
Submitted:
16 June 2023
Posted:
16 June 2023
You are already at the latest version
Abstract
Embryonic genetic mechanisms are present in the brain and ready to be placed into action upon cellular injury termed the response to injury wound healing (RTIWH) mechanism. When injured, regional brain endothelial cells initially undergo activation and dysfunction with initiation of hemostasis, inflammation (peripheral leukocytes and innate microglia and perivascular macrophage cells), proliferation (astrogliosis), remodeling, repair, and resolution phases if the injurious stimuli are removed. In conditions wherein the injurious stimuli are chronic as occurs in obesity, metabolic syndrome, and type 2 diabetes mellitus this process does not undergo resolution and there is persistent RTIWH with remodeling. Indeed, the brain is unique, in that it utilizes its neuroglia: the microglia cell along with peripheral inflammatory cells and its astroglia instead of peripheral scar-forming fibrocytes/fibroblasts. The brain undergoes astrogliosis to form a gliosis scar instead of a fibrosis scar to protect the surrounding neuropil from regional parenchymal injury. One of the unique and evolving remodeling changes in the brain is the development of enlarged perivascular spaces (EPVS), which is the focus of this brief review. EPVS are important since they serve as a biomarker for cerebral small vessel disease and also represent an impairment of the effluxing glymphatic system that is important for the clearance of metabolic waste from the interstitial fluid to the cerebrospinal fluid and disposal. Therefore, it is important to better understand how the RTIWH mechanism is involved in the development of EPVS that are closely associated with and important to the development of premature and age-related cerebrovascular and neurodegenerative diseases with impaired cognition.
Keywords:
Astrogliosis
; Brain endothelial cells
; Enlarged perivascular spaces
; Microgliosis
; Perivascular macrophages
; Wound Healing
1. Introduction
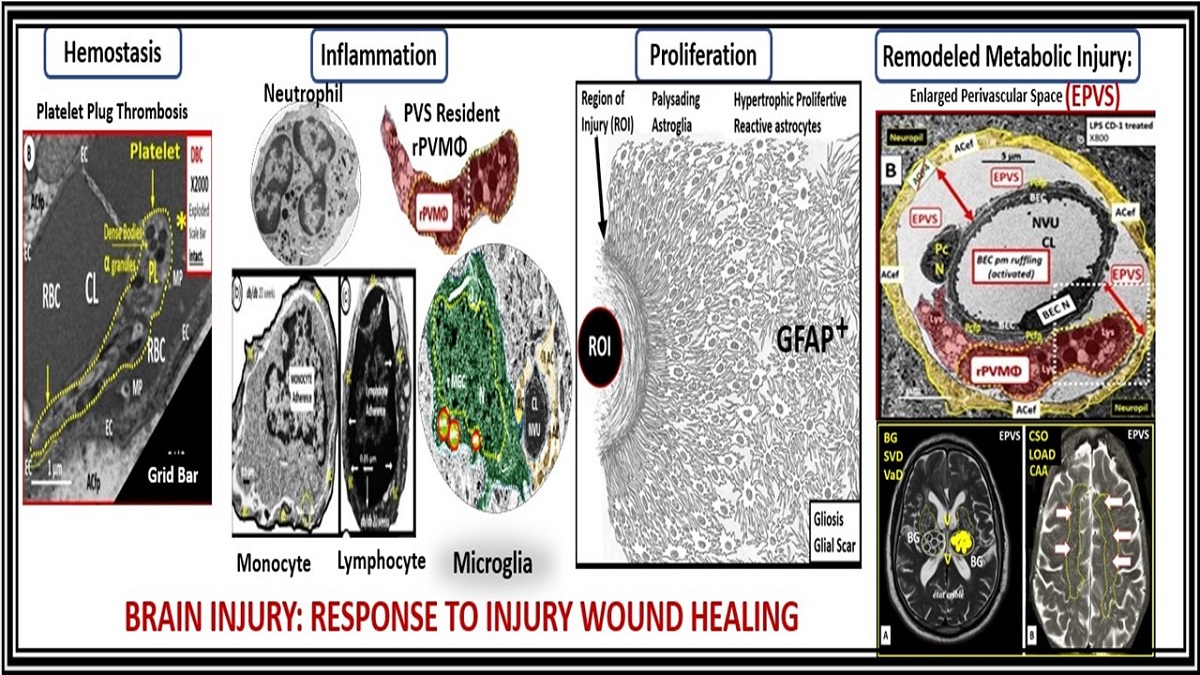
The repair and remodeling of injured brain tissue during postnatal life are associated with the upregulation of some mechanisms that are characteristic of embryonic development [1]. These embryonic genetic memory mechanisms are present in brain cells and ready to be placed into action upon cellular injury. This genetically encoded mechanism may be termed the injury, response to injury wound healing (RTIWH) mechanism [2,3,4]. This recapitulated brain RTIWH mechanism consists of four basic phases: hemostasis, inflammation, proliferation, and remodeling similar to peripheral wound healing mechanisms [5,6]. However, the inflammatory and proliferative phases are unique in the brain, in that neuroglia play an active role [6]. The innate central nervous system (CNS) microglia and perivascular macrophages become reactive and undergo polarization in addition to the peripheral monocyte and monocyte-to-macrophage transformation. Because there are no fibrocytes/fibroblasts within the brain parenchyma the astroglia become reactive and form an astroglia-derived gliosis scar instead of a fibrosis scar to protect the surrounding neuropil in injury states [6]. Additionally, brain injuries may be characterized as traumatic, radiation, metabolic, infectious, and hemodynamic stressors and in this review the focus is primarily on metabolic injurious stimuli as occur in obesity, metabolic syndrome (MetS), and type 2 diabetes mellitus (T2DM) (Figure 1 and Figure 2) [1,2,3,4,5,6].
Perivascular spaces (PVS) or Virchow-Robin spaces (VRS) are fluid-filled spaces that ensheathe the pial arteries, within the subarachnoid space (SAS) that dive deep into the white matter of the brain as ensheathed precapillary arterioles where they end in true capillaries (Figure 3) [7].
The arterial and precapillary PVS are bounded abluminally by the pia membrane that remains adherent to the pial arteries and arterioles and is adherent to the adjacent basal lamina of the astrocyte endfeet basement membranes to form the glia limitans as these structures penetrate the grey and deeper white matter regions of the parenchyma. Further, PVS serve as the delivery conduit for the CSF to admix with the interstitial fluid (ISF) and aid to promote movement within the interstitial space (ISS). The true capillaries are without a pial lining and do not have a PVS. The true capillaries transition to the postcapillary venules, which are also ensheathed by a PVS (without a pia membrane) that form the efflux conduit for the ISF and metabolic waste known as the glymphatic system [8] (abluminally lined by the basal lamina of the astrocyte endfeet) to exit the brain parenchymal ISS and travel to the SAS and hence to CSF to be delivered to the systemic circulation via the arachnoid granulations and the dural venous sinuses (Figure 3B, C) [7]. These PVS are capable of enlarging or dilating and are termed either enlarged perivascular spaces (EPVS) or dilated PVS, once they are large enough to be visualized by T-2 weighted magnetic resonance imaging (MRI) [7,9]. These EPVS are usually greater than two micrometer and average between 1-3 μm in diameter. Because EPVS can be visualized and identified by non-invasive MRI and by microscopic techniques including transmission electron microscopy (TEM) they are becoming more important clinically (Figure 4) [7,9].
EPVS have been recognized as important structural remodeling changes in various neuropathologies [10]. EPVS associate with advancing age, hypertension, lacunes, microbleeds, intracerebral hemorrhages, cerebrocardiovascular disease with transient ischemic episodes and stroke, SVD, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADISIL), cerebral amyloid angiopathy (CAA), obesity, MetS, T2DM, white matter hyperintensities (WMH), late-onset Alzheimer’s disease (LOAD), sporadic Parkinson’s disease, and non-age-related multiple sclerosis [7,9,10,11,12,13,14,15,16,17,18,19].
Cerebral small vessel disease (SVD) presents clinically as lacunar strokes that are responsible for at least 20% of ischemic strokes while representing a major cause of vascular cognitive impairment and vascular dementia (VaD). Importantly, EPVS are known to be a biomarker and feature of both SVD and VaD, which are known to be associated with lacunar stroke in addition to WMH [11,15,19,20,21]. Also, EPVS are known to be a biomarker for SVD once they are large enough to be identified by T2-weighted MRI and they are also known to associate with WMH and Lacunes [22]. Previous studies have shown that EPVS are associated with worse executive function and information processing in healthy older adults [23] and are significantly more prevalent in those with mild cognitive impairment (MCI) as compared to age-matched control subjects without MCI [24]. These findings certainly suggest that EPVS may be an early remodeling change in the development of SVD and impaired cognition [23,24].
As our global aging population continues to grow, EPVS are becoming an increasingly important structural abnormal finding, since they also relate to clinical extracranial atherosclerosis [7,25,26], neurovascular cerebromacrovascular and cerebromicrovascular disease, and age-related neurodegenerative diseases such as LOAD and sporadic PD. In older community-dwelling individuals free of clinical dementia and stroke, SVD biomarkers including EPVS, WMH, and lacunes are related to worse cognitive performance [27]. Importantly, EPVS have recently been determined to be a marker for an increased risk of cognitive decline and dementia, independent of other small vessel disease markers over a period of four years [28].
The MetS consists of a cluster of multiple interconnected risks and variables that associate with an increased risk for developing atherosclerotic cerebrocardiovascular disease (CCVD) and T2DM [25,29,30]. Importantly, visceral adipose tissue (VAT), insulin resistance (IR) and MetS are each associated with an increased risk for developing EPVS [31,32,33]. IR is a central core feature of the MetS; however, it is a risk factor that may be independent of the MetS for developing EPVS (Figure 5) [25,34].
There are four core features (the four arms of the letter X: hyperlipidemia, hyperinsulinemia, hypertension, and hyperglycemia) of the MetS and each are interconnected to the central placement of IR (Figure 5) [25,29,30,34]. Currently, there is a global increase in obesity and the MetS due to an aging population, urbanization, sedentary lifestyles, and increased caloric high fat, sucrose, fructose, and glucose diets [25, 29. 30]. Further, our global population is now considered to be one of the oldest attained in our history [35]. Tucsek et al., have recently demonstrated that aging exacerbates obesity-induced neurovascular uncoupling, cerebromicrovascular capillary rarefaction, and cognitive decline in mice [36]. Thus, it is not surprising that aging is a reason for our observations of global increases in the MetS, EPVS, and SVD [9,11,15,16,17,21,22,25,26,34,36].
Capillary rarefaction in the brain (loss of capillaries) has recently been found to be associated with an increased in obesity and the MetS [36,37,38]. Recently, Schulyatnikova and Hayden have hypothesized that capillary rarefaction may leave an empty space within the PVS that is subsequently filled with interstitial fluid [7]. This loss of capillaries within the PVS may allow for an increase in total fluid volume within the PVS when the capillary undergoes rarefaction and result in EPVS (Figure 6) [7].
Capillary rarefaction (CR) is known to occur in multiple clinical situations, including: aging, hypertension, obesity, MetS, T2DM, and LOAD. Also, there are multiple proposed mechanisms that may co-occur to result in CR, including: oxidative – redox stress, inflammation, Pc loss, BECact/dys, impaired angiogenesis (increased ration of increased antiangiogenic factors/proangiogenic factors), microvessel emboli and decreased microvessel shear stress, increased microvessel tortuosity, and in some cases increased TGF-beta [39].
While this mechanistic hypothesis for an expansion of PVS is possible, more research will be required for it to gain support as a mechanism for increased EPVS.
Obesity, MetS, and T2DM not only allow for the expansion of VAT depots to develop metainflammation and increased peripheral cytokines/chemokines (pCC) [7, 25. 34] but also provide a milieu for the development of gut microbiota dysbiosis with the secretion of pCC, sLPS, and lpsEVexos to result in the bidirectional communication between the gut and the brain ‘microbiota-gut-brain axis’ in neuroinflammation due to the excessive dual signaling of NVU BECs [7,25,34,40]. Importantly, there may be a host of metabolic functions, gut microbiota, and the innate immune system (Figure 7) [7,25,40,41,42,43].
2. Brain endothelial cell activation and dysfunction (BECact/dys)
BECs and their luminal endothelial glycocalyx (ecGCx) serve as the sentinel, gatekeeper cell of the BBB and CNS as the first effector cell that comes into direct contact with peripheral systemic solutes (ions, molecules, neurotoxins) and cells including leukocytes between the blood and the brain parenchyma (Fig.1) [44]. Importantly, BECs are key to vascular hemostasis, tone, leukocyte recruitment, hormone trafficking, and fluid movement from the blood to the parenchymal interstitial space in addition to their playing a central role in the development of EPVS [34,45]. Further, they are activated by the dual peripheral signaling from the pCC and the peripheral soluble lipopolysacchride (sLPS) and LPS-enriched small extracellular vesicles exosomes (lpsEVexos) from the regional pCC VAT depot metainflammation and the regional dysbiosis microbiota respectfully (Figure 8) [7,34].
This dual signaling of BECs are associated with the increased expression of cell-surface adhesion molecules such a VCAM-1, ICAM-1 and E selectin as defined in BEC activation. Likewise, BEC dysfunction is defined as the decreased synthesis, release, and/or activity of endothelium-derived nitric oxide (NO), which results in decreased bioavailable NO [7,34,44,46,47].
BECact/dys is induced by (1) proinflammatory cytokines, such as TNF-α and IL-6; (2) turbulent blood flow, such as occurs at bifurcations and branch points of arteries; (3) AGEs, which are elevated in hyperglycemia and aging, and (4) inflammatory stressors such as metainflammation pCC from VAT depots and plasma membrane peptides of gram-negative bacteria such as sLPS and lpsEVexos from gut microbiota dysbiotic regions. These four functions are each important mediators of ECact/dys via the activation of the nuclear transcription factor of BEC NF-κB (Figure 8). The following TEM images are examples of BEC activation [25]. BECact/dys is known to be induced by (i) proinflammatory cytokines, such as TNF-α and IL-6; (ii) turbulent blood flow, such as that which occurs at bifurcations and branch points of arteries; (iii) AGEs, which are elevated in hyperglycemia and aging, and (iv) inflammatory stressors such as metainflammation and plasma membrane peptides of gram-negative bacteria such as sLPS and lpsEVexos. These four functions are each important mediators of BECact/dys via the signaling of BEC nuclear transcription factor (NF-κB). The following TEM images are examples of BEC activation (Figure 9) [7,25,34,46].
3. Hemostasis: Phase One
The homeostatic process of hemostasis is essential for any brain injury to its vasculature with hemorrhage, such as occurs with traumatic brain injury, microbleeds, and intracerebral hemorrhage. The initial response to this injury is constriction of the injured vessel with activation of platelets to form a fibrin clot and platelet plug. The fibrin clot results in hemostasis and also provides a scaffolding for the erythrocytes and incoming leukocytes of the inflammatory phase [48].
4. Inflammation – Neuroinflammation: Phase Two
Neuroinflammation is the 2nd phase of the RTIWH mechanisms and if the CNS injurious stimuli are chronic and persistent as in obesity, MetS, and T2DM the chronic sustained peripheral inflammation with pCC, sLPS, and lpsEVexos injurious stimuli result in chronic activation of BECs with BECact/dys. This chronic BECact/dys results in chronic neuroinflammation with the generation of chronic CNS cytokines/chemokines (cnsCC) [4]. BECact/dys results in neuroinflammation, blood-brain barrier (BBB) disruption, and EPVS in obesity, insulin resistance (IR), and MetS (Fig.1, Fig.7) [34]. Importantly the systemic macrophages and the innate perivascular macrophages (PVMΦs); the innate microglia cell(s) (MGCs) are known to be the first set of effector cells in the RTIWH process and considered to be the innate immune cells of the CNS [6,49,50].
MetS, which includes obesity and T2DM is widespread globally affecting millions and known to cause the development of CNS neuroinflammation and neurodegeneration [25,51]. Indeed, the multiple components of the MetS are known to result in a wide range of effects, which include BBB disruption, neuroinflammation, EPVS, CCVD, neurodegeneration, and impaired cognition (Figure 5) [25,34,51].
PVS and EPVS have become of increasing research interest since EPVS associate early in the development of SVD and neurodegeneration that can be viewed by MRI noninvasively in addition to the important role of PVS that form the conduit structure for clearance of waste via the glymphatic system [7,8,34]. The RTIWH remodeling is important for the formation of EPVS via multiple mechanisms in the RTIWH process and entails the two-step process of neuroinflammation as well as the myriad of other remodeling mechanisms (Figure 10) [7,34,51,52,53,54].
