
Submitted:
18 June 2023
Posted:
19 June 2023
You are already at the latest version
Abstract
Mutations in the LMNA gene (encoding lamin A/C proteins) cause several human cardiac diseases, including dilated cardiomyopathies (LMNA-DCM). The main clinical risks in LMNA-DCM patients are sudden cardiac death and progressive left ventricular ejection fraction deterioration, and therefore most human and animal studies have sought to define the mechanisms through which LMNA mutations provoke cardiac alterations, with particular focus on cardiomyocytes. To investigate if LMNA mutations also cause vascular alterations that might contribute to the etiopathogenesis of LMNA-DCM, we generated and characterized Lmnaflox/floxSM22αCre mice, which constitutively lack lamin A/C in vascular smooth muscle cells (VSMCs), cardiac fibroblasts, and cardiomyocytes. Like mice with whole body or cardiomyocyte-specific lamin A/C ablation, Lmnaflox/floxSM22αCre mice recapitulated the main hallmarks of human LMNA-DCM, including ventricular systolic dysfunction, cardiac conduction defects, cardiac fibrosis, and premature death. These alterations were associated with hyperactivation of Smad3 and elevated expression of the proapoptotic protein caspase 3 in the heart. Lmnaflox/floxSM22αCre mice also exhibited perivascular fibrosis in the coronary arteries, and a switch of aortic VSMCs from the ‘contractile’ to the ‘synthetic’ phenotype. Ex vivo wire myography in isolated aortic rings revealed impaired maximum contraction capacity and an altered response to vasoconstrictor and vasodilator agents in Lmnaflox/floxSM22αCre mice. To our knowledge, our results provide the first evidence of phenotypic alterations in VSMCs that might contribute significantly to the pathophysiology of some forms of LMNA-DCM. Future work addressing the mechanisms underlying vascular defects in LMNA-DCM may open new therapeutic avenues for these diseases.
Keywords:
Lamin A/C
; laminopathies
; dilated cardiomyopathy
; vascular smooth muscle cell
; vascular dysfunction
; transgenic mice
Introduction
Nuclear A-type lamins are proteins that form type V filaments and are predominantly located underneath the inner nuclear membrane, where they are important components of the nuclear lamina found in nearly all differentiated mammalian cells [1,2,3]. There are two main A-type lamin proteins, lamins A and C, which are produced through alternative splicing of the same LMNA transcript (lamin C spans exons 1-10, while lamin A spans exons 1-12). A-type lamins play a crucial role in maintaining nuclear integrity, structure, and function. They ensure the proper spatial organization and function of chromatin, nuclear pore complexes, and other proteins that interact with nuclear lamins; additionally, A-type lamins are essential for nucleoskeleton-cytoskeleton connections, which are important for signal mechanotransduction to the nucleus [4,5,6]. As a result, lamins A and C regulate various cell functions, including cell proliferation, migration, and differentiation; signal transduction and gene expression; responses to DNA damage; and mechanosensing [4,5,6]. Interestingly, the expression level of A-type lamins correlates with tissue stiffness and the level of mechanical stress that cells experience. They are expressed at low levels in soft tissues such as fat and brain and at high levels in muscle tissues, where lamin filaments protect the nucleus from high mechanical stress [7,8].
Interest in A-type lamins has increased with the discovery of more than 400 mutations in LMNA that cause a broad range of human diseases collectively called laminopathies. Laminopathies include systemic progeroid syndromes like Hutchinson-Gilford progeria syndrome and tissue-specific diseases, such as lipodystrophies, neurological diseases, and a range of disorders affecting skeletal and/or cardiac muscle like Emery–Dreifuss muscular dystrophy, limb girdle muscular dystrophy, and dilated cardiomyopathy (DCM) [3,9]. The second most frequent DCM and the cause of more than 40% of sudden cardiac deaths is LMNA-associated DCM (LMNA-DCM), an autosomal dominant genetic disease characterized by cardiac dilation, reduced systolic function, defective atrioventricular conduction, cardiac arrythmias, extensive cardiac fibrosis, and heart failure [10,11,12,13,14,15]. Nearly 20% of LMNA-DCM patients require heart transplantation, and sudden cardiac death due to ventricular arrhythmias occurs frequently, often before the DCM becomes symptomatic [16]. There is a lack of specific therapies for LMNA-DCM, and patients are currently treated according to the standard heart failure protocol, with those with malignant arrhythmic events receiving an implantable cardioverter defibrillator to prevent sudden cardiac death [15,17]. There is therefore an urgent need for preclinical LMNA-CDM models that can be used to identify mechanisms that govern disease progression and develop specific therapies. Mouse models generated to investigate the molecular and cellular pathogenesis of LMNA-DCM include knock-in mice ubiquitously expressing Lmna mutations homologous to those that cause the disease in humans (LmnaN195K/N195K and LmnaH222P/H222P) and knock-out mice with whole body Lmna deficiency, which progressively develop cardiac fibrosis and conduction defects and DCM and die prematurely [18,19,20,21]. Similar to the whole body Lmna-null mice, Lmnaflox/floxMyh6-Cre mice with Lmna deletion restricted to cardiomyocytes develop severe cardiac dysfunction and conduction defects, ventricular arrhythmias, cardiac fibrosis, and apoptosis and die within 4 weeks of birth [22,23]. The cardiac phenotype of LMNA-DCM is also partially recapitulated in Lmnaflox/floxPdgfra-Cre mice, in which A-type lamins are absent from ~80% of cardiac fibroblasts and ~25% of cardiomyocytes [24]. Together, these mouse studies highlight the important role of cardiomyocytes in LMNA-DCM and suggest that cardiac fibroblasts also contribute to the pathogenesis of this deadly form of heart failure. However, despite the critical role of vascular smooth muscle cells (VSMCs) in cardiovascular pathophysiology, their possible involvement in LMNA-DCM has not been addressed. Here, we generated Lmnaflox/floxSM22αCre mice to evaluate the effects of combined Lmna deficiency in VSMCs, cardiomyocytes, and cardiac fibroblasts, a situation that occurs in LMNA-DCM patients and that provides a more translational model of the potential cross-talk between these cell types. In addition to developing the expected severe cardiac phenotype and dying prematurely, Lmnaflox/floxSM22Cre mice show VSMC alterations previously unrecognized in the context of LMNA-DCM that may play an important role in the etiopathogenesis of this laminopathy.
Methods
Mice.Lmnaflox/flox mice [25], and SM22αCre mice (TaglnCre, The Jackson Laboratory, stock no: 017491) [26], both on the C57BL/6J genetic background, were crossed to generate Lmnaflox/floxSM22Cre mice. All experiments were performed with 4-week-old mice and balanced numbers of males and females. Protocols for animal studies were approved by the local ethics committees and the Animal Protection Area of the Comunidad Autónoma de Madrid (PROEX 71.4/20).
Longevity studies. Animals were weighed periodically and inspected daily for health status and survival by a veterinarian blinded to genotype. Animals that met humane end-point criteria were euthanized and censored in the Kaplan Meier survival analysis. Animals sacrificed due to hydrocephalus, malocclusion, inter-male aggression, or other reasons unconnected to phenotype were excluded from the analysis.
Hematology and cardiac biochemical parameters. Blood was extracted from the submaxilar vein, and circulating blood cell populations were quantified using the PENTRA 80 hematology platform (HORIBA Medical, Madrid, Spain). Plasma was isolated by centrifugation of whole blood (2000g, 15 min at room temperature (RT)). Creatine kinase-MB and troponin were measured in plasma using DIMENSION RxL MAX chemistry analyzer (Siemens).
Histology and immunofluorescence. All mouse organs were fixed in 4% paraformaldehyde for 48 h, embedded in paraffin, and cut in 5-μm sections with a HM 355S microtome (Thermo Scientific). For immunofluorescence analysis of caspase-3 and phospho-Smad 3 (S423 + S425) (p-Smad 3) on heart sections, antigens were retrieved with 10 mM sodium citrate buffer (pH6) or TRIZMA base EDTA (pH9). Samples were then blocked and permeabilized for 1 hour at RT in PBS containing 0.3% Triton X100 (9002-93-1, Sigma), 5% bovine serum albumin (BSA, A7906, Sigma), and 5% normal goat serum (005-000-001, Jackson ImmunoResearch). Sections were then incubated overnight at 4°C with antibodies against CD31 (ab28364, Abcam, 1:100), lamin A/C (sc-376248, Santacruz, 1:100), FSP-1 (A5114, Dako, 1:200), caspase-3 (AF835, R&D Systems, 1:200), and p-Smad 3 (S423 + S425) [EP823Y] (ab52903, Abcam, 1:100). After washes, sections were incubated for 2 hours at RT with an antibody to smooth muscle α-actin (SMA) conjugated to Cy3 (C6198, Sigma, 1:20), anti-rabbit Alexa Fluor-647 secondary antibody (111-607-008, Jackson ImmunoResearch, 1:400), wheat germ agglutinin-Alexa fluor 488 (W11261, ThermoFisher, 1:300), and Hoechst 33342 or DAPI (1:1000). Sections were mounted in Fluoromount-G imaging medium (00-4958-02, ThermoFisher).
For histological studies, tissue sections were stained with hematoxylin/eosin, picrosirius red, or Masson’s trichrome; images were scanned with a NanoZoomer-RS scanner (Hamamatsu); and images were exported using NDP.view2. Immunofluorescence images were acquired with a Zeiss LSM700 confocal microscope. Images were analyzed with ImageJ Fiji software by an operator blinded to genotype. At least 3 tissue sections per animal were analysed, with removal of the immunofluorescence signals from arterioles. Mean values were used for statistical analysis.
