
Submitted:
19 June 2023
Posted:
21 June 2023
You are already at the latest version
Abstract
A simple and sensitive liquid chromatography-tandem mass spectrometry (LC– MS/MS) method has been developed for the simultaneous determination of atorvastatin (ATOR), ezetimibe (EZM) and their three metabolites including o-hydroxyl atorvastatin (o-OH ATOR), p-hydroxyl atorvastatin (p-OH ATOR), and ezetimibe-glucuronide (EZM-G) in human plasma using benzyl paraben (BP) as internal standard (IS). The analytes and IS were ionized using ESI positive ion mode (ATOR, o-OH ATOR, and p-OH ATOR), ESI negative ion mode (EZM, EZM-G, and BP), and operated in multiple reaction monitoring (MRM) mode. They were next extracted by salting-out assisted liquid–liquid extraction with acetonitrile, and then analyzed by liquid chromatography on a reversed-phase chromatographic column (50 mm × 4.6 mm; 3.5 µm), using a mixture of ac-etonitrile and an acetic acid solution (0.5%) as the mobile phase, showing high extraction effi-ciency (>70%), and a minimized matrix effect. The method was satisfactorily validated, and showed excellent linearity over wide concentration ranges of 0.06–15 ng/mL, 0.6–150 ng/mL, 0.4–100 ng/mL, 0.12–30 ng/mL, and 0.05–3 ng/mL for EZM, EZM-G, ATOR, o-OH ATOR, and p-OH ATOR, respectively.
Keywords:
atorvastatin
; ezetimibe
; o-hydroxyl atorvastatin
; p-hydroxyl atorvastatin
; ezetimibe-glucuronide
; LC-MS/MS
; SALLE
; human plasma
1. Introduction
According to the American College of Cardiology/American Heart Association (ACC/AHA)’s 2018 Guideline on the Management of Blood Cholesterol, statin therapy is the first choice of drugs to treat hypercholesterolemia. However, in patients who have not achieved their LDL-C goal despite maximal statin therapy or who are at high cardiovascular risk, ezetimibe is the first choice for combination with a statin [1]. To ensure convenience, a fixed-dose combination (FDC) of statins and ezetimibe has been marketed.
Atorvastatin, a second-generation statin, is a synthetic inhibitor of HMG-CoA reductase which affects endogenous cholesterol synthesis and subsequently increases the expression of the LDL receptor. This results in an upregulated catabolic rate for LDL-cholesterol. In the human body, atorvastatin is metabolized by the enzyme CYP3A4 to two pharmacologically active metabolites, o-OH ATOR, and p-OH ATOR [2]. In order to evaluate bioequivalence for atorvastatin products, according to the FDA, all parent compounds and metabolites must be analyzed [3]. Lots of methods were developed in this regard [4,5,6,7,8,9,10].
Until 2022, ezetimibe had been the first and only lipid-lowering drug marketed. Ezetimibe inhibits intestinal uptake of dietary and biliary cholesterol without affecting the absorption of fat-soluble nutrients. Following oral administration, in the intestines, ezetimibe is extensively metabolized (>80%) to EZM-G. Both EZM and EZM-G are rapidly absorbed and show similar bioactivity [11]. Therefore, to evaluate bioequivalence for ezetimibe products, the FDA suggests measuring both ezetimibe (unconjugated) and total ezetimibe (EZM after deconjugated) [3]. The nature of assay is direct and indirect quantification of EZM and EZM-G, respectively, through measuring ezetimibe before and after deconjugation with β-glucuronidase. As a result, the extraction efficiency was difficult to control and the measured total EZM concentration did not accurately reflect the actual concentrations of EZM and EZM-G in the human body. Despite many disadvantages, diverse studies have been conducted based on this approach [12,13,14].
Recently, a method for simultaneous determination of ezetimibe and its glucuronide metabolite in human plasma by SPE and LC-MS was developed. However, it was not validated for ATOR and its metabolites, and SPE is costly [15]. Another method using the LLE technique to assay ATOR, its two major active metabolites and EZM was published, but this method was not applied to determine EZM-G [16].
To meet high FDA requirements for evaluating bioequivalence for an FDC of atorvastatin and ezetimibe, it is essential to develop a method with only one sample preparation step for the direct simultaneous quantification of all five compounds including the two parents ATOR and EZM, and their three metabolites o-OH ATOR, p-OH ATOR, and EZM-G without deconjugation.
2. Materials and Methods
2.1. Chemicals and materials
Reference standards of EZM (99.4%), EZM-G (99.7%), o-OH ATOR and p-OH ATOR were obtained from Toronto Research chemicals (Canada), ATOR and BP were obtained from the Institute of Drug Quality Control (Ho Chi Minh city, Vietnam).
Methanol, acetonitrile (for LC-MS), ethyl acetate (for analysis) were from J.T.Baker.
Ammonium acetate (for LC-MS), ammonium formate (for LC-MS) and methyl tert-butyl ether were from Fisher.
Perchloric acid (for analysis), phosphoric acid (for analysis), acetic acid (for analysis) and formic acid (for analysis) were from Merck.
2.2. Liquid chromatography and mass spectrometry conditions
A Shimadzu HPLC system built from modules including 2 pumps (LC-30AD), an autosampler (SIL-30AC), a solvent degasser (DGU-20A3R) and a temperature-controlled compartment for columns (CTO-20AC) was used for reversed-phase chromatographic analysis.
After preparation, the sample was kept at 15 °C in an autosampler, which was kept until analyzed. The separation of analytes and IS was carried out by liquid chromatography on a reversed-phase chromatographic column Zorbax XDB C8 (50 mm × 4.6 mm, 3.5 µm), using a mixture of acetonitrile and an acetic acid solution (0.5%) as the mobile phase at 40 oC. For isocratic elution, the flow rate of the mobile phase was maintained at 1.0 mL/min, corresponding to a system pressure of 70 bar.
A triple quadrupole mass spectrometer LCMS-8040 was equipped with an electrospray ionization source. The optimized source parameters including capillary voltage, desolvation line temperature, heatblock temperature, drying gas flow, nebulizing gas flow were kept at 4500 V, 250 °C, 400 °C, 15 L/min and 3 L/min, respectively. The ESI source was operated in negative mode for EZM, EZM-G and BP, while ATOR, o-OH ATOR and p-OH ATOR were in positive mode. Thanks to being ionized by an ESI source, EZM, EZM-G, ATOR, o-ATOR and p-ATOR gave parent ions at m/z 408, 584, 559, 575 and 575, respectively. Then these ions were fragmented in a collision cell to product ions at m/z 271, 271, 440, 440 and 440, respectively.
Labsolutions version 5.82 SP1 is the software used to control all parameters of LC and MS/MS.
2.3. Calibrators and quality control samples
Calibrators and quality control (QC) samples were prepared by spiking standard solutions to plasma to get simulated samples (containing 5% solvent).
The calibrators were designed over the range of 0.06–15 ng/mL, 0.6–150 ng/mL, 0.4–100 ng/mL, 0.12–30 ng/mL and 0.05–3 ng/mL for EZM, EZM-G, ATOR, o-OH ATOR, and p-OH ATOR, respectively.
The quality control (QC) samples were prepared at four concentrations: 0.06, 0.18, 7.6, 12 ng/mL for EZM; 0.6, 1.8, 75, 120 ng/mL for EZM-G; 0.4, 1.2, 50, 80 ng/mL for ATOR; 0.12, 0.36, 15, 24 ng/mL for o-OH ATOR; and 0.05, 0.15, 1.5, 2.5 ng/mL for p-OH ATOR.
2.4. Extraction efficiency and matrix factor
To examine extraction parameters, three samples (simulated sample, spiked post-extraction sample and spiked reconstitution solvent at a concentration of 1 μg/mL) were tested. The extraction efficiency and the matrix factor were calculated using the following formulas:
where Stest, Sspiked and Ssolvent are the signals of the analyte(s) in the extracted simulated sample, the spiked post-extraction sample, and the spiked reconstitution solvent, respectively.
