
Submitted:
21 June 2023
Posted:
22 June 2023
You are already at the latest version
Abstract
Barth Syndrome, a rare X-linked disorder affecting 1:300,000 live births, results from defects in Tafazzin, an acyltransferase that remodels cardiolipin and is essential for mitochondrial respiration. Barth Syndrome patients develop cardiomyopathy, muscular hypotonia and cyclic neutropenia during childhood, rarely surviving to middle age. At present, no effective therapy exists and downstream transcriptional effects of Tafazzin dysfunction are incompletely understood. To identify novel, cell-specific, pathological pathways that mediate heart dysfunction, we performed single-nucleus RNA-sequencing (snRNA-seq) on wild-type (WT) and Tafazzin-knockout (Taz-KO) mouse hearts. We determined differentially expressed genes (DEGs) and inferred predicted cell-cell communication networks from these data. Surprisingly, DEGs were distributed heterogeneously across the cell types, with fibroblasts, cardiomyocytes, endothelial cells, macrophages, adipocytes and pericytes exhibiting the greatest number of DEGs between genotypes. One differentially expressed gene was detected for the lymphatic endothelial and mesothelial cell types, while no significant DEGs were found in the lymphocytes. A Gene Ontology (GO) analysis of these DEGs showed cell-specific effects on biological processes such as fatty acid metabolism in adipocytes and cardiomyocytes, increased translation in cardiomyocytes, endothelial cells and fibroblasts, in addition to other cell-specific processes. Analysis of ligand-receptor pair expression, to infer intercellular communication patterns, revealed the strongest dysregulated communication involved adipocytes and cardiomyocytes. For the knockout hearts, there was a strong loss of ligand-receptor pair expression involving adipocytes, and cardiomyocyte expression of ligand-receptor pairs underwent reorganization. These findings suggest that adipocyte and cardiomyocyte mitochondria may be most sensitive to mitochondrial Tafazzin deficiency and that rescuing adipocyte mitochondrial dysfunction, in addition to cardiomyocyte mitochondrial dysfunction, may provide therapeutic benefit in Barth Syndrome patients.
Keywords:
Tafazzin
; Barth Syndrome
; Single-Nucleus RNA-sequencing
; Cardiomyopathy
; Mitochondria
; Gene Expression
; Metabolism
1. Introduction
Barth Syndrome (BTHS), a rare X-linked mitochondrial disorder affecting 1:300,000 live births, is characterized by childhood onset cardiomyopathy, skeletal myopathy, cyclic neutropenia and premature death. To date, no efficacious therapy exists. The affected gene encodes Tafazzin, an inner mitochondrial membrane-associated transacylase and critical regulator of mitochondrial membrane composition. Tafazzin remodels the mitochondrial membrane phospholipid, cardiolipin, from its immature monolyso-isoform (MLCL) to its mature isoform (CL). At the organellar level, mitochondria lacking functional Tafazzin display altered cristae structure, reduced oxygen consumption, increased reactive oxygen species generation, altered substrate utilization and an elevated MLCL to CL ratio (reviewed in [1,2,3]). Failure to remodel cardiolipin to its mature form affects mitochondrial inner membrane curvature, resulting in failure to form electron transport protein supercomplexes [4,5]. Disease-causing mutations in Tafazzin are present throughout the molecule, and can affect protein localization, protein stability and enzymatic activity [6,7,8,9]. Although genomically encoded, Tafazzin is transported across the outer mitochondrial membrane and then localized to the inner mitochondrial membrane, directed by specific peptide domains [10,11]. The molecular, cellular, tissue, organ and physiological mechanisms by which Tafazzin mutations propagate dysfunction across multiscale networks are poorly understood, and a deeper understanding of the components of these networks will likely provide fundamental advances in knowledge of mitochondrially-associated disease mechanisms and the development of targeted therapies for mitochondrial disorders such as BTHS.
Single-cell RNA-sequencing methods have facilitated the analysis of cell-specific gene expression, cellular diversity and intercellular communication in a wide variety of tissues and across a wide variety of species—including human and mouse hearts—for both normal and diseased conditions [12,13,14,15,16,17,18,19,20,21]. As a first step in identifying transcriptional networks and pathological pathways that mediate the effects of Tafazzin deficiency in the heart at the single-cell level, we have performed single-nuclei transcriptomics on heart tissue extracted from a Tafazzin knockout (Taz-KO) mouse model of Barth Syndrome [22]. We have identified differentially expressed genes by genotype for specific cell populations and have found that fibroblasts, cardiomyocytes, endothelial cells, macrophages, adipocytes and pericytes demonstrate the largest numbers of DEGs. We correlated these cell-specific DEGs with potentially important biological processes through a Gene Ontology (GO) analysis and found cell-specific perturbations in metabolic pathways. We also analyzed ligand-receptor (L-R) pair gene expression to infer intercellular communication networks and identified extensive alterations in potential communication patterns, notably involving adipocytes.
2. Results
2.1. Tafazzin Deficiency is not Associated with a Disease-Specific Cell Population in the Heart
Whole hearts from 4 WT and 4 KO mice were used to generate the snRNA-seq dataset [17,18,19,20]. To identify the major cell types present in our data, we performed cell and gene-level quality control, followed by sample integration and graph-based clustering as described in Materials and Methods [23,24,25,26,27,28,29,30]. The integrated dataset, consisting of 75, 051 nuclei, revealed a total of 35 distinct clusters, represented by the Uniform Manifold Approximation and Projection (UMAP) plot shown in Supplemental Figure S1. The 35 clusters represented 12 major cell types found in the heart (Figure 1A, B).
Analysis of the distribution of these cell types per genotype revealed that all major cell types were represented by each genotype and no disease-specific cell clusters were present (S2). There were no significant differences between genotypes in the cell type distribution (Figure 1C), as determined by calculating the cell type diversity statistic (data not shown) [31]. The nuclei counts for each cell type for each genotype is listed in Supplemental Table ST1.
