
Submitted:
19 June 2023
Posted:
19 June 2023
You are already at the latest version
Abstract
KV channel-interacting proteins (KChIPs) belong to a family of Ca2+-binding EF-hand proteins that are able to bind to the N-terminus of the KV4 channel α-subunits. As the auxiliary subunit, KChIPs are critically involved in regulating the amplitude and gating properties of KV4 channels by modulating their cell surface trafficking, voltage-dependent activation, inactivation kinetics, and recovery rate from inactivation. IKs, ICa,L, and INa can also be regulated by KChIPs. KChIPs are predominantly expressed in the brain and heart, where they contribute to the maintenance of the excitability of neurons and cardiomyocytes by modulating the KV4 currents. Interestingly, all KChIPs can act as transcription factors to control the expression of genes involved in pain, memory, and circadian regulation. Altered expression of KChIPs has been implicated in the pathogenesis of many diseases, such as arrhythmia, heart failure, Alzheimer's disease, etc. In this review, we summarize the research progress of KChIPs in their structural properties, physiological functions, and pathological roles in disease progression, and provide an overview of the therapeutic potential of KChIPs as pharmacological targets for associated disorders.
Keywords:
KV channel
; KV channel-interacting proteins
; neurodegenerative disorders
; cardiovascular diseases
1. Introduction
KV channel-interacting proteins (KChIPs) are a family of Ca2+-binding EF-hand proteins consisting of four members: KChIP1, KChIP2, KChIP3 (DREAM/calsenilin), and KChIP4 (CALP), encoded by KCNIP1-4, respectively. KChIPs have high sequence similarity and share the same conserved C-terminal core domains. However, the N-termini of KChIPs are variable in sequence and length, which defines the distinctions among them. KChIP1, KChIP3, and KChIP4 are highly expressed in the brain, whereas KChIP2 is predominantly expressed in the heart [1]. Other tissues such as lung [2], kidney [3], and gastrointestinal tract [4-6] also express low levels of KChIPs.
The biological functions of KChIPs are diverse. In 1998, KChIP3, the first reported member of the KChIP family, was identified as a presenilin (PS) binding protein and was initially named "calsenilin" [7]. A year later, the transcriptional regulatory activity of KChIP3 was reported by Carrion and coworkers, as it can bind to the downstream regulatory element (DRE) on the promoter of the human prodynorphin (PDYN) gene, repressing PDYN transcription. KChIP3 is therefore coined the acronym DREAM (downstream regulatory element antagonist modulator) [8]. In 2000, An et al. used the yeast two-hybrid system to identify three proteins of 216-, 252- and 256-amino acid length that interacted with KV4 channels, and named them KChIP1, KChIP2, and KChIP3. Interestingly, they found that KChIP3 was translated from the same DNA sequence as the previously reported calsenilin and DREAM [9]. These three landmark papers marked the beginning of research into the functions of KChIPs. Academic research has uncovered KChIPs’ function revolving around these three aspects. First, KChIPs can interact directly with intracellular proteins to regulate various cellular functions through non-transcriptional mechanisms. For example, in neurons, KChIPs interact with PS to regulate the activity of γ-secretase [7,10]. In platelets, KChIP3 exerts a procoagulant effect by directly binding to and activating phosphatidylinositol 3-kinase-Iβ [11]. Second, KChIPs can also regulate physiological and pathological responses at the transcriptional level. In addition to KChIP3, other KChIPs also have transcriptional activity to modulate the transcription of genes regarding circadian rhythm, pain, memory, inflammatory response, immune response, and hormone secretion. Last but not least, KChIPs regulate the subcellular localization and gating properties of fast-inactivating KV4 channels mainly in neurons and cardiomyocytes. In addition, KChIPs are involved in the regulation of other important ion channels in the heart, including KV1.5, CaV1.2, and NaV1.5. Therefore, KChIPs play a pivotal role in controlling the electrophysiological function of cardiomyocytes. Among these, the functions of KChIPs as transcriptional regulators and auxiliary subunits of ion channels have been studied the most, which is the focus of this review.
KChIPs have been implicated in the pathogenesis of several diseases, including cardiac arrhythmias, cardiac hypertrophy, neurodegenerative diseases, and epilepsy. Small molecules acting on KChIPs have been shown to enhance or inhibit the activity of KChIPs [12], which is instructive for the development of drugs targeting KChIPs to treat related diseases. In this review, we summarized the function of KChIPs and their multiple roles in disease progression. Meanwhile, we updated the small molecule drugs targeting KChIPs to provide new guidance for future basic and translational research in this field.
2. The Molecular Properties of KChIPs
2.1. The Structure of KChIPs
KChIPs are bipolar proteins of approximately 217-270 amino acids, with a C-terminal core domain and a variable N-terminal domain containing multiple post-translational modification sites (Figure 1). The C-terminal domain is highly conserved and contains a region that interacts with potassium voltage-gated channel subfamily D member (KV4) and four EF-hand motifs [13,14]. Of the 4 EF-hands, EF-3 and EF-4 can bind Ca2+, EF-2 can bind Mg2+, EF-1 is degenerate [13,15]. KChIPs undergo conformational modifications when bound to divalent cations [16]. There are several post-translational modification sites at the N-terminus of KChIPs. For example, KChIP1 has an N-terminal myristoylation motif [17]. KChIP2 can undergo dynamic palmitoylation (Cys45 and Cys46) [18,19], namely palmitoylation and depalmitoylation. KChIP3 can undergo sumoylation (K26 and K90) [20], palmitoylation (Cys45 and Cys46) [18] and phosphorylation (Ser63 and Ser95) [21,22]. These diverse post-translational modification sites and patterns in the N-terminus determine the subcellular localization of KChIPs. Myristoylation of KChIP1, palmitoylation of KChIP2 and palmitoylation and phosphorylation (Ser95) of KChIP3 all enhance KChIPs localization to the cell membrane. In contrast, depalmitoylation of KChIP2 and sumoylation of KChIP3 increase their nuclear distribution. Furthermore, phosphorylation of KChIP3 at Ser63 inhibits its cleavage by caspase-3 [21].
KChIP4.4 (KChIP4a) [23] and KChIP3x (KChIP3b) [24], which possess the functional KV4 channel inhibitory domain (KID), an N-terminal membrane-spanning segment, are able to exert the inhibitory effect on KV4 channels. The KID contains an ER retention motif consisting of six hydrophobic and aliphatic residues 12-17, which interferes with KV4 surface expression. Residues 19-21 (VKL motif), adjacent to the ER retention motif, enhance KV4 inactivation and keep it in the closed state, thereby inhibiting channel current [14,25,26].
2.2. Regulation of KChIPs expression
2.2.1. Transcriptional Level
Some physiological functions or pathological conditions are underpinned by changes in the expression levels of KChIPs. Unfortunately, mechanistic studies on KChIPs expressional regulation are limited and have mainly focused on KChIP2. A number of signaling pathways have been identified that affect the transcription of KChIP2, including NF-κB [27], CaMKII [28], NFAT [29], MAPK [30-32] and Notch [33]. Our previous work showed that NF-κB can directly bind to the promoter region of the Kcnip2 gene and repress its transcription [27]. Several factors that downregulate KChIP2 expression, including ligands of TGF-β receptors [34], α-adrenergic receptors [35], β-adrenergic receptors [36], and C-reactive protein [37], are partially dependent on the activation of NF-κB pathway. Additionally, Ca2+ reduce the mRNA expression of KChIP2 through the Ca2+/calmodulin-dependent protein kinase II (CaMKII) [28] and the calcineurin/NFAT signaling pathway [29]. Under physiological conditions, inhibition of MEK1 increases whereas activation MEK1 decrease KChIP2 mRNA level. In phenylephrine-induced hypertrophic cardiomyocytes, inhibition of JNK1 rescues the down regulation of KChIP2. These findings suggest that mitogen-activated protein kinase (MAPK) pathways are also involved in the regulation of KChIP2 expression [30]. Notch signaling inhibits the expression of KChIP2 expression and thereby contribute to the electrophysiological differences between neonatal and adult cardiomyocytes [33]. Several transcription factors have also been reported to bind directly to the promoter region of KCNIP2 to regulate its expression. For example, cyclic AMP response element binding protein (CREB) [38], Krüppel-like factor-4 [39] and Krüppel-like factor-5 [40] are able to promote KCNIP2 transcription (Figure 2).
2.2.2. Protein Level
It is worth noting that as an integral component of the KV4 channel complex, the protein level of KChIPs is tightly coupled to KV4 and dipeptidyl peptidase-related proteins (DPPs). On the one hand, deletion of Kv4.2 significantly reduces KChIP1, KChIP2 and KChIP3 expression in the mouse brain. In particular, deletion of Kv4.2 results in reduced expression of KChIP2 and KChIP3 in the hippocampus, KChIP2 in the striatum, and KChIP1 and KChIP3 in the cerebellum [41]. Furthermore, KChIP2, KChIP3 and KChIP4 protein expression levels in cortical pyramidal neurons were extremely low in mice with a targeted deletion of either KV4.2 or KV4.3 [42]. Nerbonne et al. showed that loss of KV4.2 in cortical pyramidal neurons resulted in targeted degradation of KChIP3 protein [43]. On the other hand, DPPs also affect the expression of KChIPs. Downregulation of DPP6 reduces KV4.2 and KChIPs in CA1 hippocampal neurons [44]. Similarly, knockdown of DPP10 in the dorsal root ganglion (DRG) neurons resulted in downregulation of KChIP1 and KChIP2 [45].
In addition, members of the KChIP subfamily can have an effect on the expression of other KChIPs. For example, when KChIP3 was deficient in the cortex, the expression of other KChIPs were increased to compensate for KChIP3 deficiency [42,46]. Interestingly, there is evidence that KChIP3 can be a negative regulator of its own expression [46].
3. Biological Function of KChIPs
3.1. KChIPs are Auxiliary Subunits of KV4 Channels
3.1.1. The Interaction of KChIPs with KV4 Channels
KV4 channels are members of the KV channel superfamily. In mammals, the KV4-family consists of four members: KV4.1, KV4.2, and two splice variants of KV4.3. All KV4 channels share a functional core that is assembled as a tetramer of pore-forming α-subunits around a central pore [12]. The KV4 α-subunit contains an N-terminal cytoplasmic domain with an N-terminal hydrophobic segment, the KV channel assembly domain (T1 domain) under the tetrameric channel pore domains, a transmembrane domain with six transmembrane helices S1-S6, and the C-terminal cytoplasmic domain [47]. The KV4 channels are highly expressed in brain, heart and smooth muscle cells. The neuronal KV4 channels underlie the transient A-type current (IA), sustaining the homeostatic excitability of neurons [48]. In cardiomyocytes, KV4 channels control the early repolarization phase of the action potential by mediating the transient outward current (Ito) [49]. In gastrointestinal smooth muscle cells, KV4 currents are involved in shaping the slow wave activity and mechanical responses [4].
However, KV4 channels could not carry out normal physiological processes on their own. Two auxiliary subunits, KChIPs and DPPs, are essential for the physiological function of KV4 channels. Mechanistically, KChIP1-3 can bind to the cytoplasmic domains of KV4 α-subunits, thereby increasing total KV4 current, slowing channel inactivation, and accelerating recovery from inactivation. An et al. also found that KChIP4a, which contains the KID, can eliminate the fast inactivation of the KV4 current. Located on the intracellular side, KChIPs are anchored laterally to the T1 domains of KV4 and clamp two adjacent KV4 N-terminals in a 4:4 ratio [50]. The crystal structures of the KChIP1-KV4 complex have revealed some of the interaction sites that mediate the regulatory effects of KChIPs. In the first site, the hydrophobic pocket formed by the H10 helix of KChIP1 interacts with the KV4.3 channel N-terminal hydrophobic segment, which is responsible for KV4 inactivation. The N-terminal hydrophobic peptide of KV4.3 is sequestered in an elongated hydrophobic groove on the surface of KChIP1 and replaces the H10 helix of KChIP1 [51]. Meanwhile, KChIP1 can interact with the neighboring KV4.3 N-terminals. The second site is formed by the interaction between the KChIP1 H2 helix and the neighboring T1 domain through hydrophobic interactions and hydrogen bonds, stabilizing the KV4.3 tetramerization. In the second site, the residues 70-78 in the KV4 T1 domain forms a KChIP-specific docking loop to mediate the interaction with KChIPs. Residues R51, R58 and E63 on the KChIP1 H2 helix, which is also involved in the formation of the second site, interact with residues D39 and R60 on the KV4.3 T1N linker to form the third site, providing further stability to the KV4.3 tetrameric intracellular domain [51,52]. In addition, KChIP1 is able to capture the C-terminal cytoplasmic S6 helix on KV4.2 through the hydrophobic interactions [47].
3.1.2. KChIPs Modulate the Gating Properties of KV4 Channels
KV4 are the rapidly inactivating (A-type) KV potassium channels that generate currents at subthreshold membrane potentials. They are characterized by fast activation, fast inactivation, and fast recovery from inactivation. The inactivation of KV4 channels is classified into two types: open-state inactivation and closed-state inactivation. Closed-state inactivation is the main type, indicating that KV4 channels can be inactivated directly from the closed state [53]. Binding of KChIPs to the N-terminus of KV4 modulates the gating properties of KV4 channels. Specifically, KChIP1-3 augment KV4 currents through the following electrophysiological effects: shifting the activation midpoint of voltage activation to more negative potentials, slower inactivation, and acceleration of recovery from inactivation [9]. When co-expressed with KChIPs, the activation time of KV4 was slightly prolonged compared to KV4 alone. While the midpoint for KV4 of voltage activation significantly shifted to more hyperpolarized potentials [9]. In contrast, the modulation of KV4 gating by KChIPs is mainly manifested in the inactivation kinetics. Heterologous co-expression of KV4 and KChIPs significantly prolongs the inactivation time of KV4 channels. To be specific, KChIPs eliminate open-state inactivation and accelerate closed-state inactivation of KV4 channels [9,47,54]. However, it is still unclear about the molecular mechanism by which KChIPs control KV4 inactivation. The EF-hands were reportedly involved in the regulation of KV4.3 inactivation by sensing intracellular Ca2+ levels [55]. Recently, breakthroughs have been made in the structural basis of KChIPs that regulate the inactivation kinetics of KV4 channels. Kise et al. reported that KChIP1 is able to capture and sequester both the N-terminal hydrophobic segment and the C-terminus of KV4.2 channels. KChIP1, on one hand, binds the C-terminal intracellular S6 helix to stabilize the S6 conformation. It also binds the N-terminal hydrophobic segment and two T1 domains from the neighboring subunit of KV4.2. Together, these KChIP1-mediated structural features prevent open-state inactivation and accelerate closed-state inactivation of KV4.2 [47]. By truncating the N-terminal or C-terminal helix of KV4.2, respectively, Ye et al. demonstrated that the interactions of KChIP2 with the KV4.2 N-terminal helix play a more prominent role in modulating channel inactivation [56]. Moreover, KChIPs accelerate the rate of recovery of KV4 channels from inactivation in a Ca2+-independent manner [55].
It's interesting to note that a specific KChIP isoform KChIP4a has been reported to delay KV4.3 channel activation, abolish rapid inactivation and impedes channel closure after opening. Therefore, co-expression of KChIP4a with KV4 α-subunits converts the A-type KV4 current to a slowly inactivating delayed rectifier-type potassium current [23]. The similar suppressive effect on KV4 currents was also found for KChIP3x (KChIP3b) in subsequent research [24].
