Submitted:
20 June 2023
Posted:
20 June 2023
You are already at the latest version
Abstract
Mixed lineage leukemia 1 (MLL1) introduces 1-, 2- and 3-methylation into histone H3K4 through the evolutionarily conserved set domain. In this study, bovine embryonic stem cells (bESCs, named bESCs-F7) were established from the in vitro fertilized (IVF) embryos by Wnt signaling inhibition, while its contribution to endoderm in vivo is limited. To improve the quality of bESCs, MM-102, an inhibitor of MLL1, was applied to the culture. The results showed that MLL1 inhibition along with GSK3 and MAP2K inhibition (3i) at the embryonic stage did not affect bESCs establishment and pluripotency. MLL1 inhibition improves the pluripotency and differentiation potentials of bESCs via up-regulation of stem cell signaling pathways such as PI3K-Akt and WNT. MLL1 inhibition decreases H3K4me1 modification at promoters in bESCs and altered the distribution of DNA methylation in bESCs. In summary, MLL1 inhibition enables bESCs to acquire better pluripotency, and its application may provide high quality pluripotent stem cells for domestic animals.
Keywords:
bovine embryonic stem cells (bESCs)
; pluripotent
; H3K4me1 modification
; DNA methylation modification
1. Introduction
Mixed lineage leukemia 1 (MLL1), also known as MLL, KMT2A, HRX, HTRX and ALL1, is one of the six mixed lineage leukemia (MLL) family histone methyltransferases (HMT) in mammals (Dou et al., 2005; Milne et al., 2002; Nakamura et al., 2002). It mainly introduces 1-, 2- and 3-methylation into histone H3K4 through the evolutionarily conserved set domain. MLL1 and H3K4 methylation (H3K4me) are located in the promoter, transcription initiation site and 5’ transcription region of the target genes and promote transcription initiation, so they play an important role in transcriptional regulation, especially in the early development of zygotic gene activation (ZGA) and hematopoiesis (Guenther et al., 2005; Lauberth et al., 2013).
The H3K4 HMT activity of MLL1 is controlled by the core complex composed of MLL1 and WDR5. The activity of MLL1 alone is weak, but its H3K4 HMT activity is greatly enhanced with the formation of the core complex (Dou et al., 2006). The MLL1 core complex consists of WDR5 and MLL1 proteins (Dou and Hess, 2008; Dou et al., 2006). By disrupting the protein interaction between WDR5 and MLL1, the MLL1 core complex can be effectively dissociated and MLL1 activity can be inhibited (Patel et al., 2008). MM-102 is one of the compounds that can prevent the interaction between MLL1 and WDR5, inhibit MLL1 activity. When mouse bone marrow cells transfected with MLL1-AF9 fusion gene were cultured with MM-102, the expression of MLL1 target genes HoxA9 and Meis-1 was greatly reduced (Karatas et al., 2013). Furthermore, the down regulating H3K4me3 through MM-102 can improve ZGA and abnormal expression patterns of epigenetic chromatin modifying enzymes, pluripotency and apoptosis genes in the blastocyst stage, greatly improve the efficiency and embryo quality of porcine somatic cell nuclear transfer and make it closer to in vivo embryos (Zhang et al., 2018). Our latest research found that adding appropriate concentrations of MM-102 and 3i (MM-102, PD0325901 and CHIR99021) during the ZGA stage of mouse and bovine in vitro fertilization (IVF) embryo development can greatly improve the IVF blastocyst development without affecting the blastocyst quality (Han et al., 2020). MM-401 is another inhibitor of MLL1, which can transform mouse EpiSCs into naïve pluripotent state (Zhang et al., 2016). MLL1 inhibition cause the redistribution of H3K4me1 in enhancers, germline determinants and EpiSCs markers, to regulate the pluripotency regulatory network and regain the ability to participate in embryonic development and germline chimerism (Zhang et al., 2016). However, whether MLL1 inhibition by MM-102 or combined with other small molecule inhibitors can transform human or livestock prime ESCs into naïve state is still lack of relevant experimental study.
Cattle provide human with high-quality meat products and nutritious dairy products, which has important economic and research values. Establishment the bovine pluripotent stem cells (bPSCs) have great significance of germplasm conservation, gene editing breeding, and understanding the developmental specificity of ruminants. In 2018, Bogliotti et al. obtained bovine prime ESCs, known as CTFR-bESCs in a culture system supplemented with fibroblast growth factor 2 (FGF2) and WNT signaling pathway inhibitor IWR1, which exhibited characteristics of prime PSCs (Bogliotti et al., 2018). Recently, bovine expanded pluripotent stem cells were established from blastocysts or by reprogramming of bovine embryonic fibroblasts, which possess the embryonic and extraembryonic development potentials (Zhao et al., 2021). However, whether we can transform bovine prime ESCs to naïve state for bovine remains unsolved.
In this study, bovine prime ESCs were established from normal culture protocol IVF or MM-102 treated IVF embryos and MM-102 treated bESCs. MLL1 inhibitor enhanced the capacity of differentiation of bESCs. After addition of MM-102, bESCs could differentiate endoderm markers in vivo. And the signaling pathways of pluripotency was active by MM-102 treatment. The effects of MLL1 inhibition on H3K4 methylation were determined by RNA-sequencing, western blotting and ChIP-Seq analysis of H3K4me1. The modification pattern of H3K4me1 was altered by MM-102. In particular, we focused on the changes of H3K4me1 in the promoter region of genes, which decreased the quantity in the promoter region but significantly increased the proportion of the total modifications in the treated cells. The expression of DNMT3B was significantly increased. DNA methylation was comprehensively investigated by genome-wide DNA methylation sequencing. The analysis results showed that the pattern of DNA methylation modification was also significantly altered. The methylation level of promoter was reduced. This study will provide a new idea for stem cell state transformation by regulating epigenetic modification.
2. Materials and Methods
Animal Care and Use
All experiments with mice (generation of embryonic fibroblasts and teratoma formation) were conducted in accordance with the Guide for Care and Use of Laboratory Research Involving Animals and were approved by Inner Mongolia University’s Animal Care and Use Committee. C57 mice used in the production of mouse fibroblasts were provided by the laboratory animal breeding room of our laboratory. Teratoma nod-SCID mice used in differentiation test were purchased from Beijing Wetong Lihua Co., LTD., and tested in our laboratory temporary animal breeding room.
Bovine in vitro Fertilization
Bovine ovaries were obtained from a local abattoir, and oocytes were cultured between 22-24h after the maturation, and fertilized with the frozen cattle sperm. 6h after fertilization, the eggs transferred to the embryonic culture medium, and MM-102 was added after 48h cultivation. The embryos were cultured in fresh culture medium for 7-8 days to develop into blastocysts (Han et al., 2020).
Derivation of bESCs Cell Lines
The bovine blastocysts obtained by in vitro fertilization were removed from the zona pellucida and plated in 4-well plates, with mTeSR-E6 (05946, STEMCELL Technologies) added with 20 ng/mL FGF-2 (100-18B, STEMCELL Technologies), 2.5 μM IWR-1 (I0161, Sigma Aldrich) and 0.1328 g/mL low fatty acid BSA (219989925, MP Biomedicals NZ) and were incubated at 37 °C and 5% CO2. After 48-72 h, ICMs adhered to the feeder layer, and the medium was changed daily. Outgrowths (after 8–10 days in culture) were dissociated and passaged using TrypLE (12563011, Gibco) and were reseeded in the presence of 10 μM Rho kinase (ROCK) inhibitor Y-27632 (SCM075, Sigma). The established bESCs lines were grown in 12-wells and were passaged every 5 days at a 1:4-1:6 split ratio. To increase cell survival, the ROCK inhibitor Y-27632 (10 μM) was added to the wells 1 h before passaging and was also added to the newly prepared wells containing MEFs treated with mitomycin and fresh culture medium during the first 24h cultivation.
Alkaline Phosphatase (AP) Staining
Prior to AP staining, bESCs were cultured on feeder cells for 4 days. Following the removal of the culture medium, cells were washed with DPBS and fixed for 10 min at room temperature using 4% paraformaldehyde. The fixed cells were washed three times with DPBS and stained for 1-3 h at room temperature in the dark using the BCIP/NBT Color Development Substrate Kit (C3206, Biyuntian). Cells were washed with DPBS to terminate the staining reaction and were subsequently maintained in DPBS.
Karyotype Analysis
During the flourishing period of cell division, which is usually on the 4th day of culture, 2 mg/mL colchicine (64-86-8, Sigma) was added to the culture medium and incubated at 37°C for 2.5 h. Then, the cells were treated with 8 mL 0.075 mol KCl solution at 37°C water bath for 30 min. Collect the cells and 1mL fixing solution (acetic acid: methanol =1:3) was added to the cells and mixed. After centrifugation at 1500 rpm for 5 min, the supernatant was discarded, 10 mL fixing solution was added and mixed, and cells stood at room temperature for 15min. After three times of these fixing procedures, 50-200 μL fixing solution was added to the cells and mixed well according to the amount of precipitation. Then the cells were dropped from a height of about 1 m onto a pre-cooled slide at -20°C, and let to dry overnight. The prepared sample was immersed in 0.025% trypsin solution preheated at 37℃, digested for 10 s, then quickly washed in 0.85% NaCl solution 2 times, and dried. Dye in the Jimsa solution (48900, Sigma) for 15 min, slowly wash off the excess with water, and dry. Photographs were taken and analyzed using a cytogenetics workstation.
Quantitative Real-Time PCR (qRT-PCR)
The quantification of the mRNAs was conducted by real-time PCR using specific primers. Real-time PCR was performed using an Applied Biosystems 7500 sequence detection system (Thermo Fischer Scientific) and KAPA SYBR® FAST Universal qPCR master mix (Kapa Biosystems Pty). The PCR samples were analyzed in 96-well plates. Each reaction (20 mL) contained forward and reverse primer at 0.2 mM and 10 mL SYBR Green PCR master mix. The PCR steps included incubation for 5 min at 95℃, followed by 40 cycles of 95℃ for10 s, 60℃ for 20 s, and 72℃ for 30 s. All reactions were performed at least in triplicate, and product identity was confirmed by melting curve analysis. Relative expression levels were determined using the 2-ΔΔCT method and normalized against GAPDH levels.
RNA-seq Analysis
Transcriptome (RNA) sequencing was performed by Tianjin Nuohe Zhiyuan Biotechnology Co., LTD. PCA plot and volcano plot in this paper are all generated by ggplot2(3.3.5) in R software 4.1.0, and heatmap was generated by pheatmap (1.0.12); all tracks data (BS-seq, Chip-seq) was transformed to bigwig format used deeptools (3.4.3) (Ramirez et al., 2014) and then visualized with the Integrative Genomics Viewer (IGV,2.14.1) (Robinson et al., 2011).
Immunofluorescence Staining (IF)
Cells were cultured on the coverslips in the 4-well plate. When grown to a suitable density, the cells were fixed with 4% paraformaldehyde for 10min and then treated with Triton X-100 to penetrate the cell membrane. After three washes with PBS, the cells were incubated with the primary antibody at 4℃ overnight. Then, the cells were incubated with the second antibody at room temperature for 1 hour. After the removal of antibodies, the cells were incubated at room temperature with DAPI for 5 min. The slide was sealed after a microscopic examination. The antibodies used in this experiment include anti-OCT4 (sc-5279, Santa Cruz Biotechnology), anti-SOX2 (L1D6A2, Cell Signaling), anti-NANOG (500-P236, Peprotech), anti-SSEA-1 (MAB4301, Sigma), anti-SSEA-4 (MAB4304, Millipore), anti-TRA-1-60 (MAB4360, Millipore), anti-TRA-1-81 (MAB4381, Millipore), anti-AFP (AF5369, R&D Systems), anti-SMA (ab5694, Abcam), and anti-GFAP (I1044, DAKO).
