
Preprint
Review
Chronic Granulomatous Disease (CGD): Pathogens and Management
Altmetrics
Downloads
227
Views
59
Comments
0


Submitted:
21 June 2023
Posted:
21 June 2023
You are already at the latest version
Alerts
Abstract
Chronic granulomatous disease (CGD) is a primary immunodeficiency caused by a defect in the phagocytic function of the innate immune system owing to mutations in genes encoding the five subunits of the nicotinamide adenine dinucleotide (NADPH) oxidase enzyme complex. This review aimed to provide a comprehensive approach to the pathogens associated with chronic granulomatous disease (CGD) and its management. The topics covered included common pathogens infecting CGD patients, granulomas, and CGD-related infectious diseases, including invasive aspergillosis, Mulch pneumonitis, and liver abscesses. In addition, we also covered the differential diagnosis of CGD and its management. Patients with CGD, often children, have recurrent life-threatening infections and may develop infectious or inflammatory complications. The most common microorganisms observed in the patients with CGD are Staphylococcus aureus, Aspergillus spp, Candida spp, Nocardia spp, Burkholderia spp, Serratia spp, and Salmonella spp. Triple-dose antimicrobial prophylaxis with empiric regimens is recommended. Therefore, therapeutics against this disease must be optimised, considering the lack of curative therapies and the long-term side effects of prophylactic regimens. Consequently, allogeneic haematopoietic stem cell transplantation from a human leukocyte antigen-identical donor has been proven to cure CGD.
Keywords:
Subject: Medicine and Pharmacology - Immunology and Allergy
1. Introduction
Chronic granulomatous disease (CGD) is a rare inherited primary immunodeficiency, previously described as a “Fatal Granulomatous disease of childhood” owing to the early death of children with this condition [1]. A schematic representation of the neutrophil effector functions required to achieve an adequate primary immune defense is displayed in Figure 1 [2]. Neutrophils develop in the bone marrow; band neutrophils are released into circulation and subsequently travel to tissues and organs to fight infections using various mechanisms, including phagocytosis, production of reactive oxygen species, and release of antimicrobial peptides, to destroy pathogens [2].
CGD is caused by the impaired phagocytic function of the innate immune system cells owing to mutations in genes encoding the five subunits of the nicotinamide adenine dinucleotide (NADPH) oxidase enzyme complex[3,4,5,6]. The normal respiratory burst process is critical in killing pathogens, manifesting as CGD. CGD’s inability to produce reactive oxygen species leads to pathognomonic systemic granuloma formation, increased susceptibility to recurrent and severe opportunistic bacterial and fungal infections, unrestrained inflammation, and autoimmunity. Since the condition was first described, there have been several improvements in treatment, such as antibacterial, antifungal, immunomodulatory, and haematopoietic stem cell transplantation, which extended the life expectancy of patients [7].
The severity of the phenotypes can vary depending on the mode of genotype inheritance, which is most commonly the X-linked type, followed by the autosomal recessive type [8]. The functional NADPH oxidase complex comprises five subunits: two are localised in the cell membrane during the resting phase, and three are localised in the cytoplasm. The two membrane-bound subunits are gp91phox and p22phox (Figure 2). These proteins form a heterodimeric complex (cytochrome b558). The cell membrane’s contact with a pathogen activates the protein complex, and three cytoplasmic subunits (p47phox, p67phox, and p40phox) form a heterotrimer translocating to cytochrome b558 [3].
According to data from the United States and European nations, approximately 65 % of patients with CGD have a molecular defect in CYBB (most are hemizygous males). Autosomal recessive CGD accounts for approximately 30 % of all CGD cases. Molecular defects in any of these five genes (CYBB for gp91phox (located on the X chromosome), CYBA for p22phox, NCF1 for p47phox, NCF2 for p67phox, and NCF4 for p40phox) can occur in 90 % of patients with CGD. They harbour mutations in the CYBB (gp91phox) or NCF1 (p47phox) genes. Mutations in either the membrane or cytosolic domain disrupt the respiratory burst in phagocytes[9], as displayed in Figure 3. The clinical characteristics and rigour of the disease, as well as patient survival, depend significantly on the gene, type, and position of the mutation [3]. It was demonstrated for the first time that EROS (CYBC1/C17ORF62) regulates abundance of the gp91phox-p22phox heterodimer of the phagocyte NADPH oxidase in human cells. EROS mutations are a novel cause of chronic granulomatous disease[5]. Encoding the p40phox subunit of the phagocyte NADPH oxidase, have been described in only 1 patient. However, a report on 24 p40phox-CGD patients from families in 8 countries exist. These individuals display 8 different in-frame or out-of-frame mutations of NCF4, which are homozygous almost in the totality of the families [10].
Patients usually present with fever, malaise, or weight loss. Perirectal abscesses are also typical in patients with CGD and can persist for years despite aggressive antimicrobial treatments and intense local care. Not all pathogens cause CGD outcomes of overt pyogenic infections, as stalemates may materialise between the microorganism and the patient’s leukocytes. In these circumstances, chronic inflammatory cell responses consisting of activated lymphocytes and histiocytes evolve and arrange to form granulomas, one of the hallmarks of CGD, provoking diverse clinical manifestations of obstruction such as delayed gastric emptying, antral narrowing of the stomach, dysphagia, emesis, weight loss, biliary tract or gastrointestinal obstruction[11].
Pediatric CGD is relatively rare; this genetic condition, which has variable ethnic associations, occurs in 1 out of every 200,000–250,000 births in the United States and is often diagnosed in the first three years of life[7,12,13]. In most countries, the incidence of this condition is hindered by consanguinity, and the prevalence rates of autosomal recessive genotypes differ for the same reason[12,14,15,16,17]. A large cohort study suggested that North African/Arab and Turkish immigrants in Europe have a high prevalence of autosomal recessive CGD, reflecting the increased prevalence of consanguineous marriage in these populations[18]. Approximately 1.49 per every 10,000 live births in the Israeli Arab population and 1.05 in the Israeli Jewish population have CGD, primarily associated with an autosomal recessive inheritance, displaying increased morbidity[17]. Late presentation of CGD has been reported[19]
2. Common Pathogens
Owing to changes and the introduction of new prophylactic treatments after the initial emergence of CGD, the median age of death has increased over the last few decades, with fungal infections being the highest risk of mortality[13,20]. The five most common pathogens that infect North American patients are Staphylococcus aureus, Aspergillus spp, Nocardia spp, Burkholderia spp, and Serratia[20,21], in contrast to patients from Europe, where the five most common pathogens are Staphylococcus aureus, Aspergillus, Salmonella, Candida, and Serratia[18]. The infections can be severe or opportunistic and unusual because of the presence of fungi and bacteria that cause suppurative lymphadenitis, pneumonia, and abscesses at various locations, which will be discussed later in terms of signs, symptoms, and complications. Table 1 presents the microorganisms frequently isolated from the 284 patients with CGD and pulmonary involvement[18].
3. Granulomas
CGD derives its name from the characteristic formation of multiple granulomas in various body tissues. Granulomas often occur anywhere in the GI tract, from the mouth to the anus, with the colon and oesophagus being the most and least common sites, respectively[20]. In patients with CGD, microgranulomas, tissue eosinophilia, and brown-pigmented epithelioid histiocytes found in the lamina propria, and inflammatory changes revealed using biopsy serve as distinctive features[18,22,23,24]. Alimchandani et al. (2013) conducted a study on 87 patients with CGD and observed using GI biopsy that 74 % (64/87) of patients had prominent brown granular cytoplasmic pigmented inclusions in macrophages[22]. Multiple aseptic granulomas most frequently form in the skin[18].
4. CGD-related infectious diseases
Notably, severe recurrent bacterial and fungal infections usually present early in childhood (<5 years of age) in most patients with CGD. This is attributed to severe respiratory burst defects and the lack of ROS production. However, symptoms are delayed until adolescence and adulthood owing to the degree of residual NADPH oxidase activity[25]. In patients with CGD, osteomyelitis occurs in males aged 4–20 years. X-linked inheritance has been reported in nine patients with osteomyelitis caused by A. nidulans. Conversely, among three patients with A. fumigatus infection, osteomyelitis was associated with X-linked gp91phox in two patients and the autosomal recessive form of p67phox in one[26].
CGD patients are susceptible to a subset of catalase-positive organisms (CPO) because CPO degrades host-produced hydrogen peroxide before its conversion to hypochlorous acid by myeloperoxidase[7]. These CPOs include bacteria such as Pseudomonas spp., Staphylococcus aureus, Nocardia, Burkholderia cepacia, and Enterobacteriaceae such as Salmonella spp., Klebsiella spp., and Serratia spp.[13,18,20]. Mycobacterial infections caused by Bacillus Calmette–Guérin (BCG; in endemic countries that routinely administer vaccines) and Mycobacterium tuberculosis have been reported in Israel, Turkey, Iran, China, and Latin America. However, these patients present with a more localised disease[17,25,27,28,29].
The most frequent fungal infection is invasive aspergillosis caused by Aspergillus fumigatus, followed by Aspergillus nidulans and less common Aspergillus terreus, which has been isolated from bronchoalveolar lavage of patients with CGD [13,26,30]. Other reported species include Aspergillus niger and Aspergillus tanneri [31,32]. Fungal infections, particularly those caused by Aspergillus spp., are significant determinants of morbidity and the most common cause of mortality in patients with CGD[13,17]. The most common sites of infection are the lungs, followed by the skin, lymph nodes, liver, and gastrointestinal tract. Pneumonia is the most common pulmonary disease reported in patients with CGD (X-linked or autosomal recessive CGD), caused primarily by Aspergillus spp. and Staphylococcus aureus [33]. Mulch pneumonitis is a medical emergency and should be considered in all cases of unexplained pneumonitis, particularly in patients with acute onset and hypoxia. These patients should be treated with high-dose corticosteroids and antifungal and antibacterial agents [34,35,36]. Lung abscesses are relatively less common but potentially severe [13,18,20,25].
