Submitted:
22 June 2023
Posted:
23 June 2023
You are already at the latest version
Abstract
Proteomics in respiratory allergic diseases has such a battery of techniques and programs that one would almost think that there is nothing impossible to find, invent or mold. All these resources that we document here, are involved in solving problems in allergic diseases, both diagnostic, prognostic, treatment and immunotherapy development. The main perspectives, according to this version, are in three strands and / or lockout immunological system: 1) In blocking the diapedesis of cells involved, 2) modifications and blocking of paratopes and epitopes being understood by modifications to antibodies, antagonism or blocking them and 3) blocking FcεR1 receptors to prevent specific IgEs from sticking to mast cells and basophils. These tools and targets in the allergic landscape are, in our view, the prospects in the field.
Keywords:
Proteomics
; ELISA
; mass spectrometry
; Liquid chromatography
; Luminex
; Uniprot
; Rosseta comment
; Mascot
; allergy respiratory disease
; inflammation type 2
; diapedesis
; epitopo
1. Introduction
Proteomics is the study of proteins in cells, tissues, and organisms during homeostasis or under specific conditions. The function of proteins in altered, natural or specific conditions can be studied using proteomics. Mark Wilkins coined the term "proteome" and proteomics are used particularly in medicine and molecular biology, and these fields have made progress in identifying all known proteins and their posttranslational modifications. Proteomics has also provided information that has been uploaded to databases and serves as algorithms in free programs today [1]. In the natural sciences, chemistry, medicine and other fields of health research, proteomics has enabled the study of protein function, localization, production and modification. It has also allowed a deeper understanding of the normal functioning of an organism or, in contrast, the damage caused by its alteration. For this reason, the potential application of proteomics in biomedicine can facilitate the development of therapeutic agents, improve disease diagnosis and prognosis methods, and treat health defects (congenital or hereditary). In addition, it can be used to temporarily or permanently improve a defective protein, as in the case of cystic fibrosis [2].
Its application is sometimes confused with the expression of genes, since it is believed that expression changes will be observed with proteomics. However, the expression of a gene does not always lead to the expression of an active protein, so proteomics should not be confused with genomics or transcriptomics. Genomics studies investigate the complete genome (all of the genes in one organism) and their expression through the transcriptome, which represents the transcripts or RNA molecules present in a cell or group of cells at a certain time or in different conditions [3].
The study of proteins in living systems, especially those directed at the expression or altered structures of proteins, aids in the search for protein markers, which can result in the design of diagnostic biomarkers and the discovery of new protein disease defects and effective follow-up therapy. Therapies can consist of the replacement of defective proteins, the production of epitope antagonists that trigger opposing responses or the discovery of proteins that block receptors for cell signaling. The study of protein anomalies through software has also increased in frequency. The structure of proteins is predicted, allowing us to identify any potential alterations. At present, numerous strategies are used for the discovery, remodelling and structural analysis of proteins for diagnostic, therapeutic, biochemical, molecular and other purposes [4]. These strategies have also been used in conjunction with other techniques, such as protein engineering, pharmacogenomics, and other omics sciences. Since different disciplines are involved in proteomics, it important to collaborate on shared public health problems and work towards a common goal [5].
Proteomics identifies normal proteins and their molecular anomalies, and large amounts of biological data are quickly obtained and classified from a single sample or from the analysis of biological fluids of both healthy and sick individuals. In addition, we are seeking to improve the diagnostic methods that can be used to identify these molecular anomalies quickly and easily. The main applications of proteomics in human diseases are focused on the function of native proteins and their alterations. It has also focused on the identification of pharmacological targets, the improvement of clinical diagnosis and the modifications of such proteins by means of software prediction. Protein variations can produce nonharmful or harmful alterations or diseases such as Alzheimer's, Parkinson's, amyloidosis and even Creutzfeldt‒Jakob disease. Likewise, harmless protein modifications can affect individuals differently, as is the case of allergies [6].
2. Global characteristics of allergies for proteomics studies
Proteomics is a field of science that is approached in an interdisciplinary way and studies the complexity and dynamics of proteins in biological systems, mainly, as already said, in structure and function. Proteomics is a powerful analytical and identification technology but also has software with advanced systems to model, predict and understand the probable function of proteins within cells and organisms. Consequently, diseases that depend on the expression of proteins or have protein mediators can be thoroughly studied and analysed using proteomics.
The development of respiratory allergic diseases involves complex and multifactorial components. These components include intrinsic factors, such as genetic predisposition, sex, age and ethnicity. Recent studies have identified approximately 120 genes associated with allergic diseases [10]. The proteomic approach is vital for understanding the immunological alterations in allergies, the protein characteristics of allergens and the general characteristics of the host, as well as the characteristics of the antigen and the sequence of the epitope or epitopes of that protein. The main purpose of these approaches is to improve research tools, improve the status of patients, optimize the diagnosis, make the therapies used more efficient, improve the current ones and develop new ones. Additionally, elucidating the epidemiological aspects, such as identifying who and how they suffer from these allergic diseases, is important. By these means, we have identified which allergies are more frequent, and the age, sex, race and population with the highest incidence [11].