3.1.3. KChIPs Modulate the Trafficking of KV4 Channels
KV4 trafficking to the cell surface are vital for the maintenance of current density [9,54]. Efficient trafficking of KV4 to the cell surface depends on KChIP binding to its N-terminal domain [54]. Shibata et al. postulated that in the absence of KChIPs binding, the hydrophobic domains of multiple KV4.2 subunits will oligomerize, resulting in aggregation, misfolding, and consequently retention in the ER for degradation. Interaction of KChIP with KV4.2 leads to changes in the overall molecular properties of KV4.2 by concealing the N-terminal hydrophobic domain. These changes include an increased protein stability, phosphorylation, detergent solubility and cell surface localization [57]. Further research has shown that in the absence of KChIP1, the KV4 protein can be released from the ER but then trapped in the Golgi complex. When KV4 channels and KChIP1 are co-expressed in heterologous expression systems, their translocation to the plasma membrane is facilitated [27, 28]. In addition, KChIP2 and KChIP3 have also been shown to increase KV4.2 trafficking from the ER and Golgi complex to the plasma membrane [58]. In detail, KV4.2 is targeted by KChIP2 to cholesterol-rich lipid rafts in the cell membrane [59]. By contrast, KChIP4a, which contains the KID motif, does not have those effects on KV4.2. Meanwhile, KChIP4a suppresses KV4 trafficking by forming a ternary plasma membrane complex with KV4.2 and other KChIPs [57]. Therefore, the role of KChIPs in Kv4.2 trafficking may also contribute to its regulation on KV4 current (Figure 3).
3.1.4. KChIP Ligands Affect KChIP Regulation on KV4
Since KChIPs are essential for the control of electrophysiological processes in cells, inhibiting or enhancing their effect on KV4 channels is a promising avenue for pharmacological research in the treatment of KV4-mediated channelopathies. Several small-molecules that bind to KChIPs have been shown to modulate KV4 currents. For example, the binding of arachidonic acid to the hydrophobic C-terminus of KChIP1 accelerates KV4 inactivation and decreases current amplitude [60]. CL-888, a diaryl-urea compound, can bind to KChIP1 [61] and KChIP3 [62] to counteract their regulatory effects on KV4, reducing peak current amplitude and accelerating inactivation kinetics. IQM-PC330 and IQM-PC332, the derivatives of CL-888 modified by Lopez-Hurtado et al., have a more refined blocking effect on KV4 currents. IQM-PC330 and IQM-PC332 inhibit KV4.3 channels not only by reducing peak current amplitude and accelerating their inactivation, but also by delaying their recovery from inactivation. Mechanistically, they act as KChIP3 ligands to reverse the regulatory function of KChIP3 on KV4.3 channel gating properties. It's interesting to note that the hydrophobic combination of IQM-PC332 and KChIP3 accelerates the activation kinetics of KV4.3 current at low concentrations (0.01 to 0.1 M) and reverses its effect on channel gating at higher concentrations. However, inactivation recovery kinetics were significantly reduced at all concentrations [63]. They also identified IQM-266 as a novel KChIP3 ligand with similar inhibitory properties to IQM-PC330 and IQM-PC332 [64]. Recently, IQM-266 was reported to bind KChIP2 and increase KV4.3/KChIP2 currents [65].
The sulfonylurea compound NS5806, a ligand of KChIP3, has been demonstrated to activate KV4.3 channels in neurons. NS5806 delays the inactivation of IA and slightly reduces the maximum peak current [66]. This effect is based on the Ca2+-dependent binding of NS5806 to the hydrophobic site at the C-terminus of KChIP3, which facilitated the binding affinity between KChIP3 and KV4.3 as well as reducing their dissociation rate [67]. In cardiomyocytes, however, the control of KV4 currents by NS5806 is controversial. In canine cardiomyocytes, NS5806 functions as an Ito activator to increase the amplitude of the KV4.3/KChIP2 peak current and significantly slow the current decay [68]. While in mouse ventricular myocytes NS5806 has the opposite effect, as shown by a decrease in the amplitude of native Ito and significant acceleration of current inactivation [69]. Furthermore, in rabbit ventricular myocytes, NS5806 dramatically raised Ito amplitude, while in atrial myocytes, the Ito amplitude was repressed [70]. The mechanism by which NS5806 elicits different responses in different species and cell types remains unclear. More interestingly, repaglinide, a commonly used antidiabetic drug, can bind to KChIP3 and exert an inhibitory effect on KV4 channels [62]. Altogether, the discovery of these KChIPs ligands has improved our knowledge of the interaction between KV4 and KChIPs and has paved the way for future pharmaceutical development for the treatment of neurodegenerative and cardiovascular diseases involving KV4/KChIPs.
3.2. Role of KChIPs in Regulating Other Ion Channels
3.2.1. KV1.5
KV1.5 is another vital potassium channel characterized by rapid activation and rapid inactivation that is highly expressed in both brain and heart. In human heart, KV1.5 is expressed abundantly in atrial myocytes and mediates the ultra-rapid delayed rectifier current (IKr). In adult mouse heart, KV1.5, which encodes the slow delayed rectifier K+ current (IKs), is involved in the repolarization of ventricular myocytes [71]. Several studies have demonstrated the contribution of KChIPs in the modulation of KV1.5 channels. In contrast to their augmenting effect on KV4 currents, KChIPs negatively regulate KV1.5-encoded K+ currents. In transiently transfected HEK293 cells, KChIP1 and KChIP2 attenuate KV1.5 currents by inhibiting the trafficking of KV1.5 channels to the cell surface [72]. Moreover, in the ventricles of Kcnip2-/- mice, KV1.5 mRNA levels were significantly increased and IKs was upregulated [73]. However, the structural basis of KChIP1 and KChIP2 negative regulation of cardiomyocyte KV1.5 channels in vivo remains unclear and needs further investigation.
3.2.2. CaV1.2
The high-voltage-activated L-type Ca2+ channel, CaV1.2, located on the t-tubule sarcolemma, is the major calcium channel type in cardiomyocytes. CaV1.2 channels are macromolecular complexes that composed of α1, α2δ and β subunits [74]. The cardiac L-type Ca2+ current (ICa,L) mediated by CaV1.2 is essential for cardiomyocyte depolarization and contraction. KChIPs have a dual effect on the regulation of the cardiac L-type Ca2+ current (ICa,L). On one hand, KChIP2 regulates the ICa,L through direct interaction with the intracellular N-terminus of the CaV1.2 α1C subunit. Without raising CaV1.2 protein expression or trafficking to the plasma membrane, KChIP2 increases ICa,L current density by impeding the N-terminal inhibitory module [75]. On the other hand, KChIP2 and KChIP3 bind and repress the transcription of the Cacnb2 [76] and Cacna1c [77] genes, respectively, which encode the corresponding β2-subunit and α1C-subunit of CaV1.2 channels.
3.2.3. NaV1.5
NaV1.5, which is encoded by SCN5A, is highly expressed in heart. By mediating the rapid influx of Na+, NaV1.5 dominates the rapid depolarization phase of action potential. Previous studies suggest that Kv4.3 and NaV1.5 work coordinately and can regulate each other [78]. Deschênes et al. found that co-expression of KChIP2 with NaV1.5 increased NaV1.5 current density but had no effect on Na+ current gating properties. Whereas silencing of Kcnip2 resulted in a significant decrease in Scn5a mRNA level and complete inhibition of voltage-dependent Na+ currents [79]. However, this evidence is based on heterologous expression systems. The electrophysiological significance of the cross-regulation of KChIP2, NaV1.5 and KV4.3 in cardiomyocytes requires further study.
3.3. KChIPs are Ca2+-Dependent Transcriptional Factors
KChIP3 is the first KChIP identified to have transcriptional activity as it can bind to the downstream regulatory element (DRE) downstream of the TATA box in the human PDYN gene promoter [8]. Subsequent studies have demonstrated that all the KChIPs have Ca2+-dependent DRE binding affinities that block gene transcription in the form of homotetramer or heterotetramer. The binding of KChIP3 to DRE sequences is regulated by divalent cations [8]. Tetramer formation is required for KChIP3 in binding to DRE. Mg2+ and Ca2+ are the most important factors affecting the ability of KChIP3 to bind to DNA. The combination of Mg2+ and KChIP3 stabilizes the tertiary structure of the protein and promotes DNA binding [80]. In contrast, as intracellular Ca2+ levels increase, the Ca2+-bound KChIP3 switches to a denser dimer structure [81], preventing KChIP3-DRE interaction. Furthermore, direct interaction of nucleoprotein C-terminal binding protein [82] and αCREM [83] with KChIP3 enhances and inhibits their transcriptional repressive functions, respectively. In the nucleus, in addition to direct binding to DRE in target genes, KChIP3 also forms transcriptional regulatory complexes with nuclear proteins such as cAMP response element-binding protein [84], thyroid transcription factor 1 [85] and nuclear receptors [86]. Genes regulated by KChIP3 include but not limited to the following: Pdyn, Fos [87], Hrk [88], Bdnf [89], Ifng, Il2 and Il4 [90], Klf9 [91], Npas4, Nr4a1, Mef2c, Junb [46], Tnfaip3 [92], Tg [85], Ttf2/Foxe1, Pax8 [93], Aanat, Fra-2, Crem [94], GnRH [95], Gfap [96], Ncx3 [97], Cacna1c [77], Mid1 [98], Cant1 [99], PAX6, NRG1 [100] and GCM1 [101] (Figure 4).
Through the transcriptional regulation of the above-mentioned genes, KChIP3 has been shown to be multifunctional and is critically involved in the regulation of physiological and pathological functions of neurons, endocrine cells, immune cells, endothelial cells and hematopoietic cells. For example, in the pineal gland, KChIP1-4 are involved in the regulation of rhythmically expressed genes engaged in circadian rhythms. By binding to the DRE sites of arylalkylamine N-acetyltransferase (Aanat), inducible cAMP early repressor (Crem), and Fos-related antigen-2 (Fra-2), KChIP1-4 are able to repress the basal and induced transcription of these circadian rhythm-related genes [94]. In the pancreas, KChIP3 has been detected in islet α- and β-cells. KChIP3 represses the transcription of the Pdyn gene in a Ca2+-dependent manner and thereby affect glucagon release [102]. In the thyroid, KChIP3 inhibits the expression of thyroglobulin by binding directly to the DRE of the thyroglobulin gene and blocking thyroid-specific transcription factors such as TTF-1, TTF-2 and Pax8 [85,93]. KChIP3, as well as KChIP2, are expressed in T- and B cells to regulate immunological responses. In T lymphocytes, KChIP2 and KChIP3 act as transcriptional repressors to inhibit the expression of IFN-γ, IL-2, and IL-4 [90]. By inhibiting the transcription of the proliferation-related gene Klf9 and the protein translation-related gene Eif4g3 in B cells, KChIP2 and KChIP3 govern B cell proliferation and IgM and IgG protein synthesis [91]. In lung endothelial cells, ranulocytes, and macrophages, KChIP3 can promote the NF-κB pathway-mediated inflammatory response by suppressing the expression of TNFAIP3 gene, which encodes the anti-inflammatory deubiquitinase A20 [92,103] (Figure 4).
4. KChIPs and Diseases
4.1. Neurological Diseases
4.1.1. Epilepsy
The International League Against Epilepsy (ILAE) classification of epilepsy types includes focal epilepsy, generalized epilepsy, combined generalized and focal epilepsy, and unknown epilepsy [104]. All of these seizure types share the pathophysiological characteristic of increased neuronal excitability and synchronicity. Among them, temporal lobe epilepsy is the most common focal epilepsy that can further be subdivided into mesial temporal lobe epilepsy and lateral or neocortical temporal lobe epilepsy [105]. Adults with intractable epilepsy are most commonly affected by epilepsy of the mesial temporal lobe, which is the chronic and pharmacoresistant form of epilepsy. In this type of epilepsy, seizures originate from the hippocampus, entorhinal cortex, amygdala and parahippocampal gyrus [106]. Abundantly expressed in the hippocampus, KChIPs control the frequency of slow repetitive spike firing and attenuate action potential backpropagation by modulating the properties of KV4 channels. Thus, KChIPs play a key role in maintaining neuronal excitability [107]. To date, several studies have implicated KChIPs downregulation in temporal lobe epilepsy. For example, the hippocampus of patients with medically refractory temporal lobe epilepsy had markedly reduced KChIP3 immunoreactivity compared to normal brain [108]. By contrast, Kcnip2-/- mice showed enhanced excitability in hippocampal pyramidal neurons and increased susceptibility to epilepsy [109]. Furthermore, in models of temporal lobe epilepsy that progressed to status epilepticus, i.e., very prolonged seizures, changes in KChIPs expression were observed in specific hippocampal subfields (CA1 and CA3, but not CA2). For example, in the hippocampus of the pilocarpine-induced rat epilepsy models, loss of KChIP1 immunoreactivity in interneurons and reduction of KChIP2 in the stratum radiatum of the CA1 region were observed [110]. In the kainic acid-induced mouse epilepsy model, KChIP3 expression is reduced in the cortical area and CA3 region of the hippocampus in status epilepticus [108]. Nevertheless, the role of KChIPs in the pathogenesis of epilepsy remains an open question that requires further in-depth studies.
4.1.2. Pain
A key factor in the development and maintenance of neuropathic pain is neuronal excitability. The physiological pain circuit can be briefly summarized as follows: physicochemical signals from noxious stimuli transduces through ion channels and purinergic channels to evoke action potentials. These action potentials are amplified by Na+ channels to produce pain. The electrical signals are carried by unmyelinated C-fibres and thinly myelinated Aδ-fibres to the DRG in the body and the trigeminal ganglion (TG) in the face, where their cell bodies are located and project to the dorsal horn of the spinal cord and the medulla oblongata, respectively. After integration and processing, the pain input is transmitted via several ascending tracts to various projection sites in the brain. To process the sensory and discriminative aspects of pain, the lateral spinothalamic tract projects to the lateral thalamus. Medial projections of the spinothalamic and parabrachial tracts to the medial thalamus and limbic structures mediate the emotional and aversive components of pain. In pathological conditions such as inflammation, neuropathy and diabetes, physiological pain is converted into pathological pain, which manifests as increased sensitivity to painful stimuli (hyperalgesia) [111].
KChIP3 is critically involved in the regulation of pain. As Costigan et al. proposed: "No DREAM, no pain" [112]. Wang et al. showed that knockdown KChIP3 expression in the spine by lentivirus-mediated shRNA produced analgesic effects [113]. Cheng et al. found that Kcnip3-/- mice had significantly reduced behavioral responses to acute thermal, mechanical, chemical, and visceral pain (analgesia) [87]. Benedet et al. showed that transgenic mice expressing a constitutively active KChIP3 mutant exhibited hyperalgesia after acute trigeminal nerve stimulation, whereas Kcnip3-/- mice exhibited a hypoalgesic response [114]. The regulatory mechanism of KChIP3 in pain is extremely complex. On the one hand, KChIP3 is expressed in the neurons of the DRG [115,116] and the TG [20], where it represses the expression of genes related to pain processing, such as Pdyn [8,87], Fos [8], Bdnf [89] and Ctsl [114]. In particular, PDYN is cleaved into dynorphin which acts as an endogenous ligand of κ-opioid receptors to inhibits neuronal excitation and nociception [117]. Direct binding of KChIP3 to the DRE sequence of the prodynorphin gene inhibits the transcription of Pdyn, which promotes pain hypersensitivity. In contrast, loss of KChIP3 resulted in increased expression of PDYN and enhanced activation of κ-opioid receptors [8,102]. On the other hand, KChIP3 interacts with several receptors and ion channels involved in pain sensing and transmission. These include N-methyl-D-aspartate receptor (NMDAR) [118], transient receptor potential vanilloid 1 (TRPV1) channels [115], KV4 [119], as well as the T-type Ca2+ channels [120]. KChIP3 is therefore a potential target for the development of analgesic drugs. It has been reported that IQM-PC332, a ligand of KChIP3, can reduce mechanical hypersensitivity in mice with chronic constriction injury of the sciatic nerve. Mechanistically, IQM-PC332 binds to KChIP3 and reduces ionic currents mediated by TRPV1 channels, KV4.3 channels, low voltage-activated T-type Ca2+ channels and high voltage-activated Ca2+ channels in DRG neurons [121].
