
Submitted:
23 June 2023
Posted:
23 June 2023
You are already at the latest version
Abstract
Individual susceptibility to pulmonary oxygen toxicity (PO2tox) is highly variable and currently lacks a reliable biomarker for predicting pulmonary hyperoxic stress. As nitric oxide (NO) is involved in many respiratory system processes and functions, we aimed to determine if expired nitric oxide (FENO) levels can provide an indication of PO2tox susceptibility in humans. Eight U.S. Navy trained divers volunteered as subjects. The hyperoxic exposures consisted of six- and eight-hour hyperbaric chamber dives conducted on consecutive days in which subjects breathed 100% oxygen at 202.65 kPa. Subjects’ individual variability in pulmonary function and FENO was measured twice daily over five days and compared with their post-dive values to assess susceptibility to PO2tox. Only subjects who showed no decrements in pulmonary function following the six-hour exposure conducted the eight-hour dive. FENO decreased by 55% immediately following the six-hour oxygen exposure (n=8, p<0.0001) and by 63% following the eight-hour exposure (n=4, p<0.0001). Four subjects showed significant decreases in pulmonary function immediately following the six-hour exposure. These subjects had the lowest baseline FENO and the lowest post-dive FENO and had clinical symptoms of PO2tox. Individuals with low FENO were the first to develop PO2tox symptoms and deficits in pulmonary function from the hyperoxic exposures. These data suggest that endogenous levels of NO in the lung may protect against the development of PO2tox.
Keywords:
hyperoxia
; pulmonary function
; expired nitric oxide
; spirometry
; oxygen toxicity
; diving
; hyperbaric
1. Introduction
Pulmonary oxygen toxicity (PO2tox) results from prolonged exposure to a hyperoxic atmosphere, with the severity of symptoms increasing progressively with elevation of the inspired oxygen partial pressure (PiO2) and the duration of exposure [1]. Symptoms of PO2tox include chest pain, tightness, cough, and substernal distress that may coincide with decreases in pulmonary function, specifically a reduction in forced vital capacity (FVC) and alveolar diffusion capacity (DLCO) [1,2]. The toxic effects of oxygen are a concern for military and technical divers conducting prolonged multiday dives using oxygen rebreathers, and for patients undergoing hyperbaric oxygen therapy or aggressive oxygen therapy for respiratory insufficiency at normobaric pressure. While there are theoretical models that predict the expected level of pulmonary function deficit because of prolonged exposure to raised PiO2 that are based upon the expected decline in FVC, there is considerable individual variation in susceptibility to a uniform degree of pulmonary oxygen poisoning [3,4,5]. Currently, there are no methods to predict individual susceptibility to PO2tox. Furthermore, a sensitive non-invasive biomarker that can detect changes in lung pathology at an early stage in the oxygen toxicity process has remained elusive.
Expired nitric oxide (FENO) measurements have been studied as an exhaled marker of airway inflammation in a variety of lung diseases including asthma, lung cancer, bacterial pneumonia, pulmonary fibrosis, and idiopathic pulmonary fibrosis [6,7]. Nitric oxide (NO) in expired air is derived from nitric oxide synthase (NOS) activity from various cellular sources including neutrophils, alveolar type-II cells, endothelial cells, and airway cells, as well as from non-enzymatic sources such as s-nitrosothiols and nitrite protonation [6,8]. All three types of NOS (neuronal [nNOS], inducible [iNOS], and endothelium [eNOS]) have been identified in the human lung [9]. Endogenous NO in the lungs is thought to play an important role in host immune defenses by maintaining ciliary function, preventing the growth of bacteria and replication of viruses, modulating airway reactivity, facilitating surfactant production in the alveoli, and regulating inflammation and local blood flow in the lung [7,9].
The role of NO in the development or protection from O2 toxicity has been investigated in animal studies by several investigators [10,11] to better understand the roles of oxidative and nitrosative stress on hyperoxia-induced cell damage and acute lung injury [12,13]. Garat et al., [10] found that the survival time of hyperoxic rats treated with the NOS inhibitor NG-Nitroarginine Methyl Ester (L-NAME) was reduced compared to a hyperoxic control group, suggesting a protective effect of endogenous NO during 100% O2 breathing at normobaric pressure. Investigators at Duke University have shown that NO production may either exacerbate or mitigate the toxic effects of oxygen, depending on the NOS isoform that produces it [11,12]. These animal studies raise the intriguing possibility that individual variability of NO production in the lung may explain the large variability in individual susceptibility to PO2tox. Thus, the aim of this study was to determine if FENO levels could provide an indication of PO2tox susceptibility in humans.
2. Materials and Methods
2.1. Subjects
The current study investigated individual differences to hyperbaric oxygen (HBO) stress using a small group of healthy, well-trained divers, rather than focusing on group mean changes in a larger more diverse subject population. Consequently, the subject population was limited to qualified U.S. Navy trained divers who were fit to dive and familiar with the signs and symptoms of both pulmonary and central nervous system (CNS) oxygen toxicity. During the informed consent process all divers were reminded of the risks of pulmonary and CNS oxygen toxicity that could result from their participation in the study. Eight male U.S. Navy trained divers, aged 21–55 yrs (mean = 36.4 yrs), weighing 74.1–113.2 kg (mean = 91.8 kg), and with body stature ranging from 165–180 cm (mean = 174 cm) participated as subjects after signing informed consent. All had normal lung function, were non-smokers, and abstained from all other diving activities for the duration of the study.
2.2. Study design and hyperbaric oxygen exposure profile
The study protocol was approved by the Naval Submarine Medical Research Laboratory (NSMRL) Institutional Review Board in compliance with all applicable Federal regulations governing the protection of human subjects. The HBO exposures consisted of six-hour and eight-hour dry resting trials, breathing 100% humidified O2 at 202.65 kPa (2 ATA) in a hyperbaric chamber. The six- and eight-hour dives were performed on consecutive days. Pulmonary function and FENO were measured immediately prior to each dive, between 10- and 60-minutes post-dive, and then daily for at least three days after the dive or until complete recovery of pulmonary function. Only subjects who showed no decrements in pulmonary function following the 6 hr exposure conducted the 8 hr dive.
During each dive an inside tender who was not on oxygen accompanied the diver subjects. The hyperbaric oxygen exposure profiles were carefully selected to elicit a mild but reversible level of pulmonary oxygen toxicity in the majority of subjects while also keeping the risk of a seizure from CNS oxygen toxicity to a minimum. A single 15-min air break was incorporated during the midpoint of each HBO exposure during which the subjects ate a low nitrate/nitrite lunch. The total bottom time of the dive was adjusted for the 15-min air break to ensure that the total time breathing 100% oxygen at 2 ATA was either six or eight hours. The initial six-hour HBO exposures were conducted in two teams of three and one team of two divers. All the dives were conducted at the same time of day (initial press between 07:10 and 08:36). Compression and decompressed rates were 18.3 msw/min (60 fsw/min) and 9.1 msw/min (30 fsw/min), respectively.
Based on previous studies, the level of pulmonary oxygen toxicity induced by six hours of breathing 100% O2 at 2 ATA was predicted to cause a temporary decrease in FVC of between four and six percent in 50% of the subjects [2,3,4,14]. Extending the dive to eight hours increased the predicted decrement in FVC to 8% in 50% of the subjects [14]. Both dives were below the CNS oxygen toxicity limit (previous studies have shown no evidence of CNS oxygen toxicity in divers exposed to 2 ATA of oxygen for up to 12 hours [3]). Due to the level of uncertainty in these predictions, it was felt that the current approach of conducting two dives of increasing oxygen exposure would permit measurable but fully reversable levels of pulmonary oxygen toxicity in our subject population without exposing particularly susceptible individuals to an overly long HBO exposure.
Control exposures were conducted on two of the subjects to determine if pulmonary function or FENO were significantly affected by breathing air at surface pressure in the hyperbaric chamber on the built-in breathing system for six or eight hours. One subject completed a six-hour air exposure, and the other subject completed an eight-hour exposure.