Moreover, the second important aspect in the study of allergies is the extrinsic factors, which are factors attributed to the external environment, lifestyle and comorbidities. Exposure to harmless allergens in the case of dust, cold, rodent urine, skin from mites and other entities can cause the development of allergies. The constant exposure of key cells to allergens triggers or develops an exaggerated immune response. Moreover, exposure to tobacco smoke at any age, excessive hygiene and low exposure to microbial pathogens, especially in childhood, leads to less maturation or display of lymphocytes and less stimulation, which suppresses the development, growth and production of T-reg lymphocytes and increases the risk of developing an allergic response against unrecognized antigens that are safe in the general population [12].
Regulatory T cells (Tregs) with their active Forkhead box protein 3 (Foxp3) control homeostasis and inhibition of the immune response. In general, the main population of Foxp3+ Treg cells is generated in the thymus, and they are called tTregs, although it has become clear that they can also be generated in the periphery (pTregs). That is, an unknown percentage of Tregs can be generated from Foxp3 T-conventional cells (Tconv), both of which are considered by their nature (nTregs). In mouse models, Foxp3 T cells can be generated by stimulating nTreg cells in vitro in the presence of TGF-b, and these are called in vitro-induced Tregs or allergen-induced Tregs (iTregs). The mechanisms that induce iTregs and nTregs have been identified, and they are similar cell populations that affect different factors to elicit unique responses [13].
Immunoregulatory responses involve complex systems, both for antigen recognition and an initial response characterized by diverse immunosuppressive cell populations and immunosuppressive molecules such as IL-10 and IL-35, in addition to the activation of FOXp3 cells. These cell populations include myeloid tissue-derived suppressor cells (MDSCs), regulatory B cells (Bregs), natural killer cells (NK), immunosuppressive plasma cells (ISPCs), and a regulatory subset of innate lymphoid cells (ILCs) with interconnected functions. These populations are being widely studied with proteomic methods. This regulatory and inflammatory response also comprises two subtypes of T cells with immunosuppressive functions called regulatory T cells (Tregs).
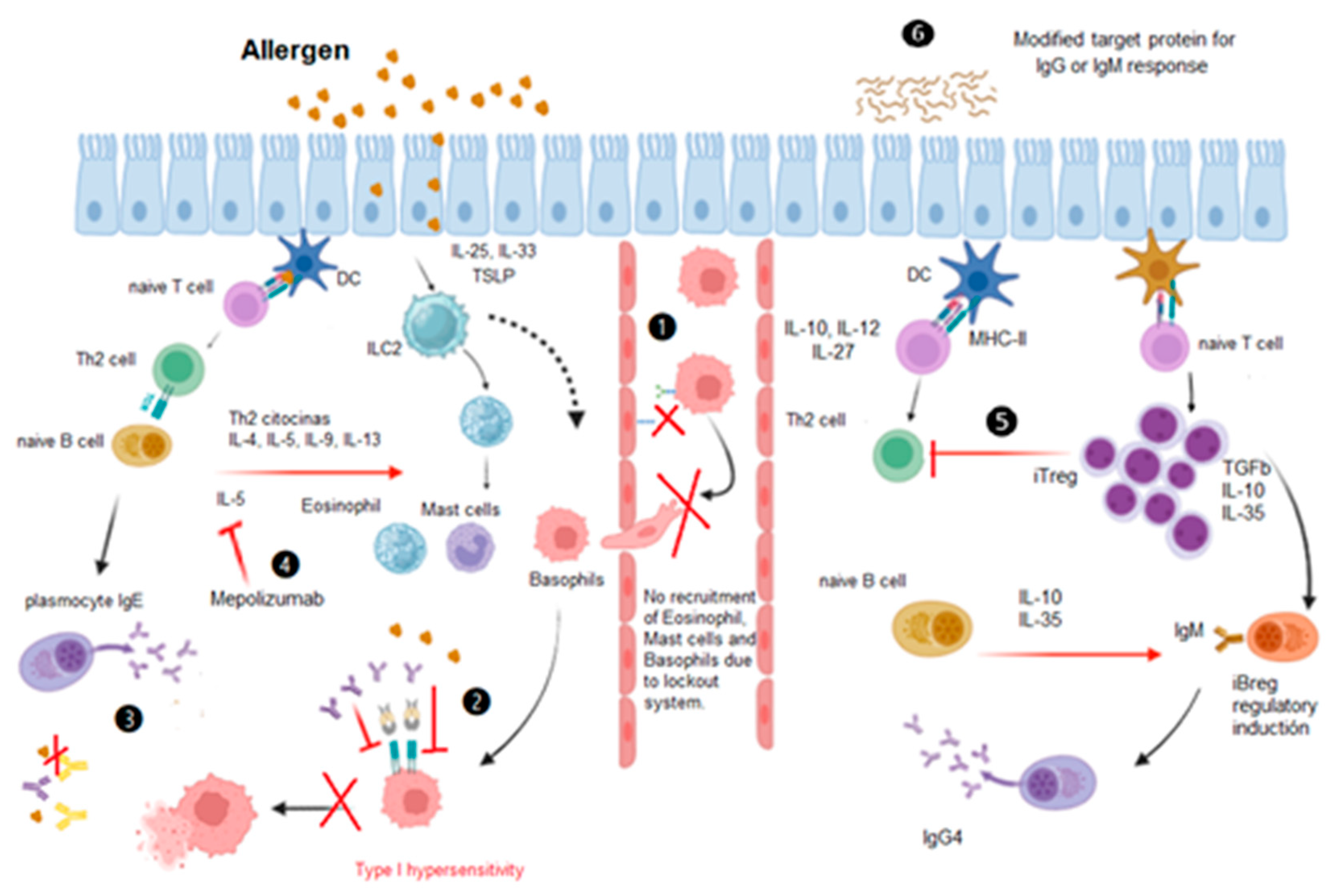
The subtypes of Tregs in the immunosuppressive response are important for the control of allergic responses and valuable for the development of new therapies. Two main groups have been classified according to their origin: natural (nTregs) and induced (iTregs). The immune response to aeroallergens begins in the respiratory epithelium of allergy patients. At the beginning, molecules such as TSLP, IL-33 or IL-25 are secreted and type 2 innate lymphoid cells (ILC2), which release interleukins IL-4, IL-5 and IL-13 and IL-19, are activated, and together with other pro-inflammatory cytokines secreted by the epithelium, among them MIP-1α and CCL19, which activate macrophages and dendritic cells (DCs) that present the allergen. Activated DCs internalize, process, and present the allergen to naive T lymphocytes, which proliferate to become Th2 cells, which secrete Th2 cytokines. From here, the recruitment of eosinophils, mast cells and basophils to the target tissue is induced. In addition, Th2 lymphocytes present allergens to naive IgM + B lymphocytes, which differentiate into allergen-specific IgE + plasma cells. The IgEs bind to mast cells, basophils and eosinophils that have already migrated to the tissue and will eventually release inflammatory mediators (see Figure 1) [14].
