
Submitted:
22 June 2023
Posted:
23 June 2023
You are already at the latest version
Abstract
MicroRNAs (miRNAs) within the exosomes are short non-coding RNAs that are associated with many diseases including cancers and are crucial players in regulating gene expression and regula-tion. Owing to their potential role as emerging biomarkers, efficient isolation, and quantification methods of miRNA from the exosomes in the cell’s supernatant are complicated and challenging. The quantitative PCR method is the gold standard for miRNA identification and estimation. Nucleic acid-based assays are expensive, labour-intensive, and require expertise. In this work, we demon-strated an aptamer-based magnetic separation of the exosomes, and quantification of the miRNA using a fluorescence switching assay. The fluorescence assay relies on the fluorescence of the fluorophore labelled cDNA quenched by carbon nanomaterials coated on magnetic beads (OFF state). When the target miRNA210 is introduced to the system, the cDNA detaches from the quenching surface. As a result, the fluorescence intensity increases (ON state) with increasing miRNA concentration within the dynamic range of 0-100 nM. This method can detect miR210 as low as 5 pM. This method does not exhibit any cross-reactivity with other closely related miRNAs. The method was validated by spiking miRNA with the standard RT-PCR method using SYBR green as a fluorescent probe. This method can be used for the minimally invasive detection of cancer biomarkers in breast cancer patients.
Keywords:
MicroRNA detection
; biosensors
; pre-concentration of exosomes
; breast cancer
1. Introduction
According to the World Health Organization (WHO), cancer is the second leading cause of death globally and early diagnosis is of paramount importance for increasing survival rates and improving treatment outcomes. Tumour biopsy is one of the most common techniques used for cancer diagnosis [1]. However, this method is highly invasive, especially when the tumour location is inaccessible. Therefore, finding alternative diagnostic methods is highly desirable. Recently, exosomes have received considerable attention as potential biomarkers for a variety of diseases including cancers [2,3]. Exosomes are found in various body fluids including blood, urine and saliva and are made up of lipid bilayer cellular vesicles with a maximum size of 150 nm. They are secreted by all types of cells and contain many important biomolecules such as DNA, RNA, microRNA, and proteins. In addition, they are transported from the parent to the recipient cells serving as an intercellular communication system [4], thus providing a wealth of information when utilized. As a major cause of tumour growth and metastasis, exosomes carry the growth-promoting genes to initiate the proliferation of cancer cells. For example, it was identified that the exosomes with epidermal growth factor receptor (EGFR) are carried to liver-specific metastasis in nude mouse models using green fluorescent protein (GFP)-tagged CD63 as a fluorescent probe [5]. Also, it is believed that exosomal miRNA can mediate and silence the downstream genes and trigger the tumorigeneses in nontumorigenic epithelial cells [6].
MicroRNAs (miRNAs) are small (18-22 nucleotides in length) non-coding molecules found in most living organisms that are involved in the post-transcriptional regulation of gene expression [7]. These miRNAs are located in various subcellular locations such as mitochondria, endomembranes and RNA granules. They are found in a variety of body fluids, and secreted outside cells through extracellular vesicles, the exosomes. The altered expression of miRNAs is also associated with many cancer progressions, including breast, lung and prostate cancers [8], thus illustrating a high potential as clinical biomarkers for non-invasive diagnosis and treatment of tumours. MiRNAs can be extracted from the exosomes and consequently be utilized in different biological assays [9,10]. Due to their stability within the exosomes, the utility of these molecules has attracted significant attention as biomarkers for the early and non-invasive diagnosis of various cancers [11,12,13].
Exosome isolation is one of the most challenging tasks due to their small size, low abundance and existence in a heterogeneous population of vesicles. A variety of exosome isolation methods have been exploited including ultrafiltration, ultracentrifugation, density gradient separation, size-exclusion chromatography, polymer precipitation and immunoaffinity [14,15] and several isolation kits are available [16]. Microfluidic-based separation technique allows extraction of biological entities from a tiny sample volume by exploiting various separation principles using intrinsic forces (e.g., fluid dynamic) or extrinsic forces (e.g., magnetic, and electric fields) and using different properties of the targeted analyte. Integration of microfluidic and magnetic separation into exosome sample preparation offers several advantages including high purity, high throughput, and low sample volume. Current procedures for exosome extraction employ antibodies as recognition elements. However, this method does not enable the non-destructive release of exosomes, which may affect the biofunction of the natural exosome and lead to false results. Aptamers are chemically synthesized oligonucleotides, which are selected by SELEX processes and can be used as an alternative to antibodies (chemical antibodies) for the isolation and separation of exosomes [17]. Aptamers can be selected to bind with high specificity to a wide range of target molecules, including proteins, small molecules, and cells. This allows for more precise targeting than antibodies, which can sometimes cross-react with other molecules. When binding to the target, the single-strand DNA or RNA of the aptamers forms a unique 3D structure under ideal physiological conditions [18,19].
In this study, aptamer-conjugated magnetic nanobeads were employed in a microfluidic-based magnetic separation system for the isolation of exosomes from cell culture media by trapping and releasing processes (Figure 1B). The exosome particle size was determined by the dynamic light scattering method. MiR210 in the exosome was detected using FAM-cDNA. The sensor was designed to quench the fluorescence of the FAM-cDNA by physical adsorption on the carbon nanomaterial (CNM) surface (OFF state) as illustrated in Figure 1A. In the presence of Mir210, the cDNA detached from the surface and fluorescence increases significantly (ON state). The limit of detection of Mir210 was estimated from the calibration curve. The detection method was compared with the standard RT-qPCR method.
2. Materials and Methods
2.1. Chemicals
Carbon-coated magnetic (CCM) beads were purchased from Turbo beads (Zurich, Switzerland). Penicillin-streptomycin was purchased from Gibco Life Technologies (Carlsbad, CA, USA). sodium chloride (NaCl), magnesium chloride (MgCl2), phosphate-buffered saline pH 7.4 (PBS), tris(hydroxymethyl)aminomethane (tris-base), boric acid, ethylenediaminetetraacetic acid (EDTA) disodium dihydrate, sodium azide and hydrochloric acid, were purchased from Sigma-Aldrich (St Louis, MO, USA; https://www.sigmaaldrich.com/united-states.html). The size exclusion chromatographic column was obtained from Izon Science Ltd (Lyon, France). Amicon Ultra Centrifugal Filter (0.5 mL) was supplied from EMD Millipore (Sigma MA USA; https://www.merckmillipore.com). DNA purification kit (Qiagen), RT2 first strand kit (Qiagen, cat No 330401). HPLC purified labeled and unlabeled ologinucleotides were purchased from Metabion International (Planegg, Germany; http://www.metabion.com) Table 1 and Table 2. Luciferase expressing 4T1 murine breast cancer cells (4T1-Luc2) were purchased form PerkinElmer and cultured as reported previously [20].
2.2. Instrumentation
Concentrations of oligonucleotides and proteins were estimated by absorption at 260 nm and 280 nm respectively using NanoDrop 2000 (Thermo Scientific, Ottawa, Canada). Fluorescence signals of fluorescein-labeled cDNA and the cDNA-miRNA duplex were monitored with the Nanodrop ND3300 fluorospectrometer (Thermo Scientific, Ottawa, Canada). Light-emitting diodes (LEDs) in the range of 470±10 nm were used as an excitation source to excite the sample. The fluorescence intensity of the samples was observed at 515 nm. All the experiments were repeated three times to get the concordant values. The measurements were carried out in a binding buffer (pH, 7.4) at room temperature. All measurements were recorded in the binding buffer at room temperature. The exosome particle size was determined by the dynamic light scattering method using Malvern pananalytical instrument (https://www.malvernpanalytical.com) with a monochromatic laser.
2.3. Exosome isolation, quantification, and particle size determination
Luciferase expressing 4T1 murine breast cancer cells (4T1-Luc2) were cultured in RPMI-1640 culture media containing 10% heat-treated fetal bovine serum (FBS), and 100 unit/mL penicillin-streptomycin (Gibco, Life Technologies, Carlsbad, CA, USA) at 37 °C in a humidified atmosphere containing 5% CO2 until 80% confluent. The cells were washed three times with PBS buffer and subsequently cultured in a serum-free medium for 48 hours. The cell culture medium was collected and centrifuged for 10 minutes at 500 g and the pellet was discarded followed by another 10 minutes of centrifugation. The supernatant was concentrated using a 100 KD cut-off amicon filter until it reached one-tenth of the original volume. The concentrated solution was then added to the Eqv column which was pre-equilibrated with PBS buffer at room temperature and the fractions were collected up to 1 mL. The collected fraction was concentrated using a 100 KD amicon filter to a final volume of 150 µL. The concentration of the total protein in the extracted exosome was estimated as described elsewhere [21] and the total amount of particles was calculated from the protein content. The particle size of the extracted exosomes was measured using Zetasizer Nano ZS equipped with a 633 nm laser (Malvern Instruments, UK). One millilitre of each diluted sample was placed in a 1 cm path-length disposable cuvette. The sample was equilibrated to room temperature before size measurements in triplicate.
