
Submitted:
20 June 2023
Posted:
21 June 2023
You are already at the latest version
Abstract
Bladder cancer (BLCA) is the sixth most common type of cancer and has a dismal prognosis if diagnosed late. To identify treatment options for BLCA, we systematically evaluated data from the Broad Institute DepMap project. We found that urothelial BLCA cell lines are among the most sensitive to microtubule assembly inhibition by paclitaxel treatment. Strikingly, we unveiled that the top dependencies in BLCA cell lines include genes encoding proteins involved in microtubule assembly, thus highlighting the importance of microtubule network dynamics as a major vulnerability in human BLCA. In cancers, such as ovarian and breast, where paclitaxel is the golden standard of care, resistance to paclitaxel treatment has been linked to p53-inactivating mutations. To study the response of BLCA to microtubule assembly inhibition and its mechanistic link with the mutational status of p53 protein, we treated a collection of BLCA cell lines with a dose range of paclitaxel and performed a detailed characterization of the response. We herein discovered that BLCA cell lines are significantly sensitive to low concentrations of paclitaxel, independently of their p53 status. Paclitaxel induced a G2/M cell cycle arrest and growth inhibition, followed by robust activation of apoptosis. Most importantly, we revealed that paclitaxel triggered a robust DNA-damage response and apoptosis program without activating the p53 pathway. Integration of transcriptomics, epigenetic and dependency data demonstrated that the response of BLCA to paclitaxel is independent of p53 mutational signatures, but strongly depends on the expression of DNA-repair genes. Our work highlights urothelial BLCA as an exceptional candidate for paclitaxel treatment and paves the way for the rational use of a combination of paclitaxel and DNA-repair inhibitors as an effective, novel therapeutic strategy.
Keywords:
Apoptosis
; Bladder
; Cancer
; Microtubule
; p53
; Paclitaxel
; Urothelium
1. Introduction
Bladder cancer (BLCA) is a devastating disease affecting over 500,000 individuals worldwide and being responsible for over 200,000 deaths on an annual base [1,2]. BLCA is classified as either low-grade, non-muscle-invasive disease, or high-grade, muscle-invasive disease, which has a higher chance of metastasis to distal organs [2,3]. Several driver mutations have been identified in low-grade papillary tumor development, such as the FGFR3, H-Ras and mTOR pathway member genes, whereas the progression to high-grade invasive urothelial carcinoma is correlated with loss of function of p53 and Rb tumor-suppressor network genes [4]. Moreover, an integrated study of 131 invasive bladder carcinomas revealed dysregulation of PI3K/Akt/mTOR and RTK/Ras/MAPK pathways in more than 40% of all tumors analyzed [5].
Current treatment regimens, such as chemo- and radio-therapy, surgery, and more recently immuno-therapy have improved survival, but in many cases are associated with severe side effects or development of secondary resistance to therapy [6,7]. Moreover, the development of new targeted therapies is a very tedious and uncertain process and can take more than 10 years for a new agent to reach the clinic. Therefore, there is a great need to develop better targeted treatments for BLCA, ideally by utilizing or repurposing FDA approved compounds.
Towards the effort to identify novel therapeutic vulnerabilities in cancer and utilize/repurpose existing drugs, the Cancer Dependency Map (DepMap) and the Profiling Relative Inhibition Simultaneously in Mixtures (PRISM) projects have made a tremendous progress [8,9,10]. DepMap utilizes the power of high-throughput genome-wide CRISPR screens, across thousands of cancer cell lines and across a wide spectrum of malignancy types, to identify genetic vulnerabilities unique to each cancer type [9], whereas the PRISM project exploits high-throughput chemical screens to repurpose and/or identify novel therapeutic compounds [10].
By investigating the data generated by the DepMap and PRISM projects, and combining with epigenetic and transcriptomics data analysis, we uncovered the importance of microtubule dynamics for survival of BLCA cells, and the therapeutic potential of perturbing this pathway.
2. Materials and Methods
2.1. Cell lines and culture conditions
The growth and manipulation of RT4, RT112, T24 and TCCSUP human urothelial BLCA cell lines used in this study have been previously described [11]. In brief, cell cultures were grown in complete DMEM medium, supplemented with 10% FBS (Thermo Fisher Scientific Inc. - Life Technologies - Gibco, Massachusetts, USA), at 37°C and 5% CO2, and were sub-cultured every 2-3 days.
2.2. Cell viability MTT assays
Cells were seeded onto 48-well plates at a confluency of ~90% and treated with different doses of paclitaxel, for 24 and 48h. Then, cells were incubated with MTT solution, for 4h, and the formazan crystals produced were dissolved in pure isopropanol. Spectrophotometric absorbance was measured in a Dynatech MR5000 ELISA microtiter plate reader (Dynatech Laboratories, Virginia, USA) at 550 nm, using 630 nm as wavelength of reference. Each assay was repeated three times, using three wells per experimental condition.
2.3. Flow cytometry for cell cycle analysis
Control and paclitaxel-treated cells were incubated with 10 μl of 7AAD solution, for 15 min, at +4°C, in the dark. All cell preparations were analyzed within 30 min by flow cytometry, using a Beckman Coulter-Cytomics FC500 cell sorter (Beckman Coulter Inc., California, USA).
2.4. Western blotting
Immunoblot experiments were performed as previously described [11]. In brief, ~40 μg of whole-cell protein extracts were separated by SDS-PAGE in 10-15% gels and subsequently transferred onto nitrocellulose membranes (Whatman-Schleicher & Schuell GmbH, Dassel, Germany). The blocking process was carried out through treatment of membranes with TBST containing 5% NFM (or BSA), for 2h, at room temperature. Each primary antibody was added at a concentration of 1:1000, for 1h, at room temperature and 16h, at +4°C, while the appropriate IgG-HRP secondary antibody (anti-rabbit or anti-mouse) was used at a dilution of 1:2000, for 2h, at room temperature. Immunoreactive bands were visualized by ECL reactions, following manufacturer’s instructions. Actin was used as protein of reference.
2.5. RT-PCR
Total RNA from control and paclitaxel-treated cells was extracted following a Trizol-based protocol (Thermo Fisher Scientific Inc. - Life Technologies - Ambion, Massachusetts, USA). RNA was reverse transcribed using an oligo-[dT]12-18 primer and the MMLV enzyme (Thermo Fisher Scientific Inc. - Life Technologies - Invitrogen, Massachusetts, USA). cDNA was amplified by sqPCR, with a Biometra T3000 Thermocycler (Biometra GmbH, Goettingen, Germany), using gene-specific oligonucleotide primers (Table S1). PCR fragments were resolved into 2-3% agarose gels, according to standard procedures. GAPDH served as gene of reference.
2.6. DepMap data analysis
Genome-scale, CRISPR-Cas9-mediated screening has been previously described [12,13]. The most recent Avana CRISPR release (DepMap public 23Q2+Score, Chronos), which includes 32 BLCA cell lines and over 1,000 lines of other lineages, screened with the Avana library, was used (data available at the DepMap Portal: https://depmap.org/portal). Gene effect difference was calculated as T-statistic from the DepMap portal as described in [13]. The primary PRISM repurposing dataset (https://depmap.org/repurposing) includes data from 4,518 compounds screened against 578 cell lines [10], whereas the secondary screen includes data from 1,448 compounds screened against 499 cell lines in an 8-step, 4-fold (4x) dilution scheme, starting from 10 μM as the highest concentration being tested [14]. The cell lines used in the PRISM screens are a subfraction of the total cell lines used in the CRISPR screen. Specifically for BLCA, only 24 out of the 32 BLCA cell lines in DepMap were successfully barcoded and included in the PRISM screen [10].
2.7. ATAC-seq. of primary human tumors
2.8. Statistical analysis
Statistical analyses for DepMap and PRISM data were generated and obtained from the DepMap portal. The statistical significance of differences in sqPCR were determined using one way ANOVA with Tukey’s test for multiple hypothesis correction, using GraphPad. p-value < 0.05 was considered as statistically significant.
3. Results
3.1. Urothelial bladder cancer cells are significantly sensitive to microtubule assembly inhibition
Given the poor prognoses of patients suffering from BLCA, we sought to identify gene dependencies and small molecules to which BLCA cells are preferentially sensitive, by interrogating the DepMap project databases [10,14,17]. We first interrogated the gene dependencies across 1,095 cell lines and focused on dependencies specific for BLCA cell lines (n=32) compared to all other cell lines in the DepMap database (n=1,063), and identified 35 statistically significant gene dependencies (Figure 1A and Table S2). The DepMap project utilizes genome-wide CRISPR screens across hundreds of cancer cell lines spanning all disease types. Each cell line is stably infected with a library of small-guide RNA (sgRNA) targeting all known human genes, plus cutting and non-cutting controls. The cells are stably infected with the lentiviral library, cultured for 2 weeks and the abundance of each sgRNA is calculated at the end of the experiment compared to the beginning (day 0: d0). The effect of each sgRNA is calculated with an algorithm taking into account the cutting efficiency of each sgRNA and CRISPR-cutting induced toxicity, and is presented as the CHRONOS score [18]. A negative log2-fold change of the CHRONOS score for a certain gene means that, at the end of the experiment, the abundance of this sgRNA was reduced compared to d0, and thus deleting that gene is toxic. The CHRONOS score is calculated for all genes for each individual cell line. When focusing on BLCA cell lines, the top dependencies included genes encoding proteins involved in microtubule dynamics, such as KATNB1 (Katanin Regulatory Subunit B1), which participates in a complex that severs microtubules in an ATP-dependent fashion (Figure 1A,B), suggesting that this pathway is a specific vulnerability in BLCA cells. Ontology analysis revealed that the genes emerging as dependencies in BLCA are enriched in pathways related to centrosomes (Figure 1C and Table S3), further highlighting the importance of microtubules assembly and dynamics for survival of BLCA cells.
To further identify therapeutic agents that could selectively target BLCA cells, we analyzed the results from the PRISM repurposing drug screens [10,14]. We focused our analysis on the primary PRISM screen, where the results are plotted as log2-fold changes in abundance of barcoded cells at day 5 (d5) compared to day 0 (d0) [14]. We first compared the sensitivity of BLCA cells to paclitaxel against cancers where paclitaxel is a standard of care, such as breast, ovarian, and lung carcinomas. We found that BLCA cells (n=24) were among the most sensitive across all the cell lines (Figure S1), and more sensitive than breast, ovarian and lung cancers (Figure 1D). Kidney cancer cells were among the least sensitive cell lines to paclitaxel (Figure 1D and Figure S1) and were used as a negative control hereafter. Representative dose response curves from a BLCA and a kidney cancer cell line showed that BLCA cells are sensitive even to very low concentrations of paclitaxel, whereas kidney cancer cells can tolerate higher doses of the drug (Figure 1E,F).