Flow Cytometry Analysis
The cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.8% Triton X-100 for 10 min. Then, the cells were incubated with antibodies in 10% goat serum for 1h at room temperature. The primary antibody was incubated at 4℃ overnight and the secondary antibody was incubated at room temperature for 1 h. The DPBS was used to wash off antibodies between each step. After incubation, the cells were screened and analyzed by flow cytometry (Cytoflex LX) according to the corresponding fluorescence intensity. The antibodies used in this experiment include anti-SSEA-1 (MAB4301, Sigma) and anti-SSEA-4 (MAB4304, Millipore).
Western Blotting (WB)
Cells were collected and protein was extracted by Mammalian Protein Extraction Reagent (CWBIO, China) according to the manufacturer’s procedure. The cracking product is added to the Loading Buffer and boiled in boiling water for 5 minutes for denaturation. Put the same amount of protein sample into the hole of SDS-PAGE gel, and maintain the constant pressure of 90 - 120 V, 30 min - 3 h. After electrophoresis and maker separation, carry out mode transfer, constant current 200 mA, 30 min-1 h. After the transfer of the required strip is completed, place it in 5% sealing solution and place it at room temperature for 1 h. Transfer the membrane to the diluent of primary antibody at 4℃ overnight, and wash the membrane with TBST solution for 3 times. The second antibody was incubated at room temperature for 1 h, and TBST washed the membrane for 3 times; Expose after treatment with indicator. The antibodies used in this experiment include anti-H3 (4620S, Cell Signaling Technology), anti-H3K4me1 (ab8895, Abcam), anti-H3K4me2 (ab32356, Abcam), anti-H3K27me3 (ab6002, Abcam), anti-GAPDH (10494-1-AP, Proteintech), anti-PRDM14 (ab187881, Abcam), anti-P300 (ab54984, Abcam), and anti-G9A (ab18894, Abcam). Images were obtained using a Tanon 5200 Multi Automatic Fluorescence and Chemiluminescence Imaging System (Tanon, China).
Embryoid Bodies (EB) Formationin vitro
When the density of cells reached 80%, the cells were digested and collected. The differentiation medium of the first stage (90% IMDM + 10% FBS) was used to suspend the cells in low adherent 35 mm culture dishes. The embryoid body forms at 6-7 days. The embryoid bodies were collected and centrifuged. Cells were resuspended with the second stage differentiation medium (90%DMEM + 10% FBS) and plated into four-well plates for adherent growth. After 3 weeks of culture, the differentiated cells were identified by immunofluorescence staining with specific antibodies of three germ layers. The antibodies used in this experiment include anti-α-SMA (ab244177, Abcam), anti-AFP (MAB1368, R&D Systems), anti-GFAP (Z0334, DAKO).
Teratoma Formation in vivo
The cells were collected and suspended in an appropriate amount of DPBS, and about 1×107 cells were injected subcutaneously into Nod-scid mouse at each site. When the tumor was visible, the teratoma was removed from mice. The resulting teratoma was paraffin embedded, sectioned and HE stained for analysis.
Statistical Analysis
The differences in transcript levels were determined by the T-test. The blastocyst rates, TE, ICM and total cells of the blastocysts subjected to different treatments were analyzed via the chi-squared test with Yates’ correction. The analyses were performed using the statistical software GraphPad PRISM 6.0 (GraphPad Software, Inc., La Jolla, California, USA), and the results are presented as the means ± SD. Differences at P < 0.05 were considered statistically significant.ChIP-seq Analysis
The Simple ChIP® Plus Enzymatic Chromatin IP Kit (Magnetic Beads) (9005, Cell Signaling) was utilized to extract DNA fragments from the cells. The sequencing was performed by Tianjin Nuohe Zhiyuan Biotechnology Co., LTD. Illumina reads were first mapped to the UCSC bosTau9 reference using bwa-mem (0.7.17) (Li and Durbin, 2009) with default parameters. Next Picard (http://broadinstitute.github.io/picard/, version 2.23.1) was used to markup PCR duplicates. And then we used macs2 (v2.2.7.1, -nomodel-broad-broad-cutoff 0.1-shift 0-gsize 2.7e9-keep-dup auto) (Zhang et al., 2008) to call peaks. H3K4me1 signals were normalized using the MA norm (1.1.4) (Shao et al., 2012) method for quantitative comparison of ChIP-seq data, and the significant differential peaks were determined as log10(p-value) <0 and M-value> 1. Subsequently, the differential peaks were annotated to UCSC bosTau9 with R package ChIP seeker (1.28.3) (Yu et al., 2015) and TxDb. Btaurus. UCSC. bosTau9. refGene (3.10.0).
Bisulfite Genomic Sequencing Analysis
All cell lines of this experiment were used to analyze the promoter methylation of OCT4 and NANOG by bisulfite-sequencing PCR. DNA treatment and methylation-specific PCR were executed using the ZYMO EZ DNA Methylation-Gold Kit (ZYMO RESEARCH) and Takara Ex Taq (Hot Start Version) according to the associated manufacturer’s protocols. The PCR products were ligated into pEASY-T1 Cloning Vector (Trans Gen Biotech) for methylation sequencing. At least 10 clones per gene were sequenced and analyzed for each sample.
Whole Genome Methylation Sequencing
DNA methylation sequencing was performed by Tianjin Nuohe Zhiyuan Biotechnology Co., LTD. R package edge R (3.34.1) (McCarthy et al., 2012) was applied to analyze the difference between treatment and control. The read counts were tested for differential expression using the `exact test`. The differentially expressed genes (DEGs) in the data set with |log2 (fold change) |≥1.5 and adjusted P ≤ 0.05 were selected for the subsequent analyses. Next cluster profiler (4.0.5) (Yu et al., 2012) package was used to annotate and enrich the GO and KEGG pathways for DEGs. P ≤ 0.05 was determined as a cut-off criterion for significant enrichment.
3. Results
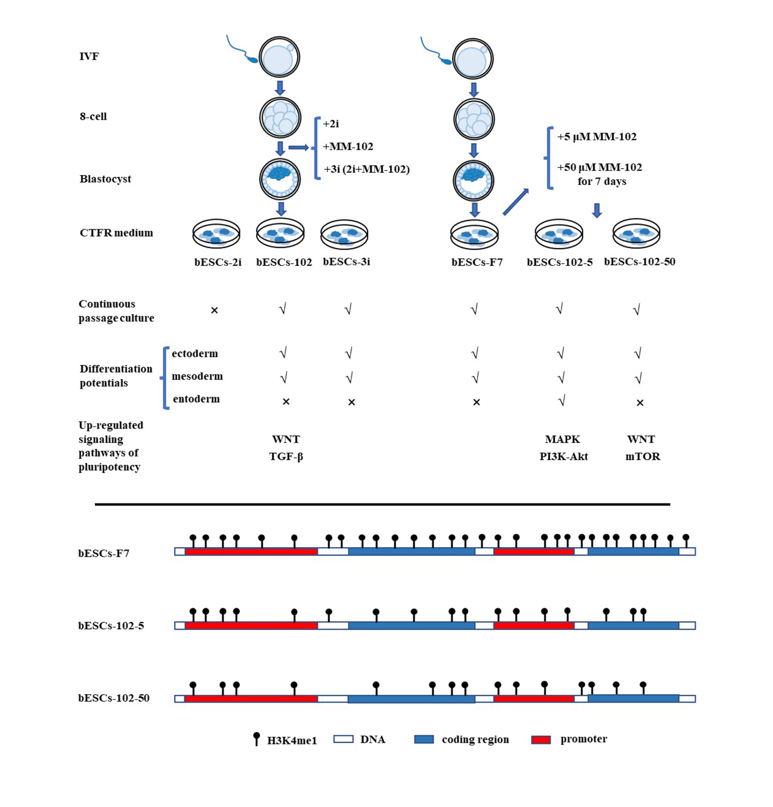
MLL1 combined with GSK3 and MAP2K inhibition (3i) at embryonic stage not affecting bESCs establishment and pluripotency
Previously, we found MLL1 combined with GSK3 and MAP2K inhibition improves the development and the quality of in vitro-fertilized embryos (Han et al., 2020). In this study, the CTFR-bESCs culture system was applied to establish bovine-ESCs from MM-102 (50μM), 2i (0.5μM PD0325901 and 3μM CHIR99021), and 3i (2i plus 30μM MM-102) treated bovine embryos at the program of IVF (Figure 1A). When embryos formed blastocysts at 7-8 days, the whole blastocysts, removed the zona pellucida, were plated to establish bESCs with mouse fetal fibroblasts (MEF) feeder cells. The obtained bESCs cell lines were named bESCs-102, bESCs-2i and bESCs-3i (Figure 1A and S1A). The establishment rate of bESCs-F7 by CTFR was 6.82%, while the rate of bESCs-2i, bESCs-102 and bESCs-3i was increased to 14.29%, 10.00% and 8.33%, respectively (Figure S1B). Although the IVF embryos which inhibited GSK3 and MAP2K generated more bESCs cell lines, the bESCs-2i cells proliferated slowly with weak alkaline phosphatase (AP) staining, and they hardly survived subculture (Figure S1C). The bESCs-102, MLL1 was inhibited, also grows slowly and AP staining was weak, but this cell line could form colonies with clear edges and survived subculture for more than 20 passages (Figure 1B). The bESCs-3i, simultaneous inhibited MLL1, GSK3 and MAP2K, was similar to bESCs-F7, exhibited strong AP positive and formed colonies with clear edges, and survived in subculture for more than 60 passages (Figure 1B). All of the cell lines maintained normal karyotype with 60 chromosomes (Figure 1B). Immunofluorescence staining (IF) analysis revealed that bESCs-F7, bESCs-102 and bESCs-3i expressing the pluripotency transcription factors OCT4, SOX2, NANOG and SSEA4 (Figure 1C). For SSEA1, which was considered as a naïve pluripotency marker, only a few positive cells could be observed in bESCs-F7, and more fluorescent cells could be found in bESCs-102 and bESCs-3i (Figure 1C). At the same time, TRA-1-60, a prime pluripotency marker, positive cells could be observed in bESCs-F7 and bESCs-3i weakly, and TRA-1-81, also a naïve pluripotency marker, positivity was observed only in bESCs-102 strongly (Figure S1D). Then, to confirm the proportion of SSEA4 and SSEA1 in bESCs to confirm the change of pluripotency by flow cytometry. The results showed that bESCs-F7 contained 98.12% SSEA4 positive cells, while the percentages in bESCs-102 and bESCs-3i were reduced to 97.23% and 85.21% (Figure 2A). bESCs-F7 contained 58.90% SSEA1 positive cells, and the percentages of bESCs-102 and bESCs-3i were reduced to 18.53% and 40.24% (Figure 2A). We performed transcriptome analyses on bESCs-F7, bESCs-102 and bESCs-3i via RNA sequencing (RNA-seq). Analysis of naïve and prime genes revealed that bESCs-F7 and bESCs-3i showed similar pluripotent gene expression patterns, and bESCs-102 showed higher expression levels in naïve and prime marker genes (Figure 2B). The expression of pluripotency genes such as OCT4, SOX2, NANOG, NCAM1, TET1, STELLA, REX1 and TEAD4 were also confirmed by quantitative real-time PCR (qRT-PCR), and no significant difference between bESCs-F7 and bESCs-3i (Figure 2C). However, the expression of these genes are slightly lower in bESCs-102 cells and the expression level of REX1 was higher in bESCs-3i (Figure 2C).
To detect the differentiation ability of the cells, in vitro embryoid body (EB) formation and in vivo teratoma formation experiments were performed. The results showed that bESCs-102 and bESCs-3i cells differentiated into three germ layer cells in vitro, the same as bESCs-F7 (Figure S1E). However, bESCs-102 and bESCs-3i cells easily formed teratoma when injected NOD-SCID mouse in vivo, the teratoma percentage was significantly higher for bESCs-102 (46.15%) and bESCs-3i (92.86%) than that for bESCs-F7 (13.33%) (Figure S1F). Furthermore, the size of the teratoma formed from bESCs-3i was bigger, and the derivatives of ectoderm and mesoderm can be easily detected (Figure 2D). Three cell lines have almost no endodermal derivatives in teratoma formation in vivo.