Major Cell Types Identified:
- Adipocytes (ADIPO);
- B-Lymphocytes (BLYMPH);
- Cardiomyocytes (CM);
- Dendritic Cells (DC);
- Endothelial Cells (EC);
- Fibroblasts (FB);
- Lymphatic Endothelial Cells (LEC);
- Macrophages (MAC);
- Mesothelial Cells (MES);
- Pericytes (PC);
- Smooth Muscle Cells (SM);
- T-Lymphocytes (TLYMPH)
2.2. Tafazzin Deficiency is Associated with Differential Expression of Genes that Varies by Cell Type and is Associated with Alterations in Multiple Biological Processes
We performed differential expression testing between Taz-KO and WT cells within each cell type using a generalized linear model framework as described in Materials and Methods [26,27,28,29,32]. Genes were considered to be differentially expressed if the absolute value of their log2fold-change was greater than 0.58 (adjusted P-value < 0.001). Surprisingly, DEGs were distributed heterogeneously, with fibroblasts, cardiomyocytes, endothelial cells, macrophages, adipocytes and pericytes exhibiting the greatest number of DEGs between genotypes; the lymphatic endothelial and mesothelial cell types each only had one differentially expressed gene detected and none were detected in the lymphocytes, dendritic cells or smooth muscle cells (Table 1). The full list of DEGs for each cell type are listed in supplemental table ST2.
To further understand the biological context of these DEGs and identify potential cell-specific dysregulated pathways associated with Tafazzin deficiency, we also performed a gene ontology over-representation test for biological process terms, on the sets of upregulated (increased expression in Taz-KO) and downregulated (decreased expression in Taz-KO) DEGs within each cell type. Over-represented terms for upregulated genes in the adipocytes largely involved lipid metabolic processes, such as acetyl-CoA and fatty acid synthesis, and were driven by the genes Acly, Fasn, Insig1, and Elovl6. Interestingly, fatty acid metabolism and specifically fatty acid beta-oxidation mapping genes such as Hadhb, Hadha, Acaa2, Acacb, and Acadm were downregulated in cardiomyocytes. Downregulated genes in adipocytes mapped only to one term—regulation of cell morphogenesis involved in differentiation—which was again conversely upregulated in the cardiomyocytes. Other terms mapping to the upregulated genes in cardiomyocytes included tRNA aminoacylation which were driven by the upregulation of aminoacyl-tRNA synthetase genes. Downregulated genes in the cardiomyocytes also mapped to circulatory system and muscle contraction related terms. Upregulated DEGs in the endothelial and fibroblast cell types mapped to the greatest number of gene ontology terms among the various cell types. As with the cardiomyocytes, upregulated terms for the fibroblasts and endothelial cells included those related to protein translation such as ribosome biogenesis. Regulation of vasculature development and Wnt signaling processes were downregulated in the endothelial cells and fibroblasts respectively. The upregulated gene set for macrophages mapped to extracellular matrix organization terms and in pericytes the upregulated genes mapped to the regulation of cell killing and protein folding, with both terms being driven by heat shock family proteins such as Hsp90ab1 and Hspa8. Zero terms mapped to the downregulated gene set in pericytes. Protein folding terms were common to the upregulated gene sets of fibroblasts, macrophages, and endothelial cells in addition to pericytes (Figure 2). The full list of GO terms over-represented by gene sets that are upregulated and downregulated for each cell type are listed in supplemental table ST3.
2.2. Tafazzin Deficiency is Associated with Alterations in General and Cell-Specific Ligand-Receptor Pair Gene Expression
To determine how Tafazzin deficiency affects potential communication between the major cell types, we performed a ligand-receptor (L-R) pair gene expression analysis comparing the two genotypes, as described in Materials and Methods [33]. Through this analysis, 9,011 dysregulated pairs were identified, with 7,237 pairs downregulated and 1,774 pairs upregulated in the Taz-KO condition. A tabular listing of the numbers of dysregulated ligands and receptors for each cell type is shown in Table 2. A graphical representation of these significantly dysregulated L-R interactions, separated by upregulation or downregulation in the Taz-KO cells, are shown in Figure 3.
Notably, adipocytes were associated with the largest number of dysregulated ligand-receptor interactions, and all significant pairs involving this cell type—as either the broadcasting or receiving cell type—were downregulated, suggesting a major disruption of cell-cell signaling involving adipocytes occurs among Tafazzin-deficient cells. Cardiomyocytes expressed the second largest number of dysregulated L-R pairs. For the remaining cell types with significantly dysregulated communication, this pattern of more downregulated than upregulated L-R pairs held, and there were no significant interactions involving B-lymphocytes, T-lymphocytes or dendritic cells.
Given that the major shares of dysregulated communication involve the adipocyte and cardiomyocyte cell types (Figure 4A), we performed another Gene Ontology over-representation analysis on the dysregulated ligands and receptors for each of the communicating cell type pairs that included adipocytes or cardiomyocytes (Figure 4). For adipocyte signaling, there was a broad downregulation of kinase activity and peptidyl-tyrosine phosphorylation with CM, EC, FB, LEC, MAC, MES, and PC cells. Phospholipase C activating signaling was downregulated in adipocyte communication with other adipocytes and PI3K communication was downregulated between adipocytes and smooth muscle cells (Figure 4B). Interestingly, for cardiomyocyte signaling the GO analysis revealed both a broad upregulation and downregulation of peptidyl-serine and peptidyl-threonine phosphorylation. This concordance among terms represented by opposing upregulated and downregulated signaling L-R pairs may be attributable to the broader nature of these biological process terms. For example, when looking at the specific up and downregulated gene sets that contributed to the GO term “Peptidyl-Serine Phosphorylation”, there was only a 34% overlap in the contributing gene lists (Figure 4C, D). Other cell-pair-specific ontology terms associated with cardiomyocyte signaling included upregulation of cell growth driven by signaling between CMs and FBs, upregulation of muscle cell proliferation driven by CM and MAC signaling, and downregulation of ossification driven by communication between cardiomyocytes and LECs, PCs, and other CMs (Figure 4C). A table of all significantly dysregulated L-R pairs can be found in supplemental table ST4.