2.3. Pulmonary Function and Expired Nitric Oxide Measurements
Pulmonary function (FVC, forced inspiratory vital capacity [FIVC], forced expiratory volume in 1 s [FEV1]), the diffusion capacity for carbon monoxide (DLCO), and FENO baseline measurements were collected from each diver twice a day (am and pm) for five consecutive days before conducting the HBO exposures. During each measurement session, subjects conducted three repetitions for each pulmonary function test that met American Thoracic Society (ATS) standards for repeatability [15,16,17,18]. All pulmonary function tests were conducted on the VMAX Encore 22 Pulmonary Function Module (Viasys Healthcare Inc., Yorba Linda, CA). FENO was measured using a chemiluminescence NO analyzer (Sievers NOA 280i, GE analytical instruments, Boulder, CO). During each measurement session FENO was measured at the following five expired flow rates: 50, 100, 150, 200, and 250 ml/s. These were used to determine alveolar NO concentration (CANO) and maximum airway wall flux of NO (J’awNO) using a two-compartment model [19]. Exhaled flow rates for on-line FENO measurements were controlled by having the subject target the desired flow rate, presented on a computer screen, while expiring against a flow restrictor. Five different flow restrictors were used to achieve the different expired flow rates. At each expired flow rate, the mean value from at least three FENO measurements that conformed to the standardized procedures recommended by the American Thoracic Society for online FENO measurement [20] were taken during each measurement session and used in the analysis. The Sievers NO analyzer and VMAX Encore 22 Pulmonary Function Module were calibrated in accordance with the manufacturer’s procedures at least twice daily (morning and afternoon) before each measurement session. During each measurement session the subjects conducted the FENO measurements before the pulmonary function tests to avoid the potential influence of the spirometry measurements on FENO. Pre-dive measurements for NO and pulmonary function were taken during the two-hour period before the dive. Post dive measurements of FENO were initiated 10 minutes after the dive had reached the surface. As DLCO measurements were always conducted after the FENO measurements, subjects breathed ambient air for at least 20 to 30 minutes following the dives before conducting their first DLCO measurement. Consequently, PAO2 levels were expected to be at normal levels during the pulmonary function test and thus no corrections were made to DLCO for PAO2.
2.4. Data Analysis
A decrement in pulmonary function for an individual was defined as outside their normal variability if one or more of their pulmonary function tests (i.e., FVC, FIVC, FEV1, or DLCO) fell more than two standard deviations (SD) below their mean baseline value for that test. A change in FENO was also considered outside normal variability if the change was greater than two SD from the individual’s mean baseline FENO value. Intra-individual variability for FENO and the various pulmonary function tests are expressed as 2x the coefficient of variation (CV) where CV = (SD/mean) x 100, to facilitate the comparison among individuals and between variables with different units and different means. All statistical analysis was carried out using Statistica software (Statsoft Inc., Tulsa, OK). Analysis on how time of day (am or pm) affected FENO across all expired flow rates was performed using a repeated measures analysis of variance (ANOVA). Repeated measures ANOVA was also used to compare group mean changes in FENO, CANO, J’awNO, and pulmonary function at the different time points. When significant main effects of time were observed, the Dunnett post-hoc test was used to explore differences between baseline and post-dive values. The relationships between FENO levels and the percent changes in pulmonary function following the six-hour dive were evaluated using linear regression and the Pearson Product moment correlation coefficient. Significance was set at p<0.05.
3. Results
Baseline individual means and 2x the coefficient of variation (2 x SD/mean x100%) for FENO (at 50 ml/s expired flow rate) and the different pulmonary function tests (i.e., FVC, FIVC, DLCO) that were derived from the twice daily measurements (am and pm) taken on five consecutive days (n=20 data points per mean per subject for each pulmonary function test) are shown in the second column of Table 1, Table 2, Table 3 and Table 4. The remaining columns in each table show the percent change in that variable from each individual’s mean baseline level following the HBO exposures. In each of the tables the subject’s data are ordered from highest (top) to lowest (bottom) baseline FENO. Additional tables showing changes in DLCO adjusted for Hb (DLCO adj), DLCO adjusted for alveolar volume (DLCO/VA), alveolar volume (VA), and FEV1 are presented in Appendix A.
As shown in Table 1 there was a threefold range (19 to 59 ppb) in the baseline FENO between subjects. Analysis of the FENO baseline data using all expired flow rates indicated a significant time of day effect, with FENO on average 10% lower in the afternoon compared to morning (p<0.001). There was, however, no difference detected between the pre-dive FENO taken on the morning before the six-hour dive and the baseline FENO (p=0.9994). Immediately following the six-hour oxygen exposure all eight subjects had significant decreases in FENO (i.e., values >2x CV less than their baseline) with the group mean change showing a 55% decrease (p<0.0001). By the morning after the dive, FENO levels had returned to normal in the majority of divers (6 out of 8).
The four subjects with the lowest baseline FENO and lowest post-dive FENO (subjects 2, 3, 4, and 9) had clinical symptoms of pulmonary O2 toxicity and showed significant decreases in pulmonary function on one or more of the pulmonary function tests immediately following the six-hour exposure (see Table 2, Table 3 and Table 4). The clinical symptoms reported included chest fullness/tightness, congestion, mild substernal burning, and tickling or cough on deep inhalation. Subjects 1, 5, 7, and 8, who had baseline FENO levels greater than the group mean of 34 ppb, showed no pulmonary function deficits or symptoms of pulmonary O2 toxicity following the six-hour HBO exposure and thus conducted the eight-hour HBO exposure the following day. Immediately following the eight-hour dive three of these subjects had pulmonary function deficits (see Table 2, Table 3 and Table 4) and all four subjects showed greater decreases in FENO than following their six-hour dive (mean ± SD FENO post-dive1 vs. post-dive 2 = 22.2 ± 3.4 ppb vs. 16.6 ± 2.7 ppb, respectively, n =4, p<0.01). Subject 5 had the highest baseline FENO and was the only subject who did not show symptoms of PO2tox or a pulmonary function deficit following the HBO exposures.
During the three days following the dives, five subjects showed significant increases in FENO (see Table 1). However, the timing of these increases and the duration of the elevated FENO was variable among the subjects. Consequently, the group analysis did not reveal any statistically significant change in the mean FENO from the pre-dive baseline during recovery days one (p= 0.8642), two (p=0.0579), or three (p=0.3358).
The pulmonary function test that demonstrated the greatest number of significant decrements following the oxygen exposures was DLCO (see Table 4). The three subjects with the lowest baseline FENO (subjects 4, 3, and 9) had the greatest relative decrements in DLCO which persisted for one to three days post exposure. Subjects 1 and 8 also showed significant decreases in DLCO during the recovery period. When DLCO was corrected for VA, all subjects except subject 5 showed significant decrements at some point during the recovery period (see Table A2 in Appendix A). Both DLCO and DLCO/VA showed a significant main effect of time (p<0.05 and p<0.01, respectively) that was predominantly due to lower values during the second day of recovery compared to baseline (see Table 4 and Table A2).
The relationship between the relative change in DLCO immediately following the six-hour dive and the immediate pre- and post-dive levels of FENO is shown in Figure 1. Regression analysis of these data found that the relative change in DLCO immediately post-dive was significantly related to the immediate post-dive FENO (r=0.948, p<0.001), as well as to the pre-dive FENO (r=0.902, p<0.01). Using the mean baseline FENO in the regression analysis instead of the pre-dive FENO slightly improved the relationship (r=0.931, p<0.001).
While some subjects had significant decrements on the spirometry tests following the dives, the group mean relative changes on the spirometry tests averaged smaller than those found for DLCO. Immediately following the six-hour dive FIVC appeared to be more affected than FVC, however, neither FIVC or FVC showed a significant main effect of time following group analysis (p=0.0658 and p=0.2176, respectively).
