
Submitted:
17 June 2023
Posted:
19 June 2023
You are already at the latest version
Abstract
Frontotemporal dementia and/or amyotrophic lateral sclerosis type 7 (FTD/ALS7) is an autosomal dominant neurodegenerative disorder characterized by the onset of ALS and/or FTD mainly in adulthood. Patients with some types of mutations, including the Thr104Asn (T104N) mutation of charged multivesicular body protein 2B (CHMP2B), have predominantly ALS phenotypes, whereas patients with other mutations have predominantly FTD phenotypes. A few patients with further other mutations have both phenotypes approximately equally; however, the reason why phenotypes differ depending on the position of the mutation is unknown. CHMP2B composes one part of the endosomal sorting complexes required for transport (ESCRT), specifically ESCRT-III, in the cytoplasm. We describe here, for the first time, that CHMP2B with the T104N mutation inhibits neuronal process elongation in the N1E-115 cell line, a model of neuronal differentiation. The inhibitory phenotype was accompanied by changes in marker protein expression. It is noteworthy that CHMP2B with the T104N mutation but not its wild-type was preferentially accumulated in the Golgi body. Of the four major Golgi stress signaling pathways currently known, the pathway through Arf4, as the small GTPase, was specifically upregulated in cells expressing CHMP2B with the T104N mutation. Conversely, knockdown of Arf4 with the cognate small interfering (si)RNA recovered the neuronal process elongation inhibited by the T104N mutation. These results suggest that the T104N mutation of CHMP2B inhibits neuronal morphological differentiation by triggering Golgi stress signaling, revealing a possible therapeutic molecular target for recovering potential molecular and cellular phenotypes underlying FTD/ALS7.
Keywords:
CHMP2B
; N1E-115 cell
; neuronal differentiation
; Arf4
; Golgi stress.
# These authors equally contributed to this study
1. Introduction
During development of the central nervous system, neuronal cells undergo continuous and dynamic morphogenesis [1,2,3,4,5,6]. Morphogenesis involves neurite outgrowth and elongation, neuronal process navigation, and synapse formation to form neuronal networks [1,2,3,4,5,6]. Neurite outgrowth and elongation are the initial steps in establishing neuronal networks; however, the whole molecular mechanism underlying a variety of neuronal cell morphological differentiation steps, including outgrowth and elongation of neuronal processes, is not yet fully understood [1,2,3,4,5,6]. In neurological diseases, neuronal cell morphogenesis can be affected at the initial steps as well as at the various other stages [1,2,3,4,5,6]
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are neurodegenerative diseases that overlap in clinical, genetic, and pathological presentation [7,8]. Therefore, FTD and ALS are now considered a single spectrum disorder. It is thought that one of the common molecular and cellular FTD/ALS pathological mechanisms is impaired proteostasis, especially for abnormal and/or inhibitory trafficking coupling endosome to lysosome [7,8]. One of the protein components that constitutes these proteostasis pathways is believed to be charged multivesicular body protein 2B (CHMP2B) [7,8,9,10]. CHMP2B is a core component of the endosomal sorting complex required for transport (ESCRT) machinery, and it coordinates the scission of intracellular membranes [7,8,9,10]. The ESCRT machinery recognizes ubiquitinated proteins at the membrane surface of endosomes or multivesicular bodies (MVBs) to couple endosomes and MVBs to lysosomes [7,8,9,10]. As expected, dysfunction of CHMP2B (e.g., due to various amino acid mutations) can result in moderate to severe neurological diseases, including the FTD/ALS spectrum [7,8,9,10].
FTD/ALS type 7 is an autosomal dominant neurodegenerative disorder characterized by onset of FTD and/or ALS primarily in adulthood, and it is caused by various positions of mutations in CHMP2B [7,8,11]. Patients with amino acid mutations including the Thr104Asn (T104N) mutation of CHMP2B have predominantly ALS phenotypes whereas other patients with other mutations have predominantly FTD phenotypes [7,8,11]. A few patients with further other mutations have both phenotypes [7,8,11]; however, the reason why phenotypes differ depending on the position of the mutation is unknown. Despite the relationship between mutations in CHMP2B and diseases, it is unclear whether and how a mutation in CHMP2B affects neuronal cells. In the present study, we describe, for the first time, that CHMP2B with the T104N mutation [12] greatly inhibits process elongation in the N1E-115 cell line, a widely used model of neuronal differentiation [13,14]. Cells expressing the T104N mutant protein caused Golgi stress, whereas a decrease in Golgi stress recovered the ability of cells to elongate processes, providing evidence of a potential pathological molecular and cellular mechanism underlying FTD/ALS7.
2. Materials and methods
2.1. Antibodies and siRNA sequences
Key antibodies used and plasmids generated in this study are listed in Table 1. Sequences of 19-mer siRNAs with dTdT (Fasmac, Kanagawa, Japan) and DNA primers (Fasmac) are described in Figure S2.
2.2. Reverse transcription-polymerase chain reaction (RT-PCR)
The cDNAs were prepared from total RNA extracted using Isogen (Nippon Gene, Tokyo, Japan) with the PrimeScript RT Master Mix kit (Takara Bio, Kyoto, Japan) in accordance with the manufacturer’s instructions. PCR amplification from reverse transcription products was performed using Gflex DNA polymerase (Takara Bio) with 30 to 36 cycles, each consisting of a denaturation reaction at 98°C (0.2 min), an annealing reaction at 56 to 65°C (0.25 min) depending on the annealing temperature, and an extension reaction at 68°C (0.5 min). The resultant PCR products were loaded onto 1% to 2% agarose gels (Nacalai Tesque, Kyoto, Japan).
2.3. Cell line culture and differentiation
Mouse neuronal N1E-115 and green monkey kidney epithelial COS-7 cells (JCRB Cell Bank/Japan Health Sciences Foundation, Osaka, Japan) were cultured on 6- or 10-cm cell culture dishes (Nunc/ThermoFisher Scientific, Waltham, MA, USA) in high-glucose Dulbecco’s modified Eagle medium (DMEM; Nacalai Tesque) containing 10% heat-inactivated fetal bovine serum (FBS) (ThermoFisher Scientific) and penicillin-streptomycin (ThermoFisher Scientific) in 5% CO2 at 37°C. COS-7 and N1E-115 cells are known to have high transfection efficiency and neuronal differentiation ability, respectively. Cell lines stably expressing the wild-type (indicated as WT in the figure) chmp2b gene or the gene with the T104N mutation were selected in the presence of G418 (Nacalai Tesque) as described previously [15] and isolated as a single clone. To induce differentiation, N1E-115 cells were cultured in DMEM and 1% FBS containing penicillin-streptomycin in 5% CO2 at 37°C for 48 hours. Cells with processes more than one cell body in length were considered to be process-bearing cells (i.e., differentiated cells) [16]. Under these conditions, attached cells incorporating trypan blue (Nacalai Tesque) were estimated to be less than 5% in each experiment.
2.4. siRNA transfection
Mouse neuronal N1E-115 cells were transfected with the respective synthesized 21-mer small interfering (si)RNAs with dTdT using the ScreenFect siRNA transfection kit (Fujifilm, Tokyo, Japan) in accordance with the manufacturer’s instructions. The medium was replaced 4 hours after transfection and was generally used for 48 hours after transfection for cell biological and biochemical experiments. Under these conditions, attached cells incorporating trypan blue were estimated to be less than 5% in each experiment.