3. Discussion
Barth Syndrome has long been known to result from mitochondrial dysfunction due to abnormal Tafazzin-dependent cardiolipin remodeling in the inner mitochondrial membrane that alters the efficiency of mitochondrial respiration (reviewed in [1,2,3]). Clinically, the disease manifests primarily in the heart, skeletal muscle and blood through the development of cardiomyopathy, skeletal myopathy, and cyclic neutropenia. Since mitochondria are present in all cells, the underlying susceptibility of specific cell types to dysfunction associated with Tafazzin deficiency has not been established–although the effects on striated muscle have been assumed to occur from the high energy requirements and increased mitochondrial numbers associated with these tissues. Here we report the first single nuclei transcriptomic analysis of hearts from Tafazzin-knockout mice that model Barth Syndrome [22,34]. As expected, Tafazzin loss of function is associated with widespread changes in cardiomyocyte gene expression, but other observed changes in adipocytes, fibroblasts, endothelial cells, macrophages, and pericytes were not anticipated. Analysis of predicted cell-cell communication networks through modeling ligand-receptor pair gene expression was notable for a marked downregulation of cell-cell interactions across all cell types in the Taz-KO condition. Given the sensitivity of this cell-cell communication analysis to differences in the product of a ligand and receptor’s expression across many cell type pairs, the contrast in the number of identified dysregulated L-R pairs compared to the number of identified DEGs per individual cell type may be expected. The predicted loss of cell-cell communication has also been noted in other conditions such as hypertrophic cardiomyopathy [17,19,20].
In a healthy heart, fatty acid metabolism serves as the primary pathway by which ATP levels, and therefore the heart’s contractile ability, is sustained; alterations in energy metabolism, and specifically fatty acid metabolism, have been observed in association with heart failure [35]. Prior studies have shown that in young BTHS patients myocardial fatty acid extraction and uptake is significantly reduced [36]. Here we report that in a mouse model of BTHS, alterations in fatty acid metabolism in the heart are cell type specific. Genes associated with fatty acid synthesis, such as those encoding the acetyl-CoA synthesis enzyme Acly, the fatty acid synthase Fasn, and the fatty acid elongase Elovl6, have increased expression in Taz-KO adipocytes, while those associated with fatty acid beta oxidation enzymes such as Hadhb, Hadha, Acaa2, Acacb, and Acadm are downregulated specifically in KO cardiomyocytes. Proteomic studies of cardiac mitochondria isolated from WT and Taz-shRNA-knockdown mice have implicated dysregulation of CoA-dependent fatty acid metabolism in BTHS through the downregulation of multiple enzymes involved in fatty acid oxidation [37]. We observed some concordance with this study in downregulation of CoA metabolism genes such as the medium-chain acyl-CoA dehydrogenase Acadm and the long-chain acyl-CoA synthetase Acsl1 in our Taz-KO cardiomyocytes [37]. The significance of the observed increase across many of the other cell types in expression of genes relating to protein translation and folding is unclear but may be related to a mitochondrial and protein homeostatic stress response in the Tafazzin deficient cells [38,39,40,41]. The common over-representation of post-translational modification and kinase activity Gene Ontology terms associated with dysregulated ligand-receptor pairs suggest phosphoproteomic studies of this BTHS model may also provide further insight to the biological pathways that are disrupted because of Tafazzin deficiency.
4. Materials and Methods
4.1. Generation of Single-Nuclei RNA-Seq Datasets from Tafazzin Knockout Mouse Hearts
Whole hearts from four WT and four Taz-KO mice were minced, cryopreserved, and then processed for snRNA-seq library generation and next generation sequencing as described previously [17,18,19,20] using commercially available reagents (10x Genomics). The Tafazzin-knockout mouse used in this study has been described previously [22,34]. All libraries were generated with tissue from 9-week-old mice.
4.2. Clustering of Cells by Gene Expression Pattern, Assignment of Cell Type Identity, and Determination of Cell Type Distribution
Sequencing reads, including intronic, were processed using Cell Ranger version 6.0.1 using the mm10-2020-A reference transcriptome [23]. Quality control measures were performed to correct for ambient RNA contamination (SoupX version 1.6.2)[24], and reduce doublets (DoubletFinder version 2.0.3)[25], followed by gene and cell-level filtering. When correcting for ambient RNA contamination, empty droplets with 2-10 transcripts (UMIs) were used to estimate the background expression profile and 0.2 was added to the estimated contamination fraction. For gene-level quality control, genes detected in fewer than 10 cells and all genes mapping to the mitochondrial genome were removed. Additionally, cells with fewer than 800 transcripts (nCount_RNA) or fewer than 250 genes (nFeature_RNA) were removed. Normalization, selection of the top 2000 highly variable genes, scaling, and linear dimension reduction (PCA) were performed on a merged dataset using the R package, Seurat 4.3.0 [26,27,28,29]. To enable the identification of shared cell types across cells from different genotypes, Harmony version 0.1.0 was used to generate a common reduced dimensional embedding from the merged dataset [30] followed by running Seurat's UMAP, Nearest Neighbor, and clustering functions using the first 15 principal components from these data [26,27,28,29]. The clustering resolution of 0.9 was chosen using the subsampling-based approach ChooseR [42]. The expression of known cell-specific gene markers was used to identify major cell types, as has been done previously [14,16,17,18,19]. To further refine cell assignments, such as distinguishing specific myeloid or lymphoid cell populations, Seurat's FindSubCluster function was utilized [26,27,28,29]. To determine whether the cell type distribution varied between WT and Taz-KO hearts, we calculated the Cell Type Diversity Statistic [31].
4.3. Differential Expression Analysis
Differentially expressed genes between WT and Taz-KO cells, for each cell type, were determined using MAST, a generalized linear model framework that uses the proportion of genes expressed in a single cell as a covariate to account for both technical and biological sources of variation [32]. The MAST framework was implemented through Seurat’s FindMarkers function, adjusting for individual mouse variation using the "latent.vars” argument [26,27,28,29]. Genes were considered differentially expressed if the absolute value of their log2fold-change was greater than 0.58 (adjusted P-value < 0.001). A biological process Gene Ontology analysis was conducted for each set of upregulated and downregualted DEGs, for each cell type, using the compareCluster function of clusterProfiler version 3.18.1[43]. All genes that passed gene-level quality control filtering were used for the background set (“universe” argument).