2.4. Magnetic bead-based isolation of exosomes (Apta-magnetic separation system)
To extract the exosomes from the cell culture supernatant, a dynamic magnetic separation separator was custom-fabricated which is based on the concept of flow-through “trapping and releasing” of magnetic beads in a microfluidic channel [22]. The separator employs a rotating magnet assembly which comprises an array of small Neodymium Iron Boron (NdFeB) disc magnets each having a diameter of 1 mm. The magnets are embedded in three cylindrical rotatable rods and arranged with alternate magnetic polar orientation i.e., the pole direction of each magnet is perpendicular to the next magnet with a spacing between adjacent magnets of 10mm. Each magnet assembly comprises two magnet sets M1 (marked in red) and M2 (marked in blue) which are perpendicular to each other (Figure 1B). The magnet assembly is positioned under a microfluidic chip, which was fabricated using 3D printing technique (Figure 1B, inset), with a long meandering channel where the supernatant sample flows at a low flow rate of 3-5 µL/min. The magnetic field due to the magnets is sufficient to attract the beads within the channel when the magnet’s pole is facing the bottom of the channel where the distance between the magnet pole and the channel is ~1 mm. The beads aggregate within the magnetic potential well (spot). But, when the magnet rotates 90° such that the pole is perpendicular to the channel, the magnetic field imposed on the beads sharply decreases and becomes not sufficient to hold the beads, therefore, the beads disaggregate and are carried in dispersed form within the fluid towards the next potential well to be trapped and aggregate again. This process is repeated 12 times corresponding to the number of magnets. Each trapping and releasing event provides a chance to release any impurity from the bead-exosome complex i.e., a washing cycle would take place. At the end of the channel, the purified bead-exosome sample will be finally trapped and collected in a reservoir (R) due to a stationary disc magnet (M) for further analysis.
2.5. RNA isolation and quantification
The total RNA was isolated from the magnetically pre-concentrated exosomes using RNeasy RNA isolation kit (Qiagen) according to the manufacturer's protocol. Briefly, 100 µl of exosomes were disturbed and homogenized in RLT buffer and lysis reagent. Then 100µl of chloroform was added and the solution was mixed and centrifuged. The aqueous layer was collected in a separate tube and 100% ethanol was added. The amount of total RNA extracted from the mixture was quantified by measuring UV absorption at 260 nm.
2.6. Reverse transcription and RT-qPCR
Purified total RNA was used for the production of cDNA of the miRNA using the RT2 first strand kit (Qiagen, cat No 330401) following the manufacture protocol. GC-rich stem-loop primer (Table 2), RT buffer, 0.25mM dNTPs, and reverse transcriptase were employed for the reverse transcription reaction. 25 µl of the mixtures were incubated in a thermocycler at 16°C, 42°C and 85°C for 30 min, 30 min and 5 min respectively. 2% agarose gel electrophoresis was used to confirm the size of the product obtained from the reverse transcription. Then the product was purified using a Qiagen DNA purification kit. The amount of cDNA was quantified by measuring UV absorption at 260 nm. The purified cDNA was aliquoted further according to the experimental requirements. Real-time RT-qPCR experiments were carried out in 25 µl using 0.2µM forward and reverse primers, 200 μM dNTP and 2 units of Taq polymerase in SYBR green PCR master mix. The PCR mixture was incubated at 94°C for 5 min, followed by 40 cycles of incubation at 94°C for 15 sec, 55°C for 30 sec, 70°C for 30 sec, and a final extension step of 10 min at 70°C. The real-time PCR amplification was followed by the fluorescence signal of SYBR green which acts as a fluorescent probe. The experiments were repeated three times for the concordant values. The threshold cycle (Ct) is the fraction of the cycle at which the fluorescence intensity crosses the threshold of the same cycle number. Finally, from the standard curve, the Ct values were converted into absolute copy numbers.
2.7. Magnetic nanobeads fluorescence sensor platform for Mir210 detection
The carbon-coated magnetic (CCM) nanobeads play a dual function as a carrier for exosome separation and a sensing substrate where the CNM is used as a fluorescence quencher which forms a major element in the developed sensor. Initially, the fluorescence-based sensing was optimized to ensure that the amount of fluorescently labeled cDNA required for the fixed amount of CNM-coated magnetic beads provides an acceptable signal-to-noise ratio. CCM nanobeads in the dynamic range of 0 to 400 µg/mL were titrated with a fixed amount of FAM-labeled cDNA of the microRNA 210. Based on the quenching efficiency of the 35 µg/mL of the CCM nanobeads (25 nM FAM-cDNA to 40 µg/mL nanobeads), the optimized concentration of the FAM-labeled cDNA concentration was fixed at 25 nM. Following the optimization, a mixture of 25 nM FAM-cDNA and 35 µg/mL nanobeads were incubated at room temperature for 30 minutes which leads to significant adsorption of the FAM-cDNA by the CNM. Then, Mir210 with a concentration within the dynamic range of 0 to 50 nM with FAM-cDNA/CCM nanobeads in binding buffer (50mM Tris.HCl +150 mM NaCl +2mM MgCl2, pH = 7.4) was incubated for 30-35 minutes. The fluorescence intensity of each sample was recorded at exciting/emission of 470±10 nm/515 nm and plotted against the concentration of FAM-cDNA.
2.8. Specificity and quantification
The specificity of the CCM nanobead-based fluorescence assay was tested against other microRNAs which are closely associated with mir210 such as mir10b, mir16 and mir191. The miRNA samples were incubated with FAM-labeled mir210 complementary DNA/nanobeads in an optimized ratio. The fluorescence signals were measured for each miRNA and the relative fluorescence values were compared for cross-reactivity.
3. Results and Discussions
3.1. Apta-magnetic separation (AMS) and particle size determination of exosomes
MicroRNAs are stabilized by the biomolecules within the exosomes and bind with proteins in the presence of transmembrane and cytosolic proteins, mRNA and DNA [23,24,25,26]. In the current separation method (Figure 1B), the magnetic particle suspension flows inside a meandering channel which is aligned with the magnet assembly. The rotation of the magnet assembly generates an array of alternate magnetic potential wells due to the two magnet sets (M1 and M2). When the magnet poles of M1 point directly to the channel, the magnetic beads will be exposed to strong magnetic forces, therefore, the beads will be trapped and aggregated. On the other side, the magnetic forces due to the set M2 have a negligible effect on the beads, therefore, they will be carried with the flow and trapped again at the next trapping. The alternate rotation of the magnets (i.e., polar orientation) generates a series of alternate “trapping and releasing” which enables a series of washing events to take place before the final trapping and concentration at the end of the channel due to a stationary magnet M.
3.2. Isolation and determination of particle size of exosome
The magnetically pre-concentrated exosomes were isolated using ssDNA complementary sequence of CEA aptamer. In the presence of the aptamer complementary sequences, the aptamer covalently conjugates to the magnetic beads to form a strong dsDNA duplex. As a result, the aptamer-bound exosomes are released into the solution [27]. The exosome size was measured using the dynamic light scattering of photon correlation spectroscopy [28]. The scattered light from the particles due to the laser beam was correlated to the particle size distribution of the particles (Figure 2).
3.3. CCM beads as a fluorescence sensing substrate
Fluorescence switching was used as a noninvasive mechanism to detect the breast cancer biomarker mir210 using the CCM nanobeads as a fluorescence quencher and the fluorescent-labelled complementary sequence of mir210 as a sensing probe. It is well known that ssDNA is effectively adsorbed on the surface of CCMs [29] due to their high surface-to-volume ratio. The CCM nanobeads act as an efficient fluorescence quencher by the fluorescence resonance energy transfer (FRET) mechanism. CNM can strongly interact with fluorescently labeled ssDNA through non-covalent interactions including, π-stacking, hydrogen bonding, hydrophobic interactions, electrostatic force of attraction, etc. [18,30,31,32]. The fluorescence of FAM-labelled DNA was significantly quenched in the presence of CCM beads. However, in the presence of the mir210, the complementary sequence will be duplexed with the target and restore the fluorescence as illustrated in Figure 1A.
3.4. Optimization of CNM/FAM-DNA ratio (fluorescence OFF state)
CCM beads were added to a fixed amount of FAM-DNA and the change in the fluorescence intensity due to quenching by the CNM was monitored. The minimum number of magnetic beads at which a significant signal-to-noise ratio (s/n) is considered the ideal condition which was found to be 25nM. 25 nM of mir210 cDNA was titrated against the variable amount of CCM beads in the dynamic range of 0 to 300 µg/mL. In the absence of the magnetic beads, a strong fluorescence of the FAM-cDNA was observed (Figure 3A). However, when the magnetic beads were added, the fluorescence intensity significantly decreased with increasing the concentration of the magnetic beads as shown in Figure 3B. The fluorescence intensity reached less than 15% at a magnetic bead concentration of 40 µg/mL. No significant reduction in intensity was observed at higher bead concentrations (Figure 3B). At this concentration, significant adsorption of mir210 cDNAs by the CCM beads was achieved. When the fluorophore conjugated to the cDNA is excited at 470±10 nm, the emitted photon energy from the fluorophore is transferred to the CNM by the fluorescence resonance energy transfer (FRET) mechanism, which leads to a drastic decrease in the signal intensity. Therefore, the optimal bead concentration at which ~80-85 % of quenching was obtained was fixed at 40 µg/mL. The optimized ratio of 25 nM of mir210 cDNA for 40 µg/mL is considered as fluorescence off state and was selected for further experiments.