Brain PVMΦs belong to a distinct population of brain-resident myeloid cells located within the perivascular space that are derived from the yolk sac similar to microglia and stain positive with CD-163 and lack the MGC specific marker P2RY12. PVMΦs also provide both structural and functional support for maintaining brain homeostasis, which includes maintenance of BBB integrity, glymphatic drainage, and mediation of immune functions including phagocytosis and antigen presentation [53]. The close ultrastructural (physical) relationship of the rPVMΦ within the postcapillary venule PVS allows for considerable cross talk between the reactive perivascular macrophage (rPVMΦ), the NVU pericyte and the astrocyte endfeet that line the abluminal PVS (Figure 11).
rPVMΦs mediate multiple mechanisms that contribute to remodeling changes of RTIWH to the PVS including: BBB disruption; increased permeability to fluids, solutes, and leukocytes; increased RONSS/RSI – oxidative/reductive stress; phagocytosis and increased phagocytic debris; CNSCC – neuroinflammation; increased MMPs (MMP2 and 9); increased proteolysis; antigen presentation; EPVS and decreased waste clearance of the PVS conduit for the glymphatic system to the CBF [53,55].
Owens et al., two-step process in the development of neuroinflammation depends heavily on the role of the postcapillary venular PVS. [52]. The PVS provides the necessary space that leukocytes enter after undergoing diapedesis across the disrupted BBB that is instigated due to peripheral metainflammation and CNS neuroinflammation injury. Once the leukocytes have entered the PVS they then have to breech the outer abluminal glia limitans of the PVS in order to arrive at their final destination the ISS of the parenchymal neuropil and neuronal regions of the brain via leukocyte matrix metalloproteinase’s (MMP2 and MMP9) [52]. The initial inflammatory cells would be the neutrophiles, followed by the monocytes and lymphocytes. The innate immune leukocytes (neutrophiles and monocytes) and the acquired immune lymphocytes would then join the resident perivascular macrophages and this combination of cells would then be capable of not only causing the accumulation of cells and phagocytic cellular debris that causes PVS obstruction and the creation of EPVS but also create breaks in the abluminal glia limitans of the now EPVS and be capable of crossing and entering the ISS of the parenchymal neurons [7,34,49,52,53,54].
There are basically two barriers to the passage of leukocytes into the parenchymal ISS of the CNS. The first barrier is the NVU BBB BECs of the postcapillary venule that is facilitated by cellular leukocyte adhesion molecules or vascular adhesion molecules including intercellular adhesion molecules (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), integrins, and selectins, which provide for the tethering, rolling, crawling, adhesion, and diapedesis via either the paracellular or transcellular routes commonly referred to as transendothelial migration [56]. The second barrier is the glia limitans of the postcapillary venule PVS [52].
Author is becoming more confident that the inflammatory phase may be the most important or significant RTIWH phase in the development of the EPVS in obesity, MetS, and T2DM. Once BECact/dys is initiated due to the constant injury by pCC, sLPS, lpsEVexos, and subsequent CNSCC, the inflammatory cells leave the postcapillary venule lumen and enter the PVS via a 2-step process as previously described [52]. These incoming leukocytes, join the rPVMΦs become excessive and along with the adaptive immune lymphocytes fill up the PVS in addition to the excessive accumulation of their phagocytic debris that contribute greatly to stalling and stagnation of flow to even the point of obstruction, which results in EPVS. This inflammatory component is in excess of the problems with impaired flow and pulsatility produced by vascular stiffening, which is another cause for impaired clearance of PVS conduit for glymphatic efflux of interstitial fluid and accumulating waste (including misfolded proteins such as amyloid beta and tau) and allowing them to accumulate and undergo deposition and growth at sites of plaque niches within the interstitium.
Once there is stalling, stagnation, and obstruction of flow within this conduit, waste products accumulate and along with proinflammatory leukocyte derived cytokines/chemokines, and ROSs increase their neurotoxicity and potential to cause even further injury to the tissue with more RTIWH being placed into play to create a vicious cycle of continuous injury and RTIWH mechanisms. In regards to LOAD, some view its early onset, development, progression, and acceleration to clinical symptomatic disease as being closely related to the vasculature as has been described with mixed dementias with both neurovascular and neurodegenerative mechanisms occurring concurrently in the acceleration of impaired cognition and neurodegeneration. Some even feel that the two primary problems in the development and progression of LOAD may be: Number one, a disrupted and leaky BBB and Number two, impaired clearance of accumulating waste, which may be now detected by non-invasive MRI studies. Thus, impaired clearance of waste via obstructed PVS with ensuing EPVS are an early remodeling change that is promoted by RTIWH.
5. Proliferation and Astrogliosis: Phase Three
Astrocyte(s) (ACs) have multiple homeostatic functions in the brain including their role as the connecting cell between the NVU and neurons creating the NVU coupling to increase cerebral blood flow when neurons increase their activity; their role as a major supplier of energy in the form of glucose and lactate via their capability of glycogen storage and glycolysis; a supplier of antioxidant reserves (glutathione and superoxide dismutase; a supplier of various growth factors such as brain-derived growth factor, transforming growth factor-beta (TGF-β) [57,58,59]. ACs also define many aspects of synapse formation, plasticity, and serve a protective function including synaptic maintenance and elimination or pruning along with microglia, since they are known to cradle the synapse [59,60]. It is important to note that human studies may not always conform and translate from rodent model studies, in that, human ACs in the neocortical regions are much larger and these cytoplasmic processes extend much further as compared to rodent models [61].
Brain injury results in reactive ACs (rACs) that are essential for early tissue protection. They are the second effector cell in the RTIWH process in the brain and are responsible for the proliferative phase and astrogliosis in the brain. Since the brain does not have fibrocytes/fibroblasts in the neuronal parenchyma as occurs in non-neuronal tissues there is no fibrosis within the parenchyma but instead the rACs are responsible for forming gliosis and the glial scar [6]. Importantly, ACs instigate the cellular proliferative phase in brain injury since neuronal cellular repair is impossible because CNS neurons are incapable of regeneration [6]. Thus, the prolifertive phase occurs in the immediate areas surrounding regions of brain injury and become reactive and undergo proliferation and hypertrophy that is termed astrogliosis or gliosis, which is responsible for astrogliosis or glial scar formation to protect the normal surrounding neuronal tissues [6, 7. 34, 61, 62, 63, 64, 65]. Further, the glial scar is known to replace the ischemic penumbra that originally surround the ischemic injury core (Figure 12) [63].
In addition to the formation of the brain injury glial scar, rACs are positioned as major regulators of vascular repair and remodeling following cerebral ischemia, stroke, and brain injury. For example, ablation of rACs disrupts vascular remodeling and worsens motor recovery following stroke by prolonging CBF deficits, increasing NVU permeability, and promoting apoptosis. Vascular remodeling, reorganization, and repair of the immediate surrounding region or penumbra of the ischemic death zone injury following stroke and the development of the glial scar is greatly dependent of rACs [66].
The TGF-beta family plays a critically important role in the formation of reactive ACs (rACs), gliosis, and the formation of the astroglia scar and TGF-beta1(TGF-β1) is primarily responsible for gliosis and glial scar formation in the brain [67,68]. In addition to being important in driving gliosis, TGF-β1 also results in microvascular basement membrane remodeling with thickening due to increased AC fibronectin production following stroke in aged individuals and this remodeling phase. Further, increased TGF-β1 results in decreased or stalling of CSF distribution within the PVS, impairs NVU coupling, and impairs neurologic recovery [68]. The predominant sources for brain TFG-β1 are the rMGCs and rPVMΦs following brain injury (stroke) and these cells including the rACs have all been shown to be capable of a marked upregulation of TGF-β1 signaling following brain injury (stroke) [69].
5.1. Astrocyte Aquaporin 4 (AQP4) Remodeling During Phase Three and Astrogliosis in Brain Injury
Among numerous roles of the ACs, they are important for controlling the volume of CNS, ISF, and PVS as well as the AC itself via its highly polarized plasma membrane water channel AQP4 especially at the ACef in contact with the vasculature including the PVS [59,67]. The bidirectional water transport function AQP4 is important in maintaining the homeostasis of water balance in the CNS. Upon forming rACs this AQP4 water channel is known to lose its ACef polarity at the plasma membrane and become maldistributed within the cytosol with loss of its normal function [59,66]. AQP4 is known to be widely expressed throughout the brain, especially at the blood-brain barrier where AQP4 is highly polarized to astrocytic foot processes in contact with blood vessels. The bidirectional water transport function of AQP4 suggests its role in cerebral water balance in the CNS (Figure 13) [59,70].
6. Remodeling: Phase Four
To remodel means to change or alter a structure or form of tissue and RTIWH remodeling implies the rebuilding, repairing, and sculpting of tissues that are injured. Just as one remodels a damaged home, you must first tear down the damaged tissue via inflammation, proteolysis via matrix metalloproteinases, and phagocytosis to clean the injured region before you can begin to rebuild the injured tissue. Once the region is completely torn down and the damaged tissues are removed, the rebuilding or remodeling process can begin. Incidently, the more tissue that is torn down the more rebuilding or remodeling will need to occur and the greater the remodeling. Interestingly, in addition to the professional phagocytes such as neutrophils and macrophages that assist in clearing the debris, rACs have been shown to be capable of phagocytosing debris including myelin [71,72].
Once the RTIWH phases have removed the damaged-injured tissues the remodeling phase begins and this entails the activation of multiple growth factors/hormones such as the vascular endothelial growth factor (VEGF) family for vascular remodeling and brain-derived neurotrophic factor (BDNF-neurotrophin that is related to the canonical nerve growth factor (NGF), important in the support and survival of existing neurons, encouraging growth and differentiation of new neurons and synapses); casein kinase (CK2), proliferation, growth cone formation, synaptic connections, and neuroprotective effects); growth-associated protein 43 (GAP-43), also known as neuromodulin, proliferation, synaptic plasticity, and growth); Kruppel-like factor-7 (KLF-7), important in neuronal migration and differentiation); TGF-B1, important in growth, proliferation, and hypertrophy for neuronal limited regeneration remodeling [73]. Overall, the remodeling phase in the CNS is considered to be quite limited as compared to the non-CNS tissues since there is no long-term remodeling changes associated with fibrosis in the CNS due to lack of fibrocytes/fibroblasts [7].
Just beyond the glial scar that surrounds the ischemic necrotic core (Figure 12) there exists an encircling ischemic penumbra following ischemic stroke that is damaged but ‘not dead yet’ and may be salvaged with thrombolytic stroke therapy [74]. Notably, this ischemic penumbra is subject to remodeling and salvage via prompt recognition of clinical stroke and administration of either intravenous or intra-arterial tissue plasminogen activator (tPA) therapy, whereby the multiple growth factors/hormones become activated and play a critically important role of neurovascular salvage in the remodeling phase once an improved cerebral blood flow has been reestablished [73,75].
Upon completion of the remodeling phase in the RTIWH mechanism there will exist multiple remodeling changes including EPVS histologically (previously presented and illustrated) and additionally, there will also remain EPVS on T2-weighted MRI located in the basal ganglia (BG) and the centrum semiovale (CSO) (Figure 14) [7,9,11,15,76].
Indeed, enlarged perivascular spaces (EPVS) may exist in an evolutionary spectrum from postcapillary venule normal PVS that are not enlarged to EPVS in BG and CSO that associate with WMH and lacunes to result in cerebral small vessel disease (SVD) vascular dementia (VaD) with neuroinflammation, impaired cognition, and neurodegeneration in conditions of aging, hypertension, obesity, MetS and T2DM [7].
Also, capillary rarefaction may be an end-point of the remodeling phase, which incorporates capillary loss especially, in obesity, MetS, and T2DM [7,37,38,77]. Importantly, during the process of capillary rarefaction an empty space will develop within the PVS and allow for an increase in the total volume of the fluid-filled spaces within the PVS and may contribute to dilated or EPVS [7].
7. Resolution: Phase Five
Because the injurious stimuli are chronic in obesity, MetS, and T2DM the RTIWH will continue and therefore, it will not be possible for the brain to enter a complete resolution phase. However, if the injurious stimuli can be dampened or partially eliminated with proper treatment the RTIWH resolution phase may ensue with limited resolution and even limited regeneration. This aspect of the RTIWH process may help to explain the improvement with diabetic end-organ remodeling with aggressive treatment to goal car plans.
8. Conclusion
In the introduction (Section 1), the first paragraph discusses brain injury and the RTIWH; the second paragraph discusses PVS and the development of EPVS as a result of the RTIWH; the third paragraph discusses the relation between obesity, MetS, T2DM, EPVS, and the RTIWH process. Section 2 discusses the central importance of BECact/dys in the development of EPVS. Section 3. begins the discussion of RTIWH with hemostasis; Section 4. discusses the inflammatory phase of the RTIWH; Section 5. discusses the proliferation phase; Section 6. discusses the remodeling phase; Section 7. discusses the relative lack of a resolution phase in the RTIWH process in obesity, MetS, and T2DM. The examination of the processes involved in the development of EPVS via the RTIWH mechanism is indeed a novel approach; however, it serves to enlighten us of this innate wound healing mechanism in all individuals that is involved in brain injury and prepared to respond to injury at a moments’ notice. Importantly, it became obvious during this review that the development of EPVS via RTIWH mechanisms there exists a bidirectional relationship between PVS and neuroinflammation as described by Owens et al. [52] and there also exists a bidirectional relation between neuroinflammation and development of EPVS (Figure 10 and Figure 15) [78].
A central theme of brain injury involves gene activation and transcription of various factors in an attempt to respond to the multiple injurious stimuli such that whenever there is injury the cells recapitulate their embryonic genetic memory in an attempt to heal through hemostasis, inflammation, proliferation growth (re-growth), hypertrophy, differentiation, development, remodeling, and limited neural regeneration. As a result of this chronically activated RTIWH mechanism, we as clinicians and researchers in this field of study must constantly review and expand our knowledge in an attempt to alter the RTIWH process in a manner to decrease the morbidity and mortality and the progressive brain injuring nature of obesity, MetS, T2DM, and the associated accelerated atherosclerosis along with their complications.
There has been increased interest in EPVS recently, since the PVS is now recognized as the essential conduit for the glymphatic system for the removal of ISF and accumulated neurotoxic waste from ISS to the CSF, which acts as a sink for the accumulation of excess toxic waste from the CNS [8,79]. This recognition has brought forth an increased interest in the CNS interstitial space(s) (ISS) and how it empties into the PVS of the postcapillary venules. Even though the ISS diameter is only approximately 20-50 nanometers in mouse brains, its total volume is known to account for up to 20% of the total CNS volume and is crucial for interstitial fluid (ISF) solute transport, signal transmission, and communication amongst neurons (Figure 16) [80].
This neurotoxic metabolic waste may include the misfolded proteins of amyloid beta (40-42), tau, and alpha-synuclein plus others that are known to accumulate in late-onset Alzheimer’s disease (LOAD) and sporadic Parkinson’s disease respectively within the interstitial fluid that is delivered to the subarachnoid space and eventually to the cerebrospinal fluid (CSF) for disposal. The finding that sleep is important in dilating the PVS to allow for improved drainage of ISF waste via the PVS – glymphatic system has recently been proposed [81,82,83]. Some have postulated that the vast majority of waste clearance occurs during sleep [82] and Zavecz et al. have recently viewed non rapid eye movement sleep as a cognitive reserve factor in the presence of LOAD [83].
The importance of a healthy non-dilated PVS and its role as a conduit for the glymphatic system to deliver toxic metabolic waste from the interstitium and the ISSs to the CSF to provide a cleansing mechanism for the CNS and the remodeled dilated EPVS -Virchow-Robins space is not to be overlooked. The ability to view these abnormal EPVS by non-invasive T-2 weighted MRIs provide an insight into the impairment of waste removal and its association with a RTIWH mechanism with impaired cognition, neuroinflammation, neurovascular and neurodegenerative remodeling diseases [28]. Indeed, EPVS are not only a biomarker for SVD and associate with WMH, lacunes, aging, hypertension, obesity, MetS, and T2DM but also according to the conclusion set forth by Paradise et al., [28] EPVS may also be considered a marker for increased risk of cognitive decline and dementia, independent of other small vessel disease markers.
Author Contributions
M.R.H. is entirely responsible for the writing, editing and submission of this manuscript.
Funding
M.R.H. has received no grants from any funding agency in the public, commercial, or not-for-profit sectors.
Institutional Review Board Statement
The tissues provided for the representative electron microscopic images utilized in this manuscript were all approved in advance by the University of Missouri Institutional Animal Care and Use Committee (No. 190), and animals were cared for in accordance with National Institutes of Health guidelines and by the Institutional Animal Care and Use Committees at the Harry S Truman Memorial Veterans Hospital and University of Missouri, Columbia, MO, USA and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH).
Informed Consent Statement
not applicable.
Data Availability Statement
Data and materials will be provided upon reasonable request.
