
Submitted:
23 June 2023
Posted:
25 June 2023
You are already at the latest version
Abstract
The process of identifying and approving a new drug is a time-consuming and expensive procedure. One of the biggest issues to overcome is the risk of hepatotoxicity, which is one of the main reasons for drug withdrawal from the market. While animal models are the gold standard in preclinical drug testing, the translation of results into therapeutic intervention is often ambiguous due to interspecies differences in hepatic metabolism. The discovery of human induced Pluripotent Stem Cells (hiPSCs) and their derivatives has opened new possibilities for drug testing. We used mesenchymal stem cells and hepatocytes both derived from hiPSC, together with endothelial cells, to miniaturize the process of generating hepatic organoids. These organoids were then cultivated in vitro using both static and dynamic cultures. Additionally, we tested spheroids composed solely by induced hepatocytes. By miniaturizing the system, we demonstrated the possibility of maintaining the organoids, but not the spheroids, in culture for up to 15 days. This timeframe may be sufficient to carry out a hypothetical pharmacological test or screening. In conclusion, we propose that the hiPSC-derived liver organoids model could complement or, in the near future, replace the pharmacological and toxicological tests conducted on animals.
Keywords:
liver
; hiPSC
; organoids
; 3D dynamic culture
; organotypic culture
1. Introduction
Modern medicine has undergone a continuous and incessant growth in recent years, thanks to the great success achieved through laboratory research, which introduced new tools in support of several disciplines including regenerative medicine, pharmacology, and tissue engineering [1]. A major breakthrough occurred in 2007 when it was discovered that adult human cells could be rejuvenated to a pluripotent state. These embryonic-like cells had the potential to be further differentiated toward any cell type of the human body [2]. Fifteen years later, the study of cells derived from iPSCs, which were generated from healthy individuals or patients affected by genetic diseases, has revealed the pathophysiology of many biochemical and molecular processes, difficult to identify using animal models. Indeed, this model has become one the most valuable tool to fulfill the European “Three Rs” (3Rs) request to reduce, refine, and replace animal testing while overcoming the limitations associated with the use of human tissue [3].
The first protocol applied to differentiate hiPSCs was similar to the one employed to differentiate embryonic stem cells. It was based on the three-dimensional (3D) self-aggregation of cells, resulting in the formation of embryoid body [4]. Further development of this concept led to the generation of the so-called “tissue-in-a-dish”, where specific differentiated cell types from a specific organ are mixed in a 3D structure that self-organizes, resembling the in vivo organogenesis process [5]. This advancement greatly increased the capacity of this model to be used as a tool for drug testing as well as for regenerative medicine [6].
Concerning liver, hepatic cells derived from hiPSCs could represent an important source of cells to be used in preclinical tests as well as in cell-based therapeutic applications such as transplantation. In addition to the difficulty of obtaining donor human hepatocytes, one of the major challenges is to preserve hepatocyte functions that are usually rapidly lost in extracorporeal conditions. Differentiation of hepatocyte-like cells from hiPSCs was already performed in several laboratories [7,8]. However, the use of hiPSCs-derived hepatocytes in assessing drug-induced hepatoxicity still need to improve their predictability. Hepatic organoids represent a potent tool for sustaining hepatic functionality also because they better mimic the complexity of the liver both from an architectural point of view and as regards the multicellular and heterotypic composition of the organ [9,10].
Even though static culture conditions favor cell expansion, they do not facilitate the diffusion of nutrients in a 3D structure, particularly during long-term cultivation. To overcome this problem, the use of continuous fluid flow in new culture systems emerges as a viable solution. Bioreactor technology was developed to address this challenge by enabling dynamic perfusion, which ensures constant nutrient renewal and continuous removal of waste metabolites. This dynamic process closely mimics the exchange of physiological body fluids in vivo [11]. Bioreactors offer the ability to create a highly controlled environment, allowing the operator to adjust various cell culture conditions such as pH, temperature, perfusion rate, and oxygen levels. Numerous studies have demonstrated that cultures maintained in bioreactors closely replicate the conditions found in living organs, thus providing the opportunity to conduct increasingly refined drug tests [9,12].
Here, we introduce a model that combines heterotypic composition of liver cells (endothelial, mesenchymal and hepatocyte cells), the 3D organization of cells (organoids in 3D matrix) as well as microfluidic simulation of sinusoidal flux within the intrahepatic microenvironment.
2. Materials and Methods
2.1. Cell culture
All cultures were kept at 37 °C in a humidified atmosphere of 5% CO2.
2.1.1. HUVECs
Human Umbilical Vein Endothelial Cells (HUVEC, Lonza) were used at early passages (I–IV) and grown on plastic petri dishes coated with 0.1% porcine gelatin (Merck) in EGM-2 (Lonza) with 5% fetal bovine serum (FBS). Cells were passaged using Trypsin-EDTA (Thermo Fisher Scientific) every 4 days.
2.1.2. Human Induced Pluripotent Stem Cells (hiPSCs)
A hiPSC line from a healthy female donor (https://hpscreg.eu/cell-line/TMOi001-A) was bought from Thermo Fisher Scientific and used for all the experiments. Either mTeSRTM or TeSR-E8TM cell media (STEMCELL Technologies) were used for maintenance of the cell line. Plates were coated with Human Biolaminin 521 LN (Thermo Fisher Scientific) for maintenance and Matrigel® hESC-qualified Matrix (Corning) for differentiation. When cells reached 70-80% of confluency they were detached using TrypLeTM Select Enzyme (Thermo Fisher Scientific) and plated in the presence of Rock inhibitor Y27632 (Stem Cell Technologies) at a seeding density of around 3x104 cells/cm2). Culture medium was refreshed daily.
2.1.3. Human Induced Mesenchymal Stem Cells (hiMSCs)
Mesenchymal progenitors were derived from the hiPSC line from healthy female donor (https://hpscreg.eu/cell-line/TMOi001-A). hiMSCs were maintained in MesenCultTM ACF Plus Medium (STEMCELL Technologies). Cells were sub-cultured with Trypsin-EDTA (Thermo Fisher Scientific) every 4 days Culture medium was refreshed daily.
2.2. hiPSC differentiation
2.2.1. Hepatic differentiation
The hepatic differentiation protocol is sought to recapitulates the in vivo changes occurring during embryogenesis. hiPSCs were differentiated toward definitive endoderm (iDE) using STEMdiffTM Definitive Endoderm Kit (STEMCELL Technologies). For hepatic lineage specification, the medium was replaced by Hepatocyte Maturation Medium consisting of RPMI 1640 + B27 Supplement + 10 ng/mL basic Fibroblast Growth Factor + 20 ng/mL Bone Morphogenetic Protein 4 (all from Thermo Fisher Scientific). From day 9 (dd9) of differentiation, the human induced hepatocytes (hiHeps) were maintained in Hepatocyte Maintenance Medium (HMM) consisting of Clonetics® HCMTM (Lonza) + 5% FBS + 100 nM Dexamethasone + 20 ng/mL Oncostatin M + 10 ng/mL Hepatocytes Growth Factor (all from STEMCELL technologies).
2.2.2. Mesenchymal differentiation and characterization
Mesenchymal progenitors were derived from the hiPSC line using STEMdiffTM Mesenchymal Progenitor Kit (STEMCELL Technologies). hiMSCs were seeded and maintained on plastic after the second passage. To verify MSC identity, several markers were tested in flow cytometry. Cells were further differentiated to adipocytes using StemPro® Complete Adipogenesis Differentiation Medium (Thermo Fisher Scientific), or to osteoblasts using StemPro® Complete Osteogenesis Differentiation Medium (Thermo Fisher Scientific). Adipocyte formation was confirmed using Oil Red O (Merck) staining, while osteoblast differentiation was revealed by Alizarin Red S (Merck) staining.