In summary, MLL1 inhibition, and MLL1 combined with GSK3 and MAP2K inhibition (3i) were not affecting bESCs establishment and their pluripotency and bESCs established from 3i treated bovine embryos had better differentiation potentials. In another way, bESCs-102 showed a greater ability to express pluripotency genes.
MLL1 inhibition improved the differentiation potential of CTFR-bESCs
Given that MM-102 treatment of embryos altered the pluripotency of established bESCs, we wondered whether MM-102 could induce similar changes in bESCs-F7 culture system. Different concentrations of MM-102 (0 μM, 10 μM, 30 μM, 50 μM and 70 μM) were added to CTFR, and the cells exhibited slow proliferation, loose colony and massive cell death when MM-102 was added over 10μM. After screening, we confirmed two protocols, one is the addition of 5μM MM-102 to CTFR in long-term cultures, in the other, bESCs were cultured for 7 days with the addition of 50μM MM-102 and then switched back to CTFR, and the derived cell lines were named bESCs-102-5 and bESCs-102-50 (Figure 3A). bESCs-102-5 and bESCs-102-50 had AP positive and correct karyotype, and clones of bESCs-102-5 had clearer edges (Figure 3B). The protein expression of OCT4, SOX2, NANOG, SSEA1 and SSEA4 can be found by IF (Figure 3C). TRA-1-60 could be detected in both bESCs-102-5 and bESCs-102-50, and TRA-1-81 was observed in bESCs-102-5 but not in bESCs-102-50 (Figure S2A). We also examined the SSEA4 and SSEA1 positive cells in bESCs-102-5 and bESCs-102-50 by flow cytometry, the proportion of SSEA4 increased from 67.72% (bESCs-F7) to 98.93% (bESCs-102-5) and 93.11% (bESCs-102-50) (Figure 4A). The proportion of SSEA1 in bESCs-F7 was 4.20%, and it got up to 65.69% in bESCs-102-5 and 39.90% in bESCs-102-50 (Figure 4A). To further investigate the pluripotency of MM-102 treated cells, we analyzed the expression of typical naïve and prime pluripotent markers identified in human ESCs after transcriptome sequencing. We found that not only most of the prime pluripotency markers, but also some of the naïve markers were upregulated in bESCs-102-5 and bESCs-102-50 (Figure 4B). qRT-PCR results showed that OCT4 and NANOG were significantly up-regulated after MM-102 was treated. However, the expression of SOX2 was decreased in bESCs-102-5 and increased in bESCs-102-50. Prime gene FGF4 was up-regulated in bESCs-102-5 and bESCs-102-50, while other prime genes, like NCAM1 and C-MYC had no significant difference or down-regulated. Meanwhile, the expression of naïve genes, such as TFCP2L1, REX1, STELLA and HOXA9, were significantly increased (Figure 4C).
We examined the differentiation ability of bESCs-102-5 and bESCs-102-50 in vitro and in vivo. bESCs-102-5 and bESCs-102-50 exhibited spontaneous differentiation ability in the formation of EBs and it can continue to differentiate ectoderm (GFAP), mesoderm (SMA) and endoderm (AFP) in vitro (Figure S2B). The teratoma formation percentage of bESCs-102-5 (26.32%) and bESCs-102-50 (29.41%) was higher than bESCs-F7 (13.33%) (Figure S2C). The teratoma formed from bESCs-102-5 and bESCs-102-50 was bigger than those from bESCs-F7, and the endoderm derivatives can be detected in bESCs-102-5 (Figure 4D).
The ability of MM-102 to improve bESC pluripotency was stronger on ESCs than on embryos. In addition, it improves the pluripotency of prime and naïve in a more comprehensive way, rather than focusing on a certain pluripotency state. Inhibition of MLL1 significantly enhanced the differentiation ability of bESCs.
Inhibition of MLL1 upregulated the expression of genes involved in stem cell-related signaling pathways
To find that MLL1 inhibition affects the pluripotency of bESCs, we dug deeply into RNA-seq data. Regarding bESCs obtained after MM-102-treated embryos, the principal component analysis (PCA) showed that bESCs-F7 and bESCs-3i are grouped together by PC2 values and bESCs-102 is slightly different from them (Figure 5A). There were among 2063 differential expressed genes, 785 genes were up-regulated and 1278 genes were down-regulated in bESCs-102 (Figure 5B, left). We enriched the up-regulated genes of bESCs-102 using KEGG and found that they were mainly concentrated in the classical singling pathways regulating pluripotency of stem cells, like WNT and TGF-beta signaling pathway (Figure 5C, up). Bioprocesses of GO analyzed showed that up-regulated genes in bESCs-102 were mainly concentrated in the biological processes related to the axon development neurogenesis, and neuron development of bESCs-102 (Figure S3A), and down-regulated genes in bESCs-102 inhibited the morphogenesis of mesoderm and endodermal related structures, such as blood vessel, skeletal system, collagen fibril organization and animal organ morphogenesis, etc. (Figure S3B). However, in bESCs-3i, 287 genes were up-regulated, and 844 genes were down-regulated only (Figure 5B, right). Most of the expression increased genes in bESCs-3i mainly participated in glycolysis/gluconeogenesis, biosynthesis of amino acids/carbon metabolism and other metabolic related signaling pathways (Figure 5C, mid). The expression of decreased genes of bESCs-3i get more involved in anatomical structure morphogenesis and regulation of the developmental process (Figure S3C). What's interesting is that the genes highly expressed in bESCs-F7 were concentrated in the PI3K-AKT signaling pathway (Figure 5C, down).
The PCA of bESCs-102-5, bESCs-102-50 and bESCs-F7 based on RNA-seq data showed they were similar, but the data repeatability of bESCs-102-5 and bESCs-102-50 were better than bESCs-F7, shows that MM-102 confers better homogeneity to bESCs (Figure 6A). The gene expression difference was examined by scatterplots, and there were 1194 genes down-regulated, and 656 genes up-regulated in bESCs-102-5 (Figure 6B, left). It also showed 818 genes were down-regulated, and 460 genes were up-regulated in bESCs-102-50 (Figure 6B, right). GO analysis of biological process showed that the down-regulated genes in bESCs-102-5 were mainly distributed in developmental process regulation, including sensory development, sensory organ development, and extracellular structure organization development (Figure S3D). KEGG analyses were performed on the highly expressed genes in bESCs-F7, bESCs-102-5 and bESCs-102-50 respectively, 234 genes in bESCs-F7 were associated with oxidative phosphorylation, inflammatory processes, RNA damage, and DNA damage (Figure 6C, up). MAPK and PI3K-Akt signaling pathway related genes, like TEK, TGFA, MAPK10, FGF8 and others, in bESCs-102-5 were highly expressed (Figure 6C, mid). The specificity of bESCs-102-50 is reflected in WNT and mTOR signaling pathway (Figure 6B, down).
MLL1 inhibition improves the pluripotency by up-regulation of the classical stem cell pluripotent signaling pathways. The WNT signaling pathway was upregulated in both bESCs-102 and bESCs-102-50. However, MAPK and PI3K-AKT signaling pathway were upregulated in bESCs-102-5, which has the strongest pluripotency
Inhibition of MLL1 increased the proportion of H3K4me1 promoter region in the bESCs
To reveal the effects of MM-102 on H3K4 modification in bESCs, the expression of related enzymes was analyzed. The expression of MLL1 was reduced by half after 50 μM MM-102 was added into the cell culture system, but 5 μM MM-102 had no significant effects on MLL1 expression (Figure 7A). However, the expression of MLL1 was almost completely inhibited by the addition of MM-102 to the embryo, but its expression was significantly increased after being combined with GSK3 and MAP2K inhibitors (3i) (Figure 7A). Meanwhile, the histone lysine methyltransferase PRDM14 is up-regulated at varying degrees by MM-102, with greater variation in bESCs-102-5 and bESCs-102-50 groups than in bESCs-102 and bESCs-3i groups (Figure 7B). Western blot results showed that protein levels of PRDM14 were increased in bESCs-102-5 and decreased in bESCs-102 compared with bESCs-F7 (Figure 7C). Protein levels of G9A, which is one of the histone methylation transferases, were lower in bESCs-102-50 and higher in bESCs-102 (Figure 7C). The protein level of histone acetyltransferase P300 was increased in bESCs-102-5 and bESCs-102-50 but decreased in bESCs-102 and bESCs-3i (Figure 7C). Methylation related genes were also analyzed in transcriptome sequencing data, and these genes in all the three MM-102 treatment groups and bESCs-3i showed high expression than bESCs-F7 (Figure S4A). Compared with bESCs-F7, the genes of ubiquitination and acetylation related enzymes were more active in bESCs-102, bESCs-102-5 and bESCs-102-50, while bESCs-3i was the same as bESCs-F7 (Figure S4B and S4C). We further explored the effects of MLL1 inhibition in bESCs on global H3K4me1 and H3K4me2 by Western blotting. H3K4me1 increased in bESCs-102-5 and bESCs-102-50, and decreased significantly in bESCs-102 and bESCs-3i. H3K4me2 significantly increased in bESCs-102-5, and slightly increased in bESCs-102 and bESCs-3i (Figure 7D). However, H3K27me3 increased in all MM-102 treated cell lines (Figure 7D). It can be concluded that treatment of MM-102 at a different stage of the establishment of bESCs have different influence on the methylation modification of bESCs.
We then focused on H3K4me1 modification changes in bESCs-102-5 and bESCs-102-50 by ChIP-seq. PCA results showed that H3K4me1 modification in bESCs and bovine fetal fibroblasts (BFF) was completely different, and the addition of MM-102 changed the distributions of H3K4me1 modification in bESCs-F7 (Figure 7E). Compared with BFF, the total amount of H3K4me1 of bESCs was lower (Figure 7F). After treatment with MM-102, the overall H3K4me1 level in bESCs-102-5 and bESCs-102-50 was decreased and more concentrated in the transcription start site (TSS) (Figure 7F). Compared with bESCs-102-5, H3K4me1 of bESCs-102-50 was more enriched in TSS, and lower in other gene regulatory regions (Figure 7F). GO analysis showed that the biological processes of genes enriched of H3K4me1 in bESCs-102-5 compared with bESCs-F7 included negative regulation of macromolecule biosynthetic process, negative regulation of gene expression, negative regulation of transcription and embryonic organ development (Figure S5A). Compared with bESCs-102-5 and bESCs-F7, the biological processes genes enriched of H3K4me1 in bESCs-102-50 include regulation of muscle system process, neurogenesis, cell-cell signaling, tube development, nervous system development and cellular response stimulus (Figure S5A). Compared with bESCs-102-5 and bESCs-102-50, the biological processes genes enriched of H3K4me1 in bESCs-F7 included circulatory system development, intracellular signal transduction, anatomical structure formation involved in morphogenesis and regulation of Animal organ morphogenesis (Figure S5A). Compared with bESCs-F7 and bESCs-102-5, KEGG analysis showed that the related signaling pathways of genes enriched of H3K4me1 in bESCs-102-50, including autophagy-animal, lysosome, protein processing in the endoplasmic reticulum and sphingolipid signaling pathway (Figure S5B). However, there are no significant differences between bESCs-102-5 and bESCs-F7 after KEGG analysis (Figure S5B).