Subcutaneous abscesses are the most common, frequently located in the perianal region. They are typically caused by S. aureus but can also be caused by Serratia spp., Aspergillus spp., and Klebsiella spp. [13,18,20,25]. Cellulitis was also reported by Winkelstein et al. (2000), although it is relatively rare [20]. Suppurative or necrotising lymphadenitis is common in patients with CGD. Individuals with autosomal recessive forms of CGD have a lower probability of suppurative lymphadenitis than those with the X-linked form, suggesting that residual oxidase, which is more frequent in patients with the autosomal recessive disease, might enhance protection against this complication [26]. Suppurative lymphadenitis can also result from region-specific medical practices. Patients with CGD are predisposed to lymphadenitis after receiving BCG vaccination; however, their disease is rarely disseminated, as mentioned previously. Frequent and chronic infections have consequences; patients with CGD reportedly present with a failure to improve owing to the long-term use of treatments [37].
Liver abscesses are a frequent complication in patients with CGD and can cause significant morbidity. However, the signs and symptoms of this complication are variable and nonspecific, with the most common being fever, malaise, weight loss, abdominal tenderness, and elevated erythrocyte sedimentation rate [38]. In cases with liver abscesses, the predominant organism isolated was Staphylococcus aureus. Liver involvement in patients with CGD is a notable concern because splenomegaly, nodular regenerative hyperplasia, noncirrhotic portal hypertension, and portal venopathy can occur [39]. Splenomegaly can subsequently cause thrombocytopenia, which has been reported to be a poor prognostic indicator in patients with CGD [40].
The gastrointestinal tract symptoms in patients with CGD are generally nonspecific and range from mild to debilitating symptoms, such as abdominal pain, bloody diarrhoea, nausea, vomiting, malabsorption, and weight loss [41]. The rate of GI involvement is much higher in the X-linked than in the autosomal recessive form, as reported in an extensive survey of patients with CGD conducted by Marciano et al. [42]. GI involvement, particularly inflammatory bowel disease (IBD), could be the first sign of undiagnosed CGD [22].
Patients with CGD tend to have an abnormally excessive inflammatory response characterised by granuloma formation. These non-caseating granulomas tend to affect the hollow viscera, most notably the stomach, colon, and bladder. These granulomas are likely unrelated to infections because microorganisms are usually not identified, and patients respond rapidly to steroids or other immunomodulators such as cyclosporine [43,44,45]. An interested data is that children with CGD had predominantly mild infection with COVID-19 among a cohort of 101 CGD patients [46]
In a multicenter collaborative study of CGD in India, where were investigated 236 patients, X-linked and AR-CGD was seen in 77 and 97, respectively. Common infections documented include pneumonia (71.6%), skin and subcutaneous abscess (23.7%), osteomyelitis (8.6%), lung abscess (2.9%), meningoencephalitis (2.5%), and splenic abscess (1.7%). Mycobacterial infection was seen 18.6 % [47].
5. Differential Diagnosis of CGD
Certain clinical disorders, notably those with abnormal enzymatic functions with clinical and similar laboratory characteristics, can be misdiagnosed as CGD. Some of these disorders are detailed below:
(i). Glucose-6-phosphate dehydrogenase deficiency: Glucose-6-phosphate dehydrogenase deficiency is an X-linked inherited disorder wherein leukocyte defect results from the deficient generation of NADPH, required as a reducing equivalent for oxidase. This deficiency reduces hexose monophosphate shunt activity and hydrogen peroxide production in leukocytes. Symptoms of acute haemolysis associated with glucose-6-phosphate dehydrogenase deficiency include anaemia, fatigue, jaundice, back or abdominal pain, and haemoglobinuria. The organisms responsible for the infection include S. aureus, S. epidermidis, Serratia marcescens, Pseudomonas, E. coli, and Aspergillus. Laboratory diagnosis is based on the presence of deficient leukocyte glucose-6-phosphate dehydrogenase. The treatment and prognosis are similar to those of CGD. Administration of rasburicase, dapsone, primaquine, and pegloticase should not be administered until a diagnostic test is performed [48].
(ii). Myeloperoxidase deficiency: Myeloperoxidase is an enzyme necessary for the normal intracellular killing of certain organisms such as Candida and S. aureus. The leukocytes of these patients exhibited regular oxygen consumption and superoxide and hydrogen peroxide production. This deficiency is characterised by the delayed intracellular killing of organisms and decreased chemiluminescence of the leukocytes. Diagnosis is based on peroxidase staining of peripheral blood, and appropriate administration of antibiotics can aid in its therapy [8].
(iii). Chediak–Higashi Syndrome (CHS): The diagnosis of CHS is based on the examination of peripheral blood and bone marrow smears for characteristic cytoplasmic giant granules in neutrophils, eosinophils, and other granulocytes. The diagnosis can be confirmed by genetic testing for mutations in CHS1/LYST. Abnormalities include elevated Epstein–Barr virus (EBV) antibody titres, the abnormal intracellular killing of microorganisms such as streptococci and pneumococci, and microorganisms found in CGD. Abnormal microtubule function, lysosomal enzyme levels, and protease deficiency have been described in granulocytes. Most patients die during childhood; however, survivors in their second and third decades have also been identified. We lack definitive treatment besides specific antibiotic therapies against causative organisms [49].
(iv). Job’s syndrome: This was initially described as a disorder of recurrent cold staphylococcal abscesses of the skin, lymph nodes, or subcutaneous tissues. Other clinical features include eczematoid skin lesions, otitis media, and chronic nasal discharge. Additional reports indicated that this disorder might be a variant of CGD. However, most patients do not undergo abnormal immunological test results. S. aureus, Candida spp, H. influenzae, S. pneumoniae, and group A Streptococci cause infections associated with Job’s syndrome. The symptoms of Job’s syndrome are similar to those of hyperimmunoglobulin E (hyper-Ig-E) syndrome (elevated immunoglobulin IgE, defective chemotaxis, eczema, and recurrent infections). These may be the same disorder, and appropriate antibiotic therapy is used [50].
6. Management of CGD
6.1. Haematopoietic stem cell transplantation (HSCT/HCT)
HCT is the principal treatment available for managing CGD with favourable results regardless of symptoms, age, sex, or mutations [51,52,53]. Transplantation therapy has an overall survival rate of > 90 % in children <14 years and has improved in the last decade, particularly with early diagnosis [51]. Additionally, HCT is associated with event-free survival rates of >80 % in patients with CGD and improves the quality of life [54].
However, the group of HCT-treated patients demonstrated excellent survival rates, although the risks and benefits still need to be assessed in individual patients. Based on the significant progress of patients with CGD treated with HCT, it is regarded as the only known curative treatment with an improved life expectancy owing to its improved implementation over time [55,56,57,58].
6.2. Drug-based treatment
Treatment using TNF-alpha inhibitors in patients with CGD could help improve the outcome of severe inflammatory complications despite the associated risk factors. This treatment could provide short-term benefits in selected patients with CGD with severe inflammatory complications awaiting HCT [59]. There is conflicting evidence regarding infliximab, a TNF-alpha inhibitor, causing rapid improvement; however, it is associated with an increased risk of severe infections and death in patients with CGD and should be strictly avoided [23,51]. In addition, corticosteroid use has proven beneficial for CGD colitis; however, their use has traditionally been contraindicated in patients with CGD and active infection. In conjunction with appropriate antimicrobials, steroids help treat hyperactive inflammatory responses [51,52]. Corticosteroids, despite their effectiveness, are associated with long-term complications such as growth retardation, osteoporosis, and an increased risk of infection [30,38,52,60,61,62,63].
To determine the optimal treatment for patients with CGD, a European study compared conventional treatments, antibacterial and antifungal prophylaxis with cotrimoxazole and azoles, and immunosuppressive therapy with HCT (Figure 4). Patients under conventional treatment and those receiving HCT treatment did not improve. Seventy-six per cent (76 %) of these patients were affected by inflammatory complications, whereas 85 % developed at least one infection even with conventional treatments, the most common being skin infection or pneumonia [64,65,66,67,68,69,70].
For inflammatory conditions, steroid treatment with immunosuppressants (such as anti-tumour necrosis factor) is adequate as second-line therapy, as they exhibit some efficacy. However, immunosuppressant (such as anti-tumor necrosis factor agents, thalidomide, and anakinra) use is still debated because of its risks, notwithstanding its benefits [55,71,72,73].
Lugo-Reyes et al. (2022) reported the outcomes of a systematic review and meta-analysis on IFN-γ’s efficacy and safety in CGD. They support the use of IFN-γ in managing patients with CGD. However, the authors did not find sufficient clinical evidence and suggested that more clinical trials are needed to assess the efficacy and long-term safety of IFN-γ. As the longevity of patients with CGD improves, a long-term and detailed assessment of the autoimmune and inflammatory complications associated with chronic IFN-γ therapy is required. For the clinicians whose patients continue to die during adolescence owing to invasive pulmonary aspergillosis, especially in Latin America, The Caribbean and other regions where resources are scant, it is imperative to ascertain the patients who are prescribed long-term use of IFN-γ and also identify the significant risks for complications [74].
