Submitted:
19 June 2023
Posted:
21 June 2023
You are already at the latest version
Abstract
Glioblastoma is considered the most aggressive primary brain tumor. Recurrence after treatment is a significant problem with a failed response to optimal therapies. The recurrence of GBM is linked to different cellular pathways and molecular pathways. Not only genetics are involved in gliomagenesis, but also epigenetics. Epigenetic mechanisms are highly involved in the pathogenesis of GBM. Histone modulation through acetylation, phosphorylation, ubiquitination, and methylation can regulate gene expression and may play a role in the pathogenesis of GBM. Preclinical and clinical studies currently target epigenetic enzymes in gliomas, including a new generation of histone deacetylase (HDAC) inhibitors. Herein, I tried to highlight the current research in glioma epigenetics, focusing on the culprit of histone modifications and the use of HDAC target therapies as a possible treatment line for glioblastoma.
Keywords:
glioma
; histone
; HDAC
; therapy
1. Introduction
Glioblastoma is considered the most aggressive primary brain tumor. Recurrence after treatment is a significant problem, with a survival rate after one year ranging about 39.7% [1]. The recurrence of GBM is linked to different cellular pathways and molecular signaling. The genetic profile of glioma is complicated, as evidenced by multi-omics studies from the landscape of GBM in the Cancer Genome Atlas Research Network (TCGA), the Chinese Glioma Genome Atlas (CGGA), and other databases [2]. 1p and 19q co-deletions (oligodendroglioma-specific), IDH gene mutations, PTEN (Phosphatase and tensin homolog) gene mutations, TP53 mutations, TERT (Telomerase reverse transcriptase) gene promoter mutations, ATRX (Alpha thalassemia/mental retardation syndrome X-linked) gene mutations, and EGFR (Epithelial growth factor receptor) gene amplification are the most prominent genetic molecules involved in gliomagenesis [3]. Not only is genetic deregulation involved in gliomagenesis, but also epigenetics. Manipulation of genetics without affecting DNA itself is a crucial role of epigenetics [4]. Therefore, more knowledge in the field of epigenetics is required to understand the biology of GBM fully. Most clinical trials failed to promote prolonged survival of glioblastoma. The only trial with the best noticeable outcome was the European Organization for Research and Treatment of Cancer (EORTC) and National Cancer Institute of Canada (NCIC) clinical trial in 2005 [5]. Since then, there were no other clinical trials with better survival chances.
Histone modulation through acetylation, phosphorylation, ubiquitination, and methylation can regulate gene expression. Histone can be modified through acetylation and deacetylation, affecting different physiological and pathological processes [6,7]. Histone acetyltransferase mediates acetylation, which is usually associated with gene activation. On the contrary, histone deacetylation is mainly associated with gene suppression Figure 1 [7,8]. The detailed general characterization of how histone modifications affect gene expression, in general, is out of this review's scope. Abnormally activated HDACs have a role in the pathogenesis of glioma. Therefore, inhibitors of that enzyme can be a therapeutic option controlling apoptosis and cellular proliferation [9,10]. HDACs inhibitors are of particular importance in glioma targeted therapy as they can pass the blood-brain barrier at variable extents [11].
2. Histone Modifications in Glioma:
2.1. Histone Deacetylation in Glioma:
Abnormal HDAC activity was identified in some cancers, but the complete mechanisms involved have not been fully elucidated [12]. Some HDACs are upregulated in certain tumors and downregulated in others [13]. HDACs have multiple types and functions, which make them a prominent target for molecular therapy. Before we can discuss the effective targeted therapy models, we should focus the light on categories of HDACs and the suggested roles they have in GBM. Studies investigating the role of HDACs in the literature are few. The types of HDACs are classified based on similarities to those found in yeast, and they vary in the cellular location and structure [12]. Classical HDACs include the following: Class I (HDAC 1-3, 8), II (HDAC 4-7, 9-10), and IV (HDAC 11), which are Zn2+ dependent, while class III is Zn2+ independent [14]. It is reported that classes II and IV are expressed at high levels in a low-grade astrocytoma [13].
HDAC1 and HDAC2 have been reported to be highly expressed in GBM cell lines, and HDAC2 knocking down increased the response to temozolomide therapy [15]. A higher expression of HDAC3 is associated with a dismal prognosis and was noticed in specific aggressive phenotypes of glioma cell lines [16]. In a specific study by Wang and his colleagues, HDAC6 was upregulated in GBM cell lines, and the response to TMZ treatment was enhanced following knocking down that enzyme [17]. Class III HDACs comprise a group of proteins named the sirtuin (SIRT) family [18]. Aberrant expression of class III HDACs was noticed in GBM cell lines, and Feng et al. noticed that SIRT1 and SIRT6 were downregulated in these cells [19] while other studies reported upregulation [20]. HDAC class IV has only one type, which is HDAC 11 [21]. It was reported that the expression of HDAC11 decreases in the more aggressive GBM tumors and has a dismal prognosis [13].
2.2. Histone Acetylation in Glioma:
Histone acetylation leads to an increased gene activity by allowing more DNA exposure for transcription complexes [22]. Acetylation is accomplished using histone acetyltransferases HATs. HATs have some role in cellular signaling, DNA damage repair, and cell cycle regulation [23,24]. HATs include the following, GNAT superfamily, MYST family, p300/CBP, nuclear receptor coactivators (SRC-1, ACTR, TIF2), TAFII250, and TFIIIC [25]. The one that was extensively studied in GBM is the p300/CBP and termed KAT2B, EP300 [26]. P300 acts as a tumor suppressor in GBM and an inhibitor for acetyltransferase P300 is highly expressed in GBM and makes the prognosis worse [26]. A group of researchers found that PI3K/Akt signaling activation, a highly involved pathway in gliomagenesis and PIK3CA expression, was recruited by H3K23 acetylation enhanced by a specific HAT called KAT6A, which belongs to the MYST family [27]. Some studies have investigated the role of HATs in GBM, as illustrated in Table 1.
2.3. Histone Methylation in Glioma:
Lysine and arginine are methylated on histones, mostly H3 and H4, by the two enzymes lysine methyltransferase (KMTs) and arginine methyltransferase (PRMTs), which include different subtypesTable 2 [28]. Various studies reported the clinical significance of aberrations related to methyltransferases and their association with different cancers [29,30,31]. A study reported that the KMT G9a is abnormally expressed in some brain tumors [32]. Moreover, a high expression rate of KMT G9a is noted to be associated with more aggressive behavior in glioma [33]. Researchers investigated the role of certain KMTs as SUV39H1 and SETDB1 in gliomagenesis, and they observed the upregulated expression in malignant glioma cell lines. Moreover, knocking down SUV39H1 and SETDB1 induced a high rate of apoptosis and a diminished migratory capacity of cells [34]. The arginine methyltransferase 2 (PRMT 2) was highly expressed in GBM and linked to an unfavorable prognosis [35]. The proposed mechanistic role of PRMT2 is thought to be through H3R8 methylation, whose function is associated with promoter enhancement and active gene expression suggesting its potential oncogenic activity [35]. We tried to list the different studies that involved KMTs and PRMTs in GBM. Table 3.