Given the high sensitivity of BLCA cells to paclitaxel, especially compared to cancers that are considered sensitive to the drug (breast, ovarian and lung), we further investigated the dependency of BLCA cells on microtubule assembly-associated proteins, compared to all other cell lines (Figure 1B and Figure 2). Using KATNB1 as a readout, the DepMap data suggest that BLCA cells are by far the most sensitive to KATNB1 perturbation compared to all other cell lines analyzed (Figure 2A).
Cancer vulnerabilities often underly dependency on a gene due to its amplification or transcriptional abundance [19,20]. We therefore hypothesized that the dependence of BLCA cells on KATNB1 will rely on a specific chromatin state that will facilitate abundant transcription of KATNB1 compared to other cancer cells. To test our hypothesis, we systematically investigated the open chromatin at the KATNB1 locus, using normalized ATAC-seq. data from a large collection of primary human cancers [15]. Aside from BLCA, we focused on breast, uterine, cervical and lung cancers, and, also, investigated kidney cancer as a negative control, similarly to our paclitaxel sensitivity analysis (Figure 2B). Our analysis demonstrates that BLCA cells have selectively open chromatin at the KATNB1 gene locus compared to the other cases analyzed (Figure 2B).
Collectively, our analysis suggests that BLCA cells are preferentially sensitive to genetic or pharmacological perturbation of microtubule dynamics.
3.2. Paclitaxel treatment induces cell cycle arrest and growth inhibition in bladder cancer cells
Our analysis of the DepMap data and chromatin accessibility, so far, suggests that BLCA cells are sensitive to microtubule dynamics perturbation. To further confirm our findings, we used a collection of BLCA cell lines deriving from different disease stages, and treated them with a dose range of paclitaxel, for 24 and 48h. RT4 cell line originates from a grade I carcinoma, RT112 from grade II, T24 from grade III and TCCSUP from grade IV. RT4 and RT112 have wild-type TP53, whereas T24 and TCCSUP carry TP53 inactivating mutations, and therefore lack the p53 protein.
Paclitaxel is known to inhibit microtubule depolymerization, which is critical for the mitotic spindle assembly. To test the effects of paclitaxel on cell cycle, we treated RT4, RT112, T24 and TCCSUP cell lines with a dose range of the drug, for 24 h, and analyzed the effects by Fluorescent Activated Cell Sorting (FACS)-based cell cycle analysis. We found that a low concentration of paclitaxel induced a robust cell cycle arrest at the G2/M phase in all cell lines analyzed, independently of their malignancy grade or p53 status (Figure 3A,B,D,E,G,H,J,K and Figure S2).
To further investigate the effects of paclitaxel on survival and growth, we treated our panel of BLCA cell lines with a dose range of the drug, for either 24 or 48h, and monitored their survival and growth using an MTT assay. Consistent with the effects of paclitaxel on cell cycle, treatment induced growth inhibition. Although the results varied among the 4 cell lines tested, all cells exhibited significant growth inhibition 48 h after treatment, especially with the higher concentrations of the drug (Figure 3C,F,I,L).
Therefore, our in vitro analysis fully recapitulated our in silico analysis and confirmed that BLCA cells are highly sensitive to microtubule dynamics inhibition.
3.3. Paclitaxel treatment induces apoptosis in bladder cancer cells
Paclitaxel treatment can promote cell death by both apoptotic and non-apoptotic pathways. To investigate the induction of caspase-mediated apoptosis in BLCA cells following exposure to paclitaxel, we treated the panel of RT4, RT112, T24 and TCCSUP cell lines with varying doses of the drug, for 24 h, and analyzed the cleavage of Caspase-8, -9, -3 and PARP, using Western immunoblotting analysis. Our results clearly demonstrate that paclitaxel treatment induced cleavage of caspase repertoire and PARP typical substrate (Figure 4A). Although the level of caspase activation varied across the cell lines used, the cleaved forms of Caspase-8, -9, -3 and PAPR could be readily detected in all the cell lines tested, strongly suggesting that paclitaxel induces apoptosis in BLCA cells independently of their malignancy grade (I-IV) or p53 functional status (wild-type or mutated/inhibited). Interestingly, the level of caspase cleavage/activation induction proved to correlate with the histological grade of the tumor each cell line derived from. The greater level of apoptosis activation was detected in the grade I RT4 cells, whereas the lower level of activation was observed in the high grade IV TCCSUP cells (Figure 4A).
To further test the induction of apoptosis in BLCA cells, in response to paclitaxel, we used semi-quantitative (sq) RT-PCR as an orthogonal assay and analyzed the expression of several genes encoding proteins involved in apoptotic cell death (Figure 4B and Figure S3). RT-sqPCR analysis showed that all the cell lines tested express high levels of apoptosis-related genes, such as FASL, FAS, CIAP-1, CIAP-2, XIAP and SURVIVIN (Figure 4B,C and Figure S3). Most importantly, paclitaxel treatment induced a significant upregulation of the FASL mRNA synthesis/accumulation in all BLCA cell lines tested, even at the lower concentrations of the drug (Figure 4B,C), thereby underpinning the activation of a FASL-dependent apoptotic program.
Overall, our immunoblot and RT-sqPCR results, so far, demonstrate that paclitaxel treatment induces a robust apoptotic program in all BLCA cell lines tested, further confirming the sensitivity of BLCA cells to microtubule dynamics perturbation.
3.4. Bladder cancer cells have selectively open chromatin at the BCL2 locus and undergo BLC2 downregulation upon paclitaxel treatment
Besides the well characterized activity of paclitaxel to bind to microtubules and inhibit their depolymerization, it has been demonstrated that it can directly bind to BCL2 and inhibit its anti-apoptotic activity [21,22,23,24]. Given that our in vitro and in silico/DepMap-based analysis revealed that BLCA cell lines are extremely sensitive to paclitaxel, we sought to investigate whether the mechanism of action involves perturbation of BCL2 expression. In cancers addicted to BCL2, there are super-enhancers formed able to maintain an open chromatin at the BCL2 locus and sustain its expression [25]. We, then, asked if there is a preferentially open chromatin at the BCL2 locus in BLCA cells that could suggest an oncogenic addiction to BCL2 expression. To answer our question, we investigated the open chromatin status in BLCA compared to breast, uterine, cervical, lung and kidney (negative control) cancer, similarly to our analysis for KATNB1 (Figure 2B). By plotting the normalized ATAC-seq. reads for each type of cancer, we found that BLCA cells have preferentially open chromatin at the BCL2 locus (Figure 5A). Besides BLCA, cervical cancer cells also exhibited strong ATAC-seq. signal at the BCL2 locus, but neither breast (and uterine) nor lung cancer demonstrated such a signal, with kidney cancer serving as negative control (Figure 5A).
To further test whether paclitaxel treatment perturbs BLC2, we analyzed the synthesis/accumulation of BCL2 mRNA, using RT-sqPCR, in our panel of BLCA cells treated with paclitaxel for 24 h. Our results unveiled that paclitaxel treatment downregulated the expression of BCL2 gene at the highest concentration tested, with RT112 and T24, but also RT4, cells presenting the most remarkable negative effect (Figure 5B,C).
Taken together, our results, so far, strongly suggest that BLCA cells are dependent on BCL2 gene activity due to a selective open chromatin status and paclitaxel treatment results in downregulation of BCL2 mRNA levels.
3.5. Paclitaxel treatment induces DNA damage without p53 activation
Microtubule dynamics disruption due to paclitaxel treatment impairs cell division and generates DNA damage, and chromosomal instability that eventually leads to cell death [26,27]. Moreover, it is well known that the DNA Damage Response (DDR) pathway can further activate p53 and induce expression of its transcriptional targets, as a downstream effect [28]. To further elucidate the mechanism of increased sensitivity of BLCA cells to microtubule dynamics perturbation, we treated our panel of BLCA cell lines with paclitaxel and analyzed the DDR and p53 activation profiles, using immunoblot for the phosphorylated forms of H2AX and p53 proteins, respectively. We uncovered a robust upregulation of phosphorylated-H2AX (p-H2AXSer139) even at the lower concentration(s) of paclitaxel used, in all the cell lines tested (Figure 6A), with total H2AX (reference/control) protein levels remaining unaffected, especially at the lower drug doses (Figure 6A). Remarkably, in the high malignancy grade cell lines T24 (III) and TCCSUP (IV) we did not detect any phospho-specific or total p53 protein forms, before and after paclitaxel administration, which confirms the lack of protein due to damaging mutations (Figure 6A). To our surprise, in the low malignancy grade cell lines RT4 (I) and RT112 (II), which have functional, wild-type, p53, there was no upregulation of phospho-p53 (Ser15) or total protein levels observed upon paclitaxel treatment (Figure 6A), thus indicating that p53 cannot be phosphorylated/activated downstream of DDR, in this setting.
To further examine whether p53 is activated, as a result of paclitaxel-induced DDR, we used RT-sqPCR as an orthogonal assay, and analyzed the mRNA expression/accumulation patterns of the p53 transcriptional targets PUMA and NOXA. Our analysis showed that there is no transcriptional upregulation of either PUMA or NOXA gene, further confirming the absence of a p53 activation program following paclitaxel treatment of BLCA cells (Figure 6B–D).
Altogether, our results, so far, suggest that microtubule dynamics inhibition in BLCA cells induces a robust DDR, possibly underlying their extreme sensitivity to paclitaxel, but there is no subsequent p53 activation.
3.6. Bladder cancer cell sensitivity to paclitaxel is independent of TP53 activity, but highly depends on the expression of DNA repair genes
Our intriguing result of DDR without p53 activation in BLCA cells following paclitaxel treatment, prompted us to revisit and further investigate the PRISM data. We first asked whether the sensitivity of BLCA cells to paclitaxel is correlated with the expression of TP53. We focused on the results of the primary PRISM screen and plotted the expression of TP53 mRNA in relation to log2-fold changes for all BLCA cell lines included in the analysis (n=24). We found no association between the expression of TP53 and the sensitivity of the cells to paclitaxel (Figure 7A). Then, we investigated the status of TP53 gene in the BLCA cell lines used in the PRISM screen, based on data from the Cancer Cell Line Encyclopedia (CCLE). We categorized the cell lines into two groups based on whether they had wild-type or mutant p53 (Table S4) and plotted the log2-fold changes of survival for each group. Again, we found no correlation between the status of TP53 and the response of BLCA cells to paclitaxel (Figure 7B), in line with our MTT and immunoblot results.
Given the robust upregulation of phospho-H2AX following paclitaxel treatment (Figure 6A), we, next, asked whether there is an association between sensitivity to the drug and expression of genes encoding for DNA repair enzymes. Since microtubule dynamics perturbation caused by paclitaxel induces mainly DNA double-strand breaks (DSBs), we focused on the expression of ATM, ATR, CHEK1 and CHEK2 kinases (Figure 7C and Figure 8A–C). Our analysis showed that lower log2-fold changes, indicating higher sensitivity to paclitaxel, correlated with lower expression of ATM, ATR, CHEK1 and CHEK2 genes (mRNA levels). Specifically, we found a strong and significant association between the expression of ATM and CHEK2 kinases and the sensitivity of BLCA cell lines (n=24) to paclitaxel (Figure 7C and Figure 8B), further highlighting the importance of the DSB repair pathway for sensitivity to paclitaxel in BLCA.