2.5. Denatured polyacrylamide electrophoresis and immunoblotting
Cells were lysed in lysis buffer (50 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, 3 mM MgCl2, 1 mM dithiothreitol, 1 mM phenylmethane sulfonylfluoride, 1 μg/mL leupeptin, 1 mM EDTA, 1 mM Na3VO4, 10 mM NaF, and 0.5% NP-40). For normal denatured conditions, cell lysates were denatured in sample buffers (Fujifilm), and samples were separated on a sodium dodecylsulfate polyacrylamide gel (Nacalai Tesque). The electrophoretically separated proteins were transferred to a polyvinylidene fluoride membrane (Fujifilm), blocked with Blocking One (Nacalai Tesque), and immunoblotted using primary antibodies, followed by peroxidase enzyme-conjugated secondary antibodies. Peroxidase-reactive bands were captured using an image scanner (Canon, Tokyo, Japan) and scanned using CanoScan software (Canon). The blots shown in the figures are representative of 3 blots. We performed some sets of experiments in immunoblotting studies and quantified other immunoreactive bands with one control’s immunoreactive band as 100% using Image J software (https://imagej.nih.gov/).
2.6. Fluorescence images
Cells on coverslips were fixed with 4% paraformaldehyde (Nacalai Tesque) or 100% cold methanol (Nacalai Tesque) and blocked with Blocking One. Slides were incubated with primary antibodies and then with Alexa Fluor fluorescent-conjugated secondary antibodies. The coverslips were mounted using the Vectashield kit (Vector Laboratories, Burlingame, CA, USA). The fluorescent images were collected and merged with microscope systems FV1200 or FV3000 equipped with a laser-scanning Fluoview apparatus and software (both from Olympus, Tokyo, Japan). The images in the figures are the representative of 3 images and were analyzed using Image J software.
2.7. Statistical analyses
Values are shown as means ± standard deviation (SD) of separate experiments. Intergroup comparisons were made using unpaired Student’s t-test in Excel (Microsoft, Redmond, WA, USA). A one-way analysis of variance (ANOVA) was followed by a Tukey’s multiple comparison test using Graph Pad Prism (GraphPad Software, San Diego, CA, USA). Differences were considered statistically significant when p<0.05.
2.8. Ethics statement
Techniques using genetically modified cells and related techniques were performed in accordance with a protocol approved by the Tokyo University of Pharmacy and Life Sciences Gene and Animal Care Committee (Approval Nos. LS28-20 and LSR3-011).
3. Results
3.1. Wild-type CHMP2B is contained in MVB-like structures whereas T104N-mutant CHMP2B forms aggregate-like structures
First, we confirmed that wild type (WT) CHMP2B protein is contained in MVB-like circular structures in COS-7 cells. Because COS-7 cells have a wide cytoplasmic region and are suitable for observing protein localization, we transfected plasmids carrying the WT CHMP2B or CHMP2B with the T104N mutation into COS-7 cells. The WT CHMP2B protein was observed in the MVB-like structure (Figure 1A), whereas CHMP2B protein with the T104N mutation was primarily distributed in aggregate-like structures in the cytoplasm (Figure 1B), suggesting that the T104N mutation makes CHMP2B protein form aggregates.
Next, to determine which organelles are colocalized with aggregate-like structures of CHMP2B with the T104N mutation, we stained cells with different organelle markers. Mutated CHMP2B was colocalized with 130 kDa Golgi membrane protein (GM130) as the Golgi body marker, whereas WT CHMP2B was not (Figure 2). The endoplasmic reticulum (ER) marker Lys-Asp-Glu-Leu (KDEL) and lysosome-specific antigen cathepsin D were not significantly colocalized with either WT CHMP2B or with the mutant’s aggregate-like structures. Compared to the localization profiles of CHMP2B with the T104N mutation in organelles, CHMP2B with mutations D148Y and Q165X (associated with the predominantly FTD phenotype) seemed unlikely to be localized in the Golgi body or in ER and lysosome (see Figure S1), suggesting specific localization of CHMP2B with the T104N mutation (associated with the predominantly ALS phenotype) in the Golgi body.
3.2. CHMP2B with the T104N mutation inhibits neuronal morphological differentiation
We explored whether CHMP2B with the T104N mutation affected neuronal morphological changes in N1E-115 cells, a differentiation model often used to study process elongation [15,16]. Cells with WT CHMP2B demonstrated process elongation. In contrast, cells harboring CHMP2B with the T104N mutation failed to undergo sufficient process elongation (Figure 3, A and B) and showed decreased expression of neuronal marker growth-associated protein 43 (GAP43) (Figure 3, C and D).
3.3. Attenuating Golgi stress recovers an inhibitory morphological differentiation phenotype
Because mutated CHMP2B protein, but not WT protein, was localized in the Golgi body, we wanted to determine which pathway related to Golgi stress was responsible for expression of the mutated protein. Golgi stress is composed of the pathways mediated by heat shock protein 47 (HSP47); transcription factor E3 (TFE3) and possible downstream targets such as structural proteins composing the Golgi body including GM130; cAMP response element binding protein (CREB) 3 and downstream Arf4; and caspase-2 [17,18,19,20,21,22,23,24]. Expression of mutated proteins led to increased levels of GM130 and Arf4 (Figure 4, A and B), indicating that these pathways may be related to Golgi stress response. In contrast, the amounts of cleaved (active) caspase-2 were decreased following expression of the mutated protein. It is possible that GM130 is a structural protein of the Golgi body and that its decreased expression could be associated with sustaining Golgi organelle structure. Because Arf4 is classified as a signal transducer molecule, its knockdown might affect Golgi stress response, more directly. As expected, knockdown of Arf4 (Figure S2) recovered the mutant-induced inhibition of process elongation (Figure 5, A and B). GAP43 expression levels were similarly recovered (Figure 5, C and D). Thus, mutated CHMP2B proteins trigger Golgi stress, at least in part through Arf4, and knockdown of Arf4 can ameliorate the mutant-induced inhibition of morphological differentiation.
4. Discussion
FTD and ALS are neurodegenerative diseases with overlapping symptoms and causes; as such, they are commonly considered a single spectrum disorder. Abnormalities in the homeostasis of biomaterials involving dysfunctional protein clearance, impaired RNA metabolism, and aberrant formation of complexes of proteins with RNA are emerging as key events underlying FTD/ALS pathogenesis. It is likely that these processes, including protein and nucleotide clearance, interact with each other at the molecular level, converging on a common molecular pathogenic pathway [25,26,27,28]. Studies on FTD/ALS7-associated CHMP2B reveal that CHMP2B preferentially participates in protein clearance [11,12]. Recent findings show that CHMP2B regulates casein kinase 1 (CK1) phosphorylation of TAR DNA-binding protein of 43kDa (TDP-43), which binds to RNAs as well as some DNAs [29]. TDP-43 is also involved in FTD/ALS [30]. Detailed analyses of the pathogenic mutations of CHMP2B will allow us to identify the relationship between CHMP2B and RNA metabolism through TDP-43. In the present study, WT CHMP2B typically displayed MVB-like structures in cells; in contrast, the T104N mutant protein exhibited aggregate-like structures; however, it is unknown whether these structures exist in the Golgi body as an aggregate-like complex containing RNA. Additionally, it is unclear whether the structure directly exhibits toxic gain-of-function in cells.