Lifelong antimicrobial and antifungal prophylaxis are the most common management routes used to minimise the incidence of infections. However, pushing antibiotics is contraindicated in healthy patients because of antibiotic resistance. Most studies suggest a link between aggressive antibiotic use for these five pathogens and preventing the spread of infection in patients with CGD [24]. Drugs such as trimethoprim-sulfamethoxazole reduce the occurrence of bacterial infections in patients with CGD but do not interfere considerably with the gut microbiome [29]. Patients with sulfamethoxazole allergy or G-6-PD deficiency have other options, such as dicloxacillin and ciprofloxacin [51]. A concern arises in pregnancy since trimethoprim is a folic acid antagonist, which increases the high risk for congenital disabilities and is discontinued during pregnancy [52]. Itraconazole considerably reduces invasive fungal infections, and newer azole drugs, such as voriconazole, posaconazole, and isavuconazole, are available, providing more options for treating these fungal infections [52]. Itraconazole should be provided as a long-term and possible lifelong treatment option to prevent fungal infections in children with CGD. However, regular monitoring of liver function is required during itraconazole therapy [29]. In cases where patients are intolerant to itraconazole, posaconazole is proven safe and effective [51]. Similarly, Aspergillus viridinutans, Neosartorya udagawae, and Sporothrix schenckii have been reported in patients with CGD treated with antifungal prophylaxis [21].
Chromobacterium violaceum, firstly described in 1927, is a facultative anaerobic gram-negative bacterium found in soil and water in tropical regions. It rarely causes infection, but in immunocompromised humans is associated with significant morbidity owing to bacteraemia, antibiotic resistance to multiple antimicrobials, disease relapse, and reinfection. It was reported the case of a previous healthy male aged 11, who was exposed to stagnant water. The patient presented skin abscesses, multiple lung and liver abscesses that were resistant to vancomycin, nafcillin, clindamycin, and ceftriaxone. Once the bacterium was identified, he received meropenem and ciprofloxacin. He was later discharged on trimethoprim-sulfamethoxazole (TMP-SMX) and ertapenem to complete a total of six months of treatment. He was then diagnosed with CGD, constituting the first report of C. violaceum in this medical condition [75,76].
6.2.1. CGD-related inflammatory responses
Patients with CGD rarely present cutaneous symptoms, except in patients with autoimmune disorders such as systemic lupus erythematosus [77]. Furthermore, there is an increased risk of autoimmune disorders such as inflammatory bowel colitis and inflammatory bowel disease among patients owing to increased activation of NF-kB, increasing the production of proinflammatory cytokines. The inflammatory manifestations of CGD are mainly observed in the GI and urogenital tracts, lungs, and eyes. Inflammation can be suppressed by blocking TNF-alpha and oral corticosteroids [73].
6.2.2. Hemophagocytic lymphohistiocytosis (HLH)
In addition, patients with CGD experience infection-triggered hemophagocytic lymphohistiocytosis (HLH), which presents as pathological hyperactive inflammation [78]. Possible pathologies, including CGD, should be considered in children with HLH because it can indicate CGD. An optimal management strategy is yet to be developed for children with CGD who manifest with HLH. Early recognition and proper management of infectious triggers and HLH are crucial to reducing mortality [79].
6.3. Gene therapy
A recent human trial involved nine patients with X-linked CGD undergoing ex-vivo autologous CD34+ haematopoietic stem-and progenitor cell-based lentiviral gene therapy following myeloablative conditioning. Two of the nine patients died during the trial; however, prophylactic antibiotic treatment was no longer required in the surviving patients. Moreover, stable vector copy numbers and no clonal dysregulation or transgene silencing were identified in six surviving an upcoming treatment option for patients with CGD. Gene therapy is only helpful if an HLA-fully matched donor is unavailable [80,81,82,83].
In contrast to the methods used to cope with CGD defects, examination revealed that the direct repair of the defect in CGD could be performed using thymosin β4 subverts. Thymosin β4 was seen to restore the body’s ability to remove damaged cells and renew healthier cells in patients with CGD by restoring autophagy and upregulating hypoxia-responsive genes in human and murine CGD. Autophagy, which may help in pathogen elimination, can prevent granuloma formation, commonly seen in CGD[84,85].
CGD leads to infections of the liver, lungs, and lymph nodes, and treatment of CGD with prophylactic drugs could be prolonged; therefore, optimal therapy must be chosen for these patients [29,51]. Current gene therapy trials have demonstrated that lentiviruses or gene editing can be used as curative therapy where HCT is inappropriate for a patient and removes the risk of graft-versus-host disease. Notably, gene therapy can be applied when human leukocyte antigen (HLA)-matched donors are difficult to identify using HCT. It is a promising method that involves the insertion of a functional copy of a defective gene into the correct cells, where success depends on viral vectors. Lentiviral systems are currently the main techniques used to deliver therapeutic genes in gene therapy for treating patients with CGD [86]. These advances in gene therapy have facilitated more accurate treatment procedures [84]. Furthermore, gene therapy as a cure for CGD is a crucial area of focus, specifically for patients with X-linked and p47 mutations [87].
There are promising future approaches for treating patients with CGD, including genome-editing technologies, such as CRISPR/Cas9 nuclease gene therapy[73,88] and SIN-lentiviral vectors. A multicentre trial should be performed in the United States and Europe to determine the feasibility of gene therapy for patients with CGD, especially for those without an HLA-identical donor [51]. Other authors have reported the use of gene therapy in CGD patients. It can provide life-saving clinical benefit to CGD patients lacking a suitable donor[89,90].
6.4. Other therapies
Results from cases where multiple granulocyte infusion was performed, although not yet evaluated in controlled studies, suggest its usefulness in treating severe bacterial and fungal infections. Adverse effects, although well tolerated, include fever, developing leucoagglutinin, and rarely pulmonary leucocytosis [52]. In patients with CGD, bacterial infections are commonly caused by Staphylococcus aureus, Aspergillus spp., Nocardia spp., Burkholderia spp., and Serratia spp. [80] and several antimicrobials have been therapeutically used[11,91,92].
7. Conclusions
In conclusion, the current management of patients with CGD involves a comprehensive multidisciplinary approach and its potential complications. Antibiotics and antifungals were once considered the most important treatment options for managing CGD. Despite starting as an experimental option, they helped achieve a high curative rate and longer life expectancy. Gene therapy may be considered a viable option to improve treatment outcomes if an HLA-identical donor is unavailable. Therefore, the treatment of CGD is progressing, from antibiotic prophylaxis developed in the 1970s to the current application of gene therapy, which is currently under investigation but can be considered for patients at high risk [93]. CGD gene therapy is a promising alternative to HSCT in absence of suitable donors. Consequently, allogeneic haematopoietic stem cell transplantation from a human leukocyte antigen-identical donor has been proven to cure CGD.