Interestingly, the "no DREAM, no pain" hypothesis does not seem to apply to rats. Kcnip3-/- rats showed a higher pain sensitivity, which is in contrast to what was found in studies with Kcnip3-/- mice. Guo et al. recently found that after global knockout of KChIP3 using CRISPR/Cas9 technology, the pain sensitivity of rats to acute and chronic pain stimuli was enhanced [122]. Part of the mechanism by which KChIP3 exerts the analgesic effect in rats has already been reported. For example, in the rat inflammatory pain model, TRPV1 in nociceptive sensory neurons undergoes functional sensitization, resulting in Ca2+ influx. Meanwhile, upregulation of KChIP3 protein expression resulted in enhanced binding of KChIP3 to the N- and C-terminus of TRPV1 via its N-terminal 31-50 fragment, which inhibits TRPV1 cell surface localization and thereby exerting an analgesic effect [115]. Unfortunately, it is still unclear what causes the discrepancy between Kcnip3-/- mice and Kcnip3-/- rats. To better understand the role of KChIP3 in pain transmission, the mechanism behind this difference needs to be further identified.
4.1.3. Memory Dysfunction
Learning is the process by which new information about the world is acquired, and memory is the process by which knowledge is stored. The cellular basis of learning and memory is thought to be the process by which synapses undergo bidirectional changes in synaptic strength, known as synaptic plasticity. Among the different types of synaptic plasticity, two opposite forms of synaptic plasticity, long-term potentiation (LTP) and long-term depression (LTD), have been the most studied [123]. LTP is the strengthening of synapses following repeated synaptic activity. Inhibition of LTP resulted in impaired learning or failure to retain memories. While LTD is the synaptic framework of weakening synaptic strengths, contributing to learning by engaging in a functional interplay with LTP [124].
Several brain regions are involved in memory, including the hippocampus, neocortex, amygdala, basal ganglia and cerebellum. Of these, the hippocampus is essential for the spatial representation of the environment and the ability to recall specific events, or 'episodic memory' [125]. KChIP3 is highly expressed in the hippocampus and is also involved in the regulation of learning and memory. Alexander et al. reported that KChIP3 knockout mice exhibited enhanced memory in a contextual fear-conditioning paradigm. They also found that translocation of KChIP3 from the membrane to the nucleus was increased in mouse hippocampal neurons following the fear conditioning training paradigm [126]. In the cell nucleus, KChIP3 functions as a transcriptional repressor to regulate the expression of genes involved in memory formation, such as Pdyn, Fos, Bdnf and Npas4 [46,126]. In addition, Fontán et al. reported that loss of KChIP3 not only enhances LTP and improves learning and memory in young mice, but also improves cognition and slows age-related brain degeneration in old age [127]. Mechanistically, KChIP3 interacts with CREB, a key transcription factor involved in memory, and negatively regulates CREB-dependent transcription. This raises the threshold for CREB activation in learning and memory. Therefore, in the Kcnip3-/- mice, the threshold for CREB phosphorylation and CREB-CBP interaction in learning and memory is lowered, and the timing of CREB phosphorylation and CBP recruitment to CREB is shortened, thereby promoting CREB-dependent transcription during learning. Consistent results were also confirmed in daDREAM (dominant active DREAM) transgenic mice. daDREAM is a Ca2+-insensitive double mutant of KChIP3 (EF-hand, leucine-charged residue-rich domains) that actively represses KChIP3 target genes and prevents the Ca2+-dependent derepression function of KChIP3. Transgenic mice expressing the daDREAM showed significant impairments in learning and memory [46].
The mechanism by which KChIP3 is involved in memory regulation also involves its protein-protein interactions. NMDAR is the major ionotropic glutamate receptor in the central nervous system and plays an important role in both synaptic transmission and plasticity [128]. NMDARs are required for LTP and LTD of synaptic transmission [129]. When Ca2+ binds to KChIP3, the interaction between KChIP3 and PSD95 is released, allowing NMDA to function as a normal receptor. The EF hands mutant KChIP3, which lacks Ca2+ binding ability, inhibits NMDAR function in mouse hippocampal CA1, impairs LTD, and thereby reduces consolidation of hippocampus-dependent contextual fear-conditioned reflexive memory [130]. In addition, KV4.2 channels play an important role in both synaptic plasticity and cognition, which in part contributes to the regulatory effect of KChIP3 on learning and memory [131].
4.1.4. Alzheimer's Disease
Amyloid beta-protein (Aβ) deposition is the main feature of AD which has toxic effects on neurons [132]. PS are the catalytic core of the high-molecular-weight enzyme complex γ-secretase, which can mediate γ-cleavage of β-amyloid precursor protein to produce Aβ [133]. KChIP3 was initially discovered as being capable of binding to the C-terminal fragment of presenilin 1 (PS1) and PS2 via the C-terminal 103 amino acids encoded by ALG3 [7]. The binding of KChIP3 to PS is dependent on the presence of Ca2+ [134]. Existing studies have shown that mutations of PS1 and PS2 lead to increased Aβ formation and apoptosis, thus causing early familial Alzheimer's disease (AD) [135]. In the brains of patients with AD, the expression of KChIP3 was increased in neurons and reactive astrocytes, which is consistent to the regions with pathological changes of AD [136]. This suggests that KChIP3 may be involved in the pathogenesis of AD. Zaidi et al. subsequently showed that KChIP3 stabilizes the structure of the PS in cerebellum and hippocampus [137]. Jo et al. reported KChIP3 as a pro-apoptotic protein that facilitates cell death induced by Aβ production [138]. Overexpression of KChIP3 increases the enzymatic activity of the presenilin-γ-secretase complex [139]. Aβ increased KChIP3 expression in cultured neurons, while blocking its expression protected neuronal cells from Aβ toxicity [136]. Repaglinide has been shown to inhibit the KChIP3-PS2 interaction, suggesting a novel avenue for future AD treatment [140].
KChIP3 also regulates the intracellular Ca2+ signaling, the dysregulation of which has been implicated in the development of AD [141]. In AD models, increased Ca2+ release from ryanodine-sensitive Ca2+ stores had been observed in neurons [142]. Lilliehook et al. showed that overexpression of KChIP3 can enhance apoptosis by increasing ER Ca2+ release in glioma cells [143]. Fedrizzi et al. reported that transient co-expression of KChIP3 with PS facilitaed Ca2+ release from the ER in neurons [144]. Mechanistically, KChIP3 binds to the DRE sequence of the isoform 3 of the Na+/Ca2+ exchanger (Ncx3) gene to repress its transcription, thereby regulating intracellular Ca2+ homeostasis [97]. In mouse hippocampal and cortical neurons, KChIP3 regulates intracellular calcium-induced Ca2+ release through direct protein-protein interaction with the ryanodine receptor [145]. Furthermore, KChIP3 appears to be involved in age-related brain degeneration [127], which is a well-known clinical symptom of AD. Although the current molecular understanding of the relationship between KChIP3 and AD pathogenesis is limited and insufficient, these findings clearly support KChIP3 as an appropriate target for further research into AD. Therefore, unravelling the diversity and complexity of KChIP3's functions would pave the way for developing more effective treatments for AD.
4.1.5. Other Neurodegenerative Disorders
In addition to AD, KChIP3 has been implicated in the pathogenesis of other neurodegenerative disorders. Recent evidence suggests that KChIP3 is closely linked to Huntington's disease (HD) [146] and amyotrophic lateral sclerosis (ASL) [147]. Firstly, the levels of KChIP3 protein are significantly reduced in the hippocampus samples of HD patients. To be specific, KChIP3 interacts with activating transcription factor 6 (ATF6) to suppress the pro-survival unfolded protein response, a common feature of neurodegenerative diseases. In the mouse model of HD, inhibition of the KChIP3-ATF6 interaction delays the onset of cognitive deficits. In addition, treatment with repaglinide has been shown to delay the onset and progression of motor and cognitive decline and extend lifespan by blocking the interaction of KChIP3 and ATF6 [62,146], suggesting that KChIP3 may be a novel target for the treatment of related diseases. Second, upregulation of KChIP3 has been observed in motor neurons and astrocytes in the spinal cord and frontal cortex of ALS patients [147]. The main feature of ALS is the relentless loss of motor neurons and increased reactive astrogliosis [148]. Cebolla and colleagues showed that KChIP3 is able to promote astrocyte differentiation of cortical precursors via cAMP-dependent calcium signaling. The neonatal cortex of Kcnip3-/- mice has a reduced number of astrocytes and an increased number of neurons [96]. Nevertheless, studies linking KChIP3 to these diseases have been limited. More evidence is needed to elucidate the mechanisms by which KChIP3 is involved in these neurodegenerative diseases.
4.2. Cardiovascular Diseases
4.2.1. Arrhythmias
Cardiac arrhythmias are classified mechanistically into two categories: focal activity due to enhanced or abnormal pulse generation and reentry due to conduction disturbances [149]. Early afterdepolarizations preceding full repolarization and delayed afterdepolarizations occurring after full repolarization are the most common causes of focal arrhythmias. Prolongation of action potentials caused by INa,L (late Na+ current), the ICa,L, or INCX, or decreases in the repolarizing potassium currents (IKr, IKs, IK1) can lead to early afterdepolarizations. As mentioned above, KChIPs have been shown to regulate a wide variety of ion channels in cardiomyocytes, including those that control the depolarization and repolarization of cardiac action potentials. Thus, KChIPs are critically involved in the pathogenesis of arrhythmias, as demonstrated by some of the available evidence, either directly or indirectly. For example, KChIP2 was upregulated in ageing porcine atria [150] and was significantly reduced in TLR4-activated inflammatory responses [151] and chronic NMDAR activation [152]. Atrial fibrillation could be induced in zebrafish hearts overexpressing KCNIP1 [153]. Consistently, Kcnip2-/- mice have an increased susceptibility to arrhythmias, manifested by a prolonged elevation in the ST segment on the electrocardiogram [154]. Mechanistically, KChIP2 on the one hand directly regulates the subcellular localization and gating properties of several ion channels as an auxiliary subunit in the cardiomyocyte. On the other hand, KChIP2 also controls the expression of ion channel subunits at the transcriptional level. In ventricular myocytes from Kcnip2-/- mice, Ito,f and INa are abolished, ICa,L is downregulated and IKs and Ito,s (the slow transient outward K+ current) are upregulated [73,79,155]. Recently, KChIP2 was reported to act as a transcriptional repressor by binding directly to the promoters of miR-34b/c, a miRNA that directly affects SCN5A (NaV1.5), SCN1B (NaVβ1), and KCND3 (KV4.3) gene expression. Inhibition of miR-34b/c can block the induction of arrhythmia [156]. Due to the multiple roles of KChIP2 in regulating ion channels in the heart, a thorough understanding of the detailed molecular mechanisms by which abnormal KChIP2 levels increase arrhythmias susceptibility is key to the development of novel therapies for the prevention and treatment of cardiac arrhythmias.
4.2.2. Cardiac Remodeling
Cardiac remodeling is defined as persistent changes in cardiac structure and function in response to physiological or pathological stimuli. Pathophysiological events that cause a decrease in contractility and/or an increase in wall stress, such as ischemia/reperfusion, myocardial infarction, pressure and volume overload, hypertension, and neuroendocrine stimulation, often result in adverse cardiac remodeling [157]. KChIP2 expression is altered in many cardiovascular events, including ischemic cardiomyopathy [158], myocarditis [159], mitral valve disease [160], inflammatory cytokine-induced myocardial injury [34,161], myocardial infarction [162], and type 2 diabetes mellitus[163]. Previous studies have shown that KChIP2 expression is significantly downregulated in hypertrophic myocardium, which is associated with reduced Ito [154,164]. Some of these mechanisms have been validated in in vitro models of hypertrophy. For example, overactivation of the JNK pathway was found to cause downregulation of KChIP2 in neonatal ventricular myocytes treated with phenylephrine, a robust inducer of hypertrophy [30]. In addition, our previous study demonstrated that the KChIP2 expression was decreased in phenylephrine-induced hypertrophic cardiomyocyte. Mechanistically, phenylephrine-activated NF-κB binds to the promoter region of the Kcnip2 gene and directly represses its transcription. Overexpression of muscle-specific mitsugumin 53 upregulates KChIP2 through inhibition of NF-κB and thereby reversed phenylephrine-induced cardiomyocyte hypertrophy [27]. In addition, Jin et al., found that adenoviral overexpression of KChIP2 in vivo significantly attenuated the development of left ventricular hypertrophy in aortic-banded rats . This protective effect of KChIP2 is achieved by inhibiting MAPK signaling activity and reducing calcineurin/NFAT expression [165].
Cardiac memory is a specific form of cardiac remodeling that is manifested by the persistence of inverted T waves after the restoration of sinus rhythm. The T wave "remembers" the QRS complex from the paced or arrhythmia phase following a short alteration in the sequence of ventricular depolarization caused by pacing or arrhythmia [166]. An important electrophysiological mechanism responsible for cardiac memory is the reduction in Ito density and its significantly prolonged recovery from inactivation [167]. The decrease expression of KChIP2 during this phase is essential for the occurrence of cardiac memory. According to Ozgen et al, left ventricular pacing induces the degradation of the transcription factor CREB by initiating the production of myocardial angiotensin II and the synthesis of reactive oxygen species, which results in the downregulation of KChIP2 expression [38].
4.2.3. Heart Failure
There is a growing body of evidence that KChIP2 also plays an important role in the pathogenesis of heart failure. The mRNA and protein expression of KChIP2 was significantly downregulated in the failing heart [168]. Meanwhile, autoantibodies against KChIP2 have been detected in patients with dilated cardiomyopathy. In vitro incubation of anti-KChIP2 antibody facilitates necrotic cell death in rat cardiomyocytes, suggesting that KChIP2 autoantibodies may be involved in the pathogenesis of dilated cardiomyopathy [169]. Candesartan, the angiotensin Ⅱ receptor blocker, attenuates KChIP2 downregulation in dilated cardiomyopathy and is involved in preventing severe electrical remodeling in inherited dilated cardiomyopathy [170]. Nassal et al. found that reduction of KChIP2 in guinea pig cardiomyocytes significantly increased ICa,L and prolonged action potentials by increasing CaV1.2 protein expression [171]. They also found that KChIP2, like the neuronal KChIP isoforms, can regulate ryanodine receptor activity by interacting with PS. Loss of KChIP2 resulted in reduced ryanodine receptor activity due to a decrease in its binding affinity to PS, which disrupted calcium-induced calcium release events. This further leads to impaired contractility of cardiomyocytes and promotes the onset of heart failure [172]. Interestingly, Speerschneider et al. showed that although KChIP2 is downregulated in heart failure, the reduction of Ito,f does not promote the development of heart failure. On the contrary, reduction of Ito exerts antiarrhythmic effects in mouse heart [173]. Likewise, Grubb et al. showed that while the downregulation of repolarization currents in heart failure was exacerbated in Kcnip2-/- mice, there was less prolongation of action potentials associated with heart failure due to compensation by upregulation of IKs and Ito,s [174]. In summary, the role of KChIP2 in heart failure remains controversial. Further research is needed to determine whether targeting KChIP2 has therapeutic implications in heart failure.