3.5. Mir210 detection using CCM beads fluorescence assay
When the labelled complementary cDNAs of mir210 become in contact with the CCM beads, it is adsorbed by the CNM through various weak interactions, such as hydrogen bonding, hydrophobic interactions, Van der Waals forces, etc. Hence, a significant quenching of fluorophore-labelled cDNA takes place. Upon the addition of mir210 within the range of 0.05 to 100 nM, to the magnetic beads-cDNA complex, the fluorescence intensity was increased with the increasing concentration of mir210 (Fig 4A). The mir210 forms a double-strand DNA duplex with FAM-cDNA, which is thermodynamically more stable than the magnetic beads-cDNA complex. Therefore, in the presence of mir210, cDNA detaches from the CNM surface and forms mir210-cDNA dsDNA with the maximum number of base pairs in the buffer solution. When FAM-cDNA is detached from the CCM beads, the fluorophore becomes at a distance from the quenching CNM thus the emitting photons from the fluorophore become highly observed, therefore, increasing the fluorescence intensity as the mir210 concentration increases. The change in the fluorescence intensities due to the presence of mir210 was plotted against the logarithmic concentration of mir210 to get the calibration curve and a linear relationship was observed as shown in Figure 4B. The limit of detection (LOD) was calculated as 3.3 SD/s, where SD is the standard deviation of the signal of blank samples, and S is the slope of the linear calibration curve. The LOD of mir210 was found to be 5 pM. The achieved sensitivity using the current method is significantly higher than that recently reported using mir21cDNA-FAM and graphene oxide which was 20 pM [33]. Mir210 present in the total RNA extracted from the exosome was quantified using the standard calibration curve. The fluorescence intensity of the sample is found to be directly proportional to the concentration of mir210 present in the sample which was further confirmed by the standard RT-PCR method in the next sections.
Zhou et al developed a G-quadruplex molecular beacon fluorescence probe for the detection of miRNA using duplex-specific nuclease (DSN) for signal amplification with a detection limit of 1 pM [34]. In another study, a fluorescence switching mechanism was also employed for the detection of miRNA using fluorescent-labelled miRNA-cDNA gold nanoparticles as a fluorescence quencher. The reported LOD of this competitive binding assay was 3.8pM [35]. Graphene oxide-based fluorescence assay has been also developed u
3.6. Reverse Transcription and RT-qPCR
The current fluorescence switching assay was compared with the RT-qPCR method which is a standard method for the quantification of miRNA. The RT-qPCR process requires two steps, namely reverse transcription and amplification by RT-qPCR. The RT primer with stem-loop structure hybridizes with mi210 and produces complementary DNA of mir210 cDNA by reverse transcription. The desired size of the reverse-transcribed cDNA was confirmed by 2% agarose gel electrophoresis (Figure 5) and purified using a PCR purification kit. The quantity of cDNA was calculated as 12 ng/µL from the UV absorption at 260 nm. The stock cDNA was aliquoted further to have the variable concentration range of the template to carry out the RT-qPCR using SYBR green as the fluorescent probe. cDNA samples with higher concentration reach the threshold value of the fluorescence intensity with a smaller number of amplification cycles whereas, more cycles of amplification are needed in the case of diluted cDNA samples (Figure 6A) which implies that the PCR mixture with more concentrated cDNA template has less threshold cycle (Ct) and vice versa. The plot of. Ct values against the log value of cDNA input in the variable concentration range of 1.2 x10-2 fM 1.2 x105 fM to (35 copies to 108 copies of the template) showed perfect linearity as depicted in Figure 6B. No change in the fluorescence of SYBR green was observed during the thermal cycles for the negative control (without mir210 or other DNA templates). The achieved sensitivity enables the detection of as low as 100 copies in the sample. The lowest detectable copies of mir210 cDNA were obtained using the formula, 3×STD/q where STD is the standard deviation in RUF value in the absence of cDNA template, q is the slope of the linear fit. To confirm that the desired product is obtained from the PCR amplification processes, the melting curve of each sample is observed from the fluorescence of the SYBR green to avoid the non-specific amplification products such as primer-dimer (Figure 6C).The sharp peaks 75.5±1.5°C (Figure 6D) in the second derivatives of the melting curves confirm that the amplified PCR products with melting temperatures in the range between 75°C and 80°C. The lowest detection amount of RT-qPCR is relatively very low in comparison with the fluorescence switching assay, however, unlike PCR, the fluorescence switching assay is a direct measurement without any further enzymatic amplification. As RT-qPCR technique is expensive, needs highly sophisticated instruments, expert technicians, suitable primer setup for the targets and storage and stability issues which makes it not suitable for point-of-care applications.
3.7. Cross-reactivity and specificity
The specific binding of mir210 with the labelled cDNA which was adsorbed onto the CCM beads was tested using the closely related miRNA sequences. The target, mir210 and other nonspecific miRNAs such as mir210, mir10b, mir16, and mir191 were incubated and the changes in the fluorescence intensity were monitored. As shown in Figure 7, mir210 showed a maximum fluorescence signal. However, there was no considerable change in the signal enhancement in the signal while treating other target sequences (mir16, mir191 and mir10b) compared to the mir210 miRNA target. Similar results were observed without target mir210 and/or scrambled DNA was used (Figure 8) which confirms the high selectivity of the current method for the target miRNA.
The method developed in the current study is simple and provides a fast assay (~30 min) without the need for signal amplification compared to the above-mentioned methods. The high signal-to-noise ratio would be multiplied by amplification or the reproduction of the probing DNA by enzymes. However, amplification methods would not be suitable for the in-vivo application. The current method would be compactable for the in-vivo diagnosis application with a slight chemical modification in the DNA backbone or 2’OMe modified RNA [37,38] or blocking the 3’end of the cDNA to protect from the nuclease digestion.3
4. Conclusions
CNM-based fluorescence switching assay was developed for the detection of mi210 biomarkers using FAM-labeled cDNAs. The aptamer-based microfluidic separation and purification of exosomes have been applied and particle size was determined. The exosomal mir210 was extracted and quantitatively detected by the fluorescence switching method using CCM beads as a fluorescence quenching platform. The limit of detection of the method was calculated to be 5 pM from the standard calibration curve. No significant cross-reactivity with the other closely related miRNAs was observed. The fluorescence method has been validated with the standard RT-qPCR amplification techniques and miRNA biomarkers screening by high-throughput methods from the exosomes for the specific disease diagnosis, prognosis, and point of care application. This method is simple and can be used for the detection of miRNA at the point of care.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Vaidyanathan R, Soon RH, Zhang P, Jiang K, Lim CT. Cancer diagnosis: From tumor to liquid biopsy and beyond. Lab Chip 2019, 19, 11–34. [CrossRef]
- Cheng J, Meng J, Zhu L, Peng Y. Exosomal noncoding RNAs in Glioma: Biological functions and potential clinical applications. Mol Cancer 2020, 19, 1–14. [CrossRef]
- Salehi M, Sharifi M. Exosomal miRNAs as novel cancer biomarkers: Challenges and opportunities. J. Cell. Physiol. 2018, 233, 6370–6380. [CrossRef]
- Zhang Y, Liu Y, Liu H, Tang WH. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci 2019, 9, 1–18. [CrossRef]
- Suetsugu A, Honma K, Saji S, Moriwaki H, Ochiya T, Hoffman RM. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv Drug Deliv Rev 2013, 65, 383–390. [CrossRef]