Acknowledgments
Author would like to acknowledge DeAna Grant Research Specialist of the Electron Microscopy Core Facility at the NexGen Precision Health Research Center, University of Missouri, Columbia, Missouri. Also, author would like to thank and acknowledge Tatyana Shulyatnikova for providing artistic illustrations and critical editing of this manuscript. Author would also like to acknowledge the William A. Banks Lab at the VA Medical Center-Seattle, Washington for their kind support.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
AC, astrocyte; ACef, astrocyte end-feet; AGE/RAGE, advanced glycation end products/receptor for advanced glycation end products; AQP4, aquaporin-4; BBB, blood–brain barrier; BEC(s), brain endothelial cell(s); BECact/dys, brain endothelial cell activation/dysfunction; BG, basal ganglia; CADASIL = cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CBF = cerebral blood flow; CCVD, cerebrocardiovascular disease; CNS, central nervous system; CSF, cerebrospinal fluid; CSO, centrum semiovale; E-selectin, endothelial–leukocyte adhesion molecule; ecGCx, endothelial cell glycocalyx; EPVS, enlarged perivascular spaces; HTN, hypertension; ISF, interstitial fluid; ICAM-1, intercellular adhesion molecule 1; ISF, interstitial fluid; ISS, interstitial space; LDL, low-density lipoprotein (LDL); LOAD, late-onset Alzheimer’s disease; LPS, lipopolysaccharide; lpsEVexos, lipopolysaccharide extracellular vesicles; MCI, mild cognitive impairment; MetS, metabolic syndrome; MMPs, matrix metalloproteinases; MRI, magnetic resonance imaging; NO, nitric oxide; NVU, neurovascular unit; Pc, pericyte; PD, Parkinson’s disease; PVS, perivascular spaces; RTIWH, response to injury wound healing; SAS, subarachnoid space; sLPS, soluble lipopolysaccharide; rPVMΦ, reactive perivascular macrophage; SVD, small vessel disease; T2DM, type 2 diabetes mellitus, TEM, transmission electron microscopy; TGF-B1, transforming growth factor beta-a; TIA, transient ischemic attack; TJ/AJ, tight and adherens junctions; VaD, vascular dementia; VAT, visceral adipose tissue; VCAM-1, vascular cell adhesion molecule 1; VRS, Virchow–Robin spaces; WMH, white matter hyperintensities.
References
- Aller MA, Arias JI, Arraez-Aybar LA, Gilsanz C, Arias J. Wound healing reaction: A switch from gestation to senescence. World J Exp Med 2014;4(2): 16-26. [CrossRef]
- Hayden MR, Tyagi SC, Kolb L, Sowers JR, Khanna R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: the emerging role of sodium thiosulfate. Cardiovasc Diabetol. 2005; 4:4. [CrossRef]
- Hayden MR, Sowers KM, Pulakat L, Joginpally T, Krueger B, Whaley-Connell A, Sowers JR. Possible Mechanisms of Local Tissue Renin-Angiotensin System Activation in the Cardiorenal Metabolic Syndrome and Type 2 Diabetes Mellitus. Cardiorenal Med. 2011;1(3):193-210. [CrossRef]
- Hayden MR. Hypothesis: Neuroglia Activation Due to Increased Peripheral and CNS Proinflammatory Cytokines/Chemokines with Neuroinflammation May Result in Long COVID. Neuroglia. 2021;2(1):7-35. [CrossRef]
- Clark RA. Cutaneous tissue repair: Basic biologic considerations. J Am Acad Dermatol. 1985;13(5 Pt 1):701-725. [CrossRef]
- Stroncek JD, Reichert WM. Overview of Wound Healing in Different Tissue Types. In: Reichert WM, editor. Indwelling Neural Implants: Strategies for Contending with the In Vivo Environment. Boca Raton (FL): CRC Press/Taylor & Francis; 2008. Chapter 1. Available from: https://www.ncbi.nlm.nih.gov/books/NBK3938/ (accessed on May 31, 2023).
- Shulyatnikova T, Hayden MR. Why are Perivascular Spaces Important? Medicina (Kaunas). 2023;59(5):917. [CrossRef]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, 556 Nedergaard M. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial 557 Solutes, Including Amyloid β. Sci Transl Med. 2012;4(147):147ra111. [CrossRef]
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, Frayne R, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013 Aug;12(8):822-38. [CrossRef]
- Francis F, Ballerini L, Wardlaw JM. Perivascular spaces and their associations with risk factors, clinical disorders and neuroimaging features: A systematic review and meta-analysis. Int J Stroke. 2019;14(4):174749301983032. [CrossRef]
- Brown R, Benveniste H, Black SE, Charpak S, Dichgans M, Joutel A, Nedergaard M, Smith KJ, Zlokovic BV, Wardlaw JM. Understanding the role of the perivascular space in cerebral small vessel disease. Cardiovasc Res. 2018; 114(11): 1462–1473. [CrossRef]
- Arba F, Quinn TJ, Hankey GJ, Lees KR, Wardlaw JM, Ali M, et al. Enlarged perivascular spaces and cognitive impairment after stroke and transient ischemic attack. Int J Stroke. 2016;13(1). [CrossRef]
- Bokura H, Kobayashi S, Yamaguchi S. Distinguishing silent lacunar infarction from enlarged Virchow-Robin spaces: a magnetic resonance imaging and pathological study. J Neurol. 1998;245(2):116-122. [CrossRef]
- Heier LA, Bauer CJ, Schwartz L, Zimmerman RD, Morgello S, Deck MD. Large Virchow-Robin spaces: MR-clinical correlation. Am J Neuroradiol. 1989;10(5):929-936.
- Doubal FN, Maclullich AMJ, Ferguson KJ, Dennis MS, Wardlaw JM. Enlarged Perivascular Spaces on MRI Are a Feature of Cerebral Small Vessel Disease. Stroke. 2010;41(3):450-454. [CrossRef]
- Bown CW, Carare RO, Schrag MS, Jefferson AL. Physiology and Clinical Relevance of Enlarged Perivascular Spaces. Neurology. 2022;98(3):107-117. [CrossRef]
- Trolli F, Cipollini V, Moci M, Morena E, Palotai M, Rinaldi V, Romano C. Ristori G, Giubilei F, Salvetti M, Orzi F, Guttmann CRG, Cavallari M. Perivascular Unit: This Must Be the Place. The Anatomical Crossroad Between the Immune Vascular and Nervous System. Front Neuroanat. 2020;14: 17. [CrossRef]
- Zhu YC, Tzourio C, Soumaré A, Mazoyer B, Dufouil C, Chabriat H. Severity of dilated Virchow-Robin spaces is associated with age, blood pressure, and MRI markers of small vessel disease: a population-based study. 2010;41(11):2483-2490. [CrossRef]
- Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, et al. Vascular dysfunction-The disregarded partner of Alzheimer's Disease. Alzheimer’s Dement. 2019;15(1):158-167. [CrossRef]
- Loos CMJ, Klarenbeek P, van Oostenbrugge RJ, Staals J. Association between Perivascular Spaces and Progression of White Matter Hyperintensities in Lacunar Stroke Patients. PLoS One. 2015; 10(9): e0137323. [CrossRef]
- Benjamin P, Trippier S, Lawrence AJ, Lambert C, Zeestraten E, Williams OA, Patel B, Morris RG, Barrick TR, MacKinnon AD, Markus HS. Lacunar Infarcts, but Not Perivascular Spaces, Are Predictors of Cognitive Decline in Cerebral Small-Vessel Disease. Stroke. 2018 Mar;49(3):586-593. [CrossRef]
- Barisano G, Law M, Custer RM, Toga AW, Sepehrband F. Perivascular Space Imaging at Ultrahigh Field MR. Imaging Magn Reson Imaging Clin N Am. 2021 Feb;29(1):67-75. [CrossRef]
- Yates KF, Sweat V, Yau PL, Turchiano MM, Convit A. Impact of Metabolic Syndrome on Cognition and Brain: A Selected Review of the Literature. Arteriosclerosis, Thrombosis, and Vascular Biology. 2012;32(9):2060-2067. [CrossRef]
- Niazi M, Karaman M, Das S, Zhou XJ, Yushkevich P, Cai K. Quantitative MRI of Perivascular Spaces at 3T for Early Diagnosis of Mild Cognitive Impairment. AJNR Am J Neuroradiol. 2018;39(9): 1622–1628. [CrossRef]
- Hayden MR. Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease. Medicina (Kaunas). 2023;59(3):561. [CrossRef]
- Gutierrez J, Rundek T, Ekind MSV, Sacco RL, Wright CB. Perivascular Spaces Are Associated with Atherosclerosis: An Insight from the Northern Manhattan Study. AJNR Am J Neuroradiol. 2013; 34(9): 1711–1716. [CrossRef]
- Passiak BS, Liu D, Kresge HA, Cambronero FE, Pechman KR, Osborn KE, et al. Perivascular spaces contribute to cognition beyond other small vessel disease markers. 2019;92(12): e1309–e1321. [CrossRef]
- Paradise M, Crawford JD, Lam BCP, Wen W, Kochan NA, Makkar S, Dawes L, Trollor J, Draper B, Brodaty H, Sachdev PS. Association of Dilated Perivascular Spaces With Cognitive Decline and Incident Dementia. Neurology. 2021;96(11):e1501-e1511. [CrossRef]
- Grundy SM, Brewer HB Jr, Cleeman JI, Smith SC Jr, Lenfant C; American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109, 433–438. [CrossRef]
- National Cholesterol Education Program (NCEP); Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA. 2002;285, 2486–2497. [CrossRef]
- Ali S, Malloci M, Safiedeen Z, Soleti R, Vergori L, Vidal-Gómez X, Besnard C, Dubois S, Le Lay S, Boursier J, Chevrollier A, Gagnadoux F, Simard S, Andriantsitohaina R, Martinez MC; Metabol study Group. LPS enriched small extracellular vesicles from metabolic syndrome patients trigger endothelial dysfunction by activation of TLR4. Metabolism. 2021;118:154727. [CrossRef]
- Qi Y, Lin M, Yang Y, Li Y. Relationship of Visceral Adipose Tissue With Dilated Perivascular Spaces. Front Neurosci. 2020; 14: 583557. [CrossRef]
- Wu D, Yang X, Liu X. Insulin Resistance Is Independently Associated With Enlarged Perivascular Space in the Basal Ganglia in Nondiabetic Healthy Elderly Population. Am J Alzheimer’s Disease and Other Dementias. 2020;35:1533317520912126. [CrossRef]
- Hayden, MR. Brain Endothelial Cells Play a Central Role in the Development of Enlarged Perivascular Spaces in the Metabolic Syndrome. Medicina. 2023;59(6): 1124. [CrossRef]
- United Nations Department of Economics and Social Affairs Populations Division Population Ageing and Sustainable Development. Available online: https://www.un.org/development/desa/pd/sites/www.un.org.development.desa.pd/files/wpp2022_summary_of_results.pdf (last accessed on 20 May 2023).
- Tucsek Z, Toth P, Tarantini S, Sosnowska D, Gautam T, Warrington JP, Giles CB, Wren JD, Koller A, Ballabh P, et al. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:1339–1352. [CrossRef]
- Paavonsalo S, Lackman MH, KaramanS. Capillary Rarefaction in Obesity and Metabolic Diseases—Organ Specificity and Possible Mechanisms. Cells. 2020;9(12): 2683. [CrossRef]
- Chantler PD, Shrader CD, Tabone LE, d’Audiffret AC, Huseynova K, Brooks SD, Branyan KW, Grogg KA, Frisbee JC. Cerebral Cortical Microvascular Rarefaction in Metabolic Syndrome is Dependent on Insulin Resistance and Loss of Nitric Oxide Bioavailability. Microcirculation 2015, 22, 435–445.
- van Dinther M, Voorter PHM, Jansen JFA, Jones EAV, van Oostenbrugge RJ, Staals J, Backes WH. Assessment of microvascular rarefaction in human brain disorders using physiological magnetic resonance imaging. J Cereb Blood Flow Metab. 2022 May; 42(5): 718–737. [CrossRef]
- Bairamian D, Sha S, Rolhion N, Sokol H, Dorothée G, Lemere CA, Krantic S. Microbiota in neuroinflammation and synaptic dysfunction: a focus on Alzheimer's disease. Mol Neurodeger. 2022;17(1):19. [CrossRef]
- Di Tommaso N Santopaolo F, Gasbarrini A, Ponziani FR. The Gut–Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases. Int. J. Mol. Sci. 2023, 24, 1470. [CrossRef]
- Tilg H, Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest. 2011;121(6):2126-32. [CrossRef]
- Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6(10):772–783. [CrossRef]
- Daneman R, Prat A. The Blood–Brain Barrier. Cold Spring Harb Perspect Biol. 2015;7(1): a020412. [CrossRef]
- Liao, JK. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. 2013;123(2):540-1. [CrossRef]
- Hayden MR. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes and coronavirus disease 2019. J Int Med Res. 2020;48(7):300060520939746. [CrossRef]
- Christ M, Bauersachs J, Liebetrau C, Heck M, Günther A, Wehling M. Glucose Increases Endothelial-Dependent Superoxide Formation in Coronary Arteries by NAD(P)H Oxidase Activation: Attenuation by the 3-Hydroxy-3-Methylglutaryl Coenzyme A. Diabetes. 2002;51(8):2648–2652. [CrossRef]
- Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound Healing: A Cellular Perspective. Physiol Rev. 2019;99(1): 665–706. [CrossRef]
- Tambuyzer BR, Ponsaerts P, Nouwen EJ. Microglia: Gatekeepers of the central nervous system immunology. J. Leukoc. Biol. 2009;85: 352–370. [CrossRef]
- Hayden MR, Grant DG, Aroor AR, DeMarco VG. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model–Part II: Microglia and Mitochondria. Neuroglia. 2018;1(2): 311-326. [CrossRef]
- Van Dyken P, Lacoste B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood–Brain Barrier. Front Neurosci. 2018;12:930. [CrossRef]
- Owens T, Bechmann I, Engelhardt BJ. Perivascular spaces and the two steps to neuroinflammation. Neuropathol Exp Neurol. 2008 Dec;67(12):1113-21. [CrossRef]
- Yang T, Guo R, Zhang F. Brain perivascular macrophages: Recent advances and implications in health and diseases. CNS Neurosci Ther. 2019;25(12):1318–1328. [CrossRef]
- Yao XY, Gao MC, Bai SW, Xie L, Song YY, Ding J. Wu YF, Xue CR, Hao Y, Zhang Y, Guan YT. Enlarged perivascular spaces, neuroinflammation and neurological dysfunction in NMOSD patients. Front Immuno. 2022;13:966781. eCollection 2022. [CrossRef]
- Esiri MM, Gay D. Immunological and neuropathological significance of the Virchow-Robin space. J Neuro Sci.1990;100(1-2):3-8. [CrossRef]
- Muller WA. Getting Leukocytes to the Site of Inflammation. Vet Pathol. 2013; 50(1): 7–22. [CrossRef]
- Hayden MR, Grant DG, Aroor AR, DeMarco VG. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia. 2018;1(1):220-244. [CrossRef]
- Verkhratsky A, Nedergaard M. Physiology of Astroglia. Physiol Rev. 2018 Jan 1;98(1):239-389. [CrossRef]
- Verkhratsky A, Butt AM. Neuroglia: Function and Pathology. 1st ed.; Academic Press. 125 London Wall, London EC2Y 5AS, United Kingdom 525 B Street. Copyright © 2023 Elsevier Inc. All rights reserved.