5. Concluding Remarks
KChIPs are a collection of multifunctional proteins involved in a wide range of physiological process and pathological diseases. However, our understanding of their structure, biological function, and regulatory mechanisms is still limited. Although several studies, including ours, have dissected the mechanisms regulating KChIPs during disease progression, this is only the tip of the iceberg. Furthermore, the role of KChIP3 in pain appears to differ among species. Small molecule compounds like NS5806, as a ligand for KChIP2, have different effects in different species or even in different parts of the same species, which will significantly hamper the development of targeted therapies. Therefore, despite the development of several small molecules targeting KChIPs, more efforts are needed to demonstrate their translational value in treating disease.
Author Contributions
Conceptualization, J.L.; Original draft preparation, L.-Y.W. and Y.-J.S.; writing—review and editing, C.-L.Z. and J.L.; visualization, L.-Y.W. and Y.-J.S.; supervision, C.-L.Z. and J.L.; funding acquisition, C.-L.Z. and J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Natural Science Foundation of China (81970250, 32271151, 82000056).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
All of the Figures were created using BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rosati, B.; Pan, Z.; Lypen, S.; Wang, H.S.; Cohen, I.; Dixon, J.E.; McKinnon, D. Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. The Journal of physiology 2001, 533, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Maniak, P.J.; Ingbar, D.H.; O'Grady, S.M. Adult alveolar epithelial cells express multiple subtypes of voltage-gated K+ channels that are located in apical membrane. American journal of physiology. Cell physiology 2003, 284, C1614–1624. [Google Scholar] [CrossRef]
- Bonne, A.; Vreede, L.; Kuiper, R.P.; Bodmer, D.; Jansen, C.; Eleveld, M.; van Erp, F.; Arkesteijn, G.; Hoogerbrugge, N.; van Ravenswaaij, C.; et al. Mapping of constitutional translocation breakpoints in renal cell cancer patients: identification of KCNIP4 as a candidate gene. Cancer genetics and cytogenetics 2007, 179, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Amberg, G.C.; Koh, S.D.; Hatton, W.J.; Murray, K.J.; Monaghan, K.; Horowitz, B.; Sanders, K.M. Contribution of Kv4 channels toward the A-type potassium current in murine colonic myocytes. The Journal of physiology 2002, 544, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Beckett, E.A.; McCloskey, C.; O'Kane, N.; Sanders, K.M.; Koh, S.D. Effects of female steroid hormones on A-type K+ currents in murine colon. The Journal of physiology 2006, 573, 453–468. [Google Scholar] [CrossRef]
- Zhang, Y.; MacLean, J.N.; An, W.F.; Lanning, C.C.; Harris-Warrick, R.M. KChIP1 and frequenin modify shal-evoked potassium currents in pyloric neurons in the lobster stomatogastric ganglion. Journal of neurophysiology 2003, 89, 1902–1909. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Choi, E.K.; Luo, Y.; Lilliehook, C.; Crowley, A.C.; Merriam, D.E.; Wasco, W. Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nature medicine 1998, 4, 1177–1181. [Google Scholar] [CrossRef]
- Carrión, A.M.; Link, W.A.; Ledo, F.; Mellström, B.; Naranjo, J.R. DREAM is a Ca2+-regulated transcriptional repressor. Nature 1999, 398, 80–84. [Google Scholar] [CrossRef]
- An, W.F.; Bowlby, M.R.; Betty, M.; Cao, J.; Ling, H.P.; Mendoza, G.; Hinson, J.W.; Mattsson, K.I.; Strassle, B.W.; Trimmer, J.S.; et al. Modulation of A-type potassium channels by a family of calcium sensors. Nature 2000, 403, 553–556. [Google Scholar] [CrossRef]
- Morohashi, Y.; Hatano, N.; Ohya, S.; Takikawa, R.; Watabiki, T.; Takasugi, N.; Imaizumi, Y.; Tomita, T.; Iwatsubo, T. Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. The Journal of biological chemistry 2002, 277, 14965–14975. [Google Scholar] [CrossRef]
- Cho, J. Downstream Regulatory Element Antagonist Modulator (DREAM), a target for anti-thrombotic agents. Pharmacological research 2017, 117, 283–287. [Google Scholar] [CrossRef]
- Cercós, P.; Peraza, D.A.; Benito-Bueno, A.; Socuéllamos, P.G.; Aziz-Nignan, A.; Arrechaga-Estévez, D.; Beato, E.; Peña-Acevedo, E.; Albert, A.; González-Vera, J.A.; et al. Pharmacological Approaches for the Modulation of the Potassium Channel K(V)4.x and KChIPs. International journal of molecular sciences 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Néant, I.; Haiech, J.; Kilhoffer, M.C.; Aulestia, F.J.; Moreau, M.; Leclerc, C. Ca(2+)-Dependent Transcriptional Repressors KCNIP and Regulation of Prognosis Genes in Glioblastoma. Frontiers in molecular neuroscience 2018, 11, 472. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Q.; Liang, P.; Zhou, J.; Lu, Y.; Lei, L.; Bian, X.; Wang, K. Auxiliary KChIP4a suppresses A-type K+ current through endoplasmic reticulum (ER) retention and promoting closed-state inactivation of Kv4 channels. The Journal of biological chemistry 2013, 288, 14727–14741. [Google Scholar] [CrossRef]
- Jerng, H.H.; Pfaffinger, P.J. Modulatory mechanisms and multiple functions of somatodendritic A-type K (+) channel auxiliary subunits. Frontiers in cellular neuroscience 2014, 8, 82. [Google Scholar] [CrossRef]
- Bähring, R. Kv channel-interacting proteins as neuronal and non-neuronal calcium sensors. Channels (Austin, Tex.) 2018, 12, 187–200. [Google Scholar] [CrossRef]
- O'Callaghan, D.W.; Hasdemir, B.; Leighton, M.; Burgoyne, R.D. Residues within the myristoylation motif determine intracellular targeting of the neuronal Ca2+ sensor protein KChIP1 to post-ER transport vesicles and traffic of Kv4 K+ channels. Journal of cell science 2003, 116, 4833–4845. [Google Scholar] [CrossRef]
- Takimoto, K.; Yang, E.K.; Conforti, L. Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. The Journal of biological chemistry 2002, 277, 26904–26911. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Workman, S.W.; Jiang, M.; Hu, J.; Sifa, I.; Bernas, T.; Tang, W.; Deschenes, I.; Tseng, G.N. Dynamic palmitoylation regulates trafficking of K channel interacting protein 2 (KChIP2) across multiple subcellular compartments in cardiac myocytes. Journal of molecular and cellular cardiology 2019, 135, 1–9. [Google Scholar] [CrossRef]
- Palczewska, M.; Casafont, I.; Ghimire, K.; Rojas, A.M.; Valencia, A.; Lafarga, M.; Mellström, B.; Naranjo, J.R. Sumoylation regulates nuclear localization of repressor DREAM. Biochimica et biophysica acta 2011, 1813, 1050–1058. [Google Scholar] [CrossRef]
- Choi, E.K.; Miller, J.S.; Zaidi, N.F.; Salih, E.; Buxbaum, J.D.; Wasco, W. Phosphorylation of calsenilin at Ser63 regulates its cleavage by caspase-3. Molecular and cellular neurosciences 2003, 23, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Gomez, A.; Mellström, B.; Tornero, D.; Morato, E.; Savignac, M.; Holguín, H.; Aurrekoetxea, K.; González, P.; González-García, C.; Ceña, V.; et al. G protein-coupled receptor kinase 2-mediated phosphorylation of downstream regulatory element antagonist modulator regulates membrane trafficking of Kv4.2 potassium channel. The Journal of biological chemistry 2007, 282, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, M.H.; Cao, J.; Hernandez-Pineda, R.; Jacobson, M.D.; Carroll, K.I.; Sung, M.A.; Betty, M.; Ge, P.; Gilbride, K.J.; Brown, M.E.; et al. Elimination of fast inactivation in Kv4 A-type potassium channels by an auxiliary subunit domain. Proceedings of the National Academy of Sciences of the United States of America 2002, 99, 1035–1040. [Google Scholar] [CrossRef]
- Jerng, H.H.; Pfaffinger, P.J. Multiple Kv channel-interacting proteins contain an N-terminal transmembrane domain that regulates Kv4 channel trafficking and gating. The Journal of biological chemistry 2008, 283, 36046–36059. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, J.; Zolles, G.; Kandias, N.G.; Neubauer, I.; Kalbacher, H.; Covarrubias, M.; Fakler, B.; Bentrop, D. NMR analysis of KChIP4a reveals structural basis for control of surface expression of Kv4 channel complexes. The Journal of biological chemistry 2008, 283, 18937–18946. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Wang, H.; Chen, H.; Cui, Y.; Gu, L.; Chai, J.; Wang, K. Structural Insights into KChIP4a Modulation of Kv4.3 Inactivation. The Journal of biological chemistry 2009, 284, 4960–4967. [Google Scholar] [CrossRef]
- Liu, W.; Wang, G.; Zhang, C.; Ding, W.; Cheng, W.; Luo, Y.; Wei, C.; Liu, J. MG53, A Novel Regulator of KChIP2 and I(to,f), Plays a Critical Role in Electrophysiological Remodeling in Cardiac Hypertrophy. Circulation 2019, 139, 2142–2156. [Google Scholar] [CrossRef]
- Wagner, S.; Hacker, E.; Grandi, E.; Weber, S.L.; Dybkova, N.; Sossalla, S.; Sowa, T.; Fabritz, L.; Kirchhof, P.; Bers, D.M.; et al. Ca/calmodulin kinase II differentially modulates potassium currents. Circulation. Arrhythmia and electrophysiology 2009, 2, 285–294. [Google Scholar] [CrossRef]
- Rossow, C.F.; Dilly, K.W.; Santana, L.F. Differential calcineurin/NFATc3 activity contributes to the Ito transmural gradient in the mouse heart. Circulation research 2006, 98, 1306–1313. [Google Scholar] [CrossRef]
- Jia, Y.; Takimoto, K. Mitogen-activated protein kinases control cardiac KChIP2 gene expression. Circulation research 2006, 98, 386–393. [Google Scholar] [CrossRef]
- Huang, H.; Tang, Y.; Wu, G.; Mei, Y.; Liu, W.; Liu, X.; Wan, N.; Liu, Y.; Huang, C. ALK7 protects against pathological cardiac hypertrophy in mice. Cardiovascular research 2015, 108, 50–61. [Google Scholar] [CrossRef]
- Ying, S.; Cao, H.; Hu, H.; Wang, X.; Tang, Y.; Huang, C. Alk7 Depleted Mice Exhibit Prolonged Cardiac Repolarization and Are Predisposed to Ventricular Arrhythmia. PloS one 2016, 11, e0149205. [Google Scholar] [CrossRef] [PubMed]
- Borghetti, G.; Eisenberg, C.A.; Signore, S.; Sorrentino, A.; Kaur, K.; Andrade-Vicenty, A.; Edwards, J.G.; Nerkar, M.; Qanud, K.; Sun, D.; et al. Notch signaling modulates the electrical behavior of cardiomyocytes. American journal of physiology. Heart and circulatory physiology 2018, 314, H68–h81. [Google Scholar] [CrossRef]
- Kaur, K.; Zarzoso, M.; Ponce-Balbuena, D.; Guerrero-Serna, G.; Hou, L.; Musa, H.; Jalife, J. TGF-β1, released by myofibroblasts, differentially regulates transcription and function of sodium and potassium channels in adult rat ventricular myocytes. PloS one 2013, 8, e55391. [Google Scholar] [CrossRef] [PubMed]
- Panama, B.K.; Latour-Villamil, D.; Farman, G.P.; Zhao, D.; Bolz, S.S.; Kirshenbaum, L.A.; Backx, P.H. Nuclear factor kappaB downregulates the transient outward potassium current I(to,f) through control of KChIP2 expression. Circulation research 2011, 108, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Panama, B.K.; Korogyi, A.S.; Aschar-Sobbi, R.; Oh, Y.; Gray, C.B.; Gang, H.; Brown, J.H.; Kirshenbaum, L.A.; Backx, P.H. Reductions in the Cardiac Transient Outward K+ Current Ito Caused by Chronic β-Adrenergic Receptor Stimulation Are Partly Rescued by Inhibition of Nuclear Factor κB. The Journal of biological chemistry 2016, 291, 4156–4165. [Google Scholar] [CrossRef]
- Xie, Y.; Mai, J.T.; Wang, F.; Lin, Y.Q.; Yuan, W.L.; Luo, N.S.; Fang, M.C.; Wang, J.F.; Chen, Y.X. Effects of C-reactive protein on K(+) channel interaction protein 2 in cardiomyocytes. American journal of translational research 2015, 7, 922–931. [Google Scholar] [CrossRef]
- Ozgen, N.; Lau, D.H.; Shlapakova, I.N.; Sherman, W.; Feinmark, S.J.; Danilo, P., Jr.; Rosen, M.R. Determinants of CREB degradation and KChIP2 gene transcription in cardiac memory. Heart rhythm 2010, 7, 964–970. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Liu, W.; Zi, M.; Li, Y.; Wang, S.; Tsui, H.; Prehar, S.; Castro, S.; Zhang, H.; Ji, Y.; et al. Stress-Activated Kinase Mitogen-Activated Kinase Kinase-7 Governs Epigenetics of Cardiac Repolarization for Arrhythmia Prevention. Circulation 2017, 135, 683–699. [Google Scholar] [CrossRef]
- Jeyaraj, D.; Haldar, S.M.; Wan, X.; McCauley, M.D.; Ripperger, J.A.; Hu, K.; Lu, Y.; Eapen, B.L.; Sharma, N.; Ficker, E.; et al. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature 2012, 483, 96–99. [Google Scholar] [CrossRef]
- Menegola, M.; Trimmer, J.S. Unanticipated region- and cell-specific downregulation of individual KChIP auxiliary subunit isotypes in Kv4.2 knock-out mouse brain. The Journal of neuroscience : the official journal of the Society for Neuroscience 2006, 26, 12137–12142. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.J.; Foeger, N.C.; Nerbonne, J.M. Interdependent roles for accessory KChIP2, KChIP3, and KChIP4 subunits in the generation of Kv4-encoded IA channels in cortical pyramidal neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010, 30, 13644–13655. [Google Scholar] [CrossRef] [PubMed]