Author Contributions
The manuscript was conceptualized by A.A.J.-V. and A.W.P. All authors wrote the initial draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgements
The authors sincerely thank The University of West Indies, the St. Augustine Campus, and The West Indian Immunology Society. The authors would like to thank Editage (www.editage.com) for English language editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bridges, R.A.; Berendes, H.; Good, R.A. A Fatal Granulomatous Disease of Childhood; the Clinical, Pathological, and Laboratory Features of a New Syndrome. AMA J. Dis. Child. 1959, 97, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.; Saffarzadeh, M.; Weber, A.N.R.; Rieber, N.; Radsak, M.; von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between Host Defence, Immune Modulation, and Tissue Injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef]
- Anjani, G.; Vignesh, P.; Joshi, V.; Shandilya, J.K.; Bhattarai, D.; Sharma, J.; Rawat, A. Recent Advances in Chronic Granulomatous Disease. Genes Dis 2020, 7, 84–92. [Google Scholar] [CrossRef]
- Roos, D. Chronic Granulomatous Disease. Br. Med. Bull. 2016, 118, 50–63. [Google Scholar] [CrossRef]
- Mollin, M.; Beaumel, S.; Vigne, B.; Brault, J.; Roux-Buisson, N.; Rendu, J.; Barlogis, V.; Catho, G.; Dumeril, C.; Fouyssac, F.; et al. Clinical, Functional and Genetic Characterization of 16 Patients Suffering from Chronic Granulomatous Disease Variants—Identification of 11 Novel Mutations in CYBB. Clin. Exp. Immunol. 2021, 203, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C. Inflammatory Consequences of Inherited Disorders Affecting Neutrophil Function. Blood 2019, 133, 2130–2139. [Google Scholar] [CrossRef] [PubMed]
- Rider, N.L.; Jameson, M.B.; Creech, C.B. Chronic Granulomatous Disease: Epidemiology, Pathophysiology, and Genetic Basis of Disease. J Pediatric Infect Dis Soc 2018, 7, S2–S5. [Google Scholar] [CrossRef]
- Mauch, L.; Lun, A.; O’Gorman, M.R.G.; Harris, J.S.; Schulze, I.; Zychlinsky, A.; Fuchs, T.; Oelschlägel, U.; Brenner, S.; Kutter, D.; et al. Chronic Granulomatous Disease (CGD) and Complete Myeloperoxidase Deficiency Both Yield Strongly Reduced Dihydrorhodamine 123 Test Signals but Can Be Easily Discerned in Routine Testing for CGD. Clin. Chem. 2007, 53, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Curnutte, J.T.; Scott, P.J.; Mayo, L.A. Cytosolic Components of the Respiratory Burst Oxidase: Resolution of Four Components, Two of Which Are Missing in Complementing Types of Chronic Granulomatous Disease. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 825–829. [Google Scholar] [CrossRef]
- Thomas, D.C.; Charbonnier, L.-M.; Schejtman, A.; Aldhekri, H.; Coomber, E.L.; Dufficy, E.R.; Beenken, A.E.; Lee, J.C.; Clare, S.; Speak, A.O.; et al. EROS/CYBC1 Mutations: Decreased NADPH Oxidase Function and Chronic Granulomatous Disease. J. Allergy Clin. Immunol. 2019, 143, 782–785.e1. [Google Scholar] [CrossRef] [PubMed]
- Roos, D. Chronic Granulomatous Disease. In NADPH Oxidases: Methods and Protocols; Knaus, U.G., Leto, T.L., Eds.; Springer New York: New York, NY, 2019; pp. 531–542. ISBN 9781493994243. [Google Scholar]
- Kuhns, D.B.; Alvord, W.G.; Heller, T.; Feld, J.J.; Pike, K.M.; Marciano, B.E.; Uzel, G.; DeRavin, S.S.; Priel, D.A.L.; Soule, B.P.; et al. Residual NADPH Oxidase and Survival in Chronic Granulomatous Disease. N. Engl. J. Med. 2010, 363, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common Severe Infections in Chronic Granulomatous Disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Wolach, B.; Gavrieli, R.; de Boer, M.; Gottesman, G.; Ben-Ari, J.; Rottem, M.; Schlesinger, Y.; Grisaru-Soen, G.; Etzioni, A.; Roos, D. Chronic Granulomatous Disease in Israel: Clinical, Functional and Molecular Studies of 38 Patients. Clin. Immunol. 2008, 129, 103–114. [Google Scholar] [CrossRef]
- Fattahi, F.; Badalzadeh, M.; Sedighipour, L.; Movahedi, M.; Fazlollahi, M.R.; Mansouri, S.D.; Khotaei, G.T.; Bemanian, M.H.; Behmanesh, F.; Hamidieh, A.A.; et al. Inheritance Pattern and Clinical Aspects of 93 Iranian Patients with Chronic Granulomatous Disease. J. Clin. Immunol. 2011, 31, 792–801. [Google Scholar] [CrossRef]
- Kutukculer, N.; Aykut, A.; Karaca, N.E.; Durmaz, A.; Aksu, G.; Genel, F.; Pariltay, E.; Cogulu, Ö.; Azarsız, E. Chronic Granulamatous Disease: Two Decades of Experience from a Paediatric Immunology Unit in a Country with High Rate of Consangineous Marriages. Scand. J. Immunol. 2019, 89, e12737. [Google Scholar] [CrossRef]
- Wolach, B.; Gavrieli, R.; de Boer, M.; van Leeuwen, K.; Berger-Achituv, S.; Stauber, T.; Ben Ari, J.; Rottem, M.; Schlesinger, Y.; Grisaru-Soen, G.; et al. Chronic Granulomatous Disease: Clinical, Functional, Molecular, and Genetic Studies. The Israeli Experience with 84 Patients. Am. J. Hematol. 2017, 92, 28–36. [Google Scholar] [CrossRef]
- van den Berg, J.M.; van Koppen, E.; Ahlin, A.; Belohradsky, B.H.; Bernatowska, E.; Corbeel, L.; Español, T.; Fischer, A.; Kurenko-Deptuch, M.; Mouy, R.; et al. Chronic Granulomatous Disease: The European Experience. PLoS One 2009, 4, e5234. [Google Scholar] [CrossRef] [PubMed]
- Barkai, T.; Somech, R.; Broides, A.; Gavrieli, R.; Wolach, B.; Marcus, N.; Hagin, D.; Stauber, T. Late Diagnosis of Chronic Granulomatous Disease. Clin. Exp. Immunol. 2020, 201, 297–305. [Google Scholar] [CrossRef]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic Granulomatous Disease. Report on a National Registry of 368 Patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Slack, M.A.; Thomsen, I.P. Prevention of Infectious Complications in Patients with Chronic Granulomatous Disease. J Pediatric Infect Dis Soc 2018, 7, S25–S30. [Google Scholar] [CrossRef]
- Alimchandani, M.; Lai, J.-P.; Aung, P.P.; Khangura, S.; Kamal, N.; Gallin, J.I.; Holland, S.M.; Malech, H.L.; Heller, T.; Miettinen, M.; et al. Gastrointestinal Histopathology in Chronic Granulomatous Disease: A Study of 87 Patients. Am. J. Surg. Pathol. 2013, 37, 1365–1372. [Google Scholar] [CrossRef]
- Peixoto, A.; Coelho, R.; Maia, T.; Sarmento, A.; Magro, F.; Macedo, G. Chronic Granulomatous Disease Mimicking Colonic Crohn’s Disease Successfully Treated with Infliximab. ACG Case Rep J 2017, 4, e46. [Google Scholar] [CrossRef]
- Garcia-Eulate, R.; Hussain, N.; Heller, T.; Kleiner, D.; Malech, H.; Holland, S.; Choyke, P.L. CT and MRI of Hepatic Abscess in Patients with Chronic Granulomatous Disease. AJR Am. J. Roentgenol. 2006, 187, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Murayama, S.; Takanashi, S.; Takahashi, K.; Miyatsuka, S.; Fujita, T.; Ichinohe, S.; Koike, Y.; Kohagizawa, T.; Mori, H.; et al. Clinical Features and Prognoses of 23 Patients with Chronic Granulomatous Disease Followed for 21 Years by a Single Hospital in Japan. Eur. J. Pediatr. 2008, 167, 1389–1394. [Google Scholar] [CrossRef] [PubMed]
- Dotis, J.; Roilides, E. Osteomyelitis due to Aspergillus Spp. in Patients with Chronic Granulomatous Disease: Comparison of Aspergillus Nidulans and Aspergillus Fumigatus. Int. J. Infect. Dis. 2004, 8, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Battersby, A.C.; Cale, A.M.; Goldblatt, D.; Gennery, A.R. Clinical Manifestations of Disease in X-Linked Carriers of Chronic Granulomatous Disease. J. Clin. Immunol. 2013, 33, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, I.P.; Smith, M.A.; Holland, S.M.; Creech, C.B. A Comprehensive Approach to the Management of Children and Adults with Chronic Granulomatous Disease. J. Allergy Clin. Immunol. Pract. 2016, 4, 1082–1088. [Google Scholar] [CrossRef] [PubMed]
- Köker, M.Y.; Camcıoğlu, Y.; van Leeuwen, K.; Kılıç, S.Ş.; Barlan, I.; Yılmaz, M.; Metin, A.; de Boer, M.; Avcılar, H.; Patıroğlu, T.; et al. Clinical, Functional, and Genetic Characterization of Chronic Granulomatous Disease in 89 Turkish Patients. J. Allergy Clin. Immunol. 2013, 132, 1156–1163.e5. [Google Scholar] [CrossRef]
- Mortaz, E.; Sarhifynia, S.; Marjani, M.; Moniri, A.; Mansouri, D.; Mehrian, P.; van Leeuwen, K.; Roos, D.; Garssen, J.; Adcock, I.M.; et al. An Adult Autosomal Recessive Chronic Granulomatous Disease Patient with Pulmonary Aspergillus Terreus Infection. BMC Infect. Dis. 2018, 18, 552. [Google Scholar] [CrossRef]
- Kaltenis, P.; Mudeniené, V.; Maknavicius, S.; Seinin, D. Renal Amyloidosis in a Child with Chronic Granulomatous Disease and Invasive Aspergillosis. Pediatr. Nephrol. 2008, 23, 831–834. [Google Scholar] [CrossRef]