2.4. Histone Demethylation in Glioma:
There is an increasing interest in the enzyme N-methyl-lysine demethylase (KDM1, also known as LSD1, AOF2, or BHC110) as a possible target for therapy in cancer [36]. Methylation and demethylation of histone's arginine side chains are of equal scientific importance as acetylation, phosphorylation, and ubiquitination. Demethylation of histone contributes to cellular development, so dysregulation of that biochemical process may result in disorganized cellular development and tumorigenesis [37]. Work is promoted to further study histone demethylases because of the evident success of demethylase inhibitors in the field of cancer [38]. Several histone demethylases are overexpressed in GBM and are suspected of having a role in TMZ resistance [39]. Sareddy et al. reported that KDM1A is expressed too much in glioma cell lines, and using pargyline to inhibit it reduced cellular proliferation [40]. Targeting KDM2A by micro RNA-3666 halted the migration of glioma cells [41]. Moreover, knocking down KDM2B blunted the numbers of glioma stem cells in primary GBM cultures suggesting that KDM2B is fundamental for GBM cell initiation and survival. [42]. A significant increase in acidic vesicular organs and autophagy-related proteins was noticed following inhibition of KDM4A using siRNA, suggesting a therapeutic role [43]. Synthetic pharmacological inhibitors against KDM4B are effective in TMZ- resistant glioma cells, suggesting a role of KDM4B in resistance to therapy [44].
2.5. Histone Ubiquitination in Glioma:
Histone ubiquitination frequently happens in two regions of histone, H2A at lysine 119 (H2AK119ub1) and H2B at lysine 120 (H2BK120ub1) [45]. Abnormal histone ubiquitination could alter tumor suppressors and oncogenes [46]. Frequent studies reported many deregulated ubiquitination enzymes in different cancer types marking ubiquitination an exciting item to study cancer [47]. Certain deubiquitinating enzymes regulate that process and include 2A-DUB, USP21, USP16, and BRCA1, and their deregulation could have a role in carcinogenesis [46]. Different research studied abnormal ubiquitin expression patterns in GBM Table 4. Ubiquitin-specific proteases as USP1,3,4,10,13 are expressed at a higher rate in GBM cells than in normal brains and are linked to poor survival [47,48,49].
2.6. Histone Sumoylation and Glioma:
Sumoylation is a type of post-translational modification attaching ubiquitin-related modifier (SUMO) groups to histones [50]. Sumoylation is reported to be involved in different cellular processes as apoptosis and signal transduction [51]. Four isoforms of SUMO have been identified and include SUMO1,2,3,4 [52]. E1 enzyme (SAE1 and SAE2/UBA2) and E2 enzyme are involved in SUMO modification [53]. SUMO-1 and SUMO-2/3 proteins were found to be expressed in both low and high-grade gliomas [54]. A study observed that SAE1 enhances glioma cells' growth via the Akt signaling pathway, which is a major pathway involved in gliomagenesis [55].
2.7. Histone Phosphorylation and Glioma:
Histone phosphorylation is one of the post-translational modifications that may play a role in cell division, apoptosis, and gene expression. However, little is known about the prognostic implication of histone phosphorylation in human cancer. Histone phosphorylation importantly occurs when DNA damage repair ensues as the phosphorylated histone H2A functions to localize the sites of DNA repair [56]. Phosphorylation of specific proteins was also linked to regulation of proliferative genes such as serine 10 and 28 of H3 and serine 32 of H2B phosphorylation which has been involved in EGF responsive gene regulation [57]. Specific phosphorylated histones’ proteins are reported to be associated with certain proto-oncogenes as c-fos, c-jun, and c-myc [58]. Research investigating phosphorylated histones’ roles in GBM is limited in the literature. The phosphorylation level of H3T3, T6, S10, S28, Y41, and T45 was analyzed in 42 GBM samples. That analysis depicted a high level of pH3T6, pH3S10, or pH3Y41 linked to poor survival [59]. Moreover, pharmacological inhibition of the phosphorylation process using enzastaurin increased GBM cells' sensitivity to irradiation/TMZ treatment [59].
2.8. Targeting Histone-Modifying Enzymes in Glioma
Despite the significant advances in molecular research focusing on GBM, the hallmark treatment proven to be the best is the classical radical surgical resection plus TMZ and radiotherapy [60]. Total excision of GBM is almost impossible due to microscopic infiltration of cells beyond radiological tumor borders, making complementary treatments a valuable tool in therapeutic strategies. What complicates treatment is the diffuse heterogeneity of the GBM microenvironment that makes GBM a notorious tumor for therapy failure. Glioma stem cells also reduce the efficacy of targeted therapies as they have an inherent capacity of self-renewal, initiating and recurring new tumor cells pool [61]. Preclinical trials testing different targeted therapies related to histone enzymes have been reported, but some only have well-established outcomes and are promoted for clinical trials Table 5. Most GBM research uses different glioma cell lines such as U87 and U251 as models for experimental trials. To some extent, GBM cell lines are different from human primary tissue samples, especially in the case of studying signaling and genetic profiling, and that eventually leads to some discrepancy in the results of experimental animals, preclinical, and clinical trials [62].
3. HDAC inhibitors (HDACi)
HDACi have been investigated as a therapy for many cancers, and candidates of this family have shown promising results in certain tumors as vorinostat for primary cutaneous T-cell lymphoma [63]. Most of the HDACi used in preclinical and clinical trials are specific for each enzyme class and are usually used as a combination therapy with other targeted therapies. HDACi work through different mechanisms as an anti-cancer therapy. HDACi can untwist chromatin's condensation, allowing TMZ and other chemotherapeutics to gain more access to DNA [64]. They may initiate autophagy through the production of complex immunomodulatory cytokines [65]. HDACi is reported to reduce the effect of VEGF, which plays a master role in hypoxia-induced angiogenesis in GBM [66]. Apoptosis can be achieved through different mechanisms using HDACi [67]. A famous HDACi called vorinostat was reported to induce DNA damage and double-strand breaks [68]. Another possible mechanism of the anticancer effect of HDACi in the production of reactive oxygen species, which help destroy tumor cells [69]. We will focus on the most prominent candidates of HDACi that were used in clinical trials.