- Nerbonne, J.M.; Gerber, B.R.; Norris, A.; Burkhalter, A. Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. The Journal of physiology 2008, 586, 1565–1579. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Maffie, J.K.; Lin, L.; Petralia, R.S.; Rudy, B.; Hoffman, D.A. DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron 2011, 71, 1102–1115. [Google Scholar] [CrossRef]
- Kuo, Y.L.; Cheng, J.K.; Hou, W.H.; Chang, Y.C.; Du, P.H.; Jian, J.J.; Rau, R.H.; Yang, J.H.; Lien, C.C.; Tsaur, M.L. K(+) Channel Modulatory Subunits KChIP and DPP Participate in Kv4-Mediated Mechanical Pain Control. The Journal of neuroscience : the official journal of the Society for Neuroscience 2017, 37, 4391–4404. [Google Scholar] [CrossRef]
- Mellström, B.; Sahún, I.; Ruiz-Nuño, A.; Murtra, P.; Gomez-Villafuertes, R.; Savignac, M.; Oliveros, J.C.; Gonzalez, P.; Kastanauskaite, A.; Knafo, S.; et al. DREAM controls the on/off switch of specific activity-dependent transcription pathways. Molecular and cellular biology 2014, 34, 877–887. [Google Scholar] [CrossRef]
- Kise, Y.; Kasuya, G.; Okamoto, H.H.; Yamanouchi, D.; Kobayashi, K.; Kusakizako, T.; Nishizawa, T.; Nakajo, K.; Nureki, O. Structural basis of gating modulation of Kv4 channel complexes. Nature 2021, 599, 158–164. [Google Scholar] [CrossRef]
- Jerng, H.H.; Pfaffinger, P.J.; Covarrubias, M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Molecular and cellular neurosciences 2004, 27, 343–369. [Google Scholar] [CrossRef]
- Patel, S.P.; Campbell, D.L. Transient outward potassium current, 'Ito', phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. The Journal of physiology 2005, 569, 7–39. [Google Scholar] [CrossRef]
- Kim, L.A.; Furst, J.; Gutierrez, D.; Butler, M.H.; Xu, S.; Goldstein, S.A.; Grigorieff, N. Three-dimensional structure of I(to); Kv4.2-KChIP2 ion channels by electron microscopy at 21 Angstrom resolution. Neuron 2004, 41, 513–519. [Google Scholar] [CrossRef]
- Wang, H.; Yan, Y.; Liu, Q.; Huang, Y.; Shen, Y.; Chen, L.; Chen, Y.; Yang, Q.; Hao, Q.; Wang, K.; et al. Structural basis for modulation of Kv4 K+ channels by auxiliary KChIP subunits. Nature neuroscience 2007, 10, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Catte, A.; Ferbel, L.; Bhattacharjee, N.; Jan Akhunzada, M.; D'Agostino, T.; Brancato, G. In silico investigation of the interaction between the voltage-gated potassium channel Kv4.3 and its auxiliary protein KChIP1. Physical chemistry chemical physics : PCCP 2019, 21, 25290–25301. [Google Scholar] [CrossRef] [PubMed]
- Fineberg, J.D.; Szanto, T.G.; Panyi, G.; Covarrubias, M. Closed-state inactivation involving an internal gate in Kv4.1 channels modulates pore blockade by intracellular quaternary ammonium ions. Scientific reports 2016, 6, 31131. [Google Scholar] [CrossRef] [PubMed]
- Bähring, R.; Dannenberg, J.; Peters, H.C.; Leicher, T.; Pongs, O.; Isbrandt, D. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. The Journal of biological chemistry 2001, 276, 23888–23894. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.P.; Campbell, D.L.; Strauss, H.C. Elucidating KChIP effects on Kv4.3 inactivation and recovery kinetics with a minimal KChIP2 isoform. The Journal of physiology 2002, 545, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Zhao, H.; Dai, Y.; Wang, Y.; Lo, Y.H.; Jan, L.Y.; Lee, C.H. Activation and closed-state inactivation mechanisms of the human voltage-gated K(V)4 channel complexes. Molecular cell 2022, 82, 2427–2442. [Google Scholar] [CrossRef]
- Shibata, R.; Misonou, H.; Campomanes, C.R.; Anderson, A.E.; Schrader, L.A.; Doliveira, L.C.; Carroll, K.I.; Sweatt, J.D.; Rhodes, K.J.; Trimmer, J.S. A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4.2 potassium channels. The Journal of biological chemistry 2003, 278, 36445–36454. [Google Scholar] [CrossRef]
- Foeger, N.C.; Marionneau, C.; Nerbonne, J.M. Co-assembly of Kv4 {alpha} subunits with K+ channel-interacting protein 2 stabilizes protein expression and promotes surface retention of channel complexes. The Journal of biological chemistry 2010, 285, 33413–33422. [Google Scholar] [CrossRef]
- Li, Y.; Duan, H.; Yi, J.; Wang, G.; Cheng, W.; Feng, L.; Liu, J. Kv4.2 phosphorylation by PKA drives Kv4.2-KChIP2 dissociation, leading to Kv4.2 out of lipid rafts and internalization. American journal of physiology. Cell physiology 2022, 323, C190–c201. [Google Scholar] [CrossRef]
- Holmqvist, M.H.; Cao, J.; Knoppers, M.H.; Jurman, M.E.; Distefano, P.S.; Rhodes, K.J.; Xie, Y.; An, W.F. Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. The Journal of neuroscience : the official journal of the Society for Neuroscience 2001, 21, 4154–4161. [Google Scholar] [CrossRef]
- Bowlby, M.R.; Chanda, P.; Edris, W.; Hinson, J.; Jow, F.; Katz, A.H.; Kennedy, J.; Krishnamurthy, G.; Pitts, K.; Ryan, K.; et al. Identification and characterization of small molecule modulators of KChIP/Kv4 function. Bioorganic & medicinal chemistry 2005, 13, 6112–6119. [Google Scholar] [CrossRef]
- Naranjo, J.R.; Zhang, H.; Villar, D.; González, P.; Dopazo, X.M.; Morón-Oset, J.; Higueras, E.; Oliveros, J.C.; Arrabal, M.D.; Prieto, A.; et al. Activating transcription factor 6 derepression mediates neuroprotection in Huntington disease. The Journal of clinical investigation 2016, 126, 627–638. [Google Scholar] [CrossRef]
- Lopez-Hurtado, A.; Peraza, D.A.; Cercos, P.; Lagartera, L.; Gonzalez, P.; Dopazo, X.M.; Herranz, R.; Gonzalez, T.; Martin-Martinez, M.; Mellström, B.; et al. Targeting the neuronal calcium sensor DREAM with small-molecules for Huntington's disease treatment. Scientific reports 2019, 9, 7260. [Google Scholar] [CrossRef] [PubMed]
- Peraza, D.A.; Cercós, P.; Miaja, P.; Merinero, Y.G.; Lagartera, L.; Socuéllamos, P.G.; Izquierdo García, C.; Sánchez, S.A.; López-Hurtado, A.; Martín-Martínez, M.; et al. Identification of IQM-266, a Novel DREAM Ligand That Modulates K(V)4 Currents. Frontiers in molecular neuroscience 2019, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- de Benito-Bueno, A.; Socuellamos, P.G.; Merinero, Y.G.; Cercos, P.; Izquierdo, C.; Daniel-Mozo, M.; Marín-Olivero, I.; Perez-Lara, A.; Gonzalez-Vera, J.A.; Orte, A.; et al. Modulation of K(V)4.3-KChIP2 Channels by IQM-266: Role of DPP6 and KCNE2. International journal of molecular sciences 2022, 23. [Google Scholar] [CrossRef]
- Witzel, K.; Fischer, P.; Bähring, R. Hippocampal A-type current and Kv4.2 channel modulation by the sulfonylurea compound NS5806. Neuropharmacology 2012, 63, 1389–1403. [Google Scholar] [CrossRef]
- Gonzalez, W.G.; Pham, K.; Miksovska, J. Modulation of the voltage-gated potassium channel (Kv4.3) and the auxiliary protein (KChIP3) interactions by the current activator NS5806. The Journal of biological chemistry 2014, 289, 32201–32213. [Google Scholar] [CrossRef]
- Calloe, K.; Soltysinska, E.; Jespersen, T.; Lundby, A.; Antzelevitch, C.; Olesen, S.P.; Cordeiro, J.M. Differential effects of the transient outward K(+) current activator NS5806 in the canine left ventricle. Journal of molecular and cellular cardiology 2010, 48, 191–200. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, H.; Wang, C.; Wang, Y.; Zou, R.; Shi, C.; Guan, B.; Gamper, N.; Xu, Y. Auxiliary subunits control biophysical properties and response to compound NS5806 of the Kv4 potassium channel complex. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2020, 34, 807–821. [Google Scholar] [CrossRef]
- Cheng, H.; Cannell, M.B.; Hancox, J.C. Differential responses of rabbit ventricular and atrial transient outward current (I(to)) to the I(to) modulator NS5806. Physiological reports 2017, 5. [Google Scholar] [CrossRef]
- Ravens, U.; Wettwer, E. Ultra-rapid delayed rectifier channels: molecular basis and therapeutic implications. Cardiovascular research 2011, 89, 776–785. [Google Scholar] [CrossRef]
- Li, H.; Guo, W.; Mellor, R.L.; Nerbonne, J.M. KChIP2 modulates the cell surface expression of Kv 1.5-encoded K(+) channels. Journal of molecular and cellular cardiology 2005, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.B.; Sosunov, E.A.; Anyukhovsky, E.P.; Ozgen, N.; Boyden, P.A.; Rosen, M.R. Deleting the accessory subunit KChIP2 results in loss of I(to,f) and increased I(K,slow) that maintains normal action potential configuration. Heart rhythm 2009, 6, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiological reviews 2014, 94, 303–326. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.B.; Wang, C.; Ozgen, N.; Wang, H.G.; Rosen, M.R.; Pitt, G.S. Accessory subunit KChIP2 modulates the cardiac L-type calcium current. Circulation research 2009, 104, 1382–1389. [Google Scholar] [CrossRef]
- Thomsen, M.B.; Foster, E.; Nguyen, K.H.; Sosunov, E.A. Transcriptional and electrophysiological consequences of KChIP2-mediated regulation of CaV1.2. Channels (Austin, Tex.) 2009, 3, 308–310. [Google Scholar] [CrossRef]
- Ronkainen, J.J.; Hänninen, S.L.; Korhonen, T.; Koivumäki, J.T.; Skoumal, R.; Rautio, S.; Ronkainen, V.P.; Tavi, P. Ca2+-calmodulin-dependent protein kinase II represses cardiac transcription of the L-type calcium channel alpha(1C)-subunit gene (Cacna1c) by DREAM translocation. The Journal of physiology 2011, 589, 2669–2686. [Google Scholar] [CrossRef] [PubMed]
- Clatot, J.; Neyroud, N.; Cox, R.; Souil, C.; Huang, J.; Guicheney, P.; Antzelevitch, C. Inter-Regulation of K(v)4.3 and Voltage-Gated Sodium Channels Underlies Predisposition to Cardiac and Neuronal Channelopathies. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Deschênes, I.; Armoundas, A.A.; Jones, S.P.; Tomaselli, G.F. Post-transcriptional gene silencing of KChIP2 and Navbeta1 in neonatal rat cardiac myocytes reveals a functional association between Na and Ito currents. Journal of molecular and cellular cardiology 2008, 45, 336–346. [Google Scholar] [CrossRef]
- Osawa, M.; Dace, A.; Tong, K.I.; Valiveti, A.; Ikura, M.; Ames, J.B. Mg2+ and Ca2+ differentially regulate DNA binding and dimerization of DREAM. The Journal of biological chemistry 2005, 280, 18008–18014. [Google Scholar] [CrossRef]
- Lusin, J.D.; Vanarotti, M.; Li, C.; Valiveti, A.; Ames, J.B. NMR structure of DREAM: Implications for Ca(2+)-dependent DNA binding and protein dimerization. Biochemistry 2008, 47, 2252–2264. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.F.; Kuplast, K.G.; Washicosky, K.J.; Kajiwara, Y.; Buxbaum, J.D.; Wasco, W. Calsenilin interacts with transcriptional co-repressor C-terminal binding protein(s). Journal of neurochemistry 2006, 98, 1290–1301. [Google Scholar] [CrossRef] [PubMed]
- Ledo, F.; Carrión, A.M.; Link, W.A.; Mellström, B.; Naranjo, J.R. DREAM-alphaCREM interaction via leucine-charged domains derepresses downstream regulatory element-dependent transcription. Molecular and cellular biology 2000, 20, 9120–9126. [Google Scholar] [CrossRef] [PubMed]
- Ledo, F.; Kremer, L.; Mellström, B.; Naranjo, J.R. Ca2+-dependent block of CREB-CBP transcription by repressor DREAM. The EMBO journal 2002, 21, 4583–4592. [Google Scholar] [CrossRef]
- Rivas, M.; Mellström, B.; Naranjo, J.R.; Santisteban, P. Transcriptional repressor DREAM interacts with thyroid transcription factor-1 and regulates thyroglobulin gene expression. The Journal of biological chemistry 2004, 279, 33114–33122. [Google Scholar] [CrossRef]
- Scsucova, S.; Palacios, D.; Savignac, M.; Mellström, B.; Naranjo, J.R.; Aranda, A. The repressor DREAM acts as a transcriptional activator on Vitamin D and retinoic acid response elements. Nucleic acids research 2005, 33, 2269–2279. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Pitcher, G.M.; Laviolette, S.R.; Whishaw, I.Q.; Tong, K.I.; Kockeritz, L.K.; Wada, T.; Joza, N.A.; Crackower, M.; Goncalves, J.; et al. DREAM is a critical transcriptional repressor for pain modulation. Cell 2002, 108, 31–43. [Google Scholar] [CrossRef]
- Sanz, C.; Mellstrom, B.; Link, W.A.; Naranjo, J.R.; Fernandez-Luna, J.L. Interleukin 3-dependent activation of DREAM is involved in transcriptional silencing of the apoptotic Hrk gene in hematopoietic progenitor cells. The EMBO journal 2001, 20, 2286–2292. [Google Scholar] [CrossRef]
- Rivera-Arconada, I.; Benedet, T.; Roza, C.; Torres, B.; Barrio, J.; Krzyzanowska, A.; Avendaño, C.; Mellström, B.; Lopez-Garcia, J.A.; Naranjo, J.R. DREAM regulates BDNF-dependent spinal sensitization. Molecular pain 2010, 6, 95. [Google Scholar] [CrossRef]
- Savignac, M.; Pintado, B.; Gutierrez-Adan, A.; Palczewska, M.; Mellström, B.; Naranjo, J.R. Transcriptional repressor DREAM regulates T-lymphocyte proliferation and cytokine gene expression. The EMBO journal 2005, 24, 3555–3564. [Google Scholar] [CrossRef] [PubMed]