2.3. Three-dimensional aggregates
2.3.1. Hepatic Spheroids formation
The cells were trypsinized, resuspended in HMM Complete Medium and then seeded (2x106 cells/well) in AggrewellTM Microwell Plates (STEMCELL Technologies), composed by approximately 1200 microwells for each of the 24-wells. Before seeding, the plates were treated with the AggrewellTM Rinsing Solution (STEMCELL Technologies) to inhibit cell attachment. After seeding the cells, the plates were centrifuged at 100g for 6 min, to distribute the cells homogeneously in the microwells. The plates were then incubated at 37 °C. Half of the medium was changed every other day. The 3D organization starts within few hours from seeding and spheroids form within 48 h.
2.3.2. Liver Organoids formation
To generate liver organoids, hiHeps at dd9 of differentiation, were mixed with HUVECs and hiMSCs according to the ratio hiHeps:HUVECs:hiMSCs = 10:7:2 [8], and resuspended in 50% HMM, 50% EGM-2 + 5% FBS. Cell suspension (2x106 cells/well) was subsequently seeded in every AggreWellTM microwell, centrifuged at 100g for 6 min, and incubated at 37 °C. Half of the medium was changed every other day. The 3D organization starts within few hours and organoids form within 48 h.
2.4. Dynamic Culture
Dynamic culture of the organoids was conducted in the bioreactor LiveBox (LB, IVTec). The bioreactor, composed of a transparent chamber, is featured with a flow inlet and outlet for the perfusion with the culture media. The system reproduces the typical volume of a 24-well plate. All the components were autoclaved before use. The content of aggregates from one AggrewellTM well was put in the LB bioreactor 48 h after seeding. In order to avoid loss of aggregates due to the dynamic flow, they were resuspended in 1.5% alginate dissolved in 20 mM HEPES-NaOH, pH 7.4, filtered through a 0.45 μm filter, and autoclaved. The aggregates were collected from the AggrewellTM, and the pellet was resuspended in 1 mL of alginate. To obtain the beads, the cell suspension was placed in a syringe provided with a 21G needle (Terumo) and dropped in a solution CaCl2 102 mM under magnetic stirring, and then left for 10 min at 4 °C. The aggregates were then cultured for 96 h at a flow rate of 330 ml/min, and half-medium was changed 48 h after the starting point.
2.5. Flow cytometry
Cells were detached and resuspended in PBS containing Ca2+/Mg2+ (PBS+), 2%BSA. Cell suspension was incubated with the indicated specific antibody (all from Biolegend) for 1 h at 4 °C. Cells were pelleted, washed with PBS/BSA and then analyzed using FACSCantoTM (BD Bioscience).
2.6. Viability Assay
The viability assay was performed using CellTiter-Glo® 3D Cell Viability Assay following manufacturer’s instructions (Promega). 30 aggregates for each condition were collected in duplicate and measured. ATP-derived luminescence was evaluated and expressed in Relative Unit (RLU). Cell viability was compared to an ATP standard curve.
2.7. RNA extraction and RT-PCR analysis
Total cellular RNA was extracted using Quick-RNATM MiniPrep Kit (Zymo Research). 3D structures were conserved in TRIzol Reagent (Thermo Fisher Scientific) and homogenized with a 1mL Tissue Grinder Potter-Elvehjem (BioSigma) for 15 minutes on ice. The sample was then passed in a 1mL insulin syringe (Therumo) for 10 times, moved in a new tube, and centrifuged 10 min at 12,000g at 4 °C. Supernatant was collected, and chloroform was added. Then, the sample was inverted 10 times, left 4 min at RT and centrifuged for 20 min at 12,000g at 4 °C. The phase containing RNA was precipitated using isopropanol and centrifuged 20 min at 12,000g at 4 °C. The pellet was washed and resuspended in 30 μl of RNAse-free water. The quantity and purity of RNA were measured using a NanoDrop spectrophotometer (Thermo Fisher Scientific). Reverse transcriptase-PCR of 1 μg of total RNA was carried out using iScriptTM cDNA Synthesis Kit (Bio-Rad). For the PCR assay, cDNA was mixed with DreamTaqTM PCR Master Mix (Thermo Scientific), specific primers, and RNase-free water (Bio-Rad).
PCR was performed on Thermocycler PCR (Bio-Rad) for 5 min at 95 °C followed by 35 cycles (95 °C for 45 sec; 60 °C for 45 sec; 72 °C for 45 sec) and 72 °C for 2 min. PCR products were visualized by agarose/Gel Red (Sigma) electrophoresis.
For the quantitative PCR (qPCR) assay, cDNA was mixed with SYBR® Select Master Mix (Thermo Fisher Scientific), specific primers, and RNase-free water (BIO-RAD). qPCR was performed on Applied Biosystem ViiATM 7 Real-Time PCR System (Thermo Fisher Scientific) for 10 min at 95 °C followed by 40 cycles (95 °C for 15 sec; 60 °C for 1 min) and melt curve stage 95 °C for 15 sec; All PCR reactions were performed in triplicate. The mean cycle threshold (Ct) values were normalized to endogenous control β-Actin and analyzed using the comparative ΔCt method. The following primers were used to assess the expression of specific genes: Oct3/4 For: 5’GGGTTTTTGGGATTAAGTTCTTCA Rev: 5’GCCCCCACCCTTTGTGTT; Nanog For: 5’AGGAAGACAAGGTCCCGGTCAA Rev: 5’TCTGGAACCAGGTCTTCACCTGT; HNF4 For: 5’TGCGACTCTCCAAAACCCTC Rev: 5’TGATGGGGACGTGTCATTGC; AFP For: 5’AAGTTTAGCTGACCTGGCTACC Rev: 5’TGCAGCAGTCTGAATGTCCG; Albumin For: 5’TCTTCTGTCAACCCCACACG Rev: 5’GCAACCTCACTCTTGTGTGC; Serpin A1 For: 5’TCCGATAACTGGGGTGACCT Rev: 5’AGACGGCATTGTCGATTCACT; ACTB For: 5’CACTCTTCCAGCCTTCCTTC Rev: 5’AGTGATCTCCTTCTGCATCCT.
2.8. Secreted Protein Quantification
Cell conditioned medium was collected every other day starting from dd5 of iDE, and subsequently from all culture conditions. The presence of alpha Fetoprotein (AFP) was quantified using the analyzer Cobas (Roche). Human alpha-1 Antitrypsin (AAT) was quantified by a sandwich ELISA. 96-well half-volume high-binding plates (Corning) were coated overnight with 2 μg/mL of polyclonal anti-mouse IgG (Sigma), saturated for 1 h at 37 °C with blocking buffer (PBS, 0.25% BSA, 0.05% Tween-20) followed by incubation with the conditioned medium of the anti-human α1-antitrypsin (AAT) monoclonal antibody 3C11 hybridoma cells, kindly provided by Prof. David Lomas (University College London, UK) [13,14]. After washing with PBS/0.05% Tween-20 (PBS-T), serial dilutions of the purified plasma AAT standard and of the cell culture media were added to the plates and incubated at 37 °C for 1 h. After washing with PBS-T, the wells were incubated for 1 h at 37 °C with sheep anti-AAT-HRP (Abcam) diluted in blocking buffer, further washed and revealed with TMB substrate (Merck). The reaction was blocked by adding 3M HCl and absorbance at 450 nm was measured using the EnSightTM plate reader (PerkinElmer).
2.9. Immunohistochemistry
For immunohistochemistry analysis, liver organoids were collected, centrifuged at 3000g x 10 min, and a compacting solution, consisting of 75% methanol (Carlo Erba), 20% chloroform (Sigma) and 5% acetic acid (Carlo Erba) was added to the pellet for 20-30 min as already described [15]. The compacting solution was subsequently discarded, the pellet was moved in a histology cassette, stained with hematoxylin and eosin, covered with 2.5% agarose and preserved in formalin until it was processed for paraffin inclusion. Endogenous peroxidase activity was blocked with 0.3% H2O2 (Sigma Aldrich) in methanol for 20 min. Antigen retrieval was performed using a microwave oven in 1 mM EDTA pH 8.0 (Carlo Erba) or in 1 mM citrate buffer pH 6 (Carlo Erba). Two-micron sections were cut, washed in TBS pH 7.4 (Carlo Erba) and incubated for 1 h with the following primary antibodies diluted in TBS/1% BSA (Merck): anti-human albumin (ThermoFisher Scientific), anti-CD90 (Abcam rabbit monoclonal) and anti-CD31 (Novocastra) antibodies. The signal was revealed using the DAKO Envision+System-HRP Labelled Polymer Anti-Mouse or Anti-Rabbit, followed by diaminobenzydine (DAB) as chromogen and hematoxylin as counterstain. Images were acquired with a Nikon DS-Ri2 camera (4908 x 3264 full-pixel) mounted on a Nikon Eclipse 50i microscope equipped with Nikon Plan lenses (x10/0.25; x20/0.40; x40/0.65; x100/1.25) using NIS-Elements 4.3 imaging software (Nikon Corporation).