- Nahand JS, Moghoofei M, Salmaninejad A, Bahmanpour Z, Karimzadeh M, Nasiri M, Mirzaei HR, Pourhanifeh MH, Bokharaei-Salim F, Mirzaei H. Pathogenic role of exosomes and microRNAs in HPV-mediated inflammation and cervical cancer: A review. Int J Cancer 2020, 146, 305–320. [CrossRef]
- Barrett LW, Fletcher S, Wilton DS. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci 2012, 69, 3613–3634. [CrossRef]
- Bottani M, Banfi G, Lombardi G. Circulating miRNAs as diagnostic and prognostic biomarkers in common solid tumors: Focus on lung, breast, prostate cancers, and osteosarcoma. J Clin Med 2019, 8, 1661. [CrossRef]
- Bayraktar R, Van Roosbroeck K, Calin GA. Cell-to-cell communication: MicroRNAs as hormones. Mol Oncol 2017, 11, 1673–1686. [CrossRef]
- Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci 2020, 101, 2087–2092. [CrossRef]
- Weber JA, Baxter DH, Zhang S, Huang DY, Huang KH, Lee MJ, Galas DJ, Wang K. The microRNA spectrum in 12 body fluids. Clin Chem. 2010, 56, 1733–1741. [CrossRef]
- Cortez MA, Bueso-Ramos C, Ferdin J, Lopez-Berestein G, Sood AK, Calin GA. MicroRNAs in body fluids—the mix of hormones and biomarkers. Nat Rev Clin Oncol 2011, 8, 467–477. [CrossRef]
- Javidi MA, Ahmadi AH, Bakhshinejad B, Nouraee N, Babashah S, Sadeghizadeh M. Cell-free microRNAs as cancer biomarkers: The odyssey of miRNAs through body fluids. Med Oncol 2014, 31, 1–11. [CrossRef]
- Chen J, Li P, Zhang T, Xu Z, Huang X, Wang R, Du L. Review on strategies and technologies for exosome isolation and purification. Front Bioeng Biotechnol 2022, 9, 811971. [CrossRef] [PubMed]
- Sidhom K, Obi PO, Saleem A. A review of exosomal isolation methods: Is size exclusion chromatography the best option? Int J Mol Sci 2020, 21, 6466. [CrossRef] [PubMed]
- Patel GM, Khan MA, Zubair H, Srivastava SK, Khushman M, Singh S, Singh AP. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci Rep 2019, 9, 1–10. [CrossRef]
- Song Z, Mao J, Barrero RA, Wang P, Zhang F, Wang T. Development of a CD63 aptamer for efficient cancer immunochemistry and immunoaffinity-based exosome isolation. Molecules 2020, 25, 5585. [CrossRef]
- Chinnappan R, Zaghloul NS, AlZabn R, Malkawi A, Rahman AA, Abu-Salah KM, Zourob M. Aptamer selection and aptasensor construction for bone density biomarkers. Talanta 2021, 224, 121818. [CrossRef]
- Alomran N, Chinnappan R, Alsolaiss J, Casewell NR, Zourob M. Exploring the Utility of ssDNA Aptamers Directed against Snake Venom Toxins as New Therapeutics for Snakebite Envenoming. Toxins 2022, 14, 469. [CrossRef]
- Al Faraj A, Shaik AS, Al Sayed B. Preferential magnetic targeting of carbon nanotubes to cancer sites: Noninvasive tracking using MRI in a murine breast cancer model. Nanomed 2015, 10, 931–948. [CrossRef]
- Gámez-Valero A, Monguió-Tortajada M, Carreras-Planella L, Franquesa M, Beyer K, Borràs FE. Size-Exclusion Chromatography-based isolation minimally alters Extracellular Vesicles’ characteristics compared to precipitating agents. Sci Rep 2016, 6, 1–9. [CrossRef]
- Ramadan Q, Christophe L, Teo W, Li SJ, Hua FH. Flow-through immunomagnetic separation system for waterborne pathogen isolation and detection: Application to Giardia and Cryptosporidium cell isolation. Anal Chim Acta 2010, 673, 101–108. [CrossRef] [PubMed]
- Chugh PE, Sin SH, Ozgur S, Henry DH, Menezes P, Griffith J, Eron JJ, Damania B, Dittmer DP. Systemically circulating viral and tumor-derived microRNAs in KSHV-associated malignancies. PLoS Pathog 2013, 9, e1003484. [CrossRef]
- Fornari F, Ferracin M, Trerè D, Milazzo M, Marinelli S, Galassi M, Venerandi L, Pollutri D, Patrizi C, Borghi A. Circulating microRNAs, miR-939, miR-595, miR-519d and miR-494, identify cirrhotic patients with HCC. PloS One 2015, 10, e0141448. [CrossRef]
- Köberle V, Pleli T, Schmithals C, Augusto Alonso E, Haupenthal J, Bönig H, Peveling-Oberhag J, Biondi RM, Zeuzem Kronenberger SB. Differential stability of cell-free circulating microRNAs: Implications for their utilization as biomarkers. PloS One 2013, 8, e75184. [CrossRef]
- Kubiczkova L, Kryukov F, Slaby O, Dementyeva E, Jarkovsky J, Nekvindova J, Radova L, Greslikova H, Kuglik P, Vetesnikova E. Circulating serum microRNAs as novel diagnostic and prognostic biomarkers for multiple myeloma and monoclonal gammopathy of undetermined significance. Haematologica 2014, 99, 511. [CrossRef]
- Zhang K, Yue Y, Wu S, Liu W, Shi J, Zhang Z. Rapid Capture and Nondestructive Release of Extracellular Vesicles Using Aptamer-Based Magnetic Isolation. ACS Sens 2019, 4, 1245–1251. [CrossRef]
- Palmieri V, Lucchetti D, Gatto I, Maiorana A, Marcantoni M, Maulucci G, Papi M, Pola R, De Spirito M, Sgambato A. Dynamic light scattering for the characterization and counting of extracellular vesicles: A powerful noninvasive tool. J. Nanoparticle Res. 2014, 16, 1–8. [CrossRef]
- Li H, Zhang Y, Wang L, Tian J, Sun X. Nucleic acid detection using carbon nanoparticles as a fluorescent sensing platform. Chem Commun 2011, 47, 961–963. [CrossRef]
- R. Chinnappan, A.A. Rahamn, R. AlZabn, S. Kamath, A.L. Lopata, K.M. Abu-Salah, M. Zourob, Aptameric biosensor for the sensitive detection of major shrimp allergen, tropomyosin. Food Chem. 2020, 314, 126133. [CrossRef]
- Chinnappan R, AlAmer A, Eissa S, Rahamn AA, Abu Salah KM, Zourob M. Fluorometric graphene oxide-based detection of Salmonella enteritis using a truncated DNA aptamer. Microchim Acta 2019, 185, 1–9. [CrossRef]
- Chinnappan R, AlZabn R, Fataftah AK, Alhoshani A, Zourob M. Probing high-affinity aptamer binding region and development of aptasensor platform for the detection of cylindrospermopsin. Anal Bioanal Chem 2020, 412, 4691–4701. [CrossRef] [PubMed]
- Ai X, Zhao H, Hu T, Yan Y, He H, Ma C. A signal-on fluorescence-based strategy for detection of microRNA-21 based on graphene oxide and λ exonuclease-based signal amplification. Anal Methods 2021, 13, 2107–2113. [CrossRef] [PubMed]
- Zhou H, Yang C, Chen H, Li X, Li Y, Fan X. A simple G-quadruplex molecular beacon-based biosensor for highly selective detection of microRNA. Biosens Bioelectron 2017, 87, 552–557. [CrossRef] [PubMed]
- Wang W, Kong T, Zhang D, Zhang J, Cheng G. Label-free microRNA detection based on fluorescence quenching of gold nanoparticles with a competitive hybridization. Anal Chem 2015, 87, 10822–10829. [CrossRef] [PubMed]
- Guo S, Yang F, Zhang Y, Ning Y, Yao Q, Zhang GJ. Amplified fluorescence sensing of miRNA by combination of graphene oxide with duplex-specific nuclease. Anal Methods 2014, 6, 3598–3603. [CrossRef]
- Ni S, Yao H, Wang L, Lu J, Jiang F, Lu A, Zhang G. Chemical modifications of nucleic acid aptamers for therapeutic purposes. Int J Mol Sci 2017, 18, 1683. [CrossRef]
- Chinnappan R, Dube A, Lemay JF, Lafontaine DA. Fluorescence monitoring of riboswitch transcription regulation using a dual molecular beacon assay. Nucleic Acids Res 2013, 41, e106. [CrossRef]
Figure 1.
(A) Schematic diagram shows the fluorescence switching mechanism in carbon nanomaterial coated on the magnetic nanobeads fluorescence assay for detection of mir210 extracted from exosome. Stage 1: The FAM-labeled cDNA of mir210 has high fluorescence signal. Stage 2: The fluorescence was quenched by the presence of carbon nanomaterial coated on the magnetic nanobeads. Stage 3: By the introduction of mir210, the cDNA detached from the carbon nanomaterial surface and duplexed with the mir210, as the results of the fluorescence intensity increased. (B) Schematic drawing of the magnetic separator showing in detail the rotating magnetic system under the microfluidic chip. A zoomed view of the beads “trapping and releasing” is shown in the inset. M is the permanent magnet used to trap the beads after purification and R is the collecting reservoir.
Figure 1.
(A) Schematic diagram shows the fluorescence switching mechanism in carbon nanomaterial coated on the magnetic nanobeads fluorescence assay for detection of mir210 extracted from exosome. Stage 1: The FAM-labeled cDNA of mir210 has high fluorescence signal. Stage 2: The fluorescence was quenched by the presence of carbon nanomaterial coated on the magnetic nanobeads. Stage 3: By the introduction of mir210, the cDNA detached from the carbon nanomaterial surface and duplexed with the mir210, as the results of the fluorescence intensity increased. (B) Schematic drawing of the magnetic separator showing in detail the rotating magnetic system under the microfluidic chip. A zoomed view of the beads “trapping and releasing” is shown in the inset. M is the permanent magnet used to trap the beads after purification and R is the collecting reservoir.