Extraction efficiency = RE = Stest/Sspiked ×100 (%)
Matrix factor = MX = Sspiked/Ssolvent ×100 (%)
2.5. Protocol for sample preparation
Before extraction, frozen samples were thawed to room temperature. To 1000 µL of the samples, 50 µL of the internal standard solution at a concentration of 1 μg/mL in acetonitrile was added and vortexed for 10 s. Further, 2 mL of acetonitrile was also added and vortexed. Thereafter, samples were centrifuged at 4000 rpm for 5 mins. The supernatant was carefully transferred to a fresh tube containing 2 mL of 2 M MgSO4 and vortexed. After being centrifuged at 4000 rpm at 0 oC for 5 mins, upper layer was evaporated to dryness under a stream of nitrogen. After that, the residues were reconstituted in 300 µL of methanol-water (8:2) and filtered through nylons filters with a pore size of 0.22 µm into vials. Finally, 7.0 µL of the eluant was injected in the chromatographic system.
2.6. Method validation
The method was validated according to the FDA’s regulatory requirements including: specification, selectivity, linearity, recovery and matrix effect, accuracy, precision, low limit of qualification, carry-over and stability.
3. Results and discussion
All five analytes are polar molecules with medium masses, so they can be detected by a TripleQuad detector using an ESI source. To minimize the matrix effect, the analytes need to be separated from extremely polar impurities in the matrix in chromatographic condition and by sample preparation. Regarding chromatographic condition, the polar impurities in the plasma matrix were mostly not retained on reversed-phase chromatographic columns. For sample preparation, most impurities are soluble in water and practically insoluble in non-polar solvents. Moreover, all analytes are acidic molecules and can be dissolved in non-polar solvents (logP >2). Thus, to prolong the retention time of the analytes (especially EZM-G), the study was directed towards preparing samples by liquid-liquid extraction following separation on reversed-phase chromatographic columns using an acidic mobile phase.
3.1. LC-ESI- MS/MS method development
It was observed that EZM and EZM-G were only ionized in negative mode, while ATOR, o-OH ATOR and p-OH ATOR were able to be ionized in both positive and negative mode (the positive showed higher responses than the negative). Thus, in this study, EZM and EZM-G were analyzed in negative ion mode, while ATOR, o-OH ATOR and p-OH ATOR were in positive ion mode for high sensitivity.
According to the scan mass spectra, the parent ions as deprotonated molecule ions [M-H]- at m/z 408 and 584 were found for EZM and EZM-G, respectively, while protonated molecule ions [M+H]+ at m/z 559, 575 and 575 were presented as parent ions for ATOR, o-OH ATOR and p-OH ATOR, respectively.
By SIM mode without chromatographic columns, the dependence of parent ions’ response on variable parameters including the buffer, acidic solutions and concentrations of acidic solutions was studied to select the most suitable water phase. With a fixed pH of 3.0, in comparison with the ammonium formate buffer, the ammonium acetate buffer showed higher responses for the groups of two EZM compounds while lower for the group of three ATOR compounds. However, the ammonium acetate buffer (pH 3.0) showed lower responses compared to a 0.1% solution of acetic acid (pH 2.6). Therefore, the acetic acid solution was further considered. After reviewing the concentration range of 0.1–0.5%, we selected 0.5% as the acetic acid solution’s concentration to ensure suitable sensibility for all analytes. Higher concentrations were not necessary, due to instrument durability.
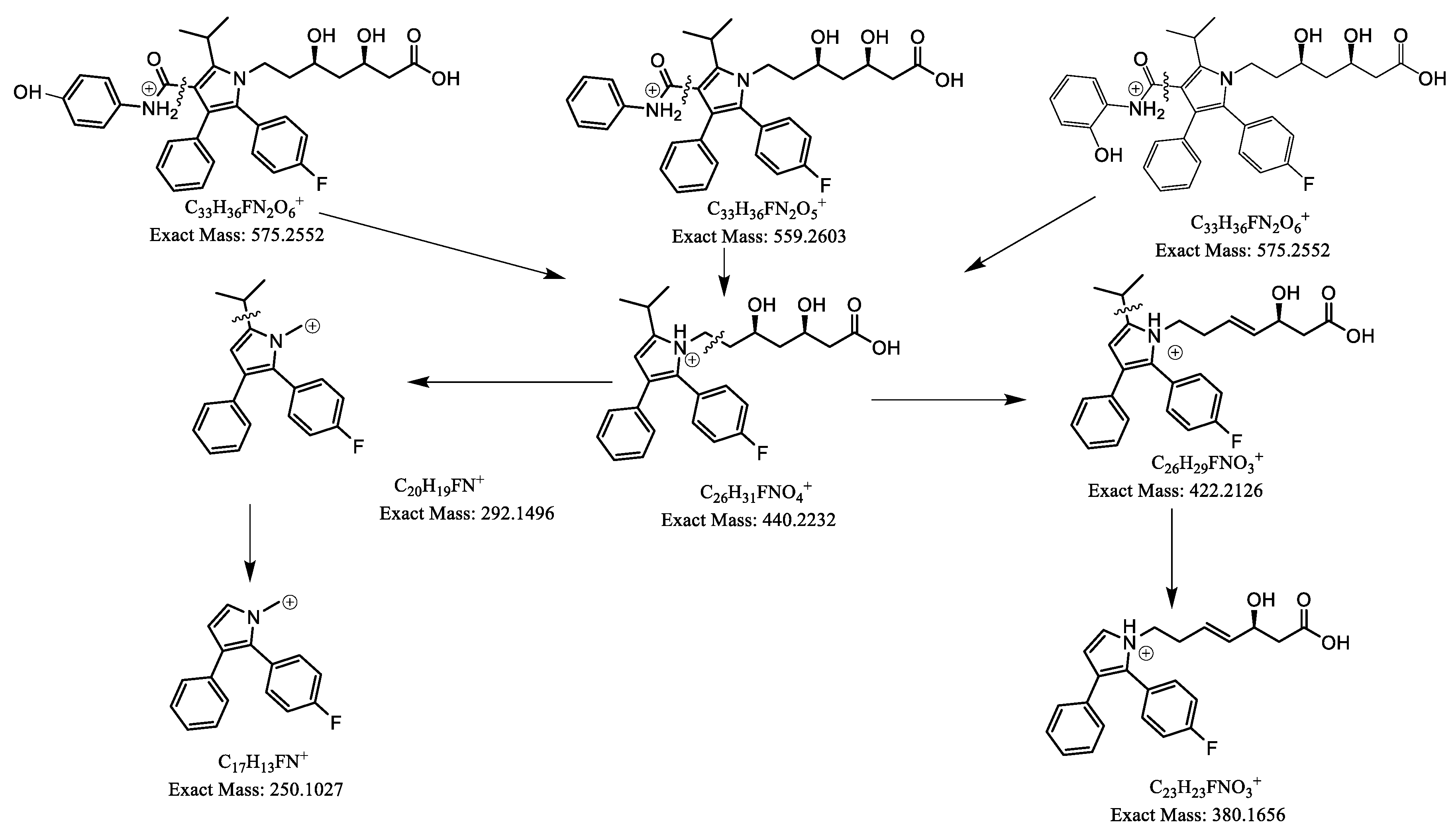
From parent ions achieved in the full scan mass spectra, the daughter ions were determined by product ion scan spectra. Fragmentations of the parent ions to daughter ions were consistent with data found on MassBank of North America (MoNA) and our proposed fragmentation mechanism (Figure 2 and Figure 3). The product ion spectra are provided in Figure 1. The parent ions and daughter ions are shown in Table 1.
Figure 1.
Production scan spectra of analytes.