Since the DepMap analysis revealed that BLCA cell lines were the most sensitive to paclitaxel, we hypothesized that the expression of DDR kinases will be overall lower in these cell lines, compared to all other cell lines in DepMap. Indeed, when analyzing the expression of DDR kinases, using the DepMap data, we found that BLCA cell lines have significantly lower expression of ATM gene compared to all other cell lines in DepMap (Figure 8D).
Altogether, our results, thus far, indicate that BLCA cells depend on DNA repair enzymes to ameliorate the toxic effects of paclitaxel-induced DNA damage, independently of the downstream activation of p53 “genotoxic sensor”.
3.7. Bladder cancer cells are highly dependent on PPARG expression for survival and growth
Based on the association between sensitivity to paclitaxel and expression of DNA repair kinases, we asked if any of these enzymes scored as dependencies in BLCA cell lines. Interestingly, although none of the DNA repair enzymes were among the significant hits in DepMap, we found that PPARG (Peroxisome Proliferator-Activated Receptor γ) scored as the top dependency (Figure 7D and Table S2). PPARG has been shown to play a role in DNA repair, and DNA damage-mediated activation of Caspase-8 and apoptosis [29]. Given that BLCA cell lines are among the most sensitive to paclitaxel, we asked if there is a differential expression of PPARG gene in BLCA cells. We found that BLCA cell lines express significantly higher levels of PPARG compared to all other cell lines tested in DepMap (Figure 8D).
Finally, to investigate if the selective dependency of BLCA cells to PPARG and its higher expression in this lineage are driven by an open chromatin state, we investigated the normalized ATAC-seq. signal of BLCA primary cancer cells compared to other cancers, at the PPARG locus. Surprisingly, when comparing BLCA to breast, uterine, cervical and lung cancer cells, known to be highly sensitive to paclitaxel, we uncovered a selectively open chromatin state at the PPARG locus of BLCA cells compared to the other cancers tested (Figure 7E; kidney cancer used as negative control). Remarkably, an open chromatin state is detected both in the coding and upstream/regulatory region of the PPARG locus (Figure 7E).
Collectively, our results, so far, strongly suggest that the dependency of BLCA cells to PPARG underlies its role in the DNA damage-mediated apoptosis and the high sensitivity of these cells to paclitaxel is, at least partially, dependent on the high expression of PPARG.
4. Discussion
In this study, we performed a comprehensive analysis of data generated by the Cancer Dependency Map (DepMap) and PRISM projects, to uncover novel genetic dependencies and sensitivities to compounds, specifically for BLCA. To our surprise, both the genetic and chemical screens converged to the importance of microtubule dynamics for the survival of BLCA cells, and the robust anticancer effects of perturbing these dynamics, specifically for BLCA.
We first focused on the genetic dependencies specific for BLCA cells and the biological processes they control. Many of the genes scored as dependencies are involved in microtubule dynamics, revealing the importance of this biological process for the survival of BLCA cells. Microtubules are dynamic structures that play a crucial role in cell homeostasis and most importantly in cell division, as they form the mitotic spindle that facilitates chromosomal segregation during mitosis and meiosis [30,31]. During cell division, microtubules are formed by polymerization of alpha and beta tubulin subunits, and at the end of metaphase they depolymerize by the reverse reaction. Numerous studies have shown the importance of microtubule dynamics for proper cell division, and that perturbing the proper and timely formation and depolymerization of mitotic spindle results in errors in cell division, and ultimately cell death [31]. Given the dependence of BLCA cells on microtubule dynamics, we next asked whether these cells are sensitive to agents that disrupt this dynamic and focused on paclitaxel (distributed and sold under the brand name Taxol), the most well characterized, and widely used, agent that perturbs microtubule dynamics.
Paclitaxel binds to beta-tubulin and blocks microtubule depolymerization, thus stabilizing microtubules and preventing the proper execution of anaphase and telophase, leading to errors in chromosomal segregation, and ultimately cell death [32]. Although paclitaxel is effective against a variety of cancers, it is currently used mainly in breast, ovarian and lung cancer. We, thus, investigated data generated by the PRISM repurposing project and asked what is the sensitivity of BLCA cells to paclitaxel, and how it is compared to other cancer types, especially breast, ovarian and lung that are treated with paclitaxel as a standard of care. In perfect alignment with the CRISPR screen/dependency data, we found that BLCA cells are among the most sensitive to paclitaxel, and significantly more sensitive than breast, ovarian and lung cancer cells. Furthermore, we confirmed these results in vitro, by treating a collection of BLCA cells with a dose range of paclitaxel and by showing robust anti-proliferative effects on all cell lines tested.
Paclitaxel treatment can cause DNA damage, and subsequently activate repair mechanisms to fix the damage and maintain genomic integrity. However, if the DDR is impaired or the DNA damage is too severe, cells can undergo apoptosis. Given the extremely high sensitivity of BLCA cells to paclitaxel, we tested markers of DDR in our system. We found a robust upregulation of the DNA DSB marker phospho-H2AX, but, to our surprise, we did not observe an activation of p53, at least in the two cell lines that are wild-type for the TP53 gene. Since we focused our analysis on the initial stages of cell response to paclitaxel (24 h), it is possible that p53 is activated at a later time-point and its status affects the later stage(s) of the response. However, since we detected activation of apoptosis, as determined by cleaved Caspase-8, -9 and -3, without transcriptional upregulation of the p53 targets PUMA and NOXA genes, it is evident that paclitaxel can induce apoptosis in BLCA cells without activation of p53 pathway. Moreover, when looking at the response to paclitaxel in the large collection of BLCA cell lines used in the PRISM project, we found no association between the response and the status of TP53 gene, further confirming the p53-independent nature of the response.
It is well established that functional p53 can effectively mediate cell death, whereas mutant p53 can help cancer cells evade cell death and, thus, provide resistance to a variety of cytotoxic agents. Although p53 is activated downstream of DDR, there are cases where DNA damage-induced apoptosis can be mediated by a p53-independent pathway, through activation of ATM and its downstream effector kinase CHEK2 [33,34]. The caspases being activated in response to DNA damage can vary depending on the type of damage and the type of cell affected [35]. However, some of the caspases commonly activated in response to DNA damage include Caspase-8, Caspase-9 and Caspase-3. Especially, Caspase-8 can be activated through the extrinsic or death receptor pathway, as well as by DDR, further activating both the extrinsic and intrinsic pathway, and subsequently activating Caspase-9 and Caspase-3. Given the lack of p53 activation, we asked if the response to paclitaxel in BLCA is associated with the expression of the DDR kinases ATM, ATR, CHEK1 and CHEK2, utilizing the transcriptomics data from the DepMap project. Our analysis clearly showed that the response to paclitaxel is strongly and significantly correlated with the expression of ATM and CHEK2, and, moreover, that BLCA cells have overall significantly lower expression of ATM compared to all other cancer cell lines, potentially contributing to their enhanced sensitivity to paclitaxel. Interestingly, paclitaxel has been shown to bind BCL2 and block its anti-apoptotic action [21,22,24]. Most importantly, an open chromatin state has been observed for BLCA at the BCL2 locus (Figure 5A) (BCL2 addiction), while a downregulation of BCL2 gene expression was detected in most BLCA cell lines treated with paclitaxel (Figure 5B,C). Therefore, it is possible that paclitaxel treatment in BLCA cells induces apoptosis by triggering a DDR-mediated activation of caspases and by inhibiting BCL2, likely both at the transcriptional and post-translational level (Figure S4).
The DepMap analysis revealed that the top dependency in BLCA was PPARG, suggesting that BLCA cells depend on its (PPARG) function for survival more than any other cancer type examined (Figure 7D and Table S2). PPARG is a transcription factor that plays diverse roles in regulating cellular responses to DNA damage and apoptosis [36]. Paclitaxel treatment induces DNA damage, leading to activation of the DDR pathway. This activation can upregulate the expression of PPARG, which can critically contribute to mediating apoptosis, in response to DNA damage. Furthermore, PPARG has proved to be involved in the regulation of paclitaxel-induced apoptosis through its ability to regulate the expression of genes controlling apoptosis, including those implicated in oxidative stress, mitochondrial dysfunction and cell cycle regulation [37,38,39,40,41]. Therefore, it is possible that the strong dependence of BLCA on PPARG underlies a crucial role for this transcription factor in maintaining genomic integrity and promoting survival of these (BLCA) cells. When BLCA cells are treated with paclitaxel, due to their low expression of ATM and high expression of PPARG, they are prone not to repair DNA damage, but rather to undergo apoptosis.
Most importantly, our analysis of publicly available ATAC-seq. data from a variety of human cancers [15], unveiled that many of the genetic dependencies, discovered in this study, are correlated with an open chromatin state for the associated genes (KATNB1, BCL2 and PPARG) (Figure 2B, Figure 5A and Figure 7E), suggesting that BLCA cells carry unique genetic and epigenetic dependencies that can be exploited for mechanistic understanding of the disease and therapeutic targeting.
The predictive value of DepMap CRISPR screens has been validated in numerous publications so far [42]. Recently, Chetverina et al., further demonstrated the power of the DepMap data in uncovering novel supertargets for cancer therapy [43]. Our study moved a step forward and combined a variety of in silico approaches, using epigenetic, transcriptional and dependency data, with hypothesis-driven experiments, in a collection of BLCA human cell lines, representing different stages of the disease. Collectively, our work demonstrates the importance of microtubule dynamics for survival and growth of BLCA cells (Figure S4), and underlies the therapeutic potential of perturbing the pathway’s integrity and functionality.
The strong correlation between sensitivity to paclitaxel and expression of DNA DSB repair enzymes, such as the ATM and CHEK2 kinases, underscores the need to further investigate the combination of paclitaxel with ATM inhibitors as a novel therapeutic regimen in BLCA, potentially allowing for lower concentrations of paclitaxel and its reduced toxic side effects. Finally, our major finding that the growth inhibitory effect of paclitaxel on BLCA cells is independent of the p53 activity strongly suggests that paclitaxel can treat a wide range of BLCA types, independently of their p53 status.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: BLCA cell lines are among the most sensitive to paclitaxel treatment; Figure S2: Paclitaxel treatment induces cell cycle arrest in BLCA cell lines; Figure S3: BLCA cell lines express high levels of pro-apoptotic genes; Figure S4: Mechanistic model; Table S1: DNA sequence and annealing/cycling parameters of PCR primers for RT-sqPCR; Table S2: Dependencies enriched in BLCA cell lines (n=32) against all other cell lines in the DepMap database (n=1,063); Table S3: Gene Ontology analysis of the 35 genes scored as dependencies in BLCA; Table S4: Mutational status of the TP53 gene in the 24 BLCA cell lines included in the PRISM analysis.