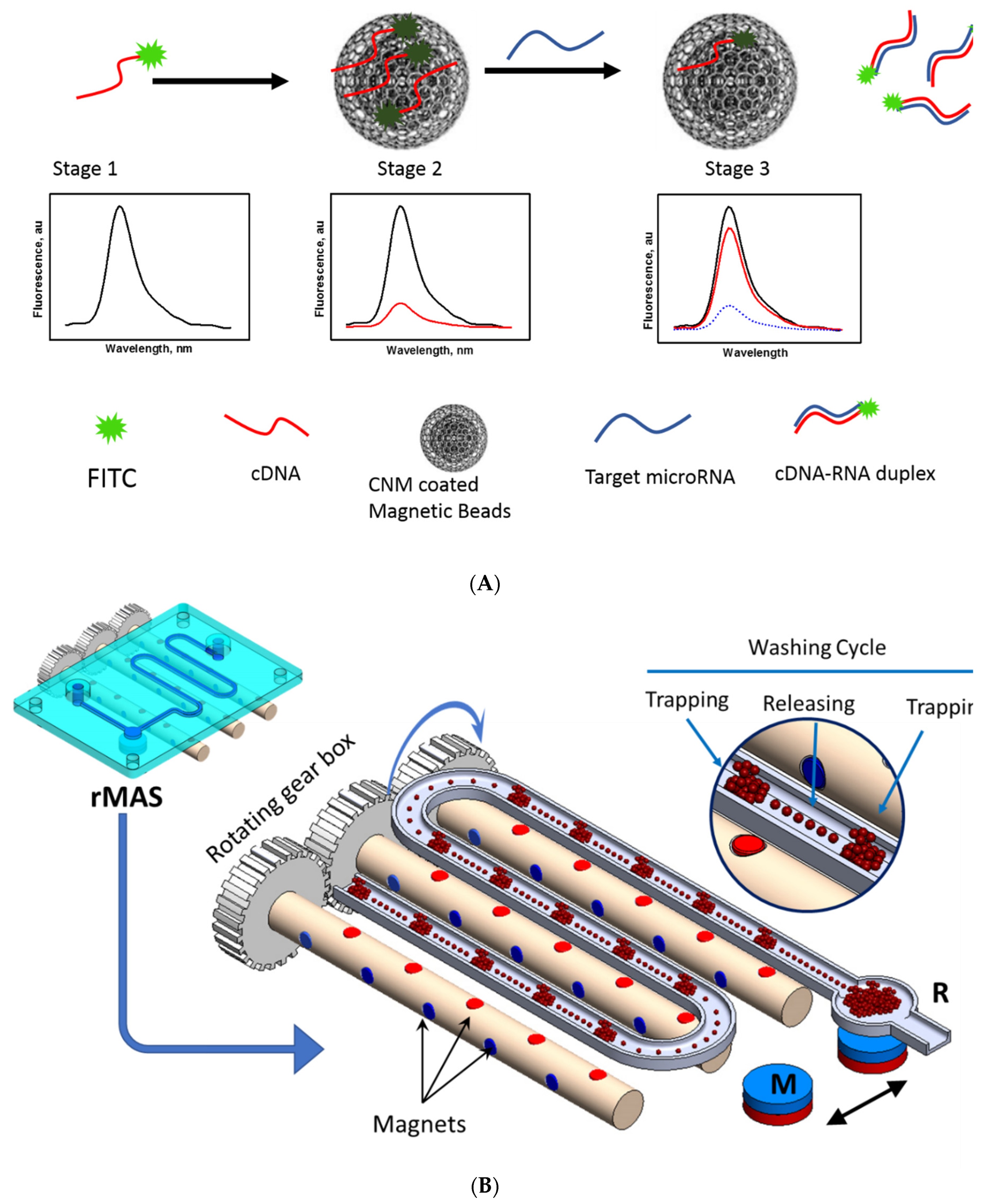
Figure 2.
Dynamic light scattering for the exosome isolated from Luc-4T1 cell line. The intensity of the scattered light by the exosome particle is monitored. 633 nm laser was used as an exciting source. The values are the average of three measurements.
Figure 2.
Dynamic light scattering for the exosome isolated from Luc-4T1 cell line. The intensity of the scattered light by the exosome particle is monitored. 633 nm laser was used as an exciting source. The values are the average of three measurements.
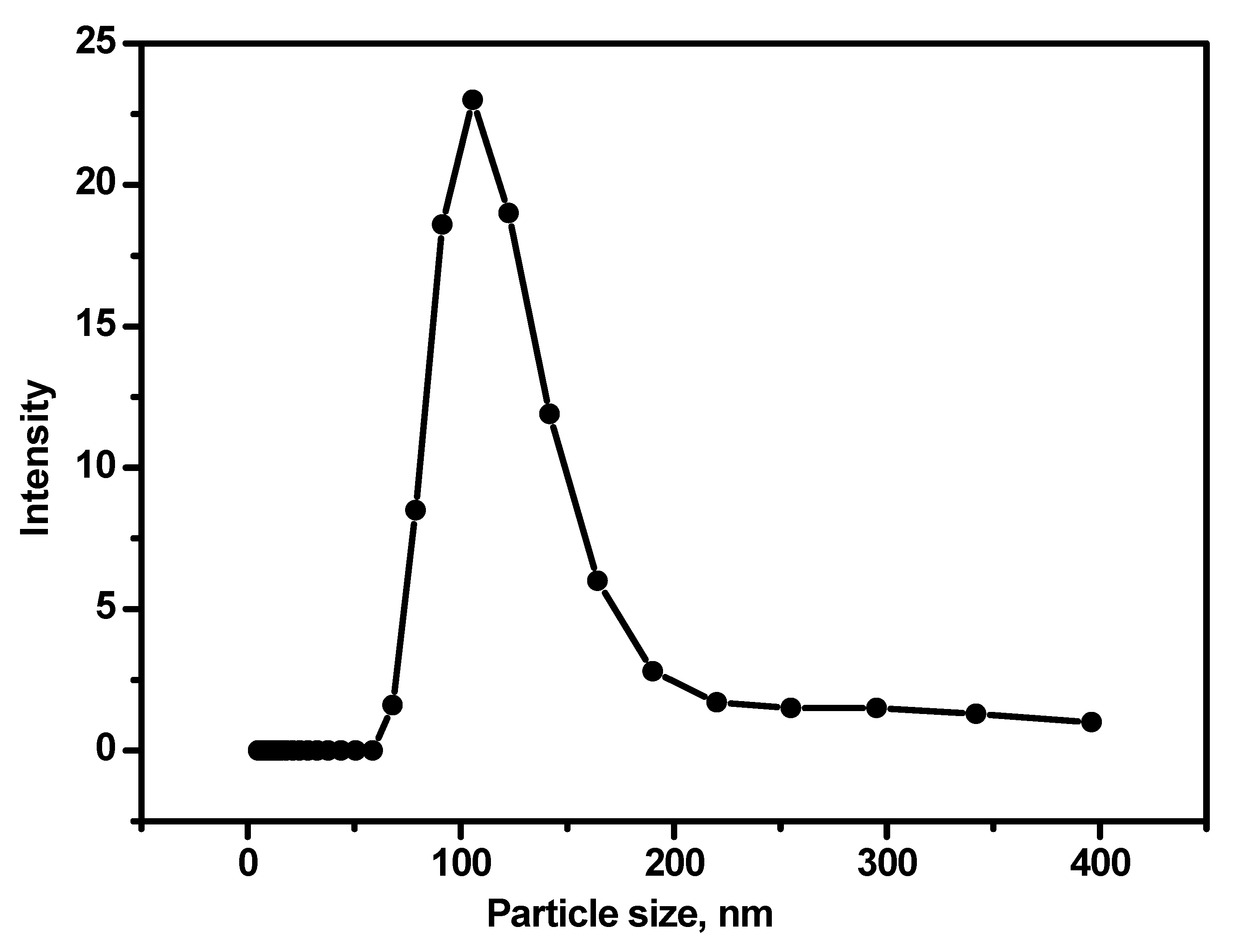
Figure 3.
(A) Fluorescence intensity of mir210 cDNA decreases with increasing the concentration of the CNM-coated magnetic nanobeads at bead concentration of 0, 10,20, 30, 40, 80,120, 200 and 300 µg/mL. (B) Magnetic nanobeads concentration vs Fluorescence intensity of the FAM-cDNA at excitation/emission of 470 ±10 nm/515 nm. The error bars represent the standard deviation of three different measurements.
Figure 3.