The study of proteomics may provide new insights into the alleviation of the allergic response. Monoclonal antibodies directed to IL-5 or its receptor (IL-5R) have been created, and these antibodies have been incorporated into asthma treatment guidelines, as in the case of mepolizumab anti-IL-5 monoclonal antibody, benralizumab antieosinophilic, humanized and afucosylated monoclonal antibody (IgG1, kappa) and others where modifications have been carried out with proteomics (see Figure 1) [15]. Moreover, other treatment alternatives, such as immunotherapy with monomers of the causative protein, blockade of the type 1 hypersensitivity pathway, and modification of the iTreg response with the production of IgGs instead of IgE [16].
Some of the main topics associated with allergic diseases that are currently being investigated with proteomic methods are 1) the antigens that trigger the response, 2) the epitopes of the immunoglobulins that recognize them, and 3) cell migration receptors. These proteins are studied because they are the responsible for the onset of hypersensitivity; without these 3 processes, there would be no allergic response. Although there are many other methods for diagnosis and treatment, markers that identify what type of allergy it is (hypersensitivity, pseudo allergy or allergies) and what protein component is the cause of the allergy in the patient are lacking.
In this case, there are important advances with proteomic methods where de novo proteins have been described in some pollens (for example, Fra e1 and Syr v 1 from Fraxinum).
Protein antigens that trigger responses with their epitopes, antibodies with their paratopes, and signalling pathways in the type 2 inflammatory response are currently being investigated. Due to their importance in the phenomenon, mutated proteins or only the portion that is recognized are given as therapy and have a diagnostic marker potential (see Figure 1: 4). Proteomics, which is based on several techniques, is probably the most informative tool that can be used to resolve the challenges associated with allergic respiratory diseases.
3. Proteomic technologies for the study of respiratory allergies.
There are approximately one million protein posttranslational modifications, although it has been calculated that the human genome codes for approximately 30,000 proteins. Under environmental conditions, protein expression is modified and sometimes altered. However, epitopes are regularly conserved and an individual can have an allergic reaction depending on the context and occasionally the immune response is increased by adjuvants [17]. In proteomics, there are multiple techniques to analyse proteins and study the quantitative and qualitative expression of proteins. The protein involved in the disease and its modifications can be studied in allergic patients, and their differences can be established between patients and healthy individuals, which guides us to know what is wrong in the individual. New proteins, membrane proteins, proteins in signal transduction pathways and phosphorylated proteins can be identified in blood or in solution, and the structure of proteins can be identified by Rx crystallography or software predictions.
Proteomics studies proteins in their different forms of expression, and they are usually detected using antibodies, but there are also techniques that separate and measure the size and composition (amino acids) of charged ions, such as chromatography and mass spectrometry. In addition to gel electrophoresis, one-dimensional (1D) and two-dimensional (2D) or 2D differential gel electrophoresis (2D-DIGE) and protein microarrays may or may not use antibodies [18]. High-throughput sampling without gel is also performed, such as multidimensional technology for proteins where specific recognition antibodies are used, such as ELISAS and Luminex. The latter use antibodies for the detection of proteins. Mass spectrometry may have some variants, some may be atomic absorption, or with isotope labelling of affinity, and isobaric labelling for relative and absolute quantification, in tandem and multidimensional protein identification technology (MudPIT) that combines it with liquid chromatography [19,20]. Additionally, in this combination, shotgun/proteomic analysis is used, which offers an indirect measurement of proteins after proteolytic digestion of intact proteins and is subjected to LC‒MSMS/MS analysis [21].
3.1. ELISA
The enzyme-linked immunosorbent assay is a technique that detects and measures the amount of a substance in a solution. Antibodies are used against particular proteins, in this case to the epitopes of the allergens. There are direct, indirect, inhibitor, competitive, sandwich and ELISPot ELISAs [22]. ELISPot is used to investigate the hypersensitivity reactions to drugs, and the confirmation of causality frequently facilitates the decision on whether to continue therapy. This technique is performed during monotherapy because the identification of the drug causing the reaction is often difficult, but this method can be used for drug challenge tests. Therefore, laboratory tests are of great interest since they can elucidate the causal diagnosis without putting the patient at risk [23].