Author Contributions
Conceptualization, Dimitrios Stravopodis; Formal analysis, Dimitrios Stravopodis; Funding acquisition, Dimitrios Stravopodis; Investigation, Dimitrios Stravopodis; Methodology, Yiannis Drosos, Eumorphia Konstantakou, Aggeliki-Stefania Bassogianni, Athanassios Velentzas and Dimitrios Stravopodis; Project administration, Dimitrios Stravopodis; Resources, Ourania Tsitsilonis and Dimitrios Stravopodis; Software, Yiannis Drosos, Dimitrios Valakos and Dimitris Thanos; Supervision, Dimitrios Stravopodis; Validation, Yiannis Drosos, Eumorphia Konstantakou, Aggeliki-Stefania Bassogianni, Konstantinos-Stylianos Nikolakopoulos, Dimitra Koumoundourou, Sophia Markaki, Ourania Tsitsilonis, Gerassimos Voutsinas, Dimitrios Valakos, Ema Anastasiadou, Dimitris Thanos, Athanassios Velentzas and Dimitrios Stravopodis; Visualization, Dimitrios Stravopodis; Writing – original draft, Yiannis Drosos; Writing – review & editing, Athanassios Velentzas and Dimitrios Stravopodis.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are contained within the article or Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J Clin 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Comperat, E.M.; Cowan, N.C.; Gakis, G.; Hernandez, V.; Linares Espinos, E.; Lorch, A.; Neuzillet, Y., et al. European Association of Urology Guidelines on Muscle-invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur Urol 2021, 79, 82-104. [CrossRef]
- Netto, G.J. Molecular biomarkers in urothelial carcinoma of the bladder: are we there yet? Nature Reviews Urology 2012, 9, 41–51. [Google Scholar] [CrossRef]
- Wu, X.-R. Urothelial tumorigenesis: a tale of divergent pathways. Nature Reviews Cancer 2005, 5, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [CrossRef]
- Kim, J.; Akbani, R.; Creighton, C.J.; Lerner, S.P.; Weinstein, J.N.; Getz, G.; Kwiatkowski, D.J. Invasive Bladder Cancer: Genomic Insights and Therapeutic Promise. Clin Cancer Res 2015, 21, 4514–4524. [Google Scholar] [CrossRef]
- Galsky, M.D.; Hahn, N.M.; Rosenberg, J.; Sonpavde, G.; Hutson, T.; Oh, W.K.; Dreicer, R.; Vogelzang, N.; Sternberg, C.N.; Bajorin, D.F., et al. Treatment of patients with metastatic urothelial cancer "unfit" for Cisplatin-based chemotherapy. J Clin Oncol 2011, 29, 2432-2438. [CrossRef]
- Boehm, J.S.; Garnett, M.J.; Adams, D.J.; Francies, H.E.; Golub, T.R.; Hahn, W.C.; Iorio, F.; McFarland, J.M.; Parts, L.; Vazquez, F. Cancer research needs a better map. Nature 2021, 589, 514–516. [Google Scholar] [CrossRef]
- Hahn, W.C.; Bader, J.S.; Braun, T.P.; Califano, A.; Clemons, P.A.; Druker, B.J.; Ewald, A.J.; Fu, H.; Jagu, S.; Kemp, C.J., et al. An expanded universe of cancer targets. Cell 2021, 184, 1142-1155. [CrossRef]
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.-L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P., et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nature Biotechnology 2016, 34, 419-423. [CrossRef]
- Konstantakou, E.G.; Voutsinas, G.E.; Velentzas, A.D.; Basogianni, A.S.; Paronis, E.; Balafas, E.; Kostomitsopoulos, N.; Syrigos, K.N.; Anastasiadou, E.; Stravopodis, D.J. 3-BrPA eliminates human bladder cancer cells with highly oncogenic signatures via engagement of specific death programs and perturbation of multiple signaling and metabolic determinants. Molecular cancer 2015, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B., et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 2016, 6, 914-929. [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S., et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 2017, 49, 1779-1784. [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A., et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nature Cancer 2020, 1, 235-248. [CrossRef]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W., et al. The chromatin accessibility landscape of primary human cancers. Science (New York, N.Y.) 2018, 362. [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat Biotechnol 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S., et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151-1161. [CrossRef]
- Dempster, J.M.; Boyle, I.; Vazquez, F.; Root, D.E.; Boehm, J.S.; Hahn, W.C.; Tsherniak, A.; McFarland, J.M. Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biology 2021, 22, 343. [Google Scholar] [CrossRef]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E., et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat Genet 2018, 50, 515-523. [CrossRef]
- Zimmerman, M.W.; Liu, Y.; He, S.; Durbin, A.D.; Abraham, B.J.; Easton, J.; Shao, Y.; Xu, B.; Zhu, S.; Zhang, X., et al. MYC Drives a Subset of High-Risk Pediatric Neuroblastomas and Is Activated through Mechanisms Including Enhancer Hijacking and Focal Enhancer Amplification. Cancer Discov 2018, 8, 320-335. [CrossRef]
- Ferlini, C.; Cicchillitti, L.; Raspaglio, G.; Bartollino, S.; Cimitan, S.; Bertucci, C.; Mozzetti, S.; Gallo, D.; Persico, M.; Fattorusso, C., et al. Paclitaxel Directly Binds to Bcl-2 and Functionally Mimics Activity of Nur77. Cancer Research 2009, 69, 6906-6914. [CrossRef]
- Rodi, D.J.; Janes, R.W.; Sanganee, H.J.; Holton, R.A.; Wallace, B.A.; Makowski, L. Screening of a library of phage-displayed peptides identifies human bcl-2 as a taxol-binding protein. J Mol Biol 1999, 285, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Rodi, D.J.; Makowski, L. Similarity between the sequences of taxol-selected peptides and the disordered loop of the anti-apoptotic protein, Bcl-2. Pac. Symp. Biocomput. 1999, 10.1142/9789814447300_0053, 532-541. [CrossRef]
- Whitaker, R.H.; Placzek, W.J. Regulating the BCL2 Family to Improve Sensitivity to Microtubule Targeting Agents. Cells 2019, 8. [Google Scholar] [CrossRef]
- Bal, E.; Kumar, R.; Hadigol, M.; Holmes, A.B.; Hilton, L.K.; Loh, J.W.; Dreval, K.; Wong, J.C.H.; Vlasevska, S.; Corinaldesi, C., et al. Super-enhancer hypermutation alters oncogene expression in B cell lymphoma. Nature 2022, 607, 808-815. [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol Cell 2015, 60, 524–536. [Google Scholar] [CrossRef]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Tsao, T.; Kornblau, S.; Safe, S.; Watt, J.C.; Ruvolo, V.; Chen, W.; Qiu, Y.; Coombes, K.R.; Ju, Z.; Abdelrahim, M., et al. Role of peroxisome proliferator-activated receptor-gamma and its coactivator DRIP205 in cellular responses to CDDO (RTA-401) in acute myelogenous leukemia. Cancer Res 2010, 70, 4949-4960. [CrossRef]
- Wittmann, T.; Hyman, A.; Desai, A. The spindle: a dynamic assembly of microtubules and motors. Nat Cell Biol 2001, 3, E28–34. [Google Scholar] [CrossRef]
- Valdez, V.A.; Neahring, L.; Petry, S.; Dumont, S. Mechanisms underlying spindle assembly and robustness. Nat Rev Mol Cell Biol 2023, 10.1038/s41580-023-00584-0. [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Yuan, Z.M.; Huang, Y.; Ishiko, T.; Nakada, S.; Utsugisawa, T.; Kharbanda, S.; Wang, R.; Sung, P.; Shinohara, A.; Weichselbaum, R., et al. Regulation of Rad51 function by c-Abl in response to DNA damage. J Biol Chem 1998, 273, 3799-3802. [CrossRef]
- Niida, H.; Katsuno, Y.; Sengoku, M.; Shimada, M.; Yukawa, M.; Ikura, M.; Ikura, T.; Kohno, K.; Shima, H.; Suzuki, H., et al. Essential role of Tip60-dependent recruitment of ribonucleotide reductase at DNA damage sites in DNA repair during G1 phase. Genes Dev 2010, 24, 333-338. [CrossRef]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging connectivity of programmed cell death pathways and its physiological implications. Nature Reviews Molecular Cell Biology 2020, 21, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Wang, G.; Cheng, S.; Xiong, Y.; Cao, R.; Qian, K.; Ju, L.; Wang, X.; Xiao, Y. The role and function of PPARγ in bladder cancer. Journal of Cancer 2020, 11, 3965–3975. [Google Scholar] [CrossRef] [PubMed]
- Chi, T.; Wang, M.; Wang, X.; Yang, K.; Xie, F.; Liao, Z.; Wei, P. PPAR-γ Modulators as Current and Potential Cancer Treatments. 2021, 11. [CrossRef]
- Smallridge, R.C.; Copland, J.A.; Brose, M.S.; Wadsworth, J.T.; Houvras, Y.; Menefee, M.E.; Bible, K.C.; Shah, M.H.; Gramza, A.W.; Klopper, J.P., et al. Efatutazone, an Oral PPAR-γ Agonist, in Combination With Paclitaxel in Anaplastic Thyroid Cancer: Results of a Multicenter Phase 1 Trial. The Journal of Clinical Endocrinology & Metabolism 2013, 98, 2392-2400. [CrossRef]
- Mierzejewski, K.; Paukszto, Ł.; Kurzyńska, A.; Kunicka, Z.; Jastrzębski, J.P.; Makowczenko, K.G.; Golubska, M.; Bogacka, I. PPARγ regulates the expression of genes involved in the DNA damage response in an inflamed endometrium. Scientific Reports 2022, 12, 4026. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Nag, A.; Gangenahalli, G.; Sharma, K. Peroxisome Proliferator Activated Receptor Gamma Sensitizes Non-small Cell Lung Carcinoma to Gamma Irradiation Induced Apoptosis. Front Genet 2019, 10, 554. [Google Scholar] [CrossRef]
- Khandekar, M.J.; Banks, A.S.; Laznik-Bogoslavski, D.; White, J.P.; Choi, J.H.; Kazak, L.; Lo, J.C.; Cohen, P.; Wong, K.K.; Kamenecka, T.M., et al. Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proceedings of the National Academy of Sciences of the United States of America 2018, 115, 561-566. [CrossRef]
- Vazquez, F.; Sellers, W.R. Are CRISPR Screens Providing the Next Generation of Therapeutic Targets? Cancer Research 2021, 81, 5806–5809. [Google Scholar] [CrossRef]
- Chetverina, D.; Vorobyeva, N.E.; Gyorffy, B.; Shtil, A.A.; Erokhin, M. Analyses of Genes Critical to Tumor Survival Reveal Potential 'Supertargets': Focus on Transcription. Cancers 2023, 15. [Google Scholar] [CrossRef]
Figure 1.
