
Submitted:
20 June 2023
Posted:
21 June 2023
You are already at the latest version
Abstract
Background: Disorders of immunity are poorly recognised in some rare multisystem genetic con-ditions. We aim to describe syndromic features and immunological defects in children with syn-dromic primary immunodeficiencies (sPIDs).
Methods: This is a retrospective descriptive study of children aged 0-18 years with sPIDs under the care of the paediatric immunology service at the Bristol Royal Hospital for Children, United Kingdom from January 2006 to September 2021.
Results: sPIDs were identified in 36 patients. Genetic diagnoses which are not commonly associ-ated with PIDs and not included in the International Union of Immunological Societies classifica-tion were present in 7/36 (19%): Trisomy 22, Arboleda-Tham syndrome, 2p16.3 deletion syn-drome, supernumerary ring chromosome 20 syndrome, Myhre syndrome, Noonan syndrome, trichothiodystrophy/Cockayne syndrome complex. Recurrent and/or severe infections were the commonest clinical features (n=33, 92%). Approximately half had combined immunodeficiency or antibody deficiency. Commonest extra-immunological manifestations include dysmorphism (72%), disorders of nervous (78%), musculoskeletal (69%), haematology/lymphatic (58%), and gastrointestinal, hepatic/pancreatic (58%) systems.
Conclusions: Patients with sPIDs often have multiorgan involvement and some are non-immunologically mediated. There should be low threshold to clinically assess and investi-gate for disorders of immunity in any patients with syndromic features especially when they pre-sent with recurrent/severe/opportunistic infections, features of immune dysregulation, autoin-flammation or lymphoproliferation.
Keywords:
Primary immunodeficiency disorder
; genetic syndromes
; syndromic children
; multisystem disorder
; inborn errors of immunity
1. Introduction
Syndromic primary immunodeficiencies (sPID) are conditions with features of inborn errors of immunity (IEI) overlapping with other multisystem clinical manifestations which are not directly associated with the immunologic deficit [1]. Well-described examples include Wiskott-Aldrich syndrome (WAS), ataxia telangiectasia (AT) and DiGeorge syndrome (DGS). The molecular aetiology of these conditions ranges from single gene defects to chromosomal anomalies, and can help understand the pathobiology underlying the immunodeficiency, organ dysfunction and dysmorphism. The increasing availability and use of next generation sequencing (NGS) technology such as whole exome/genome sequencing (WES/WGS) has resulted in the rapid molecular characterisation of previously defined and undefined genetic disorders, including those of sPIDs [2]. For example, WGS revealed IVNS1ABP haploinsufficiency, MIM #618969 (confirmed by Western blot) in 4 patients who had long-standing multiple warts, cutaneous boils, recurrent nasal polyposis and sinusitis, bronchiectasis, inflammatory colitis, protracted Epstein-Barr virus (EBV) infection, CD4 and CD8 T cell as well as B cell lymphopaenia, hypogammaglobulinaemia, coeliac disease, retinal vasculitis, as well as non-immunological features such as achalasia and hypothyroidism [3].
Cohort descriptions of sPIDs are lacking in the literature and disorders of immunity are poorly recognised in some rare multisystem genetic conditions [4,5,6,7]. In contrast, primary immunodeficiencies (PIDs) in association with infections, inflammation, autoimmunity, allergy, lymphoproliferation and malignancy are widely reported. In order to raise awareness and better describe the association between syndromic features and immunological defects, here we describe children with a wide spectrum of sPIDs diagnosed and followed up in our institution over a period of 15 years, including genetic disorders which have been rarely associated with PID.
2. Materials and Methods
We performed a retrospective case series description of all children aged 0-18 years with sPID under the care of the paediatric immunology service at the Bristol Royal Hospital for Children, United Kingdom from January 2006 to September 2021. This department serves a population of over 5 million individuals residing in Southwest region of England, United Kingdom. Clinical data (demographics, clinical characteristics, treatments and outcomes) were identified from a local clinical database that includes all children with immunodeficiency. sPID were defined as conditions with features of immunodeficiencies overlapping with other multisystem clinical manifestations that are not always associated with an immunologic deficit [1]. Disorders of immunity were classified according to European Society for Immunodeficiencies PID definitions [1].
