dependent long-term potentiation (LTP) in the hippocampal slices, both at the Schaffer collateral (SC)-CA1 synapse and the associational/commissural fiber (A/C)-CA3 synapse (Seeger et al., 2004; Zheng et al., 2012). Less clear effects on LTP at the SC-CA1 synapse were observed in the hippocampal slices from M1- and M3-KO mice. Lack of M3 receptors did not alter LTP (Shinoe et al., 2005), whereas lack of M1 receptors led to either normal or reduced LTP, depending on the induction protocol (Anagnostaras et al., 2003; Shinoe et al., 2005; Kamsler et al., 2010). Interestingly, we reported earlier that lack of M2 receptors diminished LTP at the A/C-CA3 pyramidal cells (v.s.), but enhanced NMDA receptor-independent LTP at the mossy fiber (MF)-CA3 synapse (Zheng et al., 2012), suggesting that M2 receptors can regulate the strength of the two main projections onto CA3 pyramidal cells in an opposite, input-specific fashion.
Like other excitatory synapses, MF-CA3 synapses undergo long-term depression (LTD) following prolonged low-frequency stimulation (LFS; Kobayashi et al., 1996). LTD is the counterpart of LTP, and its importance for cognitive processes is increasingly appreciated (Kemp and Manahan-Vaughan, 2007; Connor and Wang, 2015). Like LTP, LTD at MF-CA3 synapses is predominantly NMDA receptor-independent and presynaptic in origin (Kobayashi et al., 1996; Manabe, 1997; Nicoll and Schmitz, 2005). Since CA3 and dentate gyrus (DG) express appreciable levels of M1 and M3 receptors, but not M5 receptors (Vilaró et al., 1990; Levey et al., 1991; Levey, 1996), we used M1/M3 double KO (M1/M3-dKO) mice to explore how M1-type receptors shape lasting upward and downward changes in synaptic strength at the MF-CA3 synapse. Our data demonstrate that, in hippocampal slices from M1/M3-dKO mice, LTP is enhanced at the expense of LTD, which is abrogated. By contrast, elimination of M2 receptors augmented both LTP and LTD. Taken together, our data demonstrate that M1- and M2-type receptors regulate LTP and LTD at the MF-CA3 synapse in a synergistic and antagonistic fashion, respectively.
2. Materials and Methods
M1/M3-dKO mice (genetic background 129J1 × CF1) were generated as previously described (Gautam et al., 2004). In some experiments, homozygous M2 single KO (M2-KO) mice (Gomeza et al., 1999) were used for comparison. For each knockout strain, age-matched wild type (wt) mice of the matching genetic background were used in parallel as controls. Mice were housed under standard conditions. All procedures were conducted in accordance with the Animal Protection Law of Germany and the European Communities Council Directive of November 1986 /86/609/EEC), and with approval of local Franconian government.
Transverse hippocampal slices (350 µm thick) were prepared from adult male or female mice (3-7 month-old, anesthetized with sevoflurane) and maintained as described previously (Sydow et al., 2011; Zheng et al., 2012). The slices were then kept in modified artificial cerebrospinal fluid (aCSF) containing (in mM) 125 NaCl, 3 KCl, 1 CaCl2, 3 MgCl2, 1.25 NaH2PO4, 25 NaHCO3 and 10 d-glucose at room temperature for at least 2 h before being used. Individual slices were transferred to a submerged chamber perfused with normal aCSF with 1.5 mM MgCl2 and 2.5 mM CaCl2 at 31 ± 1 ºC, unless otherwise stated. All solutions were constantly gassed with 95 % O2 - 5 % CO2. Signals were filtered at 2 kHz and sampled at 20 kHz using a Multiclamp 700B amplifier together with Digidata 1440A interface and pClamp10 software (Molecular Devices, Sunnyvale, CA, USA). MiniDigi 1A and AxoScope 10 were used for low-resolution scope recording, sampled at 1 kHz. Drugs and chemicals were obtained from Tocris Bioscience (Bio-techne GmbH, Wiesbaden, Germany) and Sigma-Aldrich Chemie GmbH (Steinheim, Germany).
Whole-cell recordings of visualized CA3 pyramidal cells in dorsal hippocampal slices were performed in voltage-clamp mode with patch pipettes filled with (in mM) 135 K-gluconate, 5 HEPES, 3 MgCl2, 5 EGTA, 2 Na2ATP, 0.3 Na3GTP, 4 NaCl (pH 7.3). Cells were held at -70 mV and all potentials were corrected for liquid junction potential. Series resistance in whole-cell configuration was 5-20 MΩ and compensated by 60-80%. To monitor the excitatory synaptic drive onto CA3 pyramidal cells, spontaneously occurring excitatory postsynaptic currents (spEPSCs) were collected in the presence of the GABAA-receptor antagonist picrotoxin (100 µM). In some cases, tetrodotoxin (TTX, 1 µM) was introduced to the perfusing solution to block action potential discharge, yielding miniature EPSCs (mEPSCs). To elevate the level of ambient ACh in the slice tissue, acetylcholinesterase activity was inhibited by eserine (10 µM).
Constant-current pulses (width 0.1 ms) were delivered to a bipolar tungsten electrode located in the hilus to activate mossy fiber (MF) projection. The evoked MF EPSCs were monitored at 0.1 Hz. Stimuli were carefully adjusted at low intensities to minimize polysynaptic and/or A/C pathway activation of CA3 pyramidal cells. MF responses were characterized by their prominent feature of strong facilitation during short trains of repetitive stimulation (Nicoll and Schmitz, 2005). LTP of MF-CA3 synapses was induced by high-frequency stimulation (HFS) at 100 Hz for 1 s, repeated 3 times at an interval of 10 s. Long-term depression (LTD) of MF EPSCs was induced by low-frequency stimulation (LFS) at 1 Hz for 15 min. As LTP of A/C synapses is NMDA receptor-dependent, the NMDA receptor antagonist d-2-amino-5-phosphonopentanoic acid (d-AP5, 50 μM) was present in all experiments on MF synaptic plasticity to prevent contamination from A/C responses. Data were included only when the evoked EPSCs were reduced > 90% by the group II metabotropic glutamate receptor agonist DCG IV (2.5 μM) at the end of the experiment.
MF-LTP experiments were also performed using extracellular recording in CA3 stratum lucidum, with aCSF containing 4 mM CaCl2 and 4 mM MgCl2 to avoid multiple synaptic processes. The recording pipette for field postsynaptic potentials (fPSPs) was filled with modified aCSF, in which NaHCO3 was replaced by HEPESs to avoid pH change. LTP of CA3 MF fPSPs was induced by tetanic stimulation at 25 Hz for 5 s, in the presence of d-AP5 (50 μM; Sydow et al., 2011).
Data analysis was performed off-line with Clampfit 10.6 (Molecular Devices, CA, USA). Spontaneous events were detected using an automated event detection algorithm with an amplitude threshold set as 4* σnoise. In addition to the frequency of synaptic inputs, the amplitude and the kinetics of sp/mEPSCs were measured from averaged events which were selected only if no other event occurred during rise and decay. Rise time was measured from 10% to 90% of the peak response. The decay of averaged currents was fitted with single exponential functions using the Levenberg-Marquardt nonlinear least-squares algorithm. Tau reflects the time required for spontaneous events to decay to 37% of their peak value.
Data were expressed as means ± SEM. OriginPro 2018G (OriginLab Corporation, MA, USA) was used for statistics and figures. Statistical comparisons of data were performed using ANOVA or Student's t test as appropriate. Significance was assumed for p < 0.05.
3. Results
3.1. M1/M3 double KO reduces excitatory synaptic drive onto CA3 pyramidal cells
Initially, we examined whether M1/M3-dKO mice showed deficits in basal excitatory neurotransmission in the CA3 region using whole-cell recordings of pharmacologically isolated EPSCs from CA3 pyramidal cells voltage-clamped at -70 mV. As illustrated in Figure 1A, spontaneously occurring EPSCs (spEPSCs) in control slices exhibited a frequency of 4.49 ± 0.53 Hz (n = 24 from 10 wt mice; Figure 1B), with an averaged peak amplitude of 44.49 ± 3.38 pA (Figure 1C). M1/M3-dKO slices showed a significant reduction in both frequency (n = 34 from 13 mice, 3.30 ± 0.31 Hz; p = 0.042) and peak amplitude (32.51 ± 1.87 pA; p = 0.004) of spEPSCs, whereas spEPSC kinetics remained unchanged (Figure 1A–C). Thus, loss of M1/M3 receptors brought about a strong attenuation of the overall excitatory synaptic drive onto CA3 pyramidal cells. Interestingly, a significant reduction of spEPSC frequency was also seen in CA3 pyramidal cells of M5-deficient mice (Araya et al., 2006), although expression of M5 receptors in CA3 and DG is negligible (Vilaró et al., 1990). This seemingly paradox finding may be due to the fact that M5-KO mice suffer from constitutive constriction of cerebral arteries, leading to neuronal atrophy and impaired synaptic connectivity in hippocampus and elsewhere in the brain (Araya et al., 2006).