It is more likely that a common pathological molecular mechanism of FTD/ALS spectrum is impaired proteostasis, especially for abnormal trafficking coupling endosome to lysosome. Among protein components that constitute these proteostasis pathways, CHMP2B is a core component of the ESCRT machinery and coordinates scission of intracellular membranes such as endosome membranes. The ESCRT is composed of sequential subcomplexes, ESCRT-0 to ESCRT-III, and facilitates endosome to lysosome transport in almost all cell types, including neuronal cells [9,10]. Before and during the formation of endosomes involving MVB, ESCRT plays a key role in intracellular membrane transport and remodeling, primarily at endosomes [9,10]. The ESCRT-III subcomplex involving CHMP2B shapes their membranes, cooperates with vacuolar protein sorting-associated protein (VPS) 4 as the ATPase, and undergoes fission of the membrane neck from inside the endosome. The ESCRT-0 to -II subcomplexes mainly contribute to the formation of the ESCRT-III subcomplex [9,10]. Therefore, mutations in CHMP2B of the ESCRT-III subcomplex give rise to several neurodegenerative diseases because all ESCRT protein components are required for appropriate morphogenesis and function of neuronal cells [9,10,11,12]. Because CHMP2B protein with the T104N mutation exhibits aggregate-like structures but not MVB-like structures, this mutant protein seems unlikely to be functional within the ESCRT system. If this hypothesis is true, it is conceivable that loss of function of CHMP2B underlies the cellular basis of FTD/ALS pathogenesis.
The Golgi stress pathway is thought to mitigate the effects of specific stresses within cells or to arrest the cell cycle, as seen in the unfolded protein response (UPR) established in the ER. Increasing evidence indicates that Golgi stress is mediated by four major pathways through (1) HSP47, (2) TFE3 and possible downstream targets such as the structural proteins composing the Golgi body, (3) CREB3 and Arf4, and (4) caspase-2 [17,18,19,20,21,22,23,24]. First, HSP47 is an ER chaperone and it is likely that HSP47 is also localized in the Golgi body. HSP47 is thought to protect the Golgi body from various stresses [17,18,19,20,21,22,23,24], and expression levels of HSP47 were comparable in cells expressing WT and mutant CHMP2B. Second, TFE3 is an essential transcription factor controlling the genes that encode Golgi body structural proteins such as GM130, the intracellular vesicle transporting molecules, and Golgi-resident enzymes mediating glycosylation [17,18,19,20,21,22,23,24]. Because GM130 is upregulated in cells expressing mutated CHMP2B, it is thought that its pathway through TFE3 is responsible for Golgi stress induced by mutated CHMP2B proteins. Third, the specific targeting pathway of the transcription factor CREB3 is the small GTPase Arf4 [17,18,19,20,21,22,23,24]. Arf4 is upregulated in cells expressing mutated CHMP2B. The pathway through Arf4 may be responsible for Golgi stress induced by mutated CHMP2B proteins. Fourth, active caspase-2 mainly plays a non-apoptotic role, lacking the ability to activate effector caspases such as caspase-3. Procaspase-2 is also present in the cytoplasmic surface of the Golgi body. The prodomain of procaspase-2 is cleaved to generate active caspase-2, probably suppressing the functions of the Golgi body by cleaving proteins such as golgin-160 [17,18,19,20,21,22,23,24]; however, cleaved active caspase-2 results in a decrease in cells expressing mutated CHMP2B, indicating that the pathway through caspase-2 is not involved in Golgi stress in cells expressing mutated CHMP2B. Thus, we focused only on the pathway through CREB3 and Arf4, because the pathway through TFE3 and molecules such as GM130 significantly contribute homeostasis of the Golgi body [31]. As expected, knockdown of Arf4, possibly acting through decreasing Golgi stress, recovered morphological differentiation with process elongation in cells expressing mutated CHMP2B.
Arf4 is a small GTPase and is thought to participate in intracellular vesicle trafficking with other Arf-family small GTPases. The Arf family of small GTPases are composed of class I (Arf1 and Arf2 and/or Arf3), class II (Arf4 and Arf5), and class III (Arf6). Class I GTPases such as Arf1 regulate the vesicle transporting system around the Golgi body, and the class III GTPase Atf6 primarily controls vesicle transporting around the intracellular surface of the plasma membrane. In contrast, the precise role of the class II GTPase Arf4 remains unclear [32,33]. For example, Ezratty et al. reported that Arf4 regulates polarized exocytosis to act through the complex between the basal body and the ciliary body. It is thus likely that Arf4 regulates Notch signaling in epidermal morphological differentiation [34]. Wang et al. reported that Arf4 and the guanine-nucleotide exchange factor GBF1 synergize with the sensory receptor cargo to regulate trafficking of the ciliary membrane [35]. It is thought that the activities of Arf4 are precisely associated with the formation of the ciliary membrane [36,37]. It will be important to determine which organelles in the cognate intracellular membrane transporting are mediated by Arf4 and how this is achieved. In addition, it remains unclear how signaling around Arf4 is related to the response to Golgi stress.
The question of why and how CHMP2B is involved in promoting process elongation remains to be answered. In the initial steps of neuronal morphological differentiation, cells undergo dynamic morphogenesis such as neurite outgrowth and elongation. Dynamic morphogenesis requires synthesis of many membrane lipids and proteins to achieve neurite outgrowth and elongation. Therefore, during cell development, a quality control step of protein is required; that is, proteostasis. It is possible that CHMP2B, as the ESCRT-III subcomplex component, and other ESCRT components directly or indirectly monitor fine-tuned neurite outgrowth and elongation. Here we show that FTD/ALS7-associated mutation of CHMP2B inhibits neuronal morphological differentiation. Mutated CHMP2B is specifically localized in the Golgi body to trigger Golgi stress. In contrast, knockdown of Arf4, a Golgi stress mediator, recovers the ability of cells to differentiate. Further studies on the relationship of mutated CHMP2B with Golgi stress are needed to increase our understanding of the detailed mechanisms by which the FTD/ALS7-associated mutation of CHMP2B inhibits neuronal morphological differentiation using cells and genetically modified mice, as well as of a possible causal relationship between inhibitory differentiation and the early stages of neurodegeneration in FTD/ALS7. Additional studies will allow us to elucidate the role of Golgi stress as a potential molecular and cellular pathological mechanism underlying FTD/ALS7 and might lead to the development of therapeutic target–specific drug candidates for FTD/ALS7 and other types of FTD/ALS.
Author Contributions
Junji Yamauchi designed this study. Junji Yamauchi wrote and edited this manuscript. Remina Shirai, Mizuho Cho, Mikinori Isogai, Shoya Fukatsu, Miyu Okabe, Maho Okawa, and Tomohiro Torii performed experiments and statistical analyses. Yuki Miyamoto and Tomohiro Torii evaluated experimental and statistical data.
Funding
This work was supported by Grants-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) and Grants-in-Aid for Medical Scientific Research from the Japanese Ministry of Health, Labor and Welfare (MHLW). This work was also supported by Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Agency (JST), Takeda Science Foundation, Daiichi Sankyo Science Foundation, and Mitsubishi Tanabe Science Foundation.
Acknowledgments
We thank Drs. Takako Morimoto and Yoichi Seki (Tokyo University of Pharmacy and Life Sciences) for the insightful comments they provided throughout this study.
Conflicts of Interest
The authors have declared that no competing interests exist.