Aortic RNA extraction and real-time quantitative PCR (RT-qPCR). Mouse thoracic aortas were incubated for 10 minutes at 37°C in DMEM (Gibco) containing 2 mg/mL type I collagenase (Worthington, LS004194). Adventitia was then removed manually, and the remaining tissue was snap-frozen and stored at -80ºC until further use. Each biological sample was a pool of two aortas from mice of the same genotype and sex. RNA was isolated using the RNeasy Mini Kit (Qiagen). RNA (1 μg) was reverse-transcribed to cDNA using the High Capacity cDNA reverse Transcription kit (Applied Biosystems). Real-time quantitative PCR (RT-qPCR) was performed using the primers shown in Table 1 and Power SYBR Green PCR Master Mix (Applied Biosystems) in a C1000 Touch Thermal Cycler (Bio-Rad). All the values were normalized to the housekeeping hypoxanthine-guanine phosphoribosyl transferase (HPRT) gene. All reactions were performed in triplicate.
Western blot. Snap-frozen hearts were lysed in ice-cold 50 mM Tris-HCl buffer (pH 8.8) containing 2% SDS, 8 M Urea and 2 M thiourea using a TissueLyser (Qiagen). The primary antibodies used were anti-phospho-Smad3 (S423+S425) [EP823Y] (ab52903, Abcam, 1:1000) and rabbit polyclonal anti-Vinculin Clone hVIN-1 (V9131, SIGMA, 1:1000). Immunocomplexes were detected with species-appropriate HRP-conjugated secondary antibodies, and were visualized using HRP Western Luminata Forte (WBLUF0100, Millipore). The relative intensity of protein bands was determined by densitometry with ImageQuant software.
Echocardiography. Transthoracic echocardiography was performed by expert operators using a high-frequency ultrasound system (Vevo 2100, Visualsonics Inc., Canada) with a 40-MHz linear probe. Operators were blinded to genotype. Two-dimensional (2D) and M-mode (MM) echography scans were performed at a frame rate > 230 frames/sec, and pulse wave Doppler (PW) was acquired with a pulse repetition frequency of 40 kHz. Mice were anaesthetized with 0.5-2% isoflurane in oxygen, adjusting the isoflurane delivery to maintain the heart rate at 450±50 beats per minute (bpm). Mice were placed in supine position on a heating platform, warmed ultrasound gel was used to maintain normothermia, and eye-drop gel was used to prevent dryness. A base apex electrocardiogram (ECG) was continuously monitored. Images were analyzed off-line using the Vevo 2100 Workstation software. For left ventricular systolic function assessment, parasternal standard 2D and MM long and short axis views were acquired. Left ventricular ejection fraction (LVEF), fractional shortening, stroke volume, cardiac output, and thickness were calculated from these views. Right ventricular systolic function was indirectly estimated using tricuspid annular plane systolic excursion (TAPSE), obtained from a MM 4-chamber apical view, to measure maximum lateral tricuspid annulus movement.
Electrocardiography. Mice were anaesthetized with 0.5-1.5% isoflurane in oxygen. To avoid night-day circadian variations, ECGs were always recorded in the morning. ECG electrodes were inserted subcutaneously in the four limbs. Sequential ECG recordings were acquired at 2 kHz using a MP36R data acquisition workstation (Biopac Systems) and exported with AcqKnowledge software (Biopac Systems). Automatic analysis with custom R scripts was used to remove noise and baseline fluctuations; detect heartbeat, peaks, and waves; exclude artifacts; and calculate heart rate, ECG intervals (QRS, QT), and T-wave steepness. Lead II was selected for the study, since the signal was more stable in most experiments, allowing more robust wave identification. Heart rate was calculated as the inverse of the time difference between two consecutive R-wave peaks (RR). Due to potential noise interference with detection of the beginning of P-waves, PQ interval was measured from the P-wave peak to the beginning of the Q-wave. The end of the S-wave (J) is not evident in mice because the ST segment is absent in this species and is replaced by a J-wave corresponding to a positive segment of the T-wave. Therefore, the QRS complex was calculated from the Qs to the S-wave minimum and the QT interval from the Qs to the T-wave peak (also termed J-wave peak). T-wave morphological alterations were quantified by defining T-wave steepness as an indicator of T-wave flattening that represents the absolute value of the slope (voltage variation over time) between the T-wave peak and T90.
Wire myography. Aortas were dissected free of fat and connective tissue and placed in cold Krebs Henseleit Solution (KHS: 115 mmol/L NaCl, 25 mmol/L NaHCO3, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4·7H2O, 2.5 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 11.1 mmol/L glucose, and 0.027 mmol/L EDTA). Segments of thoracic aortas, 2 mm in length, were mounted on a wire myograph system (620M, DMT) for isometric tension recording. After a 30-min equilibration period in KHS oxygenated with a mixture of 95% O2 and 5% CO2 at 37°C and pH 7.4, diameter-tension relationships were determined by increasing the distance between the wires passing though the lumen, thus increasing its passive diameter. At each step, the force and the internal circumference of the vessel were recorded [27]. Then, segments were stretched to their optimal lumen diameter for active tension development (LabChart software, ADInstruments [27]). This was determined based on the internal circumference/wall tension ratio of the segments by setting their internal circumference, Lo, to 90% of what the vessels would have if they were exposed to a passive tension, which is equivalent to that produced by a transmural pressure of 100 mm Hg.
Contractility of the segments was tested by an initial exposure to a high K+ solution (K+-KHS, 120 mmol/L). After an equilibration period, aortic segments were precontracted with phenylephrine at ∼50% K+-KHS contraction in order to perform a concentration–response curve to acetylcholine (1 nmol/L-10 µmol/L). After washing, a concentration-response curve to phenylephrine (1 nmol/L-30 µmol/L) was performed. Finally, a concentration-response curve to diethylamine NONOate (DEA-NO, 1 nmol/L-10 µmol/L) was performed in phenylephrine pre-contracted arteries.
Statistical analysis. Quantitative data are presented as the mean ± the standard error of the mean (SEM). Statistical analysis was performed with GraphPad Prism. Normal distribution of the data was analysed using the Kolmogorov-Smirnov test. Statistical significance of differences was assessed as indicated in the figure legends. Differences were considered significant at p-values<0.05. Outliers were evaluated using the Grubbs’ test in the GraphPad outlier calculator.
Results
Lmnaflox/floxSM22αCre mice with Lmna deficiency restricted to vascular smooth muscle cells, cardiac fibroblasts, and cardiomyocytes die prematurely. We crossed Lmnaflox/flox mice [25] with SM22αCre transgenic mice [26] to generate Lmnaflox/floxSM22αCre mice with Lmna deletion restricted to VSMCs, cardiac fibroblasts, and cardiomyocytes. To examine the efficiency and specificity of lamin A/C deletion in Lmnaflox/floxSM22αCre mice, we performed immunofluorescence experiments with antibodies against lamin A/C, CD31 (to detect endothelial cells), and SMA (to detect VSMCs). Lamin A/C expression was robust in all cell types in aorta, liver, kidney, and lung in control Lmnaflox/flox mice (Figure 1A) but was undetectable in medial VSMCs in the vessels of Lmnaflox/floxSM22αCre mice, without apparent differences in other cell types (Figure 1B). While gross examination revealed widespread and robust lamin A/C expression in heart cross-sections in Lmnaflox/flox mice (Figure 2A), lamin A/C was undetectable in the coronary artery VSMCs of Lmnaflox/floxSM22αCre mice and was below-normal level in non-vascular myocardial tissue (Figure 2B). To identify which cell types in the Lmnaflox/floxSM22αCre myocardium had lamin A/C deficiency, heart cross-sections were co-stained with anti-lamin A/C antibodies, wheat germ agglutinin (WGA) to visualize cardiomyocyte cell membranes, and anti-fibroblast specific protein 1 (FSP-1) antibodies to detetct cardiac fibroblasts. These studies revealed that lamin A/C expression was undetectable in ~87% of cardiomyocytes and ~72% of cardiac fibroblasts in Lmnaflox/floxSM22αCre mice (Figure 2C).
Phenotypic characterization of Lmnaflox/flox and Lmnaflox/floxSM22αCre mice revealed no between-genotype differences in circulating blood cell populations (Figure 3A); however, Lmnaflox/floxSM22αCre mice had slightly lower body weight (16.8% lower) (Figure 3B) and dramatically reduced survival, with a median lifespan of 33 days and maximum survival of 50 days (Figure 3C).
Lmnaflox/floxSM22α-Cre mice develop cardiac fibrosis and severe systolic dysfunction and electrocardiographic alterations. Immunohistopathological studies in 4-week-old Lmnaflox/floxSM22αCre mice and Lmnaflox/flox controls showed no between-genotype differences in collagen content in tissue sections from liver, lung, and kidney (Figure 4A). However, Lmnaflox/floxSM22αCre mice showed a trend towards increased collagen content in the aortic arch and thoracic aorta (Figure 4B), which reached statistical significance in coronary arteries (Figure 4C). Moreover, Lmnaflox/floxSM22αCre hearts had significantly higher interstitial fibrosis (Figure 5A), elevated WGA staining, and above-normal expression of the profibrotic markers FSP-1 and SMA (Figure 5B). These alterations in mutant hearts were associated with higher expression of p-Smad3, the active form of the pro-fibrotic transcription factor Smad3, as revealed both by immunofluorescence experiments (Figure 6A) and by western blotting (Figure 6B). Lmnaflox/floxSM22αCre hearts also had higher expression of the pro-apoptotic protein caspase-3 (Figure 6C).