2.10. Immunofluorescence Staining
To perform immunofluorescence experiments, cells were fixed using Immunofix (Bio-Optica) for 15 min, washed and maintained in PBS+ at 4 °C. Before antibody staining, cells were permeabilized with PBS+ containing 0.5% TritonTM X-100 (Sigma) for 10 minutes and then incubated with a blocking solution (PBS+ containing 1% BSA (Sigma) and 2% Donkey Serum (Merck)) for 1 h at RT and washed with PBS+ containing 0.2% Tween 20 (Merck). At this point, samples were incubated overnight at 4 °C with the following primary antibodies diluted in blocking solution: anti-FOXA2 (rabbit polyclonal IgG, 1:400, Thermo Fisher Scientific [720061]), anti-SOX17 (mouse monoclonal IgG2b (OTI2G8), 1:400, Thermo Fisher Scientific [MA5-24891]) or anti-HNF4 (rabbit monoclonal IgG (F.674.9), 1:400, Thermo Fisher Scientific [MA5-14891]). After extensive wash, Alexa fluorophore conjugated secondary antibodies (donkey anti-rabbit IgG 594, 1:600, Thermo Fisher Scientific [A-21207]; donkey anti-mouse IgG 488, 1:600, Thermo Fisher Scientific [A-21202]) were added and incubated 1 h at RT. Nuclei were counterstained with DAPI (Thermo Fisher Scientific) for 10 min at RT. Images was acquired at x20 and x63 magnification with Zeiss Fluorescence Axiovert 200M microscope (Carl Zeiss)
3. Results
3.1. Differentiation of Mesenchymal Stem Cells from induced Pluripotent Stem Cells
We previously isolated human MSCs from bone marrow and from different adipose tissues, concluding that MSCs present distinct characteristics depending on their isolation source [16]. Since our aim was to include MSCs in liver organoids, we decided to differentiate them starting from hiPSC. As illustrated by the flow cytometry analysis in Figure 2A, hiMSCs showed a remarkable expression of the mesenchymal specific markers CD105, CD90, and CD73, while they were negative for the expression of CD34, specific for the hematopoietic lineage, and for the pluripotency genes OCT4 and NANOG (data not shown). Moreover, hiMSCs showed an osteogenic and adipogenic differentiation potential, respectively indicated by the calcium deposits highlighted with the Alizarin Red S staining (Figure 2C) and by the neutral triglycerides and lipids deposits present in adipocytes, detected by the Oil Red O staining (Figure 2D). These specific markers expression, together with cell morphology (Figure 2B), adherence to plastic as well as their proliferation for more than 10 passages categorize these cells as Mesenchymal Stem Cells.
3.2. Differentiation of hepatic cells from induced Pluripotent Stem Cells
To generate hepatic cells from hiPSC, we followed the protocol illustrated in Figure 3A that comprises preliminary steps lasting 5 days, leading to the differentiation toward hiDE, followed by the specification of the hepatic lineage. Characterization of hiDE cells is illustrated in Figure 3. In parallel to an evident change in cell morphology (Figure 3D), there is a progressive down-regulation of pluripotency genes, represented by OCT4 (Figure 3B), paralleled by an upregulation of specific markers such as HNF4 (Figure 3C), involved in the transition from endodermal cells to hepatic progenitors. Furthermore, hiDE cells were stained positive in flow cytometry for the endodermal surface markers CXCR4 and CD117 (Figure 3E), and in immunofluorescence for the endodermal markers FOXA2 and SOX17, both involved in development of the hepatic cell lineage (Figure 3F). We continued the differentiation up to dd23 and on dd21 we performed several analyses in order to determine if the hepatocyte specification had occurred. We evaluated the presence of mRNAs encoding two of the most abundant proteins produced by hepatocytes: AFP, the fetal albumin counterpart, and α1 antitrypsin (AAT), a serine proteinase inhibitor. The results shown in Figure 3G demonstrate an increase in mRNA expression of both genes starting from differentiation dd9 and more pronounced at dd21, when the expression levels are slightly higher than the levels observed in the hepatoma HepG2 cells. Moreover, the encoded proteins are correctly secreted into the culture medium (Figure 3H). AFP started to be present in the media around dd11, soon after the hepatic specification, and decreased after dd15. Also AAT appears at dd11, but shows a progressive increase over time. These trends suggest the presence of liver cells which undergo a maturation that involves the switch between AFP and albumin production.
3.3. 3D cultures
After confirming the hepatic nature of the cells, we used them to obtain two types of 3D structures: hepatic spheroids, composed only by hiHep cells (homotypic culture), and hepatic organoids, composed by hiHeps co-cultured with hiMSCs and HUVECs (heterotypic culture). Aggregates were formed in AggreWellTM 400 Microwell Plates (Figure 4).
Since the size of spheroids/organoids reaches about 200 μm, the static culture condition poorly allows nutrients’ diffusion to the core of the 3D structures. This issue is highly deleterious for the long-term culture of spheroid/organoids required in pharmacological or toxicological tests. To overcome this hurdle, we put the aggregates in the dynamic framework of the millifluidic bioreactor LiveBox (IVTech) where the aggregates can be subjected to a continuous flow of the culture medium. Moreover, these dynamic conditions better reproduce the sinusoidal blood flow impact on hepatocyte functions in the organism. To avoid the loss of samples due to medium flow, we entrapped the aggregates into 1.5% Alginate Beads (Figure 5).