4.2. Cell-Cell Communication Analysis
To identify potential cardiac cell–cell communication differences between WT and Taz-KO mouse hearts, we used scLR, a statistical method for examining dysregulated L-R interactions, between two conditions, that models the distribution of ligand and receptor expression and accounts for intersample variance and small sample size [33]. The curated set of L-R pairs used for comparison was obtained from the Omnipath database [44]. Cell communication networks were plotted using igraph version 1.3.5 [45]. Lines in our cell networks connect two cell types (circles) and represent statistically significant dysregulated ligand–receptor pairs (i.e., cell–cell communication between a broadcasting (ligand) and recipient (receptor) cell types). Line color in our networks represents the broadcasting ligand source cell type. Line thickness is proportional to the number of dysregulated ligand–receptor pairs associated between two communicating cell types. The Gene Ontology analysis of differentially expressed ligand-receptor pairs involving adipocytes or cardiomyocytes was performed using the enrichGO function of clusterProfiler [43].
5. Conclusions
Barth Syndrome cardiomyopathy has been associated with profound alterations in mitochondrial and contractile function but determination of cellular mechanisms that mediate the effects of Tafazzin deficiency beyond mitochondrial dysfunction, and the contribution of nonmyocytes to the cardiomyopathic phenotype have not been fully elucidated. Here we report that Tafazzin loss of function in a mouse model of Barth Syndrome results in distinct alterations in cardiomyocyte gene expression and we also identify adipocyte dysfunction as a potential contributor to Barth Syndrome cardiomyopathy potentially through dysregulated fatty acid metabolism.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: UMAP plot of Cells Labeled by Cluster; Figure S2: UMAP plot of Cells Labeled by Cell Type and Separated by Genotype; Table ST1: Nuclei Counts per Cell Type and Genotype; Table ST2: Differentially Expressed Genes by Cell Type; Table ST3: Gene Ontology Terms Associated with Differentially Expressed Genes; Table ST4: Dysregulated Ligand Receptor Pairs.
Author Contributions
Conceptualization, M.C.; methodology, G.P., L.P., A.L., C.C., R.B.; software, G.P., L.P., A.L., C.C., R.B.; validation, M.C. and G.P.; formal analysis, G.P. and L.P..; investigation, L.P. and A.L.; resources, M.C., D.S. and J.A.; data curation, G.P., L.P.; writing—original draft preparation, M.C..; writing—review and editing, M.C., G.P.; visualization, G.P., L.P., M.C..; supervision, M.C.; project administration, M.C., G.P., J.A.; funding acquisition, M.C. All authors have read and agreed to the published version of the manuscript.
Funding
M.C.’s research laboratory was funded by the National Institutes of Health, grant number 1 R61 HL154137-01A1. L.P. was funded by National Institutes of Health, grant number 5T32GM008448-26.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Review Board of Tufts University (protocol code B2020-40, date of approval May 28, 2020).
Data Availability Statement
The datasets used in this study are available online via the Gene Expression Omnibus database under accession number GSE235047.
Acknowledgments
We thank members of the Chin lab for helpful discussions. The authors also acknowledge the Tufts University High Performance Compute Cluster (https://it.tufts.edu/high-performance-computing) which was utilized for the research reported in this paper.
Conflicts of Interest
M.C. is founder, equity shareholder and C.E.O. of TransCellular Therapeutics, Inc., an early-stage biopharmaceutical company dedicated to developing therapeutics for Barth Syndrome. The remaining authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Clarke, Sarah LN, et al. “Barth Syndrome.” Orphanet Journal of Rare Diseases, vol. 8, no. 1, 2013. [CrossRef]
- Chin, Michael T., and Simon J. Conway. “Role of Tafazzin in Mitochondrial Function, Development and Disease.” Journal of Developmental Biology, vol. 8, no. 2, 2020, p. 10. [CrossRef]
- Thompson, Reid, et al. “Current and Future Treatment Approaches for Barth Syndrome.” Journal of Inherited Metabolic Disease, vol. 45, no. 1, 2021, pp. 17–28. [CrossRef]
- Schlame, Michael, et al. “The Physical State of Lipid Substrates Provides Transacylation Specificity for Tafazzin.” Nature Chemical Biology, vol. 8, no. 10, 2012, pp. 862–869. [CrossRef]
- McKenzie, Matthew, et al. “Mitochondrial Respiratory Chain Supercomplexes Are Destabilized in Barth Syndrome Patients.” Journal of Molecular Biology, vol. 361, no. 3, 2006, pp. 462–469. [CrossRef]
- Claypool, Steven M., J. Michael McCaffery, et al. “Mitochondrial Mislocalization and Altered Assembly of a Cluster of Barth Syndrome Mutant Tafazzins.” Journal of Cell Biology, vol. 174, no. 3, 2006, pp. 379–390. [CrossRef]
- Claypool, Steven M., Kevin Whited, et al. “Barth Syndrome Mutations That Cause TAFAZZIN Complex Lability.” Journal of Cell Biology, vol. 192, no. 3, 2011, pp. 447–462. [CrossRef]
- Whited, Kevin, et al. “Seven Functional Classes of Barth Syndrome Mutation.” Human Molecular Genetics, vol. 22, no. 3, 2012, pp. 483–492. [CrossRef]
- Lu, Ya-Wen, et al. “Defining Functional Classes of Barth Syndrome Mutation in Humans.” Human Molecular Genetics, vol. 25, no. 9, 2016, pp. 1754–1770. [CrossRef]
- Herndon, Jenny D., et al. “The Taz1p Transacylase Is Imported and Sorted into the Outer Mitochondrial Membrane via a Membrane Anchor Domain.” Eukaryotic Cell, vol. 12, no. 12, 2013, pp. 1600–1608. 1600. [CrossRef]
- Dinca, Ana A., et al. “Identification of Novel Mitochondrial Localization Signals in Human Tafazzin, the Cause of the Inherited Cardiomyopathic Disorder Barth Syndrome.” Journal of Molecular and Cellular Cardiology, vol. 114, 2018, pp. 83–92. [CrossRef]