3. Results
During the study period, we identified 36 children meeting inclusion criteria: 22 males, 61%; 14 females, 39%. Median age was 11.4 years (range 0.6-26.8 years). IEI (n=36) and genetic (n=33) diagnoses were made at a median age of 5 years (range 0.2-17 years) and 2 years (0-16 years), respectively.
A summary of the immunological disorders and their treatment alongside genetic diagnoses is displayed in Table 1. There were 17 different genetic syndromes associated with PIDs identified. Genetic diagnoses which are not commonly associated with PIDs and not included in the International Union of Immunological Societies classification were present in 7/36 (19%): Trisomy 22, Arboleda-Tham syndrome, 2p16.3 deletion syndrome, supernumerary ring chromosome 20 syndrome, Myhre syndrome, Noonan syndrome, trichothiodystrophy/Cockayne syndrome complex.
Approximately half had combined immunodeficiency or antibody deficiency (Figure 1). Recurrent/severe infections were the commonest clinical feature (33/36, 92%). Majority of patients presented with infections before one year old (20/33; 61%), 8/33 (24%) between one and five years, and 5/33 (15%) after five years. Nine (25%) patients had bacterial sepsis. Bronchiectasis occurred in 14% (5/36): DiGeorge syndrome (DGS) (n=3); Kabuki syndrome (n=1) and cartilage hair hypoplasia (CHH) (n=1). Commonest extra-immunological manifestations include dysmorphism (72%), disorders of nervous (78%), musculoskeletal (69%), haematology/lymphatic (58%), and gastrointestinal, hepatic/pancreatic (58%) systems. Genetic conditions associated with chronic diarrhoea (n=8, 22%) were trichohepatoenteric syndrome (n=2), sideroblastic anemia with B cell immunodeficiency, periodic fevers and developmental delay (SIFD) (n=3), supernumerary ring chromosome 20 syndrome (n=1), CHH (n=1) and Wiskott-Aldrich syndrome (WAS) (n=1). Lymphoma developed in two ataxia telangiectasia patients (6%).
Antibiotic prophylaxis was started in 25 (69%) patients, immunoglobulin replacement in 30 (83%) patients, and biologic modifier therapy in 10 (28%) patients. Three children (8%) underwent hematopoietic stem cell transplantation (HSCT): WAS (n=2) and SIFD (n=1). One patient with WAS had autologous gene-corrected HSCT and one DGS patient received thymic transplant. Full descriptions of clinical characteristics, treatments and outcomes is available in online supplemental materials (Tables S1 and S2).
4. Discussion
We describe diverse immunological and non-immunological manifestations (Figure 1) of a cohort of children with sPID. One fifth of our patients had genetic disorders which were very rarely associated with IEI but not included in the International Union of Immunological Societies (IUIS) classification: Trisomy 22 and Arboleda-Tham syndrome, 2p16.3 deletion syndrome, supernumerary ring chromosome 20 syndrome, Myhre syndrome, Noonan syndrome, trichothiodystrophy/Cockayne syndrome complex [2]. Our findings raise the possibility that there could be children attending specialty clinics with rare genetic conditions who have unrecognized disorders of immunity, which is of potential clinical significance to a wide range of healthcare professionals including paediatricians, subspecialists and geneticists. Patients with IEI do not only have tendency to develop infections but also autoinflammation, autoimmunity, lymphoproliferation, malignancy and allergy. [8] We advocate that such children undergo appropriate immunological investigations if clinically indicated. Immunologists should also look beyond the 485 genetic defects described in the 2022 IUIS document as some of these rare genetic disorders with immunodeficiencies were not included in the classification. [2]
Our cohort of patients with sPIDs exhibited a variety of immunological defects predisposing them to recurrent infections, autoinflammation, autoimmunity, immune dysregulation, lymphoproliferation, malignancy and allergy. These results are consistent with recent report on initial manifestations in patients with PID where majority presented with infections (77%) and well as immune dysregulation (18%) and malignancy (0.8%). [9] The authors also described that 12% of their patients with IEI presented with syndromic features. [9] In the group of syndromic combined immune deficiencies, 56% initially presented with infections and/or immune dysregulation; but importantly, 89% of the remaining patients had syndromic features as their first presentation. [9] Patients with PIDs do not only have tendency to develop infections but also non-infectious complications such as autoinflammation, autoimmunity, lymphoproliferation, malignancy and allergy. [8,10] We propose updating this schematic to include non-immunological syndromic features (Figure 2). Patients with PIDs have overlapping features and they are not dichotomised. This concept is useful in PID diagnostic workup of patients who present in any of these forms.