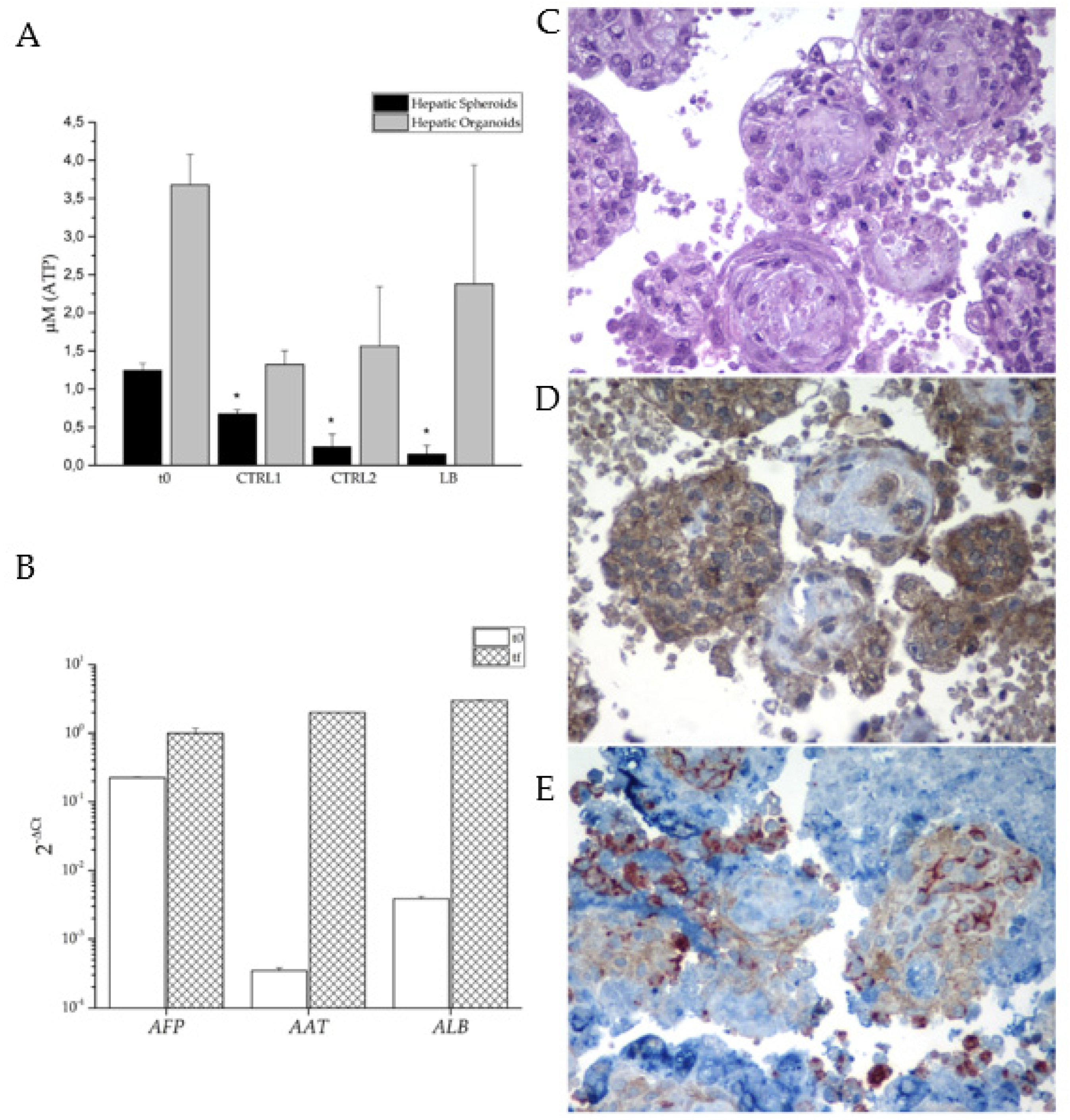
To analyze the efficiency and effectiveness of the dynamic culture condition, we compared cell viability in the static and dynamic conditions. 48 h after the aggregation, about 600 aggregates, corresponding to half a well of the AggreWellTM plate, were collected and entrapped in alginate beads and cultured in a 60 mm cell culture dish (Ctrl2) or placed in the microchamber Live Box bioreactor (LB). Samples were analyzed after 96 h (4 days).
We first analyzed cell viability using an ATP luminescent kit expressly designed for microtissues. To set up a standard curve, we measured the luminescence of 25,000 and 50,000 live trypan blue-negative cells, either hiHeps alone or mixed with HUVECs and hiMSCs in the same ratio as in organoids. Then, to reduce the sample’s variability, we quantified luciferase in 30 aggregates, in duplicate, for each experimental condition. We measured ATP-derived luminescence (RLU) at the moment of aggregate collection t0 (48 hours after seeding) and after 96 h in culture in the three different conditions (Figure 6A).
As shown, aggregates’ viability was reduced in all the conditions. However, while dynamic culture strongly reduces spheroid survival, the same condition represents the best setup for the organoid culture.
Organoids are formed by the aggregation of three cell types that could behave differently in the dynamic culture. Therefore, we must ensure that the liver cells can survive under these conditions. To better assess the characteristics of our 3D aggregates, we quantified mRNA expression levels of hepatic genes in the collected aggregates after one week of dynamic culture. All the expression levels of the hepatic genes showed a strong increase in the final sample, confirming the survival of the hepatic lineage (Figure 6B). Moreover, the large increase in the expression levels of ALB mRNA over time is particularly promising, as it strongly suggests hepatic cell maturation.
Based on these promising results, we aimed to validate the data by collecting the organoids after one week of culture in the bioreactor. The collected organoids were embedded in paraffin, sectioned, and subjected to analysis using hematoxylin-eosin (H&E) staining. Additionally, an immunohistochemical staining was performed using antibodies specific to the three cell types. The H&E staining (Figure 6C) shows the presence of viable cells throughout the center of the aggregates. Of note, a minimal presence of apoptosis/necrosis can be seen. Additionally, the organoids demonstrate ALB production, as depicted in Figure 6D. To investigate the arrangement of the three cell types within the organoids, histological sections were stained using three specific antibodies: anti-CD31 for HUVECs (stained in red), anti-CD90 for hiMSCs (stained in brown), and AFP for hiHeps (stained in blue), as shown in Figure 6E. The presence of the three colors indicates the survival of all cells type under the dynamic condition. The images suggest a structural organization of the cell types, with hiHep cells predominantly located in the surface, while hiMSCs are neatly arranged to form a structural support. Moreover, HUVECs seem to organize themselves in circular structures, probably the beginning of capillary neoformation.
4. Discussion
The process of bringing new drugs to the market starts with the discovery of new molecules, followed by several steps of safety and efficacy trials prior to their approval by national and international regulatory agencies, such as by the U.S. Food and Drugs Administration. Typically, this procedure takes several years, with a significant financial investment in range of hundreds of millions of dollars [17,18].
In drug research, the major issue is the risk associated with hepatotoxicity, which is the main reason of withdrawal of drugs from the market [17,18,19].
The animal model is considered the gold standard in drug pre-clinical testing, but it presents several issues, because animals frequently have a different hepatic metabolism compared to humans. Consequently, the transposition of results from animal studies to human therapy is difficult. These differences represent an important risk factor in preclinical trials, as the predictive level of hepatotoxicity obtained from animal tests is only around 50% [20]. Furthermore, the extended lead times and relatively high costs associated with animal models require the development of new, faster, and cheaper in vitro models capable of fulfilling the 3R principles of replacement, reduction, and refinement of animal models [3].
The discovery of hiPSC has opened new possibilities for their application as a model in drug testing. Currently, well-established protocols are described for deriving quite easily human hepatocytes from hiPSCs [21,22]. iPSC-induced hepatocytes could represent an excellent cell model for drug testing and screening, particularly because they can be derived from patients autologous cells [23]. Nevertheless, it is important to note that the liver is a highly complex organ with specific architecture, organization and functions that are correlated to the several cell types that populate it. Therefore, the self-assembling 3D structure of organoids, composed of different cell types representing the main components of the organ, could be considered as an initial step toward mimicking the complex structure of the liver and its physiologically relevant impact on hepatic functions [6,24].
In our study, we focused on deriving and characterizing both hiMSCs for the structural component, and hiHeps as the functional part of the organoids. We also incorporated HUVECs as the endothelial component. We know that endothelial cells could also be derived from hiPSC, but a robust protocol for differentiating them into liver sinusoidal endothelial cells has not been published yet.
Firstly, we successfully demonstrated the derivation of both hiMSCs and hiHeps. To confirm differentiation of hiMSCs, we observed the loss of expression of pluripotency genes, the acquisition of a new cell morphology, their ability to grow on plastic surfaces, the expression of specific markers and the potency to differentiate into both osteoblasts and adipocytes. Regarding hiHeps differentiation, we achieved the desired results as the morphological and biochemical changes recapitulated the ones that occur during hepatogenesis [25]. The hiPSCs subjected to hepatic differentiation exhibited an increased cytoplasm-nucleus ratio and assumed the peculiar polygonal morphology. Furthermore, we observed the presence, from dd5, of major key regulators of liver development such as FOXA2, SOX17 and HNF4, completely absent in undifferentiated hiPSCs. Indeed, the decrease of the pluripotency genes was paralleled by the increase of HNF4a, which is the master regulator of hepatoblastic differentiation, as well as the appearance of the DE-specific surface markers CXCR4 and CD117. The resulting hepatocyte-like cells, which emerged around dd11, just when conversion from liver progenitors to liver cells was expected, produce both AFP and AAT which are secreted into the culture medium.
The initial increase in AFP concentration, followed by its successive decrease in the culture medium, could be an indication of the maturation of liver-like cells. In physiologically conditions, AFP is progressively deactivated and replaced by ALB in adult hepatocytes. Similarly, the production of AAT, with a progressive increase along the entire differentiation, recalls the behavior observed during the physiological maturation of hepatocytes. All acquired data supported the success of the differentiation protocol, but they also showed the limitation of 2D hiHeps culture. While 2D culture is essential to obtain liver-like cells from hiPSCs, it is not sufficient to reach a stable and mature phenotype.