Echocardiography analysis detected significant systolic dysfunction in both the left and right ventricles of Lmnaflox/floxSM22αCre mice, revealed by lower EF and TAPSE, respectively (Figure 7A and supplementary videos S1-S4). Lmnaflox/floxSM22αCre mice also showed a modest but statistically significant decrease in left-ventricular thickness (Figure 7A), whereas heart weight and tibia length were similar in both genotypes (Figure 7B). ECG analysis revealed statistically significant between-genotype differences in parameters indicative of a lower repolarization rate in Lmnaflox/floxSM22αCre mice, including prolongation of the QRS and QT intervals and reduced T-wave steepness (Figure 7C). These alterations were associated with elevated plasma troponin in Lmnaflox/floxSM22α-Cre mice, with no between-genotype differences in creatine kinase-MB (Figure 7D).
Lmnaflox/floxSM22αCre mice exhibit contractile-to-synthetic phenotypic switching in VSMCs and vascular dysfunction in the aorta. To investigate possible alterations in the vasculature of Lmnaflox/floxSM22αCre mice, we performed RT-qPCR on total RNA isolated from adventitia-free aortic tissue (supplementary Figure S1A). These studies revealed significant downregulation in the expression of genes characteristic of ‘contractile’ VSMCs and upregulation of markers of ‘synthetic’ VSMCs in Lmnaflox/floxSM22αCre mice (Figure 8A), with no between-genotype differences in other genes relevant to VSMC function, including genes related to calcium homeostasis (Cam2), oxidative stress (Nox1, Sod1), and mitochondrial and sarcoplasmic reticulum function (Tfam, Calr) (supplementary Figure S1B).
Ex vivo wire myography experiments with thoracic aorta rings revealed no between-genotype differences in vessel stiffness (Figure 8B) or physiological diameter (Figure 8C). However, potassium-stimulated maximum contraction was significantly lower in Lmnaflox/floxSM22αCre mice (Figure 8D). Likewise, although aortic rings with VSMC-specific Lmna ablation contracted more than controls at phenylephrine doses < 10-7 M, their maximum contraction was lower at higher phenylephrine concentrations, with a significantly lower EC50 (phenylephrine dose giving a half-maximal response) (Figure 8E). Interestingly, lack of lamin A/C in aortic VSMCs was also associated with significantly lower endothelium-dependent vasorelaxation induced by acetylcholine (Figure 8F) and endothelium-independent vasorelaxation induced by the NO donor DEA-NO (Figure 8G).
Discussion
The major severe clinical manifestations of LMNA-DCM are sudden cardiac death and progressive LVEF deterioration [10,11,12,13,14,15,16]. Therefore, most studies have sought to define the mechanisms through which LMNA mutations provoke cardiac alterations, with particular focus on cardiomyocytes. However, A-type lamins are expressed in most differentiated cells, and it is therefore of the utmost interest to investigate the potential pathological effects of LMNA mutations on other cell types, which may cause alterations in cardiac muscle through paracrine mechanisms. In the present study, we generated and characterized Lmnaflox/floxSM22αCre mice with constitutive lamin A/C deficiency in VSMCs, cardiac fibroblasts, and cardiomyocytes, abundant cell types in the heart. Consistent with previous studies in Myh6-Cre:Lmnaf/f mice with lamin A/C deficiency exclusively in cardiomyocytes [22,23], Lmnaflox/floxSM22αCre mice recapitulated the main hallmarks of human LMNA-DCM, including cardiac fibrosis, ventricular systolic dysfunction, ECG alterations, and premature death. Importantly, the time-course and severity of disease in Lmnaflox/floxSM22αCre mice was strikingly similar to observations in whole body Lmna-null mice and the cardiomyocyte-specific Myh6-Cre:Lmnaf/f mice [19,22,23]. Indeed, our new model demonstrates that extending lamin A/C disruption to VSMCs and cardiac fibroblasts does not aggravate the lifespan reduction caused by Lmna deficiency restricted to cardiomyocytes (median lifespan ~1 month in both models). Recent studies in Pdfra-Cre:Lmnaf/f mice with lamin A/C absence in ~80% of cardiac fibroblasts and ~25% of cardiomyocytes partially recapitulated the LMNA-DCM phenotype, with a median lifespan of ~43 days [24]. Moreover, restoration of lamin A expression in ~40% of cardiomyocytes in Lmna-/-; Tg mice partially rescued ECG alterations and extended lifespan by 12% compared with controls with whole body Lmna ablation [21]. Collectively, the results in these mouse models suggest that cardiomyocyte-autonomous and non-cardiomyocyte-autonomous factors play an important role in the etiopathogenesis of LMNA-DCM. Further discrimination of the individual role of VSMCs and cardiac fibroblasts in LMNA-DCM would require the generation of new mouse models with Lmna deficiency restricted to these cell types.
Consistent with previous studies in Lmna-/- and Myh6-Cre:Lmnaf/f mice [22,23], Lmnaflox/floxSM22αCre mice showed evidence of cardiac fibrosis and apoptosis, which was accompanied by elevated fibroblast and myofibroblast markers, including WGA staining and immunostaining of FSP-1 and SMA. Previous studies have suggested that cardiomyocyte apoptosis in Lmna-null mice may result from altered gene expression, disruption of cytoskeleton tension, and defective force transmission [20]. Regarding fibrosis, it is well-known that members of the transforming growth factor β (TGFβ) superfamily trigger pro-fibrotic transcriptional programs through the activation of SMAD-dependent signaling in cardiomyocytes, fibroblasts, immune cells, and vascular cells [28]. For example, phosphorylation of SMAD3 triggers the conversion of cardiac fibroblasts into secretory pro-fibrotic myofibroblasts expressing extracellular matrix and contractile proteins (such as SMA) and integrins, thus promoting myofibroblast migration, survival, and growth arrest and scar formation [28]. Our western blot and immunofluorescence studies demonstrate elevated p-Smad3 expression in Lmnaflox/floxSM22αCre hearts. Phosphorylation (activation) of Smad proteins was also observed in the hearts of LmnaH222P/H222P mice [29], and Lmna-null cardiomyocytes isolated from Lmnaflox/floxMyh6-Cre mice exhibited high TGFβ1 mRNA and protein expression [22]. Collectively, these results suggest that lamin A/C deficiency and expression of the DCM-causing LmnaH222P protein provoke cardiac fibrosis at least in part through activation of TGFSMAD signaling. Future studies are warranted to assess if TGFSMAD signaling is abnormally activated in the heart in other mouse models of LMNA-DCM and in patients.
Uncertainty has surrounded the question of whether vascular abnormalities exist and play a role in the pathogenesis and progression of non-ischemic DCM [30]. Mathier et al. reported the presence of abnormal coronary endothelium-dependent vasodilation in the epicardium and the microcirculation at early disease stages in patients with acute-onset DCM [31]. These authors also found an association between the preservation of endothelial function and improved LVEF in this population [31]. Furthermore, non-ischemic DCM has been linked to vascular derangements and to defective vasculogenesis and angiogenesis in patients [30]. Recently, Sayed et al. reported clinical endothelial dysfunction in patients with LMNA-DCM, and human induced pluripotent stem cell-derived endothelial cells (ECs) carrying DCM-causing LMNA mutations presented hallmarks of endothelial dysfunction, including a decreased capacity to produce nitric oxide and impaired angiogenic potential in vitro [32]. To our knowledge, our current results provide the first evidence that Lmna deficiency also provokes VSMC alterations in vivo. Four-week-old Lmnaflox/floxSM22αCre mice displayed a phenotype switch in the aorta from the ‘contractile’ phenotype characteristic of ‘healthy’ VSMCs to the ‘synthetic’ VSMC phenotype that characterizes the inflamed vessel wall in various forms of vascular disease [33]. The mice also showed incipient fibrosis in the aorta, which reached statistical significance in coronary arteries. Moreover, our myograph studies show that aortic rings with VSMC-specific Lmna deficiency have impaired maximum contraction and a defective response to vasodilators and vasoconstrictors with no accompanying alterations in relaxation induced by the nitric oxide donor DEA-NO, evidencing VSMC dysfunction. VSMC injury might also contribute to the endothelial dysfunction reported in LMNA-DCM patients [32], since aortic rings lacking lamin A/C specifically in VSMCs had depressed endothelium-dependent acetylcholine-induced relaxation despite normal lamin A/C expression in ECs. Alterations in gene expression and function in lamin A/C-deficient VSMCs and ECs are likely caused by defective mechanotransduction and altered signaling, transcription, and chromatin organization, key processes that are regulated by A-type lamins [4,6]. Collectively, these studies suggest that dysfunctional ECs and VSMCs contribute to the etiopathogenesis of LMNA-DCM and that therapies to ameliorate vascular cell function may have a beneficial effect on the heart. Treatment with lovastatin has been shown to ameliorate endothelial function in cultured LMNA iPSC-ECs and in LMNA-DCM patients and also improved the functional phenotype of LMNA iPSC-derived cardiomyocytes when co-cultured with LMNA iPSC-ECs [32]. Although mouse models harboring cell-type-specific Lmna alterations have provided important insights into the mechanisms underlying LMNA-DCM, future mechanistic and therapeutic studies should focus on the use of more translational ubiquitous models that consider the crosstalk among cardiac and non-cardiac cells. More research is also warranted to identify the mechanisms that cause vascular dysfunction in LMNA-DCM and to investigate vascular pathology in LMNA-DCM patients, as these approaches may open new therapeutic avenues for the treatment of these diseases.