Our cohort shows significant amount of preventable morbidity (e.g. bronchiectasis), adding to the argument that these children would benefit from early recognition and referral to paediatric immunologist. Prompt diagnosis of IEI and appropriate management is associated with reduced morbidity and mortality. [11] A multidisciplinary approach to monitoring for complications, preventative management and definitive treatment should be appropriately instituted early with an aim to improve outcome.
5. Conclusions
Patients with sPIDs often have multiorgan involvement and some are non-immunologically mediated. There should be low threshold to clinically assess and investigate for disorders of immunity in any patients with syndromic features especially when they present with recurrent/severe/opportunistic infections, features of immune dysregulation or malignancy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, JB; methodology, KFN, FM and JB; data curation and validation, KFN, AG, FM and JB; formal analysis, KFN, AG and FM; writing—original draft preparation, KFN; writing—review and editing, KFN, AG, FM and JB; supervision, JB; project administration, KFN. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical review and approval were waived for this study because this is an observational study as part of service evaluation. No ethical approval is required according to Health Research Authority decision tool, available at http://www.hra-decisiontools.org.uk/research/index.html.
Informed Consent Statement
Patient consent was waived because this study was part of service evaluation.
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
We would like to thank our patients and their families, and the staff of the Paediatric Immunology and Infectious Diseases department of Bristol Royal Hospital for Children for looking after these patients over the years of their follow up.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Working definitions for clinical diagnosis of PID. European Society for Immunodeficiencies Registry Working Party. 2019.
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Picard C, Puel A, Puck J, Seppanen MRJ, Somech R, Su HC, Sullivan KE, Torgerson TR, Meyts I. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2022. [CrossRef]
- Thaventhiran, J.E.D.; Allen, H.L.; Burren, O.S.; Rae, W.; Greene, D.; Staples, E.; Zhang, Z.; Farmery, J.H.R.; Simeoni, I.; Rivers, E.; et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 2020, 583, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.; Revet, I. Clinical implications of the basic defects in Cockayne syndrome and xeroderma pigmentosum and the DNA lesions responsible for cancer, neurodegeneration and aging. Mech. Ageing Dev. 2008, 129, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara H, Matsumoto H, Uematsu K, Zaha K, Sekinaka Y, Miyake N, Matsumoto N, Nonoyama S Immunodeficiency in a patient with microcephalic osteodysplastic primordial dwarfism type I as compared to Roifman syndrome. Brain Dev 2021, 43, 337-342. [CrossRef]
- Tovo PA, Davi G, Fraceschini P, Delpiano A. Thymic hormone dependent immunodeficiency in an infant with partial trisomy of chromosome 22. Thymus 1986, 8, 313-318.
- Zhu T, Gong X, Bei F, Ma L, Sun J, Wang J, Qiu G, Sun J, Sun Y, Zhang Y Primary immunodeficiency-related genes in neonatal intensive care unit patients with various genetic immune abnormalities: a multicentre study in China. Clin Transl Immunology 2021, 10, e1266. [CrossRef]
- Walter JE, Ayala IA, Milojevic D. Autoimmunity as a continuum in primary immunodeficiency. Curr Opin Pediatr 2019, 31, 851-862. [CrossRef]
- Thalhammer J, Kindle G, Nieters A, Rusch S, Seppanen MRJ, Fischer A, Grimbacher B, Edgar D, Buckland M, Mahlaoui N, Ehl S, European Society for Immunodeficiencies Registry Working P. Initial presenting manifestations in 16,486 patients with inborn errors of immunity include infections and noninfectious manifestations. J Allergy Clin Immunol. 2021. [CrossRef]
- Abinun, M. An overview of infectious complications in children on new biologic response-modifying agents. Pediatr. Health 2010, 4, 509–517. [Google Scholar] [CrossRef]
- Condino-Neto, A.; Espinosa-Rosales, F.J. Changing the Lives of People With Primary Immunodeficiencies (PI) With Early Testing and Diagnosis. Front. Immunol. 2018, 9, 1439. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Clinical and immunological features of 36 children with syndromic immunodeficiencies. (A) Severe/recurrent infections. (B) Immunological features. (C) Extra-immunological clinical manifestations.
Figure 1.
Clinical and immunological features of 36 children with syndromic immunodeficiencies. (A) Severe/recurrent infections. (B) Immunological features. (C) Extra-immunological clinical manifestations.
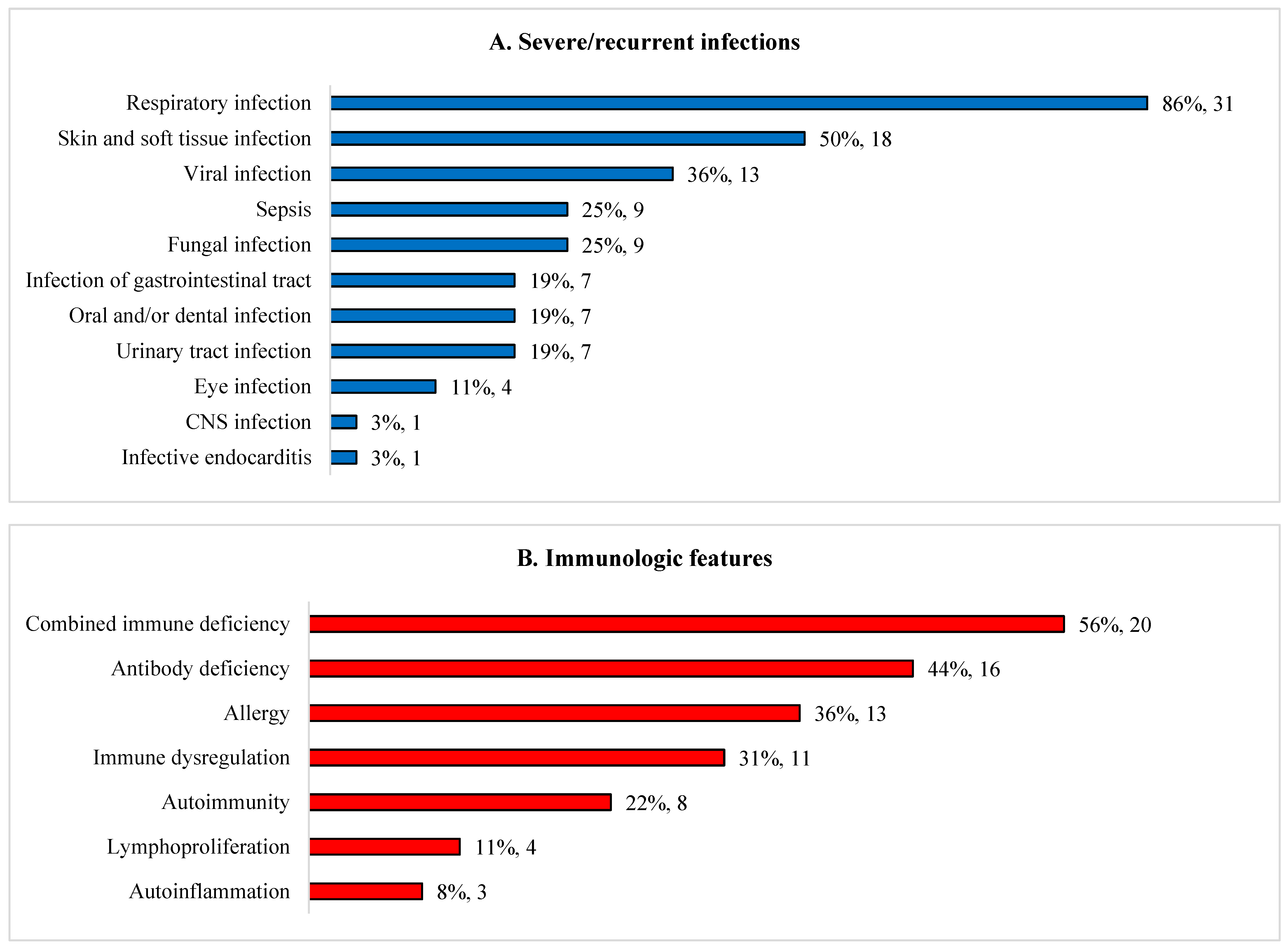
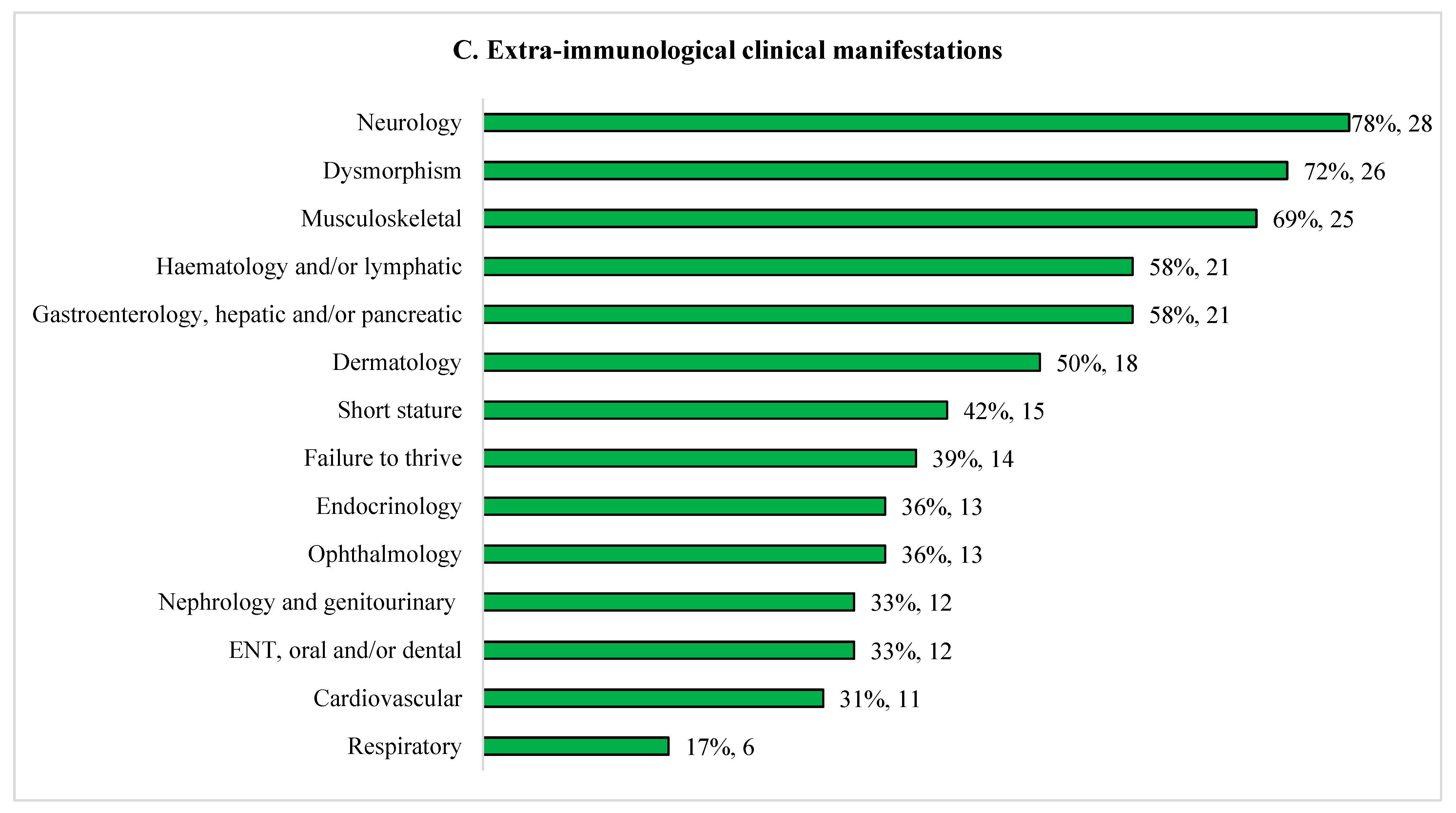
Figure 2.
