
Submitted:
20 June 2023
Posted:
21 June 2023
You are already at the latest version
Abstract
In the present study we tested the effect of small molecular weight redox molecules on collagen-induced platelet aggregation. We used N-acetylcysteine amide (AD4), the amide form of N-acetylcysteine (NAC), a thiol antioxidant with improved lipophilicity and bio-availability compared to NAC, and the thioredoxin mimetics (TXM) peptides, TXM-CB3, TXM-CB13, and TXM-CB30. All compounds significantly inhibited platelet aggregation induced by collagen, with TXM-peptides and AD4 being more effective than NAC. The levels of TxB2 and 12-HETE, the main metabolites derived from the cyclooxygenase and lipoxygenase pathways following platelet activation, were significantly reduced in the presence of AD4, TXM-peptides, or NAC, when tested at the highest concentration (0.6 mM). The effect of AD4, TXM-peptides, and NAC was tested also on the clotting time (CT) of whole blood. TXM-CB3 and TXM-CB30 showed the highest increase of CT. Furthermore, two representative compounds, TXM-CB3, and NAC, showed an increase in the an-ti-oxidant free sulfhydryl groups of plasma detected by Ellman’s method, suggesting a contribution of plasma factors to the antiaggregating effects.
Our results suggest that these small molecular weight redox peptides might become useful for the prevention and/or treatment of oxidative stress conditions associated with platelet activation.
Keywords:
platelet
; aggregation
; N-acetylcysteine
; N-acetylcysteine amide
; thioredoxin mimetic peptides
; clotting time
; TxB2
; 12-HETE
1. Introduction
The antioxidant N-acetylcysteine (NAC) a synthetic derivative of L-cysteine, and a precursor of GSH is known to protect a variety of pathological conditions involving neurodegenerative disorders, cardiovascular diseases, inflammation, and carcinogenesis [1]. NAC is available in pharmaceutical formulations for oral, intravenous, and inhalation administration, and shows low toxicity with mild adverse effects such as gastrointestinal disorders, headache, erythema, hypotension, and bronchospasm [2,3].
Although it is commonly safe and well tolerated even at high doses, the bioavailability of NAC is low (about 5%) [4,5] since at physiological pH the carboxyl group is negatively charged, preventing membrane permeability [6].
For improving bioavailability NAC was amidated to N-acetylcysteine amide (AD4, also called NACA) [6]. AD4/NACA showed enhanced antioxidant, antiapoptotic and anti-inflammatory properties, compared to its parent compound. Moreover, AD4 can replenish intracellular glutathione (GSH) through the thiol-disulfide exchange with oxidized glutathione (GSSG), or acting as a precursor of GSH [6,7,8,9,10].
Pathological conditions related to oxidative stress are often associated with the activation of stress response proteins and antioxidant enzymes including the thioredoxin reductase/thioredoxin (TrxR/Trx) system [11]. The TrxR/Trx system is one of the main cellular redox systems, which is highly conserved from yeast to mammals, and uses nicotinamide adenine dinucleotide phosphate (NADPH) as co-substrate. Through a thiol-disulfide exchange reaction, this system maintains the thiol-disulfide redox balance and protects cells from oxidative stress. In particular, the unique and highly conserved -CxxC- motif is responsible for the redox activity of thioredoxin. Tri-and tetra-peptide designed based on the CxxC motif were shown to mimic the redox activity of thioredoxin in vitro and in vivo [8,11,12,13,14,15,16,17,18,19]. These peptides, called thioredoxin mimetic (TXM) peptides, include N- and C-terminal blocked tri- and tetra-oligopeptides derived from the -CxxC- motif of thioredoxin.
The TXM peptides scavenge free radicals, display anti-inflammatory activity, prevent NF-kB nuclear translocation, and inhibit mitogen-activated protein kinases. Additionally, the TXM-peptides exhibit a higher potency compared to NAC or AD4, both in vitro and in vivo [8,11,12,13,14,15,16,17,18,19].
Recently, we showed that NAC inhibits platelet function, reducing aggregation, adhesion to collagen matrix, ROS generation, and intracellular calcium mobilization [20].
In the present study, we examined the effects of AD4 and the TXM peptides, TXM-CB3, TXM-CB13, and TXM-CB30 on platelet aggregation.
2. Materials and Methods
2.1. Blood Collection and Platelet Preparation
The study was performed according to the Declaration of Helsinki and was approved by the local institutional Ethics Committee. Twenty male and female healthy subjects (age 25-60 years) were enrolled. All the participants provided written informed consent at the time of enrollment and none of the participants had taken any anti-inflammatory drug, nor drugs known to affect platelet function, in the previous 10 days. Venous blood was drawn from the antecubital vein and collected in Vacutainer® tubes containing 0.129 mM sodium citrate. Healthy donors had a platelet count between 250-350 x103/µL. Platelet-rich plasma (PRP) was obtained by centrifugation at 100x g for 10 minutes without brake.
For washed platelet (WP) preparation, blood was collected in a Vacutainer® tube containing ACD (trisodium citrate 22 g/L, citric acid 8.0 g/L, dextrose 24.5 g/L) as an anticoagulant and PRP was obtained by blood centrifugation at 130x g for 20 minutes. After the addition of 1 µM prostaglandin E1 and 10 mM ethylenediaminetetraacetic acid (EDTA) to PRP, the platelet suspension was centrifuged at 1000x g for 10 minutes, and the pellet was resuspended in Tyrode’s HEPES buffer albumin free. The platelet count was adjusted at 300,000/µL with the same buffer. The washed platelet preparation was kept for 30 minutes at room temperature before the aggregation.
2.2. Platelet Aggregation
AD4 (NAC-amide/NACA) and thioredoxin mimetic peptides (TXM) TXM-CB3, CB13, CB30, were custom synthesized by Novetide, Ltd., Haifa, Israel. NAC (Sigma Aldrich S.r.l., Milan Italy). All compounds were dissolved in ammonium formate buffer pH 3.5 (vehicle).
Platelet aggregation was carried out in a PAP-8E BioData aggregometer (Bio/Data Corporation, Sentinel Diagnostic Milan, Italy) at 37 °C, at constant stirring (1200 rpm). Two hundred-fifty µL of PRP was pre-incubated with vehicle, NAC, AD4, or TXM peptides (all 0.1 - 0.6 mM as indicated) for 30 minutes, at 37 °C at constant stirring. After the addition of collagen (0.5 µg/mL or 8 µg/mL for PRP and washed platelets respectively), platelet aggregation was monitored for 6 minutes as the change in light transmittance. The results are expressed as the area under the curve (AUC).
2.3. Measurement of Thromboxane B2 and 12-hydroxyeicosatetraenoic Acid
At the end of the aggregation reaction, platelet aggregates were pelleted by centrifugation at 1500x g, and plasma was collected. Thromboxane A2 (TxA2) and 12-hydroxyeicosatetraenoic acid (12-HETE) are the major arachidonic acid metabolites derived from cyclooxygenase and lipoxygenase pathways respectively, and the most abundant eicosanoids released during aggregation. The levels of TxB2, the stable metabolite of TxA2, and 12-HETE were measured using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method using ExcionLC system coupled to a Qtrap 5500 mass spectrometer (Sciex, Milan, Italy) outfitted with Electrospray ionization (ESI) source and managed by Analyst software. The Multiquant software, version 3.0.2, was used to process the acquired data. A chromatographic separation was performed using Xbridge BEH C18 column (2.1 x 30 mm, particle size 2.5 µm, Waters) at 40°C. The mobile phase A was: 50 mM ammonium acetate pH=8/H2O/MeOH 4:93:3 v/v/v. The mobile phase B was: 50 mM Ammonium Acetate pH=8/ACN/MeOH 4:93:3 v/v/v. The flow rate was 0.25 mL/min, and the total run time was 12 minutes. The gradient of the mobile phase is reported in Table 1.