- Gladka, Monika M., et al. “Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation.” Circulation, vol. 138, no. 2, 2018, pp. 166–180. [CrossRef]
- Wehrens, Martijn, et al. “Single-Cell Transcriptomics Provides Insights into Hypertrophic Cardiomyopathy.” Cell Reports, vol. 39, no. 6, 2022, p. 110809. [CrossRef]
- Tucker, Nathan R., et al. “Transcriptional and Cellular Diversity of the Human Heart.” Circulation, vol. 142, no. 5, 2020, pp. 466–482. [CrossRef]
- Litviňuková, Monika, et al. “Cells of the Adult Human Heart.” Nature, vol. 588, no. 7838, 2020, pp. 466–472. [CrossRef]
- McLellan, Micheal A., et al. “High-Resolution Transcriptomic Profiling of the Heart during Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy.” Circulation, vol. 142, no. 15, 2020, pp. 1448–1463. [CrossRef]
- Codden, Christina J, et al. “Single Nucleus RNA-Sequencing Reveals Altered Intercellular Communication and Dendritic Cell Activation in Nonobstructive Hypertrophic Cardiomyopathy.” Cardiology and Cardiovascular Medicine, vol. 06, no. 04, 2022. [CrossRef]
- Larson, Amy, and Michael T. Chin. “A Method for Cryopreservation and Single Nucleus RNA-Sequencing of Normal Adult Human Interventricular Septum Heart Tissue Reveals Cellular Diversity and Function.” BMC Medical Genomics, vol. 14, no. 1, 2021. [CrossRef]
- Larson, Amy, et al. “Altered Intercellular Communication and Extracellular Matrix Signaling as a Potential Disease Mechanism in Human Hypertrophic Cardiomyopathy.” Scientific Reports, vol. 12, no. 1, 2022. [CrossRef]
- Codden, Christina J., and Michael T. Chin. “Common and Distinctive Intercellular Communication Patterns in Human Obstructive and Nonobstructive Hypertrophic Cardiomyopathy.” International Journal of Molecular Sciences, vol. 23, no. 2, 2022, p. 946. [CrossRef]
- Chaffin, Mark, et al. “Single-Nucleus Profiling of Human Dilated and Hypertrophic Cardiomyopathy.” Nature, vol. 608, no. 7921, 2022, pp. 174–180. [CrossRef]
- Tomczewski, Michelle V., et al. “Phenotypic Characterization of Male Tafazzin-Knockout Mice at 3, 6, and 12 Months of Age.” Biomedicines, vol. 11, no. 2, 2023, p. 638. [CrossRef]
- Zheng, Grace X., et al. “Massively Parallel Digital Transcriptional Profiling of Single Cells.” Nature Communications, vol. 8, no. 1, 2017. [CrossRef]
- Young, Matthew D, and Sam Behjati. “SoupX Removes Ambient RNA Contamination from Droplet-Based Single-Cell RNA Sequencing Data.” GigaScience, vol. 9, no. 12, 2020. [CrossRef]
- McGinnis, Christopher S., et al. “DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors.” Cell Systems, vol. 8, no. 4, 2019. [CrossRef]
- Hao, Yuhan, et al. “Integrated Analysis of Multimodal Single-Cell Data.” Cell, vol. 184, no. 13, 2021. [CrossRef]
- Stuart, Tim, et al. “Comprehensive Integration of Single-Cell Data.” Cell, vol. 177, no. 7, 2019. [CrossRef]
- Butler, Andrew, et al. “Integrating Single-Cell Transcriptomic Data across Different Conditions, Technologies, and Species.” Nature Biotechnology, vol. 36, no. 5, 2018, pp. 411–420. [CrossRef]
- Satija, Rahul, et al. “Spatial Reconstruction of Single-Cell Gene Expression Data.” Nature Biotechnology, vol. 33, no. 5, 2015, pp. 495–502. [CrossRef]
- Korsunsky, Ilya, et al. “Fast, Sensitive and Accurate Integration of Single-Cell Data with Harmony.” Nature Methods, vol. 16, no. 12, 2019, pp. 1289–1296. [CrossRef]
- Karagiannis, Tanya T, et al. “Cell Type Diversity Statistic: An Entropy-Based Metric to Compare Overall Cell Type Composition across Samples.” Frontiers in Genetics, vol. 13, 2022. [CrossRef]
- Finak, Greg, et al. “MAST: A Flexible Statistical Framework for Assessing Transcriptional Changes and Characterizing Heterogeneity in Single-Cell RNA Sequencing Data.” Genome Biology, vol. 16, no. 1, 2015. [CrossRef]
- Liu, Qi, et al. “Dysregulated Ligand–Receptor Interactions from Single-Cell Transcriptomics.” Bioinformatics, vol. 38, no. 12, 2022, pp. 3216–3221. [CrossRef]
- Wang, Suya, et al. “AAV Gene Therapy Prevents and Reverses Heart Failure in a Murine Knockout Model of Barth Syndrome.” Circulation Research, vol. 126, no. 8, 2020, pp. 1024–1039. [CrossRef]
- Lopaschuk, Gary D., et al. “Cardiac Energy Metabolism in Heart Failure.” Circulation Research, vol. 128, no. 10, 2021, pp. 1487–1513. [CrossRef]
- Cade, William Todd, et al. “Myocardial Glucose and Fatty Acid Metabolism Is Altered and Associated with Lower Cardiac Function in Young Adults with Barth Syndrome.” Journal of Nuclear Cardiology, vol. 28, no. 4, 2019, pp. 1649–1659. [CrossRef]
- Le, Catherine H., et al. “Tafazzin Deficiency Impairs COA-Dependent Oxidative Metabolism in Cardiac Mitochondria.” Journal of Biological Chemistry, vol. 295, no. 35, 2020, pp. 12485–12497. [CrossRef]
- Topf, Ulrike, et al. “Mitochondrial Stress-Dependent Regulation of Cellular Protein Synthesis.” Journal of Cell Science, vol. 132, no. 8, 2019. [CrossRef]
- Zhao, Q. “A Mitochondrial Specific Stress Response in Mammalian Cells.” The EMBO Journal, vol. 21, no. 17, 2002, pp. 4411–4419. [CrossRef]
- De Taffin de Tilques, Maxence, et al. “Decreasing Cytosolic Translation Is Beneficial to Yeast and Human Tafazzin-Deficient Cells.” Microbial Cell, vol. 5, no. 5, 2018, pp. 220–232. [CrossRef]
- Kim, Hyun-Eui, et al. “Lipid Biosynthesis Coordinates a Mitochondrial-to-Cytosolic Stress Response.” Cell, vol. 166, no. 6, 2016. [CrossRef]
- Patterson-Cross, Ryan B., et al. “Selecting Single Cell Clustering Parameter Values Using Subsampling-Based Robustness Metrics.” BMC Bioinformatics, vol. 22, no. 1, 2021. [CrossRef]
- Yu, Guangchuang, et al. “Clusterprofiler: An R Package for Comparing Biological Themes among Gene Clusters.” OMICS: A Journal of Integrative Biology, vol. 16, no. 5, 2012, pp. 284–287. [CrossRef]
- Türei, Dénes, et al. “Integrated Intra- and Intercellular Signaling Knowledge for Multicellular Omics Analysis.” Molecular Systems Biology, vol. 17, no. 3, 2021. [CrossRef]
- Csárdi, Gábor and Tamás Nepusz. “The igraph software package for complex network research.” (2006).