Notably, the remarkable change in synaptic input in our mutant preparations was not accompanied by significant alterations in intrinsic electrophysiological properties of the CA3 pyramidal cells, such as input resistance (wt 256.46 ± 21.24 MΩ vs M1/M3-dKO 236.47 ± 17.62 MΩ, p = 0.471) and membrane capacitance (wt 109.04 ± 5.36 pF vs. M1/M3-dKO 97.62 ± 3.59 pF, p = 0.071).
We next used tetrodotoxin (TTX, 1 µM) to silence network activity and abrogate firing-driven glutamate release. Under this condition, we observed a pronounced decrease in the frequency of the remaining miniature EPSCs (mEPSCs) in wt CA3 pyramidal cells compared to the frequency of spEPSCs before TTX was added to the bathing solution (n = 5, from 4.89 ± 1.17 Hz spEPSC to 2.11 ± 0.53 Hz mEPSCs; paired t-test, p = 0.016). To elucidate the effect of ambient acetylcholine on mEPSCs frequency and the role of M1/M3 receptors therein, we performed recordings with the acetylcholinesterase inhibitor eserine (10 µM) and the M2 receptor-preferring antagonist gallamine (20 µM) in the bath, in addition to TTX and GABAA receptor antagonist picrotoxin. We also included the GABAB receptor antagonist CGP 55845 (1 µM) to rule out putative indirect effects of GABAB receptors at the MF-CA3 pyramidal cell synapse (Vogt and Regehr, 2001). Application of eserine for 1-3 min reversibly enhanced both mEPSC frequency (from 2.75 ± 0.50 Hz to 4.66 ± 1.01 Hz, n = 6, paired t-test, p = 0.020; i.e. 163.70 ±10.79 % of control) and peak amplitude (from 43.47 ± 4.73 pA to 48.32 ± 3.68 pA, paired t-test, p = 0.049) (Figure 1D–F). To examine whether the facilitation of synaptic transmission following the eserine-induced elevation of ambient acetylcholine levels is mediated solely by M1-type receptors, we further added the nonselective nicotinic AChR (nAChR) antagonist mecamylamine (10 µM; Bacher et al., 2009) to the above cocktail of blockers. With nAChRs suppressed, eserine still caused a significant enhancement of mEPSC frequency, which increased from 2.80 ± 0.57 Hz to 3.48 ± 0.74 Hz (n = 7, paired t-test, p = 0.009) (Figure 1E–F). However, since the relative increase in mEPSC frequency in the presence of mecamylamine amounted to only 123.51 ±3.00 % of control), which was significantly lower than in the absence of this inhibitor (p = 0.003), M1-type receptors and nicotinic receptors appear to jointly promote firing-independent vesicular glutamate release.
3.2. M1/M3-dKO facilitates LTP of mossy fiber-CA3 synapses
Whereas the above recordings provided new information regarding the overall impact of M1-type receptors on the spontaneous excitatory drive experienced by CA3 pyramidal cells, they did not differentiate the synaptic events with respect to their origin, be it mossy fibers, associational/commissural fibers or perforant path. To focus on the MF input and examine how its rather unique plastic changes are modulated by mAChR subtypes, we placed a stimulation electrode into the MF pathway and monitored evoked EPSCs by means of whole-cell recordings from voltage-clamped CA3 pyramidal cells. Suppression of EPSCs after application of the metabotropic glutamate receptor agonist DCG IV at the end of the experiment served to confirm selective activation of the MF pathway (Figure 2C). MF-evoked EPSCs are distinct from other excitatory synaptic responses in that they show a very strong facilitation upon short, repetitive stimulation at relative high frequency (e.g. 4 stimuli at 20 Hz; Figure 2A, inset) – a stimulus paradigm which partially mimics firing patterns of DG granule cells in vivo (Pernía-Andrade and Jonas, 2014). To quantify this signature facilitation between genotypes, we normalized the subsequent EPSC peak amplitudes to that of the first response in the train. As summarized in Figure 2A (wt, n = 20 from 8 mice; M1/M3-dKO, n = 20 from 8 mice), the strong facilitation during the four-stimuli trains was not affected by the absence or presence of M1/M3 receptors. Likewise, another prominent feature of MF synapses, namely frequency facilitation (Nicoll and Schmitz, 2005), defined as strong facilitation during sustained low-frequency stimulation (Figure 2B), remained unchanged in the absence of M1/3 receptors (wt, n = 8 from 6 mice; M1/M3-dKO, n = 8 from 5 mice). The mean increase of evoked MF-EPSCs at 1 min of stimulation was 347.93 ± 51.33 % in wt cells and 280.14 ± 25.45 % in M1/M3-dKO cells (p = 0.256).
In striking contrast to short-term and frequency facilitation which were M1/M3 receptor-independent, these receptors came into play when we examined long-term plasticity at the MF-CA3 synapse. For induction of long-term potentiation (LTP), we used a high-frequency stimulation protocol (HFS at 100 Hz for 1 s, repeated 3 times with 10 s intervals). Lack of M1/M3 receptors led to a much larger potentiation of the evoked responses after HFS compared to the relatively modest potentiation observed in wt hippocampi (Figure 2C–F). As illustrated in Figure 2C–D, responses of MF synapses to a given stimulus varied in size, with failure rates depending on stimulation intensity in individual slices. HFS engendered a massive reduction in failure rate in both groups (wt, n = 7 from 5 mice, from 24.90 ± 6.55 % to 4.36 ± 1.85 %, n = 7, paired t-test, p = 0.020; M1/M3-dKO, n = 8 from 5 mice, from 19.37 ± 6.39 % to 1.01 ± 0.73 %, paired t-test, p = 0.018) (Figure 2C–E). In control hippocampi, HFS enhanced the averaged amplitudes of evoked MF-EPSCs to 143.73 ± 5.91 % (n = 7), measured over 16-20 min post HFS (Figure 2F). By contrast, the mean potentiation of MF-EPSCs in M1/M3-dKO hippocampi at the same time period reached 287.03 ± 32.33 % (n = 8), which was significantly different from wt hippocampi (p = 0.001; Figure 2F). Since we observed a similar pronounced increase in LTP for the same synapse in M2-deficient hippocampi (Zheng et al., 2012), both M1/M3 and M2 receptors appear to constrain mossy fiber LTP.
Consistent with the presynaptic site of MF-LTP (Nicoll and Schmitz, 2005), we observed a strong rise of spontaneous synaptic events between the three HFS stimulus trains and immediately after them (Figure 3A). Specifically, the frequency of spEPSCs in wt pyramidal cells increased by 242.21± 29.55 % (from 2.28 ± 0.46 Hz to 4.72 ± 0.96 Hz within 30 s after HFS (n = 7, paired t-test, p = 0.013; Figure 3B), accompanied by an increase in peak amplitude (from 23.80 ± 2.27 pA to 31.01 ± 4.24 pA, paired t-test, p = 0.038; Figure 3C). In M1/M3-deficient slices, the HFS-associated rise in the number of spontaneous events sky-rocketed to 1035.00 ± 215.30 % of control (1.10 ± 0.17 Hz, n = 8), with a concomitant enhancement of spEPSC amplitude (wt, 128.65 ± 9.50 %; M1/M3-dKO, 184.11 ± 15.75 % of control value 15.18 ± 1.38; p = 0.009, Figure 3C).
We asked next how the synaptic effects of M1/M3 receptors observed in single, voltage-clamped CA3 pyramidal cells would influence the collective neuronal behavior in a network with intact GABAergic inhibition. To address this issue, we performed field potential recordings in CA3 stratum lucidum in the absence of any blockers. Electrical stimulation of MFs evoked a characteristic biphasic response, in which a fiber volley (FV, reflecting synchronized action potential firing in the MF pathway) was followed by a field postsynaptic potential (fPSP, Figure 4A, inset). Again, suppression of fPSP by the metabotropic glutamate receptor agonist DCG IV demonstrated selective activation of the MF pathway. As we have previously reported (Sydow et al., 2011), tetanic stimulation of MFs at 25 Hz for 5 s produced robust LTP in control slices, with peak fPSP amplitudes at 26-30 min post-tetanus rising to 144.22 ± 7.80 % of control (0.24 ± 0.02 mV, n = 9 from 6 wt mice; Figure 4A–B). In M1/M3-dKO slices, the same protocol enhanced fPSP amplitude to 190.18 ± 22.29 % of control (0.18 ± 0.02 mV, n = 6 from mutant mice; Figure 4B), which was significantly stronger than in the wt counterparts (p = 0.037). In line with the corresponding whole-cell recordings (Figure 2A–B), fPSP recordings did not reveal significant differences between the genotypes regarding quadruple-pulse facilitation and frequency facilitation (Figure 4C–D).
3.3. M1/M3-dKO turns LTD into LTP
Like many other glutamatergic synapses in the CNS, MF synapses onto CA3 pyramidal cells undergo LTD after prolonged low-frequency stimulation (LFS at 1 Hz for 15 min). To study and compare muscarinic modulation of MF-LTD vs. -LTP at the same cellular level, we went back to whole-cell recordings of pharmacologically isolated MF-evoked EPSCs. Since MF-LTD is presynaptic in origin, as is MF-LTP, the whole-cell configuration should not interfere with either form of long-term plasticity at this particular synapse, in particular since NMDA receptors were blocked again with d-APV (see Methods). A typical example of MF-LTD in wt hippocampus is illustrated in Figure 5A. When determined 16-20 min after LFS, the averaged EPSC amplitude was reduced to 64.46 ± 9.57 % of control in wt hippocampi (n = 7 slices from 5 mice; Figure 5D), accompanied by enhanced failure rates. (Figure 5C). Note that in LTD experiments, control stimulation intensity before LFS was adjusted to obtain a low failure rate (4.31 ± 1.66 % in wt slices, n = 7). This enabled appropriate capture of the higher failure rates after LFS-induced LTD, which were significantly increased to 16.44 ± 4.81 % (paired t-test, p = 0.030; Figure 5C). In striking contrast, application of the very same stimulation protocol not only abrogated LTD in M1/M3-dKO hippocampi, but even induced LTP, with MF-EPSC amplitudes increasing to 120.74 ± 5.38 % of control value 16-20 min after LFS (n = 5 from 4 mice, p = 0.001 vs. wt; Figure 5B–D).
Since LTP of the MF-CA3 synapse in the hippocampus of M2-KO mice was augmented in a fashion similar to that reported here for M1/M3-dKO mice (Zheng et al., 2012), we wondered whether such synergism between M1- and M2-type receptors also holds for MF-LTD. To our surprise, we obtained the opposite finding: The extent of LTD in M2-KO slices was significantly enhanced, amounting to 58.40 ± 6.99 % reduction (n = 6 slices from 4 M2-KO mice), compared to 28.01 ± 6.98 % in wt slices (n = 5 from 4 mice; p = 0.031) (Figure 5E–F). LFS-induced LTD was also accompanied by higher failure rate in the mutant cells (wt, from 5.84 ± 1.69 % to 13.44 ± 4.55 %, paired t-test, p = 0.107; M2-KO, from 1.42 ± 0.90 % to 21.02 ± 6.98 %, paired t-test, p = 0.026). Thus, M1/M3 and M2 receptors exert opposite effects on LTD at the MF-CA3 pyramidal cell synapse.
4. Discussion
Muscarinic depression of LTP at the MF-CA3 pyramidal cell synapse has been reported first by Williams and Johnston in 1988 (see also Maeda et al., 1993). Since then, the peculiar electrophysiological properties of this rather unique hippocampal synapse have been studied in great detail and related to learning and memory tasks involving pattern separation and/or completion (Nicoll & Schmitz, 2005). In view of the wealth of data accumulated on the many uncommon features and functions of the MF-CA3 synapse within the hippocampal circuitry and in behavioral readouts, it is quite surprising that we still know relatively little about how and for what purposes this synapse is modulated by acetylcholine.
We report here the unexpected finding that in hippocampi of M1/M3-dKO mice, MF-LTP is significantly augmented when compared to wt hippocampi. This finding is corroborated by the fact that we observed anomalously enhanced MF-LTP in M1/M3-dKO hippocampi using two independent experimental settings with distinct induction protocols, namely (i) field potential recordings from hippocampal slices exhibiting intact network activity, and (ii) whole-cell voltage-clamp recordings from CA3 pyramidal cells, in which the GABAA receptor blocker picrotoxin was routinely added to the bathing solution to obtain unambiguous measurements of EPSCs. These experiments strongly suggest that activation of M1-type mAChRs serves to curtail MF-LTP.
Although quite obvious from the experimental evidence, this conclusion seems counterintuitive for two reasons. Firstly, as noted already by Williams and Johnston in their 1988 paper, muscarinic depression of MF-LTP would not have been predicted on the basis of the widely documented essential role of the cholinergic system in facilitating cognitive functions including hippocampus-dependent learning and memory. Common wisdom links a decline in LTP to impaired cognitive performance. This relationship holds indeed for M2-deficient mice, whose memory deficits were attributed to reduced plasticity at the Schaffer-CA1 synapse (Seeger et al., 2004). The second reason, why the above conclusion is puzzling, is based on the observations that hippocampi from both M1/M3-deficient and M2-deficient mice exhibit a strikingly similar increase in MF-LTP, as demonstrated here and in an earlier study (Zheng et al., 2012), respectively. How might signaling pathways as different as those of M2-type receptors, which couple to Gi/o proteins, and those of M1-type receptors, which couple to Gq/11 proteins, functionally converge on inhibition of MF-LTP?
In the hippocampus, M1 and M3 receptors are mainly located postsynaptically (Levey, 1996), where they target various ion conductances to enhance cell excitability and promote firing. Mechanisms include suppression of K+ currents such as M-current (Im) and a slow Ca2+-activated K+ current (IAHP), and increase of depolarizing cation currents such as the hyperpolarization-activated current (Ih) and a Ca2+-dependent nonspecific cation conductance (Icat) (Halliwell et al., 1982; Madison et al., 1987; Colino and Halliwell, 1993). Using mice lacking M1 receptors, Fisahn et al. demonstrated that M1 receptor activation depolarizes CA3 pyramidal cells by increasing Ih and Icat (Fisahn et al., 2002). Thus, reduced muscarinic excitation of presynaptic granule cells and CA3 neurons most likely accounts for the diminished spEPSC frequency that we measured in CA3 neurons from M1/M3-dKO hippocampi.
Do M1-type receptors have also a presynaptic site of action to regulate glutamate release directly? We addressed this issue by monitoring mEPSCs in the presence of TTX and pharmacological suppression of GABAA, GABAB, M2-type und nicotinic receptors. When we enhanced the level of ambient acetylcholine with the acetylcholinesterase inhibitor eserine, we observed a significant increase in mEPSC frequency, most likely mediated by presynaptic M1 receptors. In support of this notion, M1 receptors have been shown to be distributed along mossy fibers, albeit with lower density compared to those in dendrites and spines (Martinello et al., 2015). Note that, although MF-LTP is presynaptic, M1 receptor does not necessarily have to reside on terminals to regulate the strength of synaptic potentiation. An attractive candidate pathway to account for the apparent disinhibition of MF-LTP in the absence of M1/M3 receptors involves retrograde endocannabinoid signaling. Activation of postsynaptic M1 and M3 receptors during strong synaptic use may trigger release of endocannabinoids from the postsynaptic site (Ohno-Shosaku et al., 2003), which in turn bind to presynaptic CB1 receptors to suppress transmitter release (Straiker and Mackie, 2005).
Whereas it remains to be determined in future studies how postsynaptic and/or presynaptic M1/M3 receptor signaling contains MF-LTP, explaining how M2 receptor activation results in the same outcome seems more straightforward. The canonical pathway of MF-LTP comprises the following sequence (Nicoll & Schmitz, 2005): Ca2+ influx through presynaptic voltage-dependent Ca2+ channels activation of Ca2+-sensitive adenylyl cyclase 1 elevation of cAMP levels activation of PKA persistent increase in transmitter release. As discussed in more detail before, presynaptic M2 heteroreceptors on MF terminals may interfere with LTP induction through inhibition of presynaptic Ca2+ channels and/or attenuation of adenylyl cyclase activity (Zheng et al., 2012).
Whereas M1 and M2 receptors seem to use different routes to curtail MF-LTP, our study also reveals some commonalities in the way they act. Firstly, with GABAA receptors routinely blocked in our whole-cell recordings, elimination of neither mAChR subtype should have disinhibited MF-LTP through a GABAergic mechanism, where activation of presynaptic GABAA receptors facilitates MF-CA3 synaptic plasticity (Ruiz et al., 2010). Secondly, in both field potential and whole-cell recordings, we employed robust stimulation protocols to induce presynaptic MF-LTP, instead of weak stimulation protocols, which induce an unorthodox postsynaptic and NMDA receptor-mediated form of MF-LTP (Kwon and Castillo, 2008; Rebola et al., 2008). Thus, M1 and M2 subtypes should both have a presynaptic site of action to regulate LTP (including also retrograde signaling, v.s.). Thirdly, both mAChR types not only inhibit MF-LTP. They also have in common that they do not affect the unique hallmarks of MF short-term plasticity, namely quadruple-pulse facilitation and frequency facilitation.
Endowed with the latter features, MF synapses can act as a "conditional detonator" (Bischofberger et al., 2006). This particular property allows the MF synapse to assume a role as unsupervised "teacher" synapse triggering plastic changes in the connectivity pattern of CA3 neurons. In the case of place cells, such formed ensembles of CA3 pyramidal cells are important for storage and recall of spatial information (Bischofberger et al., 2006). Put simply, muscarinic inhibition of MF-LTP might thus be envisioned as a means to preserve the integrity of the "conditional detonator", which might blow up unintentionally when synaptic potentiation is not properly controlled.
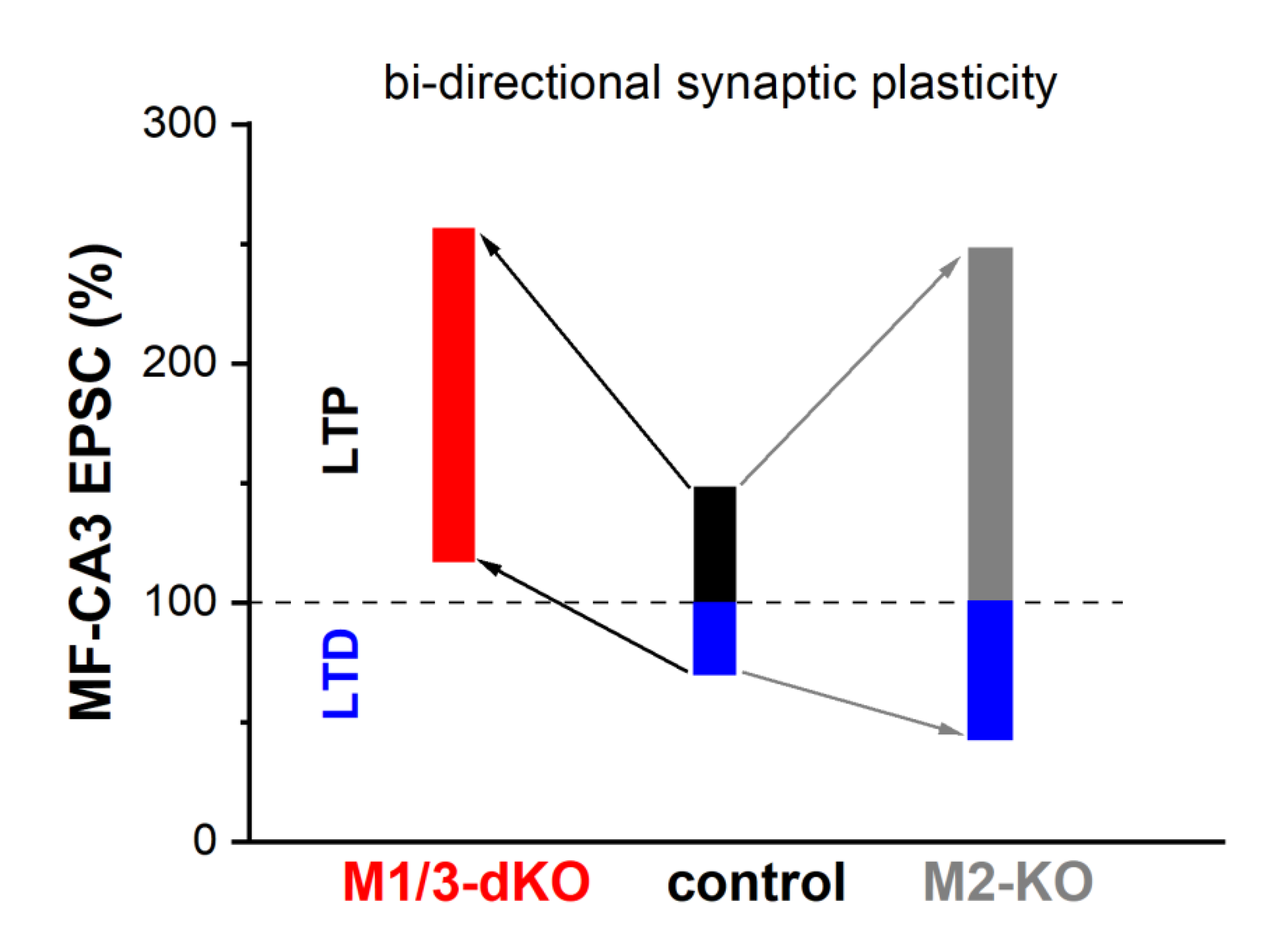
Whereas MF-LTP is synergistically capped by activation of M1- and M2- type receptors, our study demonstrates that the two receptor types exert opposite effects on MF-LTD. In M1/M3-dKO, LTD was abrogated and LFS produced even a small potentiation, whereas loss of M2 receptors augmented LTD (Fig. 6). Interestingly, a very similar shift from LTD to LTP following LFS was observed in visual cortex slices from M1/M3-dKO mice (Origlia et al., 2006). Unlike MF synapses in the hippocampus, the excitatory synapses examined in the visual cortex preparation display postsynaptic, NMDA receptor-dependent long-term plasticity. It is remarkable that, although the sites and mechanisms of induction of LTP and LTD differ substantially between hippocampal MF synapses and the synapses in visual cortex, both synapses rely on M1/M3 activation to prevent the paradoxical conversion of LTD to LTP following LFS.
Our data obtained with wt hippocampal preparations suggest that, under physiological conditions, the opposing forces that act on MF-LTD, namely M1/M3 receptor-mediated augmentation vs. M2 receptor-mediated inhibition, are matched to enable a degree LTD that is capable of counterbalancing LTP. We found indeed that long-term plasticity of the MF-CA3 pyramidal cell synapse extends almost equally in both directions, with a rather small bias in favor of LTP over LTD (Fig. 6). For several reasons, it has been postulated that in a network, where synapses undergo LTP, LTD is a necessary counterweight to enhance the overall performance in information processing, storage and recall (Rosenzweig et al., 2001). First and foremost, LTD counteracts the saturating effects that would ensue from potentiation alone. Furthermore, LTD facilitates the grouping of potentiated synapses that constitute a memory trace by suppressing synapses that do not participate in encoding this particular trace. Finally, LTD enables behavioral flexibility by weakening previously learned information that would interfere with the acquisition of new information in a changing environment.
This latter conclusion resulted from work with transgenic mice in which NMDA-dependent LTD of the Schaffer collateral-CA1 synapse was selectively disrupted (Nicholls et al., 2008). A similar approach to decipher the functional role of MF-LTD has not been reported yet. However, valuable insights come from field potential recordings in freely behaving rats demonstrating that LTD in the CA3 region encodes different aspects of a novel environment in an input-specific fashion: MF-LTD is associated with exploration of landmark objects, whereas exploration of discrete positional features of the environment facilitates AC-LTD (Hagena and Manahan-Vaughan, 2011). Whereas the full behavioral implications of MF-LTP and -LTD are only beginning to be understood, our study shows that muscarinic receptor activation confers a properly balanced bidirectional plasticity on the MF-CA3 pyramidal cell synapse, which should be important for optimal functionality and flexibility in learning and memory tasks.
Author Contributions
J.W. generated and provided the M1/M3-dKO and the M2-KO mice used in this study; FZ performed experiments and analyzed data; FZ and CA designed the research, interpreted results and wrote the manuscript, with contributions from J.W.
Acknowledgments
We thank Didier Gremelle for technical assistance. This work was supported by Deutsche Forschungsgemeinschaft Grant AL 294/9-1 (to CA). J.W. received funding from the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, NIH).
Conflicts of Interest
No conflicts of interest, financial or otherwise, are declared by the authors.
Abbreviations
aCSF: artificial cerebrospinal fluid; A/C, associational/commissural fiber; DG, dentate gyrus; EPSC, excitatory postsynaptic current; FV, fiber volley; fPSP, field postsynaptic potential; HFS, high-frequency stimulation; LFS, low-frequency stimulation; LTD, long-term depression; LTP, long-term potentiation; MF, mossy fiber; mAChR, muscarinic acetylcholine receptor; nAChR, nicotinic acetylcholine receptor; SC, Schaffer collateral; STP, short-term plasticity; TTX, tetrodotoxin.
References
- Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM and Silva AJ (2003) Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci 6:51-58. [CrossRef]
- Ananth MR, Rajebhosale P, Kim R, Talmage DA and Role LW (2023) Basal forebrain cholinergic signalling: development, connectivity and roles in cognition. Nat Rev Neurosci 24:233-251. [CrossRef]
- Araya R, Noguchi T, Yuhki M, Kitamura N, Higuchi M, Saido TC, Seki K, Itohara S, Kawano M, Tanemura K, Takashima A, Yamada K, Kondoh Y, Kanno I, Wess J and Yamada M (2006) Loss of M5 muscarinic acetylcholine receptors leads to cerebrovascular and neuronal abnormalities and cognitive deficits in mice. Neurobiol Dis 24:334-344. [CrossRef]
- Bacher I, Wu B, Shytle DR and George TP (2009) Mecamylamine - a nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert Opin Pharmacother 20:2709-2721. [CrossRef]
- Bainbridge NK, Koselke LR, Jeon J, Bailey KR, Wess J, Crawley JN and Wrenn CC (2008) Learning and memory impairments in a congenic C57BL/6 strain of mice that lacks the M2 muscarinic acetylcholine receptor subtype. Behav Brain Res 190:50-58. [CrossRef]
- Bischofberger J, Engel D, Frotscher M and Jonas P (2006) Timing and efficacy of transmitter release at mossy fiber synapses in the hippocampal network. Pflugers Arch 453:361-372. [CrossRef]
- Bymaster FP, McKinzie DL, Felder CC and Wess J (2003) Use of M1-M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem Res 28:437-442. [CrossRef]
- Colino A and Halliwell JV (1993) Carbachol potentiates Q current and activates a calcium-dependent non-specific conductance in rat hippocampus in vitro. Eur J Neurosci 5:1198-1209. [CrossRef]
- Connor SA and Wang YT (2016) A place at the Table: LTD as a Mediator of Memory Genesis. Neuroscientist 22:359-371. [CrossRef]
- Felder CC (1995) Muscarinic acetylcholine receptors: signal transduction through multiple effectors. FASEB J 9:619-625. [CrossRef]
- Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ and Wess J (2002) Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron 33:615-624. [CrossRef]
- Gautam D, Heard TS, Cui Y, Miller G, Bloodworth L and Wess J (2004) Cholinergic stimulation of salivary secretion studied with M1 and M3 muscarinic receptor single- and double-knockout mice. Mol Pharmacol 66:260-267. [CrossRef]
- Gomeza J, Shannon H, Kostenis E, Felder C, Zhang L, Brodkin J, Grinberg A, Sheng H and Wess J (1999) Pronounced pharmacologic deficits in M2 muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci USA 96:1692-1697. [CrossRef]
- Haam J and Yakel JL (2017) Cholinergic modulation of the hippocampal region and memory function. J Neurochem 142(Suppl 2):111-121. [CrossRef]
- Hagena H and Manahan-Vaughan D (2011) Learning-facilitated synaptic plasticity at CA3 mossy fiber and commissural-associational synapses reveals different roles in information processing. Cereb Cortex 21:2442-2449. [CrossRef]
- Halliwell JV and Adams PR (1982) Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res 250:71-92. [CrossRef]
- Hasselmo ME (2006) The role of acetylcholine in learning and memory. Current Opinion in Neurobiology 16:710-715. doi.org/10.1016/j.conb.2006.09.
- Henze DA, Urban NN and Barrionuevo G (2000) The multifarious hippocampal mossy fiber pathway: a review. Neuroscience 98:407-427. [CrossRef]
- Henze DA, Wittner L and Buzsáki G (2002) Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci 5:790-795. [CrossRef]
- Kamsler A, McHugh TJ, Gerber D, Huang SY and Tonegawa S (2010) Presynaptic m1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. Proc Natl Acad Sci USA 107:1618-1623. [CrossRef]
- Kemp A and Manahan-Vaughan D (2007) Hippocampal long-term depression: master or minion in declarative memory processes? Trends in Neurosciences 30:111-118. [CrossRef]
- Kobayashi K, Manabe T and Takahashi T (1996) Presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Science 273:648-650. [CrossRef]
- Kwon HB and Castillo PE (2008) Long-term potentiation selectively expressed by NMDA receptors at hippocampal mossy fiber synapses. Neuron 57:108-120. [CrossRef]
- Leaderbrand K, Chen HJ, Corcoran KA, Guedea AL, Jovasevic V, Wess J and Radulovic J (2016) Muscarinic acetylcholine receptors act in synergy to facilitate learning and memory. Learn Mem 23:631-638. [CrossRef]
- Levey AI (1996) Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci USA 93:13541-13546. [CrossRef]
- Levey AI, Kitt CA, Simonds WF, Price D and Brann MR (1991) Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci 11:3218-3226. [CrossRef]
- Madison DV, Lancaster B and Nicoll RA (1987) Voltage clamp analysis of cholinergic action in the hippocampus. J Neurosci 7:733-741. [CrossRef]
- Maeda T, Kaneko S and Satoh M (1993) Bidirectional modulation of long-term potentiation by carbachol via M1 and M2 muscarinic receptors in guinea pig hippocampal mossy fiber-CA3 synapses. Brain Res 619:324-330. [CrossRef]
- Manabe T (1997) Two forms of hippocampal long-term depression, the counterpart of long-term potentiation. Rev Neurosci 8:179-193. [CrossRef]
- Martinello K, Huang Z, Lujan R, Tran B, Watanabe M, Cooper EC, Brown DA and Shah MM (2015) Cholinergic afferent stimulation induces axonal function plasticity in adult hippocampal granule cells. Neuron 85:346-363. [CrossRef]
- Miyakawa T, Yamada M, Duttaroy A and Wess J (2001) Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci 21:5239-5250. [CrossRef]
- Nicoll RA and Schmitz D (2005) Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci 6:863-876. [CrossRef]
- Nicholls RE, Alarcon JM, Malleret G, Carroll RC, Grody M, Vronskaya S and Kandel ER (2008) Transgenic mice lacking NMDAR-dependent LTD exhibit deficits in behavioral flexibility. Neuron 58:104-117. [CrossRef]
- Ohno-Shosaku T, Matsui M, Fukudome Y, Shosaku J, Tsubokawa H, Taketo MM, Manabe T, Kano M (2003) Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoid signalling in the hippocampus. Eur J Neurosci 18:109-116. [CrossRef]
- Origlia N, Kuczewski N, Aztiria E, Gautam D, Wess J and Domenici L (2006) Muscarinic acetylcholine receptor knockout mice show distinct synaptic plasticity impairments in the visual cortex. J Physiol 577(Pt 3):829-840. [CrossRef]
- Pernía-Andrade AJ and Jonas P (2014) Theta-gamma-modulated synaptic currents in hippocampal granule cells in vivo define a mechanism for network oscillations. Neuron 81:140-152. [CrossRef]
- Poulin B, Butcher A, McWilliams P, Bourgognon JM, Pawlak R, Kong KC, Bottrill A, Mistry S, Wess J, Rosethorne EM, Charlton SJ andTobin AB (2010) The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/arrestin-dependent manner. Proc Natl Acad Sci USA 107:9440-9445. [CrossRef]
- Rebola N, Lujan R, Cunha RA and Mulle C (2008) Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron 57:121-134. [CrossRef]
- Romberg C, Bartko S, Wess J, Saksida LM and Bussey TJ (2018) Impaired object-location learning and recognition memory but enhanced sustained attention in M2 muscarinic receptor-deficient mice. Psychopharmacology (Berl) 235:3495-3508. [CrossRef]
- Rosenzweig ES, Barnes CA and McNaughton BL (2002) Making room for new memories. Nat Neurosci 5:6-8. [CrossRef]
- Ruiz A, Campanac E, Scott RS, Rusakov DA and Kullmann DM (2010) Presynaptic GABAA receptors enhance transmission and LTP induction at hippocampal mossy fiber synapses. Nat Neurosci 13:431-438. [CrossRef]
- Seeger T, Fedorova I, Zheng F, Miyakawa T, Koustova E, Gomeza J, Basile AS, Alzheimer C and Wess J (2004) M2 muscarinic acetylcholine receptor knock-out mice show deficits in behavioral flexibility, working memory, and hippocampal plasticity. J Neurosci 24:10117-10127. [CrossRef]
- Shinoe T, Matsui M, Taketo MM and Manabe T (2005) Modulation of synaptic plasticity by physiological activation of M1 muscarinic acetylcholine receptors in the mouse hippocampus. J Neurosci 25:11194-11200. [CrossRef]
- Straiker A and Mackie K (2005) Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol 569:501-517. [CrossRef]
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C and Mandelkow EM (2011) Tau-induced defects in synaptic plasticity, learning and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci 31:2511-2525. [CrossRef]
- Thiele A (2013) Muscarinic signaling in the brain. Annu Rev Neurosci 36:271-294. [CrossRef]
- Thomsen M, Sørensen G and Dencker D (2018) Physiological roles of CNS muscarinic receptors gained from knockout mice. Neuropharmacology 136(Pt C):411-420. [CrossRef]
- Tzavara ET, Bymaster FP, Felder CC, Wade M, Gomeza J, Wess J, McKinzie DL and Nomikos GG (2003) Dysregulated hippocampal acetylcholine neurotransmission and impaired cognition in M2, M4 and M2/M4 muscarinic receptor knockout mice. Mol Psychiatry 8:673-679. [CrossRef]
- Vilaró MT, Palacios JM and Mengod G (1990) Localization of m5 muscarinic receptor mRNA in rat brain examined by in situ hybridization histochemistry. Neurosci Lett 14:154-159. [CrossRef]
- Vogt KE and Regehr WG (2001) Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. J Neurosci 21:75-83. [CrossRef]
- Wess J (1996) Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol10:69-99. [CrossRef]
- Wess J, Eglen RM and Gautam D (2007) Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov 6:721-733. [CrossRef]
- Williams S and Johnston D (1988) Muscarinic depression of long-term potentiation in CA3 hippocampal neurons. Science 242:84-87. [CrossRef]
- Zheng F, Wess J and Alzheimer C (2012) M2 muscarinic acetylcholine receptors regulate long-term potentiation at hippocampal CA3 pyramidal cell synapses in an input-specific fashion. J Neurophysiol 108:91-100. [CrossRef]
Figure 1.
M1/M3-dKO reduces excitatory synaptic drive onto CA3 pyramidal cells. (A) Representative spEPSC recordings from wt neuron (black trace) and mutant neuron (red trace), respectively. Insets depict averaged synaptic events from respective cells. (B-C) Comparison of frequency and peak amplitude (B), and kinetics (C) of spEPSCs between the two genotypes. Numbers in columns indicate sample size. (D-F) Reversible increase in mEPSC frequency and amplitude during application of acetylcholinesterase inhibitor eserine (10 µM) in wt slices superfused with gallamine (20 µM) alone or in combination with mecamylamine (10 µM), in the presence of TTX (1 µM), picrotoxin and CGP 55845 (1 µM). * p < 0.05, ** p < 0.01.
Figure 1.
M1/M3-dKO reduces excitatory synaptic drive onto CA3 pyramidal cells. (A) Representative spEPSC recordings from wt neuron (black trace) and mutant neuron (red trace), respectively. Insets depict averaged synaptic events from respective cells. (B-C) Comparison of frequency and peak amplitude (B), and kinetics (C) of spEPSCs between the two genotypes. Numbers in columns indicate sample size. (D-F) Reversible increase in mEPSC frequency and amplitude during application of acetylcholinesterase inhibitor eserine (10 µM) in wt slices superfused with gallamine (20 µM) alone or in combination with mecamylamine (10 µM), in the presence of TTX (1 µM), picrotoxin and CGP 55845 (1 µM). * p < 0.05, ** p < 0.01.
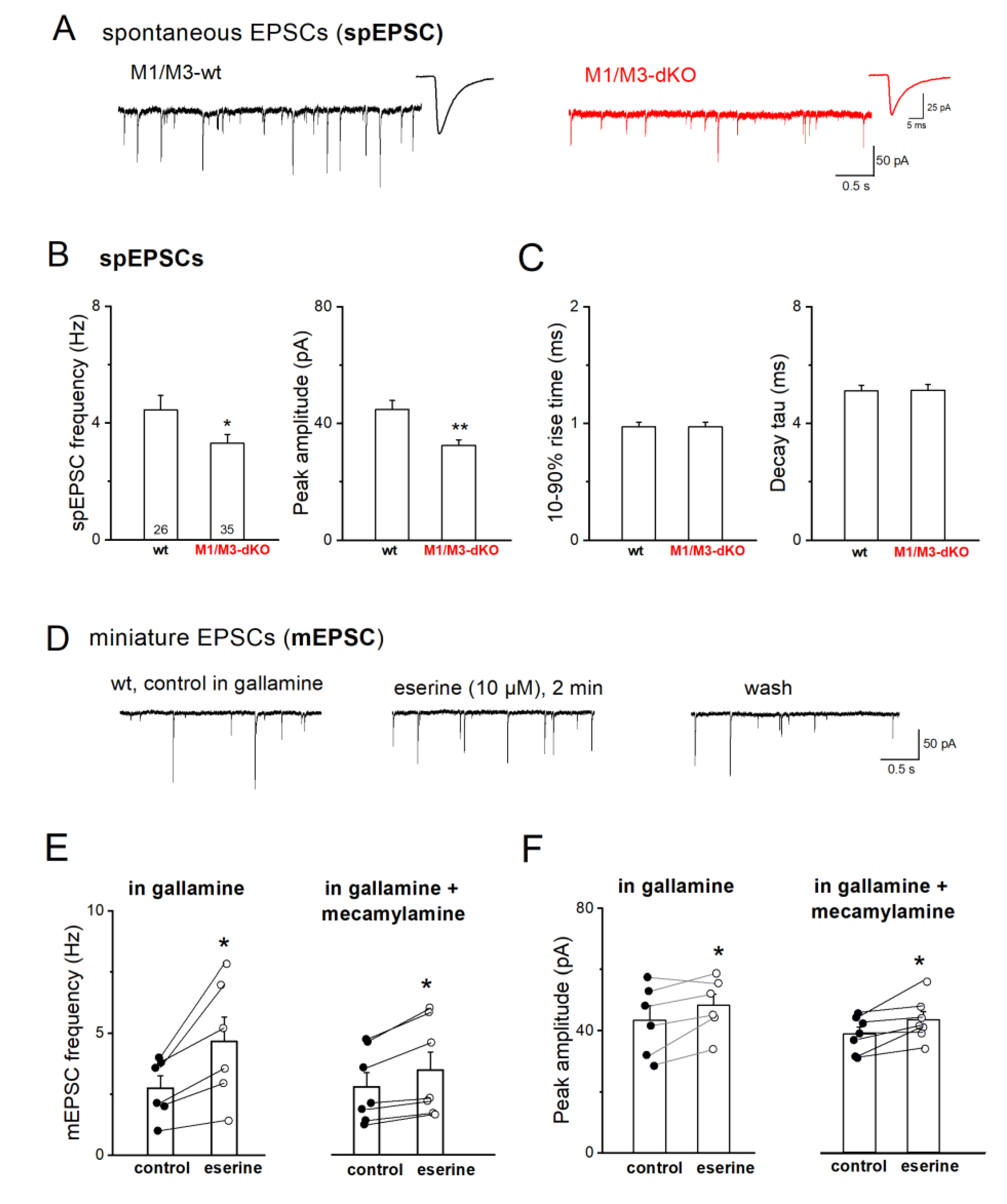
Figure 2.
M1/M3-dKO does not alter STP, but strongly facilitates LTP of the MF-CA3 pyramidal cell synapse. (A-B) No significant change in short-term plasticity (STP) in M1/M3-dKO preparation. Top current trace in A from wt slice illustrates massive synaptic facilitation during quadruple-pulse stimulation with 55 µA. Diagram below summarizes EPSC amplitudes normalized to that of 1st EPSC. (B) Frequency facilitation after switching from 0.1 Hz to 1 Hz stimulation. EPSC amplitudes were normalized for comparison. Inset shows EPSC trace from wt cell before and 1 min post 1 Hz stimulation. (C-D) Scatter plots of EPSC responses before and after HFS in wt pyramidal cell (C) and mutant pyramidal cell (D). (E) Drop in failure rates after HFS in both genotypes. (F) Comparison of HFS-induced LTP in wt cells (black symbols) and mutant cells (red symbols). * p < 0.05, ** p < 0.01.
Figure 2.
M1/M3-dKO does not alter STP, but strongly facilitates LTP of the MF-CA3 pyramidal cell synapse. (A-B) No significant change in short-term plasticity (STP) in M1/M3-dKO preparation. Top current trace in A from wt slice illustrates massive synaptic facilitation during quadruple-pulse stimulation with 55 µA. Diagram below summarizes EPSC amplitudes normalized to that of 1st EPSC. (B) Frequency facilitation after switching from 0.1 Hz to 1 Hz stimulation. EPSC amplitudes were normalized for comparison. Inset shows EPSC trace from wt cell before and 1 min post 1 Hz stimulation. (C-D) Scatter plots of EPSC responses before and after HFS in wt pyramidal cell (C) and mutant pyramidal cell (D). (E) Drop in failure rates after HFS in both genotypes. (F) Comparison of HFS-induced LTP in wt cells (black symbols) and mutant cells (red symbols). * p < 0.05, ** p < 0.01.

Figure 3.
M1/M3-dKO augments spEPSC after HFS of MF pathway. (A) Current traces from wt und M1/M3-dKO pyramidal cells before, during and after three HFS trains (arrows) to induce MF-LTP. Arrowheads indicate evoked EPSCs at 0.1 Hz. (B-C) HFS-induced changes in spEPSC frequency (B) and amplitude (C), quantified every 30 s. * p < 0.05, ** p < 0.01.
Figure 3.
M1/M3-dKO augments spEPSC after HFS of MF pathway. (A) Current traces from wt und M1/M3-dKO pyramidal cells before, during and after three HFS trains (arrows) to induce MF-LTP. Arrowheads indicate evoked EPSCs at 0.1 Hz. (B-C) HFS-induced changes in spEPSC frequency (B) and amplitude (C), quantified every 30 s. * p < 0.05, ** p < 0.01.
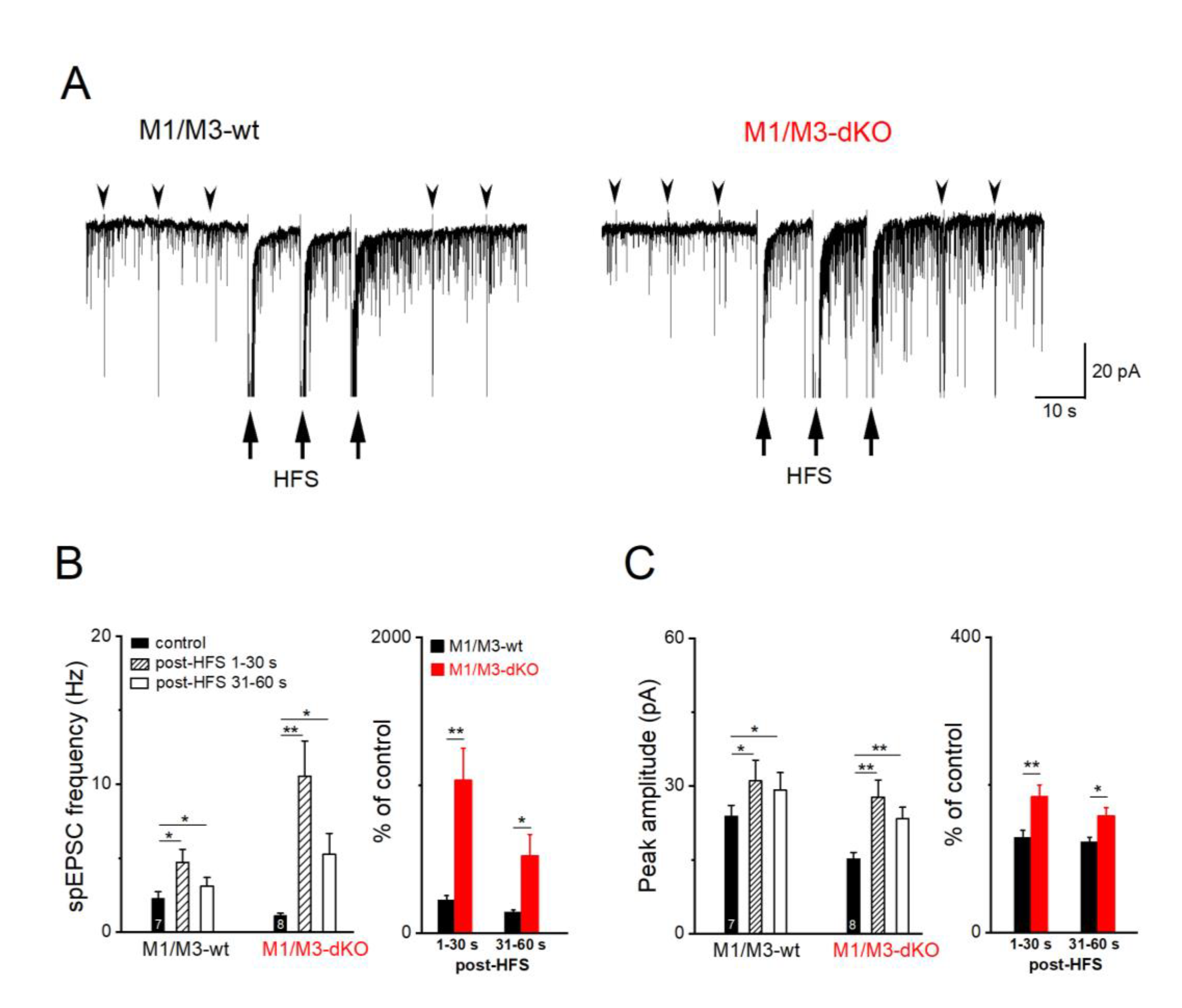
Figure 4.
M1/M3-dKO promotes MF-LTP in extracellular recordings from CA3 stratum lucidum. (A) Trajectory of fPSPs before and after LTP-inducing stimulation (25 Hz for 5 s) in wt slice. Inset above depicts fPSP traces from like-numbered time points in trajectory. Stimulus artifacts were removed from fPSP traces. Arrowhead indicates fiber volley. (B) Comparison of normalized LTP trajectories between genotypes. (C-D) M1/M3-dKO did affect neither quadruple-pulse facilitation (C) nor frequency facilitation (D) of fPSPs. * p < 0.05.
Figure 4.
M1/M3-dKO promotes MF-LTP in extracellular recordings from CA3 stratum lucidum. (A) Trajectory of fPSPs before and after LTP-inducing stimulation (25 Hz for 5 s) in wt slice. Inset above depicts fPSP traces from like-numbered time points in trajectory. Stimulus artifacts were removed from fPSP traces. Arrowhead indicates fiber volley. (B) Comparison of normalized LTP trajectories between genotypes. (C-D) M1/M3-dKO did affect neither quadruple-pulse facilitation (C) nor frequency facilitation (D) of fPSPs. * p < 0.05.
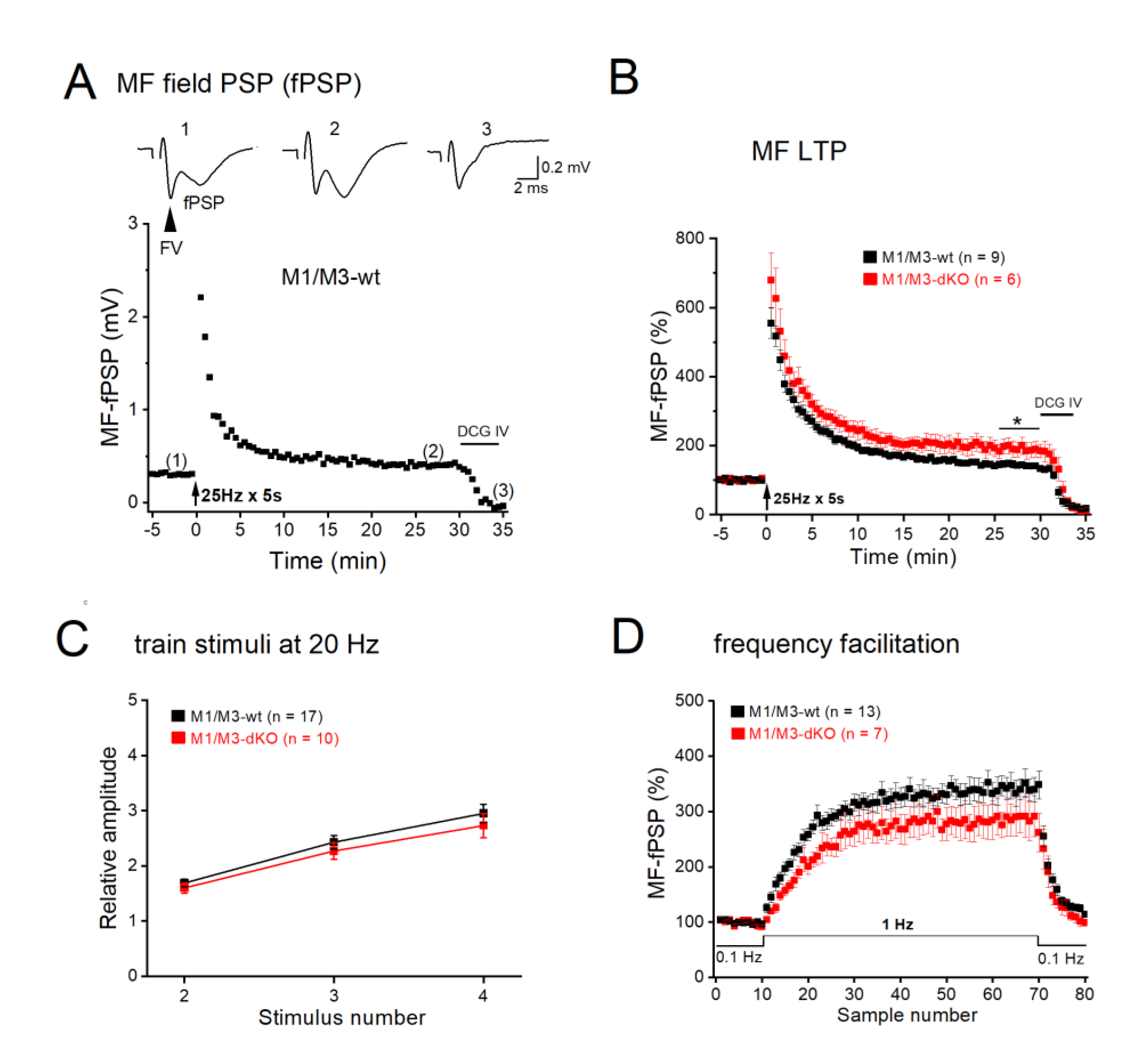
Figure 5.
M1/M3-dKO converts LFS-induced LTD into LTP of the MF-CA3 pyramidal cell synapse. (A-B) Scatter plots of EPSC before and after LFS in wt pyramidal cell (A) and M1/M3-dKO pyramidal cell (B). Insets above illustrate averaged EPSC traces from like-numbered time points. (C-D) Summary of changes in failure rates (C) and amplitudes (D) of EPSCs after LFS for either genotype. (E) Scatter plot of EPSC before and after LFS in M2-KO preparation. (F) Comparison of LFS-induced MF-LTD between wt and M2-KO hippocampi, using normalized EPSC responses. * p < 0.05, ** p < 0.01.
Figure 5.
M1/M3-dKO converts LFS-induced LTD into LTP of the MF-CA3 pyramidal cell synapse. (A-B) Scatter plots of EPSC before and after LFS in wt pyramidal cell (A) and M1/M3-dKO pyramidal cell (B). Insets above illustrate averaged EPSC traces from like-numbered time points. (C-D) Summary of changes in failure rates (C) and amplitudes (D) of EPSCs after LFS for either genotype. (E) Scatter plot of EPSC before and after LFS in M2-KO preparation. (F) Comparison of LFS-induced MF-LTD between wt and M2-KO hippocampi, using normalized EPSC responses. * p < 0.05, ** p < 0.01.
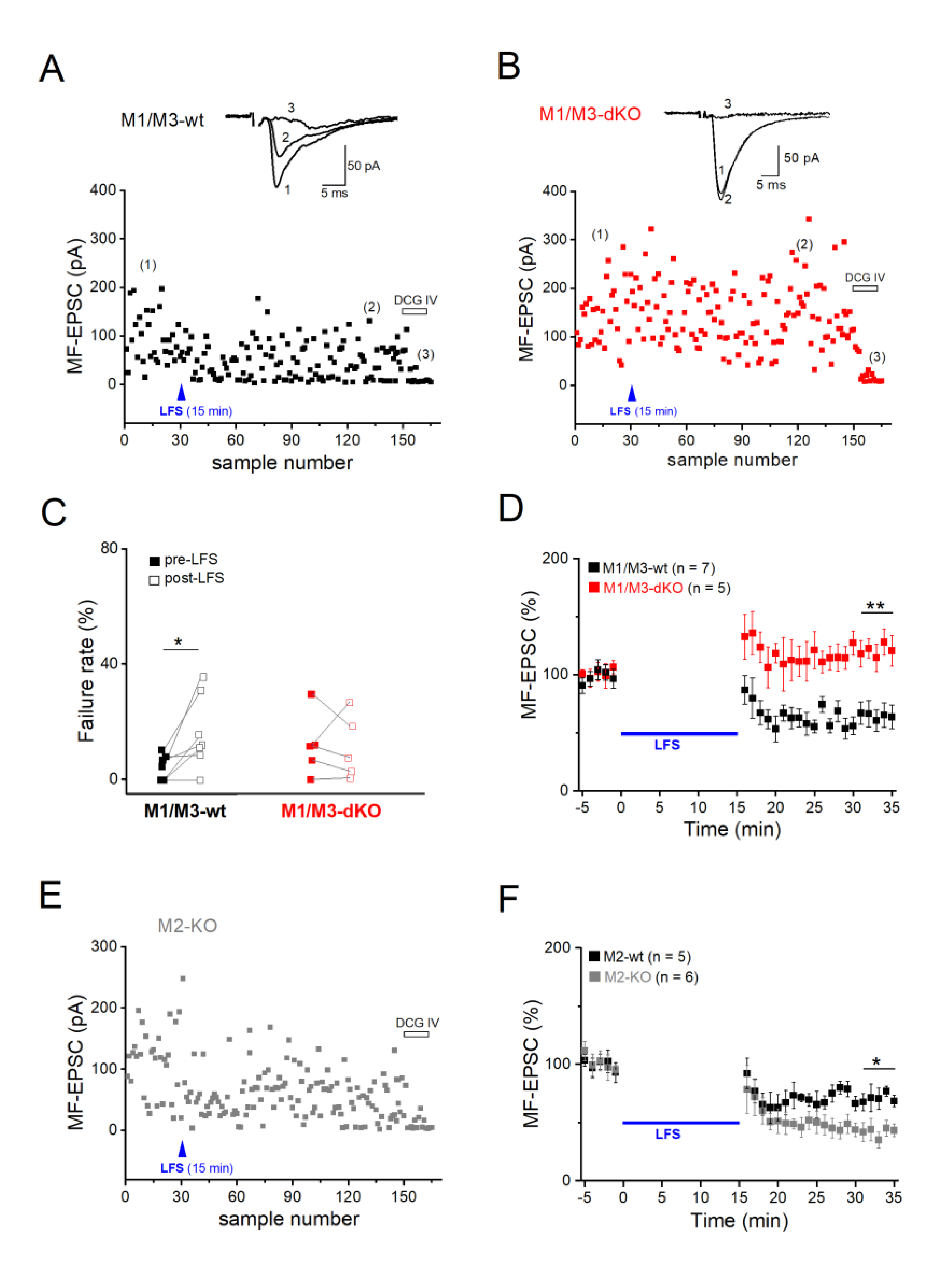
Figure 6.
Synopsis of how mAChRs shape bidirectional plasticity at the MF-CA3 pyramidal cell synapse. See text for details.
Figure 6.
Synopsis of how mAChRs shape bidirectional plasticity at the MF-CA3 pyramidal cell synapse. See text for details.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