The two-compartment model analysis of the FENO data showed a significant 58% decrease in J’awNO (mean ± SD, baseline vs. post-dive 1 = 1681 ± 747 pl/s vs. 709 ± 465 pl/s, p<0.001) with no change in CANO (mean ± SD baseline vs. post-dive 1 = 2.9 ± 1.2 ppb vs. 2.6 ± 0.6 ppb; p=0.995) immediately following dive 1. A comparison of pre and post exposure measurements for FENO and pulmonary function for the two subjects who conducted the surface control trials showed that all the dependent variables following the control exposure were within each individual’s normal daily variability (data not shown).
4. Discussion
Traditionally the “gold standard” for assessing PO2tox has been to measure changes in pulmonary function using spirometry (i.e., FVC) or DLCO. However, the sensitivity of these pulmonary function tests to assess PO2tox susceptibility has been questioned [21,22], and more recent research has explored other components in exhaled breath as potential biomarkers of PO2tox [22,23,24,25,26,27]. Since the initial discovery that NO was present in expired air [28], FENO has been one of the most widely studied exhaled breath biomarkers of pulmonary health. FENO increases significantly in a variety of inflammatory airway diseases and is now commonly used to diagnose and phenotype asthmatics [7]. It was thus originally hypothesized that FENO would increase following HBO exposure due to free oxygen radical initiation of inflammatory reactions in the lungs.
One of the first published papers on the effect of the hyperoxia on FENO reported that FENO increased with exposure to normobaric hyperoxic gas mixtures [29]. However, the 10-minute normobaric oxygen exposures in this early study were unlikely to result in inflammation of the lungs. The results of the study by Schmetterer et al., [29] are in direct contrast with our finding of a marked acute reduction in FENO following prolonged HBO exposures. One potential reason for the disparate results is that at the time that Schmetterer et al. [29] performed their study, there was no standard method for measuring FENO. Since that time, it has become clear that FENO is highly dependent on the expired flow rate and thus the recommended guidelines for FENO measurements published by the ATS in 2005 [20] have since standardized expired flow rates at 50 ml/s using a flow resistor that also prevents contamination of the FENO measurement from the high levels of NO found in the nasal cavity. Although we did observe significant increases in FENO during the recovery days in five subjects, which may be reflective of a delayed inflammatory reaction in the lungs, only one of these subjects (subject 9) exhibited consistent decrements in pulmonary function during all three recovery days that was concomitant with abnormally elevated FENO levels.
A second main finding from our study is that the duration of the HBO exposure affected the relative magnitude of the post dive decrease in FENO, with the eight-hour HBO dive resulting in significantly lower post dive FENO levels than the six-hour HBO exposure. This finding implies that the magnitude of the temporary FENO decrease following the HBO exposures may be dose dependent. Since conducting these pilot HBO dives in 2007, we have conducted a wide variety of dry human hyperoxic exposures with varying inspired oxygen partial pressures and exposure durations to determine if the FENO decreases found in the current study follow a predictable dose response relationship. Findings from these studies have been presented to the undersea and hyperbaric medical and research community at various scientific forums [30,31,32] and were summarized in preliminary form in Fothergill and Weathersby [33]. This study showed that the relative change in FENO following dry resting hyperoxic exposure follows an exponential decline that is tightly related to the hyperoxic dose of the preceding exposure [33]. In the statistical model of the changes in FENO with varying HBO exposures, Fothergill & Weathersby [33] used the following expression to define the hyperoxic dose of the HBO dives based upon the inspired partial pressure of the oxygen breathed (PiO2) and the duration of the exposure:
Hyperoxic Dose (ATA.min) = [PiO2 (ATA) x Exposure Duration (min)] – [0.21 x Exposure Duration (min)]
Other investigators have also reported acute decreases in FENO levels following HBO exposures [34,35,36,37,38,39,40]; However, the oxygen dose involved in these studies has rarely been great enough to induce changes in lung function or PO2tox symptoms noticeable enough to determine if the FENO changes are related to PO2tox susceptibility. Our study is therefore somewhat unique in that we were able to observe symptoms of PO2tox and measure significant decreases in lung function in some of our subjects and relate them to the observed changes in FENO. Based upon our observations, we found that those individuals who had the lowest pre-dive FENO levels exhibited the lowest post-dive FENO levels and were most susceptible to PO2tox.
This significant linear relationship between the pre-dive baseline levels of FENO and the relative decrease in DLCO measured immediately post dive should be taken with caution when interpreting the effects of HBO exposure on PO2tox susceptibility. In a more recent study in which healthy U.S. Navy trained divers were exposed to 6.5 h of 100% O2 at 2.0 ATA [27], one subject, who aborted the dive early due to severe PO2tox symptoms, was found to have a 15% increase in DLCO immediately post dive compared to his pre-dive base line [41]. Concomitant with the increase in DLCO was a 125% increase in total airway resistance and a 35% increase in proximal airway resistance (as measured using impulse oscillometery methodology) [41]. We surmise that the elevated DLCO post dive for this subject was an artifact caused by the increase in pulmonary resistance that resulted in a large negative interpulmonary pressure being generated during the fast inspiratory maneuver required to perform the DLCO measurement. The negative interpulmonary pressure could result in increased blood volume entering the lung before the DLCO breath hold maneuver, raising the potential sink for the inhaled carbon monoxide gas mixture and artifactually raising the DLCO level. Therefore, we hypothesize that subjects who are particularly susceptible to PO2tox might experience a narrowing of the airways, possibly due to loss of normal airway tone.
Acute changes in airway diameter can be evoked by increases in cholinergic nerve activity or withdrawal of nitrergic neural activity [42]. Interestingly, noncholinergic neurotransmitters such as NO are thought to control human airway smooth muscle and normal airway tone via nitrergic parasympathetic nerves [42]. Thus, factors that compromise normal nitrergic parasympathetic control of airway tone, such as reduced levels of NO, would act to cause narrowing of the airways. The acute post dive increase in airway resistance seen in the above PO2tox case was concomitant with an extremely low post dive FENO of 3.5 ppb [41]. This is consistent with a neurogenic PO2tox response rather than an inflammatory reaction to the HBO exposure.
Although our study was not designed to elucidate the underlying mechanisms responsible for the reduction in FENO with HBO exposure, our results are consistent with the observation from previous animal work [10,43] that suggests that endogenous levels of NO may serve to protect the lung from hyperoxic lung injury [43]. Based upon our current findings, we suspect that once FENO levels fall below a critical level the antioxidant defense and other processes in the lung that depend on NO become overwhelmed by the hyperoxic stress, resulting in changes in lung function and symptoms of PO2tox. However, as discussed in a review paper by Lui et al., [44] the role of the various NOS isoforms in the generation of NO in the face of hyperoxic stress and the impact of NO in the pathogenesis of acute lung injury is still under debate.
Several studies have attempted to ascertain the underlying mechanisms responsible for the decrease in FENO with HBO exposures [45,46,47]. The common thesis of these studies centers around the hypothesis that the decrease in FENO with hyperoxic exposures is due to decreased enzymatic generation of NO due to oxidation of tetrahydrobiopterin (BH4), which is an essential cofactor required for NO production by NOS [48]. Fismen et al. [45] found that increased O2 concentrations reduced BH4 levels in human endothelial cells in a dose-dependent manner without directly affecting the NOS enzyme. Similarly, Hesthammer et al. [46] reported that BH4 levels in human umbilical vein endothelial cells (HUVEC) decreased in a dose dependent manner. Although the latter study found that HUVEC NO production was also decreased following a 40 kPa O2 exposure, a further decrease in HUVEC NO production was not observed when the oxygen exposure was increased to 60 kPa. In a follow up study by Hesthammer et al., [47] in which BH4 was measured in venous blood samples of subjects exposed to 100% oxygen for 90 minutes at atmospheric pressure, both FENO and BH4 significantly decreased when measured 10 minutes after the exposure. Although oxidation of BH4 levels and its subsequent uncoupling/inhibitory effects on NOS on NO production appear to be a plausible reason for the reduced FENO with hyperoxic exposures, other mechanisms including the reaction of oxygen or superoxide radicals with NO to form peroxynitrite likely also contribute to the reduced FENO.