Figure 1.
Single-nucleus RNA-Sequencing of wild type and Tafazzin-knockout hearts reveals diverse cell types but no genotype-specific cell population or differences in distribution of major cell types: (A) UMAP plot of major cell types of the heart identified in the dataset. Each dot represents a single nucleus colored by its cell type identity; (B) Dot plot showing expression of known cell markers for identified cell types; (C) Distribution of cell types for each genotype.
Figure 1.
Single-nucleus RNA-Sequencing of wild type and Tafazzin-knockout hearts reveals diverse cell types but no genotype-specific cell population or differences in distribution of major cell types: (A) UMAP plot of major cell types of the heart identified in the dataset. Each dot represents a single nucleus colored by its cell type identity; (B) Dot plot showing expression of known cell markers for identified cell types; (C) Distribution of cell types for each genotype.
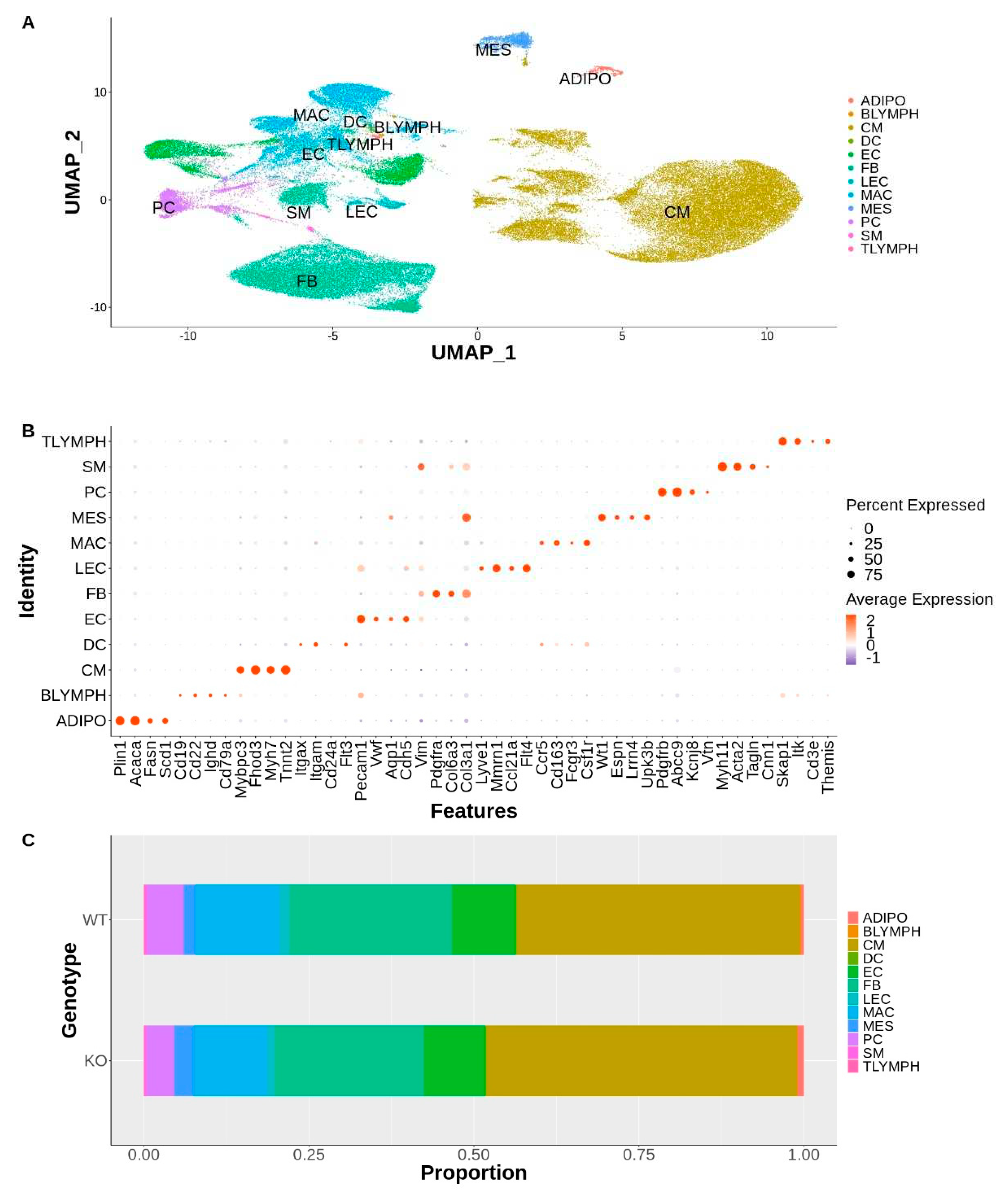
Figure 2.
A Gene Ontology analysis of differentially expressed genes reveals cell-specific differences in multiple biological processes associated with Tafazzin deficiency: (A) Dot plot of over-represented biological process terms, by cell type, for genes upregulated in Taz-KO cells. Only the top 3 terms, by count for each cell type, are displayed; (B) Dot plot of over-represented biological process terms, by cell type, for genes downregulated in Taz-KO cells. Only the top 3 terms, by count for each cell type, are displayed; (C) Heatmaps showing scaled expression of genes associated with select over-represented ontology terms. Asterisks indicate differentially expressed genes with adjusted P-values < 0.001 and |average log2 fold change| > 0.58.
Figure 2.
A Gene Ontology analysis of differentially expressed genes reveals cell-specific differences in multiple biological processes associated with Tafazzin deficiency: (A) Dot plot of over-represented biological process terms, by cell type, for genes upregulated in Taz-KO cells. Only the top 3 terms, by count for each cell type, are displayed; (B) Dot plot of over-represented biological process terms, by cell type, for genes downregulated in Taz-KO cells. Only the top 3 terms, by count for each cell type, are displayed; (C) Heatmaps showing scaled expression of genes associated with select over-represented ontology terms. Asterisks indicate differentially expressed genes with adjusted P-values < 0.001 and |average log2 fold change| > 0.58.