(A) Fluorescence intensity of mir210 cDNA decreases with increasing the concentration of the CNM-coated magnetic nanobeads at bead concentration of 0, 10,20, 30, 40, 80,120, 200 and 300 µg/mL. (B) Magnetic nanobeads concentration vs Fluorescence intensity of the FAM-cDNA at excitation/emission of 470 ±10 nm/515 nm. The error bars represent the standard deviation of three different measurements.
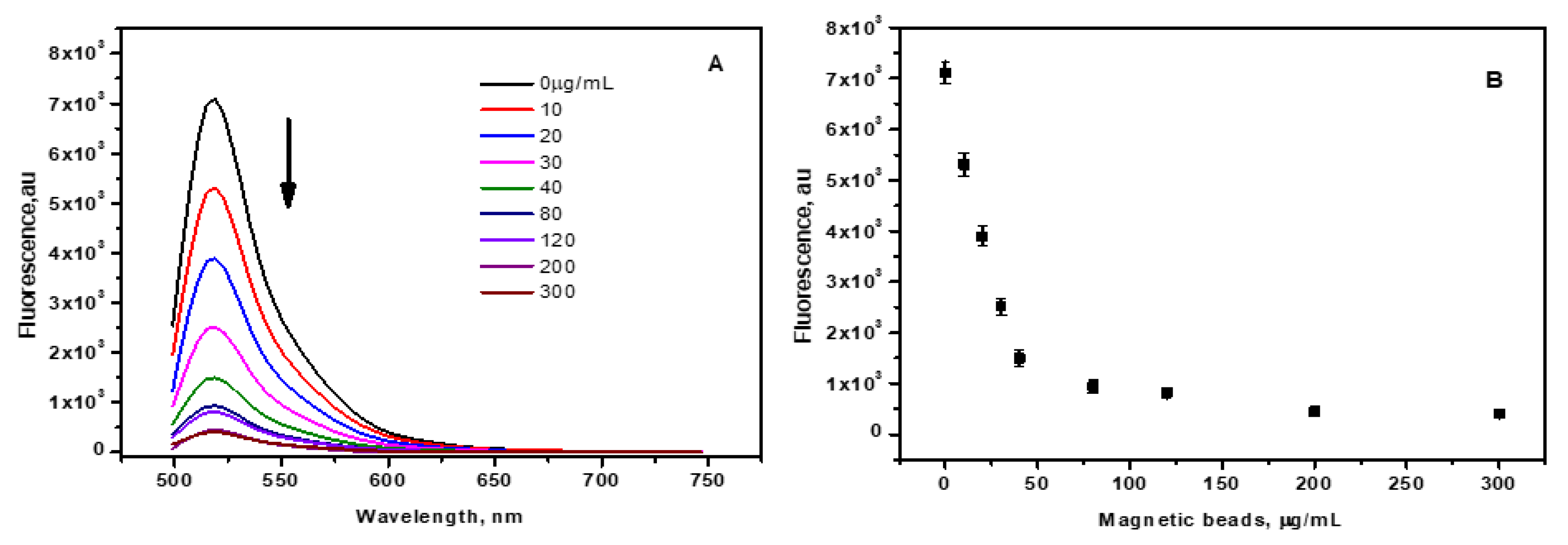
Figure 4.
(A) FAM-cDNA is released from the magnetic nanobeads surface by the addition of mir210. The fluorescence intensity increases with increasing the concentration of mir210 from 0.05 to 100 nM. The fluorescence intensity was monitored at 515 nm and plotted against the concentration of mir210. (B) The standard calibration plot of fluorescence intensity against mir210The fluorescence spectra were recorded by exciting at 470 ±10 nm and the emission intensity 515 nm. The error bars represent the standard deviation of three different measurements.
Figure 4.
(A) FAM-cDNA is released from the magnetic nanobeads surface by the addition of mir210. The fluorescence intensity increases with increasing the concentration of mir210 from 0.05 to 100 nM. The fluorescence intensity was monitored at 515 nm and plotted against the concentration of mir210. (B) The standard calibration plot of fluorescence intensity against mir210The fluorescence spectra were recorded by exciting at 470 ±10 nm and the emission intensity 515 nm. The error bars represent the standard deviation of three different measurements.
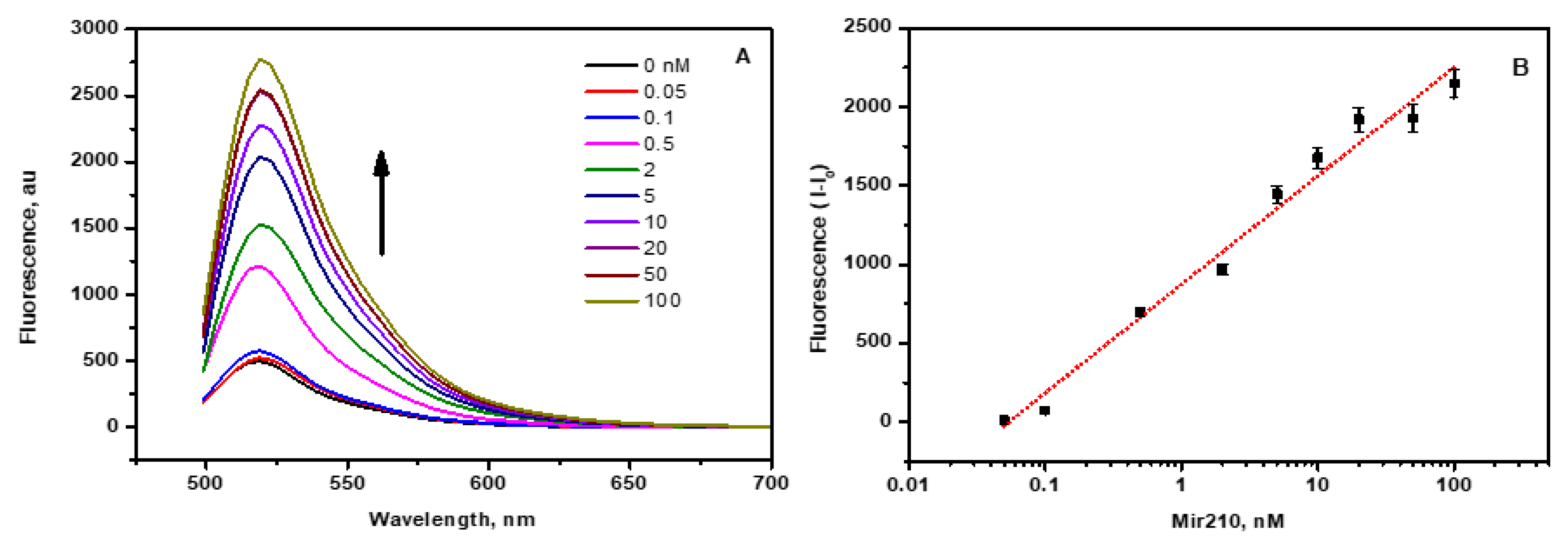
Figure 5.
Agarose gel(2%) electrophoresis of reverse transcribed product using total RNA extracted from exosome as a template and the stem-loop primers designed for mir210. The size of the cDNA from the reverse transcription is compared with the 100bp DNA ladder.
Figure 5.
Agarose gel(2%) electrophoresis of reverse transcribed product using total RNA extracted from exosome as a template and the stem-loop primers designed for mir210. The size of the cDNA from the reverse transcription is compared with the 100bp DNA ladder.
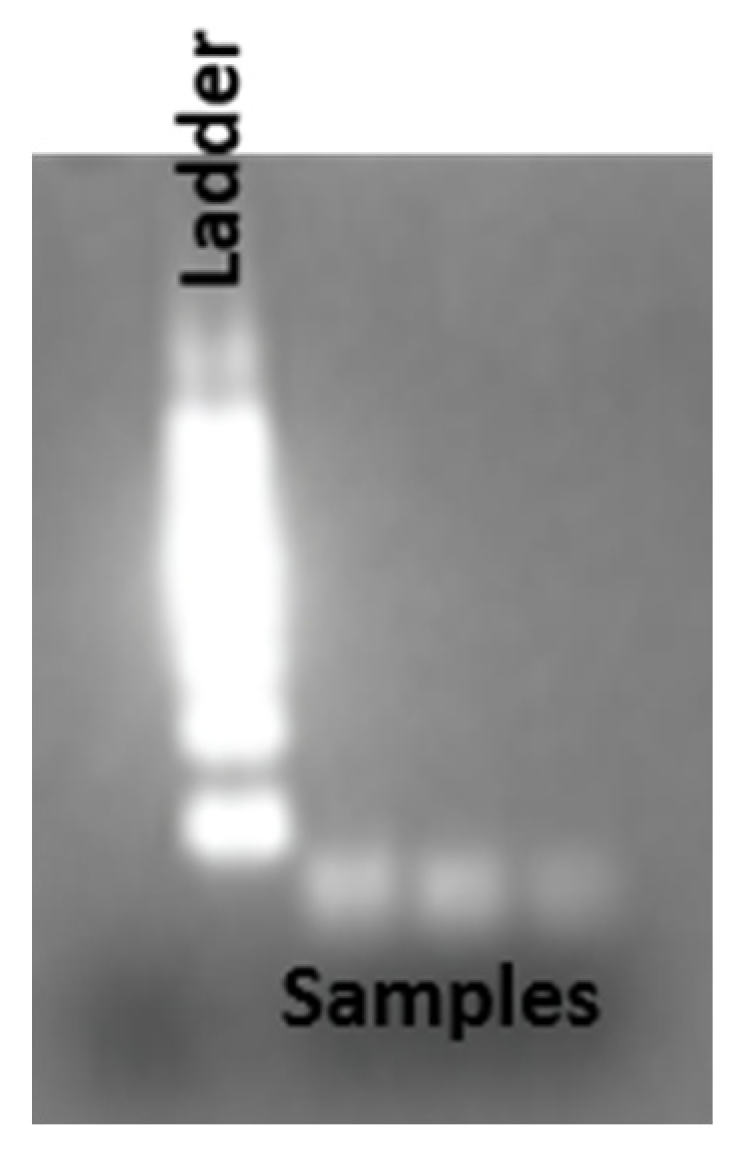
Figure 6.