4.1. Study Strengths and Limitations
To our knowledge, this is the first study that combined measurements of FENO with traditional measures of pulmonary function in healthy divers to assess PO2tox susceptibility following provocative HBO exposures that resulted in significant decrements in lung function. While the current study involved a small number of subjects, the study design incorporated multiple baseline and recovery measurements of FENO and pulmonary function to provide a robust indication of daily inter-individual variation and accurately define when these dependent variables fell significantly outside of the individual’s normal range, following the HBO exposure. The results clearly showed a wide individual variability in pulmonary function changes resulting from the HBO exposures, with half our subject population showing minimal changes in lung function following the six-hour dive and the other half showing significant decreases that were more than two standard deviations below their normal day-to-day range. While this experimental design allowed us to analyze individual susceptibility to PO2tox, the small n approach leaves group statistical analysis susceptible to type II errors from the large variability in individual responses to the HBO stress. However, given our primary aim, we felt the small n approach was ethically more defensible as a pilot study on individual PO2tox susceptibility than a larger n study with limited individual pre-dive data but a higher power to detect group level changes in pulmonary function post dive.
An additional limitation of the current study is that only two subjects completed a control (normobaric air) condition and that the study design was unblinded. This may have led to experimenter and subject bias regarding the expectation of pulmonary function decrements and PO2tox symptoms following the HBO exposures. While performing the pulmonary function measurements in accordance with the ATS recommendations [15,16,17,18] will help to reduce this potential bias, most of the spirometry measurements are dependent upon the individual performing a maximal inspiratory and/or expiratory effort to determine if pulmonary function is affected by the HBO exposure. In contrast, measurements of FENO are conducted at a fixed expired flow rate and do not require a maximum effort by the subject. FENO may thus offer an alternative or complementary assessment of pulmonary hyperoxic stress that is less prone to the subject’s effort than traditional spirometry measurements. While we acknowledged that there are many sources of NO in the lungs that can contribute to FENO, and that the underlying mechanistic role of NO in hyperoxic acute lung injury is still controversial, FENO may provide a useful noninvasive marker of the hyperbaric oxidative stress response of the lungs, and lead to new insights into individual susceptibility to PO2tox.
Author Contributions
Dr. Fothergill was the principal investigator on the study, responsible for the conceptualization and study original design, funding acquisition, data collection, formal analysis, interpretation of the results and manuscript preparation. Dr. Gertner was a co-investigator on the study who assisted in data collection, data analysis, and interpretation of the results. He also helped critically revise and approve the final content in the article. Both authors have read and agree to the published version of the manuscript.
Funding source
Sponsored by the Office of Naval Research through the In-house (Dept of Navy) Laboratory Independent Research (ILIR) Program work unit number 50707.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Naval Submarine Medical Research Laboratory Institutional Review Board (protocol # NSMRL 2007.0003, approved 5 Jan 2007) and is in compliance with all applicable Federal regulations governing the protection of human subjects.
Data Availability Statement
The data from the current study are not publicly available due to government restrictions regarding data sharing but are available from the corresponding author on reasonable request and when requirements are met.
Acknowledgements
The authors wish to thank all the U.S. Navy trained divers who volunteered as subjects for this project. This work was presented at the 35th Annual Scientific Meeting of the European Underwater and Baromedical Society in Aberdeen, Scotland, August 25th – 28th, 2009 and was reported in abbreviated form in the book of abstracts and proceedings referenced below. Fothergill, D.M. and Gertner J. Exhaled nitric oxide (NOexp) measurements as a noninvasive marker of pulmonary oxygen toxicity susceptibility in humans. In Ross, J.A. (Ed). Proceedings of the 35th Annual Scientific Meeting of the European Underwater and Baromedical Society and British Hyperbaric association Annual Meeting, The British Hyperbaric Association, Aberdeen, Scotland, August 25th – 28th. pp 94-99.
Conflicts of Interest
The authors declare no conflict of interest.
Disclaimer
The views expressed in this report are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the United States Government. The authors are employees of the U.S. Government. This work was prepared as part of their official duties. Title 17 U.S.C. §105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military Service member or employee of the U.S. Government as part of that person’s official duties.
Appendix A. Diffusion capacity for carbon monoxide measurements corrected for Hb and VA and additional spirometry measurements taken pre and post dive

Table A1.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in DLCO adjusted for Hemoglobin (DLCO adj) following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in DLCOadj from baseline.
Table A1.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in DLCO adjusted for Hemoglobin (DLCO adj) following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in DLCOadj from baseline.
| Subject | Baseline DLCO adj | Post Dive 16 hrs O2 | Post Dive 28 hrs O2 | Rec 1 | Rec 2 | Rec 3 | |
| Mean(mL/mmHg/min) | CVx2 (%) | ||||||
| 5 | 38.6 | 9.4% | +6.5% | +1.3%* | +1.3% | +1.3% | -2.6% |
| 7 | 25.5 | 6.9% | -1.6% | -5.9%* | -2.0% | -2.0% | -1.2% |
| 1 | 39.9 | 9.9% | -3.5% | -6.0%* | -18.8% | -5.8% | -3.3% |
| 8 | 31.9 | 11.8% | -1.5% | -6.2%* | -8.4% | -6.2% | +1.3% |
| 2 | 39.3 | 13.4% | -8.0%* | NA | -6.0% | -13.9% | -22.1% |
| 4 | 45.0 | 15.2% | -16.9%* | NA | -20.9% | -6.7% | -10.9% |
| 3 | 38.9 | 8.3% | -16.9%* | NA | +3.2% | -8.9% | -4.5% |
| 9 | 38.5 | 9.0% | -13.0%* | NA | -11.4%* | -30.4% | -21.1% |
| Mean | 37.2 | 10.5% | -6.9% | -4.2% | -7.9% | -9.1% | -8.1% |
* Symptoms of pulmonary O2 toxicity reported.
Table A2.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in DLCO adjusted for alveolar volume (DLCO/VA) following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in DLCO/VA from baseline.
Table A2.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in DLCO adjusted for alveolar volume (DLCO/VA) following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in DLCO/VA from baseline.
| Subject | Baseline DLCO/VA | Post Dive 16 hrs O2 | Post Dive 28 hrs O2 | Rec 1 | Rec 2 | Rec 3 | |
| Mean(mL/mmHg/min) | CVx2 (%) | ||||||
| 5 | 4.82 | 7.2% | +6.8% | +3.5%* | -1.5% | +3.7% | -1.0% |
| 7 | 4.37 | 8.9% | -2.3% | -11.0%* | -5.2% | -9.6% | -3.7% |
| 1 | 4.97 | 8.5% | -3.8% | -7.2%* | -14.5% | -7.4% | -10.3% |
| 8 | 5.10 | 7.1% | -8.0% | -6.9%* | -12.5% | -17.6% | -4.3% |
| 2 | 5.29 | 11.3% | -2.3%* | NA | -9.6% | -14.7% | -13.2% |
| 4 | 5.85 | 10.3% | -9.6%* | NA | -16.8% | -9.9% | -12.5% |
| 3 | 6.10 | 5.8% | -1.1%* | NA | -4.1% | -12.8% | -2.1% |
| 9 | 5.05 | 9.5% | +3.6%* | NA | +15.8%* | -15.6% | -11.5% |
| Mean | 5.19 | 8.6% | -2.1% | -5.4% | -6.1% | -10.5% | -7.3% |
* Symptoms of pulmonary O2 toxicity reported.
Table A3.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in alveolar volume (VA) following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in VA from baseline.