Urothelial bladder cancer (UBLCA) cells are extremely sensitive to microtubule assembly inhibition. (A) Genome-scale CRISPR-Cas9 screens of 32 BLCA cell lines. Each circle represents one gene. The x-axis represents the mean difference of the CHRONOS dependency scores in BLCA cell lines compared with all the others. Negative dependency scores indicate that BLCA cells require that gene, whereas positive scores suggest that the gene suppresses BLCA growth. Blue colored circles represent significant negative dependencies and of the top dependencies, KATNB1 is colored orange. Statistical significance was calculated as -log10 (p-value) from two-sided t tests with Benjamini–Hochberg correction. (B) Two class comparison for KATNB1; one of the most significant dependencies highlighted in (A) between the 32 BLCA cell lines compared to all other cell lines in DepMap (n=1,063). (C) Gene ontology analysis of the 49 genes enriched as statistically significant dependencies in BLCA cells in (A). Only pathways with FDR < 0.05 are shown. (D) PRISM drug sensitivity results for paclitaxel depicted as log2-fold changes in abundance of barcoded cells at d5 vs d0. The results for bladder (urinary tract) cancer cells are shown compared to breast, ovarian (fallopian tube), lung and kidney cancers. Breast, ovarian and lung cancers are among the most sensitive, and paclitaxel is currently used as a standard of care, whereas kidney cancer cells are among the least sensitive and are depicted as control. (E) and (F) Representative dose response curves for paclitaxel in BLCA (RT4) and kidney cancer (CAL54) cell lines. T-statistic and p-value were obtained from the DepMap portal.
Figure 1.
Urothelial bladder cancer (UBLCA) cells are extremely sensitive to microtubule assembly inhibition. (A) Genome-scale CRISPR-Cas9 screens of 32 BLCA cell lines. Each circle represents one gene. The x-axis represents the mean difference of the CHRONOS dependency scores in BLCA cell lines compared with all the others. Negative dependency scores indicate that BLCA cells require that gene, whereas positive scores suggest that the gene suppresses BLCA growth. Blue colored circles represent significant negative dependencies and of the top dependencies, KATNB1 is colored orange. Statistical significance was calculated as -log10 (p-value) from two-sided t tests with Benjamini–Hochberg correction. (B) Two class comparison for KATNB1; one of the most significant dependencies highlighted in (A) between the 32 BLCA cell lines compared to all other cell lines in DepMap (n=1,063). (C) Gene ontology analysis of the 49 genes enriched as statistically significant dependencies in BLCA cells in (A). Only pathways with FDR < 0.05 are shown. (D) PRISM drug sensitivity results for paclitaxel depicted as log2-fold changes in abundance of barcoded cells at d5 vs d0. The results for bladder (urinary tract) cancer cells are shown compared to breast, ovarian (fallopian tube), lung and kidney cancers. Breast, ovarian and lung cancers are among the most sensitive, and paclitaxel is currently used as a standard of care, whereas kidney cancer cells are among the least sensitive and are depicted as control. (E) and (F) Representative dose response curves for paclitaxel in BLCA (RT4) and kidney cancer (CAL54) cell lines. T-statistic and p-value were obtained from the DepMap portal.
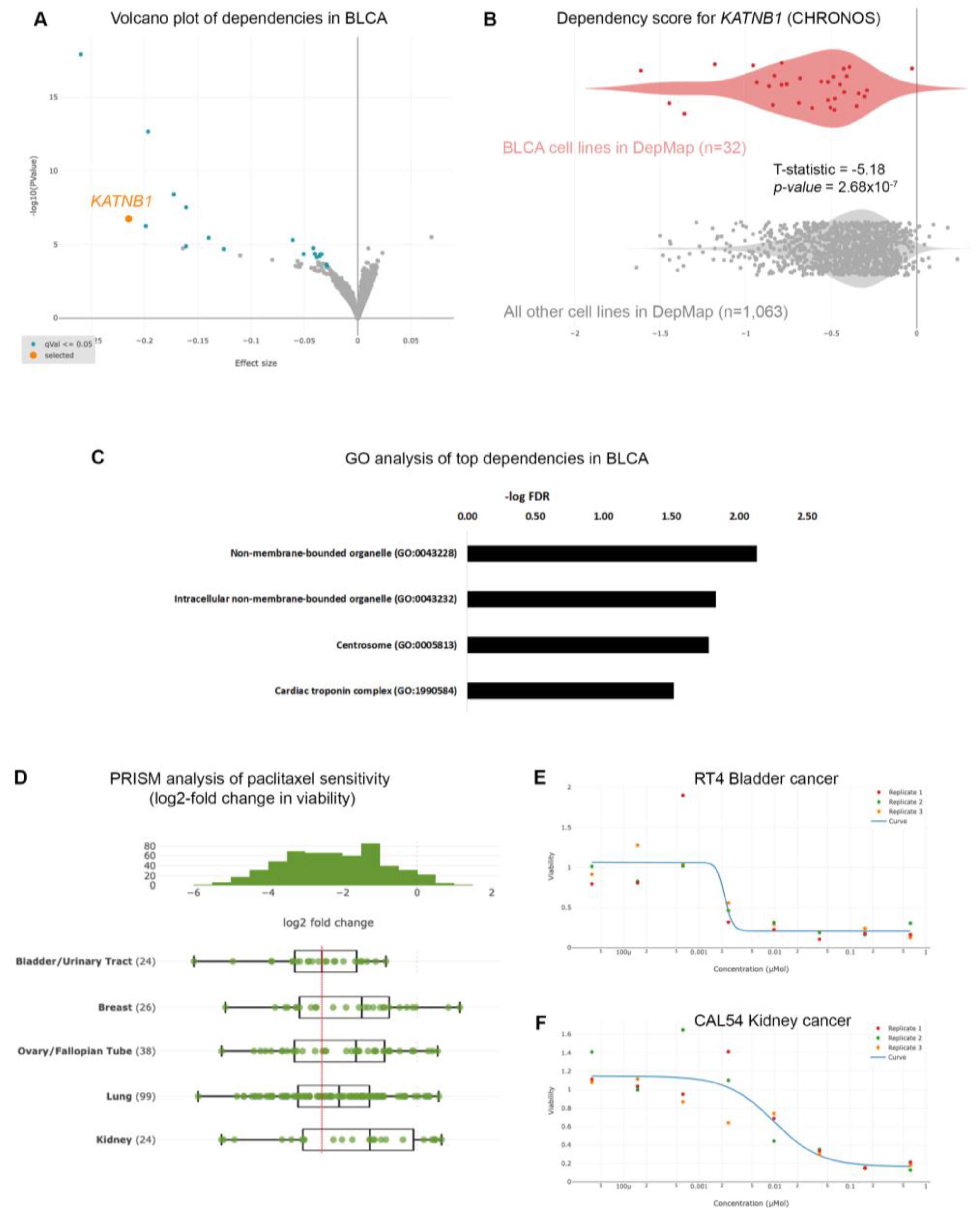
Figure 2.
Microtubule-associated proteins are vulnerabilities in BLCA cells. (A) CHRONOS score for KATNB1 gene across all cancer cell lines and types in DepMap. BLCA cells are among the most dependent cancer types. (B) Normalized ATAC-seq. tracks of the KATNB1 gene locus in 7 representative samples from various cancer types predicted to be dependent on KATNB1. Kidney cancer is used as negative control.
Figure 2.
Microtubule-associated proteins are vulnerabilities in BLCA cells. (A) CHRONOS score for KATNB1 gene across all cancer cell lines and types in DepMap. BLCA cells are among the most dependent cancer types. (B) Normalized ATAC-seq. tracks of the KATNB1 gene locus in 7 representative samples from various cancer types predicted to be dependent on KATNB1. Kidney cancer is used as negative control.
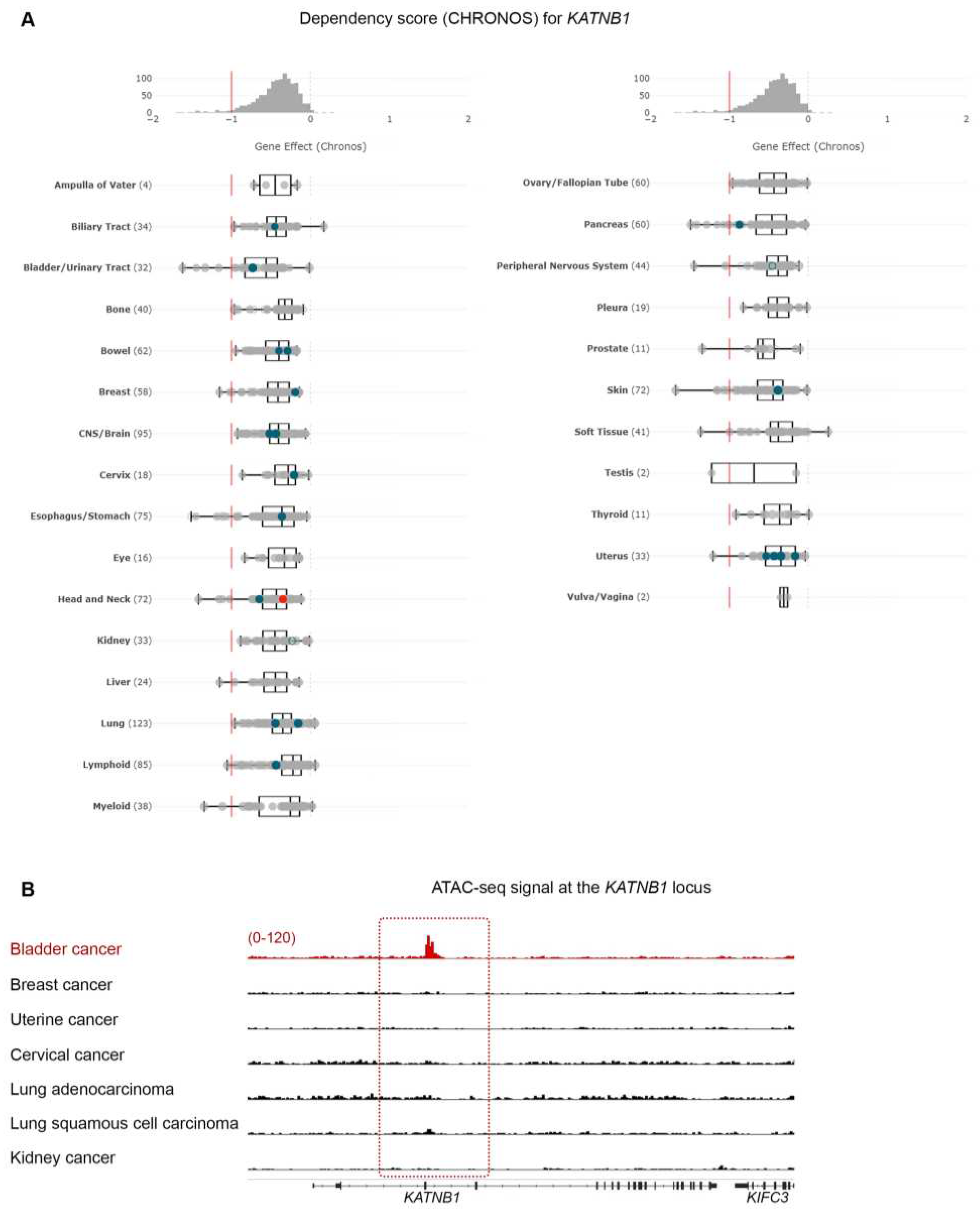
Figure 3.