Data acquisition was performed in the negative polarity mode, with the following source settings: curtain gas flow=20, collisionally-activated dissociation gas=Medium, ion spray voltage=-3500, temperature=400, gas 1=20, gas 2=30. The acquisition mode used was multiple reaction monitoring, with Unit Resolution for both quadrupole (Q1 and Q3). The transitions monitored, with their set parameters, are reported in Table 2.
TXB2 and 12-HETE standards were dissolved in methanol (10 ng/µL) and serially diluted using H2O/MeOH/ACN 80:10:10 v/v/v (Solvent MIX) to prepare the calibration standard curve. The standard curve, ranging from 3.9 to 125.0 ng/ml for both the analytes, was spiked with the deuterated standards, TXB2-d4 and 12-HETE-d8, both at 20.0 ng/mL. The concentrations of TXB2 and 12-HETE were calculated using the corresponding calibration curve based on the response ratio (peak area of unlabeled analyte/peak area of labeled internal standard).
2.4. Analysis of Platelet Function by Means of PFA-200 Analyzer
Platelet function was assessed using the platelet-function-analyzer Innovance PFA-200 (Siemens Healthcare s.r.l. Milan, Italy). Citrate blood samples collected from healthy subjects were treated with vehicle, NAC, AD4, or TXM peptides (all at 0.6 mM) for 30 minutes, with agitation at 37 °C, and loaded onto the collagen/adenosine diphosphate (coll/ADP) or collagen/epinephrine (coll/epi) cartridges (Siemens Healthcare s.r.l. Milan, Italy). The incubation of whole blood with these agonists led to platelet activation and the formation of a primary clot. The time required for the occlusion of an aperture of 147 µm of diameter was recorded by the instrument, and is reported as closure time (CT) in seconds. The maximum CT measured by PFA-200 is 300 seconds. If CT was longer than 300 seconds, it was reported as 300 seconds.
2.5. Quantification of Sulfhydryl Groups
At the end of the aggregation process, samples were centrifuged, platelet aggregates were pelleted and plasma was collected. Plasma and platelet sulfhydryl groups were detected using Ellman’s method modified by Riddles et al. [21,22]. Briefly, 50 µL of Ellman’s reagent (4 mg 5,5’-dithio-bis-(2-nitrobenzoic acid)/ml reaction buffer (0.1 M sodium phosphate, pH 8.0 containing 1 mM EDTA)) and 2.5 mL of reaction buffer were added to 250 µL of the sample. After 15 minutes of incubation at room temperature, absorbance was measured at 412 nm. Ellman’s reagent has a highly oxidizing disulfide bond, which is reduced by free sulfhydryl groups to form quantifiable, yellow-colored 2-nitro-5-thiobenzoic acid. The concentration of sulfhydryl groups in the sample was calculated using the molar extinction coefficient 14,150 M-1cm-1.
2.6. Measurement of the Plasma Antioxidant Activity
The antioxidant activity was assessed in the plasma samples obtained before and after platelet aggregation, and subsequent removal of platelets and aggregates by centrifugation. The fluorometric assay was performed using the 2’,7’-dichlorodihydrofluorescin diacetate (DCFH-DA; Sigma Aldrich S.r.l., Milan, Italy) as probe ad 2,2’-Azobis (2-amidinopropane) dihydrochloride (AAPH, Sigma Aldrich S.r.l., Milan, Italy) as the radical generator. DCFH was prepared from DCFH-DA by basic hydrolysis: 500 µL of 1 mM DCFH-DA was mixed at 4°C with 2 mL 0.01 M NaOH while protected from the light. After 20 minutes, the mixture was neutralized with 2 mL 0.01 M HCl and diluted with phosphate buffer saline (PBS; 10 µM final concentration). 20 µL of sample were loaded in triplicate in a 96-well black plate (Corning Incorporated Costar, Euroclone S.p.A., Milan, Italy) and diluted with 50 µL PBS; 20 µl AAPH (10 mM final concentration) and 10 µL DCFH (10 µM final concentration) were added. Oxidation of DCFH to 2’,7’-dichlorofluorescin (DCF) was monitored at 37 °C, setting the excitation at λ 485 nm and emission at λ 535 nm.
2.7. Statistical Analysis
Data are expressed as mean ± SD. Differences between the groups were assessed by the Student’s t-test for single comparison or by the analysis of the variance for repeated measures (ANOVA) and Dunnett’s post hoc test. A p-value <0.05 was considered significant.
3. Results
3.1. N-acetylcysteine amide (AD4), Thioredoxin-mimetics (TXM-peptides), and N-acetylcysteine (NAC) Inhibit Platelet Aggregation
PRP obtained from healthy subjects was preincubated for 30 minutes at 37 °C in the presence of the vehicle or with increasing concentrations of AD4, NAC and the corresponding TXM-peptides, TXM-CB3, TXM-CB13, and TXM-CB30. Subsequently, platelet aggregation was induced by the addition of collagen (0.5 µg/mL) and was monitored for 6 minutes. As shown, all the TXM-peptides and AD4 inhibited platelet aggregation in a concentration-dependent manner (Figure 1A–D), similar to the extent detected with NAC (Figure 1E). Inhibition of platelet aggregation by AD4 and the TXM-peptides was apparent already at 0.1mM as compared to a smaller effect of NAC at this concnetration, indicating superior efficacy.
3.2. N-acetylcysteine amide (AD4), Thioredoxin-mimetics (TXM-peptides), and N-acetylcysteine (NAC) Prevent the Generation of TXB2 and 12-HETE
Platelet activation triggers the arachidonic acid cascade, which results in generating TxA2, the main metabolite derived from the cyclooxygenase pathway, and 12-HETE, the metabolite derived from the lipoxygenase pathway. The levels of these metabolites were determined by quantitative mass spectrometry analysis. The analysis was performed on the supernatant obtained after collagen-induced platelet aggregation in the absence or in the presence of AD4, TXM-peptides, or NAC at 0.6 mM, the highest concentration. As shown in Figure 2, both AD4, NAC and the TXM-peptides, significantly reduced the synthesis of both TxB2 and 12-HETE.
3.3. N-acetylcysteine amide (AD4), Thioredoxin-mimetics and NAC Influence PFA-200 Closure Time
The PFA-200 system simulates a damaged blood vessel by aspirating blood through disposable test cartridges coated with collagen and either ADP (50 μg) or epinephrine (10 μg). Whole blood (800 μL) flows through a capillary and a microscopic aperture (147 μm) cut into the coated membrane under constant high shear rates (5000 to 6000/s). As blood contacts the membrane, platelets adhere, aggregate, and form a plug that occludes the aperture, causing blood flow to cease. The time required to occlude the aperture is automatically reported as CT. Measurements are terminated after 300 seconds. The effect of AD4, NAC, and TXM-peptides treatment in whole blood on CT was measured in the PFA-200 system. AD4, TXM-CB13 and TXM-CB30, which were tested at the highest concentration (0.6 mM), showed a slight increase in CT in the coll/epi test (147 ± 45 seconds, 131 ± 34 seconds, 216 ± 85 seconds, respectively) compared to vehicle-treated blood (111 ± 21 seconds), while TXM-CB3 and TXM-CB30 showed the highest increase in CT (> 300 seconds). NAC did not affect the CT (Figure 3A).
Similarly, in the coll/ADP test, TXM-CB3 and TXM-CB30 prolonged the CT above 300 seconds (Figure 3B).
3.4. N-acetylcysteine amide (AD4), TXM-CB3 and NAC do not Inhibit Aggregation of Washed Platelet
In contrast to PRP, preincubation of washed platelets with AD4 or TXM-CB3 as representative of the TXM-peptides, did not inhibit collagen (8 µg/mL)-induced platelet aggregation (Figure 4A,B). Similarly, the inhibitory activity of NAC on PRP aggregation was completely lost in washed platelet (Figure 4C), indicating no direct effects on platelets in the absence of plasma components.