Table A3.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in alveolar volume (VA) following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in VA from baseline.
| Subject | Baseline VA | Post Dive 16 hrs O2 | Post Dive 28 hrs O2 | Rec 1 | Rec 2 | Rec 3 | |
| Mean(L BTPS) | CVx2 (%) | ||||||
| 5 | 7.86 | 4.1% | -0.3% | -0.9%* | +1.05 | -2.3% | -3.8% |
| 7 | 5.97 | 4.2% | 0.0% | +5.7%* | +3.4% | +4.0% | +1.7% |
| 1 | 8.28 | 8.6% | +0.4% | -0.2%* | -3.2% | +0.1% | +4.0% |
| 8 | 6.50 | 5.1% | +5.4% | +2.8%* | +2.8% | +9.7% | +4.2% |
| 2 | 7.68 | 7.9% | -8.2%* | NA | +2.7% | -0.3% | +0.3% |
| 4 | 7.77 | 8.4% | -9.1%* | NA | -3.7% | +6.7% | +2.2% |
| 3 | 6.49 | 5.8% | -15.9%* | NA | +8.0% | +4.9% | -1.1% |
| 9 | 7.59 | 5.2% | -16.3%* | NA | -22.1%* | -14.4% | -10.8% |
| Mean | 7.27 | 6.2% | -5.5% | +1.9% | -1.4% | +1.1% | -0.4% |
* Symptoms of pulmonary O2 toxicity reported.
Table A4.
Mean baseline levels, 2 x coefficient of variation (CV) and percent change in FEV1 following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in FEV1 from baseline.
Table A4.
Mean baseline levels, 2 x coefficient of variation (CV) and percent change in FEV1 following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in FEV1 from baseline.
| Subject | Baseline FEV1 | Post Dive 16 hrs O2 | Post Dive 28 hrs O2 | Rec 1 | Rec 2 | Rec 3 | |
| Mean(L BTPS) | CVx2 (%) | ||||||
| 5 | 3.97 | 5.3% | -0.8% | +6.3%* | -0.5% | +1.3% | -3.3% |
| 7 | 3.08 | 6.1% | +0.6% | -2.3%* | -1.3% | -5.5% | -5.8% |
| 1 | 3.96 | 3.9% | +2.8% | -2.5%* | -1.3% | +0.3% | -2.3% |
| 8 | 3.81 | 6.4% | +2.1% | +5.5%* | +3.1% | -3.1% | -4.2% |
| 2 | 4.28 | 7.2% | +3.3%* | NA | +0.9% | -1.6% | -1.9% |
| 4 | 4.16 | 7.5% | -2.4%* | NA | -5.5% | -3.4% | -5.5% |
| 3 | 3.30 | 7.9% | -8.5%* | NA | -4.5% | +0.3% | +0.3% |
| 9 | 3.96 | 6.7% | -6.1%* | NA | -20.2%* | -9.1% | -6.3% |
| Mean | 3.82 | 6.4% | -1.1% | +1.8% | -3.7% | -2.6% | -3.6% |
* Symptoms of pulmonary O2 toxicity reported.
References
- Clark, J.M.; and Thom, S.R. Oxygen under pressure. In Brubakk, A.O.; Neuman, T.S., editors. Bennett and Elliott’s Physiology and Medicine of Diving, 5th ed.; Saunders Publishing: Philadelphia, PA; Elsevier: Amsterdam, The Netherlands, 2003; ISBN 978-0702025716. [Google Scholar]
- Clark, J.M. Pulmonary Oxygen Tolerance in Man and Derivation of Pulmonary Oxygen Tolerance Curves. Ph.D. dissertation, Institute for Environmental Medicine, University of Pennsylvania, Philadelphia, PA, USA, 1970. [Google Scholar]
- Clark, J.M.; Lambertsen, C.J. Rate of development of pulmonary O2 toxicity in man during O2 breathing at 2.0 Ata. J. Appl. Physiol. 1971 May;30(5):739-52. PMID: 4929472. [CrossRef]
- Clark, J.M.; Lambertsen, C.J.; Gelfand, R.; Flores, N.D.; Pisarello, J.B.; Rossman, M.D.; Elias, J.A. Effects of prolonged oxygen exposure at 1.5, 2.0, or 2.5 ATA on pulmonary function in men (predictive studies V). J. Appl. Physiol. (1985). 1999 Jan;86(1):243-59. PMID: 9887137. [CrossRef]
- Shykoff, B.E. Pulmonary effects of submerged oxygen breathing: 4-, 6-, and 8-hour dives at 140 kPa. Undersea Hyperb. Med. 2005 Sep-Oct;32(5):351-61. PMID: 16457084.
- Lang, J.D.; McArdle, P.J.; O'Reilly, P.J.; Matalon, S. Oxidant-antioxidant balance in acute lung injury. Chest. 2002 Dec;122(6 Suppl):314S-320S. PMID: 12475808. [CrossRef]
- Högman, M.; Lehtimäki, L. Chapter 4 - Exhaled nitric oxide physiology and modeling, Editor(s): Beauchamp, J.; Davis, C.; Pleil, J. Breathborne Biomarkers and the Human Volatilome (Second Edition), Elsevier, 2020, Pages 63-77,ISBN 9780128199671. (https://www.sciencedirect.com/science/article/pii/B9780128199671000049). [CrossRef]
- Kharitonov, S.A.; Barnes, P.J. Exhaled markers of pulmonary disease. Am. J. Respir. Crit. Care Med. 2001 Jun;163(7):1693-722. PMID: 11401895. [CrossRef]
- Singh, S.; Evans, T.W. Nitric oxide, the biological mediator of the decade: fact or fiction? Eur. Respir. J. 1997 Mar;10(3):699-707. PMID: 9073009.
- Garat, C.; Jayr, C.; Eddahibi, S.; Laffon, M.; Meignan, M.; Adnot, S. Effects of inhaled nitric oxide or inhibition of endogenous nitric oxide formation on hyperoxic lung injury. Am. J. Respir. Crit. Care Med. 1997 Jun;155(6):1957-64. [CrossRef]
- Demchenko, I.T.; Atochin, D.N.; Gutsaeva, D.R.; Godfrey, R.R.; Huang, P.L.; Piantadosi, C.A.; Allen, B.W. Contributions of nitric oxide synthase isoforms to pulmonary oxygen toxicity, local vs. mediated effects. Am. J. Physiol. Lung Cell Mol. Physiol. 2008 May;294(5): L984-90. Epub 2008 Mar 7. PMID: 18326824; PMCID: PMC2728469. [CrossRef]
- Allen, B.W.; Demchenko, I.T.; Piantadosi, C.A. Two faces of nitric oxide: implications for cellular mechanisms of oxygen toxicity. J. Appl. Physiol. (1985). 2009 Feb;106(2):662-7. Epub 2008 Oct 9. PMID: 18845774. [CrossRef]
- Dias-Freitas, F.; Metelo-Coimbra, C.; Roncon-Albuquerque, R., Jr. Molecular mechanisms underlying hyperoxia acute lung injury. Respir. Med. 2016 Oct; 119:23-28. Epub 2016 Aug 21. PMID: 27692143. [CrossRef]
- Harabin, A.L.; Homer, L.D.; Weathersby, P.K.; Flynn, E.T. An analysis of decrements in vital capacity as an index of pulmonary oxygen toxicity. J. Appl. Physiol. (1985). 1987 Sep;63(3):1130-5. PMID: 3654459. [CrossRef]
- Miller, M.R.; Crapo, R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Enright, P.; van der Grinten, C.P.; Gustafsson, P.; Jensen, R.; Johnson, D.C.; MacIntyre, N.; McKay, R.; Navajas, D.; Pedersen, O.F.; Pellegrino, R.; Viegi, G.; Wanger., J. ATS/ERS task force. General considerations for lung function testing. Eur. Respir. J. 2005 Jul;26(1):153-61. PMID: 15994402. [CrossRef]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.; Gustafsson, P.; Jensen, R.; Johnson, D.C.; MacIntyre, N.; McKay, R.; Navajas, D.; Pedersen, O.F.; Pellegrino, R.; Viegi, G.; Wanger, J.; ATS/ERS task, force. Standardization of spirometry. Eur. Respir. J. 2005 Aug;26(2):319-38. PMID: 16055882. [CrossRef]