3.1. Vorinostat:
This agent was used in stage I and II clinical trials to treat different cancers with an acceptable range of side effects [70,71]. Vorinostat was used in a phase II trial of North Central Cancer Treatment Group (NCCTG) in patients with recurrent GBM [72]. In this study, the patients receiving vorinostat showed progression-free survival at six months of about 15.2 % and median overall survival of 5.7 months [72]. Combination therapy of vorinostat, bevacizumab) anti-VEGF (erlotinib, a tyrosine kinase receptor inhibitor, bortezomib, a proteasome inhibitor, and isotretinoin with or without TMZ was tested. This combination aims at blocking all possible routes for resistance to therapy [73,74,75]. In a study conducted by the Adult Brain Tumor Consortium (ABTC), vorinostat combined with TMZ was used in patients with malignant gliomas who had previously received radiotherapy [76]. Results from this trial encouraged the ABTC and NCCTG to start a phase II trial of vorinostat with radiotherapy and TMZ in newly diagnosed GBM patients. Vorinostat combination with bevacizumab plus irinotecan was published as a phase I clinical trial [77]. HDAC inhibitors' ability to enhance the anti-tumor activity of both bevacizumab and topoisomerase I inhibitors was also supported by the preclinical data published in different studies [78,79,80]. In patients with recurrent GBM, a proteasome inhibitor bortezomib was added to vorinostat as a part of phase II clinical trial conducted by NCCTG. The median time of progression was about 1.4 months (range 0.5–5.6 months), and the median overall survival was about 2.4 months [80].
3.2. Valproic acid
The European Organization for Research and Treatment of Cancer (EORTC) and National Cancer Institute of Canada (NCIC) trials found exciting results regarding using the antiepileptic valproic acid with a distinguished effect on outcome [81]. Valproic acid (VPA) is considered a class I selective HDAC inhibitor [82]. Valproic acid is known for its antiepileptic properties, but it also has an HDAC inhibiting activity. A better survival with TMZ and radiotherapy was observed in patients receiving VPA as the only antiepileptic drug [82]. The mechanism behind the effect of VPA in prolongation of survival in GBM patients is still not fully explained. Some researchers suggested that VPA increases the bioavailability of TMZ by diminishing its clearance [83]. A phase II trial study was conducted to investigate the effect of VPA in GBM patients who are on chemo and radiotherapy [84]. This study showed a progression-free survival of about 10.5 months. In another study conducted by Deepthi Valiyaveettil et al., VPA with combined chemo and radiotherapy in GBM patients, the PFS was about ten months, and overall survival was about 16 months [84].
3.3. Romidepsin (FK228)
This agent was studied in a trial conducted by the North American Brain Tumor Consortium in patients with recurrent glioma who are on enzyme-inducing antiepileptic drugs (EIAEDs) and evaluate the antitumor efficacy of romidepsin in patients with recurrent glioblastoma who were not receiving EIAED. The resulting median PFS was only six weeks [85]. Mice treated with both romidepsin and TMZ drugs significantly reduced tumor weights and volumes compared to each drug alone [104]. Our results suggested that FK228 augmented temozolomide sensitivity in human glioma cells partially by blocking PI3K/AKT/mTOR signal pathways [104]. A study by Nguyen et al. reported the combined inhibition of TRAP1 by gamitrinib and romidepsin or panobinostat caused synergistic growth reduction of established and patient-derived xenograft (PDX) glioblastoma cells [105].
3.4. Panobinostat
FDA approved this HDACi in the treatment of multiple myeloma. It is still under investigation in an active clinical trial in children with diffuse intrinsic pontine glioma with marizomib, a proteasome inhibitor. (Source: https://clinicaltrials.gov).
3.5. Limitations of HDACi in clinical practice:
Some side effects were reported using HDACi which include neutropenia and thrombocytopenia [86]. Cardiotoxicity was noticed, which is potentially hazardous [87]. Resistance to therapy was observed in several studies, and the mechanisms of resistance are variable. One proposed mechanism is the activation of anti-apoptotic transcription factor NF-κB and other anti apoptotic proteins [88]. A slackened BBB permeability was noticed with most of HDACi [89]. The potential benefits that HDACi might offer in CNS disorders encouraged researchers to explore the brain uptake of HDAC inhibitors as potential templates for developing HDAC inhibitors fully penetrating the BBB. Brain uptake of trichostatin A (TSA)-like hydroxamates and (KB631) in the baboon brain was negligible [90,91]. On the contrary, five patients post-treatment with vorinostat exhibited an increase in histone acetylation in their post-surgical specimens, proving that vorinostat reached a sufficient concentration in the tumor [72].
4. Conclusion and future perspectives:
There is a compelling need for new studies exploring epigenetics in GBM. Therapies to prolong survival in GBM are still limited, although new molecular therapies have emerged. Some virulent features characterizing glioblastoma can be targeted using different histone deacetylase inhibitors. Recent clinical trials have demonstrated potentially effective models of therapies targeting epigenetics but failed to achieve maximum intratumoral concentration levels. Research should be directed at managing how to achieve a high CNS level of those therapies. Discovering more about the epigenetics of glioma may establish new GBM classifications that will be epigenetically based and help with the proper selection of the targeted therapy.