- Sugui, J.A.; Peterson, S.W.; Clark, L.P.; Nardone, G.; Folio, L.; Riedlinger, G.; Zerbe, C.S.; Shea, Y.; Henderson, C.M.; Zelazny, A.M.; Holland, S.M. Aspergillus Tanneri Sp. Nov., a New Pathogen That Causes Invasive Disease Refractory to Antifungal Therapy. J. Clin. Microbiol. 2020, 50, 3309–3317. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.; Kadaria, D.; Sodhi, A.; Fox, R.; Williams, G.; Threlkeld, S. Chronic Granulomatous Disease Presenting as Aspergillus Fumigatus Pneumonia in a Previously Healthy Young Woman. Am. J. Case Rep. 2017, 18, 351–354. [Google Scholar] [CrossRef]
- Maaloul, I.; Ameur, S.B.; Chabchoub, I.; Kolsi, R.; Bahloul, M.; Kamoun, T.; Bouaziz, M.; Hachicha, M. Fulminant Mulch Pneumonitis in a Previously Healthy Child. Arch. Pediatr. 2018, 25, 495–496. [Google Scholar] [CrossRef]
- Siddiqui, S.; Anderson, V.L.; Hilligoss, D.M.; Abinun, M.; Kuijpers, T.W.; Masur, H.; Witebsky, F.G.; Shea, Y.R.; Gallin, J.I.; Malech, H.L.; et al. Fulminant Mulch Pneumonitis: An Emergency Presentation of Chronic Granulomatous Disease. Clin. Infect. Dis. 2007, 45, 673–681. [Google Scholar] [CrossRef]
- Ameratunga, R.; Woon, S.-T.; Vyas, J.; Roberts, S. Fulminant Mulch Pneumonitis in Undiagnosed Chronic Granulomatous Disease: A Medical Emergency. Clin. Pediatr. 2010, 49, 1143–1146. [Google Scholar] [CrossRef]
- Norouzi, S.; Aghamohammadi, A.; Mamishi, S.; Rosenzweig, S.D.; Rezaei, N. Bacillus Calmette-Guérin (BCG) Complications Associated with Primary Immunodeficiency Diseases. J. Infect. 2012, 64, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.-S.; Lee, M.S. Concomitant Use of Corticosteroid and Antimicrobials for Liver Abscesses in Patients with Chronic Granulomatous Disease. Korean J. Pediatr. 2016, 59, 196–201. [Google Scholar] [CrossRef]
- Lublin, M.; Bartlett, D.L.; Danforth, D.N.; Kauffman, H.; Gallin, J.I.; Malech, H.L.; Shawker, T.; Choyke, P.; Kleiner, D.E.; Schwartzentruber, D.J.; et al. Hepatic Abscess in Patients with Chronic Granulomatous Disease. Ann. Surg. 2002, 235, 383–391. [Google Scholar] [CrossRef]
- Feld, J.J.; Hussain, N.; Wright, E.C.; Kleiner, D.E.; Hoofnagle, J.H.; Ahlawat, S.; Anderson, V.; Hilligoss, D.; Gallin, J.I.; Liang, T.J.; et al. Hepatic Involvement and Portal Hypertension Predict Mortality in Chronic Granulomatous Disease. Gastroenterology 2008, 134, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M.; Campos, A.; Scheffler-Mendoza, S.; León-Lara, X.; Onuma-Zamayoa, H.; Espinosa, S.; Yamazaki-Nakashimada, M.A.; Blancas-Galicia, L. [Infectious and inflammatory gastrointestinal manifestations of chronic granulomatous disease]. Rev. Alerg. Mex. 2021, 68, 198–205. [Google Scholar] [CrossRef]
- Marciano, B.E.; Rosenzweig, S.D.; Kleiner, D.E.; Anderson, V.L.; Darnell, D.N.; Anaya-O’Brien, S.; Hilligoss, D.M.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Gastrointestinal Involvement in Chronic Granulomatous Disease. Pediatrics 2004, 114, 462–468. [Google Scholar] [CrossRef]
- Chin, T.W.; Stiehm, E.R.; Falloon, J.; Gallin, J.I. Corticosteroids in Treatment of Obstructive Lesions of Chronic Granulomatous Disease. J. Pediatr. 1987, 111, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.L.; Moussa, S.L.; Villar, R.G.; Hulett, R.L. Gastrointestinal Complications of Chronic Granulomatous Disease: Case Report and Literature Review. Clin. Pediatr. 1998, 37, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Rosh, J.R.; Tang, H.B.; Mayer, L.; Groisman, G.; Abraham, S.K.; Prince, A. Treatment of Intractable Gastrointestinal Manifestations of Chronic Granulomatous Disease with Cyclosporine. J. Pediatr. 1995, 126, 143–145. [Google Scholar] [CrossRef]
- Vignesh, P.; Sharma, R.; Barman, P.; Mondal, S.; Das, J.; Siniah, S.; Goyal, T.; Sharma, S.; Pilania, R.K.; Jindal, A.K.; et al. Impact of COVID-19 Pandemic on Clinical Care of Patients and Psychosocial Health of Affected Families with Chronic Granulomatous Disease: An Observational Study from North India. J. Clin. Immunol. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rawat, A.; Vignesh, P.; Sudhakar, M.; Sharma, M.; Suri, D.; Jindal, A.; Gupta, A.; Shandilya, J.K.; Loganathan, S.K.; Kaur, G.; et al. Clinical, Immunological, and Molecular Profile of Chronic Granulomatous Disease: A Multi-Centric Study of 236 Patients From India. Front. Immunol. 2021, 12, 625320. [Google Scholar] [CrossRef]
- Agudelo-Flórez, P.; Costa-Carvalho, B.T.; López, J.A.; Redher, J.; Newburger, P.E.; Olalla-Saad, S.T.; Condino-Neto, A. Association of Glucose-6-Phosphate Dehydrogenase Deficiency and X-Linked Chronic Granulomatous Disease in a Child with Anemia and Recurrent Infections. Am. J. Hematol. 2004, 75, 151–156. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Gallin, J.I. Phagocyte Defects. Clin. Immunol. Immunopathol. 1986, 40, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Gaither, T.A.; Gallin, J.I.; Iida, K.; Nussenzweig, V.; Frank, M.M. Deficiency in C3b Receptors on Neutrophils of Patients with Chronic Granulomatous Disease and Hyperimmunoglobulin-E Recurrent Infection (Job’s) Syndrome. Inflammation 1984, 8, 429–444. [Google Scholar] [CrossRef]
- Arnold, D.E.; Heimall, J.R. A Review of Chronic Granulomatous Disease. Adv. Ther. 2017, 34, 2543–2557. [Google Scholar] [CrossRef] [PubMed]
- Yonkof, J.R.; Gupta, A.; Fu, P.; Garabedian, E.; Dalal, J. the United States Immunodeficiency Network Consortium Role of Allogeneic Hematopoietic Stem Cell Transplant for Chronic Granulomatous Disease (CGD): A Report of the United States Immunodeficiency Network. J. Clin. Immunol. 2019, 39, 448–458. [Google Scholar] [CrossRef]
- Chiesa, R.; Wang, J.; Blok, H.-J.; Hazelaar, S.; Neven, B.; Moshous, D.; Schulz, A.; Hoenig, M.; Hauck, F.; Al Seraihy, A.; et al. Hematopoietic Cell Transplantation in Chronic Granulomatous Disease: A Study of 712 Children and Adults. Blood 2020, 136, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Connelly, J.A.; Marsh, R.; Parikh, S.; Talano, J.-A. Allogeneic Hematopoietic Cell Transplantation for Chronic Granulomatous Disease: Controversies and State of the Art. J Pediatric Infect Dis Soc 2018, 7, S31–S39. [Google Scholar] [CrossRef] [PubMed]
- Magnani, A.; Mahlaoui, N. Managing Inflammatory Manifestations in Patients with Chronic Granulomatous Disease. Paediatr. Drugs 2016, 18, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Yanagimachi, M.; Kato, K.; Iguchi, A.; Sasaki, K.; Kiyotani, C.; Koh, K.; Koike, T.; Sano, H.; Shigemura, T.; Muramatsu, H.; et al. Hematopoietic Cell Transplantation for Chronic Granulomatous Disease in Japan. Front. Immunol. 2020, 11, 1617. [Google Scholar] [CrossRef]
- Marsh, R.A.; Leiding, J.W.; Logan, B.R.; Griffith, L.M.; Arnold, D.E.; Haddad, E.; Falcone, E.L.; Yin, Z.; Patel, K.; Arbuckle, E.; et al. Chronic Granulomatous Disease-Associated IBD Resolves and Does Not Adversely Impact Survival Following Allogeneic HCT. J. Clin. Immunol. 2019, 39, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Yi, E.S.; Choi, Y.B.; Lee, N.H.; Lee, J.W.; Sung, K.W.; Koo, H.H.; Kang, E.-S.; Kim, Y.-J.; Yoo, K.H. Allogeneic Hematopoietic Cell Transplantation in Patients with Primary Immunodeficiencies in Korea: Eleven-Year Experience in a Single Center. J. Clin. Immunol. 2018, 38, 757–766. [Google Scholar] [CrossRef]
- Conrad, A.; Neven, B.; Mahlaoui, N.; Suarez, F.; Sokol, H.; Ruemmele, F.M.; Rouzaud, C.; Moshous, D.; Lortholary, O.; Blanche, S.; et al. Infections in Patients with Chronic Granulomatous Disease Treated with Tumor Necrosis Factor Alpha Blockers for Inflammatory Complications. J. Clin. Immunol. 2021, 41, 185–193. [Google Scholar] [CrossRef]
- Straughan, D.M.; McLoughlin, K.C.; Mullinax, J.E.; Marciano, B.E.; Freeman, A.F.; Anderson, V.L.; Uzel, G.; Azoury, S.C.; Sorber, R.; Quadri, H.S.; et al. The Changing Paradigm of Management of Liver Abscesses in Chronic Granulomatous Disease. Clin. Infect. Dis. 2018, 66, 1427–1434. [Google Scholar] [CrossRef]
- Al Ghadeer, H.A.; Busaleh, F.N.; Al Habeeb, J.A.; Alaithan, R.M.; Almutahhar, A.E.; Bin Abd, M.M.; Aldawood, M.M. Liver Abscesses as a Sign of Chronic Granulomatous Disease in Adolescent. Cureus 2021, 13, e17467. [Google Scholar] [CrossRef]
- Mortaz, E.; Azempour, E.; Mansouri, D.; Tabarsi, P.; Ghazi, M.; Koenderman, L.; Roos, D.; Adcock, I.M. Common Infections and Target Organs Associated with Chronic Granulomatous Disease in Iran. Int. Arch. Allergy Immunol. 2019, 179, 62–73. [Google Scholar] [CrossRef]