- Wanger, J.; Clausen, J.L.; Coates, A.; Pedersen, O.F.; Brusasco, V.; Burgos, F.; Casaburi, R.; Crapo, R.; Enright, P.; van der Grinten, C.P.; Gustafsson, P.; Hankinson, J.; Jensen, R.; Johnson, D.; Macintyre, N.; McKay, R.; Miller, M.R.; Navajas, D.; Pellegrino, R.; Viegi, G. Standardization of the measurement of lung volumes. Eur. Respir. J. 2005 Sep;26(3):511-22. PMID: 16135736. [CrossRef]
- Macintyre, N.; Crapo, R.O.; Viegi, G.; Johnson, D.C.; van der Grinten, C.P.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Enright, P.; Gustafsson, P.; Hankinson, J.; Jensen, R.; McKay, R.; Miller, M.R.; Navajas, D.; Pedersen, O.F.; Pellegrino, R.; Wanger, J. Standardization of the single-breath determination of carbon monoxide uptake in the lung. Eur. Respir. J. 2005 Oct;26(4):720-35. PMID: 16204605. [CrossRef]
- Condorelli, P.; Shin, H.W.; Aledia, A.S.; Silkoff, P.E.; George, S.C. A simple technique to characterize proximal and peripheral nitric oxide exchange using constant flow exhalations and an axial diffusion model. J. Appl. Physiol. (1985). 2007 Jan;102(1):417-25. Epub 2006 Aug 3. PMID: 16888048. [CrossRef]
- American Thoracic Society; European Respiratory Society. ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005. Am. J. Respir. Crit. Care Med. 2005 Apr 15;171(8):912-30. PMID: 15817806. [CrossRef]
- Van Ooij, P.-J.A.M. Pulmonary oxygen toxicity in professional diving: Scire Est Mensurare? Ph.D. Thesis, University of Amsterdam, Amsterdam, The Netherlands, 2013. [Google Scholar]
- van Ooij, P.J.; Hollmann, M.W.; van Hulst, R.A.; Sterk, P.J. Assessment of pulmonary oxygen toxicity: relevance to professional diving; a review. Respir. Physiol. Neurobiol. 2013 Oct 1;189(1):117-28. Epub 2013 Jul 22. PMID: 23886638. [CrossRef]
- Wingelaar, T.T.; Brinkman, P.; van Ooij, P.J.A.M.; Hoencamp, R.; Maitland-van der Zee, A.H.; Hollmann, M.W.; van Hulst, R.A. Markers of pulmonary oxygen toxicity in hyperbaric oxygen therapy using exhaled breath analysis. Front. Physiology 2019, Vol. 10, https://www.frontiersin.org/articles/10.3389/fphys.2019.00475, ISSN=1664-042X. [CrossRef]
- Cronin, W.A.; Forbes, A.S.; Wagner, K.L.; Kaplan, P.; Cataneo, R.; Phillips, M.; Mahon, R.; Hall, A. Exhaled volatile organic compounds precedes pulmonary injury in a swine pulmonary oxygen toxicity model. Front. Physiol. 2019 Dec 3;10:1297. PMID: 31849689; PMCID: PMC6901787. [CrossRef]
- de Jong, F.J.M.; Wingelaar, T.T.; Brinkman, P.; van Ooij, P.A.M.; Maitland-van der Zee, A.H.; Hollmann, M.W.; van Hulst, R.A. Pulmonary oxygen toxicity through exhaled breath markers after hyperbaric oxygen treatment table 6. Front. Physiol. 2022 May 10;13:899568. PMID: 35620607; PMCID: PMC9127798. [CrossRef]
- de Jong, F.J.M.; Wingelaar, T.T.; Brinkman, P.; van Ooij, P.A.M.; Maitland-van der Zee, A.H.; Hollmann, M.W.; van Hulst, R.A. Analysis of volatile organic compounds in exhaled breath following a COMEX-30 treatment table. Metabolites. 2023 Feb 21;13(3):316. PMID: 36984755; PMCID: PMC10056109. [CrossRef]
- Fothergill, D.M.; Borras, E.; McCartney, M.M.; Schelegle, E.; Davis, C.E. Exhaled breath condensate profiles of U.S. Navy divers following prolonged hyperbaric oxygen (HBO) and nitrogen-oxygen (Nitrox) chamber exposures. J. Breath Res. 2023, 17; 037105. PMID: 37207635. [CrossRef]
- Gustafsson, L.E.; Leone, A.M.; Persson, M.G.; Wiklund, N.P.; Moncada, S. Endogenous nitric oxide is present in the exhaled air of rabbits, guinea pigs and humans. Biochem. Biophys. Res. Commun. 1991 Dec 16;181(2):852-7. PMID: 1721811. [CrossRef]
- Schmetterer, L.; Strenn, K.; Kastner, J.; Eichler, H.G.; Wolzt, M. Exhaled NO during graded changes in inhaled oxygen in man. Thorax. 1997 Aug;52(8):736-8. PMID: 9337835; PMCID: PMC1758631. [CrossRef]
- Fothergill, D.M.; Gasier, H.; Keller, M. Effect of hyperbaric oxygen (HBO) duration on expired nitric oxide (NOexp) and carbon monoxide (COexp). Abstract in Cronje, F (Ed), Program for The 17th International Congress of Hyperbaric Medicine Triennial Meeting. 16-19 March 2011, South Africa: DesignWrite, p 20.
- Fothergill, D.M.; Gasier, H.; Keller, M. Exhaled nitric oxide (NOexp) and carbon monoxide (COexp) as noninvasive markers of hyperbaric oxidative stress in humans. Undersea and Hyperbaric Medical Society Annual Scientific Meeting, Fort Worth, TX, June 15th -18th 2011. Abstract in: Undersea and Hyperbaric Medicine. 2011; 38(5), p 430. 15 June.
- Fothergill, D.M.; Gasier, H.G. Time course of changes in expired nitric oxide and carbon monoxide during acute normobaric hyperoxic exposures. Abstract in: Abstracts and proceeding from European Underwater Baromedical Society Annual Scientific Meeting, Belgrade, Serbia, Sept 12th – 15th, 2012, Specijalna bolnica za hiperbaricnu medicine, Belgrade Serbia, ISBN 978-86-915961-0-1, page 52.
- Fothergill, D.M. and Weathersby, P.K. Relationship between exhaled nitric oxide and hyperoxic stress. The FASEB Journal. 2015 Apr; 29:678-1. https://faseb.onlinelibrary.wiley.com/doi/abs/10.1096/fasebj.29.1_supplement.678.1.
- Puthucheary, Z.A.; Liu, J.; Bennett, M.; Trytko, B.; Chow, S.; Thomas, P.S. Exhaled nitric oxide is decreased by exposure to the hyperbaric oxygen therapy environment. Mediators Inflamm. 2006; 2006(5):72620. PMID: 17392577; PMCID: PMC1657071. [CrossRef]
- Taraldsøy, T.; Bolann, B.J.; Thorsen, E. Reduced nitric oxide concentration in exhaled gas after exposure to hyperbaric hyperoxia. Undersea Hyperb Med. 2007 Sep-Oct;34(5):321-7. PMID: 18019082.
- Kjelkenes, I.; Thorsen, E. Time course of the reduction in nitric oxide concentration in exhaled gas after exposure to hyperbaric hyperoxia. Diving Hyperb Med. 2009 Jun;39(2):77-80. PMID: 22753200.
- van Ooij, P.J.; Houtkooper, A.; van Hulst, R. Variations in exhaled nitric oxide concentration after three types of dives. Diving Hyperb. Med. 2010 Mar;40(1):4-7.