3.2. Luminex
Luminex is another technique that is similar to ELISA in that it broadly detects proteins. The assay is performed on well plates containing colour-coded beads and specific antibodies that capture the target analyte. Biotinylated antibodies are then added to the sample that binds to the analyte, forming an antibody-antigen-antibody sandwich through the streptavidin signal conjugated with phycoerythrin (PE). The samples are entered into the equipment, and the beads will be detected based on a dual laser flow (the analysers are Luminex 200 or FlexMap).
The duality consists of one laser classifying the bead and the other classifying the analyte to which it is bound. There are lasers that act with greater clarity depending on the equipment. This technique is most useful if the different proteins in pollen that can activate the hypersensitivity response are known so we are able to identify the several epitopes in the pollen protein that trigger a response or the cytokines involved in allergy [24].
3.3. Western blotting
Is one of the most commonly used techniques for the separation and isolation of proteins, and it can identify proteins by molecular weight and specific antibodies. In addition, proteins that have been modified can be studied with this method through the development of a specific antibody for that modification. In addition, there are antibodies that only recognize certain proteins when they are phosphorylated in tyrosine, and this method is widely used in Western blotting. SDS‒PAGE is a method that involves the use of gel electrophoresis to separate the sample proteins and then transfer them to a membrane and locate the protein by its weight or use the aforementioned antibodies to tag the protein and quantify it [25]. Additionally, immunoprecipitation is a variant of the technique used to extract proteins from ligands. Immunoprecipitation of protein complexes (Co-IP) is a technique used to locate a specific protein without an antibody for that protein. The localization of phosphorylated proteins is commonly used to locate proteins in intracellular signalling pathways since the protein activation of the cascade serves as a signal to activate transcription factors [26].
This method can be used to identify the epitopes that bind to IgEs. It is also a tool that uses high-performance liquid chromatography (HPLC) and Western blotting with specific antibodies to detect epitopes and de novo proteins in pollens, urine and the environment. Of the two-dimensional methods, 1-DE and 2-DE, the first to isolate proteins by 1-DE based on their molecular mass, can be used to verify the purity of samples, check the purification of proteins and calculate the unknown molecular weights, in the case of pollen proteins not yet described, or of foods that cause allergies and the causative proteins are unknown [27]. The difference in 2-DE is the separation of the protein; it is based on the molecular mass and the isoelectric point, identifying different forms of proteins [28].
3.4. Liquid chromatography
This method allows the isolation and purification of proteins in most samples in a production range of a few nanograms to picograms. Obtaining large amounts of protein from a sample allows more reliability and accuracy in the results. There are size exclusion chromatography (SEC) or hydrodynamic techniques that consist of a gel compound for retention of proteins by filtration and by solvents. Additionally, ion exchange chromatography (IEC) allows the separation of the molecule by the nature of the charge [29]. High-performance liquid chromatography (HPLC) is the only separation method that satisfies the standards to obtain samples without excessive degradation of the proteome or sample, helping the rapid purification and isolation of peptides. Affinity chromatography will be the protein separation process by various techniques and according to the immobilized ligand [30].
One method that does not use antibodies is mass spectrometry. It is an analytical technique that allows the study of various compounds in nature, as well as obtaining qualitative or quantitative information. In this way, it is possible to obtain information on the molecular mass of the analysed compound and its structure.
3.5. Mass spectrometry
This analysis method quickly elucidates the protein sequence and its weight. MS is mainly based on ionizing the protein, so the spectrometers have an ion source, a detector and an analyser. To ionize a molecule, there are several methods since liquid chromatography separates the molecules and spectrometry analyses it by gases or combined with liquid chromatography, as in the case of electrospray ionization (ESI).
In contrast, there is matrix-assisted laser desorption ionization (MALDI) with a matrix to which the sample adheres or absorbs and, under certain conditions, is localized by the laser beam. There is also another simpler call: a time-of-flight analyser (TOF) based on the capture of ions travelling a distance, thus increasing resolution and improving the separation of the beams, which accurately determines the mass without the need for a magnetic or electric field [31]. This method can be combined with MALDI/TOF and others, such as the LC‒MSMS/MS method, by high-resolution MRM. The latter helps us to find different molecules with the same mass, as well as identify many of the antibodies of polyclonal origin that develop in the organism. However, this technique seems to be too sophisticated for protein molecules, because there are simpler methods with the same specificity. Currently, there are used in the study and differentiation of lipids [32].
3.6. Other methods
One of the most commonly used methods with mass spectrometry is high-resolution two-dimensional electrophoresis, which separates proteins from different samples and identifies differentially expressed proteins when combined with staining and mass spectrometry. Additionally, the stable isotope labelling of two different complex mixtures for proteins is commonly used. The method consists of labelling proteins with isotopes and digesting them to produce labelled peptides [33]. Additionally, hybrid technologies use antibody-based purification of individual analytes and then mass spectrometry analysis for identification and quantification. There is the mass spectrometric immunoassay (MSIA) and the standard capture of stable isotopes with antipeptide antibodies (SISCAPA). Microarrays are also used to study proteins on a large scale, such the matrix metalloproteinase (MMP) MMP assay kit [34]. The conformational dynamics of proteins can also be studied by means of hydrogen/deuterium exchange mass spectrometry (HDX-MS), mainly in the loop-helix portions [35].