Abbreviations
| ABTC | The Adult Brain Tumor Consortium |
| EIAEDs | Enzyme-inducing antiepileptic drugs |
| EORTC | European Organization for Research and Treatment of Cancer |
| GBM | Glioblastoma |
| DNA | Deoxynucleic acid |
| EGF | Epidermal Growth Factor |
| HDAC | Histone Deacetylase |
| HDACi | Histone Deacetylase Inhibitors |
| HATs | Histone Acetyltransferases |
| NCIC | National Cancer Institute of Canada |
| USP | Ubiquitin Specific Protease |
| TMZ | Temozolomide |
References
- Sherrod, B.A.; Gamboa, N.T.; Wilkerson, C.; Wilde, H.; Azab, M.A.; Karsy, M.; Jensen, R.L.; Menacho, S.T. Effect of patient age on glioblastoma perioperative treatment costs: a value driven outcome database analysis. J. Neuro-Oncology 2019, 143, 465–473. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Mclendon, R.E.; Friedman, A.H.; Bigner, D.D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Aparna, S.; Lakshmaiah, K.; Jacob, L.; Lokanatha, D.; Saldanha, S. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Bi, G.; Jiang, G. The molecular mechanism of HDAC inhibitors in anticancer effects. Cell. Mol. Immunol. 2006, 3, 285–290. [Google Scholar] [PubMed]
- Secrist, J.P.; Zhou, X.; Richon, V.M. HDAC inhibitors for the treatment of cancer. Curr. Opin. Investig. drugs (London, Engl. : 2000) 2003, 4. [Google Scholar]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Bezecny, P. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol. 2014, 31, 985. [Google Scholar] [CrossRef]
- Adamopoulou, E.; Naumann, U. HDAC inhibitors and their potential applications to glioblastoma therapy. Oncoimmunology 2013, 2, e25219. [Google Scholar] [CrossRef]
- Sturm, D.; Bender, S.; Jones, D.T.W.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.; Hamid, A.; Hussain, A.; Majeed, R.; Qurishi, Y.; Bhat, J.A.; Najar, R.A.; Qazi, A.K.; Zargar, M.A.; Singh, S.K.; et al. Understanding Histone Deacetylases in the Cancer Development and Treatment: An Epigenetic Perspective of Cancer Chemotherapy. DNA Cell Biol. 2012, 31, S62–S71. [Google Scholar] [CrossRef] [PubMed]
- Lucio-Eterovic, A.K.; Cortez, M.A.; Valera, E.T.; Motta, F.J.; Queiroz, R.G.; Machado, H.R.; Carlotti, C.G.; Neder, L.; A Scrideli, C.; Tone, L.G. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer 2008, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- A Glozak, M.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Chen, J.; Tan, Q.; Xie, C.; Li, C.; Zhan, W.; Wang, M. Silencing of histone deacetylase 2 suppresses malignancy for proliferation, migration, and invasion of glioblastoma cells and enhances temozolomide sensitivity. Cancer Chemother. Pharmacol. 2016, 78, 1289–1296. [Google Scholar] [CrossRef]
- Zhong, S.; Fan, Y.; Wu, B.; Wang, Y.; Jiang, S.; Ge, J.; Hua, C.; Zhao, G.; Chen, Y.; Xu, H. HDAC3 Expression Correlates with the Prognosis and Grade of Patients with Glioma: A Diversification Analysis Based on Transcriptome and Clinical Evidence. World Neurosurg. 2018, 119, e145–e158. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, P.; Tang, F.; Lian, H.; Chen, X.; Zhang, Y.; He, X.; Liu, W.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016, 379, 134–142. [Google Scholar] [CrossRef]
- Guarente, L. Sirtuins, NAD+, aging, and disease: A retrospective and prospective overview. In Introductory Review on Sirtuins in Biology, Aging, and Disease (pp. 1–6); Elsevier, 2018. [Google Scholar] [CrossRef]
- Feng, J.; Yan, P.-F.; Zhao, H.-Y.; Zhang, F.-C.; Zhao, W.-H.; Feng, M. SIRT6 suppresses glioma cell growth via induction of apoptosis, inhibition of oxidative stress and suppression of JAK2/STAT3 signaling pathway activation. Oncol. Rep. 2016, 35, 1395–1402. [Google Scholar] [CrossRef]
- Chen, H.; Lin, R.; Zhang, Z.; Wei, Q.; Zhong, Z.; Huang, J.; Xu, Y. Sirtuin 1 knockdown inhibits glioma cell proliferation and potentiates temozolomide toxicity via facilitation of reactive oxygen species generation. Oncol. Lett. 2019, 17, 5343–5350. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and Functional Characterization of HDAC11, a Novel Member of the Human Histone Deacetylase Family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Sealy, L.; Chalkley, R. DNA associated with hyperacetylated histone is preferentially digested by DNase I. Nucleic Acids Res. 1978, 5, 1863–1876. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L.; Liang, J.; Shi, L.; Yang, J.; Yi, X.; Zhang, D.; Han, X.; Yu, N.; Shang, Y. Histone Acetyltransferase 1 Promotes Homologous Recombination in DNA Repair by Facilitating Histone Turnover. J. Biol. Chem. 2013, 288, 18271–18282. [Google Scholar] [CrossRef]
- Howe, L.; Auston, D.; Grant, P.; John, S.; Cook, R.G.; Workman, J.L.; Pillus, L. Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev. 2001, 15, 3144–3154. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of Histones and Transcription-Related Factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef]
- Diao P, -Y.; Li S, -X.; Peng, J.; Yang J, -H.; Pan Y, -C.; Xu X, -P., ... Huang, G.-D. Overexpression of EP300-interacting inhibitor of differentiation 3 predicts poor prognosis in patients with glioblastoma multiforme. International Journal of Clinical and Experimental Pathology 2020, 13, 979–988.
- Lv, D.; Jia, F.; Hou, Y.; Sang, Y.; Alvarez, A.A.; Zhang, W.; Gao, W.-Q.; Hu, B.; Cheng, S.-Y.; Ge, J.; et al. Histone Acetyltransferase KAT6A Upregulates PI3K/AKT Signaling through TRIM24 Binding. Cancer Res 2017, 77, 6190–6201. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. In Nature Structural and Molecular Biology (Vol. 26, issue 10, pp. 880–889); Nature publishing group:2019. [CrossRef]
- Chiang, K.; Zielinska, A.E.; Shaaban, A.M.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Zhang, J.; Francis, A.; Jones, L.J.; Smith, S.; et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep. 2017, 21, 3498–3513. [Google Scholar] [CrossRef]
- Xu, K. DNA and Histone Methylation in Prostate Cancer. In Cancer Drug Discovery and Development (Vol. 0, issue 9783319597843, pp. 489–529); Humana press Inc, 2017. [Google Scholar] [CrossRef]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef]
- Spyropoulou, A.; Gargalionis, A.; Dalagiorgou, G.; Adamopoulos, C.; Papavassiliou, K.A.; Lea, R.W.; Piperi, C.; Papavassiliou, A.G. Role of Histone Lysine Methyltransferases SUV39H1 and SETDB1 in Gliomagenesis: Modulation of Cell Proliferation, Migration, and Colony Formation. NeuroMolecular Med. 2014, 16, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Li, Q.; Yang, C.; Huo, D.; Wang, X.; Ai, C.; Kong, Y.; Sun, X.; Wang, W.; Zhou, Y.; et al. PRMT2 links histone H3R8 asymmetric dimethylation to oncogenic activation and tumorigenesis of glioblastoma. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.-H.; Zhang, W.; Chen, X.; George, J.; Ng, H.-H. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 2007, 21, 2545–2557. [Google Scholar] [CrossRef] [PubMed]
- D’Oto, A.; Tian, Q.-W.; Davidoff, A.M.; Yang, J. Histone demethylases and their roles in cancer epige-netics. Journal of Medical Oncology and Therapeutics 2016, 1, 34–40.