Funding
This study was supported by grants SAF2016-79490-R and PID2019-108489RB-I00 from the Spanish Ministerio de Ciencia e Innovación (MICIN)/Agencia Estatal de Investigación (AEI)/10.13039/501100011033, with co-funding from the European Social Fund (“The ESF invests in your future”). Microscopy was conducted at the Microscopy & Dynamic Imaging Unit, CNIC, ICTS-ReDib, co-funded by MCIN/AEI /10.13039/501100011033. A.d.M was supported by the MICIN (predoctoral contract BES-2014-067791), C.E.-E and V.F. by the Fundación “la Caixa” (predoctoral contracts LCF/BQ/DR19/1170012 and LCF/BQ/DE14/10320024, respectively), I.R.P by MICIN/AEI/10.13039/501100011033 and the European Social Fund (“The ESF invests in your future”) (predoctoral contract PRE2020-092264), and M.G.-A by MICIN (post-doctoral contract FJC 2021-047576-I). The CNIC is supported by the MCIN, the Instituto de Salud Carlos III, and the Pro-CNIC Foundation and is a Severo Ochoa Center of Excellence (grant number CEX2020-001041-S funded by MICIN/AEI/10.13039/501100011033).
Acknowledgements
We thank Yixiang Zheng for providing Lmnaflox/flox mice, David Filgueiras for advice on ECG analysis, Yaazan Blanco for body weight studies, Antonio de Molina for support with histology, Eva Santos and the CNIC Animal Facility for animal care, Marta García and Belén Ricote for help with ECG, the CNIC Microscopy Unit for support in image analysis, and Simon Bartlett for English editing.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; the collection, analysis, or interpretation of data; the writing of the manuscript; or the decision to publish the results.
References
- Gerace L, Burke B. Functional organization of the nuclear envelope. Annu Rev Cell Biol 1988;4:335–74.
- Stuurman N, Heins S, Aebi U. Nuclear lamins: Their structure, assembly, and interactions. J Struct Biol 1998;122:42–66.
- Broers JLV, Ramaekers FCS, Bonne G et al. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol Rev 2006;86:967–1008.
- Andrés V, González JM. Role of A-type lamins in signaling, transcription, and chromatin organization. Journal of Cell Biology 2009;187:945–57.
- Dechat T, Pfleghaar K, Sengupta K et al. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev 2008;22:832–53.
- Kalukula Y, Stephens AD, Lammerding J et al. Mechanics and functional consequences of nuclear deformations. Nat Rev Mol Cell Biol 2022;23:583–602.
- Swift J, Ivanovska IL, Buxboim A et al. Nuclear Lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science (1979) 2013;341:975–90.
- Carmosino M, Torretta S, Procino G et al. Role of nuclear Lamin A/C in cardiomyocyte functions. Biol Cell 2014;106:346–58.
- Benedicto I, Dorado B, Andrés V. Molecular and cellular mechanisms driving cardiovascular disease in hutchinson-gilford progeria syndrome: Lessons learned from animal models. Cells 2021;10. [CrossRef]
- Taylor MRG, Fain PR, Sinagra G et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003;41:771–80.
- Bonne G, Yaou R Ben, Béroud C et al. 108th ENMC International Workshop, 3rd Workshop of the MYO-CLUSTER project: EUROMEN, 7th International Emery-Dreifuss Muscular Dystrophy (EDMD) Workshop, 13-15 September 2002, Naarden, The Netherlands. Neuromuscular Disorders 2003;13:508–15.
- Van Berlo JH, De Voogt WG, Van Der Kooi AJ et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? J Mol Med 2005;83:79–83.
- Van Rijsingen IAW, Arbustini E, Elliott PM et al. Risk factors for malignant ventricular arrhythmias in Lamin A/C mutation carriers: A European cohort study. J Am Coll Cardiol 2012;59:493–500.
- Arbustini E, Pilotto A, Repetto A et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: A lamin A/C defect-related disease. J Am Coll Cardiol 2002;39:981–90.
- Crasto S, My I, di Pasquale E. The Broad Spectrum of LMNA Cardiac Diseases: From Molecular Mechanisms to Clinical Phenotype. Front Physiol 2020;11. [CrossRef]
- Hasselberg NE, Haland TF, Saberniak J et al. Lamin A/C cardiomyopathy: young onset,high penetrance, and frequent need forheart transplantation. Eur Heart J 2018;39:853–60.
- Zhang H, Ren L, Wu JC. New Insights Into the Therapy for Lamin-Associated Dilated Cardiomyopathy. JACC Basic Transl Sci 2022;7:1246–8.
- Zhang H, Kieckhaefer JE, Cao K. Mouse models of laminopathies. Aging Cell 2013;12:2–10.
- Sullivan, T. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999;147:913–20.
- Nikolova V, Feneley MP, Fatkin D et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A / C- deficient mice Find the latest version : and function promote dilated cardiomyopathy in lamin A / C – deficient mice. J Clin Invest 2004;113:357–69.
- Frock RL, Chen SC, Da D et al. Cardiomyocyte-specific expression of lamin A improves cardiac function in Lmna-/- mice. PLoS One 2012;7:1–9.
- Auguste G, Rouhi L, Matkovich SJ et al. BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific lamin A/C-deficient mice. Journal of Clinical Investigation 2020;130:4740–58.
- Chai RJ, Werner H, Li PY et al. Disrupting the LINC complex by AAV mediated gene transduction prevents progression of Lamin induced cardiomyopathy. Nat Commun 2021;12. [CrossRef]
- Rouhi L, Auguste G, Zhou Q et al. Deletion of the Lmna gene in fibroblasts causes senescence-associated dilated cardiomyopathy by activating the double-stranded DNA damage response and induction of senescence-associated secretory phenotype. The Journal of Cardiovascular Aging 2022;2:30.
- Kim Y, Zheng Y. Generation and characterization of a conditional deletion allele for Lmna in mice. Biochem Biophys Res Commun 2013;440:8–13.
- Lepore JJ, Cheng L, Min ML et al. High-efficiency somatic mutagenesis in smooth muscle cells and cardiac myocytes in SM22α-Cre transgenic mice. Genesis 2005;41:179–84.
- Del Campo L, Ferrer M. Wire Myography to Study Vascular Tone and Vascular Structure of . Isolated Mouse Arteries. Methods in Mouse Atherosclerosis. 2015, 255–76. [Google Scholar]
- Hanna A, Humeres C, Frangogiannis NG. The role of Smad signaling cascades in cardiac fibrosis. Cell Signal 2021;77:109826.
- Arimura T, Helbling-Leclerc A, Massart C et al. Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet 2005;14:155–69.
- Roura S, Bayes-Genis A. Vascular dysfunction in idiopathic dilated cardiomyopathy. Nature Reviews 2009;6:590–8.
- Mathier MA, Rose GA, Fifer MA et al. Coronary Endothelial Dysfunction in Patients With Acute-Onset Idiopathic Dilated Cardiomyopathy. JACC 1998;32:216–24.
- Sayed N, Liu C, Ameen M et al. Clinical trial in a dish using iPSCs shows lovastatin improves endothelial dysfunction and cellular cross-talk in LMNA cardiomyopathy. Sci Transl Med 2020;12. [CrossRef]
- Sorokin V, Vickneson K, Kofidis T et al. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front Immunol 2020;11. [CrossRef]
Figure 1.
Lamin A/C ablation in VSMCs in Lmnaflox/floxSM22αCre mice. Representative immunofluorescence images of aorta, liver, kidney, and lung from 4-week-old Lmnaflox/flox (A) and Lmnaflox/floxSM22αCre mice (B). Lamin A/C is visualized in white, endothelial cells in green (anti-CD31 antibody), VSMCs in red (anti-smooth muscle α-actin (SMA) antibody), and nuclei in blue (DAPI staining). Magnified images show vessel-containing regions (*) and vessel-free regions (**).
Figure 1.
Lamin A/C ablation in VSMCs in Lmnaflox/floxSM22αCre mice. Representative immunofluorescence images of aorta, liver, kidney, and lung from 4-week-old Lmnaflox/flox (A) and Lmnaflox/floxSM22αCre mice (B). Lamin A/C is visualized in white, endothelial cells in green (anti-CD31 antibody), VSMCs in red (anti-smooth muscle α-actin (SMA) antibody), and nuclei in blue (DAPI staining). Magnified images show vessel-containing regions (*) and vessel-free regions (**).

Figure 2.
Lamin A/C ablation in heart VSMCs, cardiomyocytes, and cardiac fibroblasts in Lmnaflox/floxSM22αCre mice. (A, B) Representative immunofluorescence images of heart tissue from 4-week-old Lmnaflox/flox (A) and Lmnaflox/floxSM22αCre mice (B). Lamin A/C is visualized in white, endothelial cells in green (anti-CD31 antibody), VSMCs in red (anti-smooth muscle α-actin (SMA) antibody), and nuclei in blue (DAPI staining). Magnifications show vessel-containing (*) and vessel-free (**) regions. (C) Representative immunofluorescence images of heart tissue from 4-week-old Lmnaflox/flox and Lmnaflox/floxSM22αCre mice. Lamin A/C is visualized in red, cell membranes in green (wheat germ agglutinin; WGA), cardiac fibroblasts in white (anti-FSP-1 antibody), and nuclei in blue (Hoechst 33342 staining). Graphs show the percentages of lamin A/C-positive nuclei in cardiomyocytes and cardiac fibroblasts. Statistical analysis was by unpaired two-tailed t-test. Data are mean±SEM. Each symbol represents one animal.