- Verkhratsky A, Nedergaard M. Astroglial cradle in the life of the synapse. Philos Trans R Soc Lond B Biol Sci. 2014 Oct 19;369(1654):20130595. [CrossRef]
- Shulyatnikova T, Verkhratsky A. Astroglia in Sepsis Associated Encephalopathy. Neurochemical Research. 2020;45(1):83-99. [CrossRef]
- Bitner C, Benjelloun-Touimi S, Dupouey P. Palisading pattern of subpial astroglial processes in the adult rodent brain: relationship between the glial palisading pattern and the axonal and astroglial organization. Brain Res.1987;465(1-2):167-178. [CrossRef]
- Choudhury GR, Ding S. Reactive astrocytes and therapeutic potential in focal ischemic stroke. Neurobiol Dis. 2016 Jan; 85: 234–244. [CrossRef]
- Wang H, Song G, Chuang H, Chiu C, Abdelmaksoud A, Ye Y, Zhao L. Portrait of glial scar in neurological diseases. Int I J Immunopathol Pharmacol. 2018;31: 2058738418801406. [CrossRef]
- von Streitberg A, Jäkel S, von Bernhardi JE, Straube C, Buggenthin F, Marr C, Dimou L. NG2-Glia Transiently Overcome Their Homeostatic Network and Contribute to Wound Closure After Brain Injury. Front Cell Dev Biol. 2021;9:662056. [CrossRef]
- Williamson MR, Fuertes CJA, Dunn A, Drew MR, Jones TA. Reactive astrocytes facilitate vascular repair and remodeling after stroke. Cell Rep. 2021;35(4):109048. [CrossRef]
- Pakyari M, Farrokhi A, Maharlooei MK, Ghahary A. Critical Role of Transforming Growth Factor Beta in Different Phases of Wound Healing. Adv Wound Care (New Rochelle). 2013;2(5): 215–224. [CrossRef]
- Howe MD, Furr JW, Munshi Y, Roy-O’Reilly MA, Maniskas ME, Koellhoffer EC, d’Aigle J, Sansing LH, McCullough LD, Urayama A. Transforming growth factor-β promotes basement membrane fibrosis, alters perivascular cerebrospinal fluid distribution, and worsens neurological recovery in the aged brain after stroke. GeroScience. 2019;41:543–559. [CrossRef]
- Doyle KP, Cekanaviciute E, Mamer LE, Buckwalter MS. TGFβ signaling in the brain increases with aging and signals to astrocytes and innate immune cells in the weeks after stroke. J Neuroinflammation. 2010;7:62. [CrossRef]
- Szu JI, Binder DK. The Role of Astrocytic Aquaporin-4 in Synaptic Plasticity and Learning and Memory. Front Integr Neurosci. 2016;10:8. [CrossRef]
- Yu G, Zhang Y, Ning B. Reactive Astrocytes in Central Nervous System Injury: Subgroup and Potential Therapy. Front Cell Neurosci. 2021;15:792764. [CrossRef]
- Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, et al. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat. Commun. 2017;8(28). [CrossRef]
- Aydeger C, Avni H. Physiopathology of Wound Healing in Central Nervous System. Hacettepe University Journal of the Faculty of Pharmacy. 2023;43(1):70-77. [CrossRef]
- Lo E. A new penumbra: transitioning from injury into repair after stroke. Nat Med. 2008;14(5):497-500. [CrossRef]
- Yoo DS, Won YD, Huh PW, Shin HE, Kim KT, Kang SG, Lee SB, Cho KS. Therapeutic Results of Intra-Arterial Thrombolysis after Full-Dose Intravenous Tissue Plasminogen Activator Administration. AJNR Am J Neuroradiol. 2010;31(8):1536-1540. [CrossRef]
- Fernandez-Miranda J, Rhoton AL, Alvarez-Linera J, Kakizawa Y, Choi C, de Oliveira EP. Three-Dimensional Microsurgical and Tractographic Anatomy of the White Matter of the Human Brain. Neurosurgery. 2008;62(6 Suppl 3):989-1026; discussion 1026-8. [CrossRef]
- Qi Y, Lin M, Yang Y, Li Y. Relationship of Visceral Adipose Tissue with Dilated Perivascular Spaces. Front Neurosci. 2021;14:583557. [CrossRef]
- Ineichen BV, Cananau C, Plattén M, Ouellette R, Moridi T, Frauenknecht KBM, Okar SV, Kulcsar Z, Kockum I, Piehl F, Reich DS, Granberga T. Dilated Virchow-Robin spaces are a marker for arterial disease in multiple sclerosis. EBioMedicine. 2023;92:104631. [CrossRef]
- Jessen NA, Munk ASF, Lundgaard I, Nedergaard M. The Glymphatic System – A Beginner's Guide. Neurochem Res. 2015; 40(12): 2583–2599. [CrossRef]
- Lei Y, Han H, Yuan F, Javeed A, Zhao Y. The brain interstitial system: Anatomy, modeling, in vivo measurement, and applications. Progress in Neurobiology. 2017;157:230-246. [CrossRef]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep Drives Metabolite Clearance from the Adult Brain. Science. 2013; 342(6156): 10.1126/science.1241224. [CrossRef]
- Reddy OC, van der Werf YD. The Sleeping Brain: Harnessing the Power of the Glymphatic System through Lifestyle Choices. Brain Sci. 2020; 10(11): 868. [CrossRef]
- Zavecz Z, Shah VD, Murillo OG, Vallat R, Mander BA, Winer JR, Jagust WJ, Walker MP. NREM sleep as a novel protective cognitive reserve factor in the face of Alzheimer's disease pathology. BMC Med. 2023;21(1):156. [CrossRef]
Figure 1.
Multiple injurious stimuli to brain endothelial cells (BECs) in obesity, metabolic syndrome (MetS), and type 2 diabetes mellitus (T2DM). This representative illustration does not depict the thin luminal endothelial glycocalyx layer covering the BECs. Ang II = angiotensin 2; AGE/RAGE = advanced glycation end products and its receptor RAGE; BEC = brain endothelial cell; BECact/dys = brain endothelial cell activation and dysfunction; LDL = low-density lipoprotein cholesterol; LPa = lipoprotein little a; LPS = lipopolysaccharide; pCC = peripheral cytokine/chemokine; ROS = reactive oxygen species and the reactive species interactome; T = transcytosis;  = radiation symbol.
= radiation symbol.
= radiation symbol.
Figure 1.
Multiple injurious stimuli to brain endothelial cells (BECs) in obesity, metabolic syndrome (MetS), and type 2 diabetes mellitus (T2DM). This representative illustration does not depict the thin luminal endothelial glycocalyx layer covering the BECs. Ang II = angiotensin 2; AGE/RAGE = advanced glycation end products and its receptor RAGE; BEC = brain endothelial cell; BECact/dys = brain endothelial cell activation and dysfunction; LDL = low-density lipoprotein cholesterol; LPa = lipoprotein little a; LPS = lipopolysaccharide; pCC = peripheral cytokine/chemokine; ROS = reactive oxygen species and the reactive species interactome; T = transcytosis; = radiation symbol.
= radiation symbol.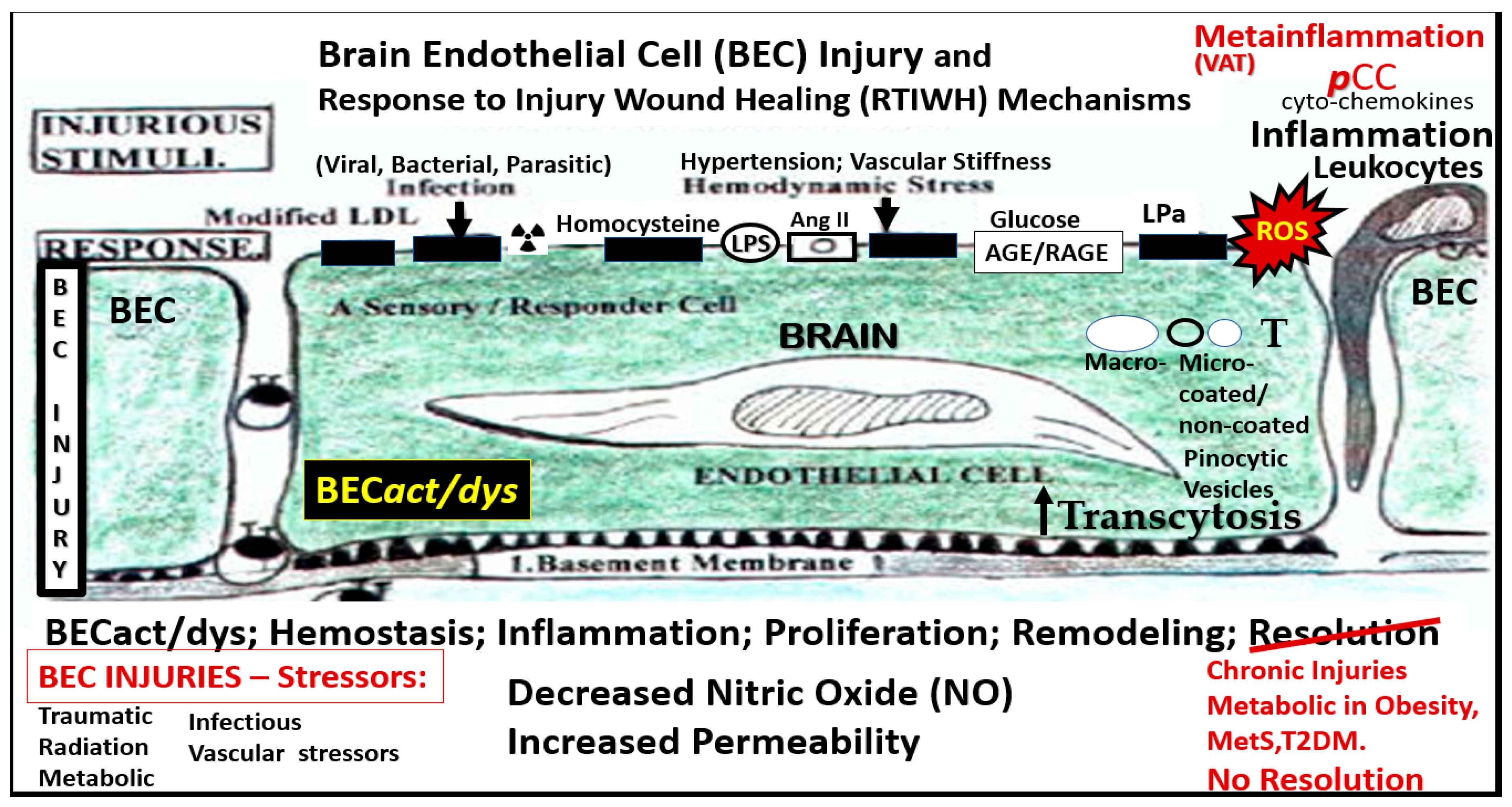
Figure 2.
Comparison of peripheral response to injury wound healing (RTIWH) to the unique brain injury RTIWH mechanisms. Panel A demonstrates the RTIWH mechanisms that occur in non-brain tissues such as in the lung as depicted in this image. Panel B depicts the RTIWH mechanisms as a result of the dual signaling by peripheral cytokines/chemokines (pCC) and soluble lipopolysaccharide (sLPS) and LPS-enriched extracellular exosomes (lpsEVexos). This dual signaling of the brain endothelial cell(s) (BECs) results in BEC activation and dysfunction (BECact/dys), which in turn results in central nervous system (CNS) neuroinflammation with increased CNSCC, reactive polarized microglia, and reactive astroglia – reactive astrogliosis, which results in astrogliosis scarring instead of fibrosis scarring since the brain does not have fibrocytes/fibroblasts in its parenchyma. Importantly, this RTIWH mechanism contributes to maladaptive remodeling and also contributes to the development of enlarged perivascular spaces (EPVS). Figure adapted with permission by CC 4.0 [4].
Figure 2.
Comparison of peripheral response to injury wound healing (RTIWH) to the unique brain injury RTIWH mechanisms. Panel A demonstrates the RTIWH mechanisms that occur in non-brain tissues such as in the lung as depicted in this image. Panel B depicts the RTIWH mechanisms as a result of the dual signaling by peripheral cytokines/chemokines (pCC) and soluble lipopolysaccharide (sLPS) and LPS-enriched extracellular exosomes (lpsEVexos). This dual signaling of the brain endothelial cell(s) (BECs) results in BEC activation and dysfunction (BECact/dys), which in turn results in central nervous system (CNS) neuroinflammation with increased CNSCC, reactive polarized microglia, and reactive astroglia – reactive astrogliosis, which results in astrogliosis scarring instead of fibrosis scarring since the brain does not have fibrocytes/fibroblasts in its parenchyma. Importantly, this RTIWH mechanism contributes to maladaptive remodeling and also contributes to the development of enlarged perivascular spaces (EPVS). Figure adapted with permission by CC 4.0 [4].

Figure 3.
Illustration of the origins of the perivascular spaces (PVS) and representative transmission electron micrograph (TEM) cross sections of the precapillary arteriole, true capillary, and postcapillary venule. Panel A illustrates the whole brain with it surface pia arteries that dive deeply into the grey matter and deeply into the white matter terminating in capillaries within the interstitial spaces (ISS). Panel B demonstrates the relationship of the perivascular spaces (PVS) to the precapillary arterioles with the vascular lumen influx of oxygen, nutrients, solutes (red arrow) and the influx of CSF via the PVS (red arrow) to true capillaries with the vascular lumen delivery of oxygen, nutrients, and solutes, and the post-capillary venules that remove metabolic waste via the vascular lumen (carbon dioxide plus others) and PVS that is responsible for the removal of the admixture of ISF and CSF, and metabolic waste (MW including neurotoxins of misfolded proteins such as toxic amyloid beta and tau neurofibrillary fibrils) (blue arrow) to the subarachnoid space (SAS). Panel C depicts how the CSF and its admixed metabolic waste from the PVS glymphatic system is transported to the dural venous sinus (DVS) and the systemic circulation. Panels D-F demonstrate representative and corresponding cross-sectional TEMs of corresponding labeled capillary images in panel B. Panel D demonstrates a precapillary arteriole with a PVS (pseudo-colored green). Panel E represents a true capillary without a PVS. Importantly, note that the pia membrane of the glia limitans abruptly ends at the true capillary and that the astrocyte endfeet directly abut the brain endothelial cell basement membrane (blue arrows). Panel F depicts a postcapillary venule that appears to have a dilated PVS and thus is considered to be an enlarged perivascular space (EPVS). Note the presence of a resident perivascular macrophage cell within the EPVS. It is important to note that the PVS provides the conduit structure for the glymphatic draining system to deliver metabolic waste products to the CSF and the systemic circulation for disposal. This adapted and modified image is provided with permission by CC 4.0 [7]. AC = astrocyte; ACef = astrocyte endfeet; ACfp = astrocyte foot process-end feet; AQP4 = aquaporin four; Cl = capillary lumen; CSF = cerebrospinal fluid; EC = brain endothelial cell; ISF = interstitial fluid; N = nucleus; Pc = pericyte; PVS = perivascular space; Mt = mitochondria; RBC = red blood cell; rMΦ = resident reactive macrophage.
Figure 3.
Illustration of the origins of the perivascular spaces (PVS) and representative transmission electron micrograph (TEM) cross sections of the precapillary arteriole, true capillary, and postcapillary venule. Panel A illustrates the whole brain with it surface pia arteries that dive deeply into the grey matter and deeply into the white matter terminating in capillaries within the interstitial spaces (ISS). Panel B demonstrates the relationship of the perivascular spaces (PVS) to the precapillary arterioles with the vascular lumen influx of oxygen, nutrients, solutes (red arrow) and the influx of CSF via the PVS (red arrow) to true capillaries with the vascular lumen delivery of oxygen, nutrients, and solutes, and the post-capillary venules that remove metabolic waste via the vascular lumen (carbon dioxide plus others) and PVS that is responsible for the removal of the admixture of ISF and CSF, and metabolic waste (MW including neurotoxins of misfolded proteins such as toxic amyloid beta and tau neurofibrillary fibrils) (blue arrow) to the subarachnoid space (SAS). Panel C depicts how the CSF and its admixed metabolic waste from the PVS glymphatic system is transported to the dural venous sinus (DVS) and the systemic circulation. Panels D-F demonstrate representative and corresponding cross-sectional TEMs of corresponding labeled capillary images in panel B. Panel D demonstrates a precapillary arteriole with a PVS (pseudo-colored green). Panel E represents a true capillary without a PVS. Importantly, note that the pia membrane of the glia limitans abruptly ends at the true capillary and that the astrocyte endfeet directly abut the brain endothelial cell basement membrane (blue arrows). Panel F depicts a postcapillary venule that appears to have a dilated PVS and thus is considered to be an enlarged perivascular space (EPVS). Note the presence of a resident perivascular macrophage cell within the EPVS. It is important to note that the PVS provides the conduit structure for the glymphatic draining system to deliver metabolic waste products to the CSF and the systemic circulation for disposal. This adapted and modified image is provided with permission by CC 4.0 [7]. AC = astrocyte; ACef = astrocyte endfeet; ACfp = astrocyte foot process-end feet; AQP4 = aquaporin four; Cl = capillary lumen; CSF = cerebrospinal fluid; EC = brain endothelial cell; ISF = interstitial fluid; N = nucleus; Pc = pericyte; PVS = perivascular space; Mt = mitochondria; RBC = red blood cell; rMΦ = resident reactive macrophage.
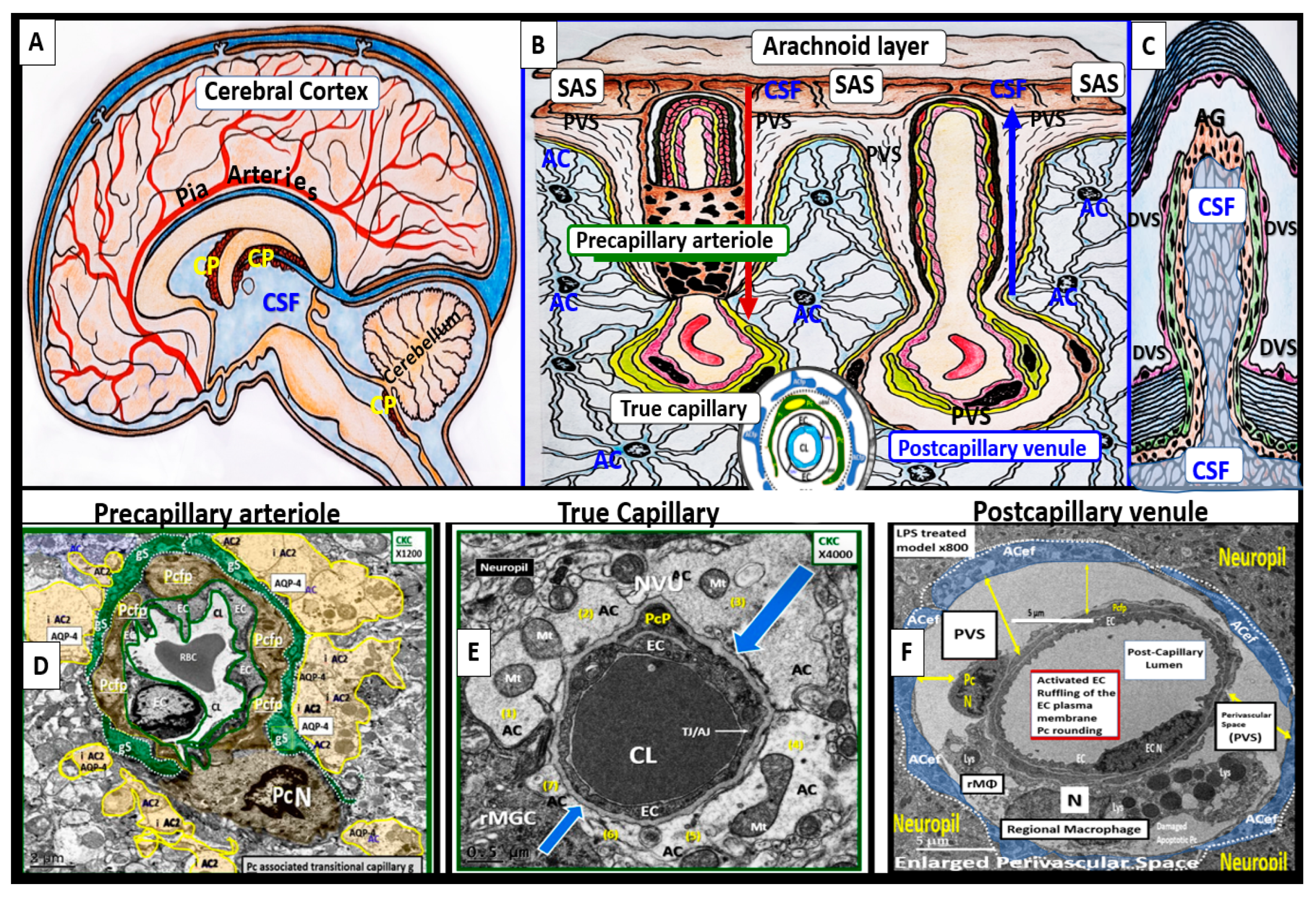
Figure 4.