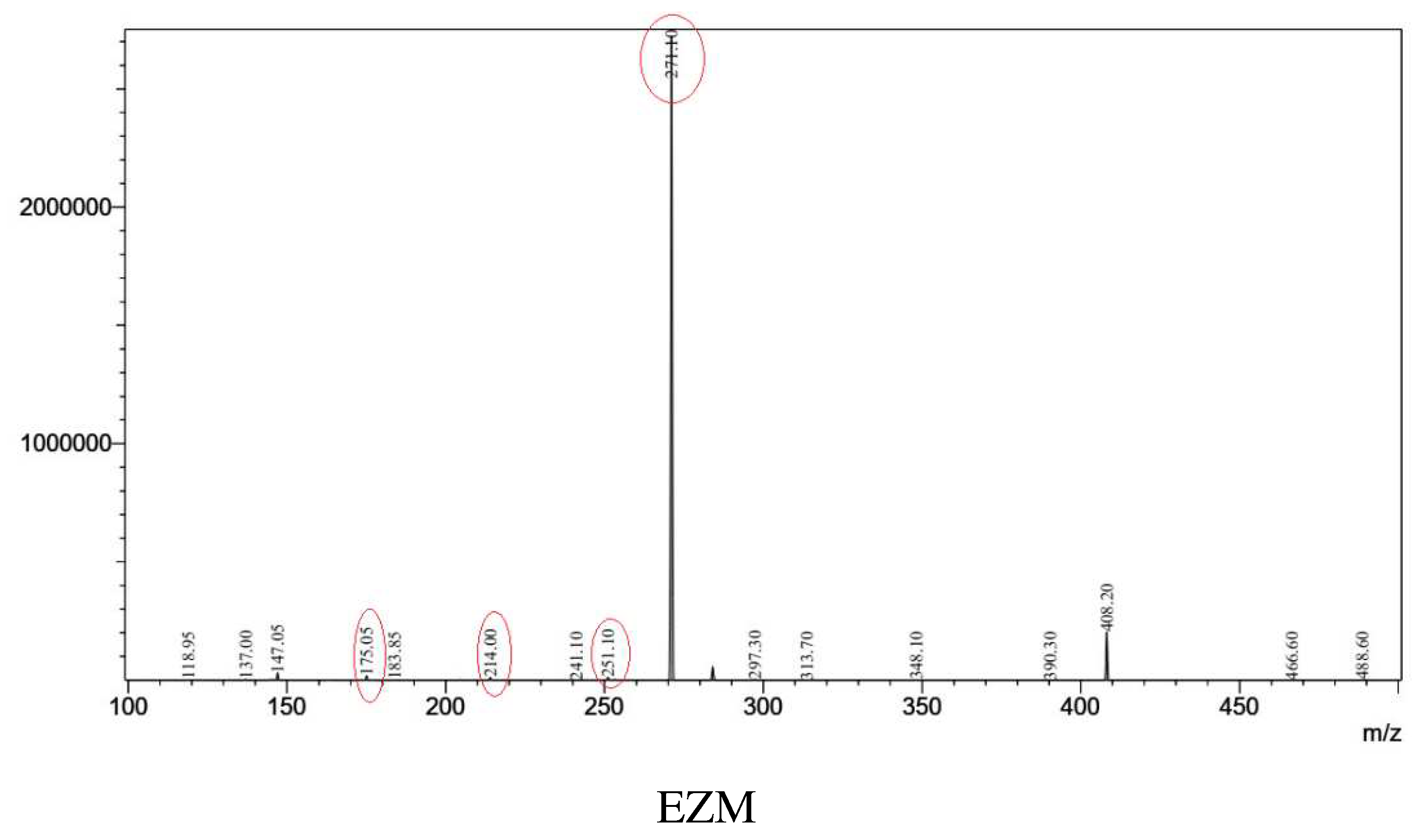
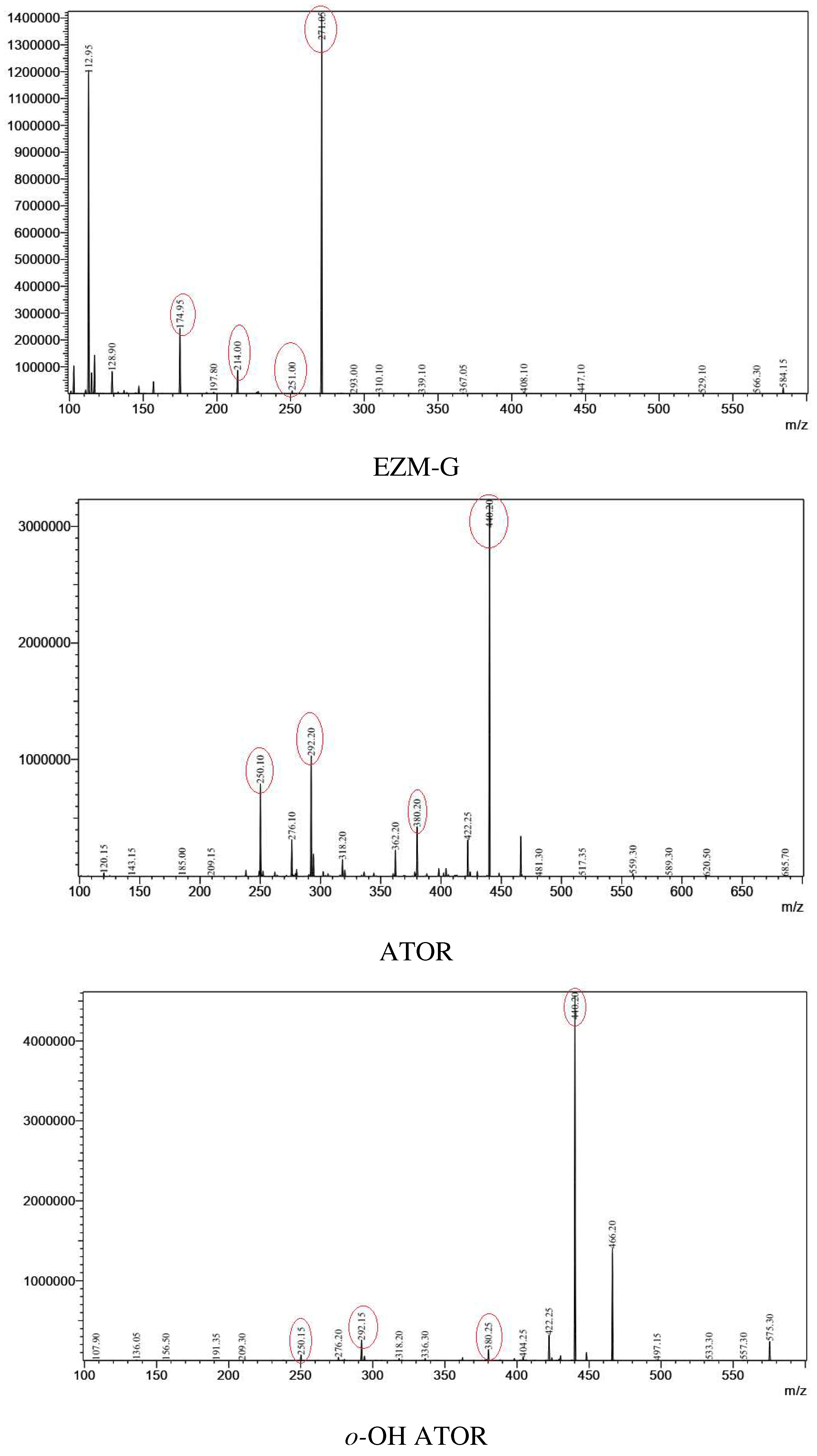
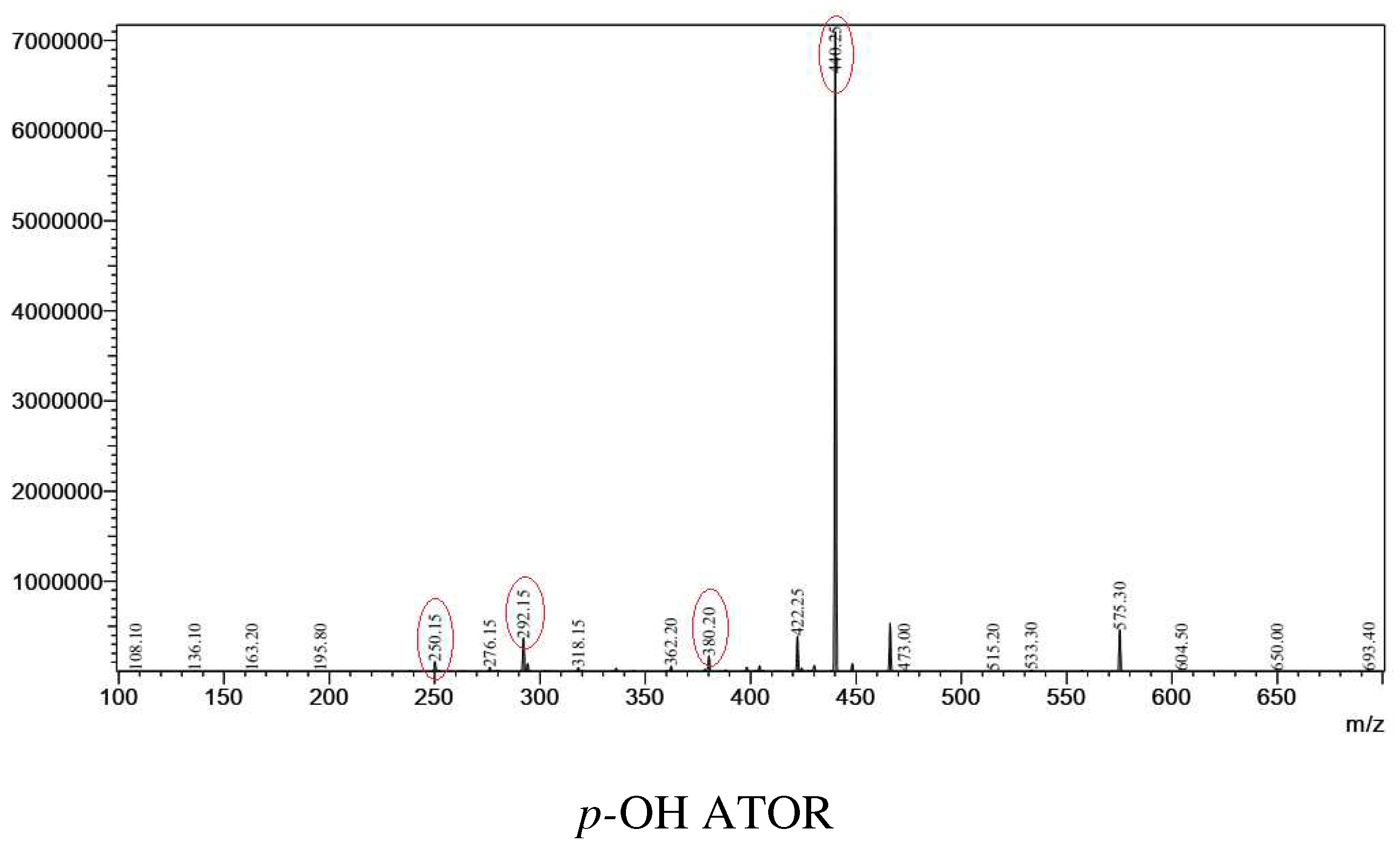

Table 1.
Critical tandem mass spectrometer parameters.
| Analytes | Sources | Parent ions (m/z) | CE (V) |
Daughter ions (m/z) | |
|---|---|---|---|---|---|
| Quantitative ions | Reference ions | ||||
| EZM | ESI (-) | 408 | 17 | 271 | 284, 214, 175 |