- Savignac, M.; Mellström, B.; Bébin, A.G.; Oliveros, J.C.; Delpy, L.; Pinaud, E.; Naranjo, J.R. Increased B cell proliferation and reduced Ig production in DREAM transgenic mice. Journal of immunology (Baltimore, Md. : 1950) 2010, 185, 7527–7536. [Google Scholar] [CrossRef] [PubMed]
- Tiruppathi, C.; Soni, D.; Wang, D.M.; Xue, J.; Singh, V.; Thippegowda, P.B.; Cheppudira, B.P.; Mishra, R.K.; Debroy, A.; Qian, Z.; et al. The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nature immunology 2014, 15, 239–247. [Google Scholar] [CrossRef] [PubMed]
- D'Andrea, B.; Di Palma, T.; Mascia, A.; Motti, M.L.; Viglietto, G.; Nitsch, L.; Zannini, M. The transcriptional repressor DREAM is involved in thyroid gene expression. Experimental cell research 2005, 305, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Link, W.A.; Ledo, F.; Torres, B.; Palczewska, M.; Madsen, T.M.; Savignac, M.; Albar, J.P.; Mellström, B.; Naranjo, J.R. Day-night changes in downstream regulatory element antagonist modulator/potassium channel interacting protein activity contribute to circadian gene expression in pineal gland. The Journal of neuroscience : the official journal of the Society for Neuroscience 2004, 24, 5346–5355. [Google Scholar] [CrossRef]
- Leclerc, G.M.; Boockfor, F.R. Calcium influx and DREAM protein are required for GnRH gene expression pulse activity. Molecular and cellular endocrinology 2007, 267, 70–79. [Google Scholar] [CrossRef]
- Cebolla, B.; Fernández-Pérez, A.; Perea, G.; Araque, A.; Vallejo, M. DREAM mediates cAMP-dependent, Ca2+-induced stimulation of GFAP gene expression and regulates cortical astrogliogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience 2008, 28, 6703–6713. [Google Scholar] [CrossRef]
- Gomez-Villafuertes, R.; Torres, B.; Barrio, J.; Savignac, M.; Gabellini, N.; Rizzato, F.; Pintado, B.; Gutierrez-Adan, A.; Mellström, B.; Carafoli, E.; et al. Downstream regulatory element antagonist modulator regulates Ca2+ homeostasis and viability in cerebellar neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience 2005, 25, 10822–10830. [Google Scholar] [CrossRef]
- Dierssen, M.; Fedrizzi, L.; Gomez-Villafuertes, R.; de Lagran, M.M.; Gutierrez-Adan, A.; Sahún, I.; Pintado, B.; Oliveros, J.C.; Dopazo, X.M.; Gonzalez, P.; et al. Reduced Mid1 Expression and Delayed Neuromotor Development in daDREAM Transgenic Mice. Frontiers in molecular neuroscience 2012, 5, 58. [Google Scholar] [CrossRef]
- Calì, T.; Fedrizzi, L.; Ottolini, D.; Gomez-Villafuertes, R.; Mellström, B.; Naranjo, J.R.; Carafoli, E.; Brini, M. Ca2+-activated nucleotidase 1, a novel target gene for the transcriptional repressor DREAM (downstream regulatory element antagonist modulator), is involved in protein folding and degradation. The Journal of biological chemistry 2012, 287, 18478–18491. [Google Scholar] [CrossRef]
- Fontán-Lozano, A.; Capilla-Gonzalez, V.; Aguilera, Y.; Mellado, N.; Carrión, A.M.; Soria, B.; Hmadcha, A. Impact of transient down-regulation of DREAM in human embryonic stem cell pluripotency: The role of DREAM in the maintenance of hESCs. Stem cell research 2016, 16, 568–578. [Google Scholar] [CrossRef]
- Baczyk, D.; Kibschull, M.; Mellstrom, B.; Levytska, K.; Rivas, M.; Drewlo, S.; Lye, S.J.; Naranjo, J.R.; Kingdom, J.C. DREAM mediated regulation of GCM1 in the human placental trophoblast. PloS one 2013, 8, e51837. [Google Scholar] [CrossRef]
- Jacobson, D.A.; Cho, J.; Landa, L.R., Jr.; Tamarina, N.A.; Roe, M.W.; Buxbaum, J.D.; Philipson, L.H. Downstream regulatory element antagonistic modulator regulates islet prodynorphin expression. American journal of physiology. Endocrinology and metabolism 2006, 291, E587–595. [Google Scholar] [CrossRef]
- Li, J.; Kumari, T.; Barazia, A.; Jha, V.; Jeong, S.Y.; Olson, A.; Kim, M.; Lee, B.K.; Manickam, V.; Song, Z.; et al. Neutrophil DREAM promotes neutrophil recruitment in vascular inflammation. The Journal of experimental medicine 2022, 219. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Maizuliana, H.; Usui, N.; Terada, K.; Kondo, A.; Inoue, Y. Clinical, semiological, electroencephalographic, and neuropsychological features of "pure" neocortical temporal lobe epilepsy. Epileptic disorders : international epilepsy journal with videotape 2020, 22, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Maillard, L.; Vignal, J.P.; Gavaret, M.; Guye, M.; Biraben, A.; McGonigal, A.; Chauvel, P.; Bartolomei, F. Semiologic and electrophysiologic correlations in temporal lobe seizure subtypes. Epilepsia 2004, 45, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Villa, C.; Combi, R. Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Frontiers in cellular neuroscience 2016, 10, 81. [Google Scholar] [CrossRef]
- Hong, Y.M.; Jo, D.G.; Lee, M.C.; Kim, S.Y.; Jung, Y.K. Reduced expression of calsenilin/DREAM/KChIP3 in the brains of kainic acid-induced seizure and epilepsy patients. Neuroscience letters 2003, 340, 33–36. [Google Scholar] [CrossRef]
- Wang, H.G.; He, X.P.; Li, Q.; Madison, R.D.; Moore, S.D.; McNamara, J.O.; Pitt, G.S. The auxiliary subunit KChIP2 is an essential regulator of homeostatic excitability. The Journal of biological chemistry 2013, 288, 13258–13268. [Google Scholar] [CrossRef]
- Monaghan, M.M.; Menegola, M.; Vacher, H.; Rhodes, K.J.; Trimmer, J.S. Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience 2008, 156, 550–562. [Google Scholar] [CrossRef]
- Kuner, R. Central mechanisms of pathological pain. Nature medicine 2010, 16, 1258–1266. [Google Scholar] [CrossRef]
- Costigan, M.; Woolf, C.J. No DREAM, No pain. Closing the spinal gate. Cell 2002, 108, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, Z.; Yu, P.; Li, J.; Bai, N.; He, Z.; Yang, S.; Guo, Q. [Construction of shRNA lentivirus vector on rat DREAM gene and its analgesic effect on CCI rats]. Zhong nan da xue xue bao. Yi xue ban = Journal of Central South University. Medical sciences 2009, 34, 723–730. [Google Scholar] [PubMed]
- Benedet, T.; Gonzalez, P.; Oliveros, J.C.; Dopazo, J.M.; Ghimire, K.; Palczewska, M.; Mellstrom, B.; Naranjo, J.R. Transcriptional repressor DREAM regulates trigeminal noxious perception. Journal of neurochemistry 2017, 141, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.X.; Xu, Y.; Yang, J.Y.; Li, L.; Sun, X.H.; Wang, Y.; Zhang, Y. KChIP3 N-Terminal 31-50 Fragment Mediates Its Association with TRPV1 and Alleviates Inflammatory Hyperalgesia in Rats. The Journal of neuroscience : the official journal of the Society for Neuroscience 2018, 38, 1756–1773. [Google Scholar] [CrossRef] [PubMed]
- Matsuyoshi, H.; Takimoto, K.; Yunoki, T.; Erickson, V.L.; Tyagi, P.; Hirao, Y.; Wanaka, A.; Yoshimura, N. Distinct cellular distributions of Kv4 pore-forming and auxiliary subunits in rat dorsal root ganglion neurons. Life sciences 2012, 91, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.Y.; Hoon, M.A. Molecular Genetics of Kappa Opioids in Pain and Itch Sensations. Handbook of experimental pharmacology 2022, 271, 255–274. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, P.; Liang, P.; Liu, T.; Liu, X.; Liu, X.Y.; Zhang, B.; Han, T.; Zhu, Y.B.; Yin, D.M.; et al. The DREAM protein negatively regulates the NMDA receptor through interaction with the NR1 subunit. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010, 30, 7575–7586. [Google Scholar] [CrossRef]
- Cheng, C.F.; Wang, W.C.; Huang, C.Y.; Du, P.H.; Yang, J.H.; Tsaur, M.L. Coexpression of auxiliary subunits KChIP and DPPL in potassium channel Kv4-positive nociceptors and pain-modulating spinal interneurons. The Journal of comparative neurology 2016, 524, 846–873. [Google Scholar] [CrossRef]
- Anderson, D.; Mehaffey, W.H.; Iftinca, M.; Rehak, R.; Engbers, J.D.; Hameed, S.; Zamponi, G.W.; Turner, R.W. Regulation of neuronal activity by Cav3-Kv4 channel signaling complexes. Nature neuroscience 2010, 13, 333–337. [Google Scholar] [CrossRef]
- Socuéllamos, P.G.; Olivos-Oré, L.A.; Barahona, M.V.; Cercós, P.; Pérez Pascual, M.; Arribas-Blázquez, M.; Naranjo, J.R.; Valenzuela, C.; Gutiérrez-Rodríguez, M.; Artalejo, A.R. IQM-PC332, a Novel DREAM Ligand with Antinociceptive Effect on Peripheral Nerve Injury-Induced Pain. International journal of molecular sciences 2022, 23. [Google Scholar] [CrossRef]
- Guo, Y.P.; Zhi, Y.R.; Liu, T.T.; Wang, Y.; Zhang, Y. Global Gene Knockout of Kcnip3 Enhances Pain Sensitivity and Exacerbates Negative Emotions in Rats. Frontiers in molecular neuroscience 2019, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Zuo, Y. Synaptic modifications in learning and memory - A dendritic spine story. Seminars in cell & developmental biology 2022, 125, 84–90. [Google Scholar] [CrossRef]
- Bin Ibrahim, M.Z.; Benoy, A.; Sajikumar, S. Long-term plasticity in the hippocampus: maintaining within and 'tagging' between synapses. The FEBS journal 2022, 289, 2176–2201. [Google Scholar] [CrossRef]
- Lisman, J.; Buzsáki, G.; Eichenbaum, H.; Nadel, L.; Ranganath, C.; Redish, A.D. Viewpoints: how the hippocampus contributes to memory, navigation and cognition. Nature neuroscience 2017, 20, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.C.; McDermott, C.M.; Tunur, T.; Rands, V.; Stelly, C.; Karhson, D.; Bowlby, M.R.; An, W.F.; Sweatt, J.D.; Schrader, L.A. The role of calsenilin/DREAM/KChIP3 in contextual fear conditioning. Learning & memory (Cold Spring Harbor, N.Y.) 2009, 16, 167–177. [Google Scholar] [CrossRef]
- Fontán-Lozano, A.; Romero-Granados, R.; del-Pozo-Martín, Y.; Suárez-Pereira, I.; Delgado-García, J.M.; Penninger, J.M.; Carrión, A.M. Lack of DREAM protein enhances learning and memory and slows brain aging. Current biology : CB 2009, 19, 54–60. [Google Scholar] [CrossRef]
- Chen, Q.Y.; Li, X.H.; Zhuo, M. NMDA receptors and synaptic plasticity in the anterior cingulate cortex. Neuropharmacology 2021, 197, 108749. [Google Scholar] [CrossRef]
- Dore, K.; Malinow, R. Elevated PSD-95 Blocks Ion-flux Independent LTD: A Potential New Role for PSD-95 in Synaptic Plasticity. Neuroscience 2021, 456, 43–49. [Google Scholar] [CrossRef]
- Wu, L.J.; Mellström, B.; Wang, H.; Ren, M.; Domingo, S.; Kim, S.S.; Li, X.Y.; Chen, T.; Naranjo, J.R.; Zhuo, M. DREAM (downstream regulatory element antagonist modulator) contributes to synaptic depression and contextual fear memory. Molecular brain 2010, 3, 3. [Google Scholar] [CrossRef]
- Lugo, J.N.; Brewster, A.L.; Spencer, C.M.; Anderson, A.E. Kv4.2 knockout mice have hippocampal-dependent learning and memory deficits. Learning & memory (Cold Spring Harbor, N.Y.) 2012, 19, 182–189. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer's disease. Handbook of clinical neurology 2019, 167, 231–255. [Google Scholar] [CrossRef]
- Robakis, N.K. Cell signaling abnormalities may drive neurodegeneration in familial Alzheimer disease. Neurochemical research 2014, 39, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Pham, K.; Miksovska, J. Molecular insight of DREAM and presenilin 1 C-terminal fragment interactions. FEBS letters 2016, 590, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science (New York, N.Y.) 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.G.; Lee, J.Y.; Hong, Y.M.; Song, S.; Mook-Jung, I.; Koh, J.Y.; Jung, Y.K. Induction of pro-apoptotic calsenilin/DREAM/KChIP3 in Alzheimer's disease and cultured neurons after amyloid-beta exposure. Journal of neurochemistry 2004, 88, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.F.; Berezovska, O.; Choi, E.K.; Miller, J.S.; Chan, H.; Lilliehook, C.; Hyman, B.T.; Buxbaum, J.D.; Wasco, W. Biochemical and immunocytochemical characterization of calsenilin in mouse brain. Neuroscience 2002, 114, 247–263. [Google Scholar] [CrossRef]
- Jo, D.G.; Chang, J.W.; Hong, H.S.; Mook-Jung, I.; Jung, Y.K. Contribution of presenilin/gamma-secretase to calsenilin-mediated apoptosis. Biochemical and biophysical research communications 2003, 305, 62–66. [Google Scholar] [CrossRef]
- Jo, D.G.; Jang, J.; Kim, B.J.; Lundkvist, J.; Jung, Y.K. Overexpression of calsenilin enhances gamma-secretase activity. Neuroscience letters 2005, 378, 59–64. [Google Scholar] [CrossRef]
- Naranjo, R.; González, P.; Lopez-Hurtado, A.; Dopazo, X.M.; Mellström, B.; Naranjo, J.R. Inhibition of the Neuronal Calcium Sensor DREAM Modulates Presenilin-2 Endoproteolysis. Frontiers in molecular neuroscience 2018, 11, 449. [Google Scholar] [CrossRef]
- Cascella, R.; Cecchi, C. Calcium Dyshomeostasis in Alzheimer's Disease Pathogenesis. International journal of molecular sciences 2021, 22. [Google Scholar] [CrossRef]
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiology of disease 2006, 23, 669–678. [Google Scholar] [CrossRef]