The diverse disease spectrum of inborn error of immunity (adapted from Walter et al) [8].
Figure 2.
The diverse disease spectrum of inborn error of immunity (adapted from Walter et al) [8].
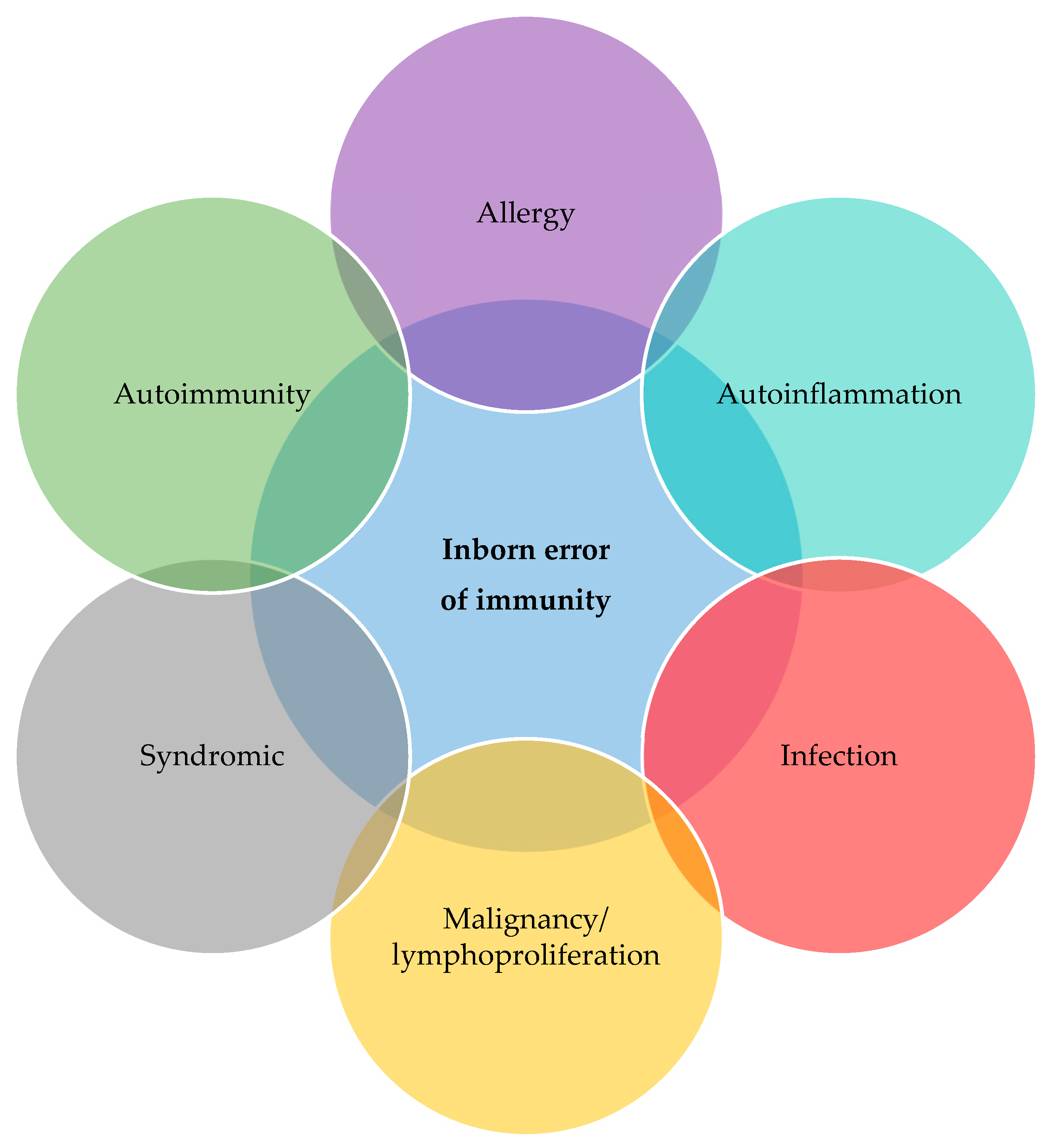

Table 1.