| EZM-G | ESI (-) | 584 | 31 | 271 | 284, 214, 175 |
| ATOR | ESI (+) | 559 | -23 | 440 | 380, 292, 250 |
| o-OH ATOR | ESI (+) | 575 | -24 | 440 | 380, 292, 250 |
| p-OH ATOR | ESI (+) | 575 | -24 | 440 | 380, 292, 250 |
Figure 2.
Proposed fragmentation mechanism for EZM and EZM-G.
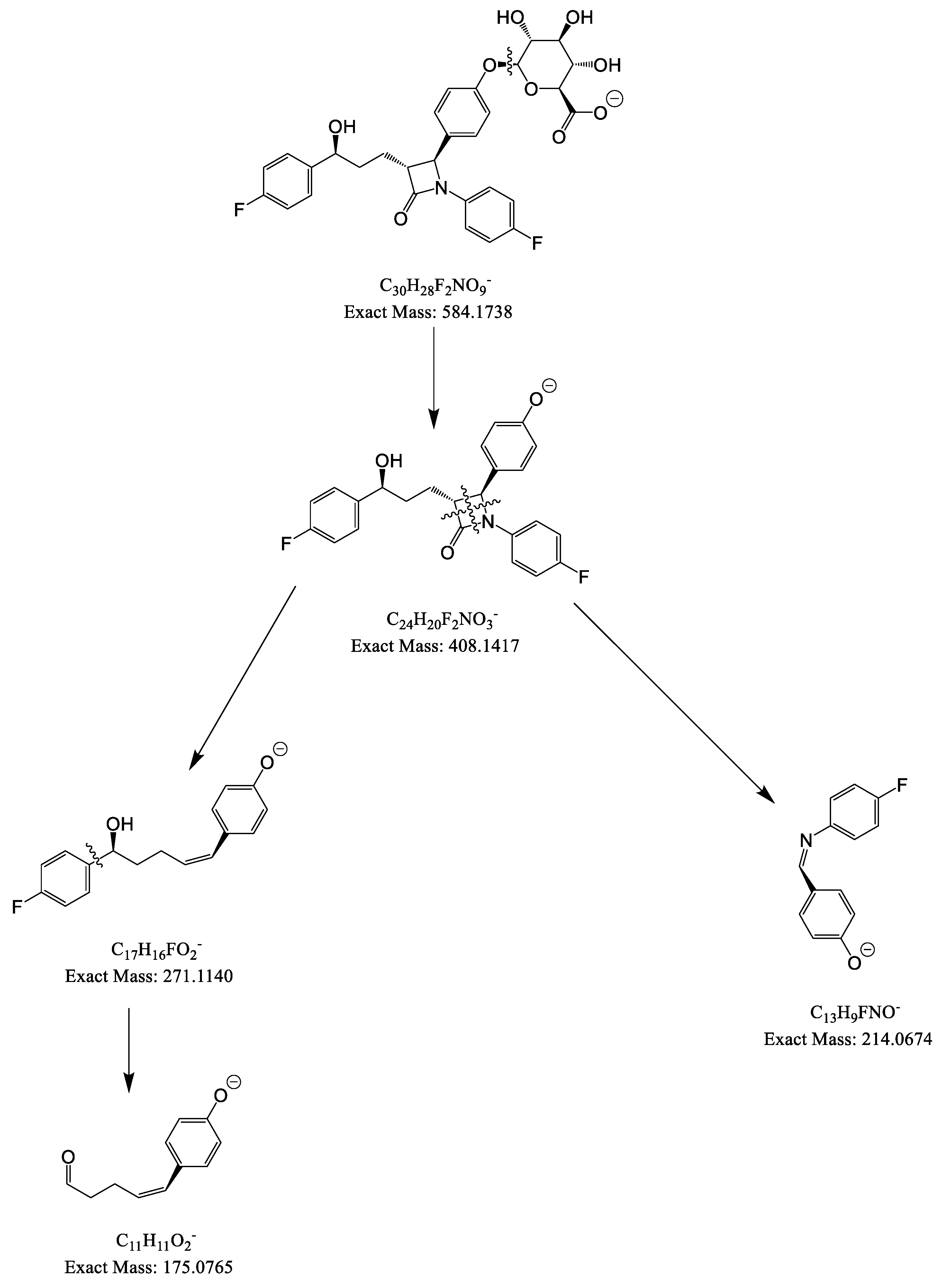
Figure 3.