To overcome the limitations of 2D cultures, and promote a more physiologically relevant model, we mixed the three cell types (hiHeps, hiMSCs and HUVECs) to create a self-assembled three-dimensional organoid with a specific architecture that mimic the liver structure. While other laboratories built their hepatic organoids with the aim of transplanting them into an organism, our goal is to use the organoids in vitro, for cell culture experiments. Our aim is to replace the use of animals to define hepatic pharmacology and toxicity, with an alternative approach.
For this purpose, we miniaturized the aggregates, seeding the cells in AggrewellTM plates, following the previously described ratio that closely recapitulates the liver composition [8]. Initially, we created spheroids using hiHep cells only, but data demonstrate that monotypic culture shows feasibility limits. For this reason, we assembled organoids, adding mesenchymal cells, also derived from hiPSCs, and endothelial cells. Furthermore, since the size of these 3D structures could hamper the perfusion of nutrients, especially in long-term cultures, we introduced the heterotypic aggregates in a bioreactor system. The bioreactor facilitates continuous recirculation of the culture medium, increasing mass transfer and ensuring sufficient nutrient supply to the center of the 3D structures. To prevent the aggregates from being carried away by the flow of the medium, we encapsulated them in alginate beads. Alginate is a compound that, by gelling, forms a stable scaffold without causing any damage to the cellular structures.
Under this culture condition, we observed higher viability of the organoids compared to spheroids. Through histological analysis and subsequent immunolocalization, we demonstrated the survival, aggregation, and structural organization of all cell types. In several organoids, we observed the presence of a fibrotic core or a ring-like structure, which is probably due to hiMSCs matrix production. This fibrotic structure appears to provide support for the hiHeps arranged in the outer area of the organoids. The endothelial cells, on the other hand, appeared to be dispersed throughout the organoids, forming small clusters, indicating the initiation of organization into sinusoidal like structures. Notably, under these conditions, the ALB expression appears to be elevated, suggesting that the dynamic culture could enhance and support the maturation of the hepatocytes involved.
Overall, this approach allows the maintenance and proper function of the organoids, providing an improved environment for their growth and development. The combination of miniaturized aggregates, incorporation of multiple cell types, and the use of a bioreactor system with encapsulation in alginate beads represents an innovative strategy to overcome challenges associated with nutrient perfusion and the maintenance of structural integrity of the organoids during long-term culture.
5. Conclusions
Based on tissue engineering concept, we developed a complex organotypic model, derived from hiPSCs, in both static and dynamic systems to discover its application benefits in research. We can conclude that 3D cell culture of heterotypic cells maintained in a dynamic environment offers a much more realistic hepatic model than the classic two-dimensional culture, thus offering us the opportunity to learn more about the physiological and pathological phenomena on which we can intervene pharmacologically. In this manuscript, we focus on more differentiated hiHeps, but this hepatic model can also be used to study hepatogenesis, since we succeeded in reproducing hiHeps from the less differentiated state of hiPSC to endoderm specific cells, hepatoblasts, and hepatocytes.
Author Contributions
Conceptualization, P.D.E., N.S., G.M., P.L.P. and S.D.; methodology, S.C., M.S. and J.B.; formal analysis, S.C., M.S., J.B., A.F., E.G., M.C., R.Ve. and R.Vi.; writing—original draft preparation, P.D.E.; writing—review and editing, S.C., M.S., J.B., A.F., E.G. and N.S.; funding acquisition, P.D.E., G.M. and S.D. All authors have read and agreed to the final version of the manuscript.
Funding
This research was funded by Ministero della Salute, grant 3R-IZSLER D.LGS 26/2014 to S.V. and P.D.E.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge Marialuisa Massardi for technical support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mao, A.S.; Mooney, D.J. Regenerative Medicine: Current Therapies and Future Directions. Proc. Natl. Acad. Sci. USA 2015, 112, 14452–14459. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Nelson, T.J.; Behfar, A.; Terzic, A. Strategies for Therapeutic Repair: The “R(3)” Regenerative Medicine Paradigm. Clin. Transl. Sci. 2008, 1, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, G. Embryoid Body Formation from Human Pluripotent Stem Cells in Chemically Defined E8 Media. StemBook 2008. [Google Scholar] [CrossRef]
- Tulloch, N.L.; Muskheli, V.; Razumova, M.V.; Korte, F.S.; Regnier, M.; Hauch, K.D.; Pabon, L.; Reinecke, H.; Murry, C.E. Growth of Engineered Human Myocardium with Mechanical Loading and Vascular Coculture. Circ. Res. 2011, 109, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345. [Google Scholar] [CrossRef] [PubMed]
- Hannan, N.R.F.; Segeritz, C.P.; Touboul, T.; Vallier, L. Production of Hepatocyte-like Cells from Human Pluripotent Stem Cells. Nat. Protoc. 2013, 8, 430–437. [Google Scholar] [CrossRef]
- Takebe, T.; Zhang, R.R.; Koike, H.; Kimura, M.; Yoshizawa, E.; Enomura, M.; Koike, N.; Sekine, K.; Taniguchi, H. Generation of a Vascularized and Functional Human Liver from an IPSC-Derived Organ Bud Transplant. Nat. Protoc. 2014, 9, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Steimberg, N.; Bertero, A.; Chiono, V.; Dell’Era, P.; Di Angelantonio, S.; Hartung, T.; Perego, S.; Raimondi, M.; Xinaris, C.; Caloni, F.; et al. IPS, Organoids and 3D Models as Advanced Tools for in Vitro Toxicology. ALTEX 2020, 37, 136–140. [Google Scholar] [CrossRef]
- Fiorotto, R.; Amenduni, M.; Mariotti, V.; Fabris, L.; Spirli, C.; Strazzabosco, M. Liver Diseases in the Dish: IPSC and Organoids as a New Approach to Modeling Liver Diseases. Biochim. Biophys. acta. Mol. basis Dis. 2019, 1865, 920–928. [Google Scholar] [CrossRef]
- Schmelzer, E.; Gerlach, J.C. Multicompartmental Hollow-Fiber-Based Bioreactors for Dynamic Three-Dimensional Perfusion Culture. Methods Mol. Biol. 2016, 1502, 1–19. [Google Scholar] [CrossRef]
- Marchesi, N.; Barbieri, A.; Fahmideh, F.; Govoni, S.; Ghidoni, A.; Parati, G.; Vanoli, E.; Pascale, A.; Calvillo, L. Use of Dual-Flow Bioreactor to Develop a Simplified Model of Nervous-Cardiovascular Systems Crosstalk: A Preliminary Assessment. PLoS One 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Ronzoni, R.; Heyer-Chauhan, N.; Fra, A.; Pearce, A.C.; Rüdiger, M.; Miranda, E.; Irving, J.A.; Lomas, D.A. The Molecular Species Responsible for A1-Antitrypsin Deficiency Are Suppressed by a Small Molecule Chaperone. FEBS J. 2021, 288, 2222–2237. [Google Scholar] [CrossRef] [PubMed]
- Laffranchi, M.; Elliston, E.L.K.; Miranda, E.; Perez, J.; Ronzoni, R.; Jagger, A.M.; Heyer-Chauhan, N.; Brantly, M.L.; Fra, A.; Lomas, D.A.; et al. Intrahepatic Heteropolymerization of M and Z Alpha-1-Antitrypsin. JCI insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Berenzi, A.; Steimberg, N.; Boniotti, J.; Mazzoleni, G. MRT Letter: 3D Culture of Isolated Cells: A Fast and Efficient Method for Optimizing Their Histochemical and Immunocytochemical Analyses. Microsc. Res. Tech. 2015, 78, 249–254. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Verardi, R.; Neva, A.; Benzoni, P.; Crescini, E.; Xia, E.; Almici, C.; Calza, S.; Dell’Era, P. Comparative Analysis of Mesenchymal Stromal Cells Biological Properties. ISRN Stem Cells 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Van Norman, G.A. Drugs, Devices, and the FDA: Part 1: An Overview of Approval Processes for Drugs. JACC. Basic to Transl. Sci. 2016, 1, 170–179. [Google Scholar] [CrossRef]
- Van Norman, G.A. Drugs, Devices, and the FDA: Part 2: An Overview of Approval Processes: FDA Approval of Medical Devices. JACC. Basic to Transl. Sci. 2016, 1, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Ruoß, M.; Vosough, M.; Königsrainer, A.; Nadalin, S.; Wagner, S.; Sajadian, S.; Huber, D.; Heydari, Z.; Ehnert, S.; Hengstler, J.G.; et al. Towards Improved Hepatocyte Cultures: Progress and Limitations. Food Chem. Toxicol. 2020, 138. [Google Scholar] [CrossRef]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W.; et al. Concordance of the Toxicity of Pharmaceuticals in Humans and in Animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef] [PubMed]
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient Differentiation of Human Embryonic Stem Cells to Definitive Endoderm. Nat. Biotechnol. 2005, 23, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Si-Tayeb, K.; Noto, F.K.; Nagaoka, M.; Li, J.; Battle, M.A.; Duris, C.; North, P.E.; Dalton, S.; Duncan, S.A. Highly Efficient Generation of Human Hepatocyte-like Cells from Induced Pluripotent Stem Cells. Hepatology 2010, 51, 297–305. [Google Scholar] [CrossRef]
- Mora, C.; Serzanti, M.; Consiglio, A.; Memo, M.; Dell’Era, P. Clinical Potentials of Human Pluripotent Stem Cells. Cell Biol. Toxicol. 2017, 33, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Olgasi, C.; Cucci, A.; Follenzi, A. IPSC-Derived Liver Organoids: A Journey from Drug Screening, to Disease Modeling, Arriving to Regenerative Medicine. Int. J. Mol. Sci. 2020, 21, 6215. [Google Scholar] [CrossRef]
- Gordillo, M.; Evans, T.; Gouon-Evans, V. Orchestrating Liver Development. Development 2015, 142, 2094–2108. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Apparatus for biodynamic culture. The bioreactor from IVTech LiveBox (LB) (A). Magnification of the culture chamber with alginate beads (upper) or alginate-containing organoids (lower) (B).