- Pilania, R.K.; Rawat, A.; Vignesh, P.; Guleria, S.; Jindal, A.K.; Das, G.; Suri, D.; Gupta, A.; Gupta, K.; Chan, K.-W.; et al. Liver Abscess in Chronic Granulomatous Disease-Two Decades of Experience from a Tertiary Care Centre in North-West India. J. Clin. Immunol. 2021, 41, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Dedieu, C.; Albert, M.H.; Mahlaoui, N.; Hauck, F.; Hedrich, C.; Baumann, U.; Warnatz, K.; Roesler, J.; Speckmann, C.; Schulte, J.; et al. Outcome of Chronic Granulomatous Disease—Conventional Treatment vs Stem Cell Transplantation. Pediatr. Allergy Immunol. 2021, 32, 576–585. [Google Scholar] [CrossRef]
- Åhlin, A.; Fugeläng, J.; de Boer, M.; Ringden, O.; Fasth, A.; Winiarski, J. Chronic Granulomatous Disease-Haematopoietic Stem Cell Transplantation versus Conventional Treatment. Acta Paediatr. 2013, 102, 1087–1094. [Google Scholar] [CrossRef]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kühlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-Linked Chronic Granulomatous Disease by Gene Therapy, Augmented by Insertional Activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Kang, E.M.; Choi, U.; Theobald, N.; Linton, G.; Long Priel, D.A.; Kuhns, D.; Malech, H.L. Retrovirus Gene Therapy for X-Linked Chronic Granulomatous Disease Can Achieve Stable Long-Term Correction of Oxidase Activity in Peripheral Blood Neutrophils. Blood 2010, 115, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.M.; Malech, H.L. Gene Therapy for Chronic Granulomatous Disease. Methods Enzymol. 2012, 507, 125–154. [Google Scholar] [CrossRef] [PubMed]
- Santilli, G.; Almarza, E.; Brendel, C.; Choi, U.; Beilin, C.; Blundell, M.P.; Haria, S.; Parsley, K.L.; Kinnon, C.; Malech, H.L.; et al. Biochemical Correction of X-CGD by a Novel Chimeric Promoter Regulating High Levels of Transgene Expression in Myeloid Cells. Mol. Ther. 2011, 19, 122–132. [Google Scholar] [CrossRef]
- Kang, H.J.; Bartholomae, C.C.; Paruzynski, A.; Arens, A.; Kim, S.; Yu, S.S.; Hong, Y.; Joo, C.-W.; Yoon, N.-K.; Rhim, J.-W.; et al. Retroviral Gene Therapy for X-Linked Chronic Granulomatous Disease: Results from Phase I/II Trial. Mol. Ther. 2011, 19, 2092–2101. [Google Scholar] [CrossRef]
- Baha, A.; Hanazay, C.; Kokturk, N.; Turktas, H. A Case of Sarcoidosis Associated with Anti-Tumor Necrosis Factor Treatment. J Investig Med High Impact Case Rep 2015, 3, 2324709615571366. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.H.; Sullivan, B.; Zerbe, C.S.; De Ravin, S.S.; Blakely, A.M.; Quezado, M.M.; Marciano, B.E.; Marko, J.; Ling, A.; Kleiner, D.E.; et al. Gastrointestinal and Hepatic Manifestations of Chronic Granulomatous Disease. J. Allergy Clin. Immunol. Pract. 2023, 11, 1401–1416. [Google Scholar] [CrossRef]
- Yu, H.-H.; Yang, Y.-H.; Chiang, B.-L. Chronic Granulomatous Disease: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2021, 61, 101–113. [Google Scholar] [CrossRef]
- Lugo Reyes, S.O.; González Garay, A.; González Bobadilla, N.Y.; Rivera Lizárraga, D.A.; Madrigal Paz, A.C.; Medina-Torres, E.A.; Álvarez Cardona, A.; Galindo Ortega, J.L.; Solís Galicia, C.; Espinosa-Padilla, S.E.; et al. Efficacy and Safety of Interferon-Gamma in Chronic Granulomatous Disease: A Systematic Review and Meta-Analysis. J. Clin. Immunol. 2023, 43, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Blancas-Galicia, L.; Santos-Chávez, E.; Deswarte, C.; Mignac, Q.; Medina-Vera, I.; León-Lara, X.; Roynard, M.; Scheffler-Mendoza, S.C.; Rioja-Valencia, R.; Alvirde-Ayala, A.; et al. Genetic, Immunological, and Clinical Features of the First Mexican Cohort of Patients with Chronic Granulomatous Disease. J. Clin. Immunol. 2020, 40, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Darmawan, G.; Kusumawardhani, R.N.Y.; Alisjahbana, B.; Fadjari, T.H. Chromobacterium Violaceum: The Deadly Sepsis. Acta Med. Indones. 2018, 50, 80–81. [Google Scholar]
- Rajani, P.S.; Slack, M.A. Papulopustular Dermatitis in X-Linked Chronic Granulomatous Disease. Front Pediatr 2018, 6, 429. [Google Scholar] [CrossRef]
- Valentine, G.; Thomas, T.A.; Nguyen, T.; Lai, Y.-C. Chronic Granulomatous Disease Presenting as Hemophagocytic Lymphohistiocytosis: A Case Report. Pediatrics 2014, 134, e1727–e1730. [Google Scholar] [CrossRef]
- Vignesh, P.; Loganathan, S.K.; Sudhakar, M.; Chaudhary, H.; Rawat, A.; Sharma, M.; Shekar, A.; Vaiphei, K.; Kumar, N.; Singh Sachdeva, M.-U.; et al. Hemophagocytic Lymphohistiocytosis in Children with Chronic Granulomatous Disease-Single-Center Experience from North India. J. Allergy Clin. Immunol. Pract. 2021, 9, 771–782. [Google Scholar] [CrossRef]
- Kohn, D.B.; Booth, C.; Kang, E.M.; Pai, S.-Y.; Shaw, K.L.; Santilli, G.; Armant, M.; Buckland, K.F.; Choi, U.; De Ravin, S.S.; et al. Lentiviral Gene Therapy for X-Linked Chronic Granulomatous Disease. Nat. Med. 2020, 26, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Rothe, M.; Santilli, G.; Charrier, S.; Stein, S.; Kunkel, H.; Abriss, D.; Müller-Kuller, U.; Gaspar, B.; Modlich, U.; et al. Non-Clinical Efficacy and Safety Studies on G1XCGD, a Lentiviral Vector for Ex Vivo Gene Therapy of X-Linked Chronic Granulomatous Disease. Hum Gene Ther Clin Dev 2018, 29, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Jofra Hernández, R.; Calabria, A.; Sanvito, F.; De Mattia, F.; Farinelli, G.; Scala, S.; Visigalli, I.; Carriglio, N.; De Simone, M.; Vezzoli, M.; et al. Hematopoietic Tumors in a Mouse Model of X-Linked Chronic Granulomatous Disease after Lentiviral Vector-Mediated Gene Therapy. Mol. Ther. 2021, 29, 86–102. [Google Scholar] [CrossRef]
- Farinelli, G.; Jofra Hernandez, R.; Rossi, A.; Ranucci, S.; Sanvito, F.; Migliavacca, M.; Brombin, C.; Pramov, A.; Di Serio, C.; Bovolenta, C.; et al. Lentiviral Vector Gene Therapy Protects XCGD Mice From Acute Staphylococcus Aureus Pneumonia and Inflammatory Response. Mol. Ther. 2016, 24, 1873–1880. [Google Scholar] [CrossRef]
- Renga, G.; Oikonomou, V.; Moretti, S.; Stincardini, C.; Bellet, M.M.; Pariano, M.; Bartoli, A.; Brancorsini, S.; Mosci, P.; Finocchi, A.; et al. Thymosin β4 Promotes Autophagy and Repair via HIF-1α Stabilization in Chronic Granulomatous Disease. Life Sci Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Renga, G.; Oikonomou, V.; Stincardini, C.; Pariano, M.; Borghi, M.; Costantini, C.; Bartoli, A.; Garaci, E.; Goldstein, A.L.; Romani, L. Thymosin β4 Limits Inflammation through Autophagy. Expert Opin. Biol. Ther. 2018, 18, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Jafarian, A.; Shokri, G.; Shokrollahi Barough, M.; Moin, M.; Pourpak, Z.; Soleimani, M. Recent Advances in Gene Therapy and Modeling of Chronic Granulomatous Disease. Iran. J. Allergy Asthma Immunol. 2019, 18, 131–142. [Google Scholar] [CrossRef]
- Kanariou, M.; Spanou, K.; Tantou, S. Long-Term Observational Studies of Chronic Granulomatous Disease. Curr. Opin. Hematol. 2018, 25, 7–12. [Google Scholar] [CrossRef]
- Flynn, R.; Grundmann, A.; Renz, P.; Hänseler, W.; James, W.S.; Cowley, S.A.; Moore, M.D. CRISPR-Mediated Genotypic and Phenotypic Correction of a Chronic Granulomatous Disease Mutation in Human iPS Cells. Exp. Hematol. 2015, 43, 838–848.e3. [Google Scholar] [CrossRef]
- Malech, H.L.; Maples, P.B.; Whiting-Theobald, N.; Linton, G.F.; Sekhsaria, S.; Vowells, S.J.; Li, F.; Miller, J.A.; DeCarlo, E.; Holland, S.M.; et al. Prolonged Production of NADPH Oxidase-Corrected Granulocytes after Gene Therapy of Chronic Granulomatous Disease. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 12133–12138. [Google Scholar] [CrossRef]
- Mukherjee, S.; Thrasher, A.J. Gene Therapy for PIDs: Progress, Pitfalls and Prospects. Gene 2013, 525, 174–181. [Google Scholar] [CrossRef]
- Lacerda-Pontes, R.; Gomes, L.N.; Albuquerque, R.S. de; Soeiro-Pereira, P.V.; Condino-Neto, A. The Extended Understanding of Chronic Granulomatous Disease. Curr. Opin. Pediatr. 2019, 31, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Chiriaco, M.; Salfa, I.; Di Matteo, G.; Rossi, P.; Finocchi, A. Chronic Granulomatous Disease: Clinical, Molecular, and Therapeutic Aspects. Pediatr. Allergy Immunol. 2016, 27, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Gennery, A.R. Progress in Treating Chronic Granulomatous Disease. Br. J. Haematol. 2021, 192, 251–264. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of normal phagocytic function reproduced from Kruger et al. (2015). Copyright: © 2015 Kruger et al. This is an open-access article distributed under the terms of the Creative Commons[2].