Proposed fragmentation mechanism for ATOR (center), o-OH ATOR (right) and p-OH ATOR (left).
Figure 3.
Proposed fragmentation mechanism for ATOR (center), o-OH ATOR (right) and p-OH ATOR (left).
As using an acidic mobile phase, all five acidic analytes were able to be retained on the reversed-phase chromatographic column. They were separated from each other and from polar impurities in the matrix due to differences in solubility and distribution. After chromatographic parameters were considered, the samples were analyzed on a Zorbax XDB C8 (50 mm × 4.6 mm; 3.5 μm) column during 4.3 mins using a mixture of acetonitrile and a 0.5% solution of acetic acid (45:55) as mobile phase at a flow rate of 1.0 mL/min. The mobile phase and the column were warmed and kept at 40 oC to reduce system pressure.
3.2. Optimization of sample preparation
To reduce the amount of unexpected impurities analyzed on chromatographic columns, thereby minimizing the matrix effect and improving sensibility, it is necessary to carry out suitable sample preparation. Protein precipitation (PPT), liquid-liquid extraction (LLE) and solid-phase extraction (SPE) are the most common biological sample preparation methods.
The expense per sample extracted by SPE is high, making it uncommon in developing countries where simple extraction at lower cost is required. As a result, SPE was not selected in this study in spite of its applicability for extracting analytes from complicated matrices and the possibility of automation.
Due to simple and fast processing, PPT with two solvents (methanol and acetonitrile) was investigated. Eliminating almost only macromolecules like proteins in plasma, PPT gave high extraction efficiency (>60%) for all analytes but the matrix effect was extremely serious. Moreover, to improve sensibility, extracts (containing water and the solvent) ought to be enriched by drying and redissolved, which was time-consuming. Thus, PPT was not selected in this study.
Past studies extracted EZM and three compounds of the ATOR group from plasma using LLE with common solvents including methyl tert-butyl ether, ethyl acetate and a mixture of them with/without acidification. In fact, it is not possible to extract the metabolite EZM-G by these non-polar solvents (recovery < 10%), due to the high polarity and high water-solubility of this glucuronide conjugate. However, it is observed that the higher the polarity of the solvent, the higher the recovery of EZM-G. Therefore, using more polar solvents than ethyl acetate can increase the extraction efficiency of EZM-G. Nevertheless, such solvents (e.g., tetrahydrofuran, isopropanol, acetonitrile) are mixed with water to a homogeneous solution so that they cannot be applied in LLE without special effects. Though known for a long time, the salting-out effect has only recently been used to assist LC-MS/MS bioanalysis: this technique is called salting-out assisted liquid-liquid extraction (SALLE). The salting-out effect is the decrease in the solubility of substances in the aqueous phase in the presence of salts, leading to three consequences: protein precipitation, a decrease in the solubility of analytes, and phase separation which would well assist LLE.
In a typical SALLE procedure for bioanalysis, initially, analytes exist in plasma (aqueous phase). After the protein precipitate is mixed with a water-miscible solvent (acetonitrile) as a homogeneous mixture, a saturated salt solution is added to cause phase separation. After that, the extraction takes place. Involving both protein precipitation and an LLE procedure, the SALLE technique shows better extraction efficiency for polar analytes from polar matrices than typical LLE, and a reduced matrix effect in comparison to PPT.
In previous SALLE studies, acetonitrile and a 2 M solution of magnesium sulfate in water were used as the most common water-miscible organic solvent and the most effective salting-out agent, respectively. Following previous studies on these agents, we tested parameters including acidification, acetonitrile-to-plasma ratios, salt solution-to-plasma ratios to get suitable extraction efficiency and a minimum matrix effect. The reconstituted solvents were also tested for best ionization.
All analytes are acidic, so acidifying agents were added to ensure that they existed as base molecules. An HClO4 solution (4%) was tested in a volume range of 0-100 μL. The results showed that acidification did not improve the extraction efficiency and the matrix effect. Thus, acidification with a strong acid (such as HClO4) was not necessary for SALLE preparation.
It was observed that the higher the acetonitrile ratio (from 2 to 4), the higher the extraction efficiency (60–80%), and the bigger the matrix effect (100–140%). The drying time was expanded while the sensibility was not improved significantly. Thus, an acetonitrile-to-plasma ratio of 2:1 was suitable for the procedure.
The effect of salt solution-to-plasma ratios was also tested. In the range of 1 to 3, the extraction efficiency and the matrix effect did not change significantly. With a ratio of 2:1 or higher, phase separation took place completely, while a ratio lower than 2:1 did not lead to the same phenomenon. Therefore, we selected 2:1 as the MgSO4 2M-to-plasma ratio.
Methanol and acetonitrile were the most common reconstitution solvents due to their solubility, but the addition of an aqueous solution to these organic solvents could result in higher ionization. Methanol gave higher responses for all analytes than acetonitrile. Using a mixture of methanol and water gave higher responses than using only methanol, and acidification with formic acid caused lower ionization. As a result, a mixture of methanol and water was chosen as reconstitution solvent in this study.
In conclusion, typical LLE with non-polar organic solvents was effective for extracting EZM, ATOR, o-OH ATOR and p-OH ATOR, but was not for EZM-G. PPT eliminated almost all proteins while keeping analyte solutes. Therefore, the recovery was high but the matrix effect was serious and analytes were diluted, which would result in reducing sensibility. SALLE, as a combination of PPT and LLE, shows efficiency in extraction, reduces the matrix effect, increases sensitivity and shortens the analysis time.