Figure 1.
Apparatus for biodynamic culture. The bioreactor from IVTech LiveBox (LB) (A). Magnification of the culture chamber with alginate beads (upper) or alginate-containing organoids (lower) (B).
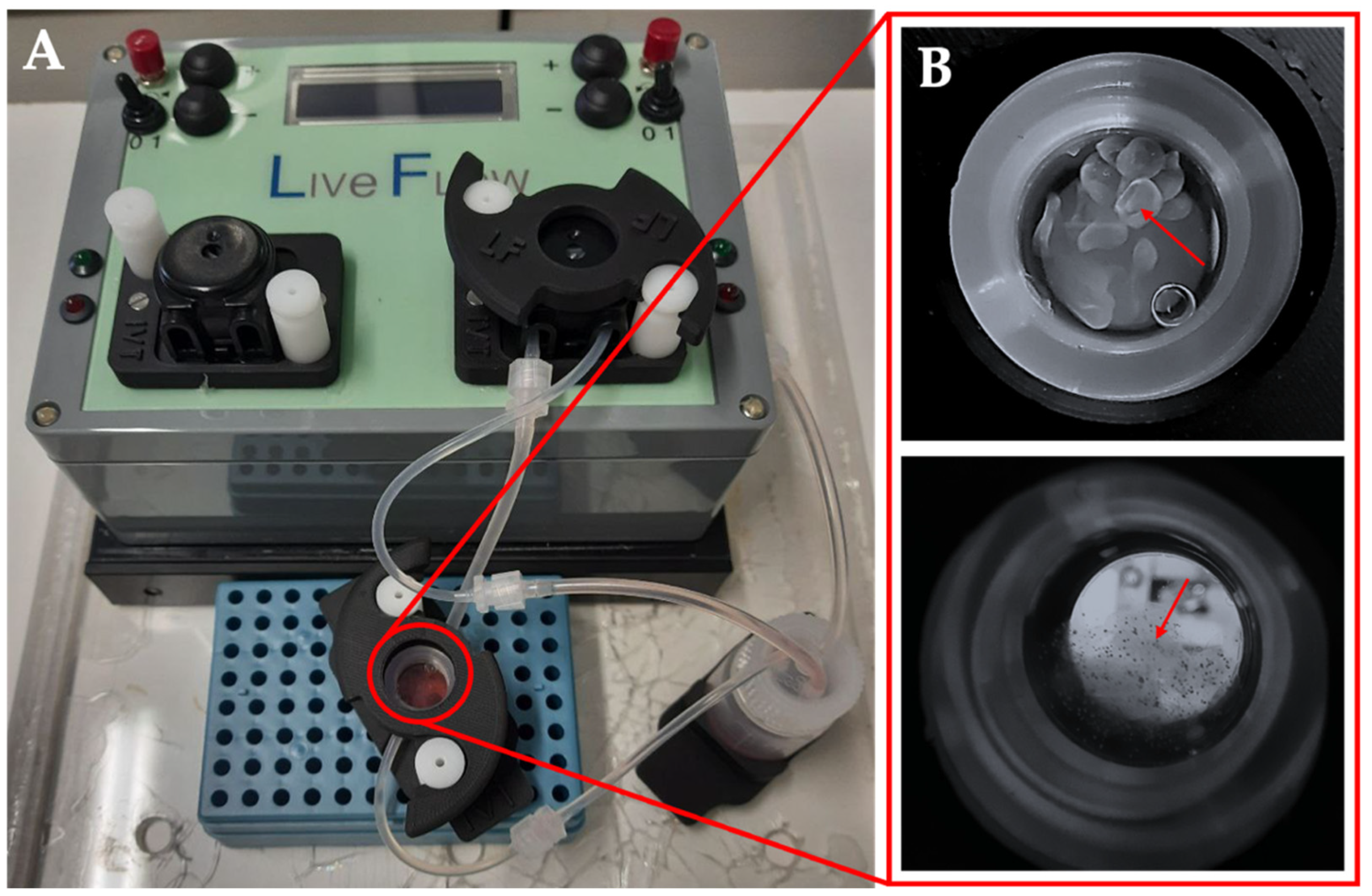
Figure 2.
Characterization of hiMSCs. Flow cytometry scatters of hiMSCs using the indicated antibodies (A). Cell morphology (B). Alizarin Red staining (C). Oil Red O staining (D).
Figure 2.
Characterization of hiMSCs. Flow cytometry scatters of hiMSCs using the indicated antibodies (A). Cell morphology (B). Alizarin Red staining (C). Oil Red O staining (D).