References
- Craig, A. M. , Banker, G. Neuronal polarity. Annu. Rev. Neurosci. 1994, 17, 267–310. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.S.C.G. Dotti. Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 2002, 3, 694–704. [Google Scholar] [CrossRef]
- Arimura, N.K.; Kaibuchi. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 2007, 8, 194–205. [Google Scholar] [CrossRef]
- Park, H.; Poo, M.-M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Bray, D. Surface Movements during the Growth of Single Explanted Neurons. Proc. Natl. Acad. Sci. USA 1970, 65, 905–910. [Google Scholar] [CrossRef]
- Rigby, M.J.; Gomez, T.M.; Puglielli, L. Glial Cell-Axonal Growth Cone Interactions in Neurodevelopment and Regeneration. Front. Neurosci. 2020, 14, 203. [Google Scholar] [CrossRef] [PubMed]
- Ugbode, C.; West, R.J.H. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 147, 105144. [Google Scholar] [CrossRef]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef]
- Henne, W. M. , Buchkovich, N. J., Emr, S. D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef]
- Sadoul, R.; Laporte, M.H.; Chassefeyre, R.; Chi, K.I.; Goldberg, Y.; Chatellard, C.; Hemming, F.J.; Fraboulet, S. The role of ESCRT during development and functioning of the nervous system. Semin. Cell Dev. Biol. 2018, 74, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in Lower Motor Neuron Predominant Amyotrophic Lateral Sclerosis (ALS). PLOS ONE 2010, 5, e9872. [Google Scholar] [CrossRef]
- Hirose, M.; Ishizaki, T.; Watanabe, N.; Uehata, M.; Kranenburg, O.; Moolenaar, W.H.; Matsumura, F.; Maekawa, M.; Bito, H.; Narumiya, S. Molecular Dissection of the Rho-associated Protein Kinase (p160ROCK)-regulated Neurite Remodeling in Neuroblastoma N1E-115 Cells. J. Cell Biol. 1998, 141, 1625–1636. [Google Scholar] [CrossRef]
- Memezawa, S.; Sato, T.; Ochiai, A.; Fukawa, M.; Sawaguchi, S.; Sango, K.; Miyamoto, Y.; Yamauchi, J. The Antiepileptic Valproic Acid Ameliorates Charcot-Marie-Tooth 2W (CMT2W) Disease-Associated HARS1 Mutation-Induced Inhibition of Neuronal Cell Morphological Differentiation Through c-Jun N-terminal Kinase. Neurochem. Res. 2022, 47, 2684–2702. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, N.; Miyamoto, Y.; Hattori, K.; Ito, A.; Harada, H.; Oizumi, H.; Ohbuchi, K.; Mizoguchi, K.; Yamauchi, J. PP1C and PP2A are p70S6K Phosphatases Whose Inhibition Ameliorates HLD12-Associated Inhibition of Oligodendroglial Cell Morphological Differentiation. Biomedicines 2020, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Yamauchi, J.; Sanbe, A.; Tanoue, A. Dock6, a Dock-C subfamily guanine nucleotide exchanger, has the dual specificity for Rac1 and Cdc42 and regulates neurite outgrowth. Exp. Cell Res. 2007, 313, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Machamer, C.M. The Golgi complex in stress and death. Front. Neurosci. 2015, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Yoshida, H. TFE3, HSP47, and CREB3 Pathways of the Mammalian Golgi Stress Response. Cell Struct. Funct. 2017, 42, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Yoshida, H. Golgi stress response and organelle zones. FEBS Lett. 2019, 593, 2330–2340. [Google Scholar] [CrossRef]
- Olsson, M.; Forsberg, J.; Zhivotovsky, B. Caspase-2: the reinvented enzyme. Oncogene 2015, 34, 1877–1882. [Google Scholar] [CrossRef]
- Reiling, J.H.; Olive, A.J.; Sanyal, S.; Carette, J.E.; Brummelkamp, T.R.; Ploegh, H.L.; Starnbach, M.N.; Sabatini, D.M. A CREB3–ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nat. Cell Biol. 2013, 15, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M. , Nadanaka, S., Tanakura, S., Sawaguchi, S., Midori, S., Kawai, Y., Yamaguchi, S., Shimada, Y., Nakamura, Y., Matsumura, Y., Fujita, N., Araki, N., Yamamoto, M., Oku, M., Wakabayashi, S., Kitagawa, H., Yoshida, H. TFE3 is a bHLH-ZIP-type transcription factor that regulates the mammalian Golgi stress response. Cell Struct. Funct. 2015, 40, 13–30. [Google Scholar]
- Miyata, S.; Mizuno, T.; Koyama, Y.; Katayama, T.; Tohyama, M. The Endoplasmic Reticulum-Resident Chaperone Heat Shock Protein 47 Protects the Golgi Apparatus from the Effects of O-Glycosylation Inhibition. PLOS ONE 2013, 8, e69732. [Google Scholar] [CrossRef]
- Kim, W.K.; Choi, W.; Deshar, B.; Kang, S.; Kim, J. Golgi Stress Response: New Insights into the Pathogenesis and Therapeutic Targets of Human Diseases. Mol. Cells 2023, 46, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Garg, N.; Park, S.B.; Vucic, S.; Yiannikas, C.; Spies, J.; Howells, J.; Huynh, W.; Matamala, J.M.; Krishnan, A.V.; Pollard, J.D.; et al. Differentiating lower motor neuron syndromes. J. Neurol. Neurosurg. Psychiatry 2017, 88, 474–483. [Google Scholar] [CrossRef]
- Scott-Solomon, E.; Boehm, E.; Kuruvilla, R. The sympathetic nervous system in development and disease. Nat. Rev. Neurosci. 2021, 22, 685–702. [Google Scholar] [CrossRef]
- Deng, X.; Sun, X.; Yue, W.; Duan, Y.; Hu, R.; Zhang, K.; Ni, J.; Cui, J.; Wang, Q.; Chen, Y.; et al. CHMP2B regulates TDP-43 phosphorylation and cytotoxicity independent of autophagy via CK1. J. Cell Biol. 2022, 221, e202103033. [Google Scholar] [CrossRef] [PubMed]
- Luan, W.; Wright, A.L.; Brown-Wright, H.; Le, S.; Gil, R.S.; Martin, L.M.S.; Ling, K.; Jafar-Nejad, P.; Rigo, F.; Walker, A.K. Early activation of cellular stress and death pathways caused by cytoplasmic TDP-43 in the rNLS8 mouse model of ALS and FTD. Mol. Psychiatry 2023, in press. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Prasad, R.; Jho, E.-H. TFEB regulates pluripotency transcriptional network in mouse embryonic stem cells independent of autophagy–lysosomal biogenesis. Cell Death Dis. 2021, 12, 343. [Google Scholar] [CrossRef]
- Jackson, C.L. Activators and Effectors of the Small G Protein Arf1 in Regulation of Golgi Dynamics During the Cell Division Cycle. Front. Cell Dev. Biol. 2018, 6, 29. [Google Scholar] [CrossRef]
- Miyamoto, Y. , Torii, T., Tago, K., Tanoue, A., Takashima, S. Yamauchi, J. BIG1/Arfgef1 and Arf1 regulate the initiation of myelination by Schwann cells in mice. Sci. Adv. 2018, 4, eaar4471. [Google Scholar] [CrossRef]
- Ezratty, E.J.; Pasolli, H.A.; Fuchs, E. A Presenilin-2–ARF4 trafficking axis modulates Notch signaling during epidermal differentiation. J. Cell Biol. 2016, 214, 89–101. [Google Scholar] [CrossRef]
- Wang, J.; Fresquez, T.; Kandachar, V.; Deretic, D. The Arf GEF GBF1 and Arf4 synergize with the sensory receptor cargo, rhodopsin, to regulate ciliary membrane trafficking. J. Cell Sci. 2017, 130, 3975–3987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, L.; Bai, B.; Li, D.; Xiao, P.; Li, Q.; Zhang, Z.; Wang, H.; Li, L.; Jiang, Q. Quantitative Proteomics Reveals Association of Neuron Projection Development Genes ARF4, KIF5B, and RAB8A With Hirschsprung Disease. Mol. Cell. Proteom. 2021, 20, 100007. [Google Scholar] [CrossRef] [PubMed]
- Pennauer, M.; Buczak, K.; Prescianotto-Baschong, C.; Spiess, M. Shared and specific functions of Arfs 1–5 at the Golgi revealed by systematic knockouts. J. Cell Biol. 2022, 221, e202106100. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Wild-type CHMP2B is present in circular MVB-like structures in cells, whereas CHMP2B with the T104N mutation forms aggregate-like structures. (A, B) COS-7 cells (indicated by white dotted lines) were transfected with the plasmid encoding wild-type (WT) CHMP2B tagged with EGFP at its N-terminus or EGFP-tagged CHMP2B with the T104N mutation. Transfected cells (green) were stained with DAPI to detect nuclear positions (blue). Cells with circular MVB-like structures are statistically depicted in the graph (** p<0.01; n = 10 fields). MVB, multivesicular bodies; WT, wild-type; EGFP, enhanced green fluorescent protein; DAPI, 4′,6-diamidino-2-phenylindole.