- Lilliehook, C.; Chan, S.; Choi, E.K.; Zaidi, N.F.; Wasco, W.; Mattson, M.P.; Buxbaum, J.D. Calsenilin enhances apoptosis by altering endoplasmic reticulum calcium signaling. Molecular and cellular neurosciences 2002, 19, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Fedrizzi, L.; Lim, D.; Carafoli, E.; Brini, M. Interplay of the Ca2+-binding protein DREAM with presenilin in neuronal Ca2+ signaling. The Journal of biological chemistry 2008, 283, 27494–27503. [Google Scholar] [CrossRef] [PubMed]
- Grillo, M.A.; Grillo, S.L.; Gerdes, B.C.; Kraus, J.G.; Koulen, P. Control of Neuronal Ryanodine Receptor-Mediated Calcium Signaling by Calsenilin. Molecular neurobiology 2019, 56, 525–534. [Google Scholar] [CrossRef]
- López-Hurtado, A.; Burgos, D.F.; González, P.; Dopazo, X.M.; González, V.; Rábano, A.; Mellström, B.; Naranjo, J.R. Inhibition of DREAM-ATF6 interaction delays onset of cognition deficit in a mouse model of Huntington's disease. Molecular brain 2018, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Larrodé, P.; Calvo, A.C.; Moreno-Martínez, L.; de la Torre, M.; Moreno-García, L.; Molina, N.; Castiella, T.; Iñiguez, C.; Pascual, L.F.; Mena, F.J.M.; et al. DREAM-Dependent Activation of Astrocytes in Amyotrophic Lateral Sclerosis. Molecular neurobiology 2018, 55, 1–12. [Google Scholar] [CrossRef]
- Singh, D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer's disease. Journal of neuroinflammation 2022, 19, 206. [Google Scholar] [CrossRef]
- Tse, G. Mechanisms of cardiac arrhythmias. Journal of arrhythmia 2016, 32, 75–81. [Google Scholar] [CrossRef]
- Kant, R.; Hu, Z.; Malhotra, J.K.; Krogh-Madsen, T.; Christini, D.J.; Heerdt, P.M.; Abbott, G.W. NHE isoform switching and KChIP2 upregulation in aging porcine atria. PloS one 2013, 8, e82951. [Google Scholar] [CrossRef]
- Monnerat-Cahli, G.; Alonso, H.; Gallego, M.; Alarcón, M.L.; Bassani, R.A.; Casis, O.; Medei, E. Toll-like receptor 4 activation promotes cardiac arrhythmias by decreasing the transient outward potassium current (Ito) through an IRF3-dependent and MyD88-independent pathway. Journal of molecular and cellular cardiology 2014, 76, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Liu, T.; Li, Y.; Qin, M.; Tang, Y.; Shen, J.Y.; Liang, J.; Yang, B.; Huang, C. Chronic N-methyl-D-aspartate receptor activation induces cardiac electrical remodeling and increases susceptibility to ventricular arrhythmias. Pacing and clinical electrophysiology : PACE 2014, 37, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.T.; Hsieh, C.S.; Chang, S.N.; Chuang, E.Y.; Ueng, K.C.; Tsai, C.F.; Lin, T.H.; Wu, C.K.; Lee, J.K.; Lin, L.Y.; et al. Genome-wide screening identifies a KCNIP1 copy number variant as a genetic predictor for atrial fibrillation. Nature communications 2016, 7, 10190. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Cheng, C.F.; Clark, R.B.; Lin, J.J.; Lin, J.L.; Hoshijima, M.; Nguyêñ-Trân, V.T.; Gu, Y.; Ikeda, Y.; Chu, P.H.; et al. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell 2001, 107, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Foeger, N.C.; Wang, W.; Mellor, R.L.; Nerbonne, J.M. Stabilization of Kv4 protein by the accessory K(+) channel interacting protein 2 (KChIP2) subunit is required for the generation of native myocardial fast transient outward K(+) currents. The Journal of physiology 2013, 591, 4149–4166. [Google Scholar] [CrossRef]
- Nassal, D.M.; Wan, X.; Liu, H.; Maleski, D.; Ramirez-Navarro, A.; Moravec, C.S.; Ficker, E.; Laurita, K.R.; Deschênes, I. KChIP2 is a core transcriptional regulator of cardiac excitability. eLife 2017, 6. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. Journal of the American College of Cardiology 2000, 35, 569–582. [Google Scholar] [CrossRef]
- Liu, X.S.; Jiang, M.; Zhang, M.; Tang, D.; Clemo, H.F.; Higgins, R.S.; Tseng, G.N. Electrical remodeling in a canine model of ischemic cardiomyopathy. American journal of physiology. Heart and circulatory physiology 2007, 292, H560–571. [Google Scholar] [CrossRef]
- Wakisaka, Y.; Niwano, S.; Niwano, H.; Saito, J.; Yoshida, T.; Hirasawa, S.; Kawada, H.; Izumi, T. Structural and electrical ventricular remodeling in rat acute myocarditis and subsequent heart failure. Cardiovascular research 2004, 63, 689–699. [Google Scholar] [CrossRef]
- Oh, S.; Kim, K.B.; Ahn, H.; Cho, H.J.; Choi, Y.S. Remodeling of ion channel expression in patients with chronic atrial fibrillation and mitral valvular heart disease. The Korean journal of internal medicine 2010, 25, 377–385. [Google Scholar] [CrossRef]
- Kawada, H.; Niwano, S.; Niwano, H.; Yumoto, Y.; Wakisaka, Y.; Yuge, M.; Kawahara, K.; Izumi, T. Tumor necrosis factor-alpha downregulates the voltage gated outward K+ current in cultured neonatal rat cardiomyocytes: a possible cause of electrical remodeling in diseased hearts. Circulation journal : official journal of the Japanese Circulation Society 2006, 70, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.Z.; Jiang, H.; Li, L.; Xie, P.; Li, J.Y.; Lu, Z.B.; He, B. Semaphorin 3A attenuates electrical remodeling at infarct border zones in rats after myocardial infarction. The Tohoku journal of experimental medicine 2011, 225, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kobayashi, T.; Kuno, A.; Miki, T.; Tanno, M.; Kouzu, H.; Itoh, T.; Ishikawa, S.; Kojima, T.; Miura, T.; et al. Type 2 diabetes induces subendocardium predominant reduction in transient outward K+ current with downregulation of Kv4.2 and KChIP2. American journal of physiology. Heart and circulatory physiology 2014, 306, H1054–1065. [Google Scholar] [CrossRef] [PubMed]
- Näbauer, M.; Kääb, S. Potassium channel down-regulation in heart failure. Cardiovascular research 1998, 37, 324–334. [Google Scholar] [CrossRef]
- Jin, H.; Hadri, L.; Palomeque, J.; Morel, C.; Karakikes, I.; Kaprielian, R.; Hajjar, R.; Lebeche, D. KChIP2 attenuates cardiac hypertrophy through regulation of Ito and intracellular calcium signaling. Journal of molecular and cellular cardiology 2010, 48, 1169–1179. [Google Scholar] [CrossRef]
- Chiale, P.A.; Etcheverry, D.; Pastori, J.D.; Fernandez, P.A.; Garro, H.A.; González, M.D.; Elizari, M.V. The multiple electrocardiographic manifestations of ventricular repolarization memory. Current cardiology reviews 2014, 10, 190–201. [Google Scholar] [CrossRef]
- Yu, H.; McKinnon, D.; Dixon, J.E.; Gao, J.; Wymore, R.; Cohen, I.S.; Danilo, P., Jr.; Shvilkin, A.; Anyukhovsky, E.P.; Sosunov, E.A.; et al. Transient outward current, Ito1, is altered in cardiac memory. Circulation 1999, 99, 1898–1905. [Google Scholar] [CrossRef]
- Radicke, S.; Cotella, D.; Graf, E.M.; Banse, U.; Jost, N.; Varró, A.; Tseng, G.N.; Ravens, U.; Wettwer, E. Functional modulation of the transient outward current Ito by KCNE beta-subunits and regional distribution in human non-failing and failing hearts. Cardiovascular research 2006, 71, 695–703. [Google Scholar] [CrossRef]
- Choudhury, S.; Schnell, M.; Bühler, T.; Reinke, Y.; Lüdemann, J.; Nießner, F.; Brinkmeier, H.; Herda, L.R.; Staudt, A.; Kroemer, H.K.; et al. Antibodies against potassium channel interacting protein 2 induce necrosis in isolated rat cardiomyocytes. Journal of cellular biochemistry 2014, 115, 678–689. [Google Scholar] [CrossRef]
- Odagiri, F.; Inoue, H.; Sugihara, M.; Suzuki, T.; Murayama, T.; Shioya, T.; Konishi, M.; Nakazato, Y.; Daida, H.; Sakurai, T.; et al. Effects of candesartan on electrical remodeling in the hearts of inherited dilated cardiomyopathy model mice. PloS one 2014, 9, e101838. [Google Scholar] [CrossRef]
- Nassal, D.M.; Wan, X.; Liu, H.; Deschênes, I. Myocardial KChIP2 Expression in Guinea Pig Resolves an Expanded Electrophysiologic Role. PloS one 2016, 11, e0146561. [Google Scholar] [CrossRef]
- Nassal, D.M.; Wan, X.; Liu, H.; Laurita, K.R.; Deschênes, I. KChIP2 regulates the cardiac Ca2+ transient and myocyte contractility by targeting ryanodine receptor activity. PloS one 2017, 12, e0175221. [Google Scholar] [CrossRef]
- Speerschneider, T.; Grubb, S.; Metoska, A.; Olesen, S.P.; Calloe, K.; Thomsen, M.B. Development of heart failure is independent of K+ channel-interacting protein 2 expression. The Journal of physiology 2013, 591, 5923–5937. [Google Scholar] [CrossRef] [PubMed]
- Grubb, S.; Speerschneider, T.; Occhipinti, D.; Fiset, C.; Olesen, S.P.; Thomsen, M.B.; Calloe, K. Loss of K+ currents in heart failure is accentuated in KChIP2 deficient mice. Journal of cardiovascular electrophysiology 2014, 25, 896–904. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram of the protein structure of the KChIP family. KChIPs are bipolar proteins of approximately 178-285 amino acids in size. Each KChIP contains an N-terminal domain and a C-terminal domain. The N-terminal domain differs between KChIPs and has multiple post-translational modification sites, whereas the C-terminal domain contains 4 conserved EF-hand Ca2+-binding motifs. Human KCNIP1 has 5 transcript variants encoding 5 different isoforms (KChIP1a, KChIP1b, KChIP1-1adetall and another 2 unnamed isoforms). KCNIP2 has 7 transcript variants and encoding 7 different isoforms (KChIP2a, KChIP2b, KChIP2s, KChIP2c, KChIP2.5 and another 2 unnamed isoforms). KCNIP3 has 3 transcript variants encoding 3 different isoforms (KChIP3.1 and KChIP3.2 and KChIP3.x). KCNIP4 has 7 transcript variants encoding 6 different isoforms (KChIP4.1, KChIP4.2, KChIP4bs, KChIP4.4 and another 2 unnamed isoforms), of which variant 3 (NM_147182.4) and variant 6 (NM_001035004.2) encode the same isoform 3 (NP_001030176.1). Functional domains are indicated by boxes with different colors. The post-translational modification sites are marked with pentagrams of different colors. The correspond protein names and accessions numbers refer to NCBI or Uniprot database. NA, not available.
Figure 1.
Diagram of the protein structure of the KChIP family. KChIPs are bipolar proteins of approximately 178-285 amino acids in size. Each KChIP contains an N-terminal domain and a C-terminal domain. The N-terminal domain differs between KChIPs and has multiple post-translational modification sites, whereas the C-terminal domain contains 4 conserved EF-hand Ca2+-binding motifs. Human KCNIP1 has 5 transcript variants encoding 5 different isoforms (KChIP1a, KChIP1b, KChIP1-1adetall and another 2 unnamed isoforms). KCNIP2 has 7 transcript variants and encoding 7 different isoforms (KChIP2a, KChIP2b, KChIP2s, KChIP2c, KChIP2.5 and another 2 unnamed isoforms). KCNIP3 has 3 transcript variants encoding 3 different isoforms (KChIP3.1 and KChIP3.2 and KChIP3.x). KCNIP4 has 7 transcript variants encoding 6 different isoforms (KChIP4.1, KChIP4.2, KChIP4bs, KChIP4.4 and another 2 unnamed isoforms), of which variant 3 (NM_147182.4) and variant 6 (NM_001035004.2) encode the same isoform 3 (NP_001030176.1). Functional domains are indicated by boxes with different colors. The post-translational modification sites are marked with pentagrams of different colors. The correspond protein names and accessions numbers refer to NCBI or Uniprot database. NA, not available.
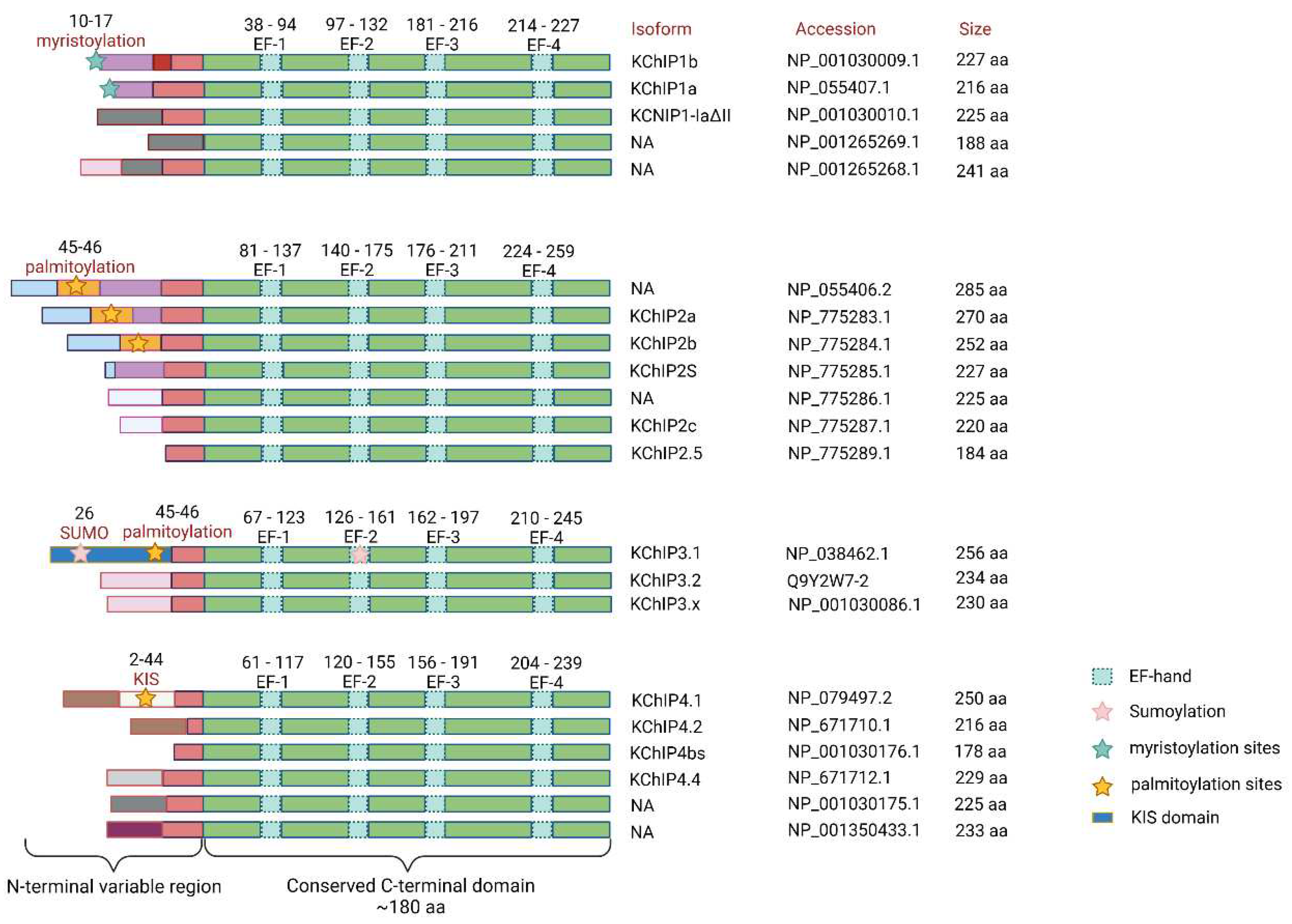
Figure 2.
Regulation of KChIP2 expression at the transcriptional level in cardiomyocytes. Positive regulation of KCNIP2 transcription includes: (1) members of the Krüppel-like factor family (KLF4 and KLF15) and (2) CREB directly bind to the promoter regions of the KCNIP2 gene. Negative regulation of KCNIP2 transcription includes: (1) Activation of TNFR, α-AR, and β-AR increases NF-κB activity and thus exerts an inhibitory effect on the KCNIP2 promoter. (2) Two branches of the MAPK signaling pathway are involved in the downregulation of KChIP2 expression: Activation of PKC phosphorylates and activates MEK, which in turn activates ERK. JNK can be phosphorylated and activated by phenylephrine (PE). Both ERK and JNK can reduce KCNIP2 mRNA levels by activating associated transcription factors. (3) The Ca2+-activated phosphatase calcineurin dephosphorylates NFAT, which then translocates to the nucleus to repress KCNIP2 transcription. (4) The Notch1 receptor is cleaved by γ-secretase to generate NICD following receptor-ligand interaction. NICD then translocates to the nucleus and interacts with transcriptional regulators to inhibit KCNIP2 expression. CREB: cAMP response element-binding protein; TNFR: tumor necrosis factor receptor; α-AR: α-adrenergic receptor; β-AR: β-adrenergic receptor; Pro: propranolol; NF-κB: nuclear factor κB; IκB: NF-κB inhibitor; IKK: IκB kinase; PKC: protein kinase C; MEK: mitogen-activated protein kinase kinase; ERK: extracellular regulated protein kinase; JNK: c-Jun N-terminal kinase; TF: Transcription factor; NFAT: nuclear factor of activated T cells; NICD: intracellular domain of the Notch receptor.
Figure 2.