Figure 1.
Schematic representation of normal phagocytic function reproduced from Kruger et al. (2015). Copyright: © 2015 Kruger et al. This is an open-access article distributed under the terms of the Creative Commons[2].
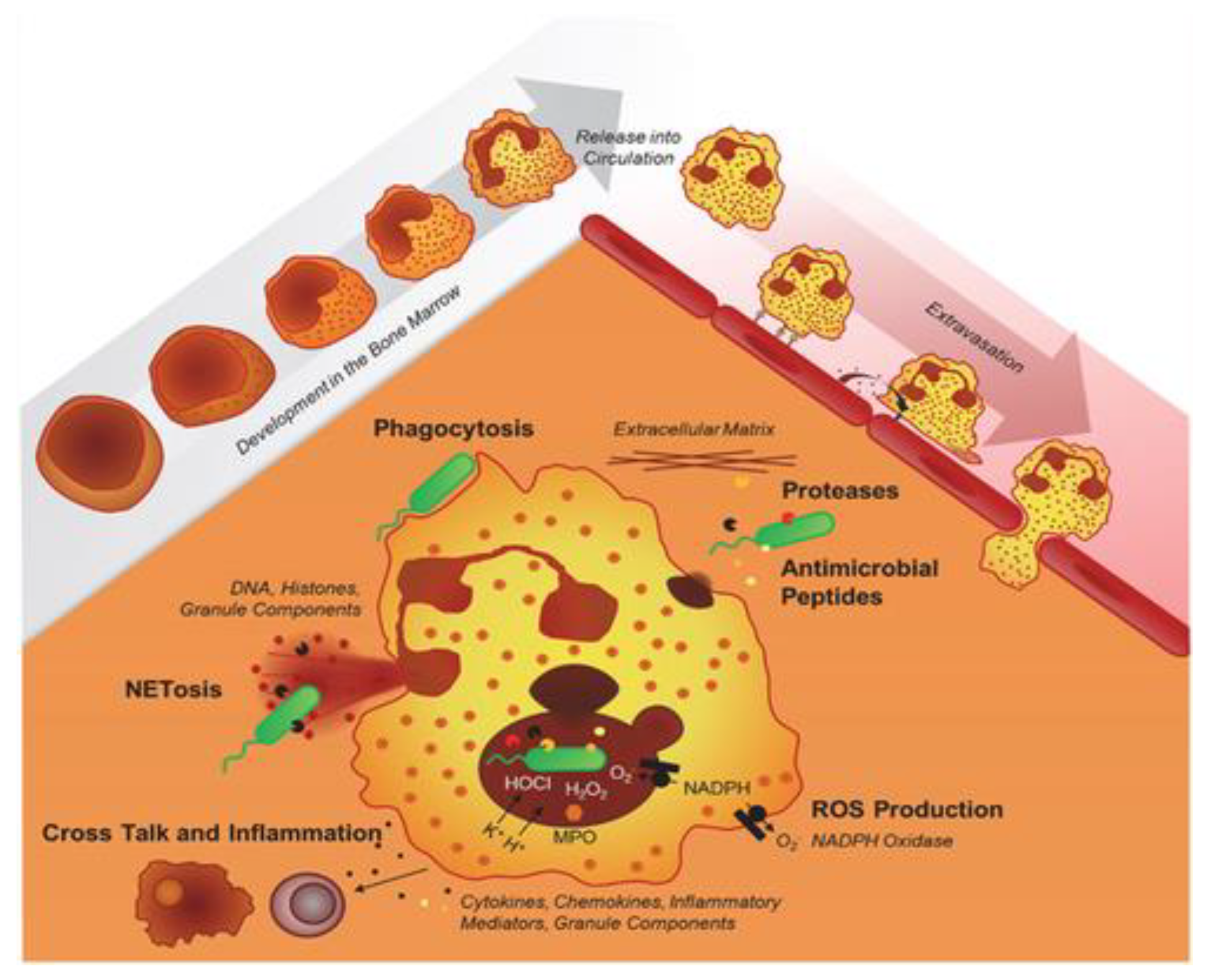
Figure 2.
Illustration of the NADPH oxidase complex’s subunits. The cytosolic components also include the molecule Rac (EROS), which interacts with other proteins. Adapted from Anjani et al. (2020)[3].
Figure 2.
Illustration of the NADPH oxidase complex’s subunits. The cytosolic components also include the molecule Rac (EROS), which interacts with other proteins. Adapted from Anjani et al. (2020)[3].
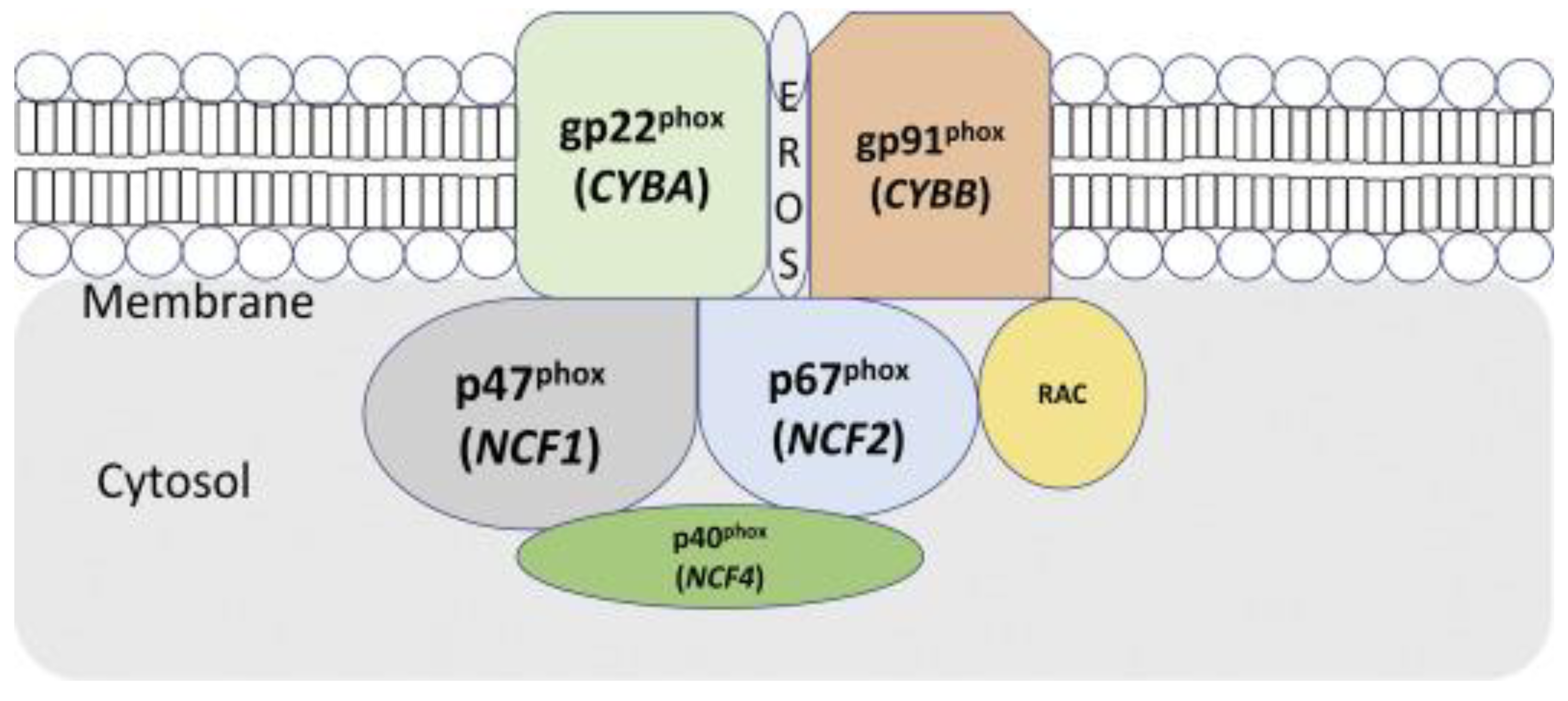
Figure 3.
Ninety per cent of patients with CGD harbour mutations in either CYBB (gp91phox) or NCF1 (p47phox). Mutations in either the membrane or cytosolic domain led to a disruption of respiratory burst in phagocytes. Adapted from [7].
Figure 3.
Ninety per cent of patients with CGD harbour mutations in either CYBB (gp91phox) or NCF1 (p47phox). Mutations in either the membrane or cytosolic domain led to a disruption of respiratory burst in phagocytes. Adapted from [7].