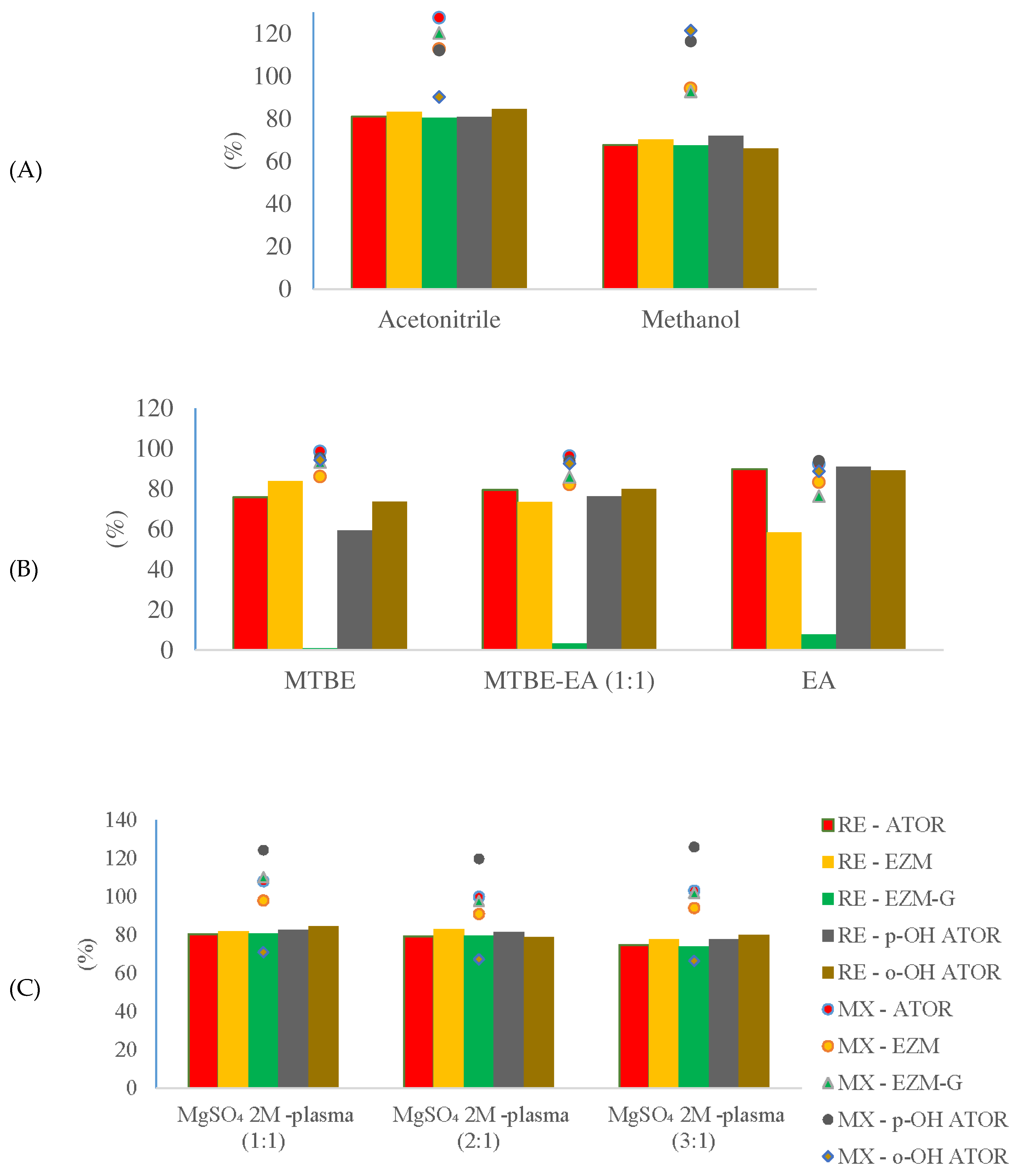
Figure 4.
Extraction efficiency (RE) and matrix factor (MX) after plasma samples were extracted by PPT (A), LLE (B) and SALLE (C).
Figure 4.
Extraction efficiency (RE) and matrix factor (MX) after plasma samples were extracted by PPT (A), LLE (B) and SALLE (C).
3.3. Method validation
3.3.1. Specifications, selectivity
All compounds in the same group (the group of EZM compounds and that of ATOR compounds) had the same quantitative ions, despite having different parent ions. Especially, o-OH ATOR and p-OH ATOR had the same parent ions and product ions. Therefore, it is essential to separate the compounds in the same group on chromatograms.
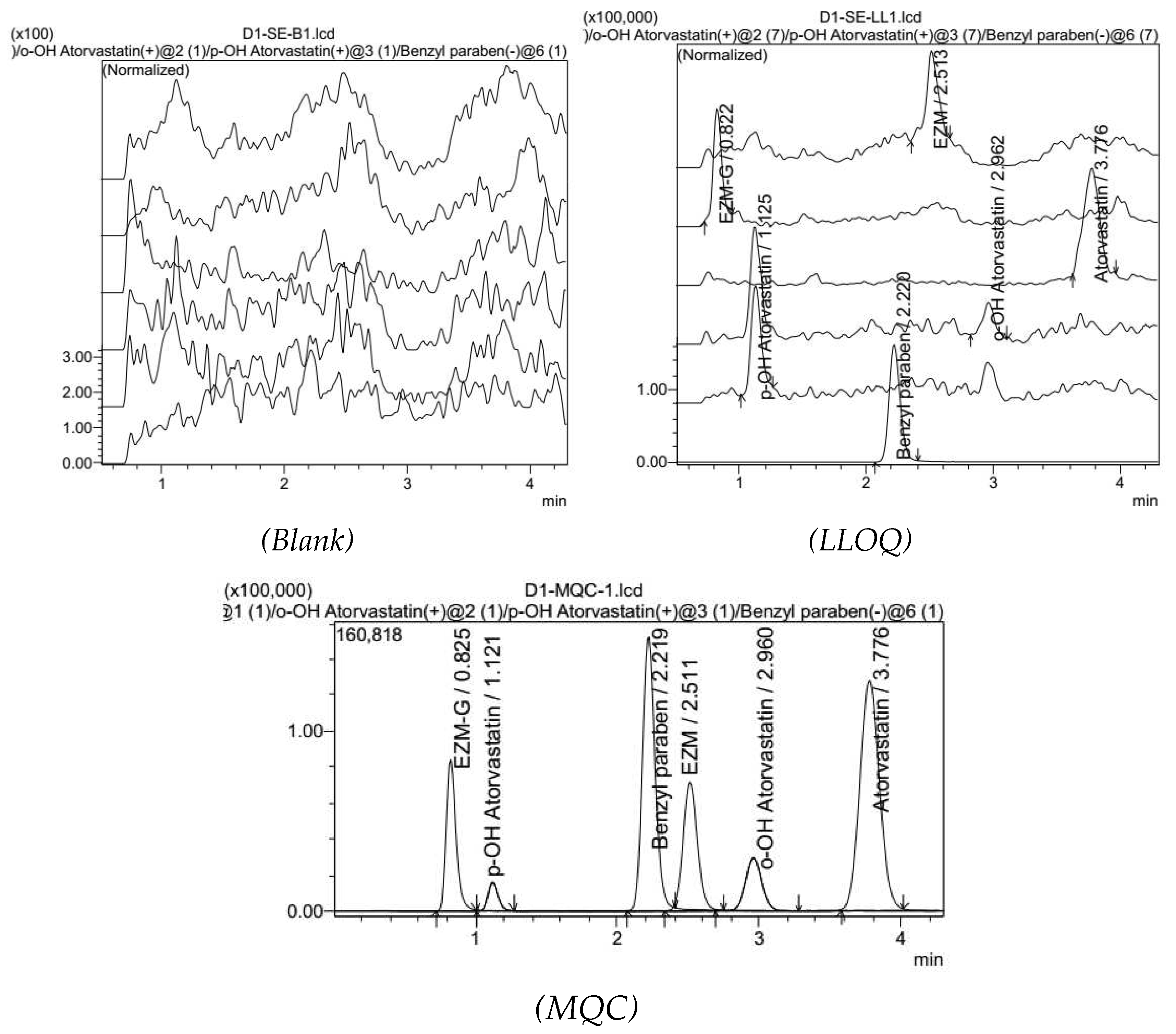
At the LLOQ concentration level, the chromatogram of our samples showed an EZM peak at Rt = 2.52, while an EZM-G peak at Rt = 0.82. Moreover, the ATOR peak was at Rt = 3.77, while those of o-OH ATOR and p-OH ATOR were at Rt = 2.97 and Rt = 1.12, respectively. All peaks were completely separated on the chromatogram.
Chromatograms of blank plasma showed no peak at the positions of our analytes and IS.
Figure 5.
Chromatograms of blank plasma sample, LLOQ and MQC level plasma samples.
3.3.2. Linearity
Calibration curves were designed to cover the range of 0.06–15 ng/mL, 0.6–150 ng/mL, 0.4–100 ng/mL, 0.12–30 ng/mL, and 0.05–3 ng/mL for EZM, EZM-G, ATOR, o-OH ATOR, and p-OH ATOR, respectively. All analytes were analyzed with linear models using a weighting factor 1/x2 that showed a strong correlation (r > 0.99). The results are provided in 0.
Table 2.