Figure 1.
Wild-type CHMP2B is present in circular MVB-like structures in cells, whereas CHMP2B with the T104N mutation forms aggregate-like structures. (A, B) COS-7 cells (indicated by white dotted lines) were transfected with the plasmid encoding wild-type (WT) CHMP2B tagged with EGFP at its N-terminus or EGFP-tagged CHMP2B with the T104N mutation. Transfected cells (green) were stained with DAPI to detect nuclear positions (blue). Cells with circular MVB-like structures are statistically depicted in the graph (** p<0.01; n = 10 fields). MVB, multivesicular bodies; WT, wild-type; EGFP, enhanced green fluorescent protein; DAPI, 4′,6-diamidino-2-phenylindole.
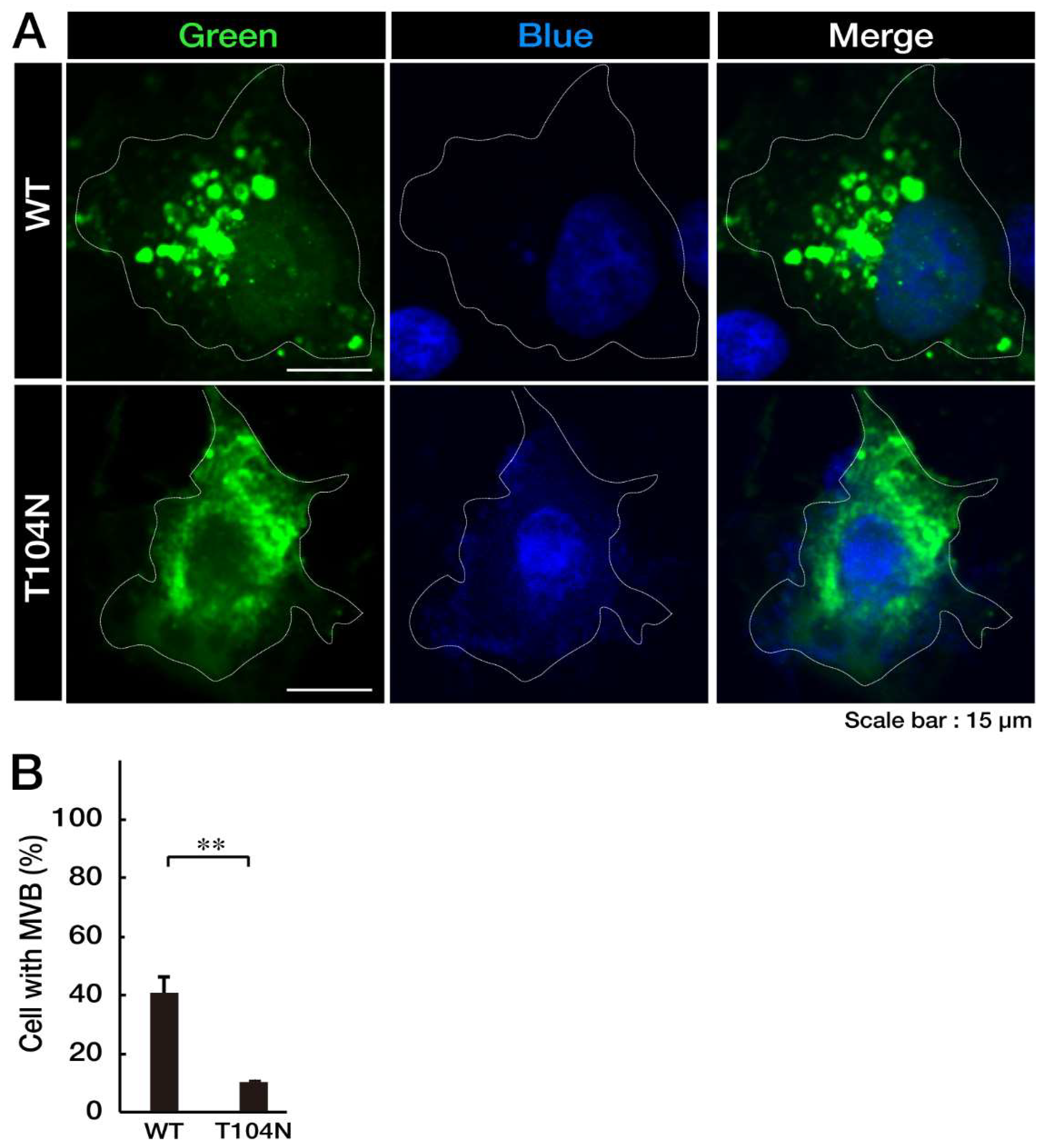
Figure 2.
CHMP2B with the T104N mutation forms aggregate-like structures in the Golgi body. (A, B) COS-7 cells were transfected with the plasmid encoding the GFP-tagged wild-type (WT) or mutated CHMP2B (T104N). Transfected cells (green) were stained with an antibody against the ER-specific antigen KDEL (red), the Golgi body-specific antigen GM130 (red), or the lysosome-specific antigen cathepsin D (CATED, red). The approximate outlines of the cells are shown by white dotted lines. Scan plots were performed along the white lines in the direction of the arrows in the green and red images. Graphs showing fluorescence intensity (F.I.; arbitrary units) along the lines in the direction of the arrows are depicted in the bottom panels. GFP, green fluorescent protein; WT, wild-type; ER, endoplasmic reticulum.
Figure 2.