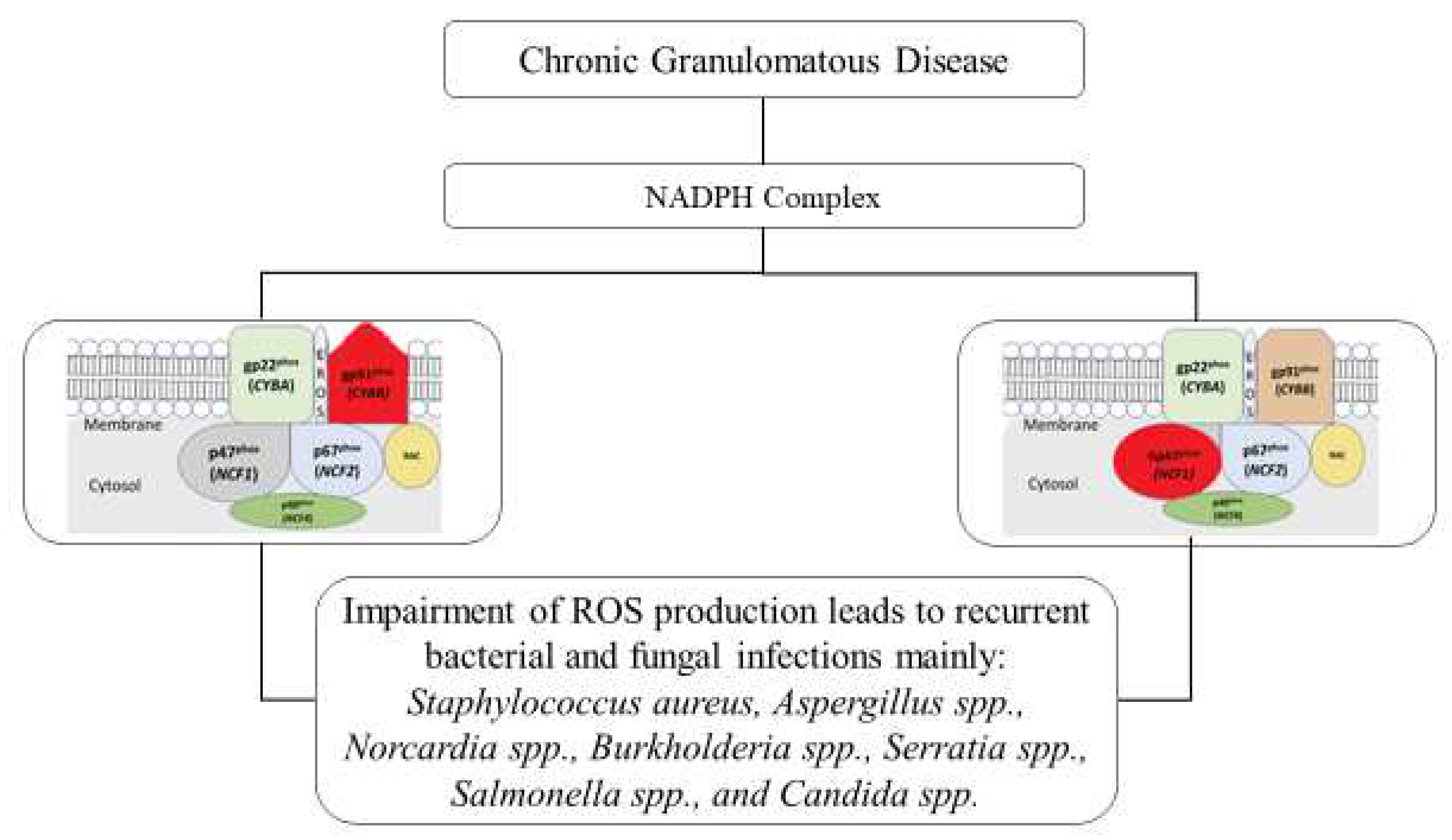
Figure 4.
Triple-dose antimicrobial prophylaxis is prescribed to newly diagnosed patients with severe CGD. IFN-γ is not part of prophylactic therapy; however, it can be added in severe cases and withdrawn after patient improvement. In many cases, mutational analysis is necessary for the initial diagnosis of CGD, particularly when there is no family history of CGD.
Figure 4.
Triple-dose antimicrobial prophylaxis is prescribed to newly diagnosed patients with severe CGD. IFN-γ is not part of prophylactic therapy; however, it can be added in severe cases and withdrawn after patient improvement. In many cases, mutational analysis is necessary for the initial diagnosis of CGD, particularly when there is no family history of CGD.
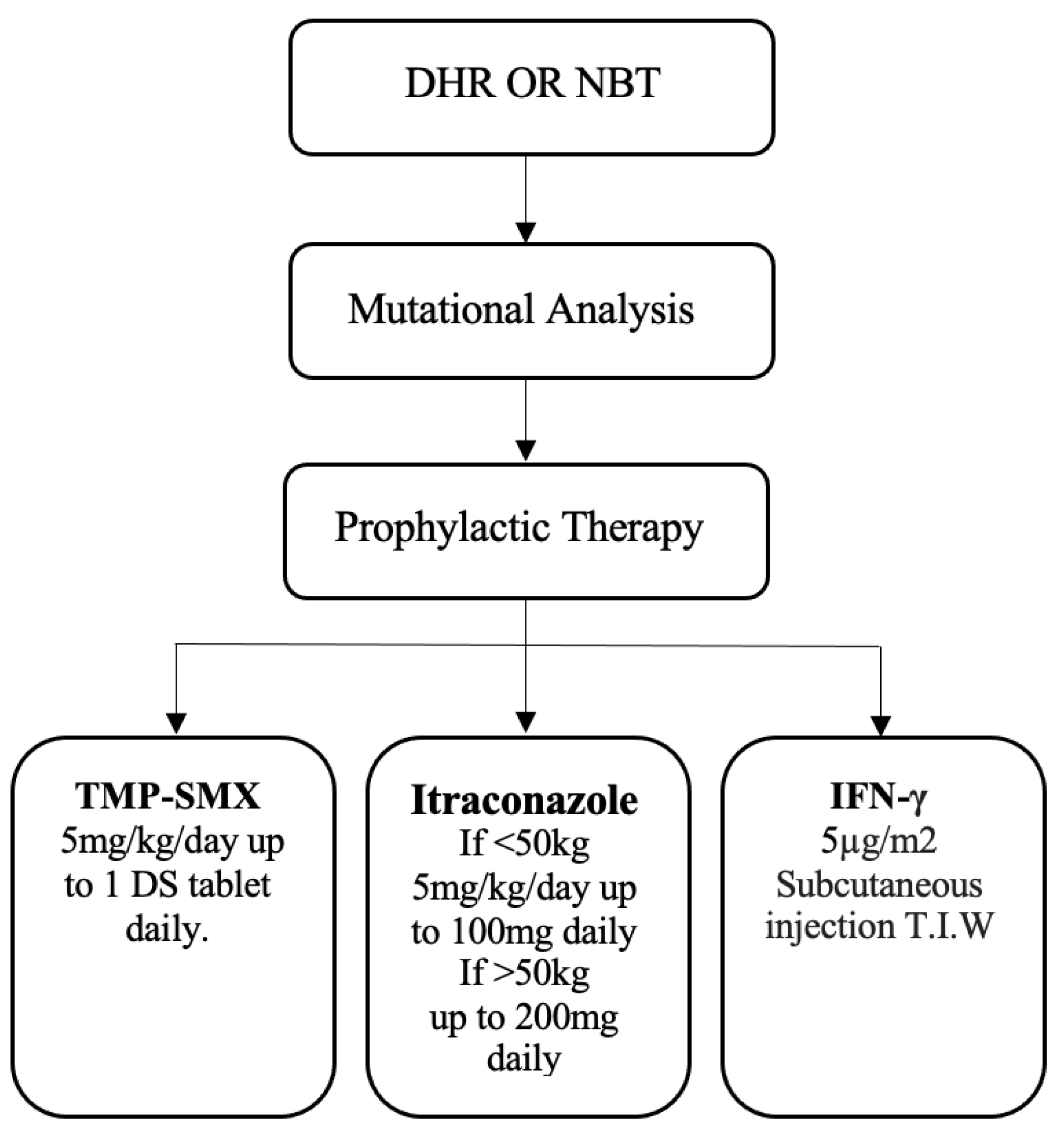

Table 1.
Pulmonary involvement and cultured microorganisms summarised from 634 episodes in 284 patients. Adapted from Van den Berg et al. [18].
Table 1.
Pulmonary involvement and cultured microorganisms summarised from 634 episodes in 284 patients. Adapted from Van den Berg et al. [18].
| Pneumonia | Lung Abscess | Granulomatous | Total | |
|---|---|---|---|---|
| Number of episodes | 597 | 26 | 11 | 634 |
| Cultured microorganism: | ||||
| Aspergillus spp. | 111 (18 %) | Klebsiella spp. | 4 (<1 %) | |
| Candida spp. | 10 (2 %) | Pseudomonas spp. | 3 (<1 %) | |
| S. aureus | 10 (2 %) | Streptococcus viridans | 3 (<1 %) | |
| H. influenzae | 6 (<1 %) | Actinomyces spp. | 2 (<1 %) | |
| M. tuberculosis | 5 (<1 %) | Acremonium spp. | 2 (<1 %) | |
| Atypical Mycobacteria | 3 (<1 %) | Other pathogens | 9 (2 %) | |
| Nocardia spp. | 4 (<1 %) | |||
| Negative culture / not done | 454 (72 %) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



MDPI Initiatives
Important Links
© 2024 MDPI (Basel, Switzerland) unless otherwise stated