Linearity for analytes in human plasma.
| Analyte | Range (ng/mL) |
Equation ŷ = ax + b, weighting factor 1/x2 | ||
|---|---|---|---|---|
| a | b | R2 | ||
| EZM | 0.06–15 | 0.0688 | 0.0007 | 0.9974 |
| EZM-G | 0.6–150 | 0.0060 | -0.0001 | 0.9941 |
| ATOR | 0.4–100 | 0.0265 | -0.0008 | 0.9951 |
| o-OH ATOR | 0.12–30 | 0.0147 | -0.0003 | 0.9959 |
| p-OH ATOR | 0.05–3 | 0.0595 | -0.0001 | 0.9932 |
3.3.3. Recovery and matrix effect
After extracted by the SALLE procedure, all analytes and IS showed high and stable extraction efficiency. Especially, the recovery of EZM-G was above 85%. The results suggest that the extraction procedure was reproducible.
It was observed that the signal of EZM-G was suppressed (MF 85.94–91.30%) and that of p-OH ATOR was enhanced (MF 129.18–129.28%). However, the matrix effect was stable over lots of blank plasma, the accuracy and precision results were reliable, suggesting that the method could be applied in practice.
3.3.4. Accuracy and precision
The intra- and the inter-day precision data are summarized in Table 3, suggesting that our results were reliable.
3.3.5. Low limit of qualification
At concentrations of 0.06, 0.600, 0.400, 0.122 and 0.050 ng/mL for EZM, EZM-G, ATOR, o-OH ATOR and p-OH ATOR, respectively, the signal-to-noise ratios were above 5.0 while results showed good accuracy and precision. Thus, these concentrations were regarded as LLOQ of the method.
3.3.6. Carry-over
The chromatogram of the blank sample after ULOQ injection showed no peak at the positions of analytes and IS.
3.3.7. Stability
The stability of analytes and IS was extensively evaluated in stock solutions, in plasma samples and in wet extract samples under different storage conditions.
Both stock solutions and plasma samples were kept stable after stressed in bench-top condition at room temperature for up to 6 hours, and after a minimum of 67 days at ultralow refrigerated temperature (<-70 °C). Plasma samples were also stable after three freeze-thaw cycles. Accuracy and precision results were reliable after the wet extract samples were kept at 15 oC in an autosampler for 45 hours.
The results of different stability experiments are shown in Table 4.
3.4. Comparison to previous analytical methods
The important characteristics advantage of proposed SALLE-LC-MS/MS method compared to some previously reported methods are shown in Table 5. The most remarkable advantage of our method is the ability to simultaneously quantify 5 analytes with only one sample preparation step without deconjugation. As a result, the method could save sample volume and sample preparation time. Moreover, all analytes showed high and stable extraction efficiency making method sensitive and reproducible.
4. Conclusions
A simple analytical procedure has been developed and validated for the direct simultaneous quantification of all five compounds including the parent compounds ATOR and EZM, and their three metabolites o-OH ATOR, p-OH ATOR and EZM-G without deconjugation. This method can be used in in vivo bioequivalence assessment of fixed-dose combination products containing atorvastatin and ezetimibe according to the FDA’s requirements.
Author Contributions
Conceptualization, Nai N. Chuong and Tuan Duc Nguyen; data curation, Nai N. Chuong.; formal analysis, T.Nguyen Nguyen Le; investigation, T.Nguyen Nguyen Le; methodology, T.Nguyen Nguyen Le.; project administration, Nai N. Chuong; supervision, Tuan Duc Nguyen.; validation, T.Nguyen Nguyen Le; writing—original draft, T.Nguyen Nguyen Le; writing—review and editing, Nai N. Chuong and Tuan Duc Nguyen. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- ACC/AHA. Clinical practice guideline: 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol. 2018.
- Lennernäs, Hans. Clinical pharmacokinetics of atorvastatin. Clinical pharmacokinetics 2003, 42, 1141–1160. [CrossRef] [PubMed]
- FDA. Draft Guidance on Atorvastatin Calcium and Ezetimibe. 2014.
- Macwan, Joyce S., et al. Development and validation of a sensitive, simple, and rapid method for simultaneous quantitation of atorvastatin and its acid and lactone metabolites by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Analytical and bioanalytical chemistry 2011, 400, 423–433. [CrossRef] [PubMed]
- Ghosh, Chinmoy, et al. Simultaneous estimation of atorvastatin and its two metabolites from human plasma by ESI-LC-MS/MS. Drug Testing and Analysis. 2011, 3, 352–362. [Google Scholar] [CrossRef] [PubMed]
- El-Zailik, Asma, et al. Simultaneous LC–MS/MS analysis of simvastatin, atorvastatin, rosuvastatin and their active metabolites for plasma samples of obese patients underwent gastric bypass surgery. Journal of pharmaceutical and biomedical analysis 2019, 164, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Hermann, Margaret, H. Christensen, and J. L. E. Reubsaet. Determination of atorvastatin and metabolites in human plasma with solid-phase extraction followed by LC–tandem MS. Analytical and bioanalytical chemistry 2005, 382, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, Ramakrishna VS, et al. Simultaneous quantification of atorvastatin and active metabolites in human plasma by liquid chromatography–tandem mass spectrometry using rosuvastatin as internal standard. Biomedical chromatography 2006, 3, 924–936. [Google Scholar] [CrossRef]
- Partani, Pankaj, et al. Simultaneous quantitation of atorvastatin and its two active metabolites in human plasma by liquid chromatography/(–) electrospray tandem mass spectrometry. Journal of pharmaceutical analysis. 2014, 4, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Ying, et al. Development and validation of a liquid chromatography–tandem mass spectrometry method for simultaneous determination of amlodipine, atorvastatin and its metabolites ortho-hydroxy atorvastatin and para-hydroxy atorvastatin in human plasma and its application in a bioequivalence study. Journal of Pharmaceutical and Biomedical Analysis 2013, 83, 101–107. [Google Scholar] [CrossRef]
- Kosoglou, Teddy, et al. Ezetimibe: a review of its metabolism, pharmacokinetics and drug interactions. Clinical pharmacokinetics 2005, 44, 467–494. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, Gholamreza, et al. Application of one-step liquid chromatography–electrospray tandem MS/MS and collision-induced dissociation to quantification of ezetimibe and identification of its glucuronated metabolite in human serum: A pharmacokinetic study. Journal of Chromatography B. 2010, 878, 2789–2795. [Google Scholar] [CrossRef]
- Bae, Jung-Woo, et al. Analytical LC-MS/MS method for ezetimibe and its application for pharmacokinetic study. Journal of liquid chromatography & related technologies. 2012, 35, 141–152. [CrossRef]
- Danafar, Hossein, and Mehrdad Hamidi. A rapid and sensitive LC–MS method for determination of ezetimibe concentration in human plasma: application to a bioequivalence study. Chromatographia 2013, 76, 1667–1675. [CrossRef]
- Guo, Lin, et al. Simultaneous determination of ezetimibe and its glucuronide metabolite in human plasma by solid phase extraction and liquid chromatography–tandem mass spectrometry. Journal of Chromatography B 2015, 986, 108–114. [CrossRef]
- Elkady, Ehab F., et al. Bioanalytical LC–MS/MS method for simultaneous estimation of atorvastatin, its major active metabolites and ezetimibe. Bioanalysis 2022. [CrossRef]
Table 3.