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- Banelli, B.; Carra, E.; Barbieri, F.; Wurth, R.; Parodi, F.; Pattarozzi, A.; Carosio, R.; Forlani, A.; Allemanni, G.; Marubbi, D.; Florio, T.; Daga, A.; Romani, M. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in 38- glioblastoma. Cell Cycle. 2015, 14, 3418–3429. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Nair, B.C.; Krishnan, S.K.; Gonugunta, V.K.; Zhang, Q.-G.; Suzuki, T.; Miyata, N.; Brenner, A.J.; Brann, D.W.; Vadlamudi, R.K. KDM1 is a novel therapeutic target for the treatment of gliomas. Oncotarget 2013, 4, 18–28. [Google Scholar] [CrossRef]
- Shou, T.; Yang, H.; Lv, J.; Liu, D.; Sun, X. MicroRNA-3666 suppresses the growth and migration of glioblastoma cells by targeting KDM2A. Mol. Med. Rep. 2019, 19, 1049–1055. [Google Scholar] [CrossRef]
- Staberg, M.; Rasmussen, R.D.; Michaelsen, S.R.; Pedersen, H.; Jensen, K.E.; Villingshøj, M.; Skjoth-Rasmussen, J.; Brennum, J.; Vitting-Seerup, K.; Poulsen, H.S.; et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol. Oncol. 2018, 12, 406–420. [Google Scholar] [CrossRef]
- Wang, B.; Fan, X.; Ma, C.; Lei, H.; Long, Q.; Chai, Y. Downregulation of KDM4A Suppresses the Survival of Glioma Cells by Promoting Autophagy. J. Mol. Neurosci. 2016, 60, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Romani, M.; Daga, A.; Forlani, A.; Pistillo, M.P.; Banelli, B. Targeting of Histone Demethylases KDM5A and KDM6B Inhibits the Proliferation of Temozolomide-Resistant Glioblastoma Cells. Cancers 2019, 11, 878. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Jeusset, L.M.-P.; McManus, K.J. Developing Targeted Therapies That Exploit Aberrant Histone Ubiquitination in Cancer. Cells 2019, 8, 165. [Google Scholar] [CrossRef]
- Fang, X.; Zhou, W.; Wu, Q.; Huang, Z.; Shi, Y.; Yang, K.; Chen, C.; Xie, Q.; Mack, S.C.; Wang, X.; et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J. Exp. Med. 2017, 214, 245–267. [Google Scholar] [CrossRef]
- Oikonomaki, M.; Bady, P.; Hegi, M.E. Ubiquitin Specific Peptidase 15 (USP15) suppresses glioblastoma cell growth via stabilization of HECTD1 E3 ligase attenuating WNT pathway activity. Oncotarget 2017, 8, 110490–110502. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, P.; Ji, W.; Yu, Z.; Chen, H.; Jiang, L. Ubiquitin-specific protease 4 promotes glioblastoma multiforme via activating ERK pathway. OncoTargets Ther. 2019, ume 12, 1825–1839. [Google Scholar] [CrossRef]
- Park, H.J.; Yun, D.-J. New Insights into the Role of the Small Ubiquitin-like Modifier (SUMO) in Plants. Int Rev Cell Mol Biol. 2013, 300, 161–209. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Han, Z.-J.; Feng, Y.-H.; Gu, B.-H.; Li, Y.-M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef]
- Yang, Y.; He, Y.; Wang, X.; Liang, Z.; He, G.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Protein SUMOylation modification and its associations with disease. Open Biol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ji, S. Inhibition of Ubc9-Induced CRMP2 SUMOylation Disrupts Glioblastoma Cell Proliferation. J. Mol. Neurosci. 2019, 69, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liang, Z.; Xia, Z.; Wang, X.; Ma, Y.; Sheng, Z.; Gu, Q.; Shen, G.; Zhou, L.; Zhu, H.; et al. SAE1 promotes human glioma progression through activating AKT SUMOylation-mediated signaling pathways. Cell Commun. Signal. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- Rossetto, D.; Avvakumov, N.; Côté, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.T.; Lee, S.-Y.; Xu, Y.-M.; Zheng, D.; Cho, Y.-Y.; Zhu, F.; Kim, H.-G.; Li, S.-Q.; Zhang, Z.; Bode, A.M.; et al. Phosphorylation of Histone H2B Serine 32 Is Linked to Cell Transformation. J. Biol. Chem. 2011, 286, 26628–26637. [Google Scholar] [CrossRef]
- Choi, H.S.; Choi, B.Y.; Cho, Y.-Y.; Mizuno, H.; Kang, B.S.; Bode, A.M.; Dong, Z. Phosphorylation of Histone H3 at Serine 10 Is Indispensable for Neoplastic Cell Transformation. Cancer Res 2005, 65, 5818–5827. [Google Scholar] [CrossRef] [PubMed]
- Pacaud, R.; Cheray, M.; Nadaradjane, A.; Vallette, F.M.; Cartron, P.-F. Histone H3 Phosphorylation in GBM: a New Rational to Guide the Use of Kinase Inhibitors in anti-GBM Therapy. Theranostics 2015, 5, 12–22. [Google Scholar] [CrossRef]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. In Nature Reviews Disease Primers. Nat Rev Dis Primers. 2015, 1, 15017. [Google Scholar] [CrossRef]
- A Azab, M.; Alomari, A.; Azzam, A.Y. Featuring how calcium channels and calmodulin affect glioblastoma behavior. A review article. Cancer Treat. Res. Commun. 2020, 25, 100255. [Google Scholar] [CrossRef]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic Changes and Gene Expression Profiles Reveal That Established Glioma Cell Lines Are Poorly Representative of Primary Human Gliomas. Mol. Cancer Res. 2008, 6, 21–30. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, R.; Braga, C.; Santos, G.; Bronze, M.R.; Perry, M.J.; Moreira, R.; Brites, D.; Falcão, A.S. Targeting Gliomas: Can a New Alkylating Hybrid Compound Make a Difference? ACS Chem. Neurosci. 2017, 8, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I Histone Deacetylase Inhibitor Entinostat Suppresses Regulatory T Cells and Enhances Immunotherapies in Renal and Prostate Cancer Models. PLOS ONE 2012, 7, e30815. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Novotny, W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem. Biophys. Res. Commun. 2005, 333, 328–335. [Google Scholar] [CrossRef]