4. Informatics for the development of protein models
Proteomics data are used to decipher and algorithmically predict proteins and to find associations between an existing protein and a modified protein, knowing its functionality and, if necessary, changing it. It can also be used for the marker discovery process, designing pharmacotherapy in immunotherapy, predicting de novo protein functions and modelling potential proteins to be used.
Sequent is a program that analyses, identifies and validates proteins of the mass spectrum in tandem individually. This program evaluates and calculates the sequences of the peptides present with respect to a database [36]. Proteomics involves the identification of proteins, peptides and amino acids through MS/MS sequences. Comet is a search engine for databases and corresponds to an open source tool that is freely available [37]. Mascot Server is a powerful free service tool that runs on the website and is ideal for identifying proteins and peptides from primary sequence databases in software or using mass spectrometry data. This tool allows the visualization of chemical and posttranslational modifications in any direction (up, down, lateral), which can be quantified by means of isobaric labelling, spectral libraries, cross-linking and intact peptides [38].
4.1. Protein Data Bank
For the design, consultation and visualization of a protein, RCSB PDB (RCSB.org) consists of a global Protein Data Bank (PDB) and contains 3D structure data for the design of large biological molecules (proteins, DNA and RNA). It is one of the first open access digital resources widely used in molecular biology and medical fields. Its capacity allows in the study of allergies by consulting 3D structure data for proteins in the environment, urine, desquamation of mites, pollen, food and others. This database allows you to find any protein structure in any organism on the planet. The forms of IgE and its probable epitopes can be determined after sequencing the target protein.
This tool has become essential for research and education in medicine, biology, biotechnology and other related science fields since access to the large quantity of 3D structure data stored in the PDB contributes significantly to the progress of the study of almost any structure [39].
4.2. UniProt
Is a high-quality tool used to access protein sequences, identification mapping, sequence alignment, peptide searches, prediction of a sequence obtained from previous studies, homology between sequences, and finding proteins with defined sequences. More than 190 million sequences in UniProtKB exist today. The database reduces redundant searches, which provides greater specificity and reliability for researchers [40].
4.3. Swiss-Prot
In a collaboration between the Swiss Institute of Bioinformatics and the European Bioinfomatics Institute (EBI) and the Protein Information Resource (PIR) in 2002, the UniProt consortium was created. Swiss-Prot together with TrEMBL, both automatic resources, also teamed up with the PIR to produce the world's most important protein catalogue, UniProt Knowledgebase. In addition, the Swiss Institute of Bioinformatics has its website Swiss-Prot, which is a biological database of protein sequences and a fully automated server for modelling the homology of protein structures [41]. ExPASy is also found here, which can analyse protein sequences and structures and 2D-PAGE. ExPASy has two important tools, "pIcarver", which is a tool to visualize theoretical distributions of the peptide by isoelectric point in a certain pH range and proposes a fractionation scheme that generates fractions with similar peptide frequencies, and MALDIPepQuant is a tool to quantify the MALDI peptides (SILAC) from Phenyx [42]. Moreover, Expasy has the Swiss Bioinformatics Resource Portal where you can find information for various techniques including genetics, metagenomics. transcriptome, and cell culture.
4.4. MODELLER
Is software that looks for the homology or comparative modelling of three-dimensional structures between proteins, which when compared, predicts the three-dimensional structure of a given sequence. This application automatically calculates a guide that has all the atoms except hydrogen. It implements the comparative modelling of protein structures by satisfying spatial restrictions according to the user and based mainly on its alignment with one or more proteins of the known structure, and it is compared with computer templates. It can also perform de novo modelling of loops in proteins, alignment of multiple sequences and/or protein structures, optimization of protein models with respect to a function, and others [43].
4.5. Rosetta commons
The Rosetta commons application contains algorithms for the modelling of protein structures and the comparison and analysis of structures. One of the characteristics of the program is the computational coupling that explains the inherent flexibility of protein monomers to simulate the conformational selection of pregenerated sets. In other words, if an amino acid moves chirally, the program detects this modification and couples it. This software has allowed remarkable advances in biology and medicine. There are reports of de novo protein design, prediction of biological macromolecule structures, enzyme design, ligand coupling, and macromolecular complexes [44].
4.6. AlphaFold Protein Structure Database
The AlphaFold Protein Structure Database from EMBL-EBI is an open access program that provides more than 200 million highly accurate protein structure predictions. Its database has expanded the programmatic and visual interaction of protein structures through atomic coordinates that can be predicted with very high prediction and confidence intervals.
It creates residual and paired models and predicts alignment errors with precision that has never been seen before in the Critical Evaluation of Protein Structure Prediction (CASP). It has been used in the fields of bioinformatics, structural biology and drug discovery and will undoubtedly widely serve the study of allergic diseases [45,46].