Figure 2.
Lamin A/C ablation in heart VSMCs, cardiomyocytes, and cardiac fibroblasts in Lmnaflox/floxSM22αCre mice. (A, B) Representative immunofluorescence images of heart tissue from 4-week-old Lmnaflox/flox (A) and Lmnaflox/floxSM22αCre mice (B). Lamin A/C is visualized in white, endothelial cells in green (anti-CD31 antibody), VSMCs in red (anti-smooth muscle α-actin (SMA) antibody), and nuclei in blue (DAPI staining). Magnifications show vessel-containing (*) and vessel-free (**) regions. (C) Representative immunofluorescence images of heart tissue from 4-week-old Lmnaflox/flox and Lmnaflox/floxSM22αCre mice. Lamin A/C is visualized in red, cell membranes in green (wheat germ agglutinin; WGA), cardiac fibroblasts in white (anti-FSP-1 antibody), and nuclei in blue (Hoechst 33342 staining). Graphs show the percentages of lamin A/C-positive nuclei in cardiomyocytes and cardiac fibroblasts. Statistical analysis was by unpaired two-tailed t-test. Data are mean±SEM. Each symbol represents one animal.
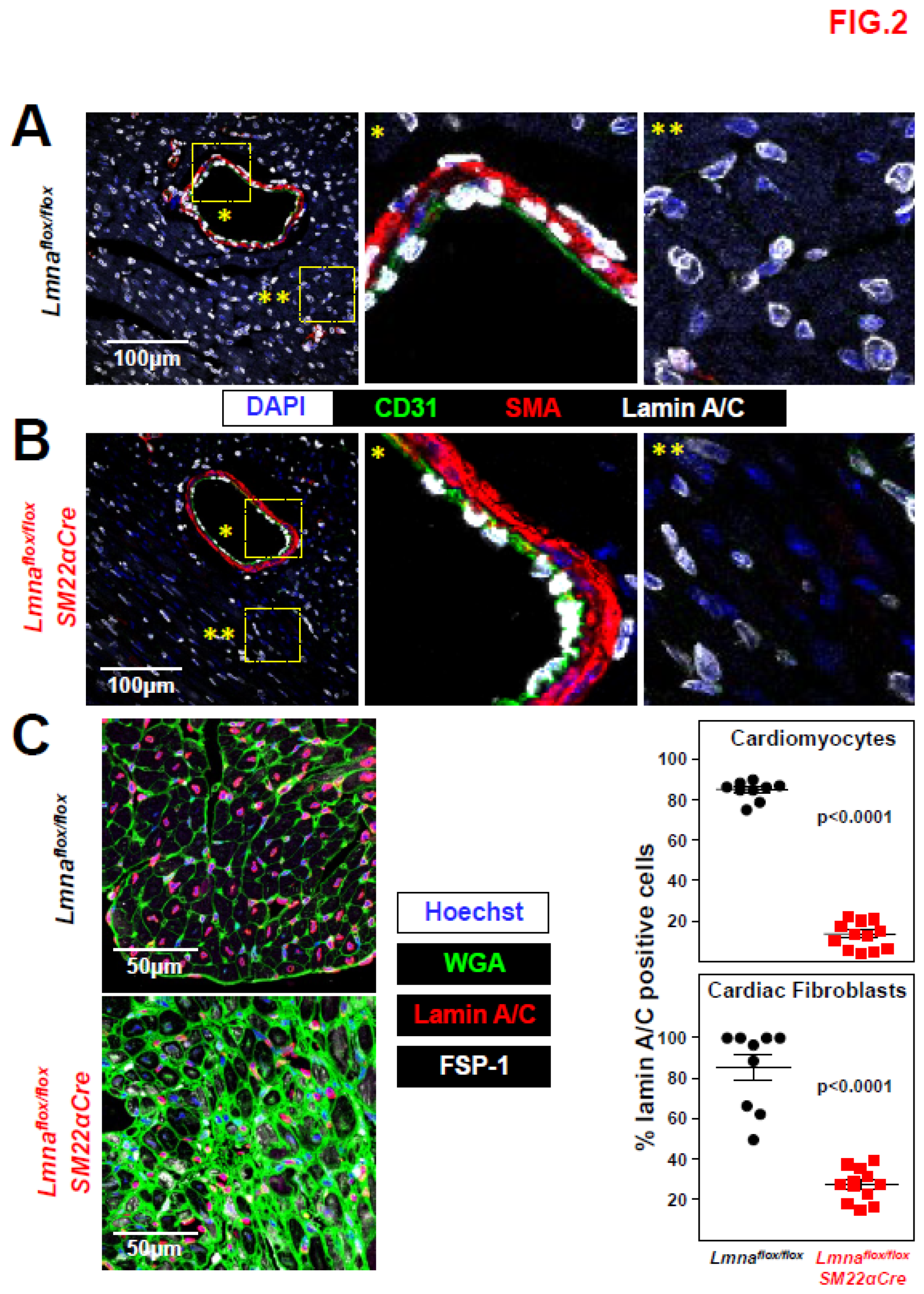
Figure 3.
Lmnaflox/floxSM22αCre mice exhibit reduced body weight and lifespan. (A) Circulating blood cell counts in 4-week-old Lmnaflox/flox and Lmnaflox/floxSM22αCre mice. (B) Representative photograph of male and female mice of both genotypes. The graph shows body weight at 4 weeks of age. (C) Kaplan-Meier survival curves of Lmnaflox/floxSM22αCre (median survival 33 days) and control Lmnaflox/flox mice (n=19 mice per genotype). Statistical analysis was by unpaired two-tailed t-test (A, B) and A log-rank (Mantel-Cox) test (C). Data are mean±SEM. Each symbol represents one animal.
Figure 3.
Lmnaflox/floxSM22αCre mice exhibit reduced body weight and lifespan. (A) Circulating blood cell counts in 4-week-old Lmnaflox/flox and Lmnaflox/floxSM22αCre mice. (B) Representative photograph of male and female mice of both genotypes. The graph shows body weight at 4 weeks of age. (C) Kaplan-Meier survival curves of Lmnaflox/floxSM22αCre (median survival 33 days) and control Lmnaflox/flox mice (n=19 mice per genotype). Statistical analysis was by unpaired two-tailed t-test (A, B) and A log-rank (Mantel-Cox) test (C). Data are mean±SEM. Each symbol represents one animal.
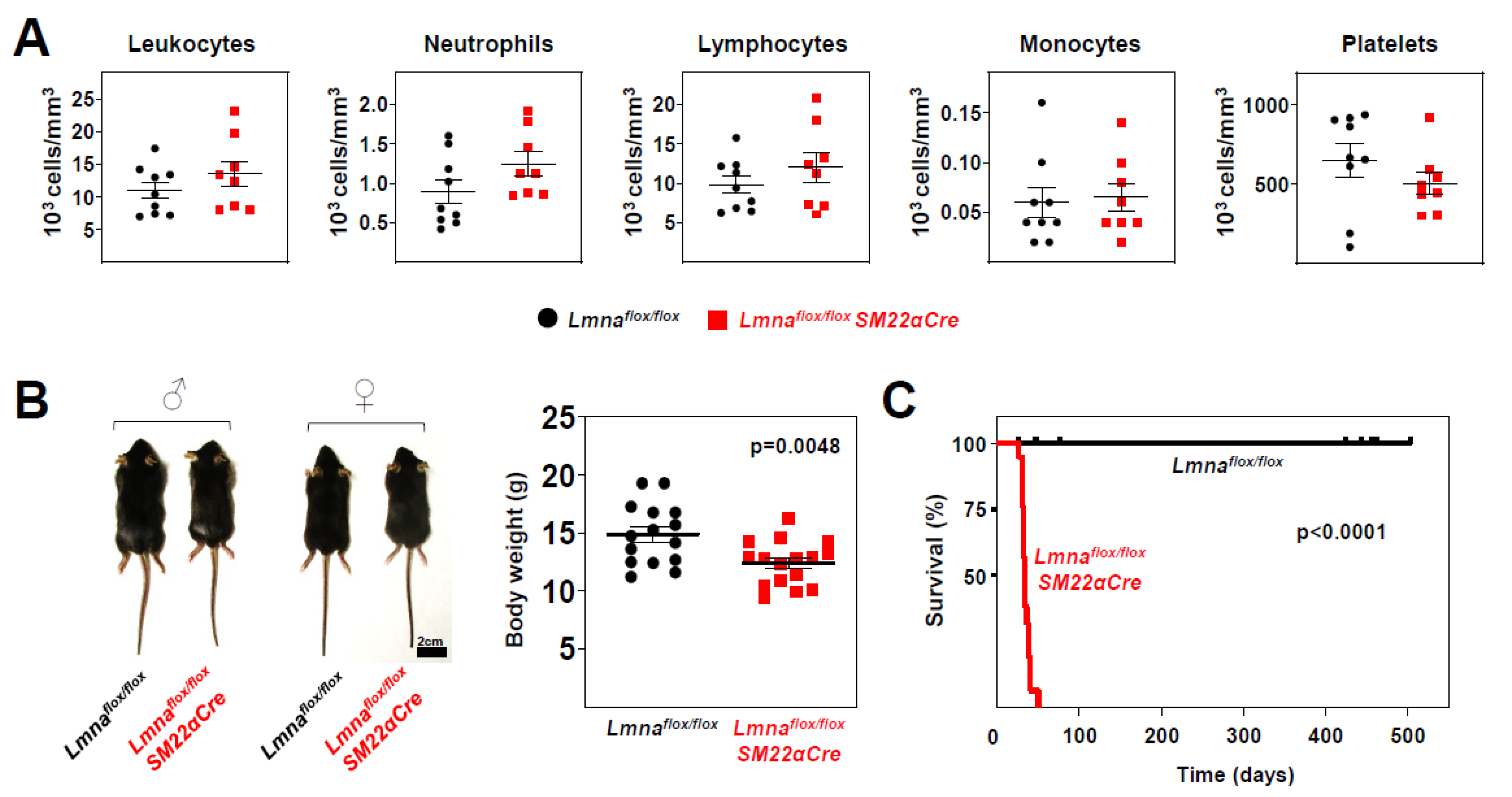
Figure 4.