Paclitaxel treatment induces cell cycle arrest and growth inhibition in BLCA cells. (A, B) Flow cytometry analysis of RT4 cells (malignancy grade I) either untreated (A) or treated with 1 μg/mL paclitaxel (B). (C) Sensitivity of RT4 cells to paclitaxel after 24 and 48 h of treatment, as assessed by an MTT assay. (D, E) Flow cytometry analysis of RT112 cells (malignancy grade II) either untreated (D) or treated with 1 μg/mL paclitaxel (E). (F) Sensitivity of RT112 cells to paclitaxel after 24 and 48 h of treatment, as assessed by an MTT assay. (G, H) Flow cytometry analysis of T24 cells (malignancy grade III) either untreated (G) or treated with 1 μg/mL paclitaxel (H). (I) Sensitivity of T24 cells to paclitaxel after 24 and 48 h of treatment, as assessed by an MTT assay. (J, K) Flow cytometry analysis of TCCSUP cells (malignancy grade IV) either untreated (J) or treated with 1 μg/mL paclitaxel (K). (L) Sensitivity of TCCSUP cells to paclitaxel after 24 and 48 h of treatment, as assessed by an MTT assay. Flow cytometry experiments were performed in triplicates and representative graphs are shown. MTT assays were performed in 3 biological replicates, with the graphs depicting the median values and error bars representing the standard error of each biological replicate.
Figure 3.
Paclitaxel treatment induces cell cycle arrest and growth inhibition in BLCA cells. (A, B) Flow cytometry analysis of RT4 cells (malignancy grade I) either untreated (A) or treated with 1 μg/mL paclitaxel (B). (C) Sensitivity of RT4 cells to paclitaxel after 24 and 48 h of treatment, as assessed by an MTT assay. (D, E) Flow cytometry analysis of RT112 cells (malignancy grade II) either untreated (D) or treated with 1 μg/mL paclitaxel (E). (F) Sensitivity of RT112 cells to paclitaxel after 24 and 48 h of treatment, as assessed by an MTT assay. (G, H) Flow cytometry analysis of T24 cells (malignancy grade III) either untreated (G) or treated with 1 μg/mL paclitaxel (H). (I) Sensitivity of T24 cells to paclitaxel after 24 and 48 h of treatment, as assessed by an MTT assay. (J, K) Flow cytometry analysis of TCCSUP cells (malignancy grade IV) either untreated (J) or treated with 1 μg/mL paclitaxel (K). (L) Sensitivity of TCCSUP cells to paclitaxel after 24 and 48 h of treatment, as assessed by an MTT assay. Flow cytometry experiments were performed in triplicates and representative graphs are shown. MTT assays were performed in 3 biological replicates, with the graphs depicting the median values and error bars representing the standard error of each biological replicate.
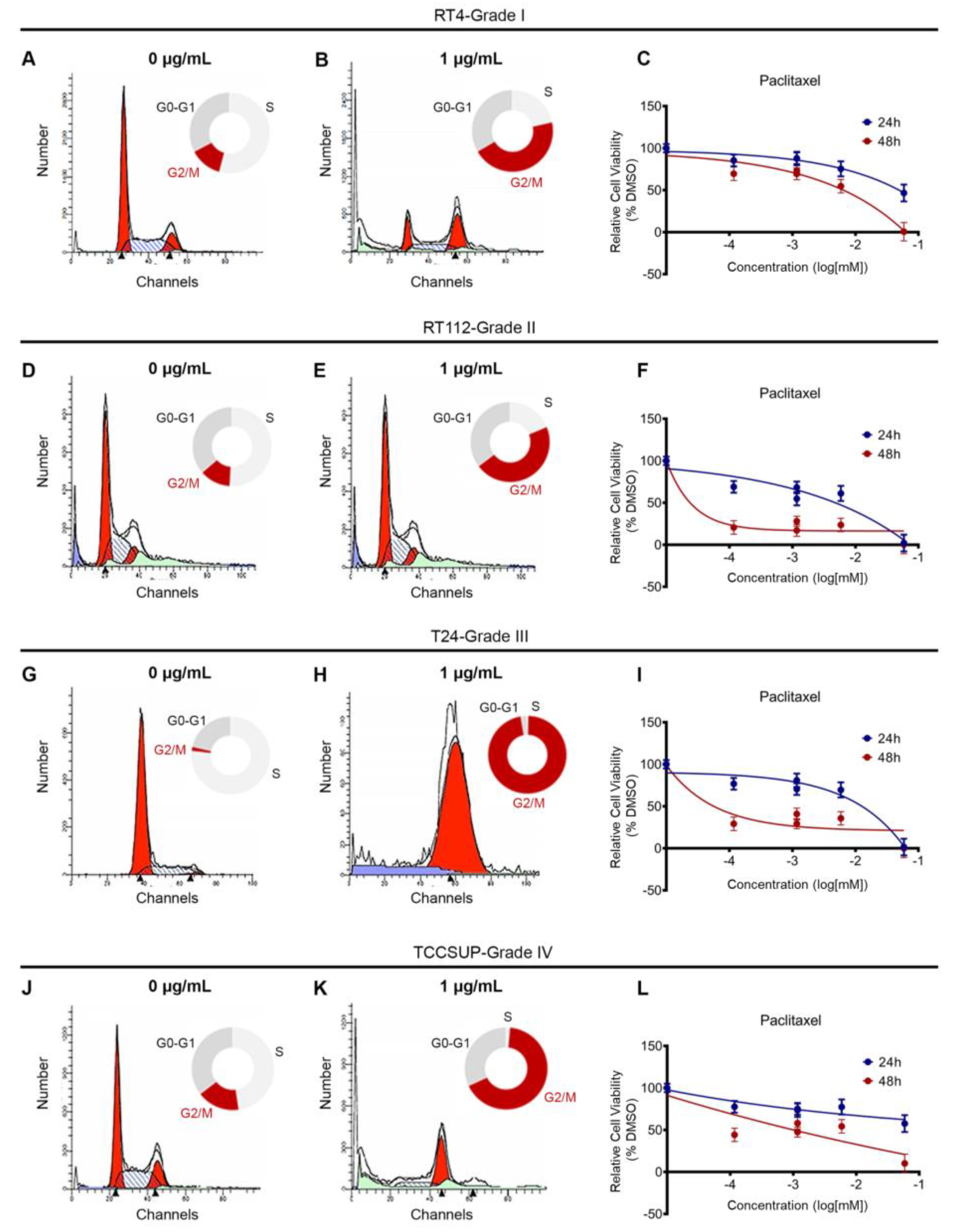
Figure 4.
Paclitaxel treatment induces apoptosis in BLCA cells. (A) Immunoblot analysis of whole-cell protein extracts obtained from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of Paclitaxel for 24 h. Protein extracts were blotted for Caspase-8, -9 and -3, and PARP to assess apoptosis, and for Pan-Actin as protein of reference (n=3 biological replicates). (B) RT-sqPCR analysis of total RNA extracted from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of paclitaxel for 24 h. Expression of FASL and FAS mRNA was analyzed to assess induction of apoptosis. GAPDH mRNA expression was used as control (n=3 biological replicates). (C) Densitometry analysis of the obtained RT-sqPCR results (B). The density of FASL and FAS RT-sqPCR bands was analyzed and normalized to the density of GAPDH mRNA for each cell line and condition, tested. Immunoblot and RT-sqPCR experiments were performed in triplicates, and representative images are shown. One-way ANOVA, with Tukey’s test for multiple hypothesis correction, ensures the statistical significance of the results. Error bars represent means ± SD (n=3 biological replicates). *p < 0.05, **p < 0.01 and ***p < 0.001.
Figure 4.
Paclitaxel treatment induces apoptosis in BLCA cells. (A) Immunoblot analysis of whole-cell protein extracts obtained from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of Paclitaxel for 24 h. Protein extracts were blotted for Caspase-8, -9 and -3, and PARP to assess apoptosis, and for Pan-Actin as protein of reference (n=3 biological replicates). (B) RT-sqPCR analysis of total RNA extracted from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of paclitaxel for 24 h. Expression of FASL and FAS mRNA was analyzed to assess induction of apoptosis. GAPDH mRNA expression was used as control (n=3 biological replicates). (C) Densitometry analysis of the obtained RT-sqPCR results (B). The density of FASL and FAS RT-sqPCR bands was analyzed and normalized to the density of GAPDH mRNA for each cell line and condition, tested. Immunoblot and RT-sqPCR experiments were performed in triplicates, and representative images are shown. One-way ANOVA, with Tukey’s test for multiple hypothesis correction, ensures the statistical significance of the results. Error bars represent means ± SD (n=3 biological replicates). *p < 0.05, **p < 0.01 and ***p < 0.001.
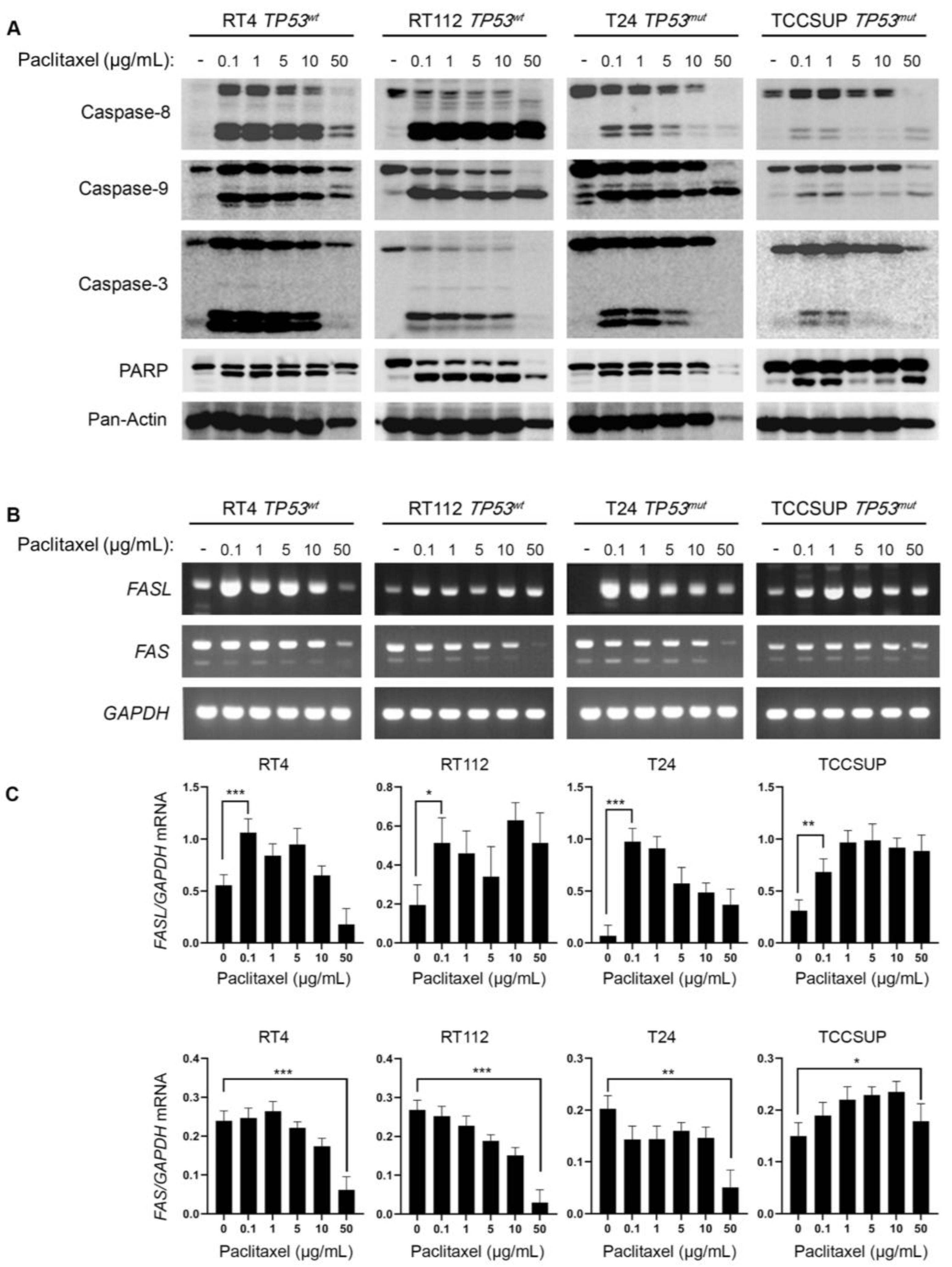
Figure 5.