Accuracy, precision, recovery and matrix effect of all analytes and IS.
| Analyte | Conc. added (ng/mL) | Accuracy (%) Mean (CV) |
Recovery (n=6) (%) |
Matrix effect (n=6) | ||
|---|---|---|---|---|---|---|
| Intraday (n=6) |
Interday (3 days, n=18) |
MF (%) | IS-normalized MFs (%) | |||
| EZM | 0.061 | 111.69 (5.86) | 98.77 (11.79) | - | - | - |
| 0.184 | 105.77 (4.69) | 103.09 (6.01) | 68.57 ± 1.91 | 106.58 ± 9.47 | 113.5 ± 9.46 | |
| 7.664 | 103.17 (1.66) | 101.49 (2.20) | 82.62 ± 3.86 | - | - | |
| 12.263 | 102.17 (1.89) | 101.28 (3.61) | 82.07 ± 3.28 | 90.98 ± 1.82 | 100.56 ± 2.19 | |
| EZM-G | 0.600 | 102.32 (6.65) | 97.09 (8.84) | - | ||
| 1.801 | 96.13 (3.33) | 99.91 (5.76) | 91.50 ± 5.19 | 85.94 ± 1.13 | 75.72 ± 5.99 | |
| 75.029 | 96.19 (2.41) | 95.29 (2.33) | 85.61 ± 4.80 | - | ||
| 120.046 | 96.42 (2.99) | 94.45 (2.78) | 87.10 ± 2.68 | 91.30 ± 1.88 | 90.79 ± 2.35 | |
| ATOR | 0.400 | 112.68 (4.05) | 104.52 (7.73) | - | - | - |
| 1.200 | 99.00 (3.86) | 103.06 (5.66) | 82.09 ± 4.23 | 104.03 ± 1.95 | 110.78 ± 2.95 | |
| 50.000 | 98.53 (1.62) | 102.79 (3.33) | 81.48 ± 4.26 | - | - | |
| 79.999 | 96.56 (2.81) | 100.98 (5.39) | 81.39 ± 3.09 | 98.24 ± 1.74 | 108.59 ± 2.25 | |
| o-OH ATOR | 0.122 | 103.51 (14.18) | 99.17 (11.50) | - | - | - |
| 0.366 | 107.71 (4.47) | 102.80 (7.43) | 74.37 ± 3.27 | 103.11 ± 3.58 | 93.07 ± 3.08 | |
| 15.261 | 113.82 (0.38) | 108.33 (6.83) | 75.37 ± 4.13 | - | - | |
| 24.418 | 111.33 (2.31) | 107.12 (5.75) | 77.36 ± 3.55 | 99.76 ± 1.73 | 91.87 ± 1.14 | |
| p-OH ATOR | 0.050 | 98.56 (5.22) | 101.98 (6.18) | - | - | - |
| 0.149 | 98.80 (4.31) | 100.76 (7.85) | 80.69 ± 4.27 | 129.18 ± 5.11 | 137.56 ± 5.67 | |
| 1.548 | 92.51 (1.81) | 101.24 (8.19) | 80.13 ± 4.80 | - | - | |
| 2.476 | 89.56 (0.75) | 101.46 (9.99) | 83.02 ± 2.11 | 129.28 ± 1.32 | 142.9 ± 0.86 | |
| IS | 50.000 | 80.01 ± 3.35* | 92.19 ± 1.93 ** | - | ||
* n = 18; ** n = 12.
Table 4.
Stability of analytes and IS in stock standard, plasma and wet extraction.
| Compound | Stock standard solution | Plasma | Wet extraction | |||||
|---|---|---|---|---|---|---|---|---|
| Conc. (μg/mL) |
Bench-top | Long-term | Conc. (ng/mL) | Bench-top | Freeze-thaw | Long-term | 45h at 15 oC in an autosampler | |
| EZM | 187.5 | 100.52 (0.82) | 100.80 (0.92) | 0.184 | 104.07 (4.45) |
106.50 (4.92) |
103.86 (6.74) | 103.78 (3.17) |
| 12.263 | 104.91 (0.75) |
104.54 (1.06) |
100.52 (0.71) | 105.45 (1.34) |
||||
| EZM-G | 100 | 94.23 (2.66) | 106.14 (0.89) | 1.801 | 93.76 (3.47) |
93.96 (5.44) |
98.06 (3.58) | 97.92 (5.02) |
| 120.046 | 93.64 (1.59) |
92.86 (1.49) |
100.70 (2.09) | 92.30 (2.43) |
||||
| ATOR | 500 | 103.16 (2.13) | 99.88 (0.67) | 1.200 | 100.87 (2.86) |
102.03 (2.98) |
98.61 (1.71) | 106.52 (3.98) |
| 79.999 | 105.99 (0.62) |
106.44 (1.03) |
99.80 (4.77) | 107.29 (0.85) |
||||
| o-OH ATOR | 60 | 98.66 (2.76) | 100.81 (0.71) | 0.366 | 94.46 (4.53) |
94.54 (3.64) |
88.88 (91.61) | 92.62 (5.47) |
| 24.418 | 97.09 (0.59) |
96.53 (1.11) |
103.76 (6.70) | 97.36 (1.35) |
||||
| p-OH ATOR | 120 | 101.50 (5.34) | 106.08 (1.36) | 0.149 | 100.57 (6.75) |
92.98 (7.24) |
100.39 (2.51) | 101.55 (8.08) |
| 2.476 | 100.04 (2.30) |
99.54 (1.04) |
108.85 (2.27) | 99.45 (2.16) |
||||
| IS | 100 | 101.88 (2.03) | 98.97 (1.00) | 50 | - | - | - | 97.47 (1.06) |
Table 5.
Comparison between the proposed SALLE-LC-MS/MS method and some reported methods.
| Ref. | Run time | Extraction technique | Analyte | LLOQ (ng/mL) | Extraction efficiency (%) |
|---|---|---|---|---|---|
| 4 | 7 mins | PPT | ATOR | 0.05 | 88.6–111 |
| o-OH ATOR | 0.05 | ||||
| p-OH ATOR | 0.05 | ||||
| 5 | 3.5 mins | SPE | ATOR | 0.05 | 66.18 |
| o-OH ATOR | 0.05 | 45.36 | |||
| p-OH ATOR | 0.05 | 54.01 | |||
| 6 | 5 mins | LLE | ATOR | 0.25 | 96.94-100.37 |
| o-OH ATOR | 0.25 | 92.15-97.71 | |||
| p-OH ATOR | 0.25 | 96.97-99.17 | |||
| 7 | 20 mins | SPE | ATOR | 0.5 | 53 – 78 |
| o-OH ATOR | 1 | ||||
| p-OH ATOR | 0.5 | ||||
| 8 | 3 mins | LLE | ATOR | 0.1 | 51.0-57.3 |
| o-OH ATOR | 0.1 | 46.8-54.3 | |||
| p-OH ATOR | 0.1 | 61.6-68.8 | |||
| 9 | 6 mins | SPE | ATOR | 0.05 | 76.3-78.0 |
| o-OH ATOR | 0.05 | 73.1-75.1 | |||
| p-OH ATOR | 0.05 | 72.6-75.7 | |||
| 10 | 6 mins | LLE | ATOR | 0.035 | 77.23-82.69 |
| o-OH ATOR | 0.02 | 76.39-81.96 | |||
| p-OH ATOR | 0.015 | 78.24-80.29 | |||
| 12 | 5 mins | PPT | EZM | 10 | Not tested |
| EZM-G | identification | Not tested | |||
| 13 | 4.5 mins | LLE | EZM | 0.075 | 61.6 |
| LLE following deconjugation | Total EZM | 1 | 42.0 | ||
| 14 | 10 mins | LLE | EZM | 0.05 | 96.21-97.27 |
| 15 | 5.5 mins | SPE | EZM | 0.1 | 65.3-72.2 |
| EZM-G | 0.5 | 58.6-61.2 | |||
| 16 | LLE | ATOR | 0.5 | 84.94 | |
| o-OH ATOR | 0.5 | 85.46 | |||
| p-OH ATOR | 0.2 | 105.46 | |||
| EZM | 0.2 | 85.2 | |||
| This study | 4.3 mins | SALLE | ATOR | 0.4 | 81.39-82.09 |
| o-OH ATOR | 0.12 | 74.37-77.36 | |||
| p-OH ATOR | 0.05 | 80.13-83.02 | |||
| EZM | 0.06 | 68.57-82.62 | |||
| EZM-G | 0.6 | 85.61-91.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