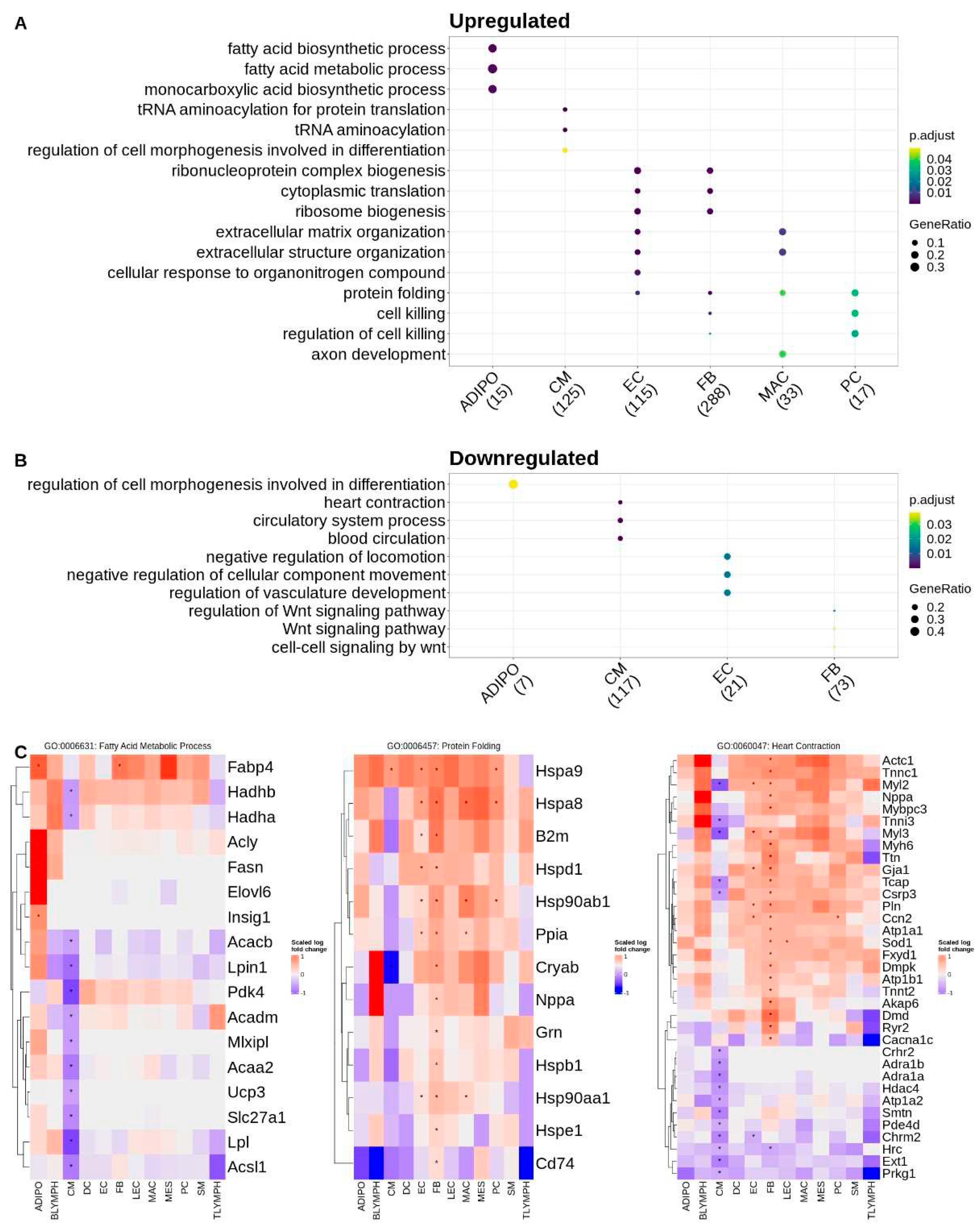
Figure 3.
A ligand-receptor pair expression analysis reveals downregulation of many predicted cell-cell interactions in Taz-KO cells. The number of significantly dysregulated ligand-receptor interactions between two cell types are graphically represented by the thickness of lines connecting two circles (cell types) and the color of a line reflects the cell type from which the ligands are being broadcasted from. Dysregulated communication that is downregulated in the Taz-KO cells are shown on the left and dysregulated communication that is upregulated in the Taz-KO cells are shown on the right.
Figure 3.
A ligand-receptor pair expression analysis reveals downregulation of many predicted cell-cell interactions in Taz-KO cells. The number of significantly dysregulated ligand-receptor interactions between two cell types are graphically represented by the thickness of lines connecting two circles (cell types) and the color of a line reflects the cell type from which the ligands are being broadcasted from. Dysregulated communication that is downregulated in the Taz-KO cells are shown on the left and dysregulated communication that is upregulated in the Taz-KO cells are shown on the right.