CHMP2B with the T104N mutation forms aggregate-like structures in the Golgi body. (A, B) COS-7 cells were transfected with the plasmid encoding the GFP-tagged wild-type (WT) or mutated CHMP2B (T104N). Transfected cells (green) were stained with an antibody against the ER-specific antigen KDEL (red), the Golgi body-specific antigen GM130 (red), or the lysosome-specific antigen cathepsin D (CATED, red). The approximate outlines of the cells are shown by white dotted lines. Scan plots were performed along the white lines in the direction of the arrows in the green and red images. Graphs showing fluorescence intensity (F.I.; arbitrary units) along the lines in the direction of the arrows are depicted in the bottom panels. GFP, green fluorescent protein; WT, wild-type; ER, endoplasmic reticulum.
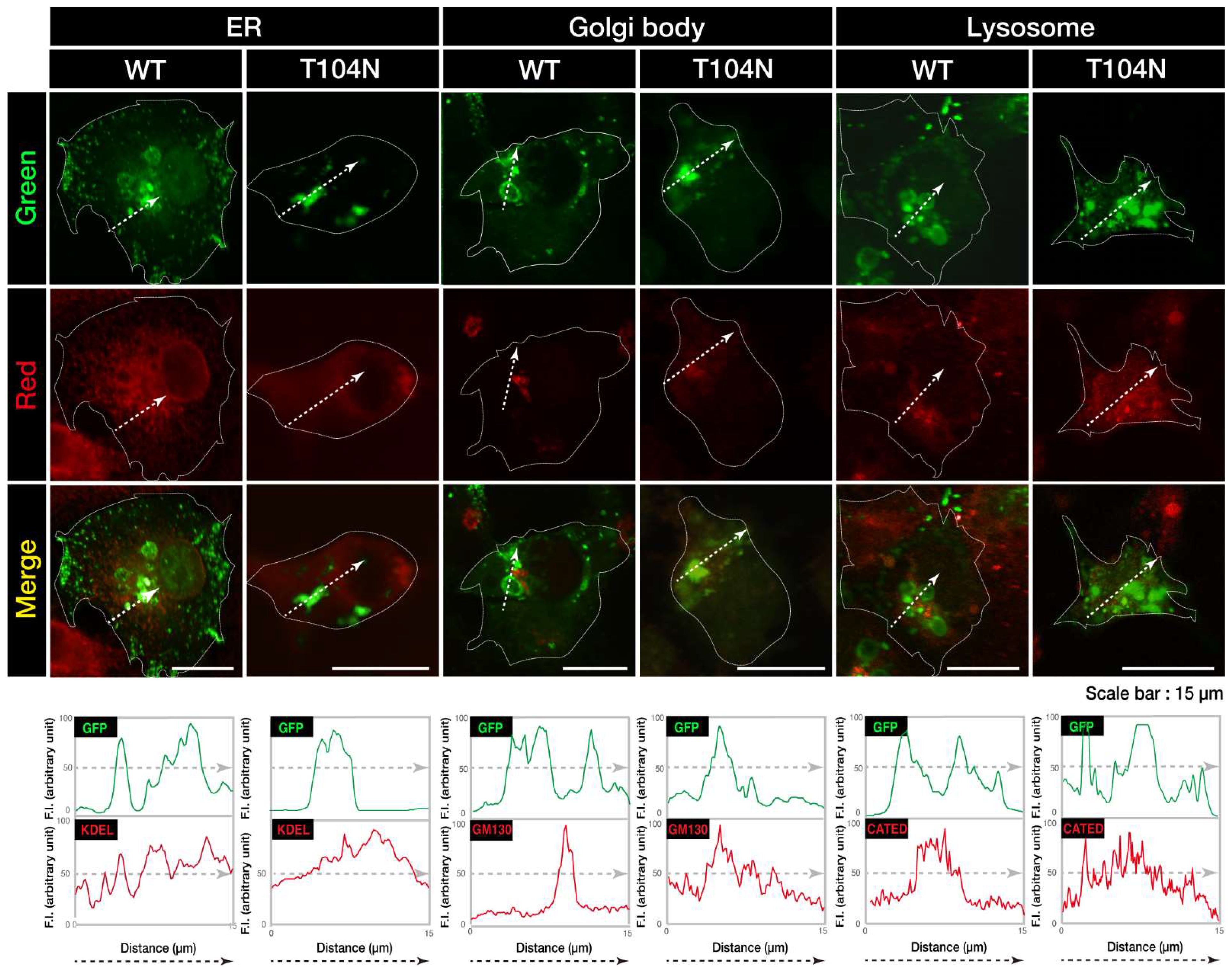
Figure 3.
CHMP2B with the T104N mutation inhibits neuron-like process extension. N1E-115 cells with WT or mutated CHMP2B (T104N) were allowed to differentiate for 48 hours. Typical cell images after 48 hours are shown in (A). (B) Cells with processes with a body length more than one cell were counted as cells with neurites and statistically shown in the graph (** p<0.01; n = 10 fields). (C, D) The lysates of cells following the induction of differentiation (48 hours) were immunoblotted with an antibody against neuron-specific marker GAP43 and actin as the internal marker protein, and their immunoreactive band intensities are statistically depicted (D) (** p<0.01; n = 3 blots). WT, wild-type.
Figure 3.
CHMP2B with the T104N mutation inhibits neuron-like process extension. N1E-115 cells with WT or mutated CHMP2B (T104N) were allowed to differentiate for 48 hours. Typical cell images after 48 hours are shown in (A). (B) Cells with processes with a body length more than one cell were counted as cells with neurites and statistically shown in the graph (** p<0.01; n = 10 fields). (C, D) The lysates of cells following the induction of differentiation (48 hours) were immunoblotted with an antibody against neuron-specific marker GAP43 and actin as the internal marker protein, and their immunoreactive band intensities are statistically depicted (D) (** p<0.01; n = 3 blots). WT, wild-type.