We further analyzed the H3K4me1 modification patterns in BFF, bESCs-F7, bESCs-102-5 and bESCs-102-50. bESCs-102-5 showed similar H3K4me1 modification to bESCs-F7 in the 10-100kb region upstream and downstream of the TSS, and bESCs-102-50 is similar to BFF (Figure S5C). The distribution of H3K4me1 in each gene regulatory region was also analyzed (Figure 8A). The percentage of H3K4me1 in the promoter region was slightly increased in bESCs-102-50 compared with bESCs-F7, but it was significantly increased in bESCs-102-5 (Figure 8A). Although the distribution of H3K4me1 at promoters was increased by MLL1 inhibition, the total amount of H3K4me1 enriched at the promoter position decreased, and bESCs-102-50 was more obvious than bESCs-102-5 (Figure 8B). A volcanic map was drawn for the genes enriched of H3K4me1 in the promoter region, and the difference between bESCs-102-50 and bESCs-F7 (1469 different genes) was greater than that between bESCs-102-5 and bESCs-F7 (603 different genes) (Figure 8C). Among them, 327 genes with increased enrichment and 276 genes with down-regulation in bESCs-102-5 compared with bESCs-F7, and 729 genes with increased enrichment and 740 genes with down-regulation in bESCs-102-50 compared with bESCs-F7 (Figure 8C). Analyses of the peak patterns of promoter regions of OCT4, DUSP5, KLF4 and PRDM14 showed a decrease in the number of peaks clustered in bESCs-102-5 and bESCs-102-50 (Figure 8D and S5D). The enrichment of H3K4me1 in the whole gene of NANOG, OCT4, KLF4 and DUSP5 was increased in bESCs-102-5 and bESCs-102-50 and showed obvious differences in distribution (Figure 8E). Heat maps of the enriched naive and primed related genes showed high similarity between bESCs-F7 and bESCs-102-50 (Figure S5E and S5F). In bESCs-102-5, the H3K4me1 modification of naive genes CD9, FGF4 and DPPA3 was increased (Figure S5E), and the H3K4me1 modification of primed genes DUSP6, ZIC2, C-MYC and HOXB3 was also increased (Figure S5F). In summary, MLL1 inhibition decreased and changed the pattern of H3K4me1 modification in bESCs, which may lead to the pluripotency improvement of bESCs.
MLL1 inhibition down-regulated mCG levels in the promoter region of bESCs
To further investigate the influences of the MLL1 inhibition on bESCs, we analyzed the alteration of DNA methylation in MM-102 treated cells. At first, the expression of DNMT family genes related to DNA methylation was detected by qRT-PCR, after MLL1 inhibition, the level of DNMT3A was down-regulated, but DNMT3B was up-regulated. Interestingly, DNMT1 and DNMT3L in bESCs-102-5 and bESCs-102-50 were down-regulated, while bESCs-102 and bESCs-3i were opposite (Figure 9A). The protein of DNMT3A was down-regulated and DNMT3B was up-regulated in bESCs-102-5 and bESCs-102-50. In bESCs-102, both DNMT3A and DNMT3B were up-regulated, whereas in bESCs-3i, both were down-regulated (Figure 9B).
Then, the whole genome methylation sequencing (WGMS) of the cells was performed. The results showed that the methylated regions of bESCs-102-5 and bESCs-102-50 are more widely than those of bESCs-102 and bESCs-3i (Figure 9C). Moreover, at the methylation level, bESCs-102 and bESCs-3i were more strongly correlated with bESCs-F7, while bESCs-102-5 and bESCs-102-50 showed strong differences with F7 (Figure 9C and 9D). The methylation regions of bESCs-102-5 and bESCs-102-50 are more concentrated than those of bESCs-F7. bESCs-102 and bESCs-3i have a portion of the less methylated regions compared with bESCs-F7 (Figure S6A). DMR analysis of the methylation of CpG showed that MLL1 inhibition changed the methylation distribution of bESCs, and compared with bESCs-F7, all cell lines treated with MM-102 showed different patterns of CpG DMR methylation, but bESCs-102-5 was similar to bESCs-102-50, bESCs-102 was similar to bESCs-3i (Figure S6B). Heat maps showed that MLL1 inhibition showed higher levels of methylation in the CpG region than bESCs-F7 (Figure S6C). Notably, MM-102 decreased mCG levels in the upstream regulatory region and downstream regulatory region of bESCs, on the gene body, the degree of methylation of bESCs-102-50 and bESCs-102 was almost the same as that of bESCs-F7, while bESCs-102-5 was higher than bESCs-F7 and bESCs-3i was lower than bESCs-F7 (Figure S6D). We further analyzed the changes in DNA methylation levels in each gene region, the methylation levels of CpG island (CGI), promoter and exon in bESCs-102-5 and bESCs-102-50 were decreased compared with bESCs-F7, but the methylation levels of CGI shore, intron and repeat regions was increased (Figure 9E). About bESCs-102 and bESCs-3i, the methylation level of the promoter was also decreased, the difference is that except for the promoter, the methylation level of bESCs-102 did not change, but the exon, intron, 3’-untranslated region (3’-UTR, utr3) and repeat regions of bESCs-3i decreased (Figure 9E). mCG in the promoter region of OCT4 and NANOG decreased upon MLL1 inhibition by bisulfite genomic sequencing of the promoter regions (Figure 9F). In short, inhibition of MLL1 down-regulated the expression level of DNMT3A, up-regulated the expression level of DNMT3B, and changed the distribution of DNA methylation modification, which was mainly reflected in the increase of the overall CpG methylation level, the decrease of mCG in the upstream and downstream regions of the genes, and the decrease of mCG in the promoter region. These changes may be one of the reasons why MM-102 changes the pluripotency of bESCs.
4. Discussion
Epigenetics is an important type of gene regulation, which has a critical effect on all aspects of life activity. Given the highly accessible and hyperactive chromatin structures in ESCs, it is generally assumed that H3K4me plays an important ‘‘housekeeping’’ role in ESCs and is necessary for ESCs to maintain self-renewal and unlimited differentiation potential (De Los Angeles et al., 2015). Previous research shows that the mixed-lineage leukemia 1 (MLL1)/WD-40 repeat protein 5 (WDR5) complex is responsible for the H3K4me3 (Dou et al., 2006; van Nuland et al., 2013). Hui et al. found that H3K4me1 was significantly different between ESCs and EpiSCs, but not H3K4me3. Interestingly, inhibition of MLL1 led to genome-wide change of H3K4me1 in EpiSCs and global redistribution of H3K4me1 at enhancers and represses lineage determinant factors and EpiSC markers, which indirectly regulate the transcription of mouse ESCs (Zhang et al., 2016). This is basically consistent with our conclusions. After MLLI inhibitor MM-102 was added into bESCs culture system, the expression of MLL1 in bESCs-102-50 was significantly down-regulated, and ChIP results showed that H3K4me1 in bESC was down-regulated and lower than that in bESCs-102-5, while MLL1 in 102-5 was not inhibited (Figure 7A, 7D and 7F). But bESCs-102-5 shows higher pluripotency than bESCs-102-50 (Figure 4 and S2A), and a higher proportion of H3K4me1 in the promoter region was found in bESCs-102-5 (Figure 8A), and more H3K4me1 modifications were found in the promoter sites of the representative pluripotent genes DUSP5, KLF4, PRAM14 (Figure 8D and S5D). In conclusion, the reduction of the total amount of H3K4me1 modification was related to the inhibition of MLL1, but the proportion of H3K4me1 in promoter was more important for the influence of pluripotent. The appropriate addition of MM-102 improved the pluripotency of bESCs by increasing H3K4me1 on the promoter of pluripotent genes.
In addition, in our results, the expression of histone lysine methyltransferase PRDM14 was significantly up-regulated after MM-102 treatment, although H3K4me1 was decreased. PRDM14 is also a common marker of primordial germ cells and pluripotent embryonic stem cells (Nady et al., 2015). In hESCs, knockdown studies have shown that loss of PRDM14 leads to the rapid downregulation of OCT4 and differentiation of hESCs (Chia et al., 2010; Tsuneyoshi et al., 2008). Accordingly, the overexpression of PRDM14 in hESCs prevents the upregulation of differentiation markers, including GATA6, GATA4, SOX7 (endoderm), T, MIXL1, FOXF1 (mesoderm), and PAX6 (ectoderm) (Tsuneyoshi et al., 2008). In our previous study, PRDM14 was highly expressed in embryos from 8-cell to blastocyst stage (Han et al., 2020). The up-regulation of PRDM14 by MM-102 may also be one of the reasons for the change of H3K4me1 distribution structure, and PRDM14 is closely related to the increase of pluripotency, which may be the reason for the change of bESCs status.
DNA methylation is a heritable and reversible enzyme-mediated modification of DNA. In vertebrate genomic DNA, the cytosine residues at the fifth position in CpG sequences are often methylated (F Antequera, 1993). Most CpG sequences are located in non-genic repeated sequences, whereas some CpGs are located in the promoter regions of housekeeping genes and tissue-specific genes, where they regulate gene expression (Bird, 1987; Gardiner-Garden and Frommer, 1987). Although there are some methods for controlling gene expression, CpG methylation is one of the epigenetic hallmarks determining chromatin structure and controlling gene expression (Horii and Hatada, 2016). DNA methyltransferases (DNMTs) have different functions. DNMT1 is mainly responsible for the maintenance of DNA methylation during replication, and DNMT3A and DNMT3B are also involved in the de novo methylation of unmethylated DNA (Jackson et al., 2004). DNMT3B is the primary driver of de novo DNA methylation on actively transcribed genes, while DNMT3A plays a minimal role in ESCs (Baubec et al., 2015). In zygotes, DNA is typically highly methylated but, during preimplantation, DNA methylation is erased globally. Then, at the start of post-implantation development in mouse embryos, DNA again becomes dramatically hypermethylated (Kubiura-Ichimaru et al., 2021). Increased expression of DNMT3B in bESCs is associated with increased DNA methylation (CpG) (Figure 9A and S6C).
In conclusion, MLL1 inhibition enhances the pluripotency of bovine embryonic stem cells, which enriched the promoter of pluripotency genes with H3K4me1 to increase the expression of pluripotency genes, and MM-102 altered the global DNA methylation distribution of bESCs.
Supplemental Information
Supplemental information can be found in the document, named supplement materials.
Author Contributions
Xueling Li designed the project and reviewed this paper. Chen Li and Xuejie Han performed experiments and collected and analyzed the data. Jing Wang performed bioinformatics analyses. Other authors helped during the experiment. Chen Li analyzed the results and wrote the manuscript.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (32160172), the Science and Technology Major Project of the Inner Mongolia Autonomous Region of China (2020ZD0007), the Major Program of the Inner Mongolia Natural Science Foundation of China (2020ZD10), the development plan for young scientific and technological talents in colleges and universities of Inner Mongolia Autonomous Region of China (NMGIRT2204), the National Transgenic Project of China (2016ZX0801000-002 and 2016ZX08010005001), and the Science and Technology Major Project of the Inner Mongolia Autonomous Region of China to the State Key Laboratory of Reproductive Regulation and Breeding of Grassland Livestock (zdzx2018065, 2021ZD0048).
Conflicts of Interest
The authors declare no competing interests.
References
- Baubec, T. , Colombo, D.F., Wirbelauer, C., Schmidt, J., Burger, L., Krebs, A.R., Akalin, A., and Schubeler, D. (2015). Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520, 243-247. [CrossRef]
- Bird, A.P.J.T.i.G. (1987). CpG islands as gene markers in the vertebrate nucleus. Trends in Genetics 3, 342-347. [CrossRef]
- Bogliotti, Y.S. , Wu, J., Vilarino, M., Okamura, D., Soto, D.A., Zhong, C., Sakurai, M., Sampaio, R.V., Suzuki, K., Izpisua Belmonte, J.C., and Ross, P.J. (2018). Efficient derivation of stable primed pluripotent embryonic stem cells from bovine blastocysts. Proc Natl Acad Sci U S A 115, 2090-2095. [CrossRef]
- Chia, N.Y. , Chan, Y.S., Feng, B., Lu, X., Orlov, Y.L., Moreau, D., Kumar, P., Yang, L., Jiang, J., Lau, M.S., et al. (2010). A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 468, 316-320. [CrossRef]
- De Los Angeles, A. , Ferrari, F., Xi, R., Fujiwara, Y., Benvenisty, N., Deng, H., Hochedlinger, K., Jaenisch, R., Lee, S., Leitch, H.G., et al. (2015). Hallmarks of pluripotency. Nature 525, 469-478. [CrossRef]
- Dou, Y. , and Hess, J.L. (2008). Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int J Hematol 87, 10-18. [CrossRef]
- Dou, Y. , Milne, T.A., Ruthenburg, A.J., Lee, S., Lee, J.W., Verdine, G.L., Allis, C.D., and Roeder, R.G. (2006). Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 13, 713-719. [CrossRef]
- Dou, Y. , Milne, T.A., Tackett, A.J., Smith, E.R., Fukuda, A., Wysocka, J., Allis, C.D., Chait, B.T., Hess, J.L., and Roeder, R.G. (2005). Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121, 873-885. [CrossRef]
- F Antequera, A.B.-B.V., Basel (1993). CpG islands in DNA methylation in Molecular Biology and Biological Significance.