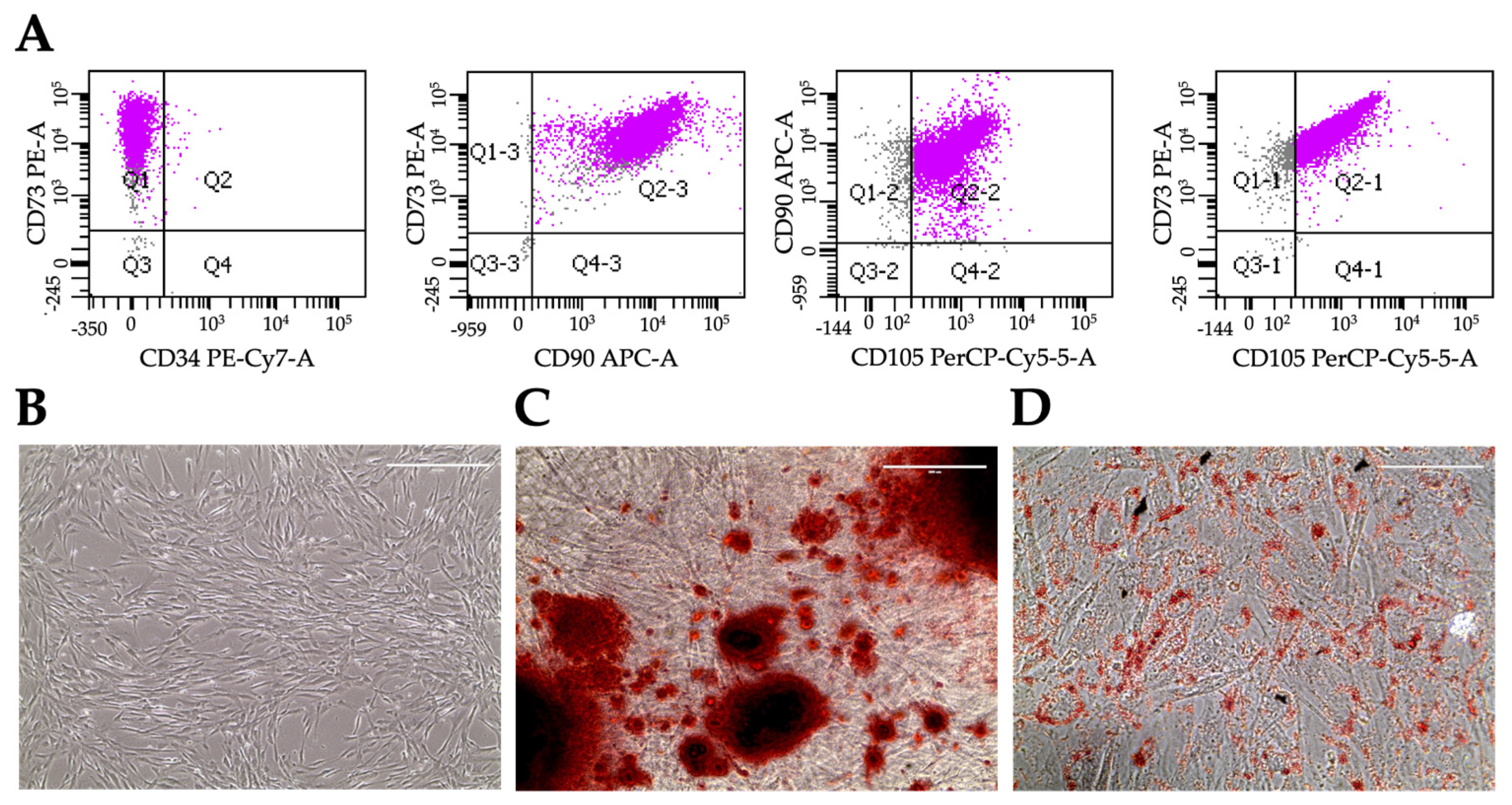
Figure 3.
Characterization of human-induced hepatic-like cells. Differentiation protocol (A). Expression of pluripotency gene OCT4 (B). Expression of hepatic lineage marker HNF4 (C). Progressive change in cell morphologies at dd0, dd5, dd9 respectively (D). Flow cytometry (E) and Immunocytochemistry of endodermal markers (F). Hepatic gene expression (G). Hepatic protein secretion (H).
Figure 3.
Characterization of human-induced hepatic-like cells. Differentiation protocol (A). Expression of pluripotency gene OCT4 (B). Expression of hepatic lineage marker HNF4 (C). Progressive change in cell morphologies at dd0, dd5, dd9 respectively (D). Flow cytometry (E) and Immunocytochemistry of endodermal markers (F). Hepatic gene expression (G). Hepatic protein secretion (H).
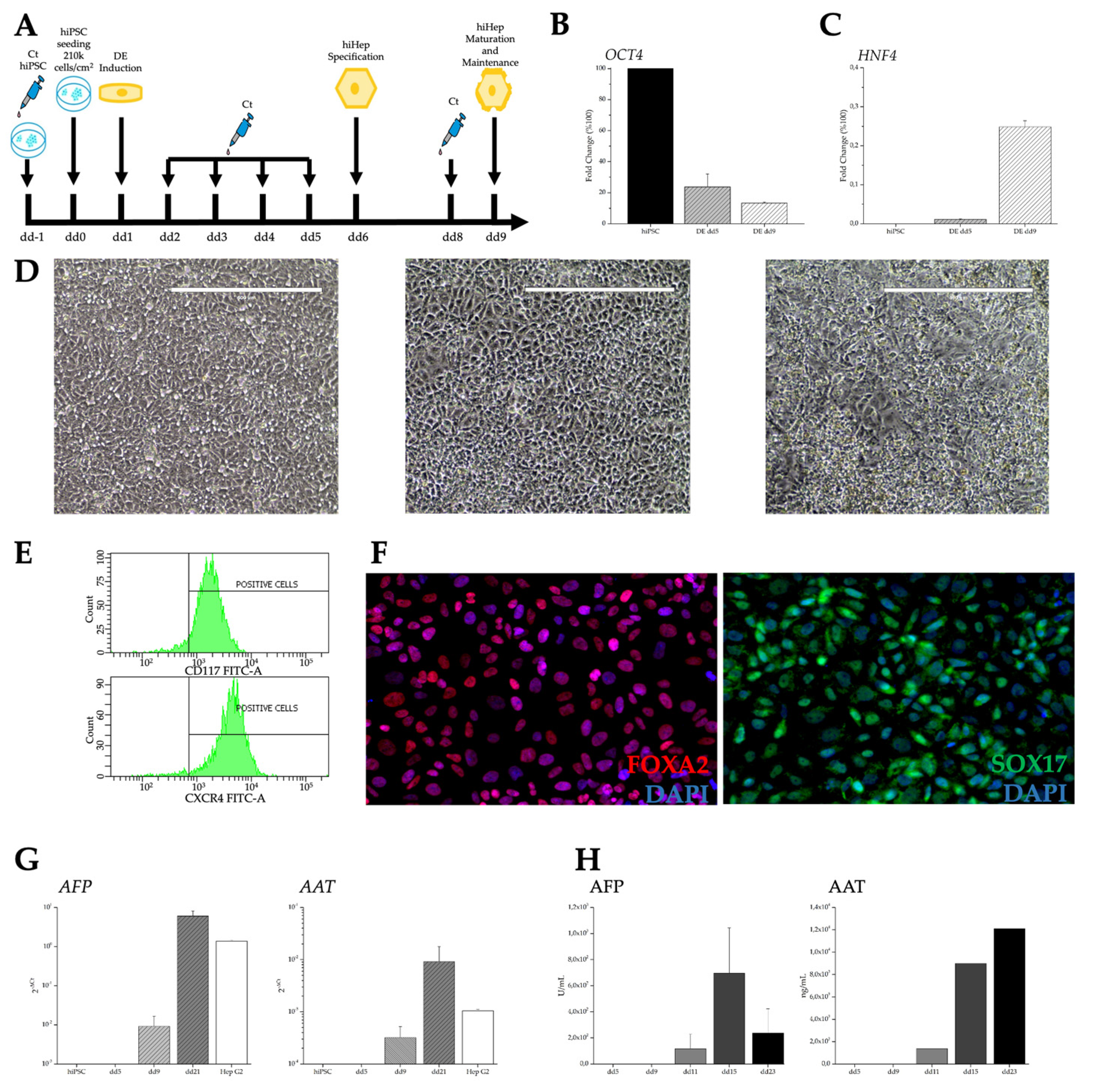
Figure 4.
Aggregate formation. Representative microscopic images overviewing the aggregates’ formation starting from single cell seeding (A), after 48 h (B) and after a 7 days culture at a lower (C) or a higher (D) magnification.
Figure 4.
Aggregate formation. Representative microscopic images overviewing the aggregates’ formation starting from single cell seeding (A), after 48 h (B) and after a 7 days culture at a lower (C) or a higher (D) magnification.