- Carew, J.S.; Giles, F.J.; Nawrocki, S.T. Histone deacetylase inhibitors: Mechanisms of cell death and promise in combination cancer therapy. Cancer Lett. 2008, 269, 7–17. [Google Scholar] [CrossRef]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. 2010, 107, 20003–20008. [Google Scholar] [CrossRef]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.-S.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. 2005, 102, 673–678. [Google Scholar] [CrossRef]
- Siegel, D.; Hussein, M.; Belani, C.; Robert, F.; Galanis, E.; Richon, V.M.; Garcia-Vargas, J.; Sanz-Rodriguez, C.; Rizvi, S. Vorinostat in solid and hematologic malignancies. J. Hematol. Oncol. 2009, 2, 31–31. [Google Scholar] [CrossRef]
- Modesitt, S.C.; Sill, M.; Hoffman, J.S.; Bender, D.P. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Jaeckle, K.A.; Maurer, M.J.; Reid, J.M.; Ames, M.M.; Hardwick, J.S.; Reilly, J.F.; Loboda, A.; Nebozhyn, M.; Fantin, V.R.; et al. Phase II Trial of Vorinostat in Recurrent Glioblastoma Multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2009, 27, 2052–2058. [Google Scholar] [CrossRef]
- Galanis, E.; Anderson, S.K.; Lafky, J.M.; Uhm, J.H.; Giannini, C.; Kumar, S.K.; Kimlinger, T.K.; Northfelt, D.W.; Flynn, P.J.; Jaeckle, K.A.; et al. Phase II Study of Bevacizumab in Combination with Sorafenib in Recurrent Glioblastoma (N0776): A North Central Cancer Treatment Group Trial. Clin. Cancer Res. 2013, 19, 4816–4823. [Google Scholar] [CrossRef] [PubMed]
- Ghiaseddin, A.; Reardon, D.; Massey, W.; Mannerino, A.; Lipp, E.S.; Herndon, J.E.; McSherry, F.; Desjardins, A.; Randazzo, D.; Friedman, H.S.; et al. Phase II Study of Bevacizumab and Vorinostat for Patients with Recurrent World Health Organization Grade 4 Malignant Glioma. Oncol. 2018, 23, 157–e21. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.B.; Lipp, E.S.; Miller, E.; Herndon, J.E.; McSherry, F.; Desjardins, A.; Reardon, D.A.; Friedman, H.S. Phase I/II trial of vorinostat, bevacizumab, and daily temozolomide for recurrent malignant gliomas. J. Neuro-Oncol. 2018, 137, 349–356. [Google Scholar] [CrossRef]
- Lee, E.Q.; Puduvalli, V.K.; Reid, J.M.; Kuhn, J.G.; Lamborn, K.R.; Cloughesy, T.F.; Chang, S.M.; Drappatz, J.; Yung, W.K.A.; Gilbert, M.R.; et al. Phase I Study of Vorinostat in Combination with Temozolomide in Patients with High-Grade Gliomas: North American Brain Tumor Consortium Study 04-03. Clin. Cancer Res. 2012, 18, 6032–6039. [Google Scholar] [CrossRef]
- Chinnaiyan, P.; Chowdhary, S.; Potthast, L.; Prabhu, A.; Tsai, Y.-Y.; Sarcar, B.; Kahali, S.; Brem, S.; Yu, H.M.; Rojiani, A.; et al. Phase I trial of vorinostat combined with bevacizumab and CPT-11 in recurrent glioblastoma. Neuro-Oncology 2012, 14, 93–100. [Google Scholar] [CrossRef]
- Qian, D.Z.; Wang, X.; Kachhap, S.K.; Kato, Y.; Wei, Y.; Zhang, L.; Atadja, P.; Pili, R. The Histone Deacetylase Inhibitor NVP-LAQ824 Inhibits Angiogenesis and Has a Greater Antitumor Effect in Combination with the Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor PTK787/ZK222584. Cancer Res 2004, 64, 6626–6634. [Google Scholar] [CrossRef] [PubMed]
- Sarcar, B.; Kahali, S.; Chinnaiyan, P. Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor SN38 in glioblastoma cell lines. J Neurooncol. 2010, 99, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Friday, B.B.; Anderson, S.K.; Buckner, J.; Yu, C.; Giannini, C.; Geoffroy, F.; Schwerkoske, J.; Mazurczak, M.; Gross, H.; Pajon, E.; et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro-Oncology 2012, 14, 215–221. [Google Scholar] [CrossRef]
- Weller, M.; Gorlia, T.; Cairncross, J.G.; Bent, M.J.v.D.; Mason, W.; Belanger, K.; Brandes, A.A.; Bogdahn, U.; Macdonald, D.R.; Forsyth, P.; et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology 2011, 77, 1156–1164. [Google Scholar] [CrossRef]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Wen, P.Y.; Schiff, D. Valproic acid as the AED of choice for patients with glioblastoma?: The jury is out. Neurology 2011, 77, 1114–1115. [Google Scholar] [CrossRef] [PubMed]
- Valiyaveetti, D.; Malik, M.; Joseph, D.M.; Ahmed, S.F.; Kothwal, S.A.; Vijayasaradhi, M. Effect of valproic acid on survival in glioblastoma: A prospective single-arm study. South Asian J. Cancer 2018, 07, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, F.M.; Lamborn, K.R.; Kuhn, J.G.; Wen, P.Y.; Yung, W.K.A.; Gilbert, M.R.; Chang, S.M.; Lieberman, F.S.; Prados, M.D.; Fine, H.A. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neuro-Oncology 2011, 13, 509–516. [Google Scholar] [CrossRef]
- Hirata, Y.; Sasaki, T.; Kanki, H.; Choong, C.-J.; Nishiyama, K.; Kubo, G.; Hotei, A.; Taniguchi, M.; Mochizuki, H.; Uesato, S. New 5-Aryl-Substituted 2-Aminobenzamide-Type HDAC Inhibitors with a Diketopiperazine Group and Their Ameliorating Effects on Ischemia-Induced Neuronal Cell Death. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.T.; Wen, P.Y. Novel Chemotherapeutic Approaches in Adult High-Grade Gliomas. In Cancer Treatment and Research. Cancer Treat Res. 2015, 163, 117–142. [Google Scholar] [CrossRef]
- Mayo, M.W.; Denlinger, C.E.; Broad, R.M.; Yeung, F.; Reilly, E.T.; Shi, Y.; Jones, D.R. Ineffectiveness of Histone Deacetylase Inhibitors to Induce Apoptosis Involves the Transcriptional Activation of NF-κB through the Akt Pathway. J. Biol. Chem. 2003, 278, 18980–18989. [Google Scholar] [CrossRef]