Regulation of KChIP2 expression at the transcriptional level in cardiomyocytes. Positive regulation of KCNIP2 transcription includes: (1) members of the Krüppel-like factor family (KLF4 and KLF15) and (2) CREB directly bind to the promoter regions of the KCNIP2 gene. Negative regulation of KCNIP2 transcription includes: (1) Activation of TNFR, α-AR, and β-AR increases NF-κB activity and thus exerts an inhibitory effect on the KCNIP2 promoter. (2) Two branches of the MAPK signaling pathway are involved in the downregulation of KChIP2 expression: Activation of PKC phosphorylates and activates MEK, which in turn activates ERK. JNK can be phosphorylated and activated by phenylephrine (PE). Both ERK and JNK can reduce KCNIP2 mRNA levels by activating associated transcription factors. (3) The Ca2+-activated phosphatase calcineurin dephosphorylates NFAT, which then translocates to the nucleus to repress KCNIP2 transcription. (4) The Notch1 receptor is cleaved by γ-secretase to generate NICD following receptor-ligand interaction. NICD then translocates to the nucleus and interacts with transcriptional regulators to inhibit KCNIP2 expression. CREB: cAMP response element-binding protein; TNFR: tumor necrosis factor receptor; α-AR: α-adrenergic receptor; β-AR: β-adrenergic receptor; Pro: propranolol; NF-κB: nuclear factor κB; IκB: NF-κB inhibitor; IKK: IκB kinase; PKC: protein kinase C; MEK: mitogen-activated protein kinase kinase; ERK: extracellular regulated protein kinase; JNK: c-Jun N-terminal kinase; TF: Transcription factor; NFAT: nuclear factor of activated T cells; NICD: intracellular domain of the Notch receptor.
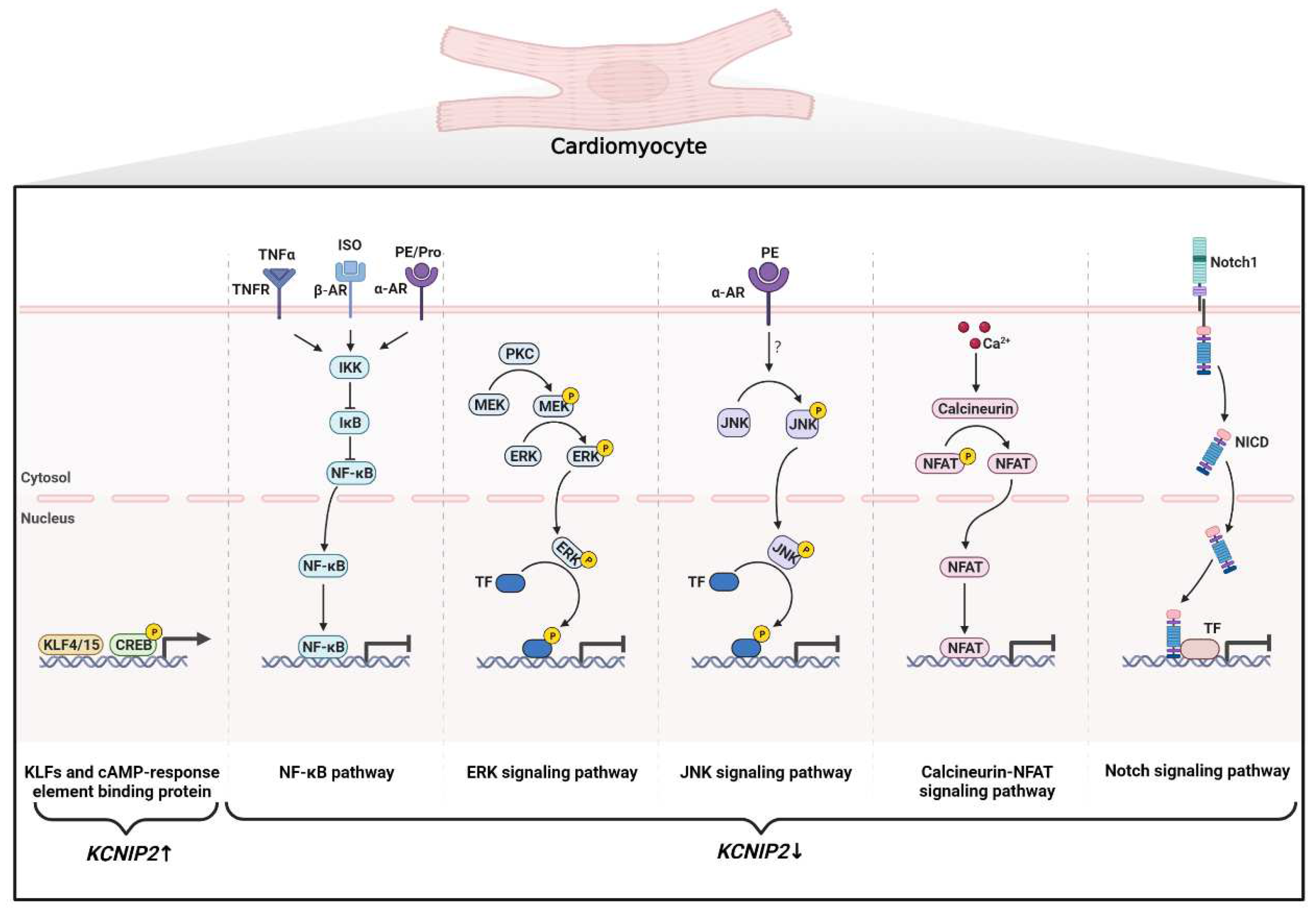
Figure 3.
KChIPs allow KV4 trafficking to the cell surface. KV4 channels cannot be trafficked to the cell membrane on their own because they are aggregated, misfolded, and retained in the ER for degradation. Co-expression of KChIP1-3 releases KV4 channels from ER retention and redistributes them to the cell surface.
Figure 3.
KChIPs allow KV4 trafficking to the cell surface. KV4 channels cannot be trafficked to the cell membrane on their own because they are aggregated, misfolded, and retained in the ER for degradation. Co-expression of KChIP1-3 releases KV4 channels from ER retention and redistributes them to the cell surface.
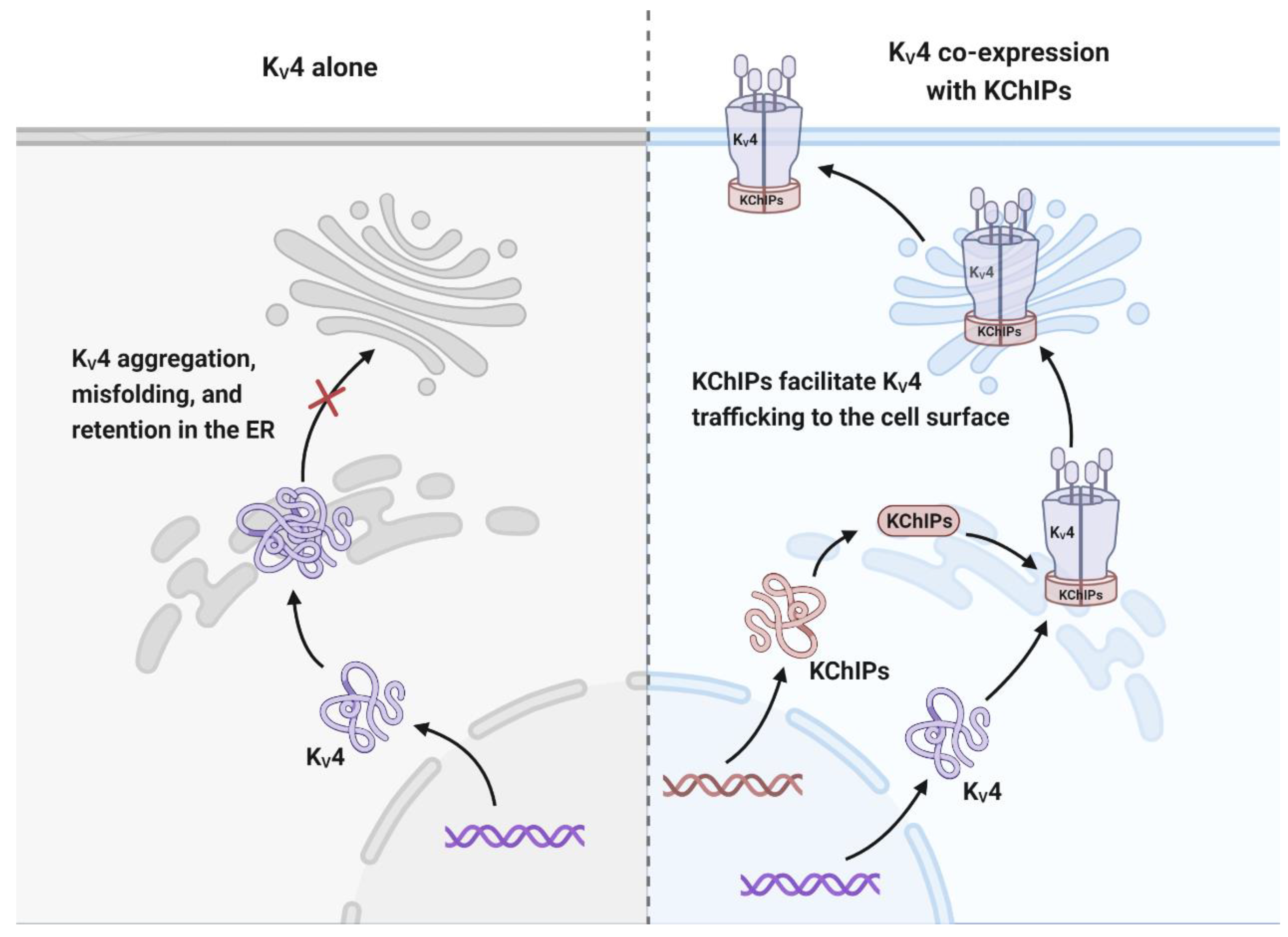
Figure 4.
Function of KChIP3 in neuron. Altered KChIP3 expression is associated with several nervous system disorders. For example, KChIP3 is downregulated in epilepsy and Huntington's disease (HD), and upregulated in Alzheimer's disease (AD) and amyotrophic lateral sclerosis (ALS). KChIP3 has physiological functions in both the cytoplasm and the nucleus. Sumoylated KChIP3 translocates to the nucleus to repress the transcription of associated genes by interacting with the DRE sequences. This interaction can be enhanced by Mg2+ and C-terminal binding protein (CtBP) and inhibited by Ca2+ and cyclic AMP-responsive element modulator α (αCREM). KChIP3 can also repress target genes transcription by interacting with the transcription factor CREB to interfere with CREB phosphorylation and CREB-binding protein (CBP) recruitment. Palmitoylation and phosphorylation of KChIP3 increase its location in the cytoplasm where it interacts with presenilin (PS) to regulate the enzymatic activity of the γ-secretase. In the cytoplasm KChIP3 regulates intracellular K+, Ca2+ and Na+ currents by interacting with KV4, N-methyl-D-aspartate receptor (NMDAR), transient receptor potential vanilloid 1 (TRPV1) and the ryanodine receptor.
Figure 4.
Function of KChIP3 in neuron. Altered KChIP3 expression is associated with several nervous system disorders. For example, KChIP3 is downregulated in epilepsy and Huntington's disease (HD), and upregulated in Alzheimer's disease (AD) and amyotrophic lateral sclerosis (ALS). KChIP3 has physiological functions in both the cytoplasm and the nucleus. Sumoylated KChIP3 translocates to the nucleus to repress the transcription of associated genes by interacting with the DRE sequences. This interaction can be enhanced by Mg2+ and C-terminal binding protein (CtBP) and inhibited by Ca2+ and cyclic AMP-responsive element modulator α (αCREM). KChIP3 can also repress target genes transcription by interacting with the transcription factor CREB to interfere with CREB phosphorylation and CREB-binding protein (CBP) recruitment. Palmitoylation and phosphorylation of KChIP3 increase its location in the cytoplasm where it interacts with presenilin (PS) to regulate the enzymatic activity of the γ-secretase. In the cytoplasm KChIP3 regulates intracellular K+, Ca2+ and Na+ currents by interacting with KV4, N-methyl-D-aspartate receptor (NMDAR), transient receptor potential vanilloid 1 (TRPV1) and the ryanodine receptor.
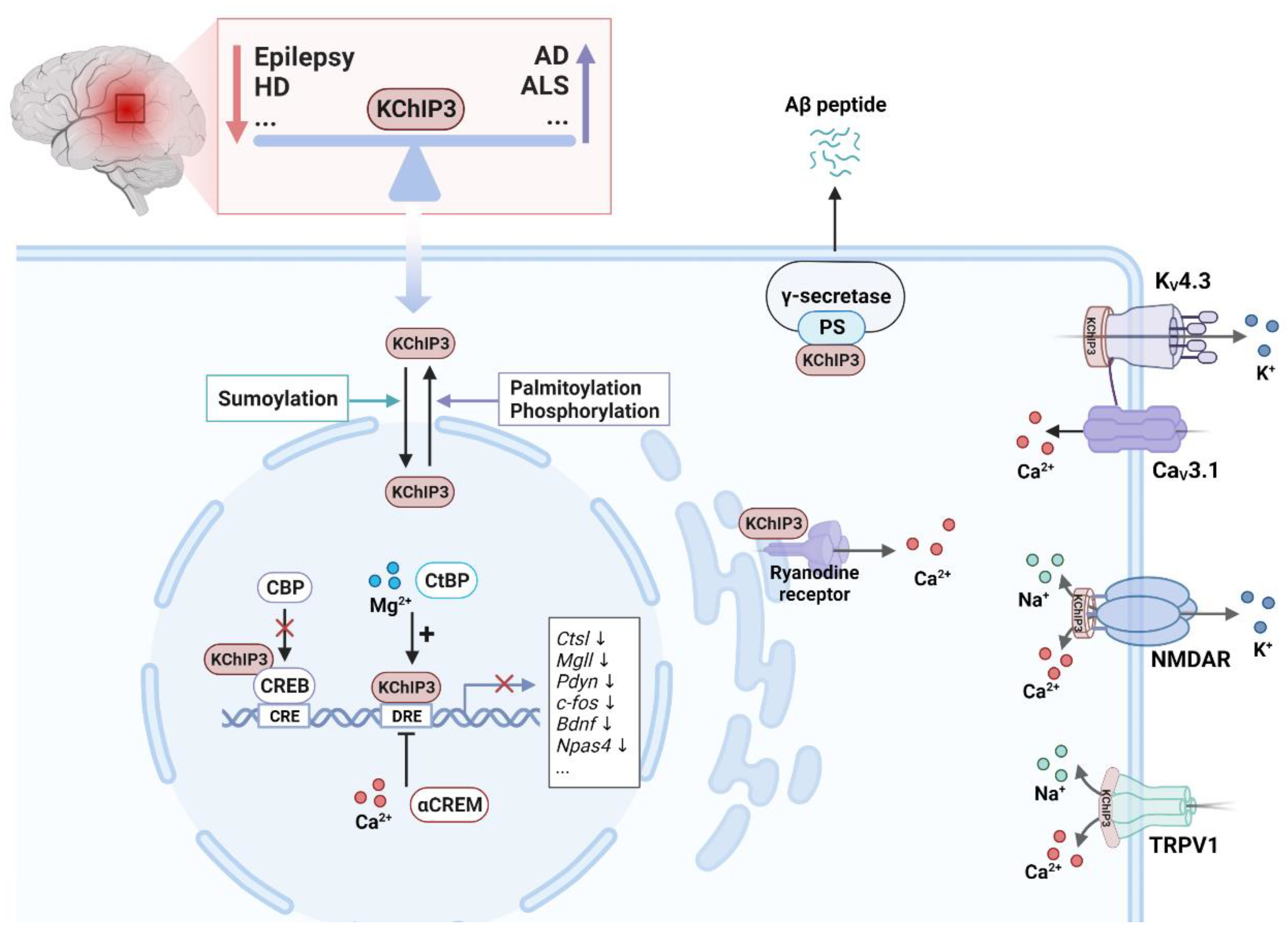
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