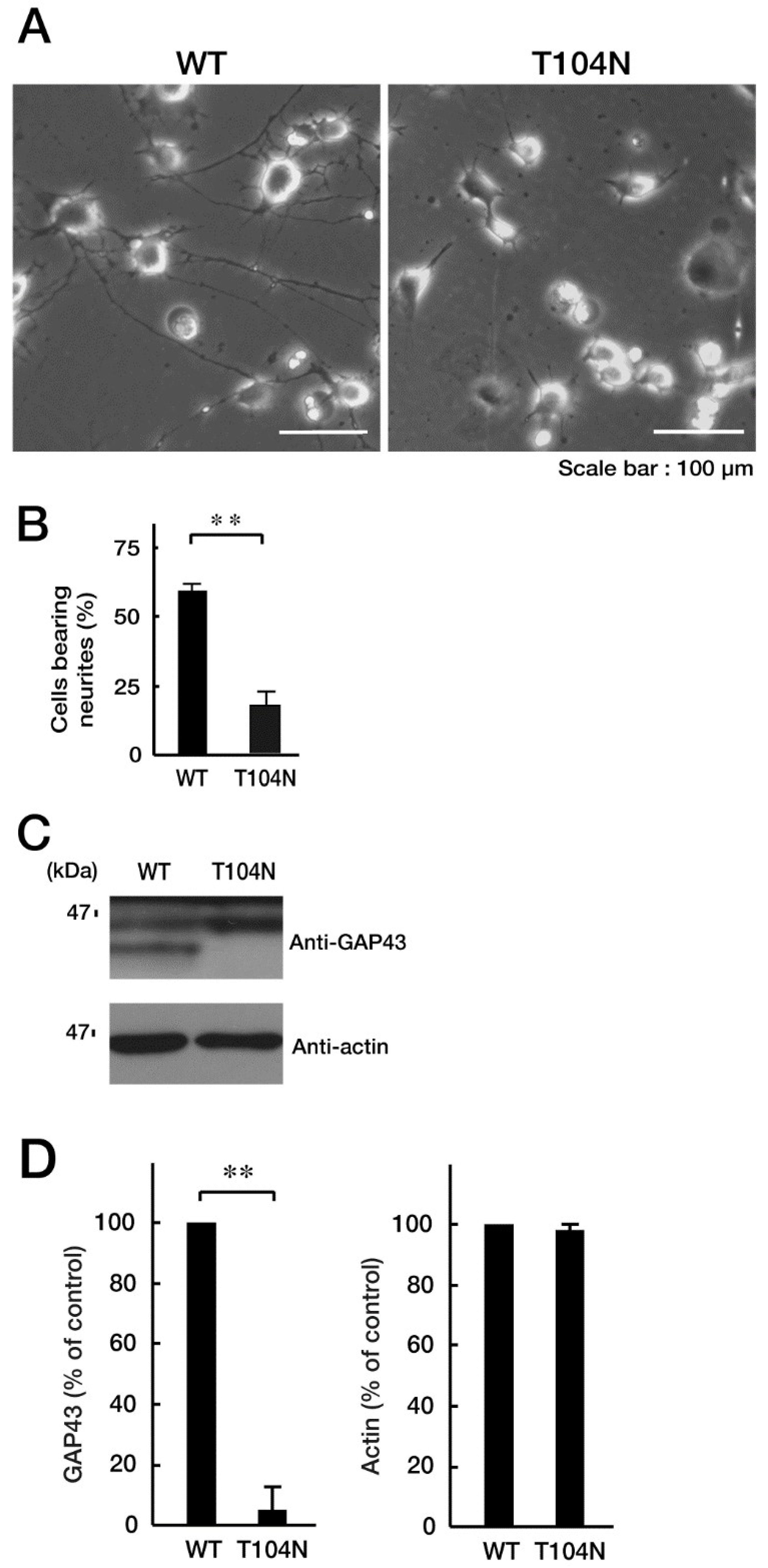
Figure 4.
CHMP2B with the T104N mutation upregulates Golgi stress signal. (A, B) N1E-115 cells with WT or mutated CHMP2B (T104N) were allowed to differentiate for 48 hours and lysed. The lysates were immunoblotted with an antibody against Golgi stress marker Hsp47, GM130, Arf4, cleaved caspase-2 (indicated with **; pro-caspase-2 indicated with *), or control actin, and their immunoreactive band intensities are statistically depicted in the graph (** p<0.01; n = 3 blots). WT, wild-type.
Figure 4.
CHMP2B with the T104N mutation upregulates Golgi stress signal. (A, B) N1E-115 cells with WT or mutated CHMP2B (T104N) were allowed to differentiate for 48 hours and lysed. The lysates were immunoblotted with an antibody against Golgi stress marker Hsp47, GM130, Arf4, cleaved caspase-2 (indicated with **; pro-caspase-2 indicated with *), or control actin, and their immunoreactive band intensities are statistically depicted in the graph (** p<0.01; n = 3 blots). WT, wild-type.
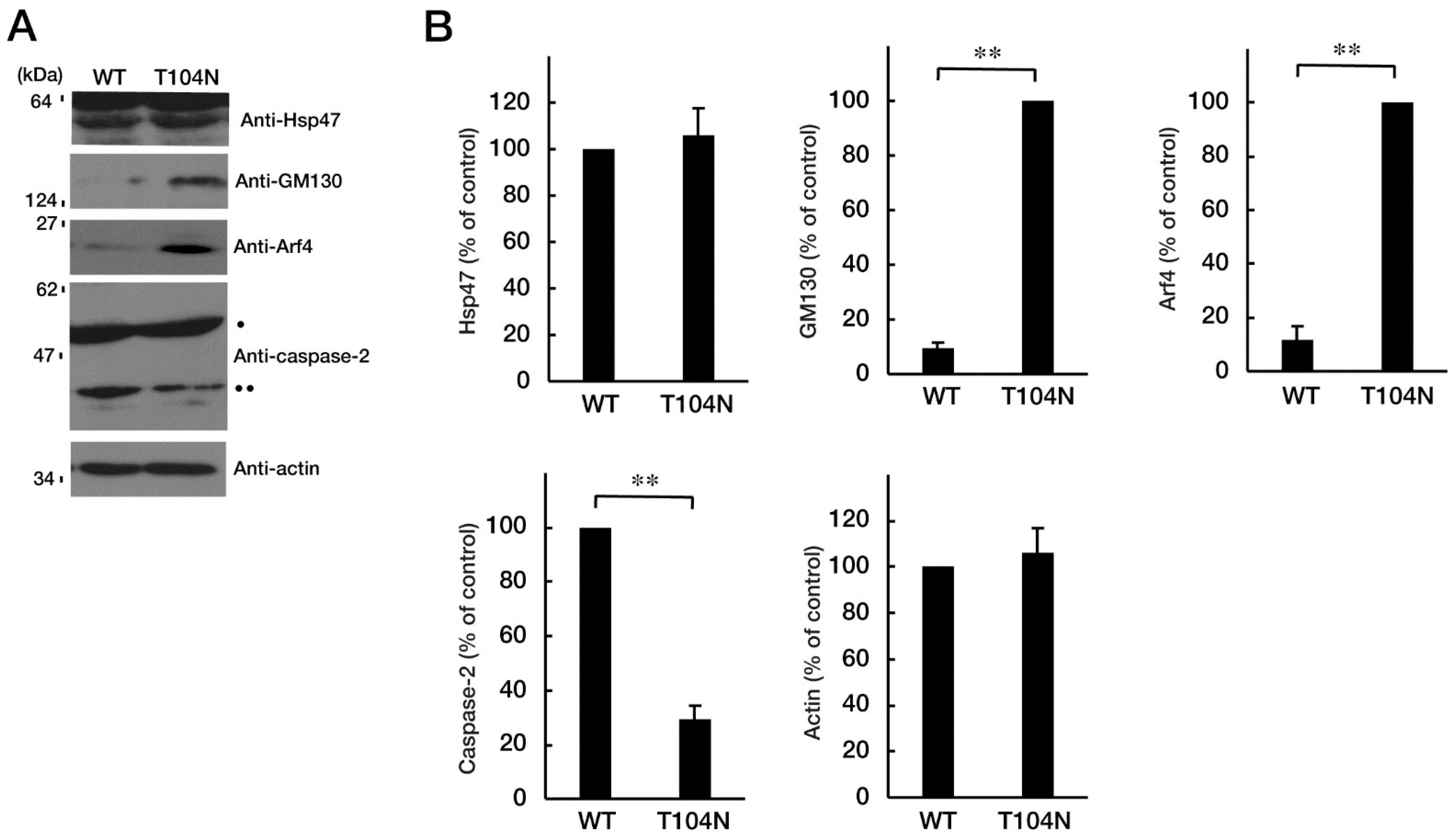
Figure 5.