4.7. X-ray cryptography
Another Among the older technologies that continue to be used and that were the pioneers of proteomics, X-ray cryptography stands out. It provides not only the three-dimensional structure but also a deeper understanding of protein structure and function. Almost 86% of the data in the common databases have been generated by this technique. In general, there are two methods for this technique: 1) vapour diffusion, which consists mainly of the protein and a precipitating mixture that is dehydrated to concentrate the protein crystal with balanced osmolarity in a nucleus and then radiological study is applied. 2) the batch method consists of bringing the protein directly to the nucleation zone combined with a precipitant until it is crystallized with oil [47]. The interest of looking for proteins that are involved in chemotaxis and the inhibition of the immune response by the cells involved in the allergic reaction. Several cell types must migrate from the peripheral blood to the zone of stimulation or to the zone of extravasation during contact with the allergen, which is represented by a rash. The inhibition or blocking of cell migration is an approach that this group has for the control of the type two inflammatory response, type one hypersensitivity, the Th2 response and anaphylaxis.
5. Discussion
Proteins in respiratory allergy diseases are of paramount importance, and we believe that the allergic response can be controlled at 3 points called the immune lockout systems. The first step consists of identifying with proteomic methods the specific paratope and epitope of the given allergy problem, with the intention of blocking or occupying it, either with an antagonist or an antibody. This process is already happening, as we mentioned above, with the antibodies benralizumab, dupilumab, mepolizumab, omalizumab and reslizumab, which are used in asthma. B) Another blocking system that consists of preventing cell migration of mast cells, basophils and eosinophils to the site of attachment to the epithelium or endothelium [48].
In addition, to understand the modifications of the inflammatory system response, the activation of the different subtypes of T cells with immunosuppressive function generically called regulatory T cells (Tregs) should be investigated. The activation of the two main groups, natural (nTregs) or induced (iTregs), is important for the inflammatory response. The latter have been described in humans and play a key role in allergen tolerance. In contrast, regDCs present peptides to naive T cells, which differentiate into induced regulatory T cells (iTregs) that can be activated to promote tolerance. ITreg cells secrete IL-10, TGFβ and IL-35, which are the most important cytokines that control and decrease the response and that specifically increase IL-10 and IL-35 levels, promote the maturation of naive B cells to iBreg cells, which secrete more IL-35, in an inhibitory autocrine immune signalling response.
IL-35 is a cytokine responsible for the maintenance of the immune system and the inhibition of immune responses since it promotes the expansion of regulatory T cells (Tregs) and regulatory B cells (Bregs) while suppressing T effector cells, Th1 cells, Th17 cells and macrophages.
It is hypothesized that naive B cells also differentiate into IgG4+ plasma cells and that they secrete blocking IgG4 antibodies with the ability to block the specific IgE response. The stimulation and monitoring of this phenomenon should be studied, and changes in plasma cells that produce IgG4 would be reversed if immunotherapy in animals works as well as in humans. The administration of the complete allergen to iTreg cells produced by immunotherapy can prevent the proliferative response of Th2 cells [14]. We believe that both the control and the activation of the entire path of the type 1 hypersensitivity response can be controlled in different phases.
However, although there are models for the study of migration and for the blocking of some molecules, the blocking of some sites has not yet been studied. Type 1 hypersensitivity is more sensitive to inhibitors. The purpose of this study was to use proteomics to reveal various proteins involved in the different processes of the allergen inflammatory response.
Author Contributions
M.Á.G.-M. All authorship belongs to the author of the manuscript.
Funding
This work was supported by the National Institute of Respiratory Diseases “Ismael Cosío Villegas,” Mexico City. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I thank CONAHCYT for its support.
Conflicts of Interest
Not applicable.
References
- Wilkins, MR. ; Christian, P. ; Ron, D.A; Ou, Keli; Golaz, Olivier; Sanchez, Jean-Charles; Yan, Jun X.; Gooley, Andrew. A.; Hughes, Graham; Humphery-Smith, Ian; Williams, Keith L. Denis FH. "From Proteins to Proteomes: Large Scale Protein Identification by Two-Dimensional Electrophoresis and Arnino Acid Analysis". Nat Biotechnol.1996;1,61–65.
- Al-Amrani, S.; Al-Jabri, Z.; Al-Zaabi, A.; Alshekaili, J.; Al-Khabori, M. Proteomics: Concepts and applications in human medicine. World J Biol Chem. 2021, 5, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Humphery-Smith, I. The 20th anniversary of proteomics and some of its origins. Proteomics. 2015, 11, 1776. [Google Scholar] [CrossRef] [PubMed]