Mir210 RT-qPCR assay. (A) PCR amplification plot of mir210 cDNA over seven orders of magnitude from 0.012 fM to 1.2x105 fM. (B) Standard calibration curve of mir210 cDNA. (C) Melting curve of the amplified PCR product in the temperature range between 05 to 95 C. (D) First derivative of the melting curve. The Tm of the products is close to 75.5±1.5 ºC.
Figure 6.
Mir210 RT-qPCR assay. (A) PCR amplification plot of mir210 cDNA over seven orders of magnitude from 0.012 fM to 1.2x105 fM. (B) Standard calibration curve of mir210 cDNA. (C) Melting curve of the amplified PCR product in the temperature range between 05 to 95 C. (D) First derivative of the melting curve. The Tm of the products is close to 75.5±1.5 ºC.
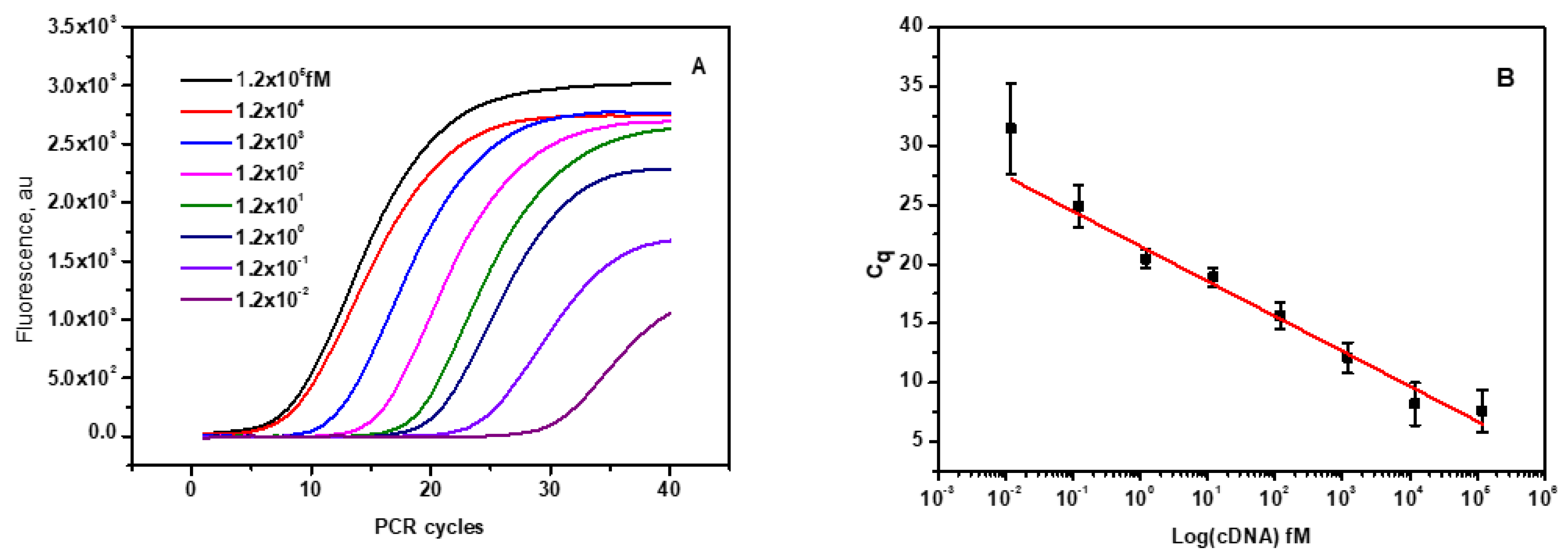
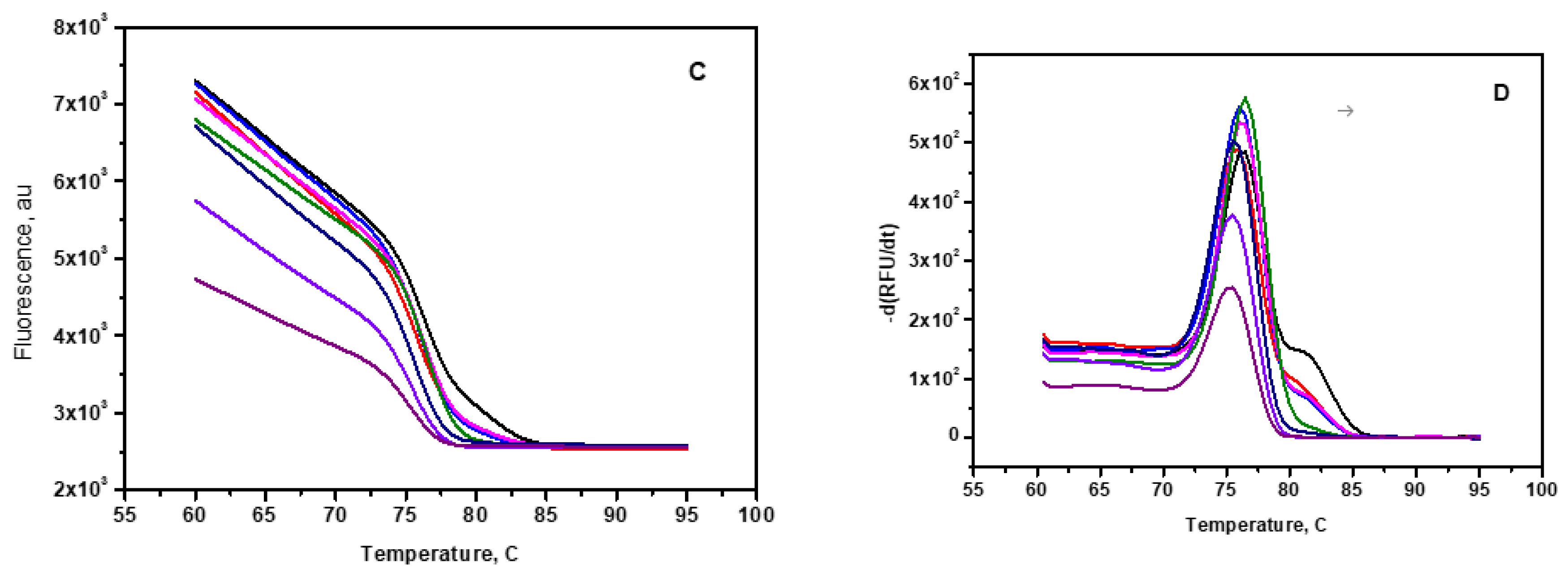
Figure 7.
Change in the fluorescence signal by the addition of different miRNA to the cDNA adsorbed on the CNM surface. The concentration of the mir210, mir10b, mir16 and mir191 are 50 nM in the binding buffer. The fluorescence signals were recorded by exciting at 470 ±10 nm and monitoring emission intensity at 515 nm. The error bars represent the standard deviation of three different measurements.
Figure 7.
Change in the fluorescence signal by the addition of different miRNA to the cDNA adsorbed on the CNM surface. The concentration of the mir210, mir10b, mir16 and mir191 are 50 nM in the binding buffer. The fluorescence signals were recorded by exciting at 470 ±10 nm and monitoring emission intensity at 515 nm. The error bars represent the standard deviation of three different measurements.