BLCA cells have selectively open chromatin at the BCL2 locus and undergo BLC2 downregulation upon paclitaxel treatment. (A) Normalized ATAC-seq. tracks of the BCL2 locus in 7 representative samples from various cancer types predicted to be dependent on BCL2 and sensitive to paclitaxel. Kidney cancer is used as negative control. (B) RT-sqPCR analysis of total RNA extracted from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of paclitaxel for 24 h. Expression of BCL2 and BCL-XL mRNA was analyzed to assess induction of apoptosis. GAPDH mRNA expression was used as control (n=3 biological replicates). (C) Densitometry analysis of the obtained RT-sqPCR results (B). The density of BCL2 and BCL-XL RT-sqPCR bands was analyzed and normalized to the density of GAPDH mRNA for each cell line and condition, examined. One-way ANOVA, with Tukey’s test for multiple hypothesis correction, ensures the statistical significance of the results. Error bars represent means ± SD (n=3 biological replicates) (n.s.: non-significant). *p < 0.05, **p < 0.01 and ***p < 0.001.
Figure 5.
BLCA cells have selectively open chromatin at the BCL2 locus and undergo BLC2 downregulation upon paclitaxel treatment. (A) Normalized ATAC-seq. tracks of the BCL2 locus in 7 representative samples from various cancer types predicted to be dependent on BCL2 and sensitive to paclitaxel. Kidney cancer is used as negative control. (B) RT-sqPCR analysis of total RNA extracted from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of paclitaxel for 24 h. Expression of BCL2 and BCL-XL mRNA was analyzed to assess induction of apoptosis. GAPDH mRNA expression was used as control (n=3 biological replicates). (C) Densitometry analysis of the obtained RT-sqPCR results (B). The density of BCL2 and BCL-XL RT-sqPCR bands was analyzed and normalized to the density of GAPDH mRNA for each cell line and condition, examined. One-way ANOVA, with Tukey’s test for multiple hypothesis correction, ensures the statistical significance of the results. Error bars represent means ± SD (n=3 biological replicates) (n.s.: non-significant). *p < 0.05, **p < 0.01 and ***p < 0.001.
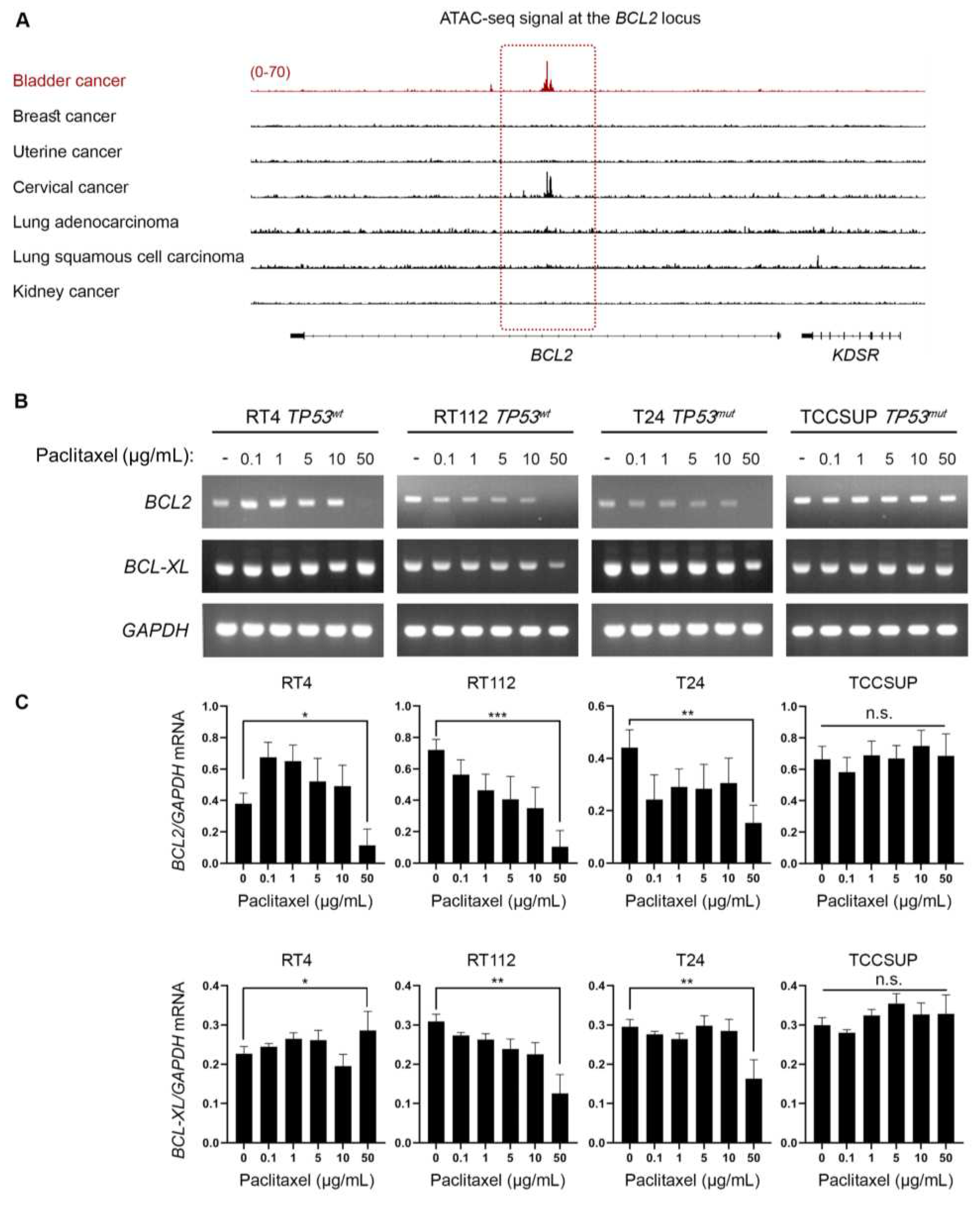
Figure 6.
Paclitaxel treatment induces DNA damage without p53 activation. (A) Immunoblot analysis of whole-cell protein extracts obtained from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of paclitaxel for 24 h. Protein extracts were blotted for total and phospho-H2AX (Ser139) to assess DNA Damage Response (DDR), and for total and phospho-p53 (Ser15) to assess p53 activation. Pan-Actin was used as protein of reference (n=3 biological replicates). (B) RT-sqPCR analysis of total RNA extracted from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of paclitaxel for 24 h. Expression of the known p53 targets, PUMA and NOXA genes (mRNA levels), was analyzed to assess p53 activation. GAPDH mRNA expression was used as control (n=3 biological replicates). (C) Densitometry analysis of the obtained RT-sqPCR results. The density of PUMA and NOXA RT-sqPCR bands was analyzed and normalized to the density of GAPDH mRNA for each cell line and condition, tested. One-way ANOVA, with Tukey’s test for multiple hypothesis correction, was engaged to examine the statistical significance of the results. Error bars represent means ± SD (n=3 biological replicates) (n.s.: non-significant).
Figure 6.
Paclitaxel treatment induces DNA damage without p53 activation. (A) Immunoblot analysis of whole-cell protein extracts obtained from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of paclitaxel for 24 h. Protein extracts were blotted for total and phospho-H2AX (Ser139) to assess DNA Damage Response (DDR), and for total and phospho-p53 (Ser15) to assess p53 activation. Pan-Actin was used as protein of reference (n=3 biological replicates). (B) RT-sqPCR analysis of total RNA extracted from RT4, RT112, T24 and TCCSUP cells, seeded at ~60% confluency, and exposed to the indicated doses (0, 0.1, 1, 5, 10 and 50 μg/mL) of paclitaxel for 24 h. Expression of the known p53 targets, PUMA and NOXA genes (mRNA levels), was analyzed to assess p53 activation. GAPDH mRNA expression was used as control (n=3 biological replicates). (C) Densitometry analysis of the obtained RT-sqPCR results. The density of PUMA and NOXA RT-sqPCR bands was analyzed and normalized to the density of GAPDH mRNA for each cell line and condition, tested. One-way ANOVA, with Tukey’s test for multiple hypothesis correction, was engaged to examine the statistical significance of the results. Error bars represent means ± SD (n=3 biological replicates) (n.s.: non-significant).