- Caspersen, C.; Stensrud, T.; Thorsen, E. Bronchial nitric oxide flux and alveolar nitric oxide concentration after exposure to hyperoxia. Aviat. Space Environ. Med. 2011 Oct;82(10):946-50. PMID: 21961398. [CrossRef]
- Uusijärvi, J.; Eriksson, K.; Larsson, A.C.; Nihlén, C.; Schiffer, T.; Lindholm, P.; Weitzberg, E. Effects of hyperbaric oxygen on nitric oxide generation in humans. Nitric Oxide. 2015 Jan 30; 44:88-97. Epub 2014 Dec 11. PMID: 25498903. [CrossRef]
- Castagna, O.; Bergmann, C.; Blatteau, J.E. Is a 12-h Nitrox dive hazardous for pulmonary function? Eur. J. Appl. Physiol. 2019 Dec;119(11-12):2723-2731. Epub 2019 Nov 1. PMID: 31676994. [CrossRef]
- Fothergill, D.M. and Ross, W. Impulse oscillometery and spirometry indices of pulmonary function in a diver with severe symptoms of pulmonary oxygen toxicity. Abstract in: Abstract & Conference Book TRICON2018, Editors Bennett, M and Germonpre, P. 144th Annual Meeting of the European Underwater and Baromedical Society (EUBS), 47th Annual Scientific Meeting of the South Pacific Underwater Medical Society (SPUMS), Conference of the South African Underwater and Hyperbaric Medical Association, 23-29 Sep 2018, p 68.
- Canning, B.J.; Woo, A.; Mazzone, S.B. Neuronal modulation of airway and vascular tone and their influence on nonspecific airways responsiveness in asthma. J. Allergy (Cairo). 2012; 2012:108149. Epub 2012 Oct 23. PMID: 23150736; PMCID: PMC3485909. [CrossRef]
- Turanlahti, M.; Pesonen, E.; Lassus, P.; Andersson, S. Nitric oxide and hyperoxia in oxidative lung injury. Acta Paediatr. 2000 Aug;89(8):966-70. PMID: 10976840. [CrossRef]
- Liu, W.W.; Han, C.H.; Zhang, P.X.; Zheng, J.; Liu, K.; Sun, X.J. Nitric oxide and hyperoxic acute lung injury. Med. Gas Res. 2016 Jul 11;6(2):85-95. PMID: 27867474; PMCID: PMC5110127. [CrossRef]
- Fismen, L.; Eide, T.; Hjelde, A.; Svardal, A.M.; Djurhuus, R. Hyperoxia but not ambient pressure decreases tetrahydrobiopterin level without affecting the enzymatic capability of nitric oxide synthase in human endothelial cells. Eur J Appl Physiol. 2013 Jul;113(7):1695-704. Epub 2013 Feb 6. PMID: 23385656. [CrossRef]
- Hesthammer, R.; Eide, T.; Thorsen, E.; Svardal, A.M.; Djurhuus, R. Decrease of tetrahydrobiopterin and NO generation in endothelial cells exposed to simulated diving. Undersea Hyperb. Med. 2019 Mar-Apr-May;46(2):159-169. PMID: 31051061.
- Hesthammer, R.; Dahle, S.; Storesund, J.P.; Eide, T.; Djurhuus, R.; Svardal, A.M.; Thorsen, E. Nitric oxide in exhaled gas and tetrahydrobiopterin in plasma after exposure to hyperoxia. Undersea Hyperb. Med. 2020 Second Quarter;47(2):197-202. PMID: 32574435. [CrossRef]
- McNeill, E.; Channon, K.M. The role of tetrahydrobiopterin in inflammation and cardiovascular disease. Thromb. Haemost. 2012 Nov;108(5):832-9. Epub 2012 Oct 10. PMID: 23052970; PMCID: PMC5238931. [CrossRef]
Figure 1.
Relationship between the change in DLCO immediately following six hours of breathing 100% oxygen at 202.65 kPa and the immediate pre-dive FENO (circles) and post-dive FENO (squares). FENO was measured at 50 ml/s expired flow rate. The numbers next to the data points are subject number identifiers. Subjects 3, 4, and 9 all showed significant decrements in DLCO immediately post-dive (see Table 4). Regression equations for the solid line and dashed line are: Percent change in DLCO = -0.2265 + 0.0043*pre-dive FENO; (r = 0.9018, p = 0.0022; r2 = 0.8132); Percent change in DLCO = -0.2286 + 0.0096*post-dive FENO (r = 0.9485, p = 0.0003; r2 = 0.8996).
Figure 1.
Relationship between the change in DLCO immediately following six hours of breathing 100% oxygen at 202.65 kPa and the immediate pre-dive FENO (circles) and post-dive FENO (squares). FENO was measured at 50 ml/s expired flow rate. The numbers next to the data points are subject number identifiers. Subjects 3, 4, and 9 all showed significant decrements in DLCO immediately post-dive (see Table 4). Regression equations for the solid line and dashed line are: Percent change in DLCO = -0.2265 + 0.0043*pre-dive FENO; (r = 0.9018, p = 0.0022; r2 = 0.8132); Percent change in DLCO = -0.2286 + 0.0096*post-dive FENO (r = 0.9485, p = 0.0003; r2 = 0.8996).
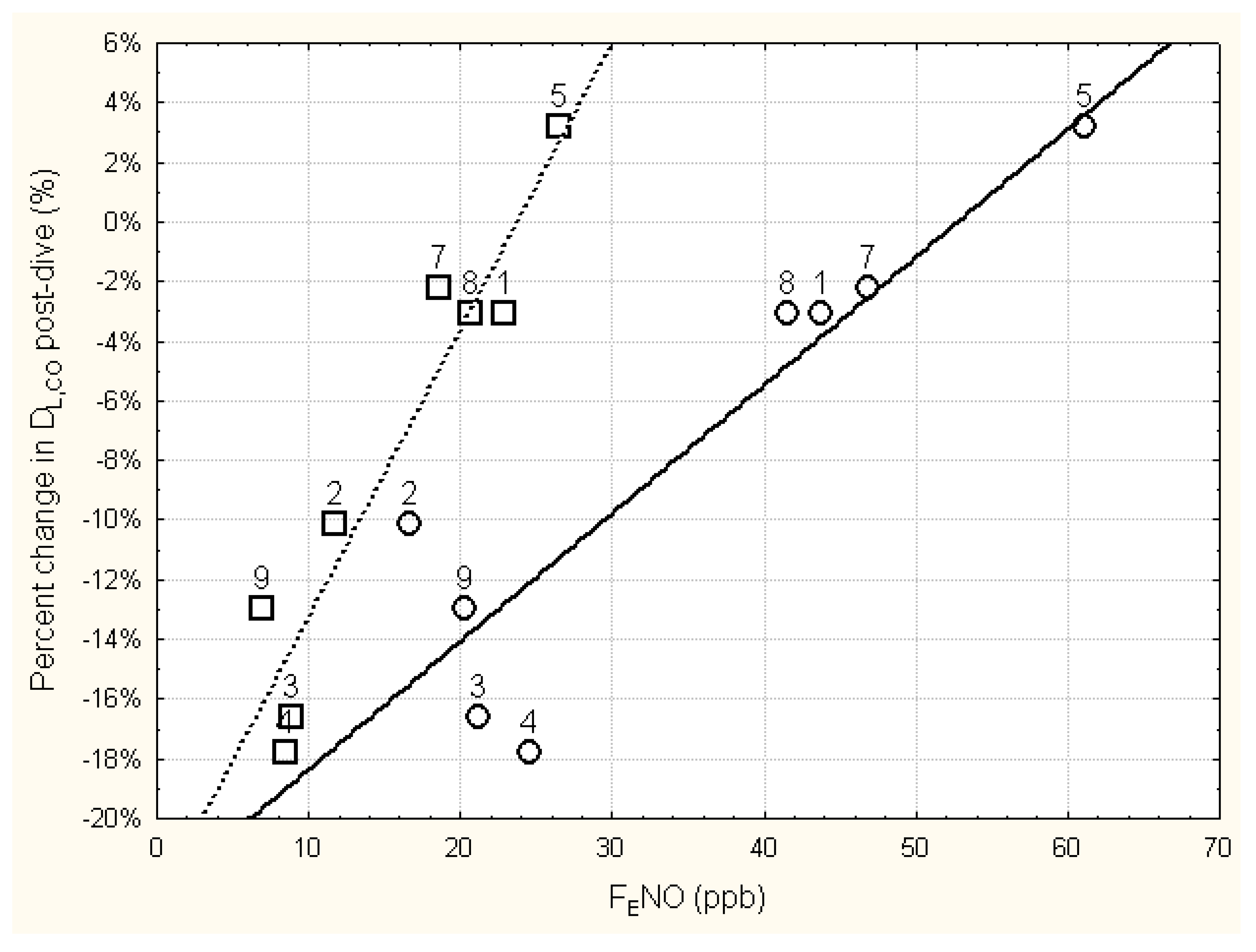
Table 1.
Mean baseline levels, 2x coefficient of variation (CV), and percent change in FENO (expired flow rate = 50 ml/s) following the 202.65 kPa HBO exposures. The subject data (rows) are ordered from highest to lowest baseline FENO. Cells highlighted in grey indicate the time points where significant decrements in pulmonary function were observed (see Table 2, Table 3 and Table 4). The baseline CV was determined from 10 measurements taken over five consecutive days before the dive (see methods). Post Dive 1 and post Dive 2 measurements were taken between 15 minutes and one hour post dive. Recovery measurements (Rec 1, 2, 3) were taken 1-, 2- and 3-days post-dive.
Table 1.
Mean baseline levels, 2x coefficient of variation (CV), and percent change in FENO (expired flow rate = 50 ml/s) following the 202.65 kPa HBO exposures. The subject data (rows) are ordered from highest to lowest baseline FENO. Cells highlighted in grey indicate the time points where significant decrements in pulmonary function were observed (see Table 2, Table 3 and Table 4). The baseline CV was determined from 10 measurements taken over five consecutive days before the dive (see methods). Post Dive 1 and post Dive 2 measurements were taken between 15 minutes and one hour post dive. Recovery measurements (Rec 1, 2, 3) were taken 1-, 2- and 3-days post-dive.
| Subject | Baseline FENO | Post Dive 16 hrs O2 | Post Dive 28 hrs O2 | Rec 1 | Rec 2 | Rec 3 | |
| MeanPpb | CVx2 (%) | ||||||
| 5 | 59 | 24% | -55% | -65% | -11% | 0% | -6% |
| 7 | 44 | 32% | -58% | -67% | +5% | 0% | +22% |
| 1 | 41 | 16% | -44% | -61% | -5.7% | +28% | -4% |
| 8 | 38 | 30% | -46% | -60% | +14% | +76% | +36% |
| 2 | 24 | 22% | -51% | NA | +37% | +3% | +15% |
| 4 | 24 | 34% | -65% | NA | +9% | +40% | +10% |
| 3 | 21 | 34% | -57% | NA | -5% | +2% | +4% |
| 9 | 19 | 20% | -64% | NA | +55% | +34% | +66% |
| Mean | 34 ppb | 26.5% | -55%† | -63% † | +12% | +23% | +18% |
† Group mean FENO significantly different from baseline (p<0.0001). NA =Not applicable.
Table 2.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in FVC following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in FVC from the individual’s mean baseline.
Table 2.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in FVC following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in FVC from the individual’s mean baseline.
| Subject | Baseline FVC | Post Dive 16 hrs O2 | Post Dive 28 hrs O2 | Rec 1 | Rec 2 | Rec 3 | |
| Mean(L BTPS) | CVx2 (%) | ||||||
| 5 | 5.61 | 4.9% | -0.8% | +1.9%* | -1.5% | -0.4% | -0.6% |
| 7 | 4.22 | 6.3% | +1.9% | -0.5%* | +0.9% | -2.8% | -1.9% |
| 1 | 5.99 | 2.2% | +4.3% | -2.8%* | -0.5% | -0.7% | -4.2% |
| 8 | 4.91 | 6.8% | +4.7% | +3.3%* | +3.3% | -0.4% | -0.8% |
| 2 | 5.58 | 7.0% | +3.6%* | NA | +2.9% | -1.6% | -1.3% |
| 4 | 5.49 | 6.6% | -3.5%* | NA | -2.2% | -0.4% | -2.7% |
| 3 | 4.78 | 5.3% | -3.1%* | NA | +1.3% | -5.2% | -0.8% |
| 9 | 5.49 | 5.3% | -9.3%* | NA | -17.3%* | -7.7% | -5.4% |
| Mean | 5.26 L | 5.6% | -0.3% | +0.5% | -1.6% | -2.4% | -2.2% |
* = Symptoms of PO2tox reported. NA =Not applicable.
Table 3.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in FIVC following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in FIVC from baseline.
Table 3.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in FIVC following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in FIVC from baseline.
| Subject | Baseline FIVC | Post Dive 16 hrs O2 | Post Dive 28 hrs O2 | Rec 1 | Rec 2 | Rec 3 | |
| Mean(L BTPS) | CVx2 (%) | ||||||
| 5 | 6.15 | 5.5% | -0.7% | +0.5%* | -2.4% | -0.8% | -0.2% |
| 7 | 4.57 | 4.4% | +2.0% | +3.9%* | -1.8% | -0.4% | +2.6% |
| 1 | 6.93 | 3.3% | -2.6% | -0.7%* | -0.3% | +0.7% | -0.3% |
| 8 | 5.35 | 3.2% | +4.9% | +4.9%* | +5.8% | +6.5% | +1.9% |
| 2 | 6.43 | 4.4% | -6.8%* | NA | -4.0% | -1.4% | -1.7% |
| 4 | 6.61 | 6.0% | -3.4%* | NA | +0.5% | -2.1% | +5.6% |
| 3 | 5.43 | 2.7% | -18.8%* | NA | -5.0% | -0.9% | -5.0% |
| 9 | 6.15 | 5.9% | -15.1%* | NA | -12.5%* | -12.5% | -10.4% |
| Mean | 5.95 L | 4.5% | - 5.1% | +2.2% | -2.5% | -1.4% | -0.9% |
* Symptoms of PO2tox reported.
Table 4.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in DLCO following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in DLCO from baseline.
Table 4.
Mean baseline levels, 2x coefficient of variation (CV) and percent change in DLCO following the 202.65 kPa HBO exposures. Cells highlighted in grey indicate significant decrements in DLCO from baseline.
| Subject | Baseline DLCO | Post Dive 16 hrs O2 | Post Dive 28 hrs O2 | Rec 1 | Rec 2 | Rec 3 | |
| Mean(mL/mmHg/min) | CVx2 (%) | ||||||
| 5 | 37.9 | 8.7% | +3.2% | +2.6%* | +0.3% | +1.3% | -5.0% |
| 7 | 26.1 | 7.0% | -2.2% | -5.6%* | -2.2% | -5.6% | -2.2% |
| 1 | 41.1 | 9.5% | -3.1% | -7.3%* | -17.0% | -6.5% | -6.8% |
| 8 | 33.1 | 11.2% | -3.1% | -4.3%* | -10.1% | -9.8% | -0.4% |
| 2 | 40.6 | 13.9% | -10.1%* | NA | -7.7% | -15.1% | -21.7% |
| 4 | 45.5 | 15.4% | -17.8%* | NA | -20.0% | -4.0% | -10.7% |
| 3 | 39.6 | 6.9% | -16.6%* | NA | +3.6% | -8.5% | -3.2% |
| 9 | 38.3 | 8.7% | -13.0%* | NA | -9.6%* | -27.6% | -20.8% |
| Mean | 37.8 | 10.2% | -7.8% | -3.7% | -7.8% | -9.5% | -8.9% |
* Symptoms of PO2tox reported.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