- Gardiner-Garden, M. , and Frommer, M. (1987). CpG islands in vertebrate genomes. J Mol Biol 196, 261-282. [CrossRef]
- Guenther, M.G. , Jenner, R.G., Chevalier, B., Nakamura, T., Croce, C.M., Canaani, E., and Young, R.A. (2005). Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci U S A 102, 8603-8608. [CrossRef]
- Han, X. , Xiang, J., Li, C., Wang, J., Wang, C., Zhang, Y., Li, Z., Lu, Z., Yue, Y., and Li, X. (2020). MLL1 combined with GSK3 and MAP2K inhibition improves the development of in vitro-fertilized embryos. Theriogenology 146, 58-70. [CrossRef]
- Horii, T. , and Hatada, I. (2016). Regulation of CpG methylation by Dnmt and Tet in pluripotent stem cells. J Reprod Dev 62, 331-335. [CrossRef]
- Jackson, M. , Krassowska, A., Gilbert, N., Chevassut, T., Forrester, L., Ansell, J., and Ramsahoye, B. (2004). Severe global DNA hypomethylation blocks differentiation and induces histone hyperacetylation in embryonic stem cells. Mol Cell Biol 24, 8862-8871. [CrossRef]
- Karatas, H. , Townsend, E.C., Cao, F., Chen, Y., Bernard, D., Liu, L., Lei, M., Dou, Y., and Wang, S. (2013). High-affinity, small-molecule peptidomimetic inhibitors of MLL1/WDR5 protein-protein interaction. J Am Chem Soc 135, 669-682. [CrossRef]
- Kubiura-Ichimaru, M. , Ito, T., Lefebvre, L., and Tada, M. (2021). Cyclic DNA remethylation following active demethylation at euchromatic regions in mouse embryonic stem cells. Chromosome Res 29, 145-157. [CrossRef]
- Lauberth, S.M. , Nakayama, T., Wu, X., Ferris, A.L., Tang, Z., Hughes, S.H., and Roeder, R.G. (2013). H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152, 1021-1036. [CrossRef]
- Li, H. , and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754-1760. [CrossRef]
- McCarthy, D.J. , Chen, Y., and Smyth, G.K. (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288-4297. [CrossRef]
- Milne, T.A. , Briggs, S.D., Brock, H.W., Martin, M.E., Gibbs, D., Allis, C.D., and Hess, J.L. (2002). MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 10, 1107-1117. [CrossRef]
- Nady, N. , Gupta, A., Ma, Z., Swigut, T., Koide, A., Koide, S., and Wysocka, J. (2015). ETO family protein Mtgr1 mediates Prdm14 functions in stem cell maintenance and primordial germ cell formation. Elife 4, e10150. [CrossRef]
- Nakamura, T. , Mori, T., Tada, S., Krajewski, W., Rozovskaia, T., Wassell, R., Dubois, G., Mazo, A., Croce, C.M., and Canaani, E. (2002). ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 10, 1119-1128. [CrossRef]
- Patel, A. , Vought, V.E., Dharmarajan, V., and Cosgrove, M.S. (2008). A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J Biol Chem 283, 32162-32175. [CrossRef]
- Ramirez, F. , Dundar, F., Diehl, S., Gruning, B.A., and Manke, T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187-191. [CrossRef]
- Robinson, J.T. , Thorvaldsdottir, H., Winckler, W., Guttman, M., Lander, E.S., Getz, G., and Mesirov, J.P. (2011). Integrative genomics viewer. Nat Biotechnol 29. [CrossRef]
- Shao, Z. , Zhang, Y., Yuan, G.C., Orkin, S.H., and Waxman, D.J. (2012). MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13, R16. [CrossRef]
- Tsuneyoshi, N. , Sumi, T., Onda, H., Nojima, H., Nakatsuji, N., and Suemori, H. (2008). PRDM14 suppresses expression of differentiation marker genes in human embryonic stem cells. Biochem Biophys Res Commun 367, 899-905. [CrossRef]
- van Nuland, R. , Smits, A.H., Pallaki, P., Jansen, P.W., Vermeulen, M., and Timmers, H.T. (2013). Quantitative dissection and stoichiometry determination of the human SET1/MLL histone methyltransferase complexes. Mol Cell Biol 33, 2067-2077. [CrossRef]
- Yu, G. , Wang, L.G., Han, Y., and He, Q.Y. (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284-287. [CrossRef]
- Yu, G. , Wang, L.G., and He, Q.Y. (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382-2383. [CrossRef]
- Zhang, H. , Gayen, S., Xiong, J., Zhou, B., Shanmugam, A.K., Sun, Y., Karatas, H., Liu, L., Rao, R.C., Wang, S., et al. (2016). MLL1 Inhibition Reprograms Epiblast Stem Cells to Naive Pluripotency. Cell Stem Cell 18, 481-494. [CrossRef]
- Zhang, Y. , Liu, T., Meyer, C.A., Eeckhoute, J., Johnson, D.S., Bernstein, B.E., Nusbaum, C., Myers, R.M., Brown, M., Li, W., and Liu, X.S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [CrossRef]
- Zhang, Z. , Zhai, Y., Ma, X., Zhang, S., An, X., Yu, H., and Li, Z. (2018). Down-Regulation of H3K4me3 by MM-102 Facilitates Epigenetic Reprogramming of Porcine Somatic Cell Nuclear Transfer Embryos. Cell Physiol Biochem 45, 1529-1540. [CrossRef]
- Zhao, L. , Gao, X., Zheng, Y., Wang, Z., Zhao, G., Ren, J., Zhang, J., Wu, J., Wu, B., Chen, Y., et al. (2021). Establishment of bovine expanded potential stem cells. Proceedings of the National Academy of Sciences 118. [CrossRef]
Figure 1.
The generation of bovine blastocysts and establishment of bESCs in CTFR. (A) Schematic diagram of embryos culture with different combinations of MM-102, CHIR99021 and PD0325901, and establishment of bESCs from blastocyst by CTFR culture system. (B) The morphology, alkaline phosphatase staining and karyotyping analysis of bESCs-F7, bESCs-102 and bESCs-3i (Scale bar, 200μm). (C) Immunofluorescence staining of pluripotency marker OCT4, SOX2 and NANOG, primed pluripotency marker SSEA4 and naïve pluripotency marker SSEA1 (Scale bar, 100μm).
Figure 1.
The generation of bovine blastocysts and establishment of bESCs in CTFR. (A) Schematic diagram of embryos culture with different combinations of MM-102, CHIR99021 and PD0325901, and establishment of bESCs from blastocyst by CTFR culture system. (B) The morphology, alkaline phosphatase staining and karyotyping analysis of bESCs-F7, bESCs-102 and bESCs-3i (Scale bar, 200μm). (C) Immunofluorescence staining of pluripotency marker OCT4, SOX2 and NANOG, primed pluripotency marker SSEA4 and naïve pluripotency marker SSEA1 (Scale bar, 100μm).
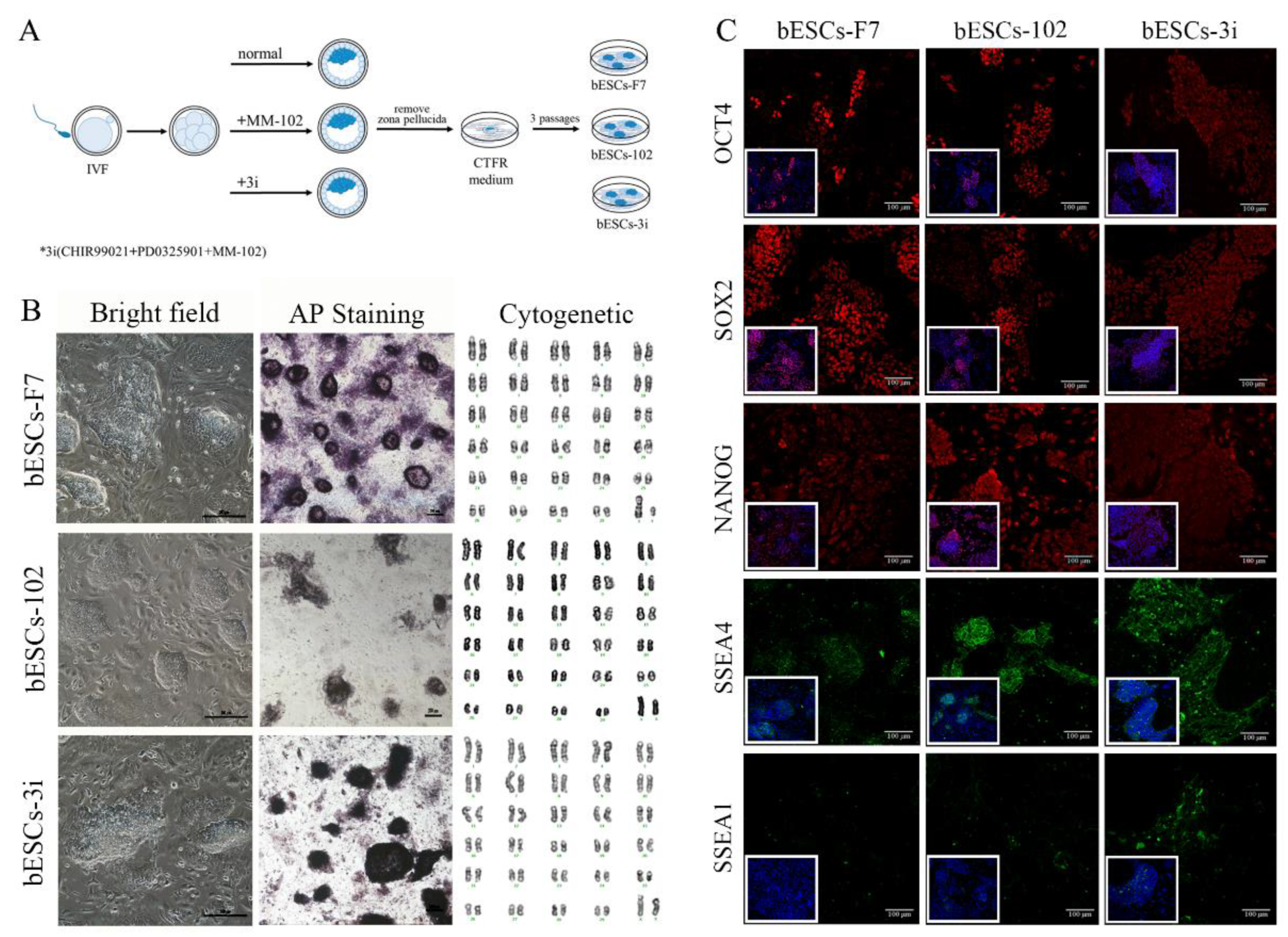
Figure 2.
bESCs-102 and bESCs-3i have higher pluripotency marker expression than bESCs-F7. (A) Flow cytometry results showing expression of SSEA4 and SSEA1 in bESCs-F7, bESCs-102 and bESCs-3i, the negative control consisted of a mixture of three cell lines. (B) Transcriptome analysis of selected naïve and primed pluripotency markers in bESCs-F7, bESCs-102 and bESCs-3i. RNA-seq was performed, and RPKM values were used to define up-regulated expressed genes (RPKM≥1, red) and down-regulated expressed genes (RPKM <1, blue). (C) The relative expression level of pluripotency genes in bESCs-F7, bESCs-102 and bESCs-3i by qRT-PCR. qRT-PCR data, normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) Representative images showing H&E staining of histological sections derived from teratomas generated by bESCs-F7, bESCs-102 and bESCs-3i. Derived teratomas contained tissues of two germ lineages: ectoderm and mesoderm (Magnification 100×).
Figure 2.
bESCs-102 and bESCs-3i have higher pluripotency marker expression than bESCs-F7. (A) Flow cytometry results showing expression of SSEA4 and SSEA1 in bESCs-F7, bESCs-102 and bESCs-3i, the negative control consisted of a mixture of three cell lines. (B) Transcriptome analysis of selected naïve and primed pluripotency markers in bESCs-F7, bESCs-102 and bESCs-3i. RNA-seq was performed, and RPKM values were used to define up-regulated expressed genes (RPKM≥1, red) and down-regulated expressed genes (RPKM <1, blue). (C) The relative expression level of pluripotency genes in bESCs-F7, bESCs-102 and bESCs-3i by qRT-PCR. qRT-PCR data, normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) Representative images showing H&E staining of histological sections derived from teratomas generated by bESCs-F7, bESCs-102 and bESCs-3i. Derived teratomas contained tissues of two germ lineages: ectoderm and mesoderm (Magnification 100×).
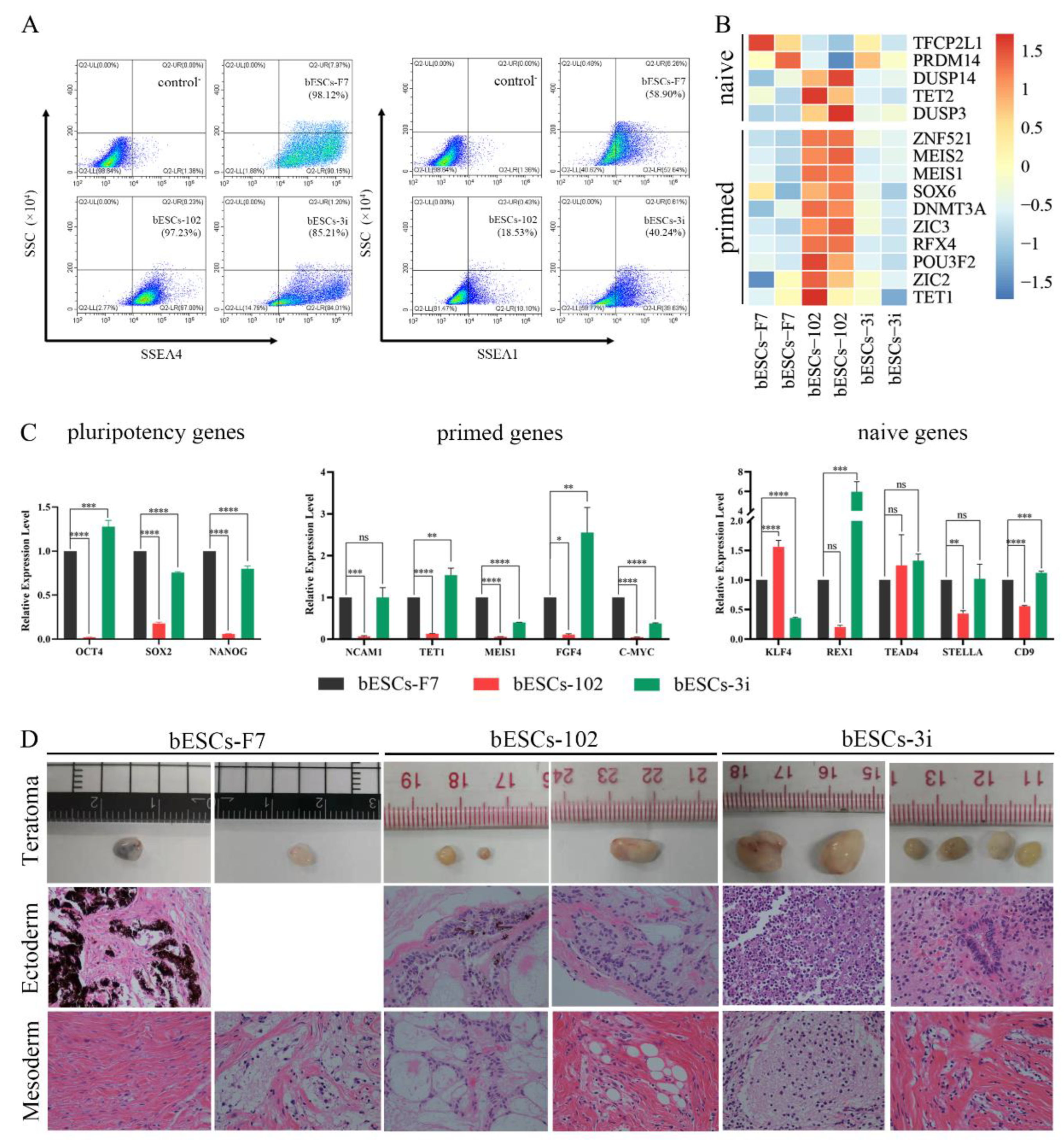
Figure 3.
The establishment of MLL1 inhibited bESCs. (A) Schematic diagram of the establishment of MLL1 inhibited cell lines bESCs-102-5 and bESCs-102-50. (B) The morphology, alkaline phosphatase staining and karyotyping analysis of bESCs-102-5 and bESCs-102-50 (Scale bar, 200μm). (C) Immunofluorescence staining of pluripotent marker OCT4, SOX2 and NANOG, primed pluripotent marker SSEA4 and naïve pluripotent marker SSEA1 (Scale bar, 100μm).
Figure 3.
The establishment of MLL1 inhibited bESCs. (A) Schematic diagram of the establishment of MLL1 inhibited cell lines bESCs-102-5 and bESCs-102-50. (B) The morphology, alkaline phosphatase staining and karyotyping analysis of bESCs-102-5 and bESCs-102-50 (Scale bar, 200μm). (C) Immunofluorescence staining of pluripotent marker OCT4, SOX2 and NANOG, primed pluripotent marker SSEA4 and naïve pluripotent marker SSEA1 (Scale bar, 100μm).
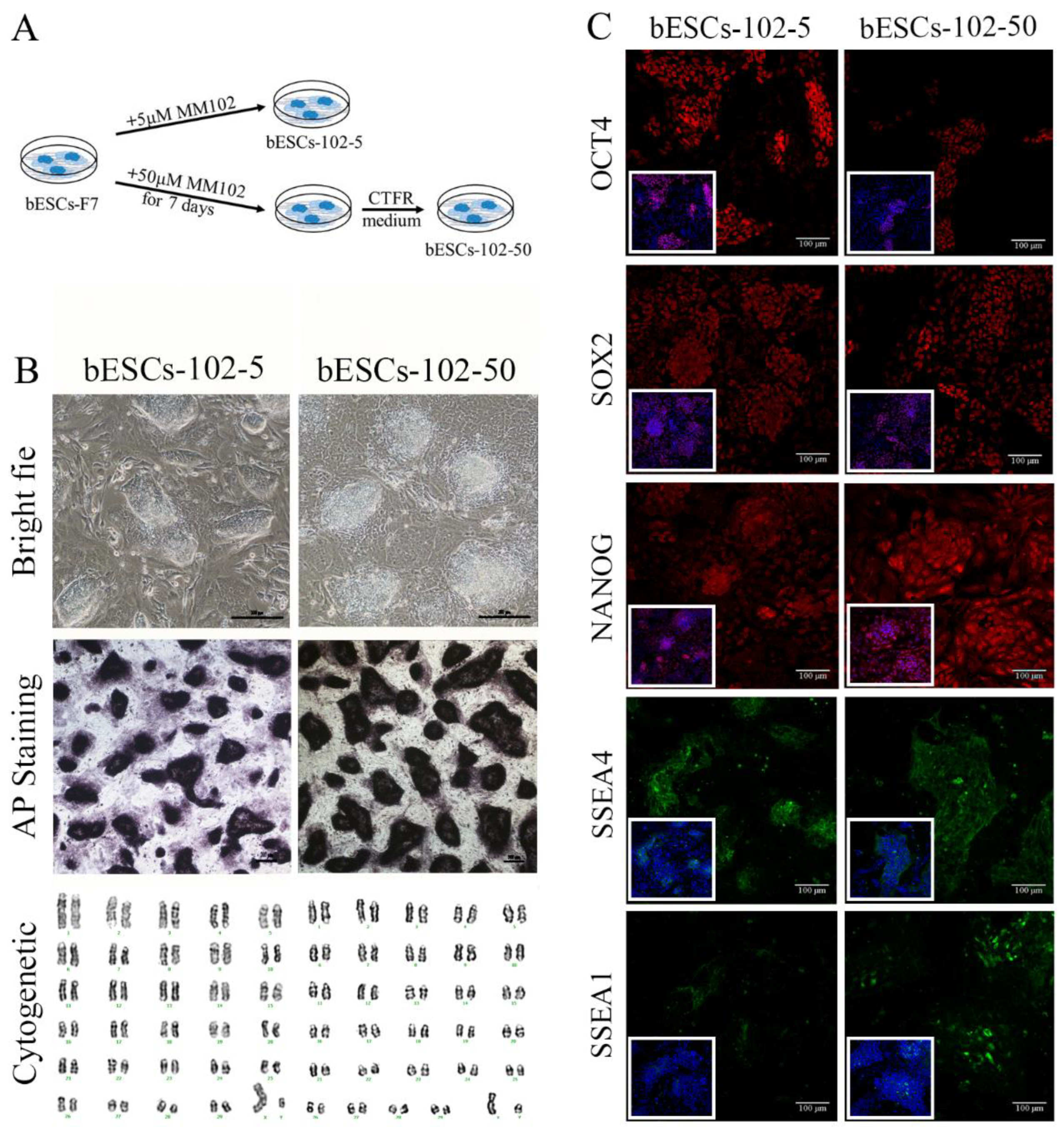
Figure 4.
MLL1 inhibition improves the pluripotency of bESCs. (A) Flow cytometry results showing expression of SSEA4 and SSEA1 in bESCs-F7, bESCs-102-5 and bESCs-102-50. The negative control consisted of a mixture of three cell lines. (B) Transcriptome analysis of selected naive and primed pluripotent markers in different bESCs. RNA-seq was performed, and RPKM values were used to define up-regulated expressed genes (RPKM≥1; red) and down-regulated expressed genes (RPKM <1; blue). (C) The relative expression level of pluripotent genes in bESCs-F7, bESCs-102-5 and bESCs-102-50 by qRT-PCR. qRT-PCR data normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) H&E staining of histological sections derived from teratomas generated by bESCs-102-5 and bESCs-102-50. Derived teratomas contained tissues of all three germ layer lineages: ectoderm, mesoderm and endoderm (Magnification 100×).
Figure 4.
MLL1 inhibition improves the pluripotency of bESCs. (A) Flow cytometry results showing expression of SSEA4 and SSEA1 in bESCs-F7, bESCs-102-5 and bESCs-102-50. The negative control consisted of a mixture of three cell lines. (B) Transcriptome analysis of selected naive and primed pluripotent markers in different bESCs. RNA-seq was performed, and RPKM values were used to define up-regulated expressed genes (RPKM≥1; red) and down-regulated expressed genes (RPKM <1; blue). (C) The relative expression level of pluripotent genes in bESCs-F7, bESCs-102-5 and bESCs-102-50 by qRT-PCR. qRT-PCR data normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) H&E staining of histological sections derived from teratomas generated by bESCs-102-5 and bESCs-102-50. Derived teratomas contained tissues of all three germ layer lineages: ectoderm, mesoderm and endoderm (Magnification 100×).