Lmnaflox/floxSM22αCre mice show increased vascular collagen content. Representative images of Masson’s trichrome staining in (A) liver, lung, kidney, (B) aortic arch, thoracic aorta, and (C) coronary arteries of 4-week-old Lmnaflox/flox and Lmnaflox/floxSM22αCre mice. Graphs show collagen content calculated relative to the content in Lmnaflox/flox mice (=1). Statistical analysis was by unpaired two-tailed t-test. Data are mean±SEM. Each symbol represents one animal.
Figure 4.
Lmnaflox/floxSM22αCre mice show increased vascular collagen content. Representative images of Masson’s trichrome staining in (A) liver, lung, kidney, (B) aortic arch, thoracic aorta, and (C) coronary arteries of 4-week-old Lmnaflox/flox and Lmnaflox/floxSM22αCre mice. Graphs show collagen content calculated relative to the content in Lmnaflox/flox mice (=1). Statistical analysis was by unpaired two-tailed t-test. Data are mean±SEM. Each symbol represents one animal.
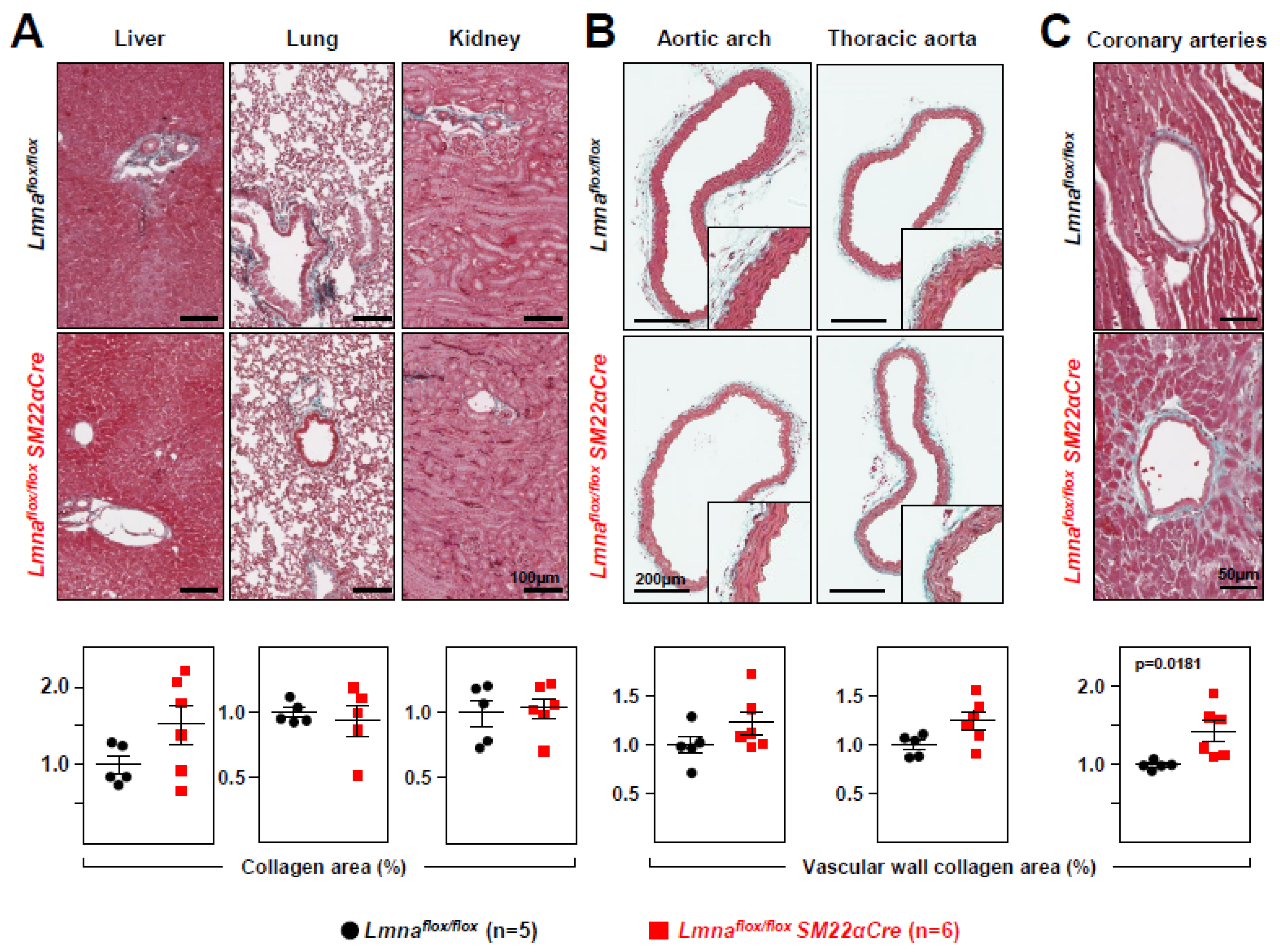
Figure 5.
Lmnaflox/floxSM22αCre mice develop cardiac fibrosis.Lmnaflox/flox and Lmnaflox/floxSM22αCre mice were examined at 4 weeks of age. (A) Representative images of Masson’s trichrome and Sirius red staining; graphs show collagen content in heart vessel-free regions, calculated relative to the content in Lmnaflox/flox mice (=1). One outlier identified with the GraphPad outlier calculator in the Lmnaflox/floxSM22αCre group was eliminated. (B) Representative immunofluorescence images of heart tissue stained with wheat germ agglutinin to visualize cell membranes (WGA, green), anti-FSP-1 antibody to identify cardiac fibroblasts (white), and anti-smooth muscle actin antibody to identify fibrogenic activated fibroblasts (SMA, red). Graphs show the positive area relative to the total area of tissue. Statistical analysis was by unpaired two-tailed t-test. Data are mean±SEM. Each symbol represents one animal.
Figure 5.
Lmnaflox/floxSM22αCre mice develop cardiac fibrosis.Lmnaflox/flox and Lmnaflox/floxSM22αCre mice were examined at 4 weeks of age. (A) Representative images of Masson’s trichrome and Sirius red staining; graphs show collagen content in heart vessel-free regions, calculated relative to the content in Lmnaflox/flox mice (=1). One outlier identified with the GraphPad outlier calculator in the Lmnaflox/floxSM22αCre group was eliminated. (B) Representative immunofluorescence images of heart tissue stained with wheat germ agglutinin to visualize cell membranes (WGA, green), anti-FSP-1 antibody to identify cardiac fibroblasts (white), and anti-smooth muscle actin antibody to identify fibrogenic activated fibroblasts (SMA, red). Graphs show the positive area relative to the total area of tissue. Statistical analysis was by unpaired two-tailed t-test. Data are mean±SEM. Each symbol represents one animal.