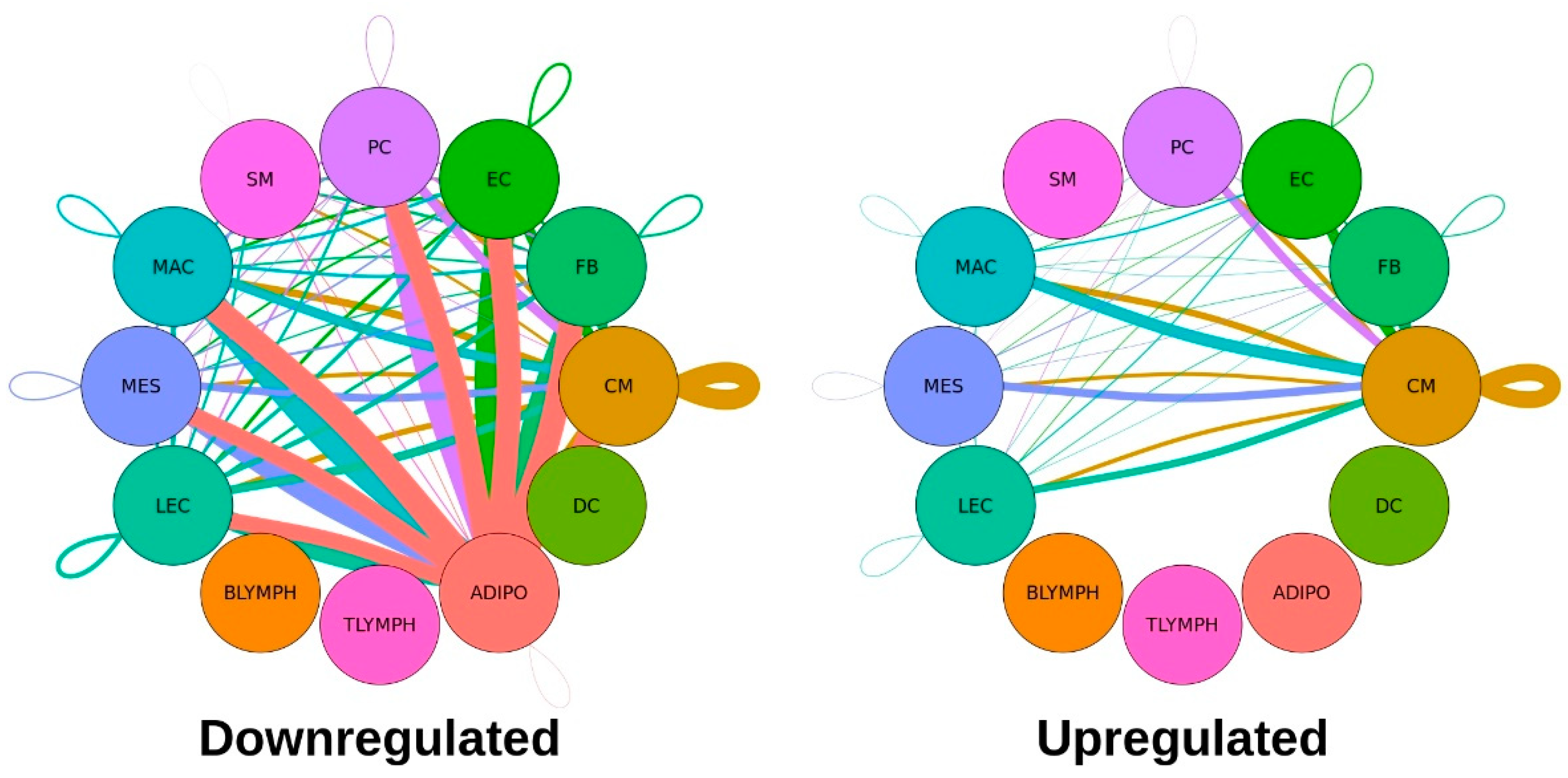
Figure 4.
A biological process gene ontology analysis of dysregulated ligand-receptor pairs reveals differences in communication associated with Tafazzin deficiency: (A) Number of dysregulated pairs between two communicating cell types; (B) Dot plot of over-represented biological process terms, by cell type, for all genes contributing to dysregulated signaling involving adipocytes. Only the top 3 terms, by count for each cell type pair, are displayed; (C) Dot plot of over-represented biological process terms, by cell type, for all genes contributing to up or downregulated signaling involving cardiomyocytes. Only the top 3 terms, by count for each cell type pair, are displayed; (D) Venn diagram showing overlap of genes of dysregulated ligand-receptor pairs that contribute to the GO term “Peptidyl-Serine Phosphorylation” in up and down regulated L-R pairs involving cardiomyocytes.
Figure 4.
A biological process gene ontology analysis of dysregulated ligand-receptor pairs reveals differences in communication associated with Tafazzin deficiency: (A) Number of dysregulated pairs between two communicating cell types; (B) Dot plot of over-represented biological process terms, by cell type, for all genes contributing to dysregulated signaling involving adipocytes. Only the top 3 terms, by count for each cell type pair, are displayed; (C) Dot plot of over-represented biological process terms, by cell type, for all genes contributing to up or downregulated signaling involving cardiomyocytes. Only the top 3 terms, by count for each cell type pair, are displayed; (D) Venn diagram showing overlap of genes of dysregulated ligand-receptor pairs that contribute to the GO term “Peptidyl-Serine Phosphorylation” in up and down regulated L-R pairs involving cardiomyocytes.

Table 1.
Number of Differentially Expressed Genes Detected in Each Cell Type.
| Cell Type | Upregulated | Downregulated | Total |
|---|---|---|---|
| Adipocyte | 16 | 8 | 24 |
| B-Lymphocyte | 0 | 0 | 0 |
| Cardiomyocyte | 154 | 141 | 295 |
| Dendritic | 0 | 0 | 0 |
| Endothelial | 121 | 21 | 142 |
| Fibroblast | 296 | 75 | 371 |
| Lymphatic Endothelial | 1 | 0 | 1 |
| Macrophage | 33 | 0 | 33 |
| Mesothelial | 1 | 0 | 1 |
| Pericyte | 17 | 5 | 22 |
| Smooth Muscle | 0 | 0 | 0 |
| T-Lymphocyte | 0 | 0 | 0 |
1 Number of significantly up or downregulated genes in Taz-KO cells with adjusted P-values < 0.001 and |average log2fold-change | > 0.58.
Table 2.
Number of Significantly Dysregulated Ligands and Receptors by Cell Type.
| Cell Type | Downregulated | Upregulated | ||
| Ligands | Receptors | Ligands | Receptors | |
| Adipocyte | 2009 | 2289 | 0 | 0 |
| B-Lymphocyte | 0 | 0 | 0 | 0 |
| Cardiomyocyte | 1162 | 1295 | 676 | 974 |
| Dendritic | 0 | 0 | 0 | 0 |
| Endothelial | 749 | 622 | 204 | 217 |
| Fibroblast | 680 | 679 | 226 | 143 |
| Lymphatic Endothelial | 661 | 541 | 142 | 114 |
| Macrophage | 716 | 635 | 213 | 132 |
| Mesothelial | 555 | 452 | 157 | 95 |
| Pericyte | 620 | 476 | 156 | 99 |
| Smooth Muscle | 85 | 248 | 0 | 0 |
| T-Lymphocyte | 0 | 0 | 0 | 0 |
| Total | 7237 | 7237 | 1774 | 1774 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