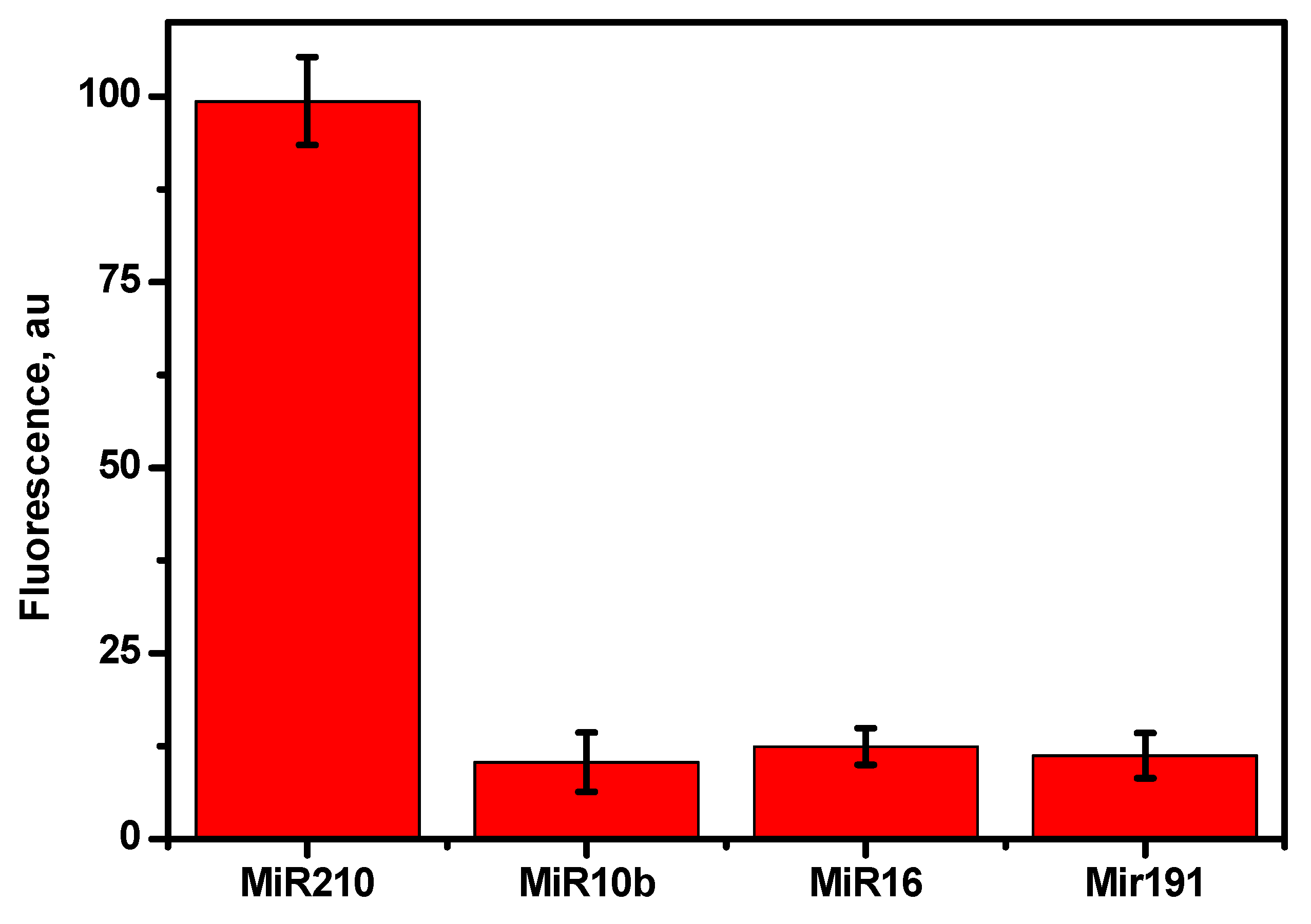
Figure 8.
Change in the fluorescence signal in the presence of mir210, without target and non-specific scrambled DNA. The fluorescence signals were recorded by exciting at 470 ±10 nm and monitoring emission intensity at 515 nm. The error bars represent the standard deviation of three different measurements.
Figure 8.
Change in the fluorescence signal in the presence of mir210, without target and non-specific scrambled DNA. The fluorescence signals were recorded by exciting at 470 ±10 nm and monitoring emission intensity at 515 nm. The error bars represent the standard deviation of three different measurements.
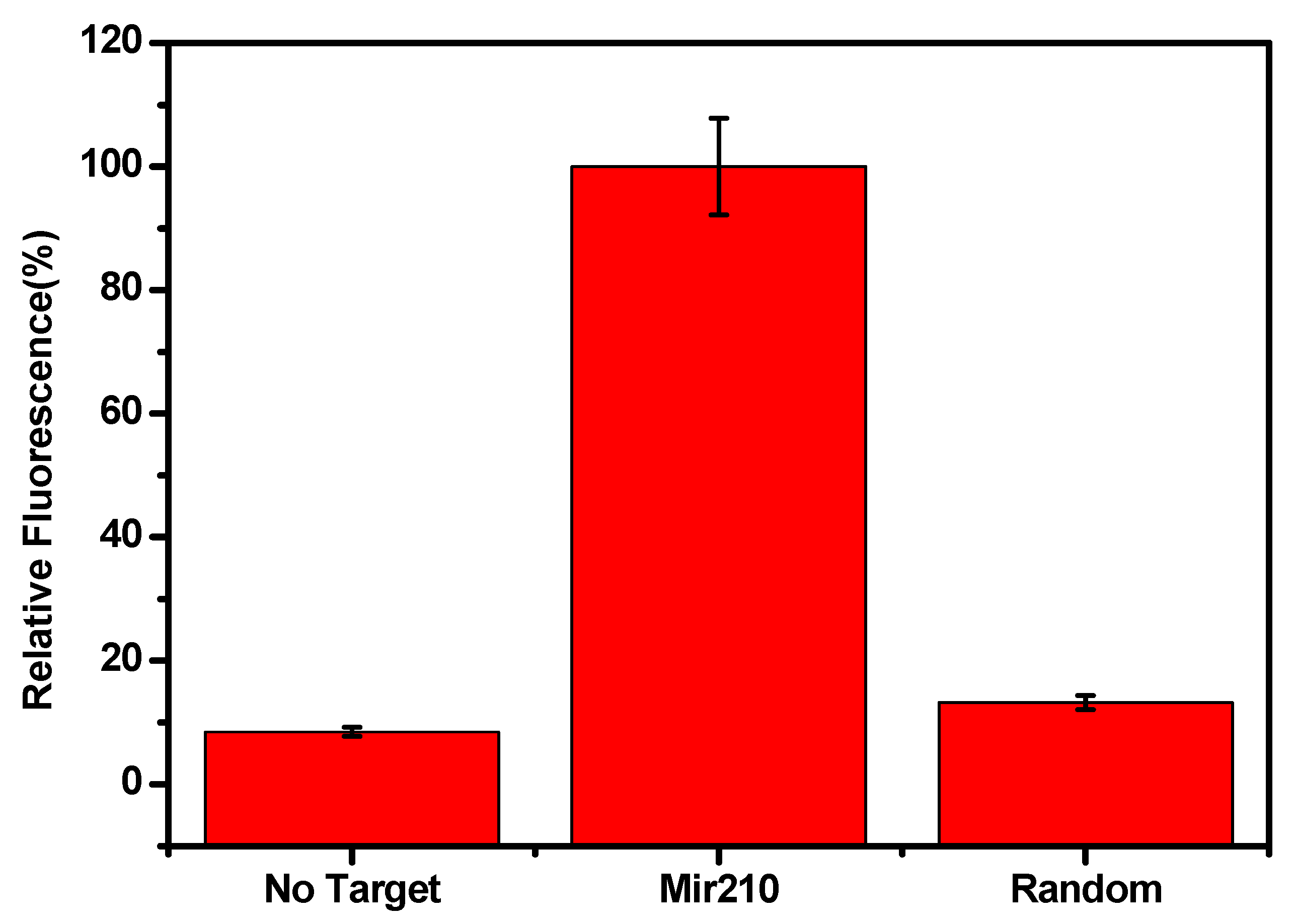

Table 1.
Synthetic Mir210 and other Oligonucleotides used in this work.
| Oligonucleotides (5’-3’) used |
|---|
| Mir210: CUGUGCGUGUGACAGCGGCUGA Mir210 cDNA: GACACGCACACTGTCGCCGACT Reverse complement: FAM-TCAGCCGCTGTCACACGCACAG Anti-CEA Aptamer: H2NTCGCGCGAGTCGTCTGGGGAACCATCGAGTTACACCGACCTTCTATGTGCGGCCCCCCGCATCGTCCTCCC Reverse complementary of CEA aptamer: GGGAGGACGATGCGGGGGGCCGCACATAGAAGGTCGGTGTAACTCGATGGTTCCCCAGACGACTCGCGCGA |
Table 2.
Reverse transcription primers and final product.
| MiRNA-210; Reverse transcription primers and the final product |
|---|
| Reverse transcription primer: GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCAGCC Forward primer: GTATACCTGTGCGTGTGACAG Reverse primer: GTGCAGGGTCCGAGGT Final product: 5'-GTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCAGCCGCTGTCACACGCACAGGTATAC-3' 3’-CACGTCCCAGGCTCCATAAGCGTGACCTATGCTGAGTCGGCGACAGTGTGCGTGTCCATATG-5' |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