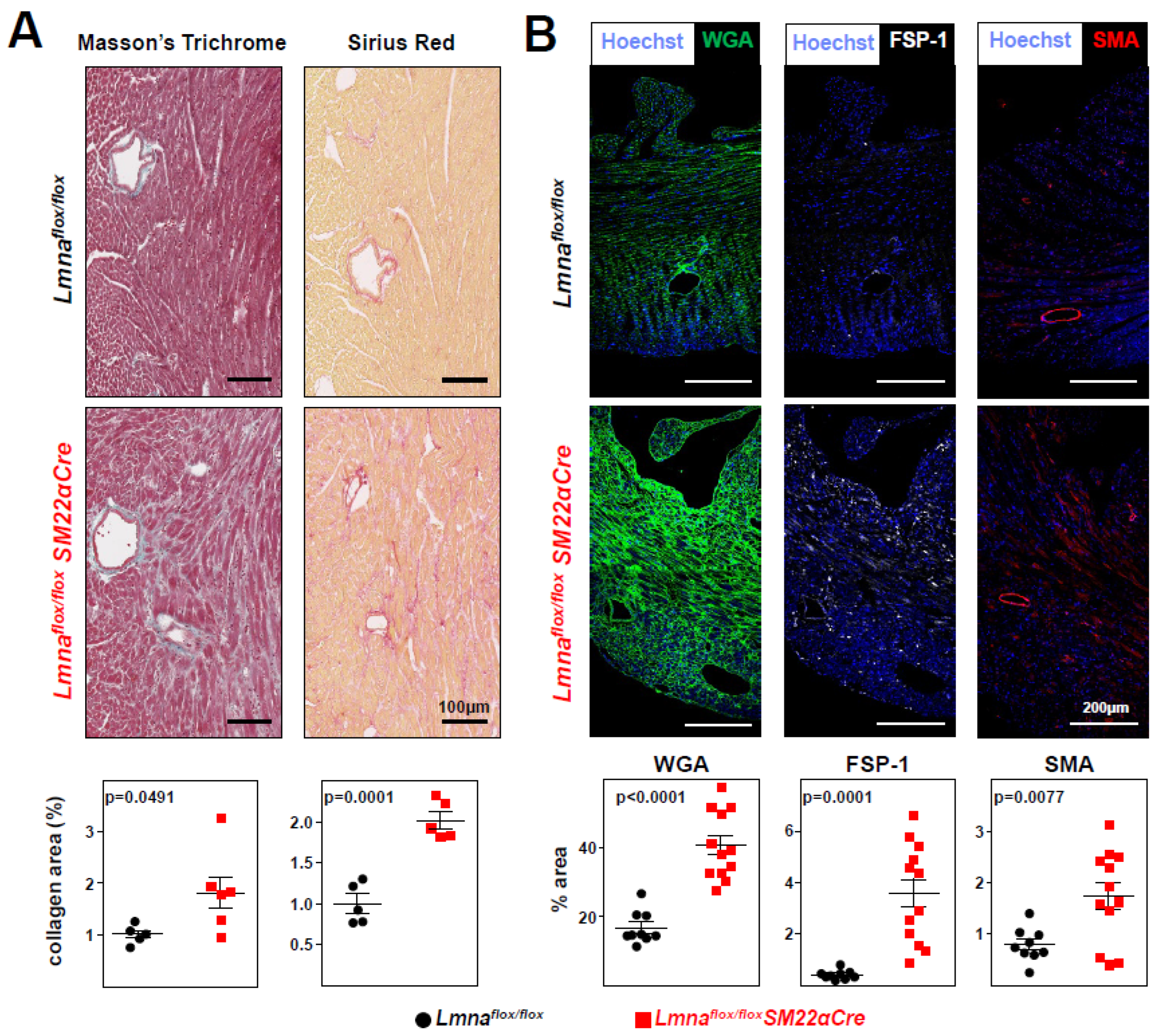
Figure 6.
Increased P-SMAD3 and caspase 3 protein levels in Lmnaflox/floxSM22αCre mice.Lmnaflox/flox and Lmnaflox/floxSM22αCre mice were examined at 4 weeks of age. (A) Representative immunofluorescence images of heart tissue showing phosphorylated SMAD3 (S423+S425; P-SMAD3; white) and nuclei (stained with Hoechst 33342; blue). The graphs show P-SMAD3-positive nuclei and median intensity fluorescence (MIF) of P-SMAD3-positive nuclei. (B) Representative western blot image of heart protein lysates from Lmnaflox/flox and Lmnaflox/flox SM22αCre mice probed with anti-P-SMAD3 antibody and anti-vinculin antibody as a housekeeping loading control. Each lane corresponds to heart tissue from one mouse. The graph shows the fold changes in all samples used in the western blot. One outlier identified with GraphPad outlier calculator in the Lmnaflox/floxSM22αCre group was eliminated. (C) Representative immunofluorescence images of caspase-3 (white) and nuclei (Hoechst 33342; blue). The graph shows caspase-3 positive area as a percentage of the total area of tissue. Statistical analysis was byunpaired two-tailed t-test. Data are mean±SEM. Each symbol represents one animal.
Figure 6.
Increased P-SMAD3 and caspase 3 protein levels in Lmnaflox/floxSM22αCre mice.Lmnaflox/flox and Lmnaflox/floxSM22αCre mice were examined at 4 weeks of age. (A) Representative immunofluorescence images of heart tissue showing phosphorylated SMAD3 (S423+S425; P-SMAD3; white) and nuclei (stained with Hoechst 33342; blue). The graphs show P-SMAD3-positive nuclei and median intensity fluorescence (MIF) of P-SMAD3-positive nuclei. (B) Representative western blot image of heart protein lysates from Lmnaflox/flox and Lmnaflox/flox SM22αCre mice probed with anti-P-SMAD3 antibody and anti-vinculin antibody as a housekeeping loading control. Each lane corresponds to heart tissue from one mouse. The graph shows the fold changes in all samples used in the western blot. One outlier identified with GraphPad outlier calculator in the Lmnaflox/floxSM22αCre group was eliminated. (C) Representative immunofluorescence images of caspase-3 (white) and nuclei (Hoechst 33342; blue). The graph shows caspase-3 positive area as a percentage of the total area of tissue. Statistical analysis was byunpaired two-tailed t-test. Data are mean±SEM. Each symbol represents one animal.
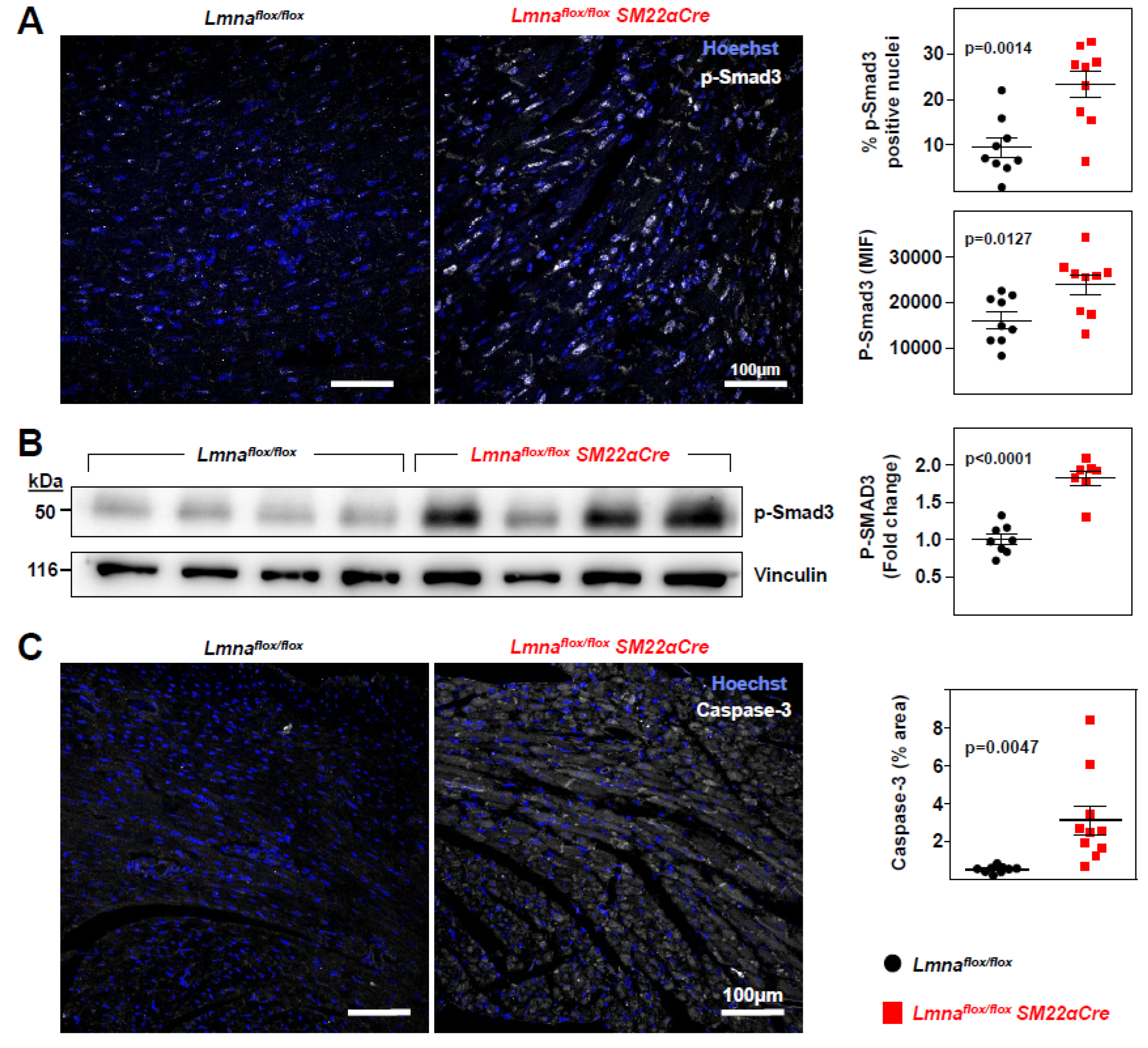
Figure 7.
Lmnaflox/floxSM22αCre mice show severe loss of cardiac function and electrocardiographic defects.Lmnaflox/flox and Lmnaflox/floxSM22αCre mice were examined at 4 weeks of age. (A) Representative echocardiography images (sagittal plane) and quantification of left ventricle (LV) function (EF, ejection fraction), right ventricle (RV) function (TAPSE, tricuspid annular plane systolic excursion), and LV wall thickness. See sagittal and longitudinal planes in supplementary videos S1-S4. (B) Representative images of hearts and tibia bones, and quantification of tibia length and heart weight. (C) Quantification of PQ, QRS, and QT intervals and T-wave steepness obtained by electrocardiography. (D) Plasma levels of creatine kinase MB isoform and troponin. Statistical analysis was by unpaired two-tailed t-test. For troponin analysis, Mann Whitney test was performed. Data are mean±SEM. Each symbol represents one animal.
Figure 7.