- Seo, Y.J.; Kang, Y.; Muench, L.; Reid, A.; Caesar, S.; Jean, L., ... Kim, S.W.(2014). Image guided synthesis reveals potent blood-brain barrier permeable histone deacetylase inhibitors. ACS Chemical Neuroscience 5(7), 588–596. [CrossRef]
- Seo, Y.J.; Muench, L.; Reid, A.; Chen, J.; Kang, Y.; Hooker, J.M.; Volkow, N.D.; Fowler, J.S.; Kim, S.W. Radionuclide labeling and evaluation of candidate radioligands for PET imaging of histone deacetylase in the brain. Bioorganic Med. Chem. Lett. 2013, 23, 6700–6705. [Google Scholar] [CrossRef]
- Hooker, J.M.; Kim, S.W.; Alexoff, D.; Xu, Y.; Shea, C.; Reid, A.; Volkow, N.; Fowler, J.S. Histone Deacetylase Inhibitor MS-275 Exhibits Poor Brain Penetration: Pharmacokinetic Studies of [11C]MS-275 using Positron Emission Tomography. ACS Chem. Neurosci. 2010, 1, 65–73. [Google Scholar] [CrossRef]
- Saidi, D.; Cheray, M.; Osman, A.M.; Stratoulias, V.; Lindberg, O.R.; Shen, X.; Blomgren, K.; Joseph, B. Glioma-induced SIRT1-dependent activation of hMOF histone H4 lysine 16 acetyltransferase in microglia promotes a tumor supporting phenotype. Oncoimmunology 2018, 7, e1382790. [Google Scholar] [CrossRef]
- Heddleston, J.M.; Wu, Q.; Rivera, M.; Minhas, S.; Lathia, J.D.; E Sloan, A.; Iliopoulos, O.; Hjelmeland, A.B.; Rich, J.N. Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell tumorigenic potential. Cell Death Differ. 2012, 19, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Fontebasso, A.M.; Schwartzentruber, J.; Khuong-Quang, D.-A.; Liu, X.-Y.; Sturm, D.; Korshunov, A.; Jones, D.T.W.; Witt, H.; Kool, M.; Albrecht, S.; et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013, 125, 659–669. [Google Scholar] [CrossRef]
- Bozek, D.; Wang, A.; Hao, X.; Johnston, M.; Luchman, H.A.; Weiss, S. STEM-28. DOT1L EPIGENETICALLY REGULATES GBM BRAIN TUMOR STEM CELLS. Neuro-Oncology 2017, 19, vi231–vi232. [Google Scholar] [CrossRef]
- Wang, S.; Tan, X.; Yang, B.; Yin, B.; Yuan, J.; Qiang, B.; Peng, X. The role of protein arginine-methyltransferase 1 in gliomagenesis. BMB Rep. 2012, 45, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Xu, Y. Effects of Enhancer of Zeste Homolog 2 (EZH2) Expression on Brain Glioma Cell Proliferation and Tumorigenesis. Experiment 2018, 24, 7249–7255. [Google Scholar] [CrossRef]
- Yan, F.; Alinari, L.; Lustberg, M.E.; Martin, L.K.; Cordero-Nieves, H.M.; Banasavadi-Siddegowda, Y.; Virk, S.; Barnholtz-Sloan, J.; Bell, E.H.; Wojton, J.; et al. Genetic Validation of the Protein Arginine Methyltransferase PRMT5 as a Candidate Therapeutic Target in Glioblastoma. Cancer Res 2014, 74, 1752–1765. [Google Scholar] [CrossRef]
- Kong, Y.; Ai, C.; Dong, F.; Xia, X.; Zhao, X.; Yang, C.; Kang, C.; Zhou, Y.; Zhao, Q.; Sun, X.; et al. Targeting of BMI-1 with PTC-209 inhibits glioblastoma development. Cell Cycle 2018, 17, 1199–1211. [Google Scholar] [CrossRef]
- Fan, L.; Chen, Z.; Wu, X.; Cai, X.; Feng, S.; Lu, J.; Wang, H.; Liu, N. Ubiquitin-Specific Protease 3 Promotes Glioblastoma Cell Invasion and Epithelial–Mesenchymal Transition via Stabilizing Snail. Mol. Cancer Res. 2019, 17, 1975–1984. [Google Scholar] [CrossRef]
- Grunda, J.M.; Nabors, L.B.; Palmer, C.A.; Chhieng, D.C.; Steg, A.; Mikkelsen, T.; Diasio, R.B.; Zhang, K.; Allison, D.; Grizzle, W.E.; et al. Increased Expression of Thymidylate Synthetase (TS), Ubiquitin Specific Protease 10 (USP10) and Survivin is Associated with Poor Survival in Glioblastoma Multiforme (GBM). J. Neuro-Oncology 2006, 80, 261–274. [Google Scholar] [CrossRef]
- Wang, Z.; Song, Q.; Xue, J.; Zhao, Y.; Qin, S. Ubiquitin-specific protease 28 is overexpressed in human glioblastomas and contributes to glioma tumorigenicity by regulating MYC expression. Exp. Biol. Med. 2016, 241, 255–264. [Google Scholar] [CrossRef]
- Lee, E.Q.; Reardon, D.A.; Schiff, D.; Drappatz, J.; Muzikansky, A.; Grimm, S.A.; Norden, A.D.; Nayak, L.; Beroukhim, R.; Rinne, M.L.; et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro-Oncology 2015, 17, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dong, L.; Bao, S.; Wang, M.; Yun, Y.; Zhu, R. FK228 augmented temozolomide sensitivity in human glioma cells by blocking PI3K/AKT/mTOR signal pathways. Biomed. Pharmacother. 2016, 84, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Zhang, Y.; Shang, E.; Shu, C.; Quinzii, C.M.; Westhoff, M.-A.; Karpel-Massler, G.; Siegelin, M.D. Inhibition of HDAC1/2 Along with TRAP1 Causes Synthetic Lethality in Glioblastoma Model Systems. Cells 2020, 9, 1661. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Histone undergoes certain post-translational modifications that control gene expression. Histone acetylation promotes relaxation of the DNA, while deacetylation makes histones more compact with resulting gene repression. Methylation of CpG islands is associated with more condensed heterochromatin, while demethylation favors euchromatin and exposure of DNA to the action of RNA-polymerases and transcription factors.
Figure 1.
Histone undergoes certain post-translational modifications that control gene expression. Histone acetylation promotes relaxation of the DNA, while deacetylation makes histones more compact with resulting gene repression. Methylation of CpG islands is associated with more condensed heterochromatin, while demethylation favors euchromatin and exposure of DNA to the action of RNA-polymerases and transcription factors.