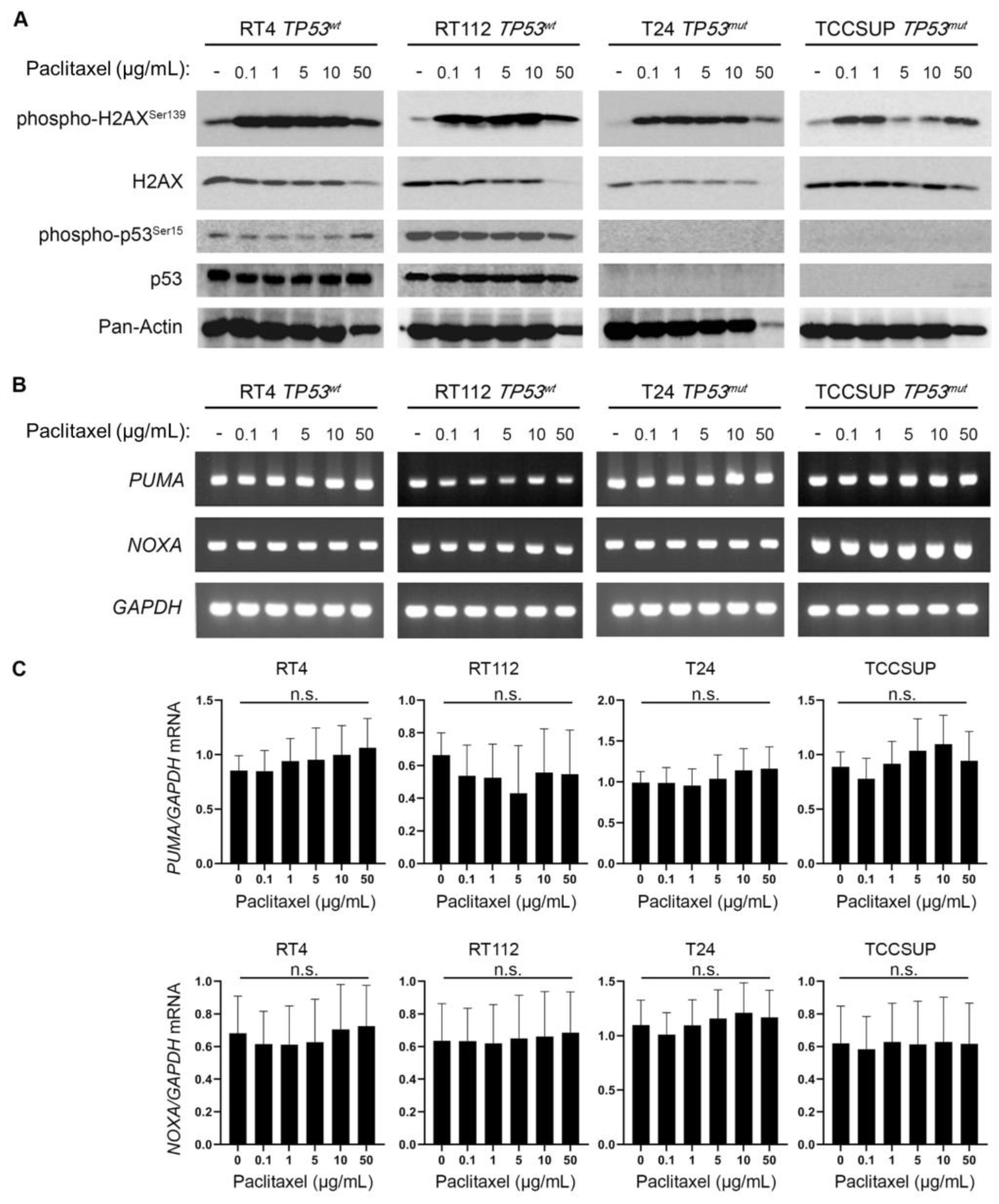
Figure 7.
BLCA sensitivity to paclitaxel is independent of p53 activity, but highly depends on the expression of ATM and PPARG. (A) PRISM analysis for the sensitivity of BLCA cells to paclitaxel (log2 fold change in abundance of barcoded cells at d5 vs d0) plotted against the expression of TP53 mRNA (log2 TPM+1). (B) BLCA cell lines in PRISM analysis were grouped based on their TP53 (mutational, or not) status and the median sensitivity to paclitaxel was plotted. (C) PRISM analysis for the sensitivity of BLCA cells to paclitaxel] (log2 fold change in abundance of barcoded cells at d5 vs d0) plotted against the expression of ATM mRNA (log2 TPM+1). (D) Two-class comparison of CHRONOS score for PPARG between DepMap BLCA cell lines (n=32) and all other cell lines in the database (n=1,063). (E) Normalized ATAC-seq. tracks of the PPARG gene locus in 7 representative samples from various cancer types predicted to be dependent on PPARG and sensitive to paclitaxel (also, see Figure 2B and Figure 5A). Kidney cancer is used as negative control. FC: Fold Change.
Figure 7.
BLCA sensitivity to paclitaxel is independent of p53 activity, but highly depends on the expression of ATM and PPARG. (A) PRISM analysis for the sensitivity of BLCA cells to paclitaxel (log2 fold change in abundance of barcoded cells at d5 vs d0) plotted against the expression of TP53 mRNA (log2 TPM+1). (B) BLCA cell lines in PRISM analysis were grouped based on their TP53 (mutational, or not) status and the median sensitivity to paclitaxel was plotted. (C) PRISM analysis for the sensitivity of BLCA cells to paclitaxel] (log2 fold change in abundance of barcoded cells at d5 vs d0) plotted against the expression of ATM mRNA (log2 TPM+1). (D) Two-class comparison of CHRONOS score for PPARG between DepMap BLCA cell lines (n=32) and all other cell lines in the database (n=1,063). (E) Normalized ATAC-seq. tracks of the PPARG gene locus in 7 representative samples from various cancer types predicted to be dependent on PPARG and sensitive to paclitaxel (also, see Figure 2B and Figure 5A). Kidney cancer is used as negative control. FC: Fold Change.
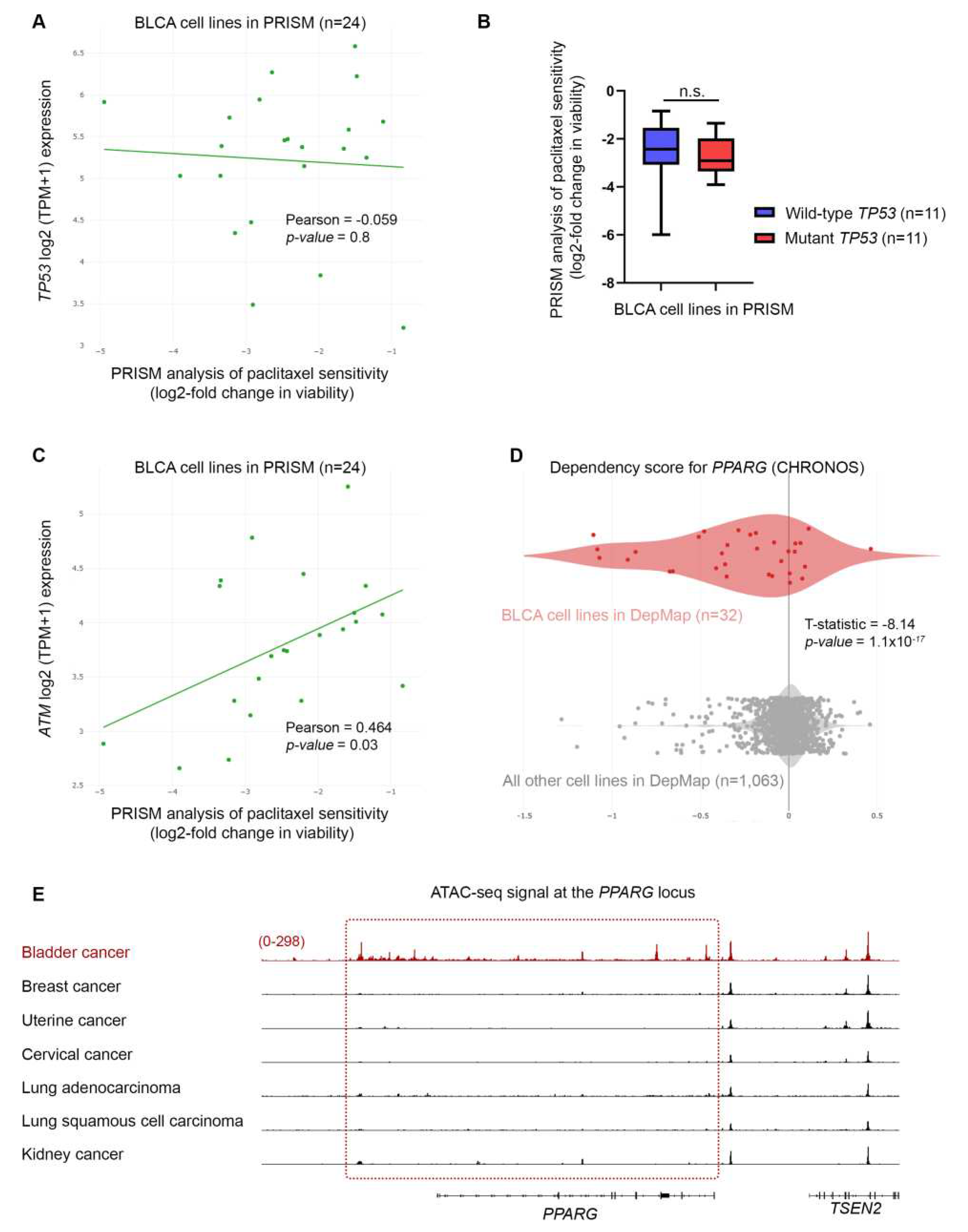
Figure 8.
BLCA sensitivity to paclitaxel depends on the expression of CHEK2 DNA repair genes. (A) PRISM analysis of the sensitivity of BLCA cells to paclitaxel [Log2 (TPM+1) = Log2-FC] plotted against the expression of ATR mRNA (RNA-seq.). (B) PRISM analysis of the sensitivity of BLCA cells to paclitaxel [Log2 (TPM+1) = Log2-FC] plotted against the expression of CHEK2 mRNA (RNA-seq.). (C) PRISM analysis of the sensitivity of BLCA cells to paclitaxel [Log2 (TPM+1) = Log2-FC] plotted against the expression of CHEK1 mRNA (RNA-seq.). (D) Two-class comparison of mRNA expression (RNA-seq.) for ATM gene between DepMap BLCA cell lines (n=32) and all other cell lines in the database (n=1,063). (E) Two-class comparison of mRNA expression (RNA-seq.) for PPARG gene between DepMap BLCA cell lines (n=32) and all other cell lines in the database (1,063). FC: Fold Change.
Figure 8.
BLCA sensitivity to paclitaxel depends on the expression of CHEK2 DNA repair genes. (A) PRISM analysis of the sensitivity of BLCA cells to paclitaxel [Log2 (TPM+1) = Log2-FC] plotted against the expression of ATR mRNA (RNA-seq.). (B) PRISM analysis of the sensitivity of BLCA cells to paclitaxel [Log2 (TPM+1) = Log2-FC] plotted against the expression of CHEK2 mRNA (RNA-seq.). (C) PRISM analysis of the sensitivity of BLCA cells to paclitaxel [Log2 (TPM+1) = Log2-FC] plotted against the expression of CHEK1 mRNA (RNA-seq.). (D) Two-class comparison of mRNA expression (RNA-seq.) for ATM gene between DepMap BLCA cell lines (n=32) and all other cell lines in the database (n=1,063). (E) Two-class comparison of mRNA expression (RNA-seq.) for PPARG gene between DepMap BLCA cell lines (n=32) and all other cell lines in the database (1,063). FC: Fold Change.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