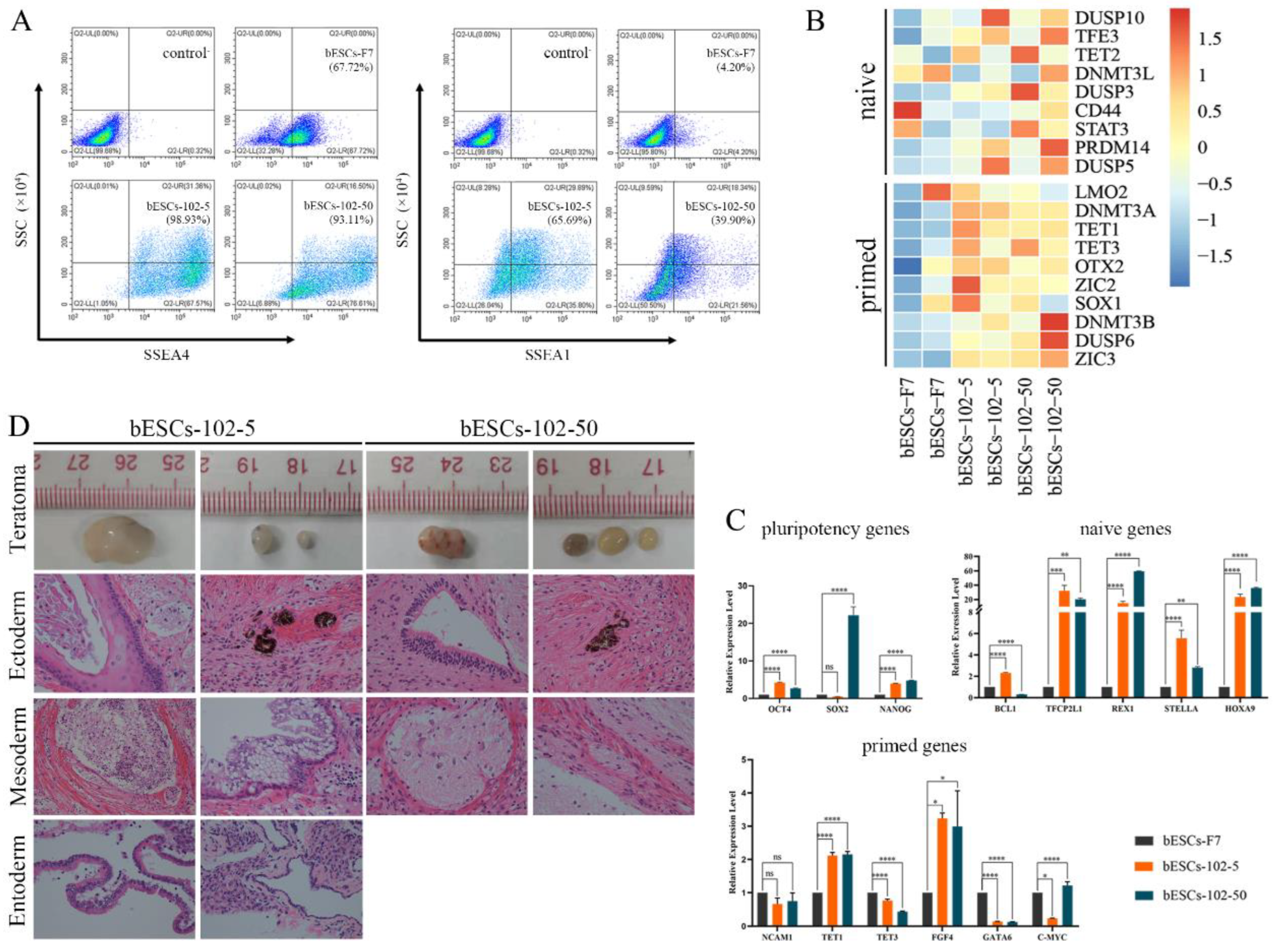
Figure 5.
Comparison of the pluripotency of bESCs-102 and bESCs-F7, and bESCs-3i and bESCs-F7. (A) PCA based on RNA-seq data of bESCs-F7, bESCs-102 and bESCs-3i. (B) Scatterplots showing significantly up-regulated (in red) and down-regulated (in blue) genes between bESC-F7 and bESC-102, and bESC-F7 and bESC-3i. Genes not differentially expressed are presented in gray. (C) Heat maps and KEGG analysis of highly expressed genes in bESCs-F7, bESCs-102 and bESCs-3i, respectively. RNA-seq was performed, and RPKM values were used to define up-regulated expressed genes (RPKM≥1, red) and down-regulated expressed genes (RPKM <1, blue).
Figure 5.
Comparison of the pluripotency of bESCs-102 and bESCs-F7, and bESCs-3i and bESCs-F7. (A) PCA based on RNA-seq data of bESCs-F7, bESCs-102 and bESCs-3i. (B) Scatterplots showing significantly up-regulated (in red) and down-regulated (in blue) genes between bESC-F7 and bESC-102, and bESC-F7 and bESC-3i. Genes not differentially expressed are presented in gray. (C) Heat maps and KEGG analysis of highly expressed genes in bESCs-F7, bESCs-102 and bESCs-3i, respectively. RNA-seq was performed, and RPKM values were used to define up-regulated expressed genes (RPKM≥1, red) and down-regulated expressed genes (RPKM <1, blue).
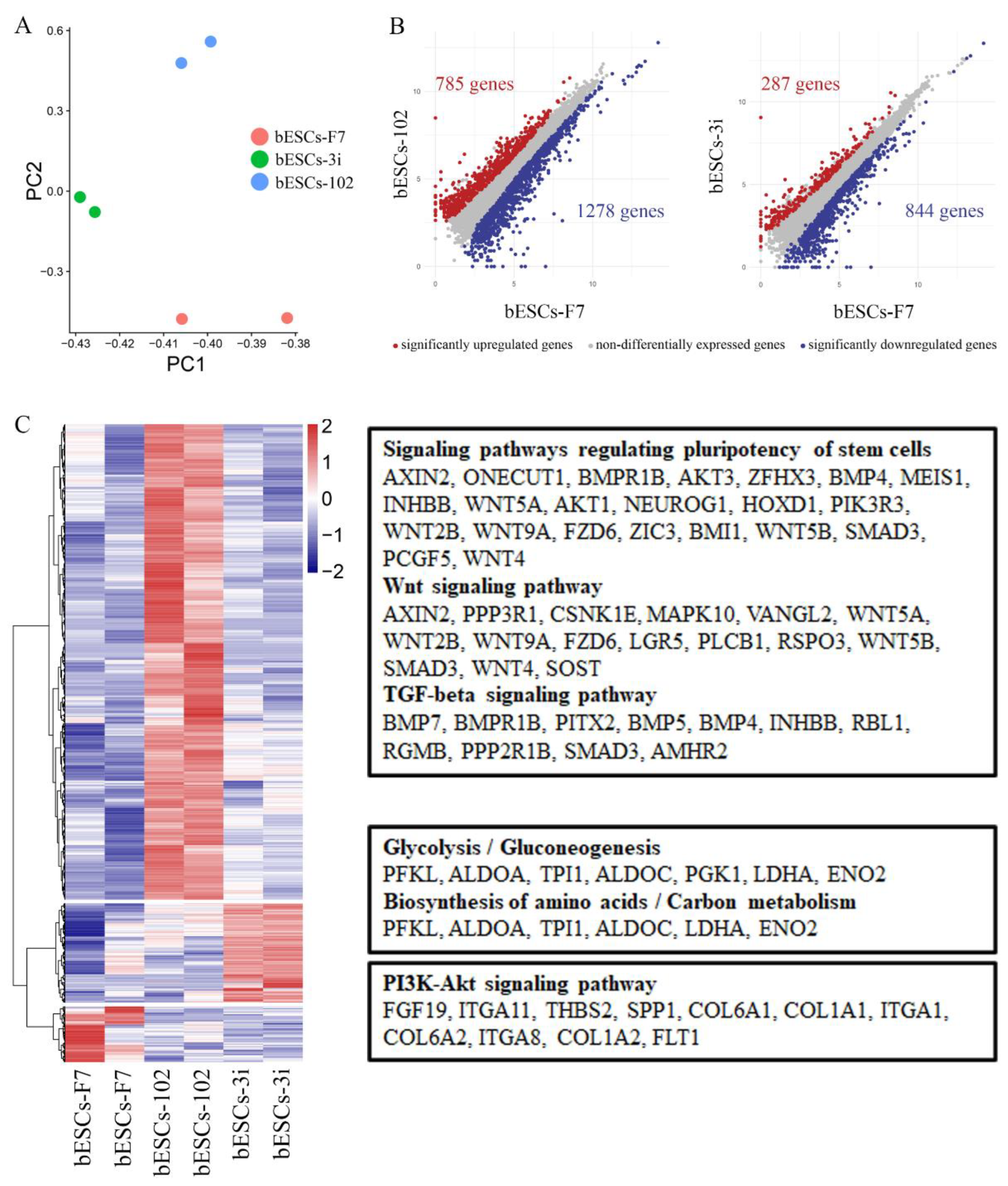
Figure 6.
MLL1 inhibition enhanced stem cell pluripotent related pathways in bESCs-102-5 and bESCs-102-50. (A) PCA based on RNA-seq data in bESCs-F7, bESCs-102-5 and bESCs-102-50. (B) The scatterplots show significantly up-regulated (in red) and down-regulated (in blue) genes between bESC-F7 and bESC-102-5, and bESC-F7 and bESC-102-50. Genes not differentially expressed are presented in gray. (C) Heat maps and KEGG analysis of highly expressed genes in bESCs-F7, bESCs-102-5 and bESCs-102-50, respectively. RNA-seq was performed, and RPKM values were used to define up-regulated expressed genes (RPKM≥0.5, red) and down-regulated expressed genes (RPKM <0.5, blue).
Figure 6.
MLL1 inhibition enhanced stem cell pluripotent related pathways in bESCs-102-5 and bESCs-102-50. (A) PCA based on RNA-seq data in bESCs-F7, bESCs-102-5 and bESCs-102-50. (B) The scatterplots show significantly up-regulated (in red) and down-regulated (in blue) genes between bESC-F7 and bESC-102-5, and bESC-F7 and bESC-102-50. Genes not differentially expressed are presented in gray. (C) Heat maps and KEGG analysis of highly expressed genes in bESCs-F7, bESCs-102-5 and bESCs-102-50, respectively. RNA-seq was performed, and RPKM values were used to define up-regulated expressed genes (RPKM≥0.5, red) and down-regulated expressed genes (RPKM <0.5, blue).
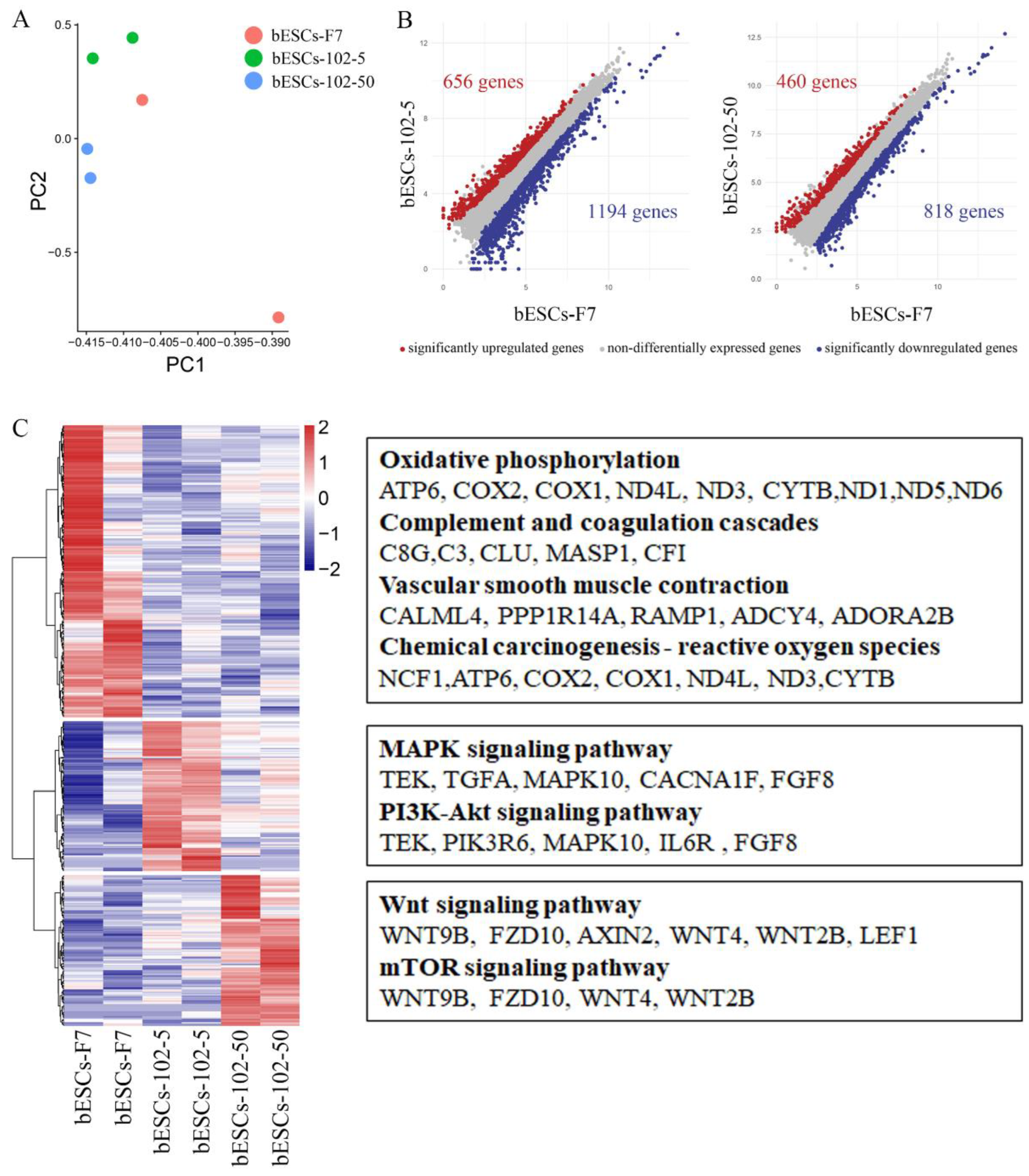
Figure 7.
The histone modification of bESCs-F7 was changed by MLL1 inhibition. (A) The relative expression level of MLL1 in bESCs-F7, bESCs-102-5, bESCs-102-50 (left), bESCs-102 and bESCs-3i (right) by qRT-PCR. qRT-qPCR data, normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (B) The relative expression level of PRDM14 in bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i by qRT-qPCR. qRT-qPCR data, normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (C) Western blotting showing protein level of PRDM14, P300 and G9A in bESCs-F7, bESCs-102-5, bESCs-102-50 (left), bESCs-102 and bESCs-3i (right). (D) Western blotting showing protein level of H3K3me1, H3K4me2 and H3K27me3 in bESCs-F7, bESCs-102-5, bESCs-102-50 (left), bESCs-102 and bESCs-3i (right). (E) PCA based on ChIP-seq data of H3K4me1 in BFF, bESCs-F7, bESCs-102-5 and bESCs-102-50. (F) Heatmap of ChIP-seq of global genes of BFF, bESCs-F7, bESCs-102-5 and bESCs-102-50. RPKM values were used to define up-regulated expressed genes (RPKM≥1, red) and down-regulated expressed genes (RPKM <1, blue).
Figure 7.
The histone modification of bESCs-F7 was changed by MLL1 inhibition. (A) The relative expression level of MLL1 in bESCs-F7, bESCs-102-5, bESCs-102-50 (left), bESCs-102 and bESCs-3i (right) by qRT-PCR. qRT-qPCR data, normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (B) The relative expression level of PRDM14 in bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i by qRT-qPCR. qRT-qPCR data, normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (C) Western blotting showing protein level of PRDM14, P300 and G9A in bESCs-F7, bESCs-102-5, bESCs-102-50 (left), bESCs-102 and bESCs-3i (right). (D) Western blotting showing protein level of H3K3me1, H3K4me2 and H3K27me3 in bESCs-F7, bESCs-102-5, bESCs-102-50 (left), bESCs-102 and bESCs-3i (right). (E) PCA based on ChIP-seq data of H3K4me1 in BFF, bESCs-F7, bESCs-102-5 and bESCs-102-50. (F) Heatmap of ChIP-seq of global genes of BFF, bESCs-F7, bESCs-102-5 and bESCs-102-50. RPKM values were used to define up-regulated expressed genes (RPKM≥1, red) and down-regulated expressed genes (RPKM <1, blue).
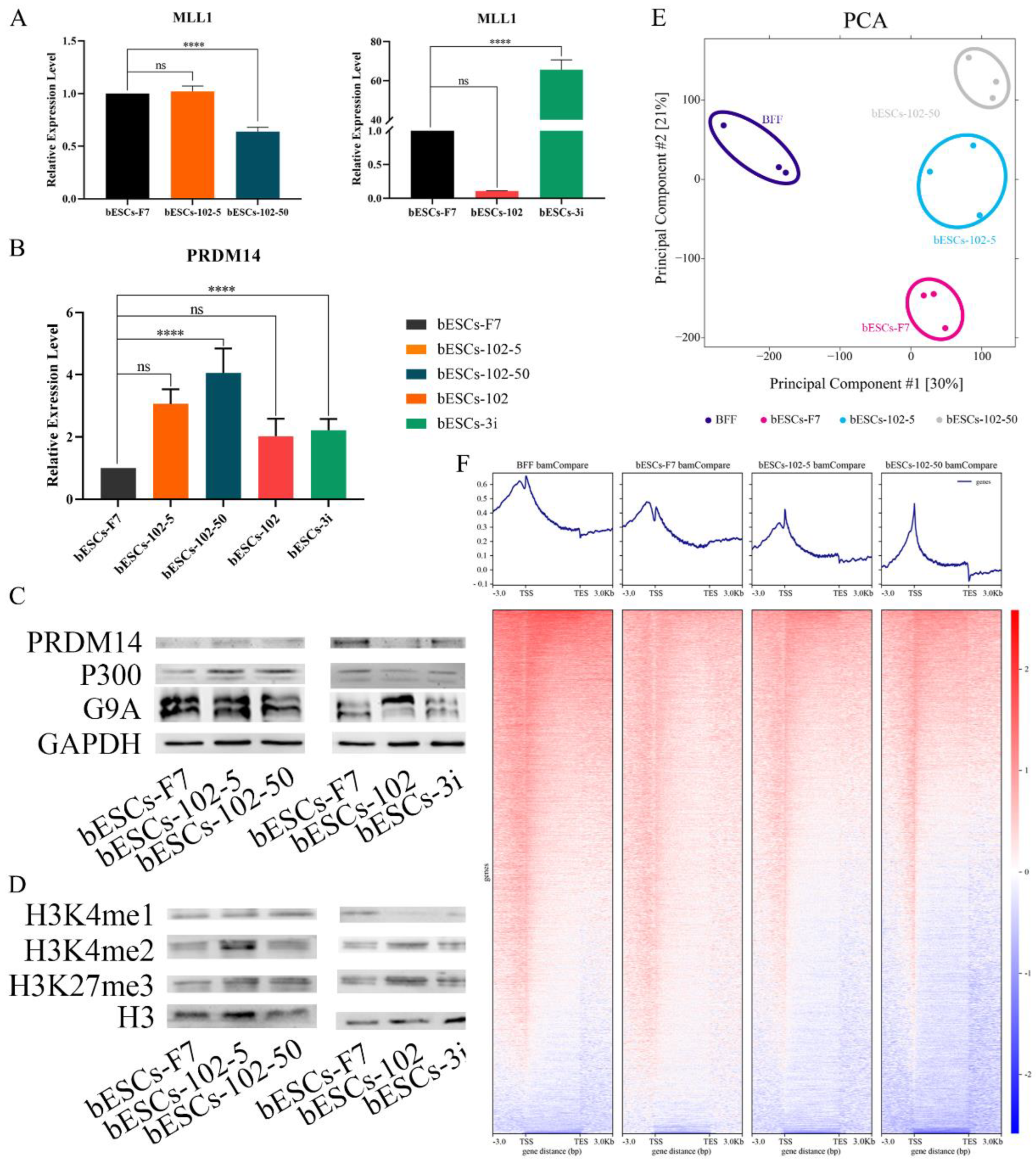
Figure 8.
MLL1 inhibition changed the distribution of H3K4me1 at the promoter region. (A) Ratios of H3K4me1 at gene regulatory regions based on H3K4me1 ChIP-seq of bESCs-F7, bESCs-102-5, bESCs-102-50 and BFF. (B) Comparison of H3K4me1 modification levels on the promoters of bESCs-F7, bESCs-102-5 and bESCs-102-50. (C) Volcano plot displaying the H3K4me1 modification on the promoters of bESCs-F7, bESCs-102-5, and bESCs-102-50. Red and blue dots indicate up-regulated and down-regulated genes. (D) Peak map of H3K4me1 enrichment in the promoter region of DUSP5 and OCT4 of bESCs-F7, bESCs-102-5, and bESCs-102-50 (pink regions). (E) Integrative Genomics Viewer (IGV) Genome Browser views showing H3K4me1 tracks of pluripotent genes in bESCs-F7, bESCs-102-5 and bESCs-102-50.
Figure 8.
MLL1 inhibition changed the distribution of H3K4me1 at the promoter region. (A) Ratios of H3K4me1 at gene regulatory regions based on H3K4me1 ChIP-seq of bESCs-F7, bESCs-102-5, bESCs-102-50 and BFF. (B) Comparison of H3K4me1 modification levels on the promoters of bESCs-F7, bESCs-102-5 and bESCs-102-50. (C) Volcano plot displaying the H3K4me1 modification on the promoters of bESCs-F7, bESCs-102-5, and bESCs-102-50. Red and blue dots indicate up-regulated and down-regulated genes. (D) Peak map of H3K4me1 enrichment in the promoter region of DUSP5 and OCT4 of bESCs-F7, bESCs-102-5, and bESCs-102-50 (pink regions). (E) Integrative Genomics Viewer (IGV) Genome Browser views showing H3K4me1 tracks of pluripotent genes in bESCs-F7, bESCs-102-5 and bESCs-102-50.
Figure 9.
MLL1 inhibition altered DNA methylation in bESCs. (A) The relative expression level of DNMT family in bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i by qRT-PCR. qRT-qPCR data, normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (B) Western blotting showing decreased expression of DNMT3A and DNMT3B in bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i. (C) Correlation plot of methylated sites in bESCs-F7 versus either bESCs-102-5, bESCs-102-50, bESCs-102 or bESCs-3i. The Red line represents fit based on linear regression modeling (off-center best fit indicates lower correlation); the blue line is based on LOESS weighted regression modeling (curved best-fit line indicates non-linear correlation). (D) Pearson correlation of bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i. (E) Comparison of DNA methylation of bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i showed the differences in the overall distribution of CG methylation levels on gene functional elements. (F) Bisulfite genomic sequencing of the promoter regions of OCT4 and NANOG in bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i.
Figure 9.
MLL1 inhibition altered DNA methylation in bESCs. (A) The relative expression level of DNMT family in bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i by qRT-PCR. qRT-qPCR data, normalized to GAPDH. nsp < 0.1, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (B) Western blotting showing decreased expression of DNMT3A and DNMT3B in bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i. (C) Correlation plot of methylated sites in bESCs-F7 versus either bESCs-102-5, bESCs-102-50, bESCs-102 or bESCs-3i. The Red line represents fit based on linear regression modeling (off-center best fit indicates lower correlation); the blue line is based on LOESS weighted regression modeling (curved best-fit line indicates non-linear correlation). (D) Pearson correlation of bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i. (E) Comparison of DNA methylation of bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i showed the differences in the overall distribution of CG methylation levels on gene functional elements. (F) Bisulfite genomic sequencing of the promoter regions of OCT4 and NANOG in bESCs-F7, bESCs-102-5, bESCs-102-50, bESCs-102 and bESCs-3i.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