Lmnaflox/floxSM22αCre mice show severe loss of cardiac function and electrocardiographic defects.Lmnaflox/flox and Lmnaflox/floxSM22αCre mice were examined at 4 weeks of age. (A) Representative echocardiography images (sagittal plane) and quantification of left ventricle (LV) function (EF, ejection fraction), right ventricle (RV) function (TAPSE, tricuspid annular plane systolic excursion), and LV wall thickness. See sagittal and longitudinal planes in supplementary videos S1-S4. (B) Representative images of hearts and tibia bones, and quantification of tibia length and heart weight. (C) Quantification of PQ, QRS, and QT intervals and T-wave steepness obtained by electrocardiography. (D) Plasma levels of creatine kinase MB isoform and troponin. Statistical analysis was by unpaired two-tailed t-test. For troponin analysis, Mann Whitney test was performed. Data are mean±SEM. Each symbol represents one animal.
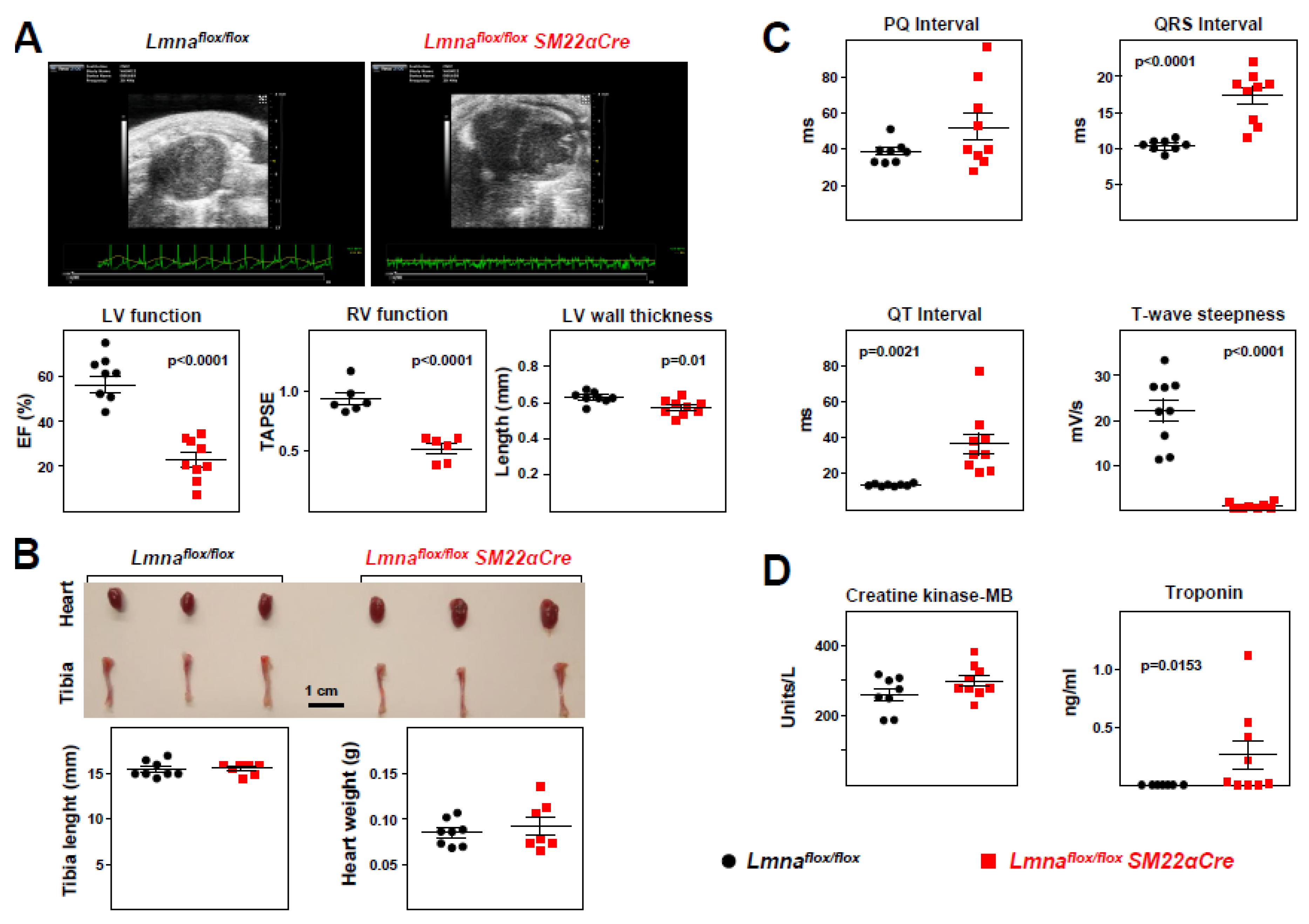
Figure 8.
Lmnaflox/floxSM22αCre mice exhibit contractile-to-synthetic phenotypic switching in vascular smooth muscle cells and vascular dysfunction in aorta. Lmnaflox/flox and Lmnaflox/floxSM22αCre mice were examined at 4 weeks of age. (A) RT-qPCR analysis of adventitia-free thoracic aorta, examininb the expression of vascular smooth muscle cell ‘contractile’ and ‘synthetic’ genes (n=5). Each replicate contained the thoracic aortas from two mice of the same genoypte and sex. Data are presented as the ΔCt fold change relative to control samples. Hprt was used as the housekeeping gene. The heatmap shows the log2 of the fold change relative to control Lmnaflox/flox mice. (B-G) Thoracic aorta rings were mounted in a wire myograph system to examine the following parameters (n=10 each genotype): diameter–force relationship and its linear regression slope (B); estimated aortic ring diameter at 100 mmHg (C); maximum response induced by 120 mmol/L KCl (D); concentration-response curves to phenylephrine, and the concentration of phenylephrine giving the half-maximal response (EC50) (E); endothelium-dependent vasodilation induced by increasing concentrations of acetylcholine; (F) and endothelium-independent vasodilation induced by increasing concentrations of diethylamine NONOate (DEA-NO) (G). Statistical differences were analyzed by unpaired two-tailed t-test in A, B (right graph), C, D, and E (right graph) or by two-way ANOVA and Fisher’s LSD multiple comparisons test in the dose-response curves in E, F, and G. Data are mean±SEM.
Figure 8.
Lmnaflox/floxSM22αCre mice exhibit contractile-to-synthetic phenotypic switching in vascular smooth muscle cells and vascular dysfunction in aorta. Lmnaflox/flox and Lmnaflox/floxSM22αCre mice were examined at 4 weeks of age. (A) RT-qPCR analysis of adventitia-free thoracic aorta, examininb the expression of vascular smooth muscle cell ‘contractile’ and ‘synthetic’ genes (n=5). Each replicate contained the thoracic aortas from two mice of the same genoypte and sex. Data are presented as the ΔCt fold change relative to control samples. Hprt was used as the housekeeping gene. The heatmap shows the log2 of the fold change relative to control Lmnaflox/flox mice. (B-G) Thoracic aorta rings were mounted in a wire myograph system to examine the following parameters (n=10 each genotype): diameter–force relationship and its linear regression slope (B); estimated aortic ring diameter at 100 mmHg (C); maximum response induced by 120 mmol/L KCl (D); concentration-response curves to phenylephrine, and the concentration of phenylephrine giving the half-maximal response (EC50) (E); endothelium-dependent vasodilation induced by increasing concentrations of acetylcholine; (F) and endothelium-independent vasodilation induced by increasing concentrations of diethylamine NONOate (DEA-NO) (G). Statistical differences were analyzed by unpaired two-tailed t-test in A, B (right graph), C, D, and E (right graph) or by two-way ANOVA and Fisher’s LSD multiple comparisons test in the dose-response curves in E, F, and G. Data are mean±SEM.

Table 1.
Primer sequences used for real-time quantitative PCR.
| Forward (5' → 3') | Reverse (5' → 3') | |
|---|---|---|
| Acta2 | AAGAGGAAGACAGCACAGCC | AGCGTCAGGATCCCTCTCTT |
| Calr | CCAGAAATTGACAACCCTGAA | CCTTAAGCCTCTGCTCCTCAT |
| Cam2 | AAGTTGATGAAATGATCAGGGAAG | TGAAGTCCTAATTACTATACATGCATA |
| Cnn1 | TGGGAGTCAAGTATGCAGAG | CTGACTGGCAAACTTGTTGG |
| Hprt | CCTAAGATGAGCGCAAGTTGAA | CCACAGGACTAGAACACCTGCTAA |
| Klf4 | TTGTGACTATGCAGGCTGTG | TAGTGCCTGGTCAGTTCATC |
| Lum | TTCACTGGGCTGCAATACC | TCCCAGGATCTTACAGAAGC |
| Mmp2 | ACCTTGACCAGAACACCATC | AGCATCATCCACGGTTTCAG |
| Nox1 | CAACAGCACTCACCAATGCC | ACATCCTCACTGACTGTGCC |
| Prkg1 | ACTGCATGTGTGGTAGAAGC | GCCAGTCAGAAGCTCATACATC |
| Smtn | AGAACACCATCACCCACATC | TCTTGTCCAGGACTCCTTCG |
| Sod1 | TGGGTTCCACGTCCATCAGTA | ACCGTCCTTTCCAGCAGTCA |
| Sox9 | AGAACAAGCCACACGTCAAG | GTCTCTTCTCGCTCTCGTTC |
| Spp1 | GGTGATAGCTTGGCTTATGG | TGGGCAACAGGGATGACATC |
| Tagln | CCCAGACACCGAAGCTACTC | GACTGCACTTCTCGGCTCAT |
| Tfam | CAGGAGGCAAAGGATGATTC | CCAAGACTTCATTTCATTGTCG |
| Vcam1 | TCAAGGGTGACCAGCTCATG | TCGTTGTATTCCTGGGAGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