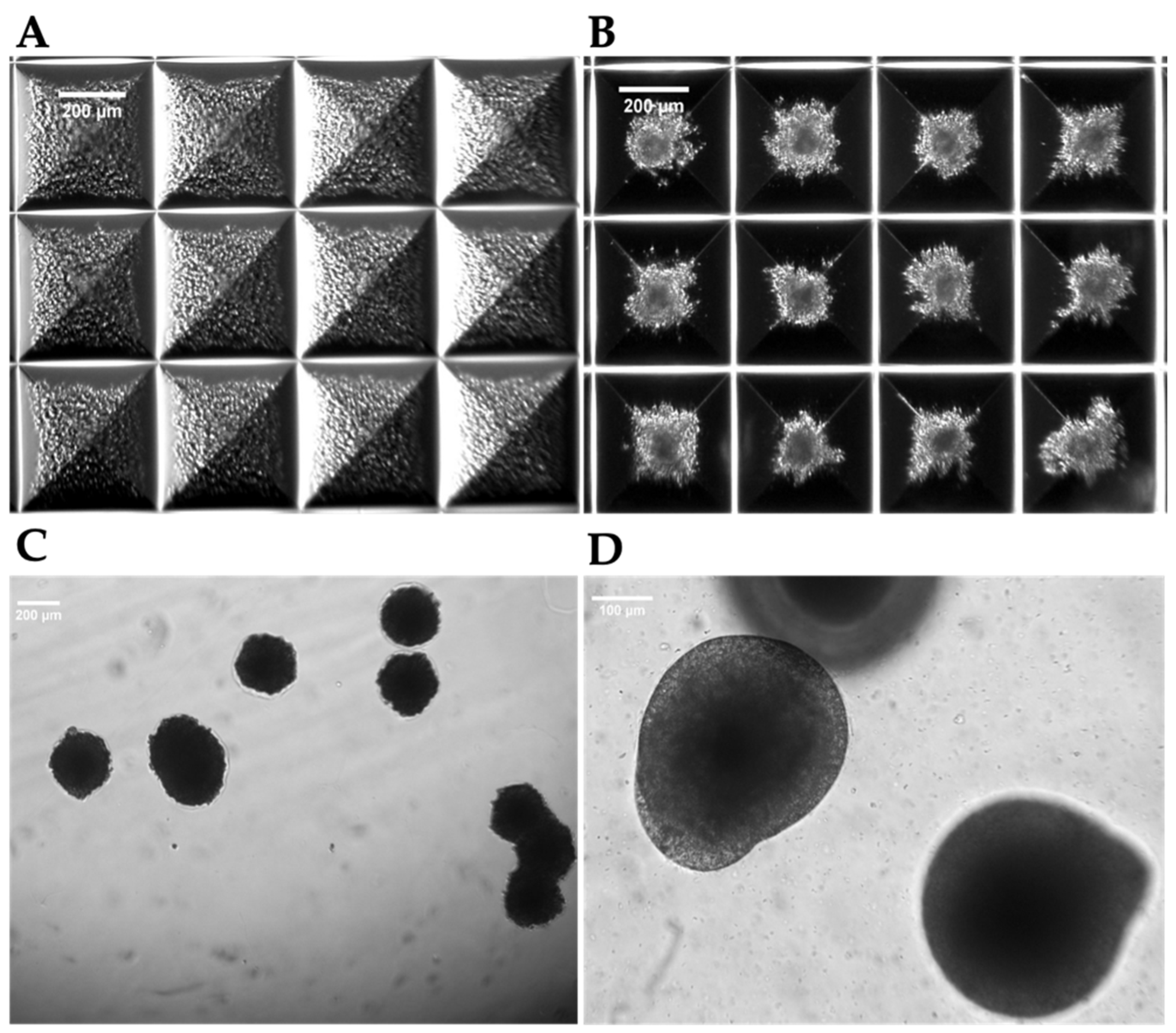
Figure 5.
Organoids in alginate beads. Representative microscopic images of organoids entrapped in 1.5% alginate beads at lower (A) or higher (B) magnification.
Figure 5.
Organoids in alginate beads. Representative microscopic images of organoids entrapped in 1.5% alginate beads at lower (A) or higher (B) magnification.
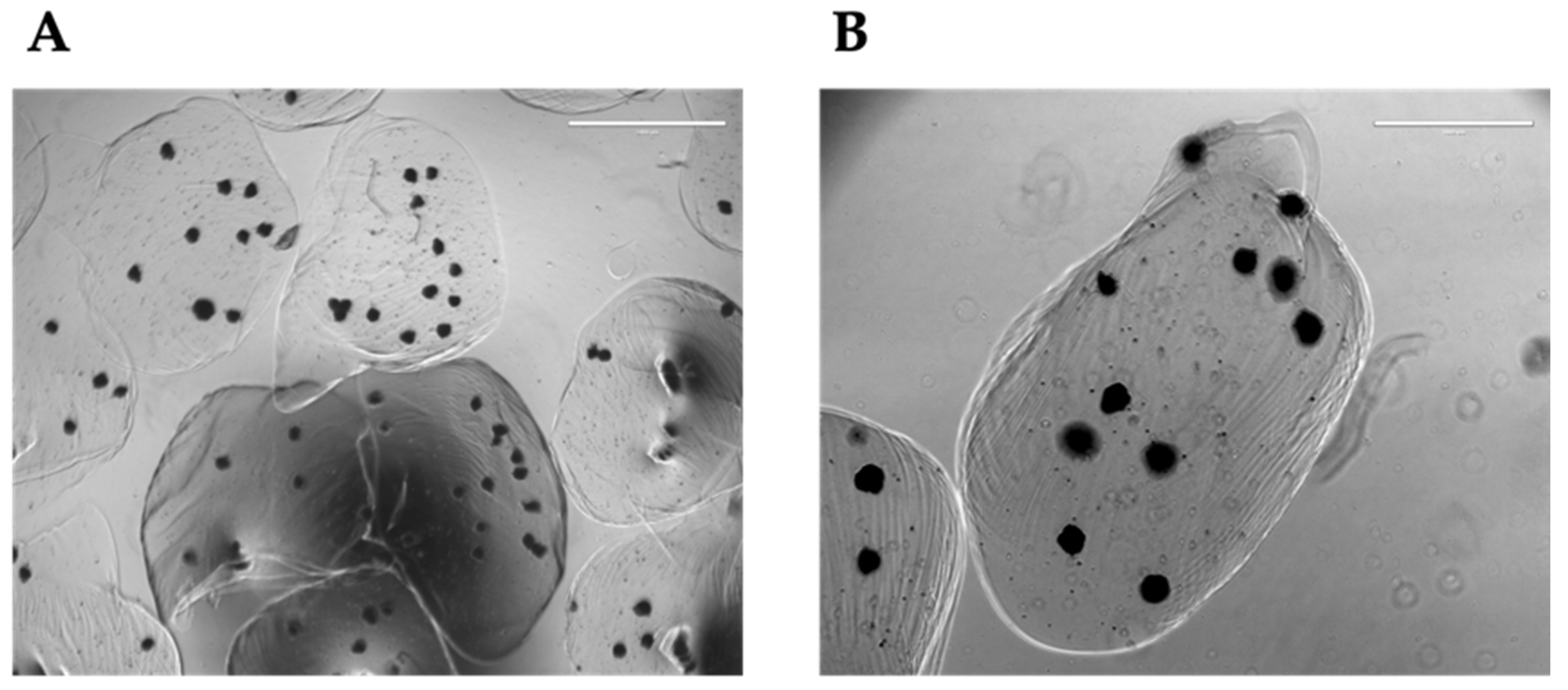
Figure 6.
Organoid characterization. Viability assay showing the amount of cellular ATP in the newly formed aggregates; t0 correspond to 48 hours after seeding and CTRL1, CTRL2, and LB correspond to the end of the different experimental conditions. * p < 0.05 (A). Hepatic Genes Expression in Liver Organoids (B). H&E staining of 3D organoids cultured in dynamic conditions for one week (C). Albumin immunolocalization (D). Immunolocalization of the three cell types using antibodies to CD31 (red), CD90 (brown) and AFP (blue) (E).
Figure 6.
Organoid characterization. Viability assay showing the amount of cellular ATP in the newly formed aggregates; t0 correspond to 48 hours after seeding and CTRL1, CