
Submitted:
22 June 2023
Posted:
23 June 2023
Read the latest preprint version here
Abstract
Abstract: In the last decade, gypsogenin has widely grabbed the attention of medicinal chemists by virtue of its prominent anti-cancer potential. Despite its late identification, gypsogenin proved itself as a new an-ti-proliferative player battling for a front position among other classic pentacyclic triterpenes such as oleanolic acid, glycyrrhetinic acid, ursolic acid, betulinic acid, and celastrol. Herein, we present insights into the anti-cancer activity of gypsogenin and its semisynthetic derivatives and go further by introducing our perspective to judiciously guide the prospect rational design. The present article opens a new venue for better exploitation of gypsogenin chemical entity as a lead compound in cancer chemotherapy. To the best of our knowledge, this is the first review article exploring the anti-cancer activity of gypsogenin derivatives.
Keywords:
pentacyclic triterpenes
; gypsogenin
; anti-cancer
1. Introduction
Cancer is the second major worldwide cause of mortality preceded with cardiovascular diseases [1,2,3,4]. Medicinal chemists are continuously urged to innovate new chemical entities to overcome resistance, reduce side effects, and enhance the efficacy of commercial drugs in the hard-fought battle against cancer [5,6,7,8,9,10]. Many natural products have provided skeletons and structural references for the invention of modern drugs [11,12,13,14]. Found in higher plants, pentacyclic triterpenes (PTs) are bio-nutrient phytochemicals endowed with diverse range of bioactivities such as hepatoprotective [15,16,17], anti-inflammatory [18,19,20], anti-hypertensive [17,21,22,23], anti-atherosclerotic [21,22], anti-viral [24,25,26], anti-fibrosis [27,28,29], and anti-ulcer effect [18,30,31]. In particular, PTs have ubiquitous applications in terms of anti-cancer drug discovery [32,33,34,35,36,37].
The literature is loaded with a plenty of success stories linking PTs derivatives with a prominent role in the prevention of cancer initiation, promotion, angiogenesis, and progression through disrupting different intermittent mechanisms and path ways. The number of scientific publications and citations linking PTs and cancer has been soaring over the past twenty years according to Web of Science database (Figure 1). PTs are generally non cytotoxic albeit minor derivatizations can lead to dramatic changes in activity.
PTs comprise four main chemical skeletons namely oleanane, ursane, lupane, and friedelane. Oleanolic acid, from the oleanane type, is one of the most extensively studied PTs in terms of medicinal chemistry. Oleanolic acid suppresses proliferation of hepatocellular carcinoma [38,39], human bladder cancer [40], breast cancer [41,42], lung carcer [43], and colon cancer [44,45]. To have such diverse activities, oleanolic acid modulates multiple cell signaling pathways [46]. Two oleanolic acid derivatives, CDDO and CDDO-Me, have entered clinical trials for treatment of solid tumors and lymphoma making it on the throne of pentacyclic triterpenes in terms of chemotherapy [47,48]. Glycyrrhetinic acid is another representative of oleanane-type triterpenoids with ubiquitous anti-cancer activities [49,50,51,52,53,54]. Ursolic acid [44,55,56,57,58], betulinic acid [59,60,61,62,63], and celastrol [64,65,66,67], representing ursane, lupane and friedelane type triterpenoids, respectively, were reported to possess multifaceted anti-cancer properties
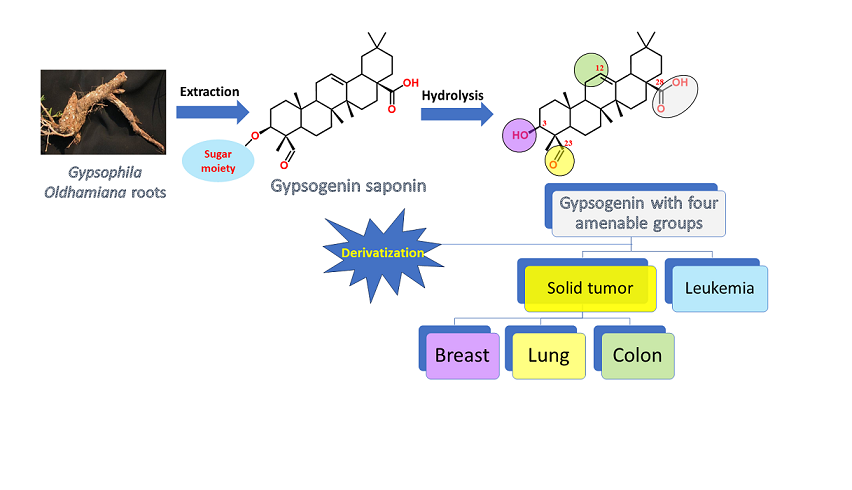
Gypsogenin (3-hydroxy-23-oxoolean-12-en-28-oic acid), a less-explored PT, extracted from Gypsophila oldhamiana in a saponin form and generated by hydrolysis (Figure 2) [68]. It has an oleanane type skeleton and possesses four active sites, C-3 hydroxyl, ring C double bond, C-23 aldehyde group and C-28 carboxylic acid, which are amenable for a wide range of chemical transformation. The aldehyde group is unique as other classic triterpenes lack such group which represents a structural alert for the medicinal chemistry community [69]. Recently, gypsogenin proved itself as an outstanding entity that can enter the competition between PTs for a front position as a lead anticancer agent. Stunningly, the first carboxamide derivative of gypsogenin came out in 2018 which points out the shortage of enough structure-activity relationship (SAR) studies on this precious PT [68]. Moreover, it was not there gypsogenin derivatives with modified ring C before 2023.
Several PTs exhibit limited water solubility and low bioavailability, which can be addressed by derivatization [70]. Derivatization not only optimizes triterpenes pharmacokinetics but also their pharmacodynamics. Herein, we compiled the previous success stories of gypsogenin and its semi-derivatives that showed promise for cancer therapy. We generated SAR for gypsogenin and its derivatives against leukemia, breast cancer and lung cancer. We present our recommendations for prospective work and the missing information that should be addressed. Our study represents a cornerstone reference for any future research linking and cancer. We believe that extensive SAR studies of gypsogenin will advance it to a front position in position in the pentacyclic triterpenes game of thrones on anti-cancer therapy.
2. Anti-cancer effect of gypsogenin, gypsogenic acid, and their semisynthetic derivatives
2.1. Anti-leukemic effect
In 2007, gypsogenic acid, the 4-carboxyl analogue of gypsogenin (Figure 2), didn't show observable activity against chronic myeloid leukemia (K562) and acute myeloid leukemia (HL-60) where its IC50 exceeded 100 µM for both cells [71]. Another study was in accordance with this where gypsogenic acid IC50 against K562 was 227.6 µM, however HL-60 was more sensitive (IC50 61.1 µM) [72]. The latter value is in discrepancy with the previous report by Lee group [71]. Gypsogenic acid demonstrated low activity against other lymphoid leukemias SKW-3 and BV-173 (IC50 79.1 and 41.4 µM, respectively) [72].
Later on, we found that gypsogenin highly outperforms gypsogenic acid with IC50 12.7 µM against K562 highlighting the crucial role of 4-aldehyde group [73]. Simultaneously, Emirdag et al., revealed that gypsogenin has anti-proliferative effect on HL-60 (IC50 10.4 µM) by inducing apoptosis [74,75]. Acetylation of the 3-OH group has almost no impact on activity against HL-60 (1b IC50 10.77 µM). Gypsogenin activity is increased by oximation of the 4-aldehyde group (compound 1a IC50 3.9 µM). Mutually, the 3-acetylated oxime analogue 1d surpassed the activity of 1b (IC50 5.9 µM) [74]. Gypsogenin benzyl ester 1c has IC50 8.1 µM whereas its 3-acetylated analogue 1h has IC50 6.7 µM [74] (Figure 3).
By virtue of its notable apoptotic effect, 1c was further benchmarked for its effect on K562 cell line where it showed moderate activity (IC50 9.3 µM) [76]. However, this study represented a turning point for a better understanding of gypsogenin’s molecular target. 1c inhibited ABL1 tyrosine kinase with IC50 8.71 µM. This is assumed to be the main target for its cytotoxic effect on K562. It is needless to say that the presence of other off targets cannot be excluded. Concomitantly, 1c inhibited other kinases such as C-terminal Src kinase (CSK) and Lyn kinase isoform B; LYN B (IC50 1.5 µM and 2.9 µM, respectively) [76]. It is clear that oximation of 1c is detrimental for its activity on both K562 and HL-60 as the respective IC50 value of 1f is 21.3 µM and 10.6 µM [76].
Ciftci et al. moved forward with a structure activity relationship study of 1c and succeeded in enhancing its activity [73]. As mentioned above, the free aldehyde group is crucial for activity against leukemia. Therefore, Ciftci et al. came up with phenyl substituted esters of 1c keeping a free 4-aldehyde group [73]. Compounds GP2 and GP5 have IC50 4.7 and 3.1 µM, respectively, against K562 cells. Additionally, IC50 of GP2 and GP5 for ABL1 tyrosine kinase was 7.1 µM and 6.1 µM, respectively. Both compounds have induced an explicit apoptosis effect, especially GP2 whose apoptosis induction was clearer than imatinib; a gold standard ABL1 kinase inhibitor for CML therapy. Concomitantly, GP2 suppressed the downstream signaling of extracellular signal-regulated kinase (ERK) phosphorylation [73]. In a similar vein, both compounds exhibited moderate activity on MT-2 and Jurkat cells. Of interest, the IC50 of GP5 for MT-2 and Jurkat was 7.2 µM and 4.8 µM, respectively. The authors evaluated both compounds for their effect on peripheral blood mononuclear cells (PBMC) and calculated the selectivity index as the ratio of the IC50 between PBMC and K562 cells. The higher selectivity index of GP2, 11.0, than GP5, 8.0, reflects a more satisfactory safety profile of GP5.
A recent report by Ulusoy et al. showed that reductive amination of the 4-aldehyde group with different aromatic and alicyclic amines has either reduction or complete abrogation of anti-K562 activity [77]. The hit compound in this study, 6l, has IC50 11.3 µM which is even less active than the parent compound, gypsogenin [77]. Furthermore, 6l inhibited ABL1 kinase in a moderate fashion (IC50 value of 13.0 µM). This is another evidence of the crucial role of the 4-aldehyde group for anti-K562 activity (Figure 4). In addition, 6l has less effect on MT-2 and Jurkat than those of GP-2 and GP-5. Compound 6l has moderate effect on a panel of kinases at 30 µM of drug concentration specially BRK, BTK, LYN B, and SRC. Compound 6j with the more hydrophobic 4-isopropyl substitution exhibited less activity (IC50 23.8 µM), whereas presence of a bulky N-piperazinyl benzyl moiety abolished activity as for 6c (IC50 > 100 µM). The activity is also abolished in presence of electron donating substitution as for 6i (IC50 > 100 µM).
Surprisingly, there is no report linking between gypsogenin or gypsogenic acid carboxamides and leukemia till now. This is the same case for gypsogenin derivatives with modified ring C (Figure 3). In a word, gypsogenin benzyl esters are the most active derivatives against K562 and HL-60 leukemias till now. The SAR pertaining to activity against K562 and HL-60 is depicted in Figure 4.
2.2. Anti-breast cancer activity
Gypsogenin has moderate cytotoxic activity on MCF-7 (IC50 9.0 µM) however its benzyl ester derivative 1c has IC50 5.1 µM [74]. Surprisingly, substituted benzyl esters such as GP2 and GP2 showed less activity than gypsogenin with respective IC50 51.58 µM and 15.3 µM. Notably, the 3-acetyl analogues of gypsogenin and 1c, namely 1b and 1h, possess less activity (IC50 20.5 µM and 65.1 µM, respectively). However, oximation of gypsogenin and 1c slightly improves their cytotoxic effect as seen for 1a and 1f. The exact mechanism of action is not elucidated [74].
Wu et al. found that gypsogenic acid has weak antiproliferative effect on MCF-7 (IC50 26.8 µM) which also highlight the role of 4-aldehyde group. The authors highly enhanced gypsogenin and gypsogenic acid activity through mono-and bisamidation [78]. Gypsogenin carboxamide with imidazole, compound 9, has IC50 3.7 µM which is similar to gypsogenic acid mono-amide of C28 with pyrazole, compound 14c, with IC50 3.8 µM. Gypsogenic acid bisamide of both C23 and C28, compound, 8f demonstrated a pronounced activity (IC50 4.1 µM). The favorable safety profile of those carboxamides is shown through measuring their activity on human umbilical vein endothelial cells (HUVEC cells). 8f possesses the highest selectivity index (24.0) among the mentioned active compounds.
Another evidence of the efficiency of gypsogenin amides was disclosed this year by Sun et al [79]. Two strong amides namely, 5 and 8 possess IC50 5.7 µM and 13.8 µM, respectively, towards MCF-7. They also synthesized compound 9a which is afforded by reductive amination using methylamine; its IC50 is 11.3 µM which is more than gypsogenin (IC50 9.0 µM). The selectivity index of 5, 8, and 9a exceeds 30 when related to their effect on HUVEC.
Ring C oxidized gypsogenin derivatives have been recently developed (Figure 3) [79]. The epoxide derivative (2) has IC50 26.6 µM on MCF-7. In parallel with this, the 11-keto derivative (3) has a similar activity (IC50 25.3 µM) implying that modification of this ring by oxidation reduces MCF-7 sensitivity. Conclusively, gypsogenin carboxamides are the best cytotoxic entities against MCF-7 when compared to other derivatives (Figure 5).
2.3. Anti-lung cancer activity
Gypsogenin can inhibit the growth and metastasis of Lewis lung cancer through inhibition of tumor angiogenesis and induction of apoptosis [80]. Different molecular targets were implicated in this mechanism. Gypsogenin downregulated mutant P53 and vascular endothelial growth factor (VEGF). It reduces the expression of Bcl-2 protein and raises Bax expression, promoting tumor apoptosis. The anti-proliferative effect of gypsogenin, 3-acetyl gypsogenin, (1b), and 3-acetyl gypsogenic acid against A549 lung cancer cells is moderate (IC50 19.6, 30.8, and 23.7 µM) [68,78]. Oximation of gypsogenin and 1b keeps the activity without significant change [68]. 2,4-dinitrophenyl)hydrazono derivative of gypsogenin (4) demonstrated strong cytotoxic effect on A549 cells (IC50 3.1 µM) [68]. In accordance, the amino product (9a) exhibited stronger cytotoxic effect (IC50 1.5 µM) [79].
The two carboxamides 9 and 14c showed a bit higher activity than compound 4 (IC50 2.5 and 2.8 µM, respectively) [78]. Both compounds destroyed the cell membranes and increase their permeability, which led to the outflow of intracellular nucleic acid, but they weakly induce apoptosis and arrest the cell cycle [78]. Another anti-lung cancer hit derivative is gypsogenic acid bisamidation product of (8f) whose IC50 is 2.0 µM. However, it is noteworthy that mono-amidation products 9 and 14c surpass (8f) activity but with lower selectivity index for HUVEC.
Concomitantly, compounds 5 and 8 showed sub-micromolar effect on A549 (IC50 0.5 µM and 0.9 µM, respectively), induced both apoptosis through damaging the cell membrane and cell cycle arrest. Combining in silico and in vitro tools defined VEGF1 as gypsogenin target. Remarkably, compound 5 showed higher binding affinity to VEGF1 than the parent compound which is in accordance with the cytotoxicity results. Gypsogenin esters showed disappointing results as for GP2 whose IC50 exceeds 100 µM and GP5 which is less active than the parent compound (IC50 24.5 µM).
The epoxide analogue (2) has almost same activity as the parent compound (IC50.18.7 µM) whereas the 11-keto derivative (3) has a slightly better activity (IC50.13.5 µM)). In conclusion, gypsogenin carboxamides are the most active anti-proliferative entities against A549 (Figure 6).
2.4. Other anti-cancer activities
A batch of gypsogenin derivatives demonstrated other notable anti-cancer effects. In this regard, we will focus only on compounds with at least single digit micromolar IC50. Compound 1a has a remarkable anti-proliferative activity against SaoS-2 cells (osteosarcoma) and HeLa cells (cervical cancer). Its 3-acetylated derivative (1d) also has a similar effect on SaoS-2 but not on HeLa. It is noteworthy that gypsogenin has IC50 7.8 against SaoS-2 which is better than 1a and 1d, and 3-acetyl gypsogenin (1b). On the other hand, 1d is distinguished by its high activity against HT-29 cells (colorectal adenocarcinoma) [74] (Table 1).
Other study showed that gypsogenin suppressed gastric cancer cells NCI-N87 proliferation via targeting VEGF and MM-9 and promoting the expression of caspase-3 and Bax proteins [80]. Compounds 4 and 7g were reported mainly for targeting colon cancer cells (LOVO) through strong induction of apoptosis and dose-dependent S-phase arrest in cells pre-treated with either of them. Both compounds exhibited moderate effect on SKOV3 (ovarian cancer) and HepG2 cells (Hepatocellular carcinoma) [68]. The amino compound 9a also exhibited notable activity against LOVO. Compounds 2 and 3 showed no or moderate activity towards LOVO [79]. The most active compound against LOVO cells is compound 8 with submicromolar cytotoxicity implying that gypsogenin carboxamides are usually on the top among other derivatives [79] (Table 1).
Three amides were reported by Wu et al., 9, 14c, and 8f with outstanding activities against HepG2, TE-1 (esophageal cancer), and MC3-8 (colon cancer) cells [78]. Gypsogenin 28-COOH ester GP5 showed better activity on HeLa cells than GP2 [73]. Ciftci et al., revealed new derivatives that suppress glioma proliferation through EGFR inhibition. The amino derivative compound 10 has the strongest effect against EGFR and glioma cells U251, T98G, and U87, consequently (Table 1). The titled compound clearly induced apoptosis in U251 in a comparable fashion to cisplatin. The study revealed that gypsogenin benzyl esters were less effective than 10 on glioma cells [81] (Table 1). Furthermore, at 30 µM concentration, compound 10 showed moderate inhibition for a panel of other kinases including ABL1 tyrosine kinase.
3. Conclusion and Future Directions
Befitting with its anticancer promise, we presented a critical review of gypsogenin and its derivatives. Gypsogenin carboxamides demonstrated high cytotoxic activity against breast and lung cancer. The bisamides of gypsogenic acid possess prominent activity as well. However, their anti-leukemic activity is yet to be explored by medicinal chemists. Gypsogenin benzyl esters showed pronounced activity against CML. Ring C modified gypsogenin derivatives are weak antiproliferative agents against lung and breast cancer, but they haven’t been tested for their anti-leukemic effect. Gypsogenin and its derivatives were reported to target kinases such as ABL1 and VEGF. The selectivity index of some active compounds is high reflecting their potential safety. Further medicinal chemistry studies on gypsogenin are urgently needed to afford more active hits and elucidate other plausible off targets.
Acknowledgments
The authors would like to thank the Deanship of scientific research at Umm Al-Qura University for supporting this work by grant code (23UQU4340520DSR009).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Santucci, C.; Carioli, G.; Bertuccio, P.; Malvezzi, M.; Pastorino, U.; Boffetta, P.; Negri, E.; Bosetti, C.; La Vecchia, C. Progress in cancer mortality, incidence, and survival: a global overview. Eur. J. Cancer Prev. 2020, 29, 367–381. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, Y.; Weng, S.; Xu, H.; Li, L.; Han, X. A New Trend in Cancer Treatment: The Combination of Epigenetics and Immunotherapy. Front. Immunol. 2022, 13, 809761. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Billaud, M.; Almeida, R. RNA-Binding Proteins in Cancer: Old Players and New Actors. Trends Cancer 2017, 3, 506–528. [Google Scholar] [CrossRef]
- Bhinder, B.; Gilvary, C.; Madhukar, N.S.; Elemento, O. Artificial Intelligence in Cancer Research and Precision Medicine. Cancer Discov. 2021, 11, 900–915. [Google Scholar] [CrossRef]
- Radwan, M.O.; Toma, T.; Arakaki, Y.; Kamo, M.; Inoue, N.; Koga, R.; Otsuka, M.; Tateishi, H.; Fujita, M. New insight into the bioactivity of substituted benzimidazole derivatives: Repurposing from anti-HIV activity to cell migration inhibition targeting hnRNP M. Bioorganic Med. Chem. 2023, 86, 117294. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- AlQathama, A.; Umm Al-Qura University; Yonbawi, A. R.; Shao, L.; Bader, A.; Abdalla, A.N.; Gibbons, S.; Prieto, J.M.; King Abdulaziz University; University College London; et al. The in vitro cytotoxicity against human melanoma cells, tyrosinase inhibition and antioxidant activity of Grewia tenax leaves extracts. Boletin Latinoam. Caribe Plantas Med. Aromat. 2022, 22, 268–276. [Google Scholar] [CrossRef]
- Bader, A.; Abdalla, A.N.; Obaid, N.A.; Youssef, L.; Naffadi, H.M.; Elzubier, M.E.; Almaimani, R.A.; Flamini, G.; Pieracci, Y.; El-Readi, M.Z. In Vitro Anticancer and Antibacterial Activities of the Essential Oil of Forsskal’s Basil Growing in Extreme Environmental Conditions. Life 2023, 13, 651. [Google Scholar] [CrossRef]
- Abo-Elghiet, F.; Ibrahim, M.H.; El Hassab, M.A.; Bader, A.; Abdallah, Q.M.; Temraz, A. LC/MS analysis of Viscum cruciatum Sieber ex Boiss. extract with anti-proliferative activity against MCF-7 cell line via G0/G1 cell cycle arrest: An in-silico and in-vitro study. J. Ethnopharmacol. 2022, 295, 115439. [Google Scholar] [CrossRef]
- Bader, A.; Bkhaitan, M.M.; Abdalla, A.N.; Abdallah, Q.M.A.; Ali, H.I.; Sabbah, D.A.; Albadawi, G.; Abushaikha, G.M. Design and Synthesis of 4-O-Podophyllotoxin Sulfamate Derivatives as Potential Cytotoxic Agents. Evidence-Based Complement. Altern. Med. 2021, 2021, e6672807. [Google Scholar] [CrossRef]
- Gutiérrez-Rebolledo, G.A.; Siordia-Reyes, A.G.; Meckes-Fischer, M.; Jiménez-Arellanes, A. Hepatoprotective properties of oleanolic and ursolic acids in antitubercular drug-induced liver damage. Asian Pac. J. Trop. Med. 2016, 9, 644–651. [Google Scholar] [CrossRef]
- Xu, G.-B.; Xiao, Y.-H.; Zhang, Q.-Y.; Zhou, M.; Liao, S.-G. Hepatoprotective natural triterpenoids. Eur. J. Med. Chem. 2018, 145, 691–716. [Google Scholar] [CrossRef]
- Ayeleso, T.B.; Matumba, M.G.; Mukwevho, E. Oleanolic Acid and Its Derivatives: Biological Activities and Therapeutic Potential in Chronic Diseases. Molecules 2017, 22, 1915. [Google Scholar] [CrossRef]
- Aly, A.M.; Al-Alousi, L.; Salem, H.A. Licorice: A possible anti-inflammatory and anti-ulcer drug. AAPS PharmSciTech 2005, 6, E74–E82. [Google Scholar] [CrossRef]
- Tsai, S.-J.; Yin, M.-C. Antioxidative and Anti-Inflammatory Protection of Oleanolic Acid and Ursolic Acid in PC12 Cells. J. Food Sci. 2008, 73, H174–H178. [Google Scholar] [CrossRef]
- Radwan, M.O.; Ismail, M.A.; El-Mekkawy, S.; Ismail, N.S.; Hanna, A.G. Synthesis and biological activity of new 18β-glycyrrhetinic acid derivatives. Arab. J. Chem. 2016, 9, 390–399. [Google Scholar] [CrossRef]
- Somova, L.; Shode, F.; Ramnanan, P.; Nadar, A. Antihypertensive, antiatherosclerotic and antioxidant activity of triterpenoids isolated from Olea europaea, subspecies africana leaves. J. Ethnopharmacol. 2003, 84, 299–305. [Google Scholar] [CrossRef]
- Somova, L.; Nadar, A.; Rammanan, P.; Shode, F. Cardiovascular, antihyperlipidemic and antioxidant effects of oleanolic and ursolic acids in experimental hypertension. Phytomedicine 2003, 10, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Wang, X.; Tian, Z.; Qi, D.; Li, Y.; Jiang, H. Antihypertensive activity of oleanolic acid is mediated via downregulation of secretory phospholipase A2 and fatty acid synthase in spontaneously hypertensive rats. Int. J. Mol. Med. 2020, 46, 2019–2034. [Google Scholar] [CrossRef]
- Pompei, R.; Laconi, S.; Ingianni, A. Antiviral properties of glycyrrhizic acid and its semisynthetic derivatives. Mini-Reviews Med. Chem. 2012, 9, 996–1001. [Google Scholar] [CrossRef]
- Sun, Z.-G.; Zhao, T.-T.; Lu, N.; Yang, Y.-A.; Zhu, H.-L. Research Progress of Glycyrrhizic Acid on Antiviral Activity. Mini-Reviews Med. Chem. 2019, 19, 826–832. [Google Scholar] [CrossRef]
- Tohmé, M.; Giménez, M.; Peralta, A.; Colombo, M.; Delgui, L. Ursolic acid: A novel antiviral compound inhibiting rotavirus infection in vitro. Int. J. Antimicrob. Agents 2019, 54, 601–609. [Google Scholar] [CrossRef]
- Yang, Y.; Huang, Y.; Huang, C.; Lv, X.; Liu, L.; Wang, Y.; Li, J. Antifibrosis effects of triterpene acids of Eriobotrya japonica (Thunb.) Lindl. leaf in a rat model of bleomycin-induced pulmonary fibrosis. J. Pharm. Pharmacol. 2012, 64, 1751–1760. [Google Scholar] [CrossRef]
- Lee, M.K.; Lee, K.Y.; Jeon, H.Y.; Sung, S.H.; Kim, Y.C. Antifibrotic activity of triterpenoids from the aerial parts ofEuscaphis japonicaon hepatic stellate cells. J. Enzym. Inhib. Med. Chem. 2009, 24, 1276–1279. [Google Scholar] [CrossRef]
- Xiang, H.; Han, Y.; Zhang, Y.; Yan, W.; Xu, B.; Chu, F.; Xie, T.; Jia, M.; Yan, M.; Zhao, R.; et al. A New Oleanolic Acid Derivative against CCl4-Induced Hepatic Fibrosis in Rats. Int. J. Mol. Sci. 2017, 18, 553. [Google Scholar] [CrossRef]
- Farina, C.; Pinza, M.; Pifferi, G. Synthesis and anti-ulcer activity of new derivatives of glycyrrhetic, oleanolic and ursolic acids. Il Farm. 1998, 53, 22–32. [Google Scholar] [CrossRef]
- Somensi, L.B.; Costa, P.; Boeing, T.; Mariano, L.N.B.; Longo, B.; Magalhães, C.G.; Duarte, L.P.; e Silva, A.T.M.; de Souza, P.; de Andrade, S.F.; et al. Gastroprotective properties of Lupeol-derived ester: Pre-clinical evidences of Lupeol-stearate as a potent antiulcer agent. Chem. Interactions 2020, 321, 108964. [Google Scholar] [CrossRef]
- Chudzik, M.; Korzonek-Szlacheta, I.; Król, W. Triterpenes as Potentially Cytotoxic Compounds. Molecules 2015, 20, 1610–1625. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-Y.; Li, Y.; Tang, Y.-T.; Ma, X.-D.; Tang, Z.-Y. Anticancer activity of oleanolic acid and its derivatives: Recent advances in evidence, target profiling and mechanisms of action. Biomed. Pharmacother. 2022, 145, 112397. [Google Scholar] [CrossRef]
- Salvador, J.A.R.; Leal, A.S.; Valdeira, A.S.; Gonçalves, B.M.F.; Alho, D.P.S.; Figueiredo, S.A.C.; Silvestre, S.M.; Mendes, V.I.S. Oleanane-, ursane-, and quinone methide friedelane-type triterpenoid derivatives: Recent advances in cancer treatment. Eur. J. Med. Chem. 2017, 142, 95–130. [Google Scholar] [CrossRef]
- Laszczyk, M.N. Pentacyclic Triterpenes of the Lupane, Oleanane and Ursane Group as Tools in Cancer Therapy. Planta Medica 2009, 75, 1549–1560. [Google Scholar] [CrossRef]
- Ghante, M.H.; Jamkhande, P.G. Role of Pentacyclic Triterpenoids in Chemoprevention and Anticancer Treatment: An Overview on Targets and Underling Mechanisms. J. Pharmacopunct. 2019, 22, 55–67. [Google Scholar] [CrossRef]
- Shaheen, U.; Ragab, E.A.; Abdalla, A.N.; Bader, A. Triterpenoidal saponins from the fruits of Gleditsia caspica with proapoptotic properties. Phytochemistry 2018, 145, 168–178. [Google Scholar] [CrossRef]
- Liese, J.; Abhari, B.A.; Fulda, S. Smac mimetic and oleanolic acid synergize to induce cell death in human hepatocellular carcinoma cells. Cancer Lett. 2015, 365, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bai, H.; Zhang, X.; Liu, J.; Cao, P.; Liao, N.; Zhang, W.; Wang, Z.; Hai, C. Inhibitory effect of oleanolic acid on hepatocellular carcinoma via ERK–p53-mediated cell cycle arrest and mitochondrial-dependent apoptosis. Carcinog. 2013, 34, 1323–1330. [Google Scholar] [CrossRef]
- Mu, D.-W.; Guo, H.-Q.; Zhou, G.-B.; Li, J.-Y.; Su, B. Oleanolic acid suppresses the proliferation of human bladder cancer by Akt/mTOR/S6K and ERK1/2 signaling. Int. J. Clin. Exp. Pathol. 2015, 8, 13864–13870. [Google Scholar]
- Amara, S.; Zheng, M.; Tiriveedhi, V. Oleanolic Acid Inhibits High Salt-Induced Exaggeration of Warburg-like Metabolism in Breast Cancer Cells. Cell Biochem. Biophys. 2016, 74, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, B.; Maurya, R.; Siddiqui, J.A.; Bid, H.K.; Rajendran, S.; Yadav, P.P.; Konwar, R. In vitro anti-breast cancer activity of ethanolic extract of Wrightia tomentosa: Role of pro-apoptotic effects of oleanolic acid and urosolic acid. J. Ethnopharmacol. 2012, 142, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, M.; Li, D. Oleanolic acid suppresses the proliferation of lung carcinoma cells by miR-122/Cyclin G1/MEF2D axis. Mol. Cell. Biochem. 2015, 400, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Furtado, R.A.; Rodrigues. P.; Araújo, F.R.R.; Oliveira, W.L.; Furtado, M.A.; Castro, M.B.; Cunha, W.R.; Tavares, D.C. Ursolic Acid and Oleanolic Acid Suppress Preneoplastic Lesions Induced by 1,2-Dimethylhydrazine in Rat Colon. Toxicol. Pathol. 2008, 36, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Janakiram, N.B.; Indranie, C.; Malisetty, S.V.; Jagan, P.; Steele, V.E.; Rao, C.V. Chemoprevention of Colon Carcinogenesis by Oleanolic Acid and Its Analog in Male F344 Rats and Modulation of COX-2 and Apoptosis in Human Colon HT-29 Cancer Cells. Pharm. Res. 2008, 25, 2151–2157. [Google Scholar] [CrossRef]
- Žiberna, L.; Šamec, D.; Mocan, A.; Nabavi, S.F.; Bishayee, A.; Farooqi, A.A.; Sureda, A.; Nabavi, S.M. Oleanolic Acid Alters Multiple Cell Signaling Pathways: Implication in Cancer Prevention and Therapy. Int. J. Mol. Sci. 2017, 18, 643. [Google Scholar] [CrossRef]
- Yadav, V.R.; Prasad, S.; Sung, B.; Kannappan, R.; Aggarwal, B.B. Targeting Inflammatory Pathways by Triterpenoids for Prevention and Treatment of Cancer. Toxins 2010, 2, 2428–2466. [Google Scholar] [CrossRef]
- Borella, R.; Forti, L.; Gibellini, L.; De Gaetano, A.; De Biasi, S.; Nasi, M.; Cossarizza, A.; Pinti, M. Synthesis and Anticancer Activity of CDDO and CDDO-Me, Two Derivatives of Natural Triterpenoids. Molecules 2019, 24, 4097. [Google Scholar] [CrossRef]
- Hsu, Y.-C.; Hsieh, W.-C.; Chen, S.-H.; Li, Y.-Z.; Liao, H.-F.; Lin, M.-Y.; Sheu, S.-M. 18β-glycyrrhetinic Acid Modulated Autophagy is Cytotoxic to Breast Cancer Cells. Int. J. Med Sci. 2023, 20, 444–454. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.-Q.; Song, J.; Liu, Q.-M.; Wang, C.; Huang, Z.; Chu, L.; Liang, H.-F.; Zhang, B.-X.; Chen, X.-P. 18β-Glycyrrhetinic-acid-mediated unfolded protein response induces autophagy and apoptosis in hepatocellular carcinoma. Sci. Rep. 2018, 8, 9365. [Google Scholar] [CrossRef]
- Sun, Y.; Dai, C.; Yin, M.; Lu, J.; Hu, H.; Chen, D. Hepatocellular carcinoma-targeted effect of configurations and groups of glycyrrhetinic acid by evaluation of its derivative-modified liposomes. Int. J. Nanomed. 2018, ume 13, 1621–1632. [Google Scholar] [CrossRef]
- Lee, C.S.; Kim, Y.J.; Lee, M.S.; Han, E.S.; Lee, S.J. 18β-Glycyrrhetinic acid induces apoptotic cell death in SiHa cells and exhibits a synergistic effect against antibiotic anti-cancer drug toxicity. Life Sci. 2008, 83, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Noshita, T.; Yu, T.; Kidachi, Y.; Kamiie, K.; Umetsu, H.; Ryoyama, K. Novel effects of glycyrrhetinic acid on the central nervous system tumorigenic progenitor cells: Induction of actin disruption and tumor cell-selective toxicity. Eur. J. Med. Chem. 2010, 45, 2943–2948. [Google Scholar] [CrossRef]
- Roohbakhsh, A.; Iranshahy, M.; Iranshahi, M. Glycyrrhetinic Acid and Its Derivatives: Anti-Cancer and Cancer Chemopreventive Properties, Mechanisms of Action and Structure- Cytotoxic Activity Relationship. Curr. Med. Chem. 2016, 23, 498–517. [Google Scholar] [CrossRef] [PubMed]
- Zafar, S.; Khan, K.; Hafeez, A.; Irfan, M.; Armaghan, M.; Rahman, A.U.; Gürer, E.S.; Sharifi-Rad, J.; Butnariu, M.; Bagiu, I.-C.; et al. Ursolic acid: a natural modulator of signaling networks in different cancers. Cancer Cell Int. 2022, 22, 399. [Google Scholar] [CrossRef] [PubMed]
- Raphael, T.; Kuttan, G. Effect of Naturally Occurring Triterpenoids Ursolic Acid and Glycyrrhizic Acid on the Cell-Mediated Immune Responses of Metastatic Tumor-Bearing Animals. Immunopharmacol. Immunotoxicol. 2008, 30, 243–255. [Google Scholar] [CrossRef]
- Kim, D.K.; Baek, J.H.; Kang, C.M.; A Yoo, M.; Sung, J.W.; Chung, H.Y.; Kim, N.D.; Choi, Y.H.; Lee, S.H.; Kim, K.W. Apoptotic activity of ursolic acid may correlate with the inhibition of initiation of DNA replication. Int. J. Cancer 2000, 87. [Google Scholar] [CrossRef]
- Liu, X.-S.; Jiang, J. Induction of Apoptosis and Regulation of the MAPK Pathway by Ursolic Acid in Human Leukemia K562 Cells. Planta Medica 2007, 73, 1192–1194. [Google Scholar] [CrossRef]
- Pisha, E.; Chai, H.; Lee, I.-S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.; Fong, H.H.; Kinghorn, A.D.; Brown, D.M.; et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef]
- Hordyjewska, A.; Ostapiuk, A.; Horecka, A.; Kurzepa, J. Betulin and betulinic acid: triterpenoids derivatives with a powerful biological potential. Phytochem. Rev. 2019, 18, 929–951. [Google Scholar] [CrossRef]
- Fulda, S. Betulinic acid: A natural product with anticancer activity. Mol. Nutr. Food Res. 2009, 53, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Selzer, E.; Pimentel, E.; Wacheck, V.; Schlegel, W.; Pehamberger, H.; Jansen, B.; Kodym, R. Effects of Betulinic Acid Alone and in Combination with Irradiation in Human Melanoma Cells. J. Investig. Dermatol. 2000, 114, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Debatin, K.-M. Sensitization for Anticancer Drug-Induced Apoptosis by Betulinic Acid. Neoplasia 2005, 7, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Ong, P.S.; Wang, L.; Goel, A.; Ding, L.; Wong, A.L.-A.; Ho, P.C.-L.; Sethi, G.; Xiang, X.; Goh, B.C. Celastrol in cancer therapy: Recent developments, challenges and prospects. Cancer Lett. 2021, 521, 252–267. [Google Scholar] [CrossRef]
- Yang, H.; Chen, D.; Cui, Q.C.; Yuan, X.; Dou, Q.P. Celastrol, a Triterpene Extracted from the Chinese “Thunder of God Vine,” Is a Potent Proteasome Inhibitor and Suppresses Human Prostate Cancer Growth in Nude Mice. Cancer Res 2006, 66, 4758–4765. [Google Scholar] [CrossRef]
- Nagase, M.; Oto, J.; Sugiyama, S.; Yube, K.; Takaishi, Y.; Sakato, N. Apoptosis Induction in HL-60 Cells and Inhibition of Topoisomerase II by Triterpene Celastrol. Biosci. Biotechnol. Biochem. 2003, 67, 1883–1887. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Manu, K.A.; Chen, L.; Li, F.; Rajendran, P.; Subramaniam, A.; Lam, P.; Kumar, A.P.; Sethi, G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis Int. J. Program. Cell Death 2011, 16, 1028–1041. [Google Scholar] [CrossRef]
- Zhang, H.; Mu, Y.; Wang, F.; Song, L.; Sun, J.; Liu, Y.; Sun, J. Synthesis of gypsogenin derivatives with capabilities to arrest cell cycle and induce apoptosis in human cancer cells. R. Soc. Open Sci. 2018, 5, 171510. [Google Scholar] [CrossRef]
- Gampe, C.; Verma, V.A. Curse or Cure? A Perspective on the Developability of Aldehydes as Active Pharmaceutical Ingredients. J. Med. Chem. 2020, 63, 14357–14381. [Google Scholar] [CrossRef]
- Furtado, N.A.J.C.; Pirson, L.; Edelberg, H.; Miranda, L.M.; Loira-Pastoriza, C.; Preat, V.; Larondelle, Y.; André, C.M. Pentacyclic Triterpene Bioavailability: An Overview of In Vitro and In Vivo Studies. Molecules 2017, 22, 400. [Google Scholar] [CrossRef]
- Lee, I.; Yoo, J.K.; Na, M.; Min, B.S.; Lee, J.; Yun, B.S.; Jin, W.; Kim, H.; Youn, U.; Chen, Q.C.; et al. Cytotoxicity of Triterpenes Isolated from Aceriphyllum rossii. Chem. Pharm. Bull. 2007, 55, 1376–1378. [Google Scholar] [CrossRef]
- Krasteva, I.; Yotova, M.; Yosifov, D.; Benbassat, N.; Jenett-Siems, K.; Konstantinov, S. Cytotoxicity of gypsogenic acid isolated from Gypsophila trichotoma. Pharmacogn. Mag. 2014, 10, 430–S433. [Google Scholar] [CrossRef] [PubMed]
- Ciftci, H.I.; Radwan, M.O.; Ozturk, S.E.; Ulusoy, N.G.; Sozer, E.; Ellakwa, D.E.; Ocak, Z.; Can, M.; Ali, T.F.; Abd-Alla, H.I.; et al. Design, Synthesis and Biological Evaluation of Pentacyclic Triterpene Derivatives: Optimization of Anti-ABL Kinase Activity. Molecules 2019, 24, 3535. [Google Scholar] [CrossRef] [PubMed]
- Emirdağ-Öztürk, S.; Karayıldırım, T.; Çapcı-Karagöz, A.; Alankuş-Çalışkan, Ö.; Özmen, A.; Poyrazoğlu-Çoban, E. Synthesis, antimicrobial and cytotoxic activities, and structure–activity relationships of gypsogenin derivatives against human cancer cells. Eur. J. Med. Chem. 2014, 82, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Emirdağ-Öztürk, S.; Babahan, I.; Özmen, A. Synthesis, characterization and in vitro anti-neoplastic activity of gypsogenin derivatives. Bioorganic Chem. 2014, 53, 15–23. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Ozturk, S.E.; Ali, T.F.S.; Radwan, M.O.; Tateishi, H.; Koga, R.; Ocak, Z.; Can, M.; Otsuka, M.; Fujita, M. The First Pentacyclic Triterpenoid Gypsogenin Derivative Exhibiting Anti-ABL1 Kinase and Anti-chronic Myelogenous Leukemia Activities. Biol. Pharm. Bull. 2018, 41, 570–574. [Google Scholar] [CrossRef]
- Ulusoy, N.G.; Emirdağ, S.; Sözer, E.; Radwan, M.O.; Çiftçi, H.; Aksel, M.; Bölükbaşı, S. .; Özmen, A.; Yaylı, N.; Karayıldırım, T.; et al. Design, semi-synthesis and examination of new gypsogenin derivatives against leukemia via Abl tyrosine kinase inhibition and apoptosis induction. Int. J. Biol. Macromol. 2022, 222, 1487–1499. [Google Scholar] [CrossRef]
- Wu, G.; Chu, H.; Wang, J.; Mu, Y.; Sun, J. Synthesis of gypsogenin and gypsogenic acid derivatives with antitumor activity by damaging cell membranes. New J. Chem. 2019, 43, 18898–18914. [Google Scholar] [CrossRef]
- Sun, K.; Zhao, T.; Liu, L.; Mu, X.; Sun, J. Anticancer Structure-activity Relationships and Potential Target Exploration of the Natural Product Gypsogenin. Chemistryselect 2023, 8, e202300072. [Google Scholar] [CrossRef]
- Tian, G.; Zhou, L.; Zhong, Y.; Xu, W.; Bai, H.; Liu, L.; Cui, S. Experimental studies of the therapeutic effect of Gypsophila oldhamiana gypsogenin on Lewis lung cancer in mice. Chin. J. Clin. Oncol. 2008, 5, 206–210. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Radwan, M.O.; Sever, B.; Hamdy, A.K.; Emirdağ, S.; Ulusoy, N.G.; Sozer, E.; Can, M.; Yayli, N.; Araki, N.; et al. EGFR-Targeted Pentacyclic Triterpene Analogues for Glioma Therapy. Int. J. Mol. Sci. 2021, 22, 10945. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Number of citations and scientific publications containing research on anti-cancer triterpenes over period 2000 – 2023. Data was obtained from the Web of Science database by searching the keywords triterpene cancer.
Figure 1.
Number of citations and scientific publications containing research on anti-cancer triterpenes over period 2000 – 2023. Data was obtained from the Web of Science database by searching the keywords triterpene cancer.
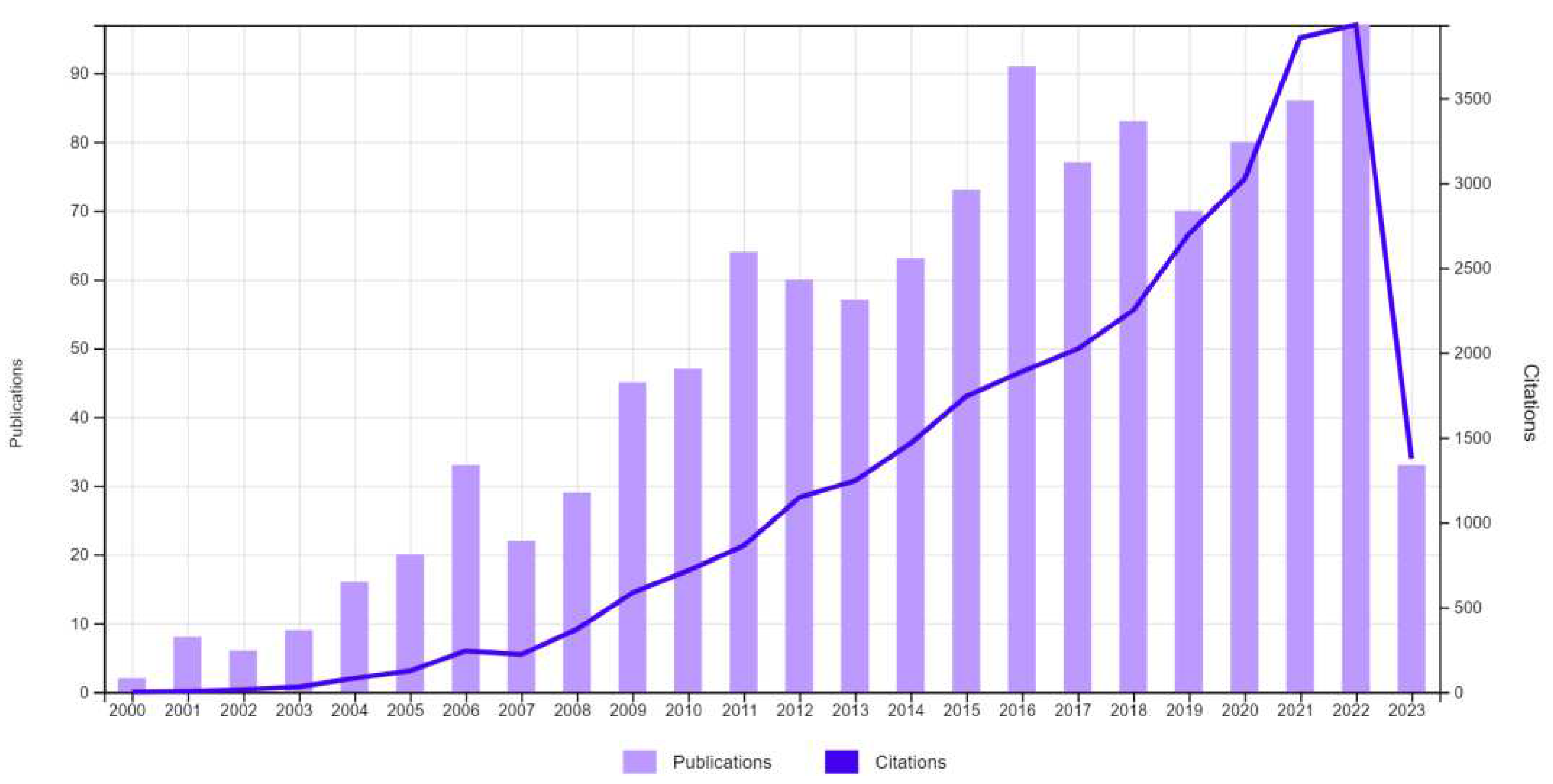
Figure 2.
Structure of gypsogenin, gypsogenic acid, and their bioactive derivatives by reacting with 3-OH, C-23 -CHO or -COOH, and C28 -COOH.
Figure 2.
Structure of gypsogenin, gypsogenic acid, and their bioactive derivatives by reacting with 3-OH, C-23 -CHO or -COOH, and C28 -COOH.
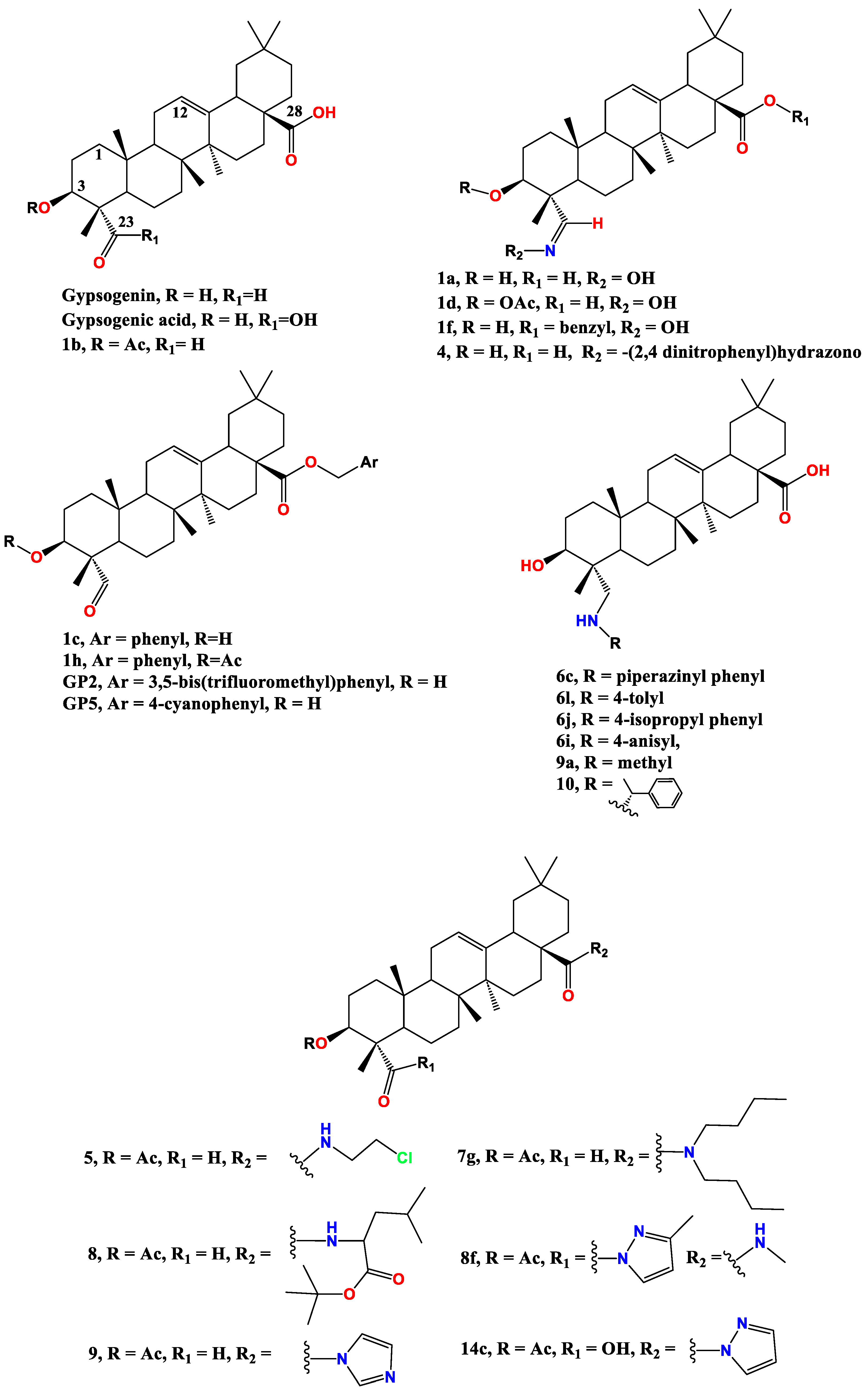
Figure 3.
Gypsogenin derivatives with modified ring C.
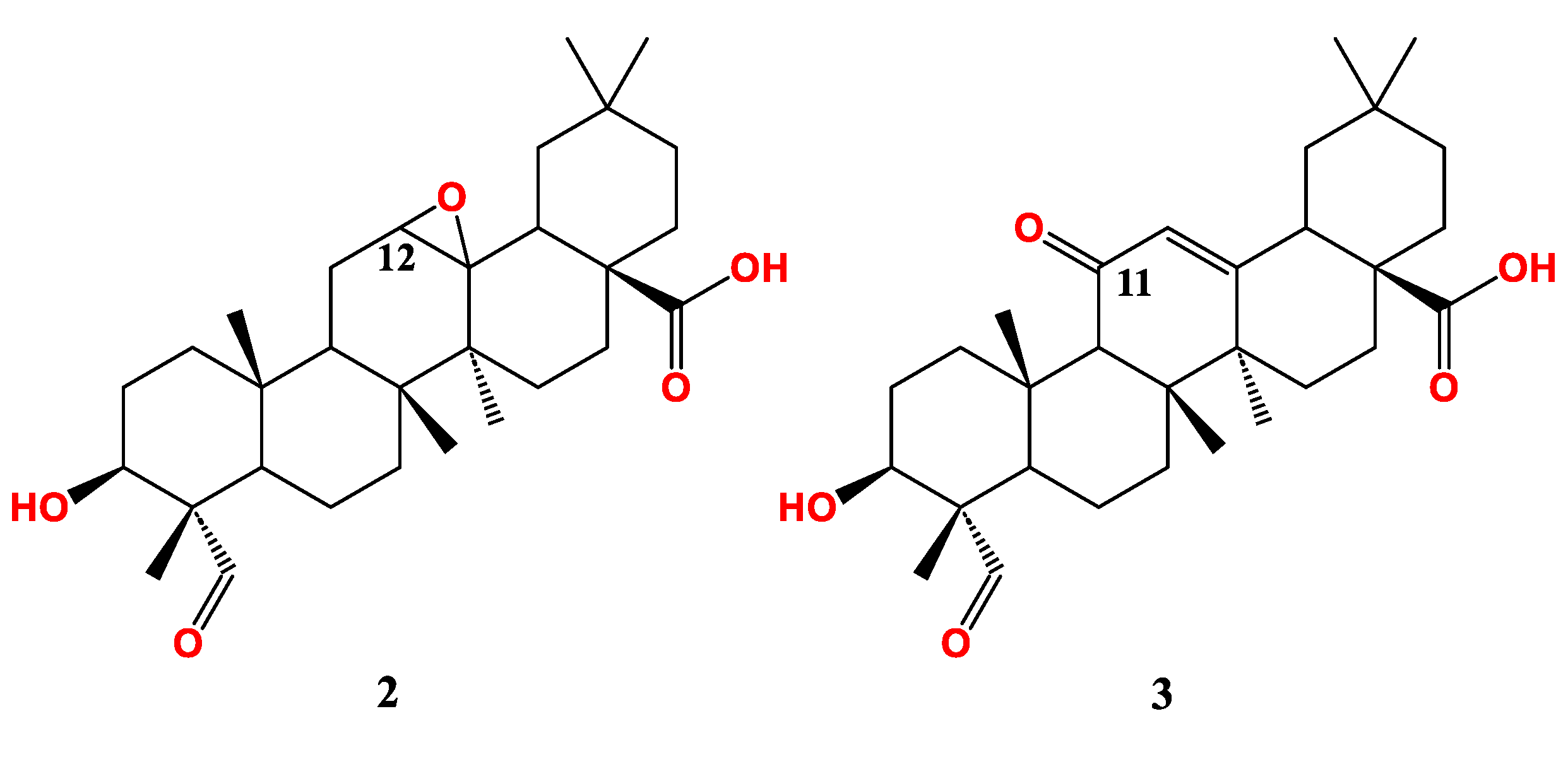
Figure 4.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against K562 and HL-60 cells.
Figure 4.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against K562 and HL-60 cells.
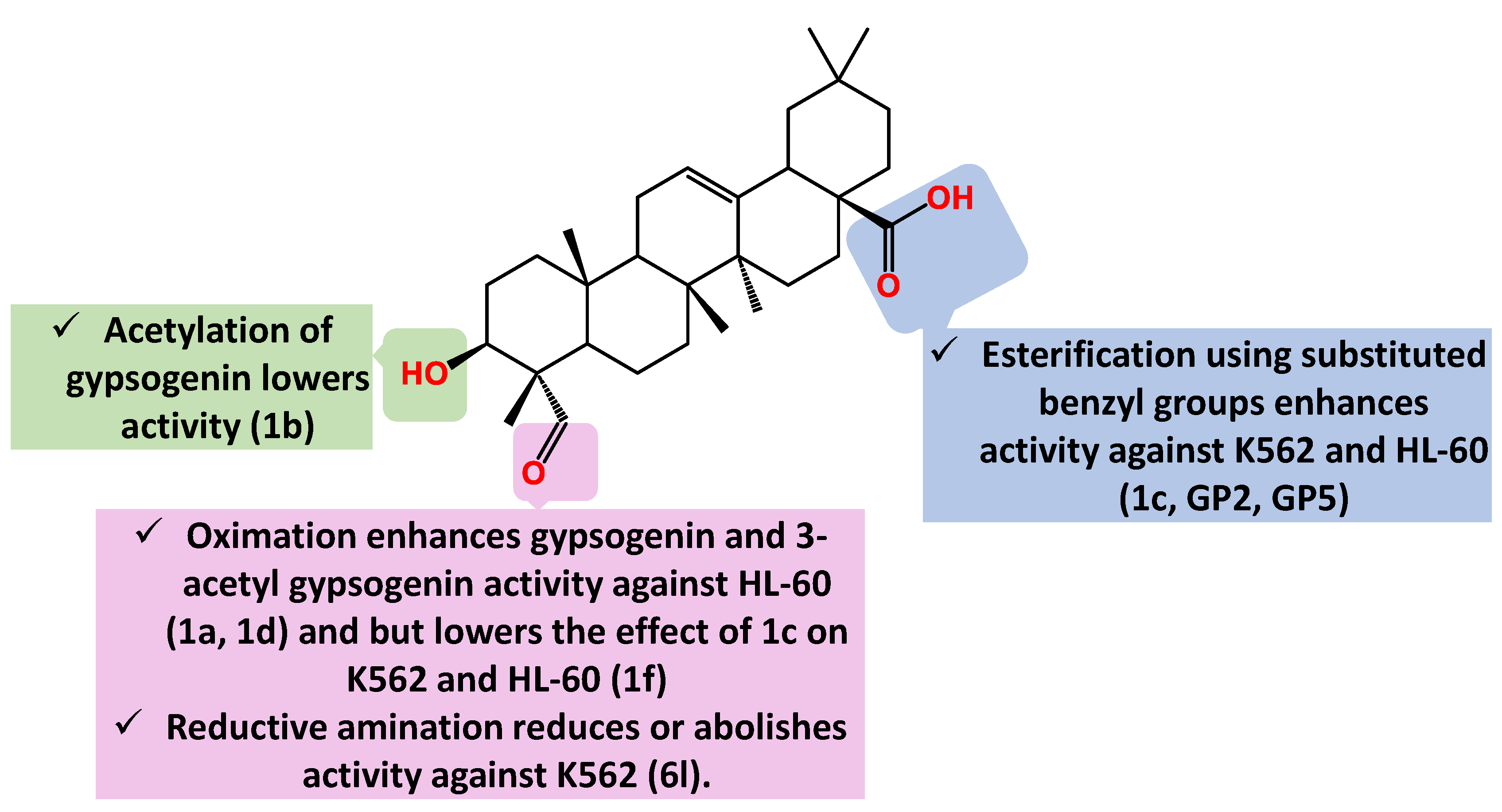
Figure 5.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against breast cancer cells.
Figure 5.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against breast cancer cells.
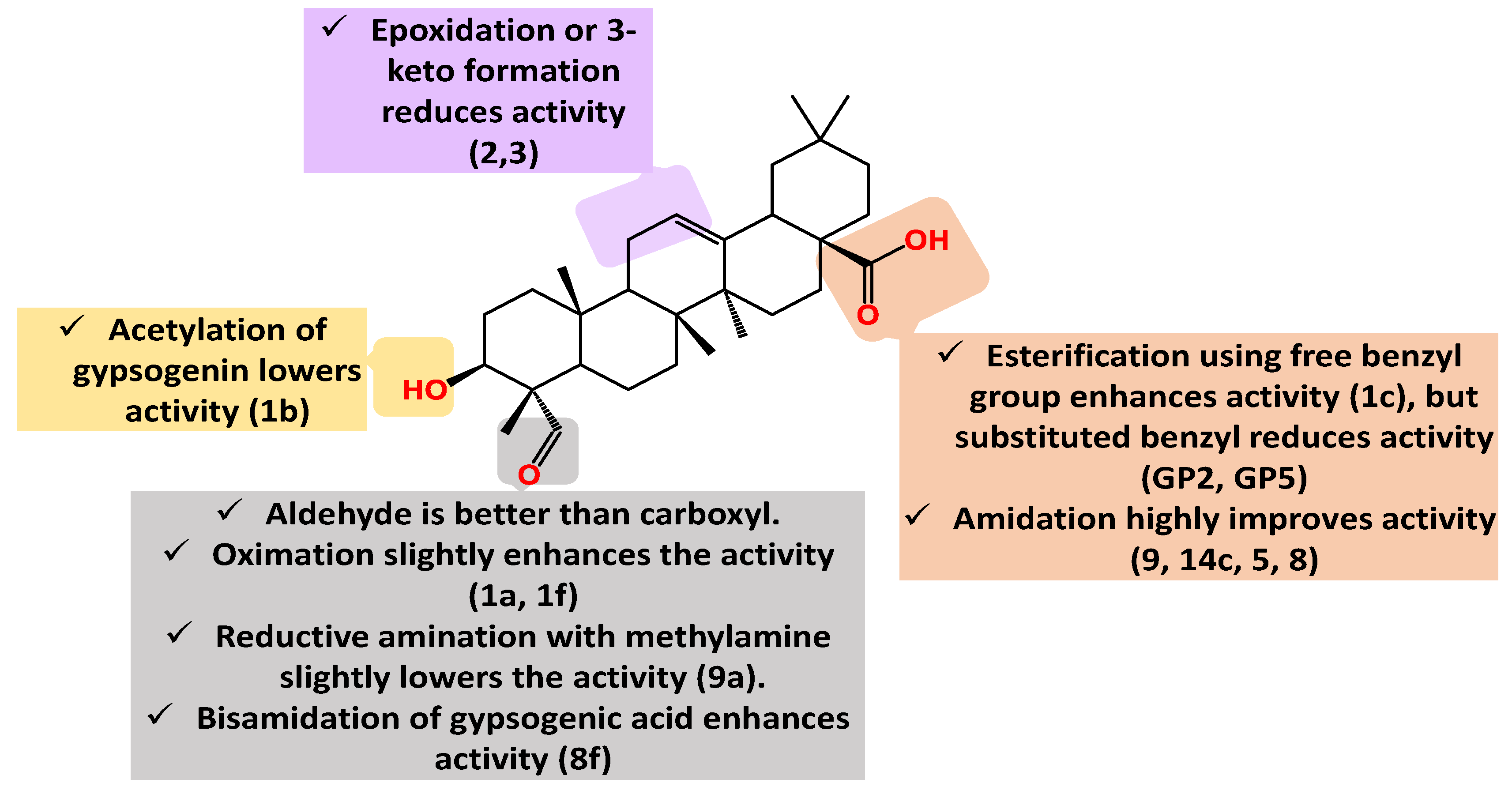
Figure 6.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against lung cancer cells.
Figure 6.
Summary of gypsogenin derivatives SAR pertaining to cytotoxicity against lung cancer cells.
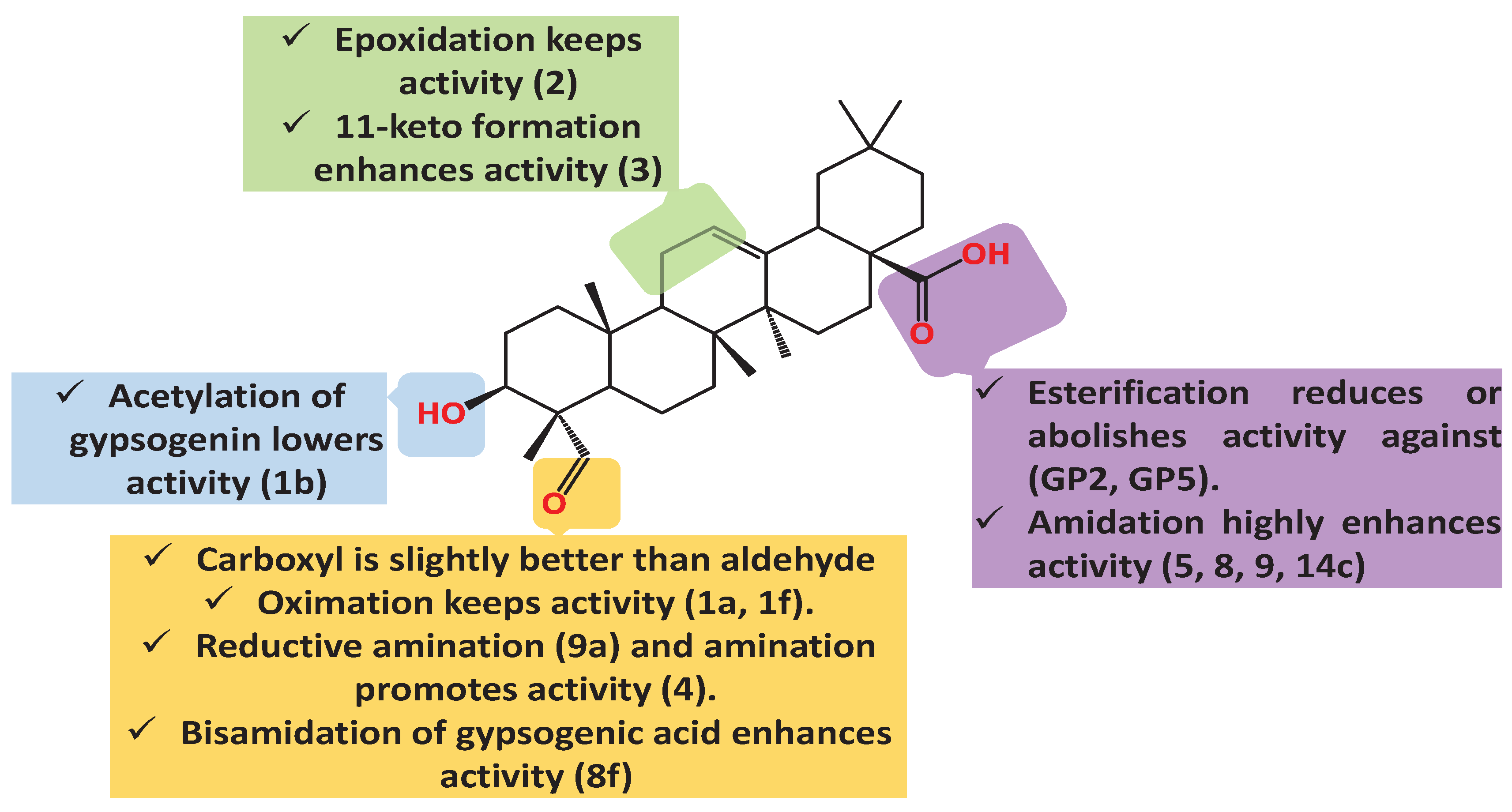

Table 1.
Gypsogenin derivatives with different cytotoxic activities.
| Compound Cell line name and IC50 µM | |||
| HT-29 [74] | Saos-2 [75] | HeLa [74] | |
| Gypsogenin | 10.4 | 7.8 | 22.4 |
| 1a | 10.8 | 7.9 | 8.7 |
| 1b | 11.1 | 8.2 | 35.0 |
| 1d | 6.7 | 8.9 | >100 |
| LOVO [79] | |||
| 2 | > 30 | ||
| 3 | 17.8 | ||
| 5 | 7.2 | ||
| 8 | 0.8 | ||
| 9a | 5.8 | ||
| LOVO [68] | HePG2 [68] | SKOV3 [68] | |
| 4 | 2.9 | 10.0 | 9.7 |
| 7g | 3.5 | 12.5 | 13.1 |
| HepG2 [78] | TE-1[78] | MC3-8 [78] | |
| 8f | 3.6 | 5.4 | 4.8 |
| 9 | 4.0 | 4.7 | 2.9 |
| 14c | 2.2 | 4.2 | 2.6 |
| HeLa [73] | |||
| GP2 | 35.2 | ||
| GP5 | 5.6 | ||
| U251 | T98G | U87 | |
| 10 [81] | 5.8 | 8.1 | 17.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