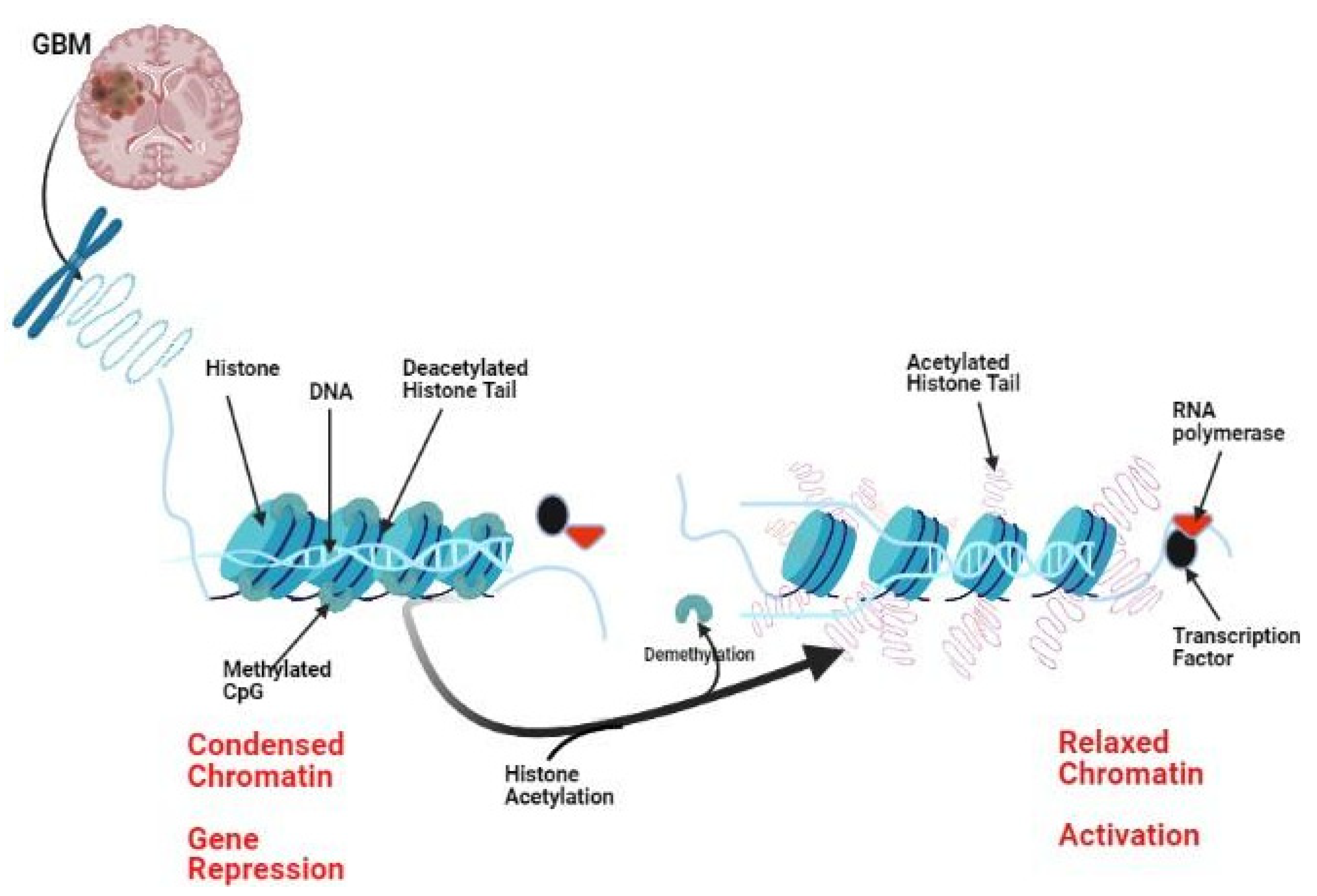

Table 1.
Studies Involving HATs in Glioma.
| HAT involved | Mechanism involved | Reference |
|---|---|---|
| KAT6A/MYST3 | Glioma cell-induced proliferation through H3K23ac/TRIM24-PI3K/AKT pathway | 27 |
| KAT8 | Manipulation of the H4K16 acetylation level in microglia, using the intrinsic H4K16 acetyltransferase activities, adjusted the microglia's tumor-supporting function. | 92 |
| KAT3B | An inhibitor for KAT3B acetyltransferase is highly expressed in GBM and correlates with a dismal prognosis. | 26 |
Table 2.
Lysine and arginine methyltransferases subtypes.
| Methyltransferases | ||
|---|---|---|
| KAMTs | PRMTs | |
| Type 1 | Type 2 | |
| SET1 | PRMT1 | PRMT5 |
| SET2 | PRMT2 | PRMT7 |
| SMYD | PRMT3 | PRMT9 |
| SUV4-20 | PRMT6 | |
| SET7/9 | PRMT8 | |
| SUV39 | PRMT4 | |
Table 3.
Studies Involving Methyltransferases in Glioma.
| HATs | Cell line used | Effect | Reference |
|---|---|---|---|
| KMT1A | Glioma cell lines (GOS-3, 1321N1, T98G, U87MG) | Positive correlation with aggressive tumors | 34 |
| KMT2A | Cell lines isolated from primary human GBM | Glioma stem cells were blunted following silencing of KMT2A | 93 |
| KMT3A | Patient-derived tumor cells | Expressed in High-grade pediatric glioma | 94 |
| KMT4 | Xenograft models | Inhibition of KMT4 reduced stem cell expression of stemness markers | 95 |
| KMT6 | Patient-derived GBM cultures | Reduced expression levels of KMT6 are associated with low expression of oncogenes as c-myc. | 97 |
| PRMT1 | T98G, U87MG, and A172 cell lines and mouse xenografts | Highly expressed in glioma cell lines | 96 |
| PRMT2 | U87 and T98G cell lines | Expressed in high-grade gliomas and associated with poor prognosis. | 35 |
| PRMT5 | U373MG and LN229 cell lines | The expression is high in the high-grade glioma | 98 |
Table 4.
Studies Involving Ubiquitin Specific Enzymes in Glioma.
| Ubiquitin specific enzymes | Preclinical study | Reference |
|---|---|---|
| USP1 | USP1 is overexpressed in glioma stem cells. Inhibition of USP1 increased radiosensitivity of GBM cells | 99 |
| USP3 | USP3 is highly expressed in GBM and correlates with poor prognosis. | 100 |
| USP4 | USP4 is highly expressed in GBM cells | 49 |
| USP 10 | USP 10 is overexpressed and linked to poorsurvival in GBM patients | 101 |
| USP 13 | USP13 is highly expressed in GBM and is required by glioma stem cells to maintain its stemness features. | 47 |
| USP 15 | USP15 attenuates the WNT pathway mediated by stabilization of HECTD1, supporting a tumor-suppressing role of USP15 in GBM cells. | 48 |
| USP28 | USP 28 is overexpressed in GBM cell lines andis associated with a high grade of glioma. | 102 |
Table 5.
Certain Clinical Trials Involving HDACi in Glioma.
| HDAC inhibitor | Combination therapy | Tumor type | Result | Sponsor | Reference |
|---|---|---|---|---|---|
| Vorinostat | Temozolomide + Isotretinoin, bortezomib |
Recurrent GBM | Still active | ||
| Bevacizumab | Recurrent GBM | No change in overall survival or progression-free survival compared to bevacizumab therapy. | Duke University Durham | 74 | |
| Temozolomide + Bevacizumab |
Recurrent GBM | Progression-free survival for six months was not affected. | Duke University Durham | 75 | |
| Bevacizumab + Irinotecan, Temsirolimus |
Diffuse intrinsic pontine glioma | Active | |||
| Radiotherapy | High –grade glioma and anaplastic astrocytoma | Active | National Cancer Institute | ||
| Valproic acid | VPA, temozolomide, and radiotherapy | Newly diagnosed GBM in adults | Active | National Cancer Institute |
|
| Romidepsin | Recurrent GBM | Completed and showed that romidepsin is ineffective in the treatment of recurrent GBM. | 85 | ||
| Panobinostat | Pediatric intrinsic pontine glioma |
Active | |||
| Bevacizumab | Recurrent GBM | Adding this agent to bevacizumab did not improve the outcome compared to bevacizumab alone. | 103 | ||
| Convection-enhanced delivery (CED) |
Diffuse pontine glioma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