Comparison of normal true capillary and precapillary arteriole to postcapillary venules with enlarged perivascular spaces (EPVS). Panel A demonstrates a true capillary with the astrocyte endfeet (ACef) abutting the shared basement membrane of the neurovascular unit (NVU) brain endothelial cell and pericyte without a pia mater membrane. Panel B demonstrates a precapillary arteriole with only a thinned perivascular space (PVS) (pseudo-colored green). Note that the abluminal perivascular space is still ensheathed by the pia matter and basal lamina of the adjacent ACef. Panels C and D each depict two postcapillary venules with enlarged perivascular spaces (EPVS) between 2 and 6.5 μm. Image provided with permission by CC 4.0 [7]. Magnification x1000; x2000; scale bars = 2 and 1μm in panels C and D respectively. AC astrocyte; ACef = astrocyte endfeet; AQP 4 = aquaporin 4; Cl = capillary lumen; EC = brain endothelial cell; Pc = pericyte; Pc N = pericyte nucleus; Pcfp; = pericyte foot process - endfeet; RBC = red blood cell.
Figure 4.
Comparison of normal true capillary and precapillary arteriole to postcapillary venules with enlarged perivascular spaces (EPVS). Panel A demonstrates a true capillary with the astrocyte endfeet (ACef) abutting the shared basement membrane of the neurovascular unit (NVU) brain endothelial cell and pericyte without a pia mater membrane. Panel B demonstrates a precapillary arteriole with only a thinned perivascular space (PVS) (pseudo-colored green). Note that the abluminal perivascular space is still ensheathed by the pia matter and basal lamina of the adjacent ACef. Panels C and D each depict two postcapillary venules with enlarged perivascular spaces (EPVS) between 2 and 6.5 μm. Image provided with permission by CC 4.0 [7]. Magnification x1000; x2000; scale bars = 2 and 1μm in panels C and D respectively. AC astrocyte; ACef = astrocyte endfeet; AQP 4 = aquaporin 4; Cl = capillary lumen; EC = brain endothelial cell; Pc = pericyte; Pc N = pericyte nucleus; Pcfp; = pericyte foot process - endfeet; RBC = red blood cell.

Figure 5.
Metabolic syndrome (MetS), enlarged perivascular spaces (EPVS), and cerebral small vessel disease (cSVD). Note how the accumulation of visceral adipose tissue (VAT), obesity, and hyperlipidemia (atherogenic dyslipidemia) appear to drive the MetS and the other three arms of the letter X, which includes insulin resistance (IR) and the associated hyperinsulinemia, hypertension; vascular stiffening, and hyperglycemia with or without manifest type 2 diabetes mellitus (T2DM). The global increase in obesity and the accumulation of visceral obesity is thought to be a major driver of the MetS that is related to the excessive metainflammation arising from VAT depots. Therefore, note the prominent closed red arrows emanating from VAT to cerebrocardiovascular disease (CCVD), SVD, TIA, Stroke, and cerebral microbleeds and hemorrhages. Ensuing brain endothelial cell(s) (BEC) activation and dysfunction (BECact/dys) with its proinflammatory and prooxidative properties result in endothelial nitric oxide synthesis (eNOS) uncoupling with increased superoxide (O2 • –) and decreased nitric oxide (NO) bioavailability. Also, note that obesity, MetS, and decreased bioavailable NO interact to result in capillary rarefaction that may allow the PVS to undergo expansion and dilation to become enlarged perivascular spaces (EPVS), which are biomarkers for cerebral small vessel disease (SVD). Figure adapted with permission by CC 4.0 [7,25] AGE = advanced glycation end-products; RAGE = receptor for AGE; AGE/RAGE = advanced glycation end-products and its receptor interaction; βcell = pancreatic islet insulin-producing beta cell; FFA = free fatty acids – unsaturated long chain fatty acids; IGT = impaired glucose tolerance; LOAD = late-onset Alzheimer’s disease; ROS = reactive oxygen species; RSI = reactive species interactome; Sk = skeletal: TG Index = triglyceride/glucose index; TIA = transient ischemia attack.
Figure 5.
Metabolic syndrome (MetS), enlarged perivascular spaces (EPVS), and cerebral small vessel disease (cSVD). Note how the accumulation of visceral adipose tissue (VAT), obesity, and hyperlipidemia (atherogenic dyslipidemia) appear to drive the MetS and the other three arms of the letter X, which includes insulin resistance (IR) and the associated hyperinsulinemia, hypertension; vascular stiffening, and hyperglycemia with or without manifest type 2 diabetes mellitus (T2DM). The global increase in obesity and the accumulation of visceral obesity is thought to be a major driver of the MetS that is related to the excessive metainflammation arising from VAT depots. Therefore, note the prominent closed red arrows emanating from VAT to cerebrocardiovascular disease (CCVD), SVD, TIA, Stroke, and cerebral microbleeds and hemorrhages. Ensuing brain endothelial cell(s) (BEC) activation and dysfunction (BECact/dys) with its proinflammatory and prooxidative properties result in endothelial nitric oxide synthesis (eNOS) uncoupling with increased superoxide (O2 • –) and decreased nitric oxide (NO) bioavailability. Also, note that obesity, MetS, and decreased bioavailable NO interact to result in capillary rarefaction that may allow the PVS to undergo expansion and dilation to become enlarged perivascular spaces (EPVS), which are biomarkers for cerebral small vessel disease (SVD). Figure adapted with permission by CC 4.0 [7,25] AGE = advanced glycation end-products; RAGE = receptor for AGE; AGE/RAGE = advanced glycation end-products and its receptor interaction; βcell = pancreatic islet insulin-producing beta cell; FFA = free fatty acids – unsaturated long chain fatty acids; IGT = impaired glucose tolerance; LOAD = late-onset Alzheimer’s disease; ROS = reactive oxygen species; RSI = reactive species interactome; Sk = skeletal: TG Index = triglyceride/glucose index; TIA = transient ischemia attack.
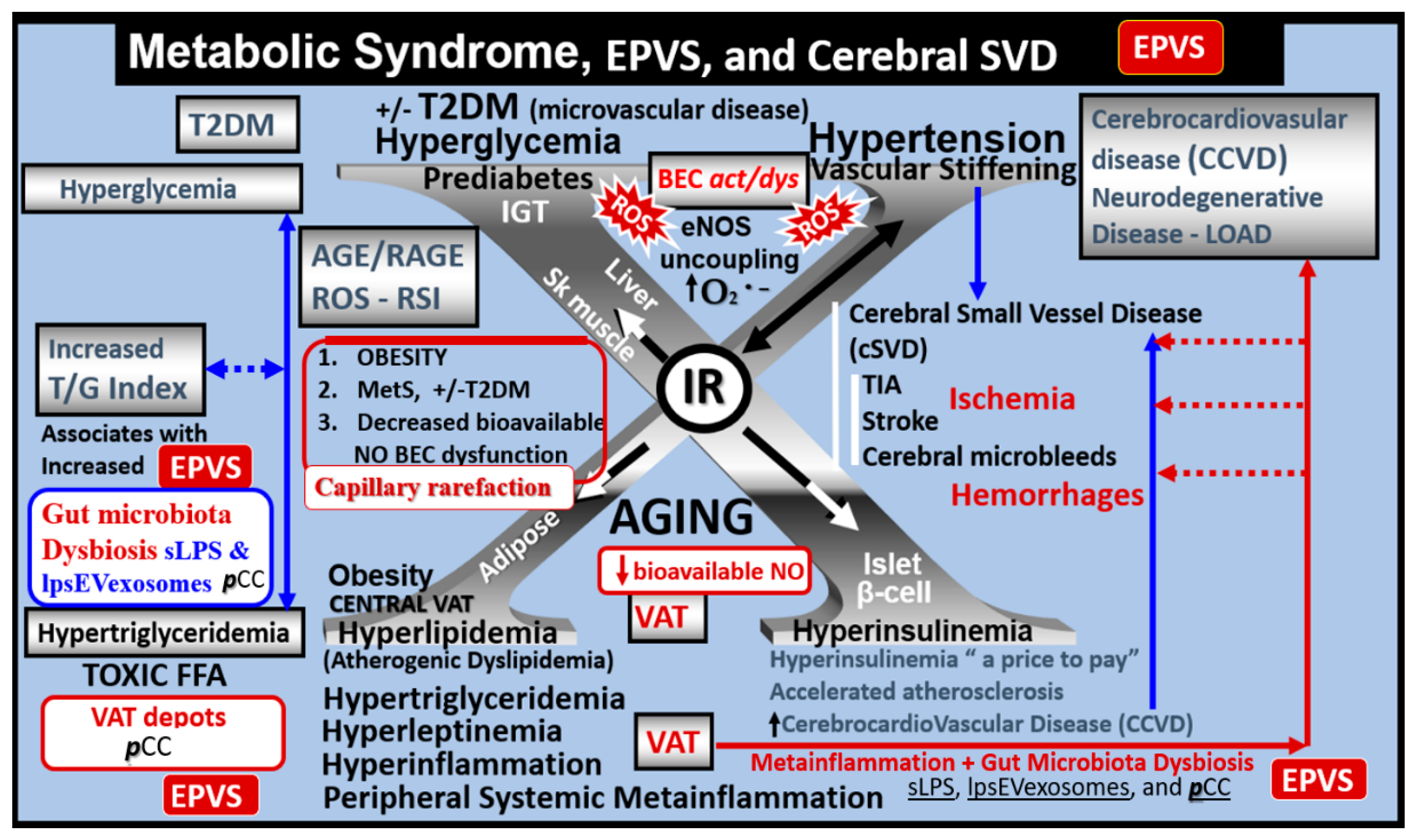
Figure 6.
Cross and longitudinal sections representitive of pre- and postcapillary arterioles and venules with an encompassing surrounding perivascular space (PVS). Panel A depicts a cross section of a capillary surrounded by a PVS (solid double red arrows) and its increase in total volume to become an enlarged perivascular space (EPVS) (dashed double red arrows), which represents capillary rarefaction. Panel B demonstrates a control longitudinal capillary that runs through an encompassing PVS (light blue). Panel C depicts capillary rarefaction in a longitudinal view and note how the volume of the PVS increases it total volume once the capillary has undergone rarefaction. Panel D depicts the progression of a normal precapillary arteriole and postcapillary venule PVS to an EPVS once the capillary has undergone rarefaction allowing for an increase total volume of the PVS (1-3). Panels B and C provided by CC 4.0 [7]. ACef = astrocyte endfeet; AQP4 = aquaporin 4; BEC = brain endothelial cells; BECact/dys = brain endothelial cell activation and dysfunction; CL =capillary lumen; EC = endothelial cell; lpsEVexos = lipopolysaccharide extracellular vesicle exosomes; NVU = neurovascular unit; Pcef = pericyte endfeet.
Figure 6.
Cross and longitudinal sections representitive of pre- and postcapillary arterioles and venules with an encompassing surrounding perivascular space (PVS). Panel A depicts a cross section of a capillary surrounded by a PVS (solid double red arrows) and its increase in total volume to become an enlarged perivascular space (EPVS) (dashed double red arrows), which represents capillary rarefaction. Panel B demonstrates a control longitudinal capillary that runs through an encompassing PVS (light blue). Panel C depicts capillary rarefaction in a longitudinal view and note how the volume of the PVS increases it total volume once the capillary has undergone rarefaction. Panel D depicts the progression of a normal precapillary arteriole and postcapillary venule PVS to an EPVS once the capillary has undergone rarefaction allowing for an increase total volume of the PVS (1-3). Panels B and C provided by CC 4.0 [7]. ACef = astrocyte endfeet; AQP4 = aquaporin 4; BEC = brain endothelial cells; BECact/dys = brain endothelial cell activation and dysfunction; CL =capillary lumen; EC = endothelial cell; lpsEVexos = lipopolysaccharide extracellular vesicle exosomes; NVU = neurovascular unit; Pcef = pericyte endfeet.
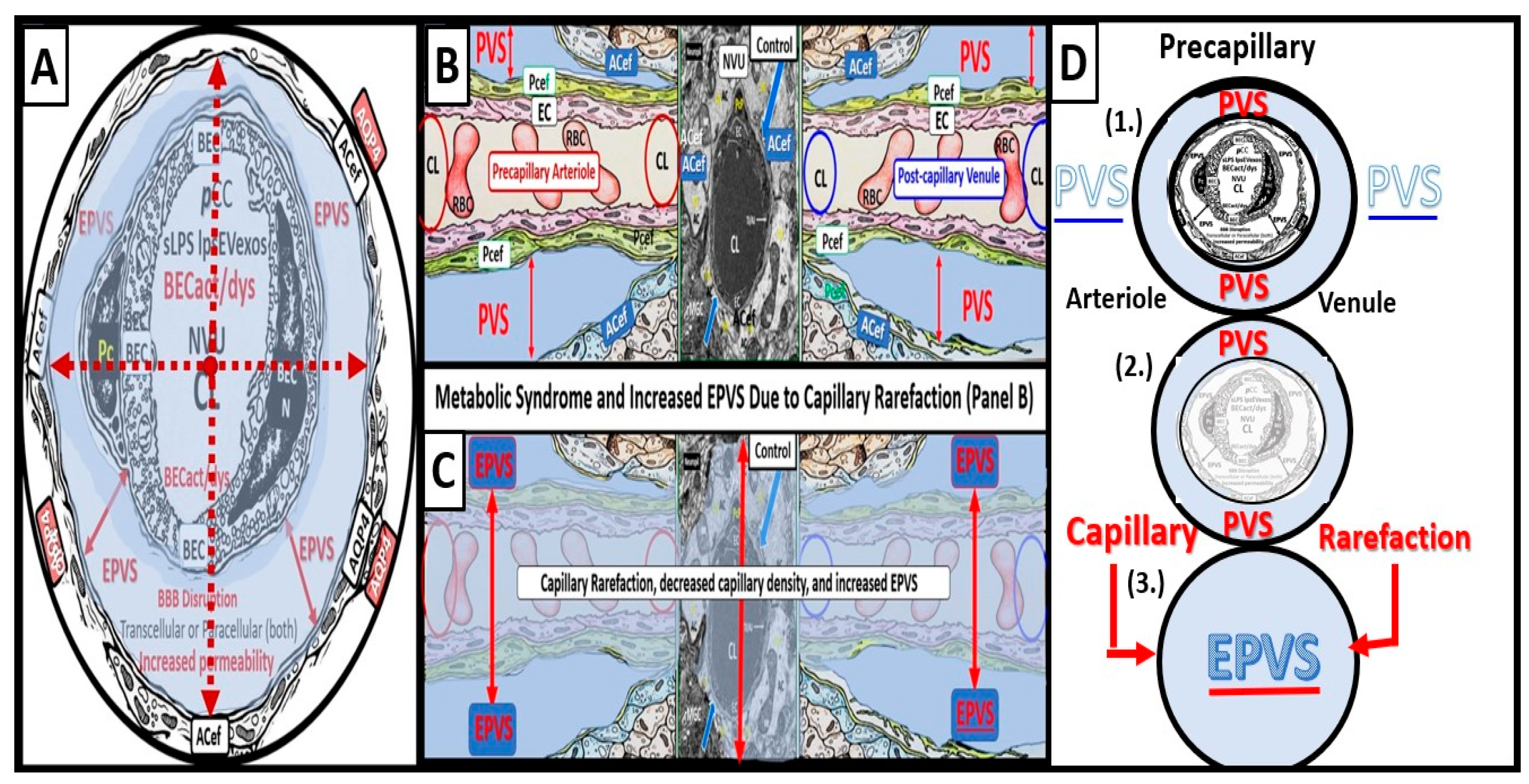
Figure 7.
The triangulation of metabolic function, gut dysbiosis, and the metabolic syndrome to the innate immune system, peripheral, proinflammatory cytokines and chemokines (pCC) to signal the neurovascular unit (NVU) brain endothelial cell(s) (BECs) resulting in BEC activation and dysfunction (BECact/dys) to result in neuroinflammation. This schematic demonstrates that gut microbiota dysbiosis, metabolic dysfunction, and an activated proinflammatory innate immune system are associated with obesity and metabolic syndrome (lower-left red box). Importantly, this triangulation also associates with the metainflammation that is produced in the obese visceral adipose tissue depots (upper-right red box). These two distinct sites of metainflammation (red boxes) and dual signaling of BECs will each signal the central nervous system brain endothelial cells to result in neuroinflammation to result in BECact/dys and contribute to enlarged perivascular spaces EPVS. Importantly, this dual signaling by the gut and visceral adipose tissue (VAT) of BECs may be synergistic in obesity and the MetS. Image provided by CC 4.0 [34]. db/db = obese, insulin resistance diabetic genetic mouse model; DIO = diet-induced obesity; BTBRob/ob = black and tan brachyuric ob/ob mouse model of obesity and diabetes; IR = insulin resistance; LR = leptin resistance; sLPS = soluble lipopolysaccharide; lpsEVexos = lipopolysaccharide extracellular vesicle exosomes; MΦ = macrophage; MetS = metabolic syndrome; pCC = peripheral cytokines/chemokines; T2DM = type 2 diabetes mellitus.
Figure 7.
The triangulation of metabolic function, gut dysbiosis, and the metabolic syndrome to the innate immune system, peripheral, proinflammatory cytokines and chemokines (pCC) to signal the neurovascular unit (NVU) brain endothelial cell(s) (BECs) resulting in BEC activation and dysfunction (BECact/dys) to result in neuroinflammation. This schematic demonstrates that gut microbiota dysbiosis, metabolic dysfunction, and an activated proinflammatory innate immune system are associated with obesity and metabolic syndrome (lower-left red box). Importantly, this triangulation also associates with the metainflammation that is produced in the obese visceral adipose tissue depots (upper-right red box). These two distinct sites of metainflammation (red boxes) and dual signaling of BECs will each signal the central nervous system brain endothelial cells to result in neuroinflammation to result in BECact/dys and contribute to enlarged perivascular spaces EPVS. Importantly, this dual signaling by the gut and visceral adipose tissue (VAT) of BECs may be synergistic in obesity and the MetS. Image provided by CC 4.0 [34]. db/db = obese, insulin resistance diabetic genetic mouse model; DIO = diet-induced obesity; BTBRob/ob = black and tan brachyuric ob/ob mouse model of obesity and diabetes; IR = insulin resistance; LR = leptin resistance; sLPS = soluble lipopolysaccharide; lpsEVexos = lipopolysaccharide extracellular vesicle exosomes; MΦ = macrophage; MetS = metabolic syndrome; pCC = peripheral cytokines/chemokines; T2DM = type 2 diabetes mellitus.
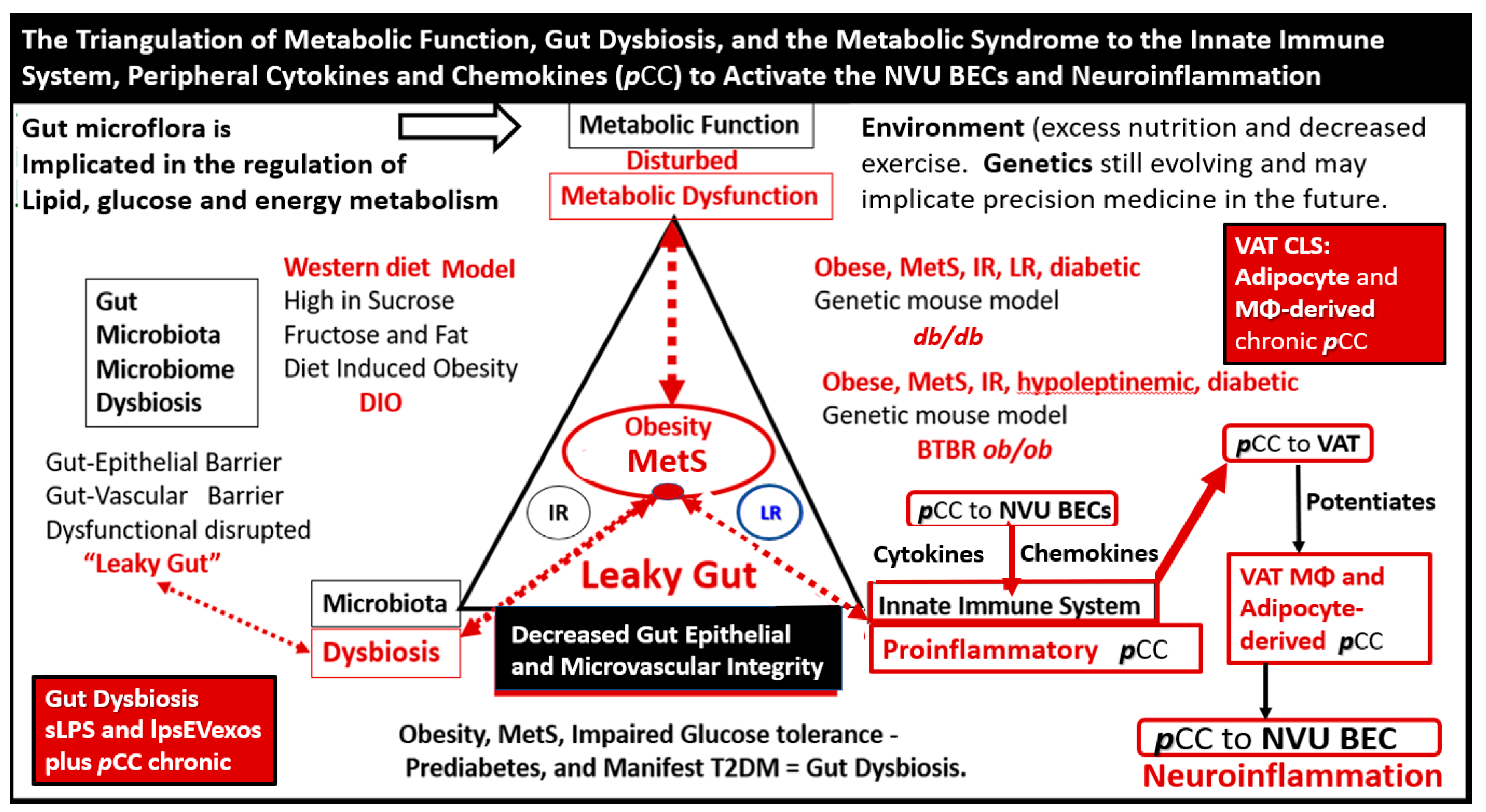
Figure 8.
Dual Signaling of BECs by peripheral cytokines/chemokines (pCC) and soluble lipopolysaccharide (sLPS) and LPS-enriched small extracellular vesicle exosome (lpsEVexos) to result in brain endothelial activation and dysfunction (BECact/dys). Note the insert lower-left, which depicts a small EVexo liberating multiple LPS-enriched vesicles and the metabolic signaling of BECs to result in central nervous system cytokines chemokines (cnsCC) and attracted microglia cells (atMGCs) via CCL5 (RANTES). BECact/dys results in neuroinflammation, blood-brain barrier (BBB) disruption, and enlarged perivascular spaces (EPVS) in obesity, insulin resistance (IR), and the metabolic syndrome (MetS). This modified image is presented with permission by CC4.0 [34]. atMGC = attracted microglia cell; CCL5 = chemokine (C-C motif) ligand 5 (chemoattractant for microglia cells); CD14 = cluster of differentiation 14; CCR5 = C-C chemokine receptor type 5, also known as CD195; CL = capillary lumen; EC = endothelial cell, brain endothelial cells; ecGCx = endothelial glycocalyx; EV=extracellular vesicles; EVMp =EVmicroparticles or microvesicles; EVexo = EVexosomes; IL1-β = interleukin-1 beta; IL-6 = interleukin-6; LPS = lipopolysaccharide; LBP = lipopolysaccharide-binding protein; MAPK = mitogen-activated protein kinase; Mp = microparticles; N = nucleus; PI3K/AKT = phosphatidylinositol 3-kinase / protein kinase B; PM = plasma membrane; RANTES = regulated on activation, normal T cell expressed and secreted; TNFα = tumor necrosis alpha.
Figure 8.
Dual Signaling of BECs by peripheral cytokines/chemokines (pCC) and soluble lipopolysaccharide (sLPS) and LPS-enriched small extracellular vesicle exosome (lpsEVexos) to result in brain endothelial activation and dysfunction (BECact/dys). Note the insert lower-left, which depicts a small EVexo liberating multiple LPS-enriched vesicles and the metabolic signaling of BECs to result in central nervous system cytokines chemokines (cnsCC) and attracted microglia cells (atMGCs) via CCL5 (RANTES). BECact/dys results in neuroinflammation, blood-brain barrier (BBB) disruption, and enlarged perivascular spaces (EPVS) in obesity, insulin resistance (IR), and the metabolic syndrome (MetS). This modified image is presented with permission by CC4.0 [34]. atMGC = attracted microglia cell; CCL5 = chemokine (C-C motif) ligand 5 (chemoattractant for microglia cells); CD14 = cluster of differentiation 14; CCR5 = C-C chemokine receptor type 5, also known as CD195; CL = capillary lumen; EC = endothelial cell, brain endothelial cells; ecGCx = endothelial glycocalyx; EV=extracellular vesicles; EVMp =EVmicroparticles or microvesicles; EVexo = EVexosomes; IL1-β = interleukin-1 beta; IL-6 = interleukin-6; LPS = lipopolysaccharide; LBP = lipopolysaccharide-binding protein; MAPK = mitogen-activated protein kinase; Mp = microparticles; N = nucleus; PI3K/AKT = phosphatidylinositol 3-kinase / protein kinase B; PM = plasma membrane; RANTES = regulated on activation, normal T cell expressed and secreted; TNFα = tumor necrosis alpha.
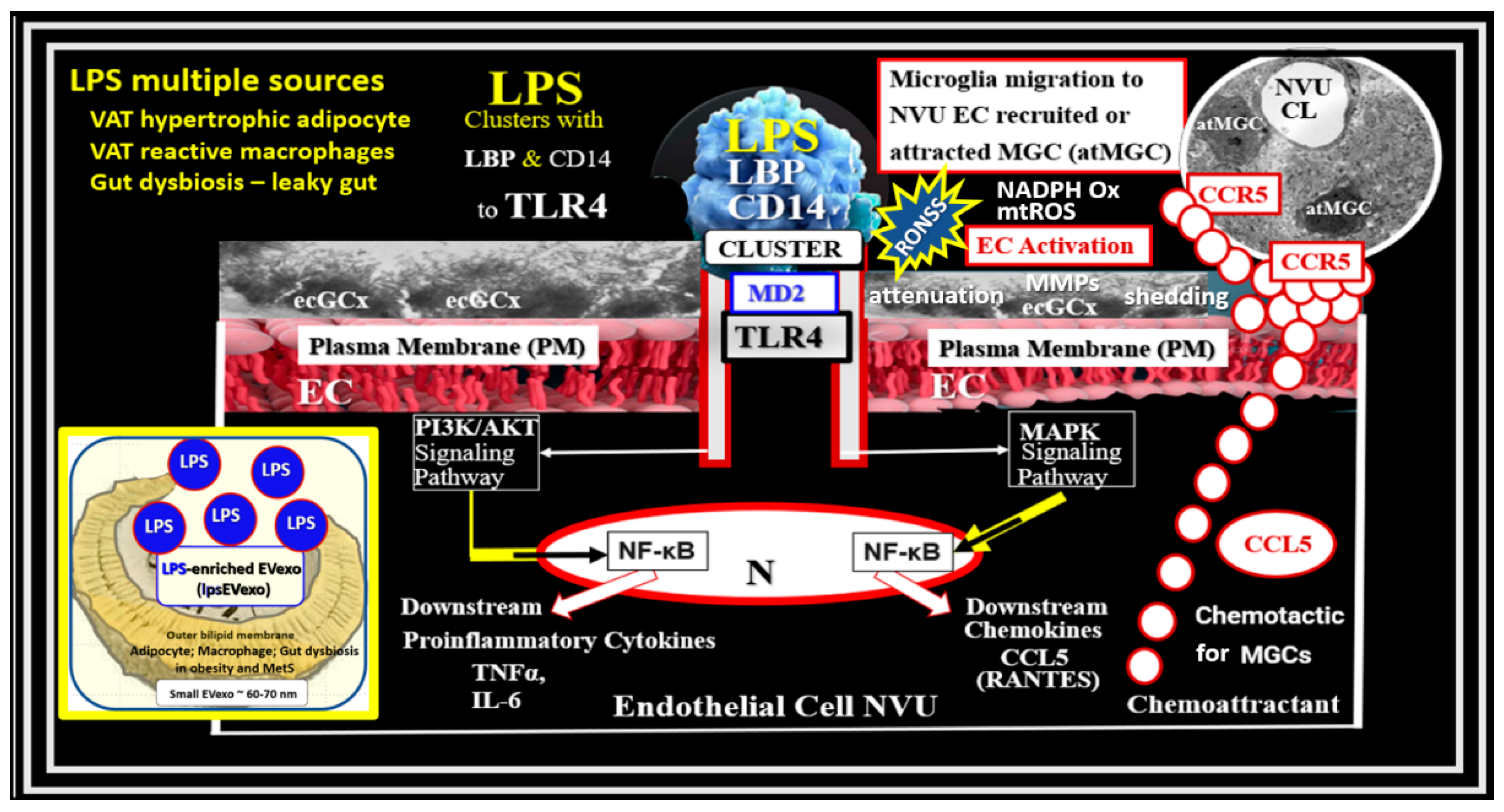
Figure 9.
Representative transmission electron microscopic (TEM) images of endothelial cell activation (ECact). Panel A demonstrates a control BEC. Panel B depicts the thickened electron-lucent areas (red arrows) of ECact as compared to control models in panel A. Panel C depicts basement membrane (BM) thickening with increased vacuoles (V) and vesicles (v). Panels D and E depict monocyte (D) and lymphocyte (E), platelet (F) and red blood cell (RBC) adhesion (G) to activated ECs. Adhesions sites in panels E, F, and G are denoted by closed arrows. Magnifications and scale bars vary and images are modified with permission of CC 4.0 [25]. Images (Panels C–G). Images are reproduced and modified with permission by CC 4.0 [25]. ACfp = astrocyte foot processes; Cl, capillary lumen; EC = endothelial cells, brain endothelial cells; MP = microparticle of the platelet.
Figure 9.
Representative transmission electron microscopic (TEM) images of endothelial cell activation (ECact). Panel A demonstrates a control BEC. Panel B depicts the thickened electron-lucent areas (red arrows) of ECact as compared to control models in panel A. Panel C depicts basement membrane (BM) thickening with increased vacuoles (V) and vesicles (v). Panels D and E depict monocyte (D) and lymphocyte (E), platelet (F) and red blood cell (RBC) adhesion (G) to activated ECs. Adhesions sites in panels E, F, and G are denoted by closed arrows. Magnifications and scale bars vary and images are modified with permission of CC 4.0 [25]. Images (Panels C–G). Images are reproduced and modified with permission by CC 4.0 [25]. ACfp = astrocyte foot processes; Cl, capillary lumen; EC = endothelial cells, brain endothelial cells; MP = microparticle of the platelet.
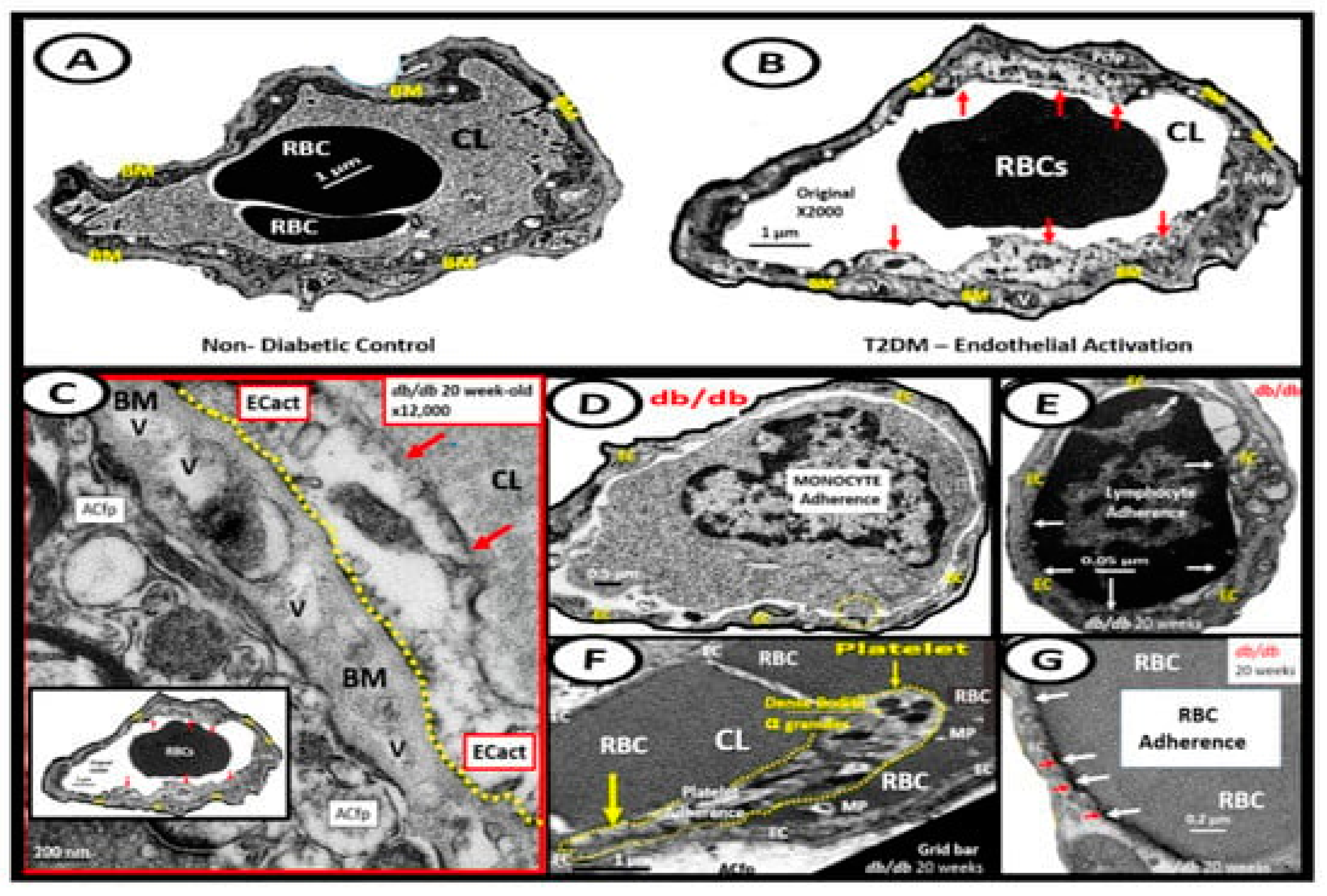
Figure 10.
Response to injury wound healing (RTIWH) remodeling, neuroinflammation, and enlarged perivascular spaces (EPVS). Panel A illustrates the progression of the perivascular space (PVS) as it undergoes transformation – RTIWH remodeling to EPVS in a linear fashion from left to right. Panel B demonstrates a transmission electron microscopic (TEM) image of a true capillary in a control 20-week-old female model. Panel C depicts a TEM image of an EPVS with a resident, reactive, migratory perivascular macrophage (rPVMΦ) in a 20-week-old female lipopolysaccharide-treated model. Panels D-F depicts leukocytes that undergo diapedesis across the neurovascular unit (NVU) brain endothelial cell(s) (BECs) disrupted blood-brain barrier (BBB), which enters the perivascular space(s) (PVS). Along with the rPVMΦ, these leukocytes are capable of creating excessive phagocytic cellular debris. This increase of leukocytes and rPVMΦs along with their excessive cellular debris result in obstruction of the PVS and results in the dilation of the PVS proximal to the obstruction creating the EPVS observed on magnetic resonance imaging (MRI) in the basal ganglia and centrum semiovale structures that appear early-on in the development of cerebral small vessel disease and neurodegenerative diseases. While panels A-F depict step one of a two-step process, step two incorporates the reactive oxygen species and the matrix metalloproteinases (MMP2, MMP9) that are capable of digesting the abluminal glia limitans of the EPVS, which allow for the transmigration of leukocytes to enter the interstitial spaces of the parenchymal neurons to complete the neuroinflammation remodeling mechanism in obesity, metabolic syndrome, and type 2 diabetes mellitus.
Figure 10.
Response to injury wound healing (RTIWH) remodeling, neuroinflammation, and enlarged perivascular spaces (EPVS). Panel A illustrates the progression of the perivascular space (PVS) as it undergoes transformation – RTIWH remodeling to EPVS in a linear fashion from left to right. Panel B demonstrates a transmission electron microscopic (TEM) image of a true capillary in a control 20-week-old female model. Panel C depicts a TEM image of an EPVS with a resident, reactive, migratory perivascular macrophage (rPVMΦ) in a 20-week-old female lipopolysaccharide-treated model. Panels D-F depicts leukocytes that undergo diapedesis across the neurovascular unit (NVU) brain endothelial cell(s) (BECs) disrupted blood-brain barrier (BBB), which enters the perivascular space(s) (PVS). Along with the rPVMΦ, these leukocytes are capable of creating excessive phagocytic cellular debris. This increase of leukocytes and rPVMΦs along with their excessive cellular debris result in obstruction of the PVS and results in the dilation of the PVS proximal to the obstruction creating the EPVS observed on magnetic resonance imaging (MRI) in the basal ganglia and centrum semiovale structures that appear early-on in the development of cerebral small vessel disease and neurodegenerative diseases. While panels A-F depict step one of a two-step process, step two incorporates the reactive oxygen species and the matrix metalloproteinases (MMP2, MMP9) that are capable of digesting the abluminal glia limitans of the EPVS, which allow for the transmigration of leukocytes to enter the interstitial spaces of the parenchymal neurons to complete the neuroinflammation remodeling mechanism in obesity, metabolic syndrome, and type 2 diabetes mellitus.
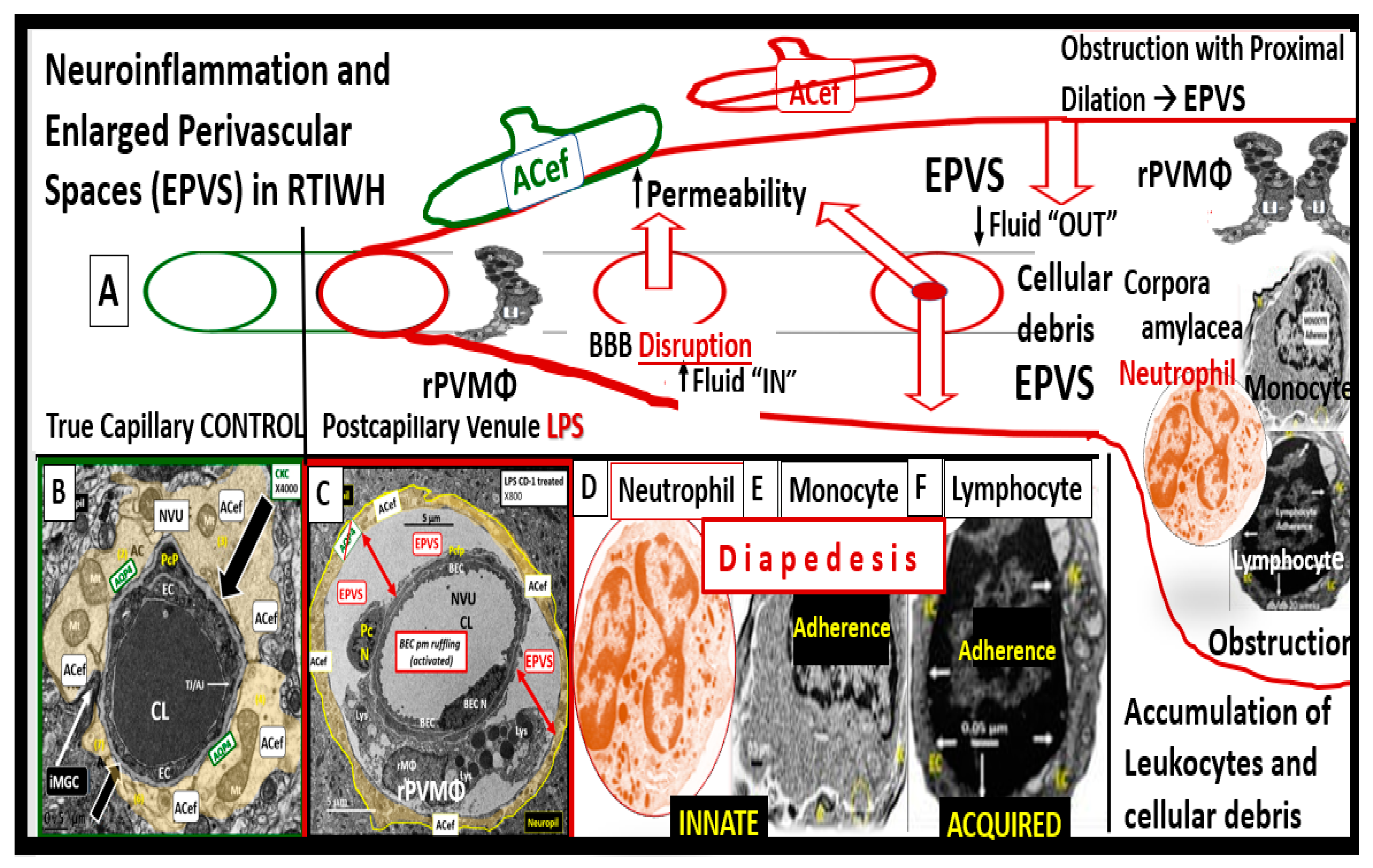
Figure 11.
Enlarged Perivascular Spaces (EPVS), reactive perivascular macrophages (rPVMΦs), and crosstalk with pericytes and foot processes (Pcfps) and PVS astrocyte endfeet (ACef). Panel A demonstrates a normal true capillary in a 20-week-old female C57B6/J control model and note how the ACef tightly abut the shared basement membrane of the brain endothelial cell (BEC) and Pericyte. Panel B depicts an EPVS with an aberrant polarized rPVMΦ in a 20-week-old lipopolysaccharide (LPS)-treated male model and note how the ACef are markedly separated from the capillary (red double arrows). Panel C depicts the communication crosstalk via rPVMΦs contacting the Pcfps and the EPVS abluminal lining ACfps. AQP4 = aquaporin 4; Lys = lysosomes; Mt = mitochondria; N = nucleus; NVU = neurovascular unit; V =vacuoles; ves = vesicles.
Figure 11.
Enlarged Perivascular Spaces (EPVS), reactive perivascular macrophages (rPVMΦs), and crosstalk with pericytes and foot processes (Pcfps) and PVS astrocyte endfeet (ACef). Panel A demonstrates a normal true capillary in a 20-week-old female C57B6/J control model and note how the ACef tightly abut the shared basement membrane of the brain endothelial cell (BEC) and Pericyte. Panel B depicts an EPVS with an aberrant polarized rPVMΦ in a 20-week-old lipopolysaccharide (LPS)-treated male model and note how the ACef are markedly separated from the capillary (red double arrows). Panel C depicts the communication crosstalk via rPVMΦs contacting the Pcfps and the EPVS abluminal lining ACfps. AQP4 = aquaporin 4; Lys = lysosomes; Mt = mitochondria; N = nucleus; NVU = neurovascular unit; V =vacuoles; ves = vesicles.
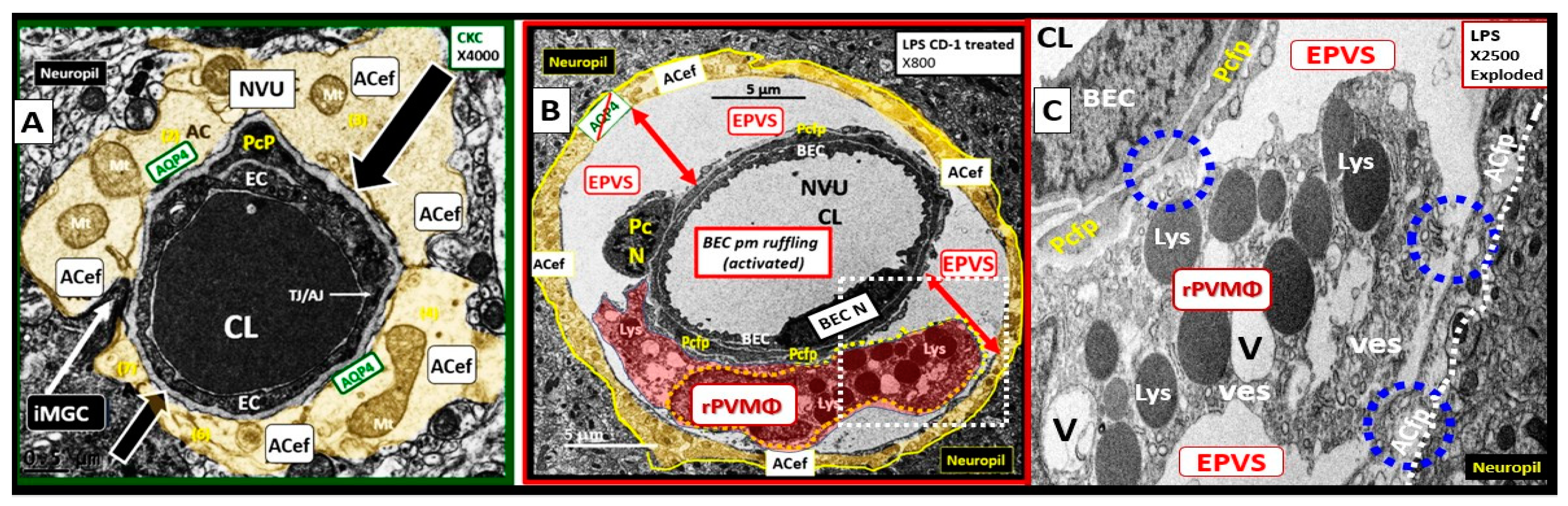
Figure 12.
Response to injury wound healing (RTIWH) at regions of injury in the brain and formation of the glial scar. This illustration depicts the RTIWH at regions of brain injury that become surrounded with palysading astroglia that progress to hypertrophic, proliferative, and reactive astrocytes following brain injury from left to right. Further, this gliosis scar serves to protect the surrounding uninjured brain structures from ongoing injury. This modified image is provided with permission by CC 4.0 [61].
Figure 12.
Response to injury wound healing (RTIWH) at regions of injury in the brain and formation of the glial scar. This illustration depicts the RTIWH at regions of brain injury that become surrounded with palysading astroglia that progress to hypertrophic, proliferative, and reactive astrocytes following brain injury from left to right. Further, this gliosis scar serves to protect the surrounding uninjured brain structures from ongoing injury. This modified image is provided with permission by CC 4.0 [61].
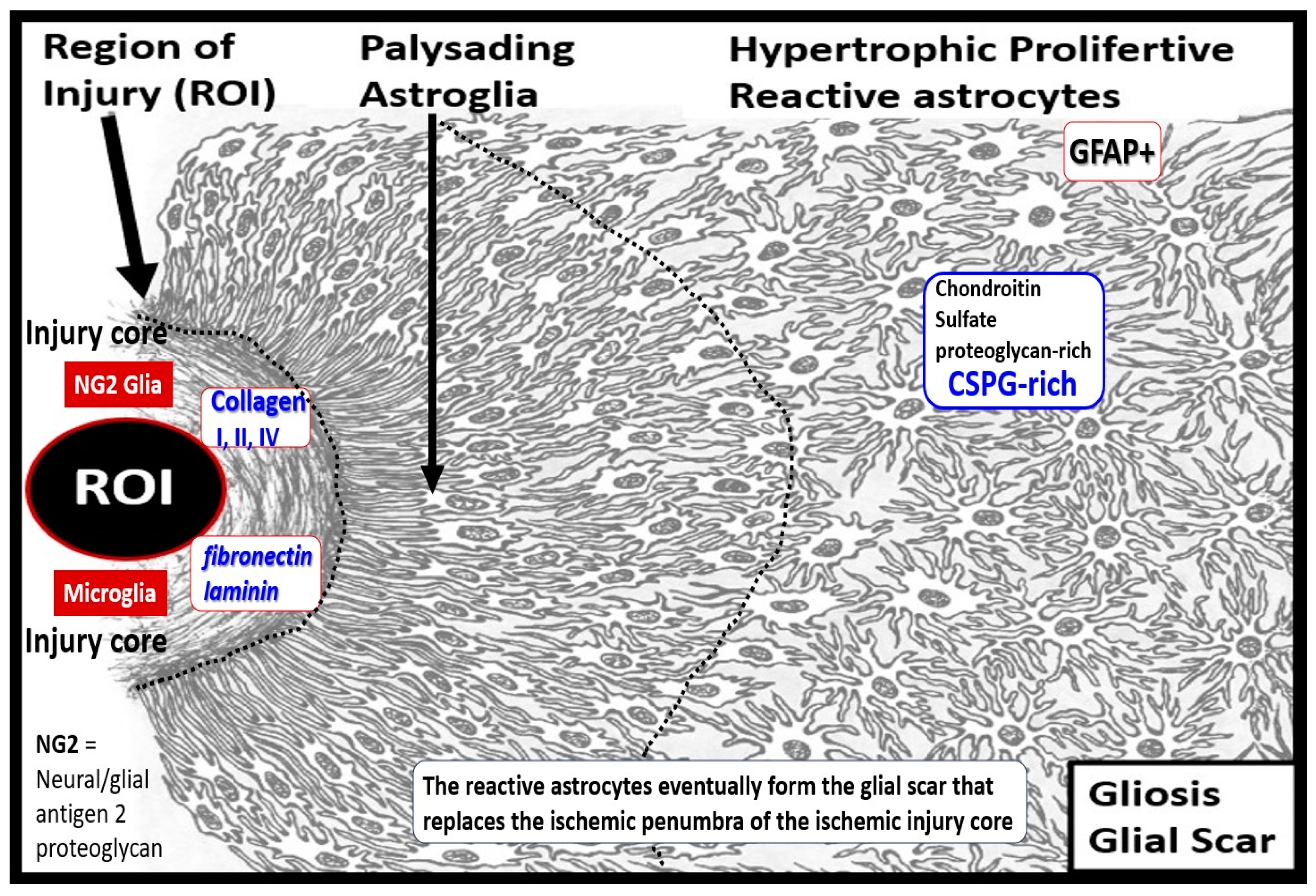
Figure 13.