Immunologic features and treatment of 36 children with syndromic immunodeficiencies.
| Syndromic immunodeficiencies (n), genetic defect (OMIM number) |
Immunological and clinical manifestations (n) [1; 2] | Treatment (n) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Combined immune deficiency | Antibody deficiency | Innate immune deficiency | Recurrent / severe Infections | Autoimmunity | Auto-inflammation | Immune dysregulation | Lympho-proliferation | Malignancy | Allergy | Ig replacement | Antibiotic prophylaxis | Immuno-suppression | |
| AT (n=5), ATM (#208900) | 4 | 1 | 2 | 2 | 2 | 1 | 4 | 2 | 2 | ||||
| CHH (n=1), RMRP (#250250) | 1 | 1 | 1 | 1 | 1 | 1 | |||||||
| 2p16.3 deletion (n=1) (#614332) a | 1 | 1 | 1 | ||||||||||
| DiGeorge (n=5), 22q11.2 (#188400) | 5 | 5 | 5 | 2 | 2 | 5 | 5 | 4 | |||||
| Supernumerary ring chromosome 20 (n=1) a, b | 1 | 1 | 1 | 1 | 1 | ||||||||
| STAT3 deficiency (n=4), STAT3 (#147060) | 4 | 4 | 3 | 3 | 3 | ||||||||
| Kabuki (n=3), KMT2D (#147920) | 3 | 3 | 1 | 2 | |||||||||
| Arboleda-Tham (n=1), KAT6A (#616268) a | 1 | 1 | 1 | ||||||||||
| Roifman (n=2), RNU4ATAC (#616651) | 2 | 2 | 1 | 1 | 1 | 2 | |||||||
| MIRAGE (n=1), SAMD9 (#617053) | 1 | 1 | 1 | ||||||||||
| Myhre (n=1), SMAD4 (#139210) a | 1 | 1 | 1 | 1 | |||||||||
| Noonan (n=1), PTPN11 (#163950) a | 1 | 1 | 1 | 1 | 1 | ||||||||
| SIFD (n=3), TRNT1 (#616084) | 3 | 3 | 2 | 2 | 1 | 3 | 1 | 3 | |||||
| THES (n=2), SKIV2L (#614602)/TTC37 (#222470) | 1 | 1 | 2 | 2 | 2 | 2 | 1 | 2 | |||||
| Trisomy 22 (n=1) a, b | 1 | 1 | 1 | 1 | |||||||||
| TTD/CS (n=1), ERCC2 (#601675) a | 1 | 1 | 1 | 1 | |||||||||
| WAS (n=3), WAS (#30100) | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 1 | |||||
AT, ataxia telangiectasia; CHH, cartilage hair hypoplasia; Ig, immunoglobulin; MIRAGE (myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, enteropathy); OMIM, Online Mendelian Inheritance in Man; SIFD, sideroblastic anaemia with B cell immunodeficiency, periodic fevers and developmental delay; STAT3, signal transducer and activator of transcription 3; THES, trichohepatoenteric syndrome; TTD/CS, trichothiodystrophy/Cockayne syndrome complex; WAS, Wiskott-Aldrich syndrome. a Genetic condition not included in the 2022 International Union of Immunological Societies (IUIS) classification [2]; b OMIM number not available.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