- Badotti, F.; Barbosa, A.S.; Reis, A.L.M.; do Valle, I.F.; Ambrósio, L.; Bitar, M. Comparative modeling of proteins: A method for engaging students’ interest in bioinformatics tools. Biochem Mol Biol Educ. 2014, 1, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Karlsson, MJ.; Hober, A.; Svensson, AS.; Scheffel, J.; Kotol, D. The human secretome. Sci Signal. 2019, 609, eaaz0274. [Google Scholar] [CrossRef]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 1, 50. [Google Scholar] [CrossRef]
- Akbar, R.; Robert, P.A.; Pavlović, M.; Jeliazkov, J.R.; Snapkov, I.; Slabodkin, A.; et al. A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep. 2021;34, 11,108856. [CrossRef]
- Galván-Morales, M.A.; Montero-Vargas, J.M.; Vizuet-de-Rueda, JC.; Teran, LM. New Insights into the Role of PD-1 and Its Ligands in Allergic Disease. Int J Mol Sci. 2021, 21, 11898. [Google Scholar] [CrossRef]
- Han, X.; Krempski, J.W.; Nadeau, K. Advances and novel developments in mechanisms of allergic inflammation. Allergy. 2020, 12, 3100–3111. [Google Scholar] [CrossRef]
- Demoly, P. Respiratory allergic disease genes. Rev Pneumol Clin. 2003, 2 Pt 1, 67–75. [Google Scholar] [CrossRef]
- Portelli, M.A.; Hodge, E.; Sayers, I. Genetic risk factors for the development of allergic disease identified by genome-wide association. Clin Exp Allergy. 2015, 1, 21–31. [Google Scholar] [CrossRef]
- Campbell, D.E.; Boyle, R.J.; Thornton, C.A.; Prescott, S.L. Mechanisms of allergic disease - environmental and genetic determinants for the development of allergy. Clin Exp Allergy. 2015, 5, 844–858. [Google Scholar] [CrossRef]
- Shevach, E.M.; Thornton, A.M. tTregs, pTregs, and iTregs: similarities and differences. Immunol Rev. 2014, 259, 1, 88–102. [Google Scholar] [CrossRef]
- Calzada, D.; Cremades-Jimeno, L.; López-Ramos, M.; Cárdaba, B. Peptide Allergen Immunotherapy: A New Perspective in Olive-Pollen Allergy. Pharmaceutics. 2021, 7, 1007. [Google Scholar] [CrossRef] [PubMed]
- Agache, I.; Beltran, J.; Akdis, C.; Akdis, M.; Canelo-Aybar, C:; Canonica, GW:; et al. Efficacy and safety of treatment with biologicals (benralizumab, dupilumab, mepolizumab, omalizumab and reslizumab) for severe eosinophilic asthma. A systematic review for the EAACI Guidelines - recommendations on the use of biologicals in severe asthma. Allergy 2020, 5, 1023–1042. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Mariotti-Ferrandiz, M.E.; Wang, Y.; Malissen, B.; Waldmann, H.; Hori, S. Heterogeneity of natural Foxp3+ T cells: A committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc. Natl. Acad. Sci. USA. 2009, 106, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Shumate, A.; Pertea, G.; Varabyou, A.; Breitwieser, F.P.; Chang, Y.C.; Madugundu, A.K.; Pandey, A.; Salzberg, SL. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018, 1, 208. [Google Scholar]
- Mortuaire, G.; Marchetti, P.; Formstecher, P.; Danzé, P.M. Micro-array based technologies to study the proteome: technological progress and applications. Ann Biol Clin. 2004, 2, 139–48. [Google Scholar]
- Link, A.J.; Washburn, M.P. Analysis of protein composition using multidimensional chromatography and mass spectrometry. Curr Protoc Protein Sci. 2014, 78, 23.1.1–23.1.25. [Google Scholar] [CrossRef]
- Yoon, J.H.; Seo, J.; Shin, S.K. Multi-functional MBIT for peptide tandem mass spectrometry. Mass Spectrom Rev. 2015, 2, 209–218. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M,C.; Yates, J.R. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem Rev. 2013, 4, 2343–2394. [Google Scholar]
- Porebski, G.; Piotrowicz-Wojcik, K.; Spiewak, R. ELISpot assay as a diagnostic tool in drug hypersensitivity reactions. J Immunol Methods. 2021, 495, 113062. [Google Scholar] [CrossRef]
- Saff, R.R. Skin testing as a biomarker in drug allergy. Ann Allergy Asthma Immunol. 2023, 2, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.C.; Wang, K.; Wadhwa, P.D.; Culhane, J.F.; Nelson, E.L. Validation and comparison of luminex multiplex cytokine analysis kits with ELISA: determinations of a panel of nine cytokines in clinical sample culture supernatants. J Reprod Immunol. 2005, 2, 175–191. [Google Scholar]
- Rognon, B.; Reboux, G.; Roussel, S.; Barrera, C.; Dalphin, J.C.; Fellrath, J.M.; Monod, M.; Millon, L. Western blotting as a tool for the serodiagnosis of farmer's lung disease: validation with Lichtheimia corymbifera protein extracts. J Med Microbiol. 2015, 64 Pt 4, 359–368. [Google Scholar]
- Bakalarski, C.E.; Kirkpatrick, D.S. A Biologist's Field Guide to Multiplexed Quantitative Proteomics. Mol Cell Proteomics. 2016, 5, 1489–97. [Google Scholar] [CrossRef]
- Brunelle, J.L.; Green, R. One-dimensional SDS-polyacrylamide gel electrophoresis (1D SDS-PAGE). Methods Enzymol 2014, 541, 151–159. [Google Scholar] [PubMed]
- Gargan, E.; Ohlendieck, K. Sample Preparation and Protein Determination for 2D-DIGE Proteomics. Métodos Mol Biol. 2023, 2596, 325–337. [Google Scholar]
- Coskun, O. Separation techniques: Chromatography. North Clin Istanb. 2016, 2, 156–160. [Google Scholar]