Knockdown of Arf4 recovers phenotypes of cells with CHMP2B with the T104N mutation. N1E-115 cells with mutated CHMP2B were transfected with control luciferase (Luc) or Arf4 siRNA and allowed to differentiate for 48 hours. Typical cell images after 48 hours are shown in (A). (B) Cells with processes of more than one cell body length were counted as cells with neurites and are statistically depicted in the graph (* p<0.05; n = 10 fields). (C, D) The lysates were immunoblotted with an antibody against GAP43 or control actin, and their immunoreactive band intensities are statistically depicted in the graph (** p<0.01; n = 3 blots).
Figure 5.
Knockdown of Arf4 recovers phenotypes of cells with CHMP2B with the T104N mutation. N1E-115 cells with mutated CHMP2B were transfected with control luciferase (Luc) or Arf4 siRNA and allowed to differentiate for 48 hours. Typical cell images after 48 hours are shown in (A). (B) Cells with processes of more than one cell body length were counted as cells with neurites and are statistically depicted in the graph (* p<0.05; n = 10 fields). (C, D) The lysates were immunoblotted with an antibody against GAP43 or control actin, and their immunoreactive band intensities are statistically depicted in the graph (** p<0.01; n = 3 blots).
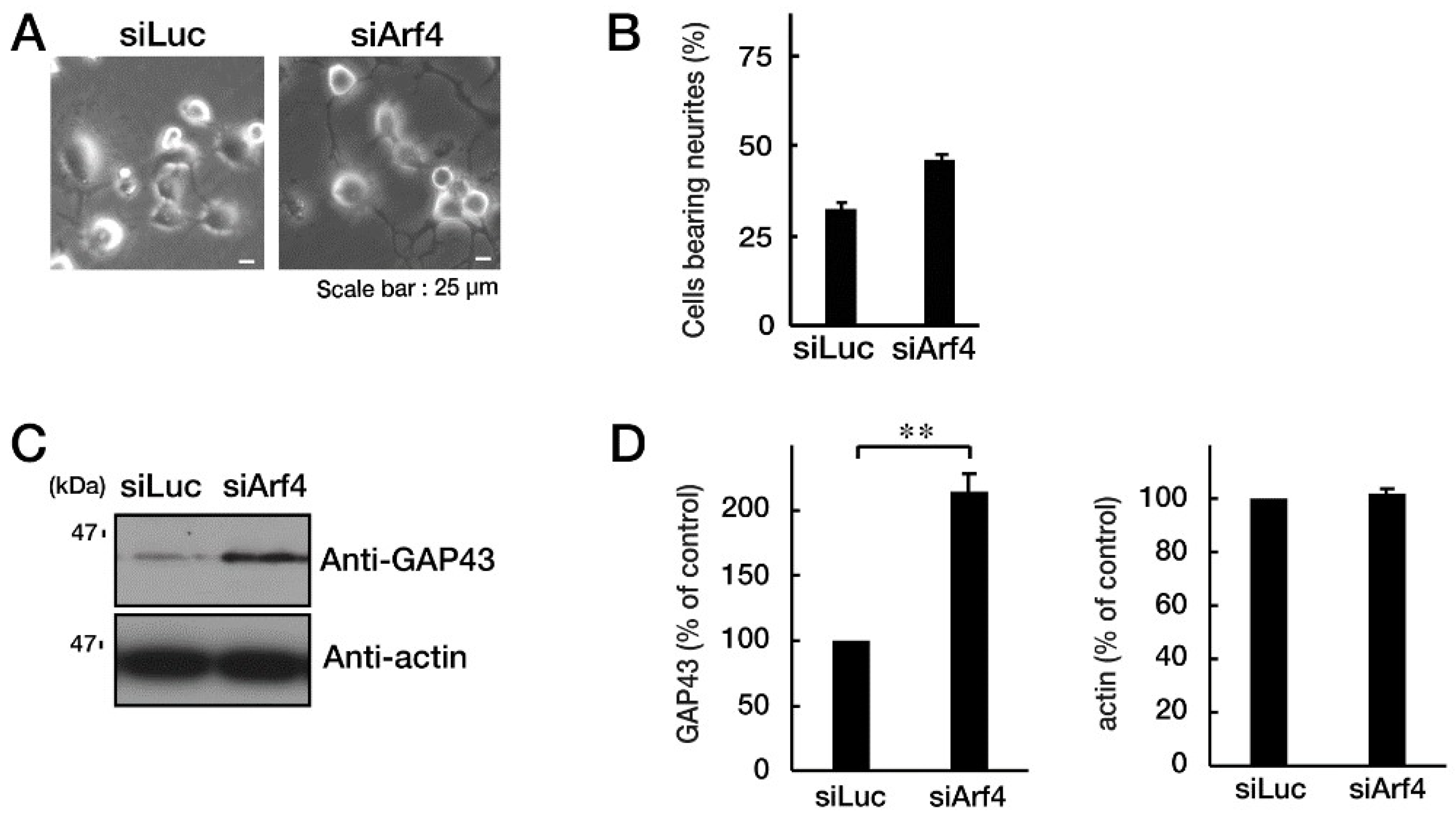

Table 1.
Key antibodies and plasmids used in this study.
| Reagent or material | Company or source | Cat. No. | Lot. No. | Concentration used |
|---|---|---|---|---|
| Antibody | ||||
| Anti-heat shock protein (HSP) 47 | Santa Cruz Biotechnology | sc-5293 | I2118 | Immunoblotting (IB), 1/200 |
| Anti-Arf4 | Proteintech | 11673-1-AP | 00048284 | IB, 1/1,000 |
| Anti-caspase-2 | Abcam | ab179520 | GR209449-2 | IB, 1/1,000 |
| Anti-actin | MBL | M177-3 | 007 | IB, 1/5,000 |
| Ant-growth-associated protein 43 (GAP43) | Santa Cruz Biotechnology | sc-17790 | J0920 | IB, 1/5,000 |
| Anti-Lys-Asp-Glu-Leu (KDEL) | MBL | M181-3 | 004 | IF, 1/200 |
| Anti-130 kDa Golgi membrane protein (GM130) | BD Biosciences | 610823 | 8352796 | IB, 1/500 and IF, 1/200 |
| Anti-cathepsin D | Abcam | ab75852 | GR260148-33 | IF, 1/200 |
| Anti-IgG (H+L chain) (Rabbit) pAb-HRP | MBL | 458 | 353 | IB, 1/5,000 |
| Anti-IgG (H+L chain) (Mouse) pAb-HRP | MBL | 330 | 365 | IB, 1/5,000 |
| Goat pAb to Ms IgG (Alexa Fluor 488 conjugate) | abcam | ab150113 | GR173498-1 | IF, 1/500 |
| Alexa Fluor TM 594 goat anti-mouse IgG (H+L) | invitrogen | A11005 | 226-8383 | IF, 1/500 |
| Alexa Fluor TM 488 goat anti-rabbit IgG (H+L) | invitrogen | A11008 | 075-1094 | IF, 1/500 |
| Alexa Fluor TM 594 goat anti-rabbit IgG (H+L) | invitrogen | A11012 | 201-8240 | IF, 1/500 |
| Recombinant DNA | ||||
| pEGFP-C1-human CHMP2B | Generated in this study | Not applicable | 1.25 µg of DNA per 6 cm dish | |
| pEGFP-C1-human CHMP2B with the T104N mutation | Generated in this study | Not applicable | 1.25 µg of DNA per 6 cm dish |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