3.5. Thioredoxin-mimetic TXM-CB3 and NAC Increase the Free Sulfhydryl Groups in Plasma but not in Platelets
We then assessed whether the anti-aggregany effects are dependent on the regeneration of free sulphydryl groups. We measured free -SH groups in the plasma enviroment surrounding platelets before and after aggregation using Ellman’s reagent. Compared to the basal condition, collagen-induced aggregation further decreases the levels of free sulphydryl groups in plasma proteins (Figure 3A). Preincubation with TXM-CB3 and AD4, significantly increased the free sulphydryl groups, which were detected in the plasma after PRP collagen stimulation, and subsequently removal of platelet aggregates (Figure 5B,C).
Collagen stimulation of PRP induced a modest non-significant decrease in free sulphydryl groups in platelets (36.53 ± 8.09 µM under basal condition, and 25.72 ± 9.81 µM after collagen-induced aggregation; n = 5). No change was observed in the levels of free sulphydryl groups following preincubation of PRP with TXM-CB3 or NAC, compared to collagen-stimulated PRP (31.74 ± 4.12 µM and 29.58 ± 5.16 µM for TXM-CB3 and NAC, respectively vs 25.72 ± 9.81 µM for collagen-stimulated PRP; n =5). These results support lack of a direct effects on platelets by these compounds.
3.6. Thioredoxin-mimetic TXM-CB3 and NAC Increase the Total Plasma Antioxidant Activity
As regeneration of free thiols is one of the main protective mechanisms against oxidative stress, which ultimately leads to excessive platelet activation, we evaluated the antioxidant activity within the plasma samples before and after collagen-induced platelet aggregation. After the addition of AAPH as radical generator, a lag time was observed and the propagation phase started after ~ 30 minutes (Figure 6). After 70 minutes of incubation, a marked increase in DCFH oxidation was detected in collagen-treated samples. In contrast, preincubation of PRP with TXM-CB3, or NAC, completely prevented DCFH oxidation and the total antioxidant activity measured in these samples was similar to that detected in unstimulated PRP samples (Figure 6).
4. Discussion
Inhibition of platelet aggregation by antioxidant/anti-inflammatory agents may prove to be efficacious in the treatment of cardiovascular diseases. Here we show that the amino acids-based compounds NAC-amide (AD4) and the thioredoxin mimetic peptides TXM-CB3, TXM-CB13, and TXM-CB30 inhibit collagen-induced platelet aggregation. Compared to NAC, all these thiol-containing peptides exhibited a significantly higher antiaggregant activity. Platelet activation involves the mobilization of arachidonic acid from phospholipids of the plasma membrane, which is rapidly converted to thromboxane and prostaglandins from cyclooxygenase and hydroxyeicosatetraenoic acid from lipoxygenase enzymes. In platelets, the major metabolites derived from these two metabolic pathways are TxA2 and 12-HETE synthesized from cyclooxygenase and lipoxygenase, respectively [23]. While it is known that TxA2 is a potent mediator of platelet activation and contributes to sustaining platelet aggregation through the activation of the TxA2 receptor on platelets, the role of 12-HETE in platelet function remains less clear, but it has been suggested to contribute to both platelet adhesion and aggregation [24,25]. The mechanism(s) by which all these compounds inhibit the synthesis of the arachidonic acid metabolites is unknown, however, it has been shown that NAC reduced the mobilization of arachidonic acid in macrophages with concomitant inhibition of the release of the inflammatory lipid mediators, prostaglandin E2 and TxB2 [26]. The present results demonstrate a marked inhibition of both TxB2, a stable metabolic product of TxA2, and 12-HETE after treatment of PRP with the TXM-peptides, NAC, and AD4.
The PFA-200 system was designed as a global assay for platelet-dependent primary hemostasis, however, besides detection of platelet dysfunction, it may be also be used for monitoring anti-platelet therapy by assessing the contribution of systemic inflammation onto platelet function [27,28]. By this ex vivo test, we found that all compounds prolong CT in the PFA-200 system. Among them, the most marked effect was exerted by TXM-CB3 and TXM-CB30 both in coll/ADP and coll/epi tests, suggesting that they interfere with the binding of epinephrine and ADP to the corresponding platelet receptors.
It is known that the effect of ADP on platelets is mediated by 3 purinergic receptors, designated P2Y1, P2Y12, and P2X1 [29]. While the P2X1 receptor is an ion channel that upon activation induces calcium influx and doesn't lead to aggregation by itself, the activation of the other two purinergic receptors results in platelet aggregation. Also, the activation of P2Y1 receptor leads to changes in the shape of platelets, TxA2 generation, adhesion to fibrinogen, and to aggregation. Similarly, P2Y12 activation sustains the aggregation and potentiates the platelet activation induced by other physiological agonists including collagen, von Willebrand and TxA2. The importance of this receptor is linked to the use of antithrombotic drugs such as clopidogrel, ticlopidine, and prasugrel, which through blocking the P2Y12 receptor inhibit platelet activation and reduce the risk of heart attack and stroke [29]. In contrast to the P2Y1 receptor, which has 2 disulfide bridges in the extracellular domain, the P2Y12 receptor shows 2 free cysteines (Cys17 and Cys270), essential for its activity. Therefore, it has been proposed that clopidogrel as well as other thiol reagents, inactivate P2Y12 receptor through the formation of disulfide bridges [29].
With regard to the effect on coll/epi, it is known that the catecholamines, epinephrine and norepinephrine, induce platelet aggregation through binding to the α-adrenergic receptor and inhibition of adenylate cyclase activity [30]. It has been demonstrated that stimulation of downstream targets of the α-adrenergic receptor requires an oxidative modification of thiols [31]. It led to suggest that reducing the activity of the compounds is likely to affect mediator(s) involved in platelet activation.
The lack of an effect on the clotting time by NAC, as shown in the present study can be explained by the use of a lower concentration compared to previous studies (> 2 mM). The authors suggested a reduction of disulfide bonds of von Willebrand factor, a multimeric plasma protein that mediates adhesion and aggregation of platelets at the site of vascular injury [32].
In contrast to what was detected in PRP, no inhibitory effect of the thiol compounds was observed on collagen-induced aggregation in washed platelets suspended in albumin-free Tyrode’s HEPES buffer. These results led us to hypothesize that the factors present in the plasma are likely to be affected. Previously, we have demonstrated that NAC inhibits platelet activation through regeneration of the anti-oxidant form of plasma albumin [20]. Further studies are therefore required to understand the mechanism(s), including the role of albumin, by which the thiol-based reagents AD4 and TXM-peptides modulate platelet activation.
It is known that reactive oxygen species (ROS) affect platelet function not only by causing aggregation, but also by modulating intracellular signaling pathways [33,34]. Furthermore, in addition to being targeted by ROS, platelet activation itself induces ROS formation, promoting platelet aggregation [35,36,37].
We have previously demonstrated that collagen induced stimulation of platelets resulted in a marked increase in ROS generation [20]. Here we show that the protecting effect of the thiol reagents is mainly due to an increase of free sulfhydryl groups of proteins. This novel finding of an increase of free sulfhydryl groups by NAC and TXM-CB3 indicates regeneration in the plasma compartment after collagen-induced platelet aggregation. The sulfhydryl groups of proteins are mainly responsible for their antioxidant properties and have been demonstrated to contribute ~ 53% to the total antioxidant capacity of serum in healthy subjects [38]. Under our experimental conditions, platelet stimulation with collagen led to a significant reduction of the free sulfhydryl group measured in the plasma after the removal of platelet aggregates. Preincubation of PRP with TXM-CB3 appeared to prevent the reduction in free sulfhydryl groups, suggesting that by reducing oxidized proteins the TXM peptide was able to maintain the plasma redox status and protect platelets from oxidative stress.
The plasma compartment represents an important defense system against ROS that are continuously generated in the body, including the redox enzymes e.g superoxide dismutase, glutathione peroxidases and catalase, protein thiols, and uric acid, and exogenous antioxidants such as ascorbic acid, vitamin E, tocopherols and carotenoids [39,40]. We measured the total antioxidant capacity of plasma obtained after activation of platelets with collagen and removal of aggregates, using the radical generator AAPH, and the hydrophilic oxidable substrate DCFH. After a short lag-time, a marked increase in the generation of DCF, the oxidized form of DCFH was observed in the collagen-treated samples compared to unstimulated samples, and was completely prevented in the presence of TXM-CB3 or NAC. Although the antioxidant activity of the TXM peptides has been shown to be significantly more potent than NAC [19], both TXM-CB3 and NAC showed similar antioxidant activity. Further concentration-dependent studies are required to evaluate this discrepancy.
5. Conclusions
This study shows for the first time the antiaggregating activity of N-acetylcysteine amide (AD4), the amide form of N-acetylcysteine (NAC), a thiol antioxidant with improved lipophilicity and bioavailability compared with NAC, and the thioredoxin mimetic (TXM) peptides, TXM-CB3, TXM-CB13, and TXM-CB30. The findings show that they act by inhibiting the generation of arachidonic acid metabolites following platelet activation through interfering with whole blood clotting, and regenerating the plasma anti-oxidant free sulfhydryl groups. Overall, these data suggest that small molecular weight redox peptides might represent useful tools for preventing and/or treatment of oxidative stress conditions associated with platelet activation.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, C.B. and D.A.; methodology, S.E. and M.M.; validation S.E. and M.M.; formal analysis, S.E. and M.M.; investigation, S.E.; resources, C.B. and D.A.; data curation, S.E.; writing—original draft preparation, S.E. and C.B.; writing—review and editing, S.E., M.M., D.A. and C.B.; visualization, S.E.; supervision, D.A. and C.B.; project administration, C.B.; funding acquisition, C.B. All authors have read and agreed to the published version of the manuscript. Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
This work was supported by the Italian Ministry of Health, Italy (Ricerca Corrente)
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Centro Cardiologico Monzino (protocol code 1626, date of approval: 2 December 2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Data collected in the study will be made available using the data repository Zenodo (https://zenodo.org) with restricted access upon request to direzione.scientifica@ccfm.it. Any remaining information can be obtained from the corresponding Author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Samuni, Y.; Goldstein, S.; Dean, O.M.; Berk, M. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta 2013, 1830, 4117–4129. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A. N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr Opin Pharmacol 2007, 7, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, M.; Graciliano, N.G.; Moura, F.A.; Oliveira, A.C.M.; Goulart, M.O.F. N-Acetylcysteine (NAC): Impacts on Human Health. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Borgstrom, L.; Kagedal, B.; Paulsen, O. Pharmacokinetics of N-acetylcysteine in man. Eur J Clin Pharmacol 1986, 31, 217–222. [Google Scholar] [CrossRef]
- Olsson, B.; Johansson, M.; Gabrielsson, J.; Bolme, P. Pharmacokinetics and bioavailability of reduced and oxidized N-acetylcysteine. Eur J Clin Pharmacol 1988, 34, 77–82. [Google Scholar] [CrossRef]
- Offen, D.; Gilgun-Sherki, Y.; Barhum, Y.; Benhar, M.; Grinberg, L.; Reich, R.; Melamed, E.; Atlas, D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. J Neurochem 2004, 89, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, L.; Fibach, E.; Amer, J.; Atlas, D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic Biol Med 2005, 38, 136–145. [Google Scholar] [CrossRef]
- Atlas, D. Emerging therapeutic opportunities of novel thiol-amides, NAC-amide (AD4/NACA) and thioredoxin mimetics (TXM-Peptides) for neurodegenerative-related disorders. Free Radic Biol Med 2021, 176, 120–141. [Google Scholar] [CrossRef]
- Ates, B.; Abraham, L.; Ercal, N. Antioxidant and free radical scavenging properties of N-acetylcysteine amide (NACA) and comparison with N-acetylcysteine (NAC). Free Radic Res 2008, 42, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Sunitha, K.; Hemshekhar, M.; Thushara, R.M.; Santhosh, M.S.; Yariswamy, M.; Kemparaju, K.; Girish, K.S. N-Acetylcysteine amide: a derivative to fulfill the promises of N-Acetylcysteine. Free Radic Res 2013, 47, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic Biol Med 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kutner, M.; Khomsky, L.; Trus, M.; Aisner, Y.; Niv, M.Y.; Benhar, M.; Atlas, D. Thioredoxin-mimetic peptides (TXM) reverse auranofin induced apoptosis and restore insulin secretion in insulinoma cells. Biochem Pharmacol 2013, 85, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kutner, M.; Khomsky, L.; Trus, M.; Ben-Yehuda, H.; Lenhard, J.M.; Liang, Y.; Martin, T.; Atlas, D. Thioredoxin-mimetic peptide CB3 lowers MAPKinase activity in the Zucker rat brain. Redox Biol 2014, 2, 447–456. [Google Scholar] [CrossRef]
- Kronenfeld, G.; Engelman, R.; Weisman-Shomer, P.; Atlas, D.; Benhar, M. Thioredoxin-mimetic peptides as catalysts of S-denitrosylation and anti-nitrosative stress agents. Free Radic Biol Med 2015, 79, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Baratz-Goldstein, R.; Deselms, H.; Heim, L.R.; Khomski, L.; Hoffer, B.J.; Atlas, D.; Pick, C.G. Thioredoxin-Mimetic-Peptides Protect Cognitive Function after Mild Traumatic Brain Injury (mTBI). PLoS One 2016, 11, e0157064. [Google Scholar] [CrossRef] [PubMed]
- Lejnev, K.; Khomsky, L.; Bokvist, K.; Mistriel-Zerbib, S.; Naveh, T.; Farb, T.B.; Alsina-Fernandez, J.; Atlas, D. Thioredoxin-mimetic peptides (TXM) inhibit inflammatory pathways associated with high-glucose and oxidative stress. Free Radic Biol Med 2016, 99, 557–571. [Google Scholar] [CrossRef]
- Hemling, P.; Zibrova, D.; Strutz, J.; Sohrabi, Y.; Desoye, G.; Schulten, H.; Findeisen, H.; Heller, R.; Godfrey, R.; Waltenberger, J. Hyperglycemia-induced endothelial dysfunction is alleviated by thioredoxin mimetic peptides through the restoration of VEGFR-2-induced responses and improved cell survival. Int J Cardiol 2020, 308, 73–81. [Google Scholar] [CrossRef]
- Canesi, F.; Mateo, V.; Couchie, D.; Karabina, S.; Negre-Salvayre, A.; Rouis, M.; El Hadri, K. A thioredoxin-mimetic peptide exerts potent anti-inflammatory, antioxidant, and atheroprotective effects in ApoE2.Ki mice fed high fat diet. Cardiovasc Res 2019, 115, 292–301. [Google Scholar] [CrossRef]
- Bachnoff, N.; Trus, M.; Atlas, D. Alleviation of oxidative stress by potent and selective thioredoxin-mimetic peptides. Free Radic Biol Med 2011, 50, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Eligini, S.; Porro, B.; Aldini, G.; Colli, S.; Banfi, C. N-Acetylcysteine Inhibits Platelet Function through the Regeneration of the Non-Oxidative Form of Albumin. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L. Tissue sulfhydryl groups. Arch Biochem Biophys 1959, 82, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Riddles, P.W.; Blakeley, R.L.; Zerner, B. Reassessment of Ellman's reagent. Methods Enzymol 1983, 91, 49–60. [Google Scholar] [CrossRef]
- Willoughby, S.; Holmes, A.; Loscalzo, J. Platelets and cardiovascular disease. Eur J Cardiovasc Nurs 2002, 1, 273–288. [Google Scholar] [CrossRef]
- Maderna, P.; Caruso, D.; Tremoli, E.; Galli, G. Differential effects of oral administrations to human volunteers of acetylsalicylic acid, sodium salicylate and indomethacin on 12-hydroxyeicosatetraenoic acid formation by stimulated platelets. Thromb Res 1988, 52, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, M.R.; Butt, R.W.; Hirsh, J.; Markham, B.A.; Nazir, D.J. Role of lipoxygenase metabolism in platelet function: effect of aspirin and salicylate. Prostaglandins Leukot Med 1986, 21, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Fritsch-Decker, S.; Both, T.; Mulhopt, S.; Paur, H.R.; Weiss, C.; Diabate, S. Regulation of the arachidonic acid mobilization in macrophages by combustion-derived particles. Part Fibre Toxicol 2011, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Kottke-Marchant, K.; Powers, J.B.; Brooks, L.; Kundu, S.; Christie, D.J. The effect of antiplatelet drugs, heparin, and preanalytical variables on platelet function detected by the platelet function analyzer (PFA-100). Clin Appl Thromb Hemost 1999, 5, 122–130. [Google Scholar] [CrossRef]
- Homoncik, M.; Blann, A.D.; Hollenstein, U.; Pernerstorfer, T.; Eichler, H.G.; Jilma, B. Systemic inflammation increases shear stress-induced platelet plug formation measured by the PFA-100. Br J Haematol 2000, 111, 1250–1252. [Google Scholar] [CrossRef]
- Murugappa, S.; Kunapuli, S.P. The role of ADP receptors in platelet function. Front Biosci 2006, 11, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.B.; Lefkowitz, R.J. Agonist interactions with alpha-adrenergic receptors. J Cardiovasc Pharmacol 1982, 4 Suppl 1, S14–18. [Google Scholar] [CrossRef]
- Kuster, G.M.; Pimentel, D.R.; Adachi, T.; Ido, Y.; Brenner, D.A.; Cohen, R.A.; Liao, R.; Siwik, D.A.; Colucci, W.S. Alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes is mediated via thioredoxin-1-sensitive oxidative modification of thiols on Ras. Circulation 2005, 111, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Reheman, A.; Gushiken, F.C.; Nolasco, L.; Fu, X.; Moake, J.L.; Ni, H.; Lopez, J.A. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest 2011, 121, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol 2018, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Krotz, F.; Sohn, H.Y.; Gloe, T.; Zahler, S.; Riexinger, T.; Schiele, T.M.; Becker, B.F.; Theisen, K.; Klauss, V.; Pohl, U. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood 2002, 100, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Gazzaniga, P.P.; Violi, F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood 1998, 91, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Krotz, F.; Sohn, H.Y.; Pohl, U. Reactive oxygen species: players in the platelet game. Arterioscler Thromb Vasc Biol 2004, 24, 1988–1996. [Google Scholar] [CrossRef] [PubMed]
- Erel, O. A novel automated direct measurement method for total antioxidant capacity using a new generation, more stable ABTS radical cation. Clin Biochem 2004, 37, 277–285. [Google Scholar] [CrossRef]
- Wayner, D.D.; Burton, G.W.; Ingold, K.U.; Barclay, L.R.; Locke, S.J. The relative contributions of vitamin E, urate, ascorbate and proteins to the total peroxyl radical-trapping antioxidant activity of human blood plasma. Biochim Biophys Acta 1987, 924, 408–419. [Google Scholar] [CrossRef]
- Yeum, K.J.; Russell, R.M.; Krinsky, N.I.; Aldini, G. Biomarkers of antioxidant capacity in the hydrophilic and lipophilic compartments of human plasma. Arch Biochem Biophys 2004, 430, 97–103. [Google Scholar] [CrossRef]
Figure 1.
Inhibition of platelet aggregation by N-acetylcysteine amide (AD4), thioredoxin-mimetics (TXM-CB3, TXM-CB13, TXM-CB30), and N-acetylcysteine (NAC). PRP was incubated with the vehicle or the different compounds for 30 minutes at 37 °C with constant stirring. Subsequently, platelets were stimulated with collagen (0.5 µg/mL). Concentration-dependent inhibition of platelet aggregation induced by collagen (0.5 µg/ml) after pretreatment with (A) AD4, n =3; (B) TXM-CB3, n =4; (C) TXM-CB13, n =4; (D) TXM-CB30, n =4; (E) NAC, n =7. *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 vs collagen-stimulated PRP for ANOVA and Dunnett’s post hoc test.
Figure 1.
Inhibition of platelet aggregation by N-acetylcysteine amide (AD4), thioredoxin-mimetics (TXM-CB3, TXM-CB13, TXM-CB30), and N-acetylcysteine (NAC). PRP was incubated with the vehicle or the different compounds for 30 minutes at 37 °C with constant stirring. Subsequently, platelets were stimulated with collagen (0.5 µg/mL). Concentration-dependent inhibition of platelet aggregation induced by collagen (0.5 µg/ml) after pretreatment with (A) AD4, n =3; (B) TXM-CB3, n =4; (C) TXM-CB13, n =4; (D) TXM-CB30, n =4; (E) NAC, n =7. *p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 vs collagen-stimulated PRP for ANOVA and Dunnett’s post hoc test.
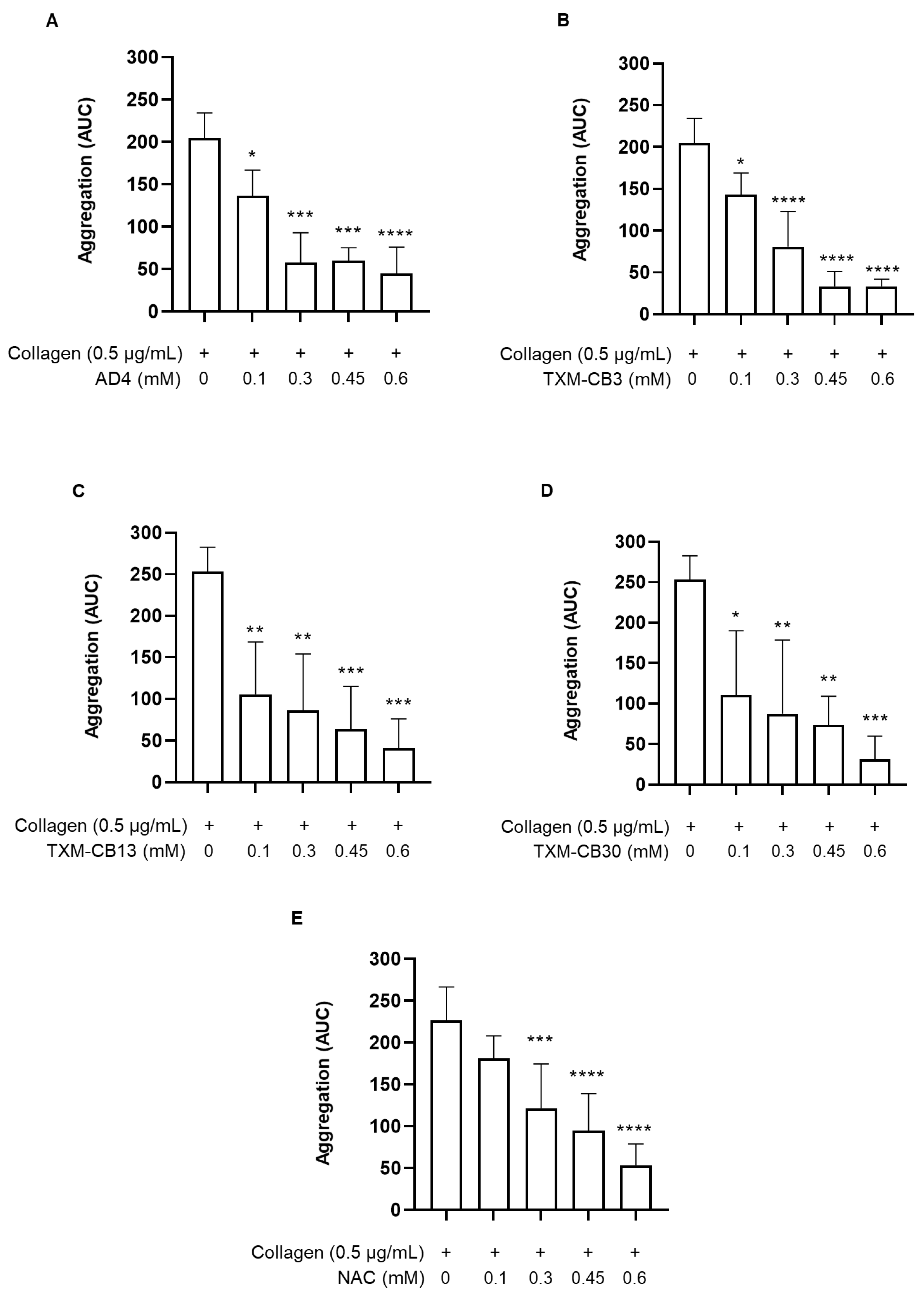
Figure 2.
Inhibition of TxB2 and 12-HETE synthesis by N-acetylcysteine amide (AD4), thioredoxin-mimetics (TXM-CB3, TXM-CB13, TXM-CB30), and N-acetylcysteine (NAC). PRP was incubated with the vehicle or the different compounds for 30 minutes at 37 °C with constant stirring. Subsequently, platelets were stimulated with collagen (0.5 µg/mL). At the end of aggregation, platelet aggregates were pelleted and the levels of (A) TxB2 and (B) 12-HETE were simultaneously measured. n =3. *p < 0.05, ** p < 0.01, vs collagen-stimulated PRP for ANOVA and Dunnett’s post hoc test.
Figure 2.
Inhibition of TxB2 and 12-HETE synthesis by N-acetylcysteine amide (AD4), thioredoxin-mimetics (TXM-CB3, TXM-CB13, TXM-CB30), and N-acetylcysteine (NAC). PRP was incubated with the vehicle or the different compounds for 30 minutes at 37 °C with constant stirring. Subsequently, platelets were stimulated with collagen (0.5 µg/mL). At the end of aggregation, platelet aggregates were pelleted and the levels of (A) TxB2 and (B) 12-HETE were simultaneously measured. n =3. *p < 0.05, ** p < 0.01, vs collagen-stimulated PRP for ANOVA and Dunnett’s post hoc test.
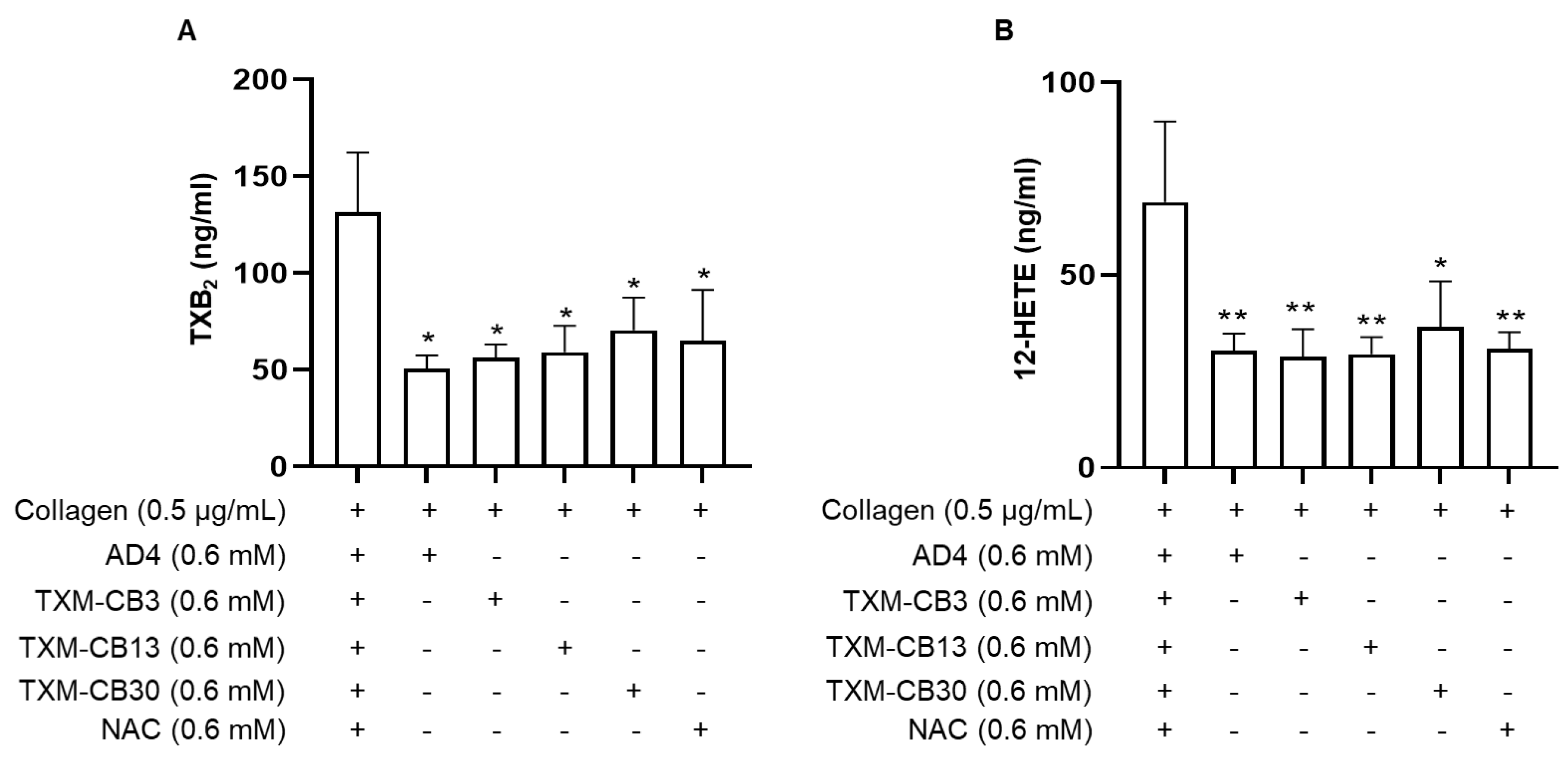
Figure 3.
N-acetylcysteine amide (AD4), thioredoxin-mimetics (TXM-CB3, TXM-CB13, TXM-CB30), and N-acetylcysteine (NAC) prolonged the closure time (CT) in PFA-200 analysis. Whole blood was incubated for 30 minutes at 37 °C and perfused in a (A) coll/epi or (B) coll/ADP cartrige of the PFA-200 system, and the CT was recorded. n =4. *p < 0.05, **** p < 0.0001, vs vehicle-incubated blood for ANOVA and Dunnett’s post hoc test.
Figure 3.
N-acetylcysteine amide (AD4), thioredoxin-mimetics (TXM-CB3, TXM-CB13, TXM-CB30), and N-acetylcysteine (NAC) prolonged the closure time (CT) in PFA-200 analysis. Whole blood was incubated for 30 minutes at 37 °C and perfused in a (A) coll/epi or (B) coll/ADP cartrige of the PFA-200 system, and the CT was recorded. n =4. *p < 0.05, **** p < 0.0001, vs vehicle-incubated blood for ANOVA and Dunnett’s post hoc test.
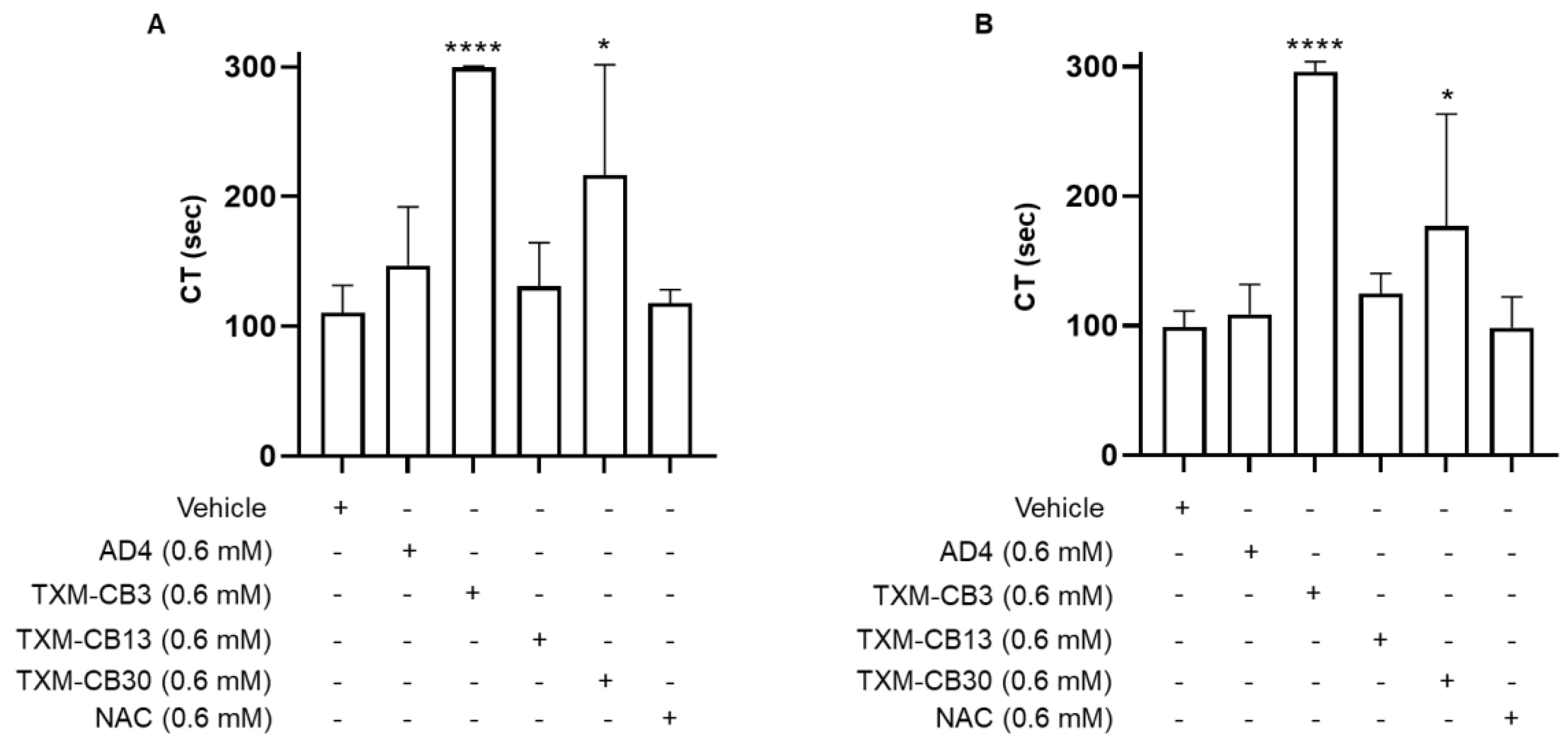
Figure 4.
N-acetylcysteine amide (AD4), TXM-CB3, and NAC did not inhibit aggregation of washed platelets. Washed platelets were preincubated with the vehicle or (A) AD4, (B) TXM-CB3, (C) NAC for 30 minutes at 37 °C with constant stirring, and subsequently stimulated with collagen (8 µg/ml). Aggregation was monitored for 6 minutes. n =7.
Figure 4.
N-acetylcysteine amide (AD4), TXM-CB3, and NAC did not inhibit aggregation of washed platelets. Washed platelets were preincubated with the vehicle or (A) AD4, (B) TXM-CB3, (C) NAC for 30 minutes at 37 °C with constant stirring, and subsequently stimulated with collagen (8 µg/ml). Aggregation was monitored for 6 minutes. n =7.
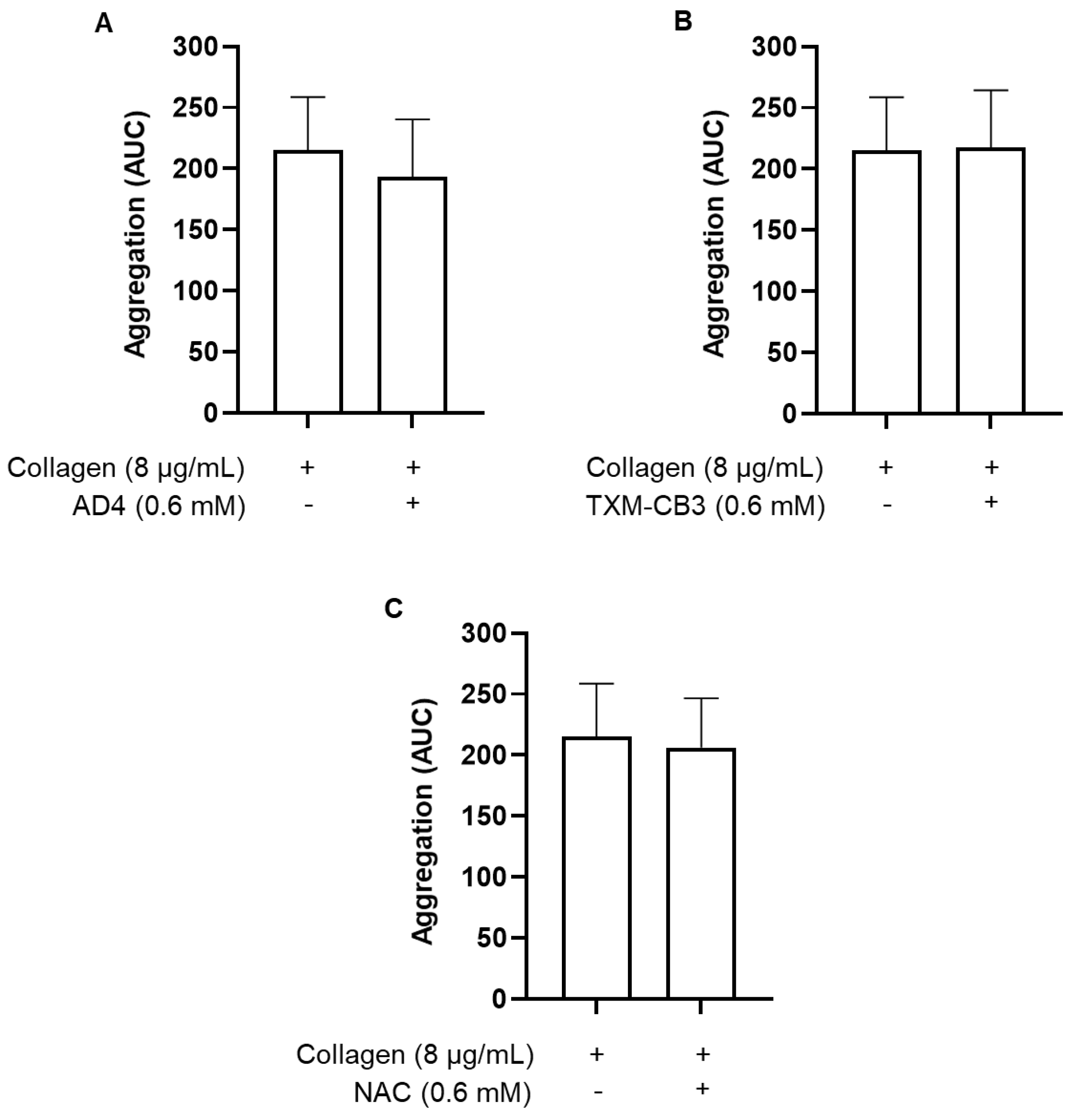
Figure 5.
Quantitation of sulfhydryl groups. PRP was preincubated with the (A) vehicle or (B) thioredoxin-mimetics (TXM-CB3), or (C) N-acetylcysteine (NAC), for 30 minutes at 37 °C with constant stirring. Platelets were stimulated with collagen (0.5 µg/mL). After 6 minutes plasma was separated by centrifugation and free sulfhydryl groups were quantified using Ellman’s reagent. n =5. * p < 0.05; *** p < 0.001; **** p < 0.0001 vs collagen-stimulated PRP.
Figure 5.
Quantitation of sulfhydryl groups. PRP was preincubated with the (A) vehicle or (B) thioredoxin-mimetics (TXM-CB3), or (C) N-acetylcysteine (NAC), for 30 minutes at 37 °C with constant stirring. Platelets were stimulated with collagen (0.5 µg/mL). After 6 minutes plasma was separated by centrifugation and free sulfhydryl groups were quantified using Ellman’s reagent. n =5. * p < 0.05; *** p < 0.001; **** p < 0.0001 vs collagen-stimulated PRP.
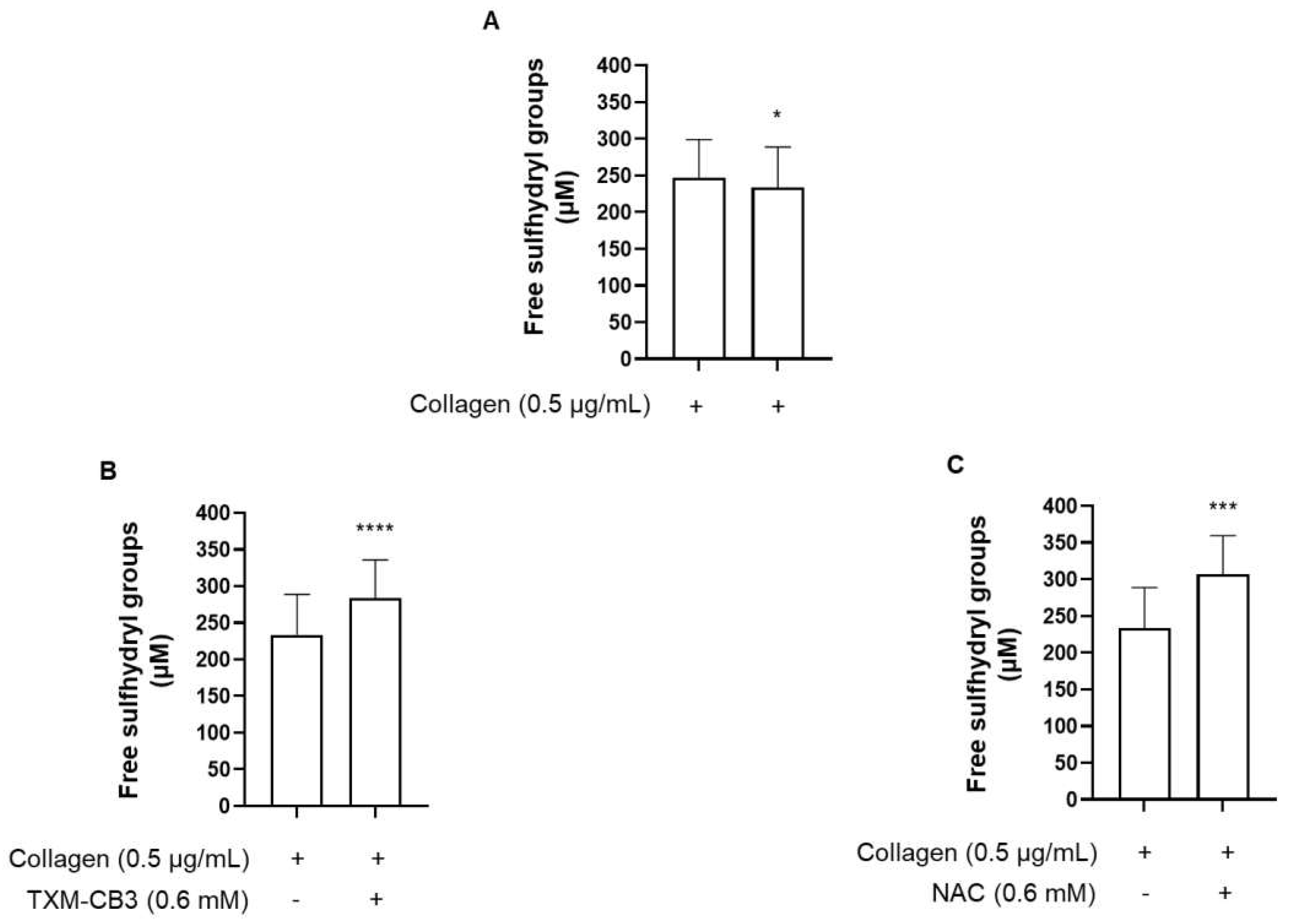
Figure 6.
Total antioxidant activity. PRP was preincubated with vehicle, TXM-CB3, or NAC for 30 minutes at 37 °C with constant stirring. Platelets were stimulated with 0.5 µg/mL collagen. After 6 minutes plasma was separated by centrifugation and antioxidant activity was detected by DCF fluorescence. Data are expressed as fluorescence units of DCF. n =5. * p < 0.05 vs unstimulated PRP (70 minutes).
Figure 6.
Total antioxidant activity. PRP was preincubated with vehicle, TXM-CB3, or NAC for 30 minutes at 37 °C with constant stirring. Platelets were stimulated with 0.5 µg/mL collagen. After 6 minutes plasma was separated by centrifugation and antioxidant activity was detected by DCF fluorescence. Data are expressed as fluorescence units of DCF. n =5. * p < 0.05 vs unstimulated PRP (70 minutes).


Table 1.
Mobile phase gradient for the determination of TxB2 and 12-HETE.
| Time (min) | % mobile phase A 50 mM Ammonium Acetate pH=8/H2O/MeOH 4:93:3 v/v/v |
% mobile phase B 50 mM Ammonium Acetate pH=8/ACN/MeOH 4:93:3 v/v/v |
|---|---|---|
| 0.0 | 85 | 15 |
| 0.2 | 85 | 15 |
| 2.0 | 65 | 35 |
| 2.5 | 15 | 85 |
| 6.0 | 15 | 85 |
| 6.5 | 85 | 15 |
| 12.0 | 85 | 15 |
Table 2.
Mass transitions.
| Q1 (m/z) | Q3 (m/z) | Name |
|---|---|---|
| 369.2 | 169.1 | TXB2 Quant |
| 369.2 | 195.2 | TXB2 Qual |
| 373.2 | 173.0 | TXB2-d4 Quant |
| 373.2 | 199.2 | TXB2-d4 Qual |
| 319.1 | 179.0 | 12HETE Quant |
| 319.1 | 301.2 | 12HETE Qual |
| 327.2 | 184.2 | 12HETE-d8 Quant |
| 327.2 | 309.3 | 12HETE-d8 Qual |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