- Barderas, R.; Purohit, A.; Rodríguez, R.; Pauli, G.; Villalba, M. Isolation of the main allergen Fra e 1 from ash (Fraxinus excelsior) pollen: comparison of the natural and recombinant forms. Ann Allergy Asthma Immunol. 2006, 4, 557–563. [Google Scholar]
- Hsiao, J.T.; Chen, K.H.; Sheu, F. Determination of the soybean allergen Gly m 6 and its stability in food processing using liquid chromatography-tandem mass spectrometry coupled with stable-isotope dimethyl labelling. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2022, 6, 1033–1046. [Google Scholar] [CrossRef]
- Acunha, T.; Nardini, V.; Ferranti-Peti, A.P.; Borges-Prado, M.K.; Beraldo-Moraes, L.A.; Faccioli, L.H. Targeted analysis of eicosanoids derived from cytochrome P450 pathway by high-resolution multiple-reaction monitoring mass spectrometry. J Mass Spectrom. 2021, 7, e4769. [Google Scholar] [CrossRef]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the Concept and Practice of Two-Dimensional Gel Electrophoresis in the Analysis of Proteomes at the Proteoform Level. Proteomes. 2019, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Burk, J.; Sassmann, A.; Kasper, C.; Nimptsch, A.; Schubert, S. Extracellular Matrix Synthesis and Remodeling by Mesenchymal Stromal Cells Is Context-Sensitive. Int J Mol Sci. 2022, 3, 1758. [Google Scholar] [CrossRef] [PubMed]
- Uhrik, L.; Henek, T.; Planas-Iglesias, J.; Kucera, J.; Damborsky, J.; Marek, M.; Hernychova, L. Study of Protein Conformational Dynamics Using Hydrogen/Deuterium Exchange Mass Spectrometry. Methods Mol Biol. 2023, 2652, 293–318. [Google Scholar] [CrossRef]
- Adkins, J.N.; Varnum, S.M.; Auberry, K.J.; Moore, R.J.; Angell, N.H.; Smith, R.D.; Springer, D.L.; Pounds, J.G. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Mol Cell Proteomics. 2002, 12, 947–55. [Google Scholar] [CrossRef]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source MS/MS sequence database search tool. Proteomics. 2013, 1, 22–24. [Google Scholar] [CrossRef]
- Bouyssié, D.; Gonzalez de Peredo, A.; Mouton, E.; Albigot, R.; Roussel, L.; et al. Mascot file parsing and quantification (MFPaQ), a new software to parse, validate, and quantify proteomics data generated by ICAT and SILAC mass spectrometric analyses: application to the proteomics study of membrane proteins from primary human endothelial cells. Mol Cell Proteomics. 2007, 9, 1621–1637. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016, 12, 2301–2319. [Google Scholar] [CrossRef]
- Peng, J.; Chan, C.; Meng, F.; Hu, Y.; Chen, L.; Lin, G.; Zhang, S.; Wheeler, A.R. Comparison of Database Searching Programs for the Analysis of Single-Cell Proteomics Data. J Proteome Res. 2023, 4, 1298–1308. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, D1, D480-D489.
- Krajaejun, T.; Reamtong, O.; Lohnoo, T.; Yingyong, W.; Thammasudjarit, R. Assessment of temperature-dependent proteomes of Pythium insidiosum by using the SWISS-PROT database. Med Mycol. 2019, 7, 918–921. [Google Scholar] [CrossRef]
- Vaezzadeh, A.R.; Hernandez, C.; Vadas, O.; Deshusses, J.J.; Lescuyer, P.; Lisacek, F.; Hochstrasser, D.F. PICarver: a software tool and strategy for peptides isoelectric focusing. J Proteome Res. 2008, 10, 4336–45. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics. 2014, 47, 5.6.1–32. [Google Scholar] [CrossRef] [PubMed]
- Marze, N.A.; Roy-Burman, S.S.; Sheffler, W.; Gray, J.J. Efficient flexible backbone protein-protein docking for challenging targets. Bioinformatics 2018, 20, 3461–3469. [Google Scholar] [CrossRef]
- Varadi, M.; Velankar, S. The impact of AlphaFold Protein Structure Database on the fields of life sciences. Proteomics. 2022, e2200128. [Google Scholar] [CrossRef]
- Bertoline, L.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An overview of protein structure prediction. Front Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef]
- Dessau, M.A.; Modis, Y. Protein crystallization for X-ray crystallography. J Vis Exp. 2011, 47, 2285. [Google Scholar] [CrossRef]
- Gaudet, A.; Portier, L.; Prin, M.; Copin, M.C.; Tsicopoulos, A.; Mathieu, D.; Lassalle, P.; De Freitas-Caires, N. Endocan regulates acute lung inflammation through control of leukocyte diapedesis. J Appl Physiol. 2019, 3, 668–678. [Google Scholar] [CrossRef]
- Ye, L.; Fan, J.; Shi, X.; Tao, Q.; Ye, D.; Xian, Z.; Zeng, X.; Li, Y.; Feng, M.; Ju, D. Tumor necrosis therapy antibody interleukin-2 fusion protein elicits prolonged and targeted antitumor effects in vivo. Appl Microbiol Biotechnol. 2014, 9, 4053–61. [Google Scholar] [CrossRef]
Figure 1.
Lockout system. 1) Migration, 2) FcεR1 Lockout. 3) Specific antigen-epitope or paratopoid-specific blockade. 4) anti-interleukin antibody blockade. 5) Recombinant cytokines. 6) Stimulation and monitoring of different proteins. Created with BioRender.com (accessed on 12th Jun 2023).
Figure 1.
Lockout system. 1) Migration, 2) FcεR1 Lockout. 3) Specific antigen-epitope or paratopoid-specific blockade. 4) anti-interleukin antibody blockade. 5) Recombinant cytokines. 6) Stimulation and monitoring of different proteins. Created with BioRender.com (accessed on 12th Jun 2023).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