Representative illustrations of water channel aquaporin 4 (AQP4), and representative immunohistochemistry (IHC) staining of thalamus AQP4 in models. Panel A demonstrates an illustration of the AQP4 water channel localized to the astrocyte end-feet (ACef) at the plasma membrane that extends to the enlarged perivascular spaces (EPVS). Panel B illustrates an elongated capillary in the region of the thalamus in a control model and note the mild staining of the AQP4 via IHC staining with the AQP4 antibody. Panel C depicts marked heavily stained AQP4 water channels with IHC AQP4 antibody staining in a model with acetaminophen-induced liver failure and hepatic encephalopathy. AQP4 is known to be upregulated in aging and in ammonia-induced brain injury; however, it becomes maldistributed and there is a loss of plasma membrane (pm) polarity associated with its maldistribution and dysfunction. Panel D is a representative illustration of the AQP4 water channel that is polarized to the ACef pm (pseudo-colored green outlined by yellow line) that undergoes a loss of ACef polarization to become maldistributed in the cytosol of ACef in panel E. Panel E depicts the maldistribution of AQP4 IHC staining (pseud-colored red) and note the increased condensation as compared to more flocculent IHC staining panel D. Representative illustrations in panels A D, and E were created in Microsoft 3-D paint program. BM = basement membrane; CL = capillary lumen; DAPI = 4′,6-diamidino-2-phenylindole fluorescent stain; EC/Pc = endothelial cell/pericyte; H20 = water; pm = plasma membrane.
Figure 13.
Representative illustrations of water channel aquaporin 4 (AQP4), and representative immunohistochemistry (IHC) staining of thalamus AQP4 in models. Panel A demonstrates an illustration of the AQP4 water channel localized to the astrocyte end-feet (ACef) at the plasma membrane that extends to the enlarged perivascular spaces (EPVS). Panel B illustrates an elongated capillary in the region of the thalamus in a control model and note the mild staining of the AQP4 via IHC staining with the AQP4 antibody. Panel C depicts marked heavily stained AQP4 water channels with IHC AQP4 antibody staining in a model with acetaminophen-induced liver failure and hepatic encephalopathy. AQP4 is known to be upregulated in aging and in ammonia-induced brain injury; however, it becomes maldistributed and there is a loss of plasma membrane (pm) polarity associated with its maldistribution and dysfunction. Panel D is a representative illustration of the AQP4 water channel that is polarized to the ACef pm (pseudo-colored green outlined by yellow line) that undergoes a loss of ACef polarization to become maldistributed in the cytosol of ACef in panel E. Panel E depicts the maldistribution of AQP4 IHC staining (pseud-colored red) and note the increased condensation as compared to more flocculent IHC staining panel D. Representative illustrations in panels A D, and E were created in Microsoft 3-D paint program. BM = basement membrane; CL = capillary lumen; DAPI = 4′,6-diamidino-2-phenylindole fluorescent stain; EC/Pc = endothelial cell/pericyte; H20 = water; pm = plasma membrane.

Figure 14.
Magnetic resonance imaging (MRI) comparisons of basal ganglia (BG) enlarged perivascular spaces (EPVS) to centrum semiovale (CSO) EPVS. Panel A depicts the paired EPVS within the BG that are traced in open on the left and masked yellow on the right BG. Note the white spaces within the paired dashed lines just above the paired BG structures. Panel B depicts the paired elongated oval structures outlined by yellow dashed lines to enclose multiple white enlarged perivascular spaces. Note the open white arrows outlined in red pointing to prominent EPVS. Importantly, note that BG EPVS are strongly associated with cerebral small vessel disease (SVD) in panel A and that CSO EPVS are strongly associated with late-onset Alzheimer’s disease and cerebral amyloid angiopathy (CAA) in panel B.
Figure 14.
Magnetic resonance imaging (MRI) comparisons of basal ganglia (BG) enlarged perivascular spaces (EPVS) to centrum semiovale (CSO) EPVS. Panel A depicts the paired EPVS within the BG that are traced in open on the left and masked yellow on the right BG. Note the white spaces within the paired dashed lines just above the paired BG structures. Panel B depicts the paired elongated oval structures outlined by yellow dashed lines to enclose multiple white enlarged perivascular spaces. Note the open white arrows outlined in red pointing to prominent EPVS. Importantly, note that BG EPVS are strongly associated with cerebral small vessel disease (SVD) in panel A and that CSO EPVS are strongly associated with late-onset Alzheimer’s disease and cerebral amyloid angiopathy (CAA) in panel B.
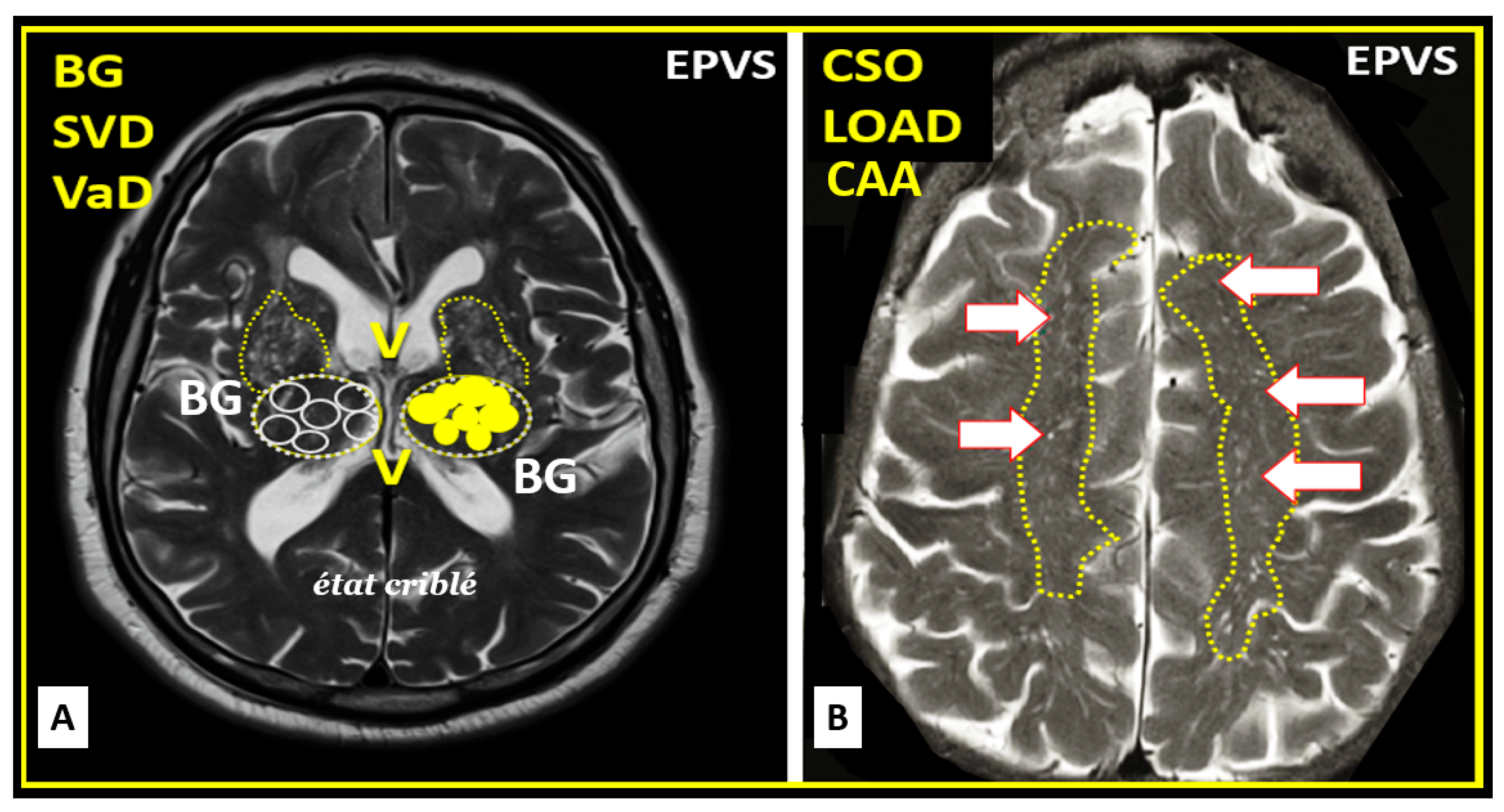
Figure 15.
Brain injury and the response to injury wound healing (RTIWH) facilitates brain endothelial cell (BEC) activation, neuroinflammation, and enlarged perivascular spaces (EPVS). Panel A illustrates the bidirectional relationship between perivascular spaces (PVS) and neuroinflammation in relation to EPVS. Panel B demonstrates the contribution and mechanisms to the development of EPVS. The brain is exposed to many injurious stimuli over time and in multiple diseases and syndromes such as obesity, metabolic syndrome, and type 2 diabetes mellitus. These injurious stimuli result in BEC activation and dysfunction (BECact/dys), which acts as a pivotal point in the RTIWH and these mechanisms also contribute to multiple wound healing phases (hemostasis, inflammation-neuroinflammation, proliferation, remodeling, and resolution only if the injurious stimuli are damped down or removed. Since these injurious stimuli are chronic, the brain remains under the constant influence of the wound healing phases and chronic remodeling of essential brain structures and the development of EPVS. Herein we propose a sequence of events in the development of EPVS as follows: Brain injury, BECact/dys, neuroinflammation with RTIWH, obstruction of PVS due to accumulation of inflammatory cell leukocytes within and around the neurovascular unit and postcapillary venular PVS and inflammatory-derived cellular debris, vascular stiffening with deceased pulsatility, and neuronal degeneration with neuronal loss and brain atrophy.
Figure 15.
Brain injury and the response to injury wound healing (RTIWH) facilitates brain endothelial cell (BEC) activation, neuroinflammation, and enlarged perivascular spaces (EPVS). Panel A illustrates the bidirectional relationship between perivascular spaces (PVS) and neuroinflammation in relation to EPVS. Panel B demonstrates the contribution and mechanisms to the development of EPVS. The brain is exposed to many injurious stimuli over time and in multiple diseases and syndromes such as obesity, metabolic syndrome, and type 2 diabetes mellitus. These injurious stimuli result in BEC activation and dysfunction (BECact/dys), which acts as a pivotal point in the RTIWH and these mechanisms also contribute to multiple wound healing phases (hemostasis, inflammation-neuroinflammation, proliferation, remodeling, and resolution only if the injurious stimuli are damped down or removed. Since these injurious stimuli are chronic, the brain remains under the constant influence of the wound healing phases and chronic remodeling of essential brain structures and the development of EPVS. Herein we propose a sequence of events in the development of EPVS as follows: Brain injury, BECact/dys, neuroinflammation with RTIWH, obstruction of PVS due to accumulation of inflammatory cell leukocytes within and around the neurovascular unit and postcapillary venular PVS and inflammatory-derived cellular debris, vascular stiffening with deceased pulsatility, and neuronal degeneration with neuronal loss and brain atrophy.
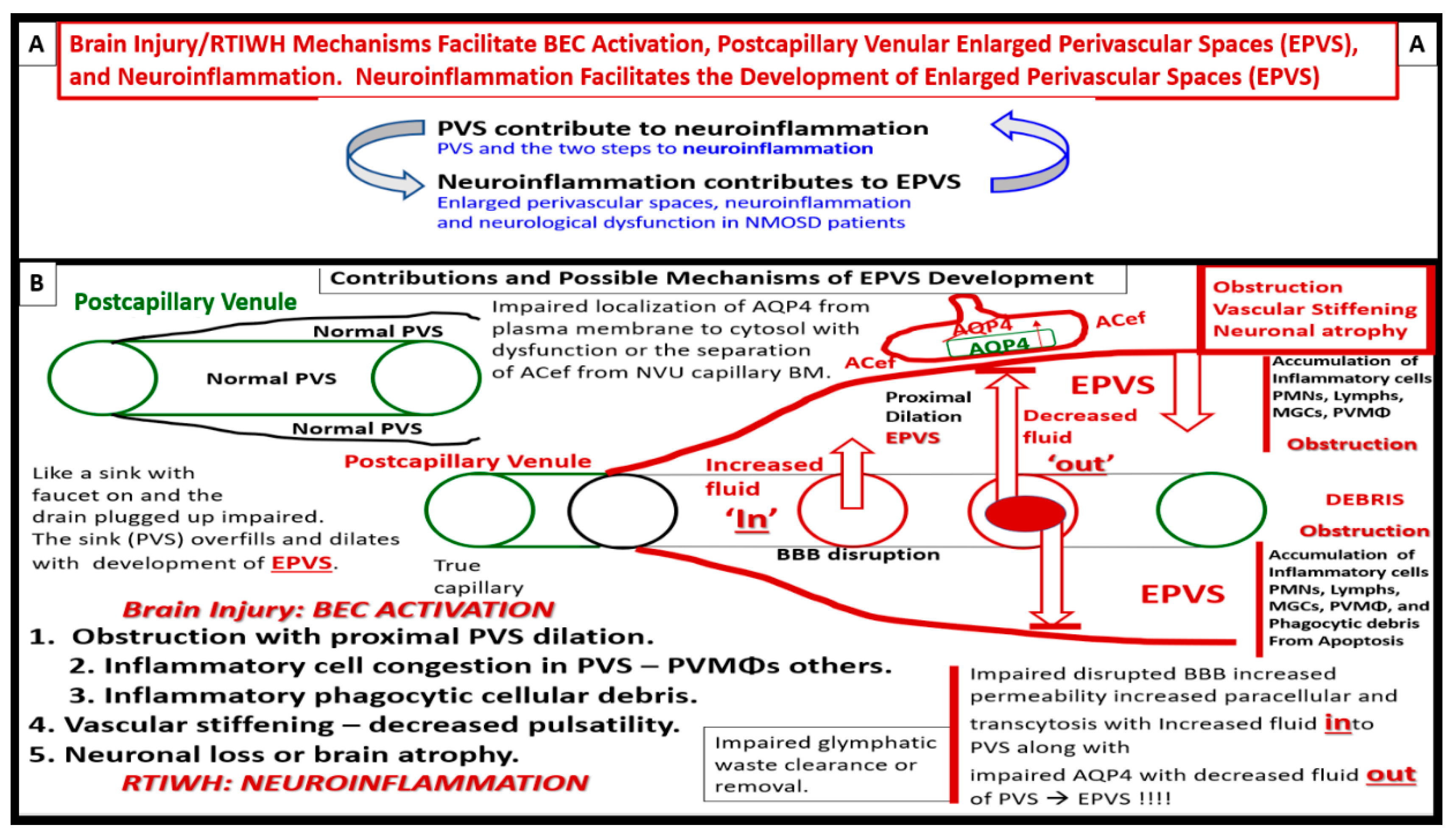
Figure 16.
Interstitial spaces (ISS) are nanometer structures; however, they combine to create a total of 20 percent of total brain volume and empty their interstitial fluid contents and metabolic wastes into the perivascular conduit spaces of the glymphatic system (GS) to be delivered to the cerebrospinal fluid (CSF). Panel A demonstrates the nanosized diameter of ISS containing the ISF in a control 20-week-old mouse model from the cortical grey matter. Panel B depicts how the ISS containing the ISF empties into the post-capillary venule perivascular space (PVS) or the conduit of the GS, which is responsible for toxic waste clearance. AC = astrocyte; ACef = astrocyte endfeet; EC = endothelial cell; •= solutes.
Figure 16.
Interstitial spaces (ISS) are nanometer structures; however, they combine to create a total of 20 percent of total brain volume and empty their interstitial fluid contents and metabolic wastes into the perivascular conduit spaces of the glymphatic system (GS) to be delivered to the cerebrospinal fluid (CSF). Panel A demonstrates the nanosized diameter of ISS containing the ISF in a control 20-week-old mouse model from the cortical grey matter. Panel B depicts how the ISS containing the ISF empties into the post-capillary venule perivascular space (PVS) or the conduit of the GS, which is responsible for toxic waste clearance. AC = astrocyte; ACef = astrocyte endfeet; EC = endothelial cell; •= solutes.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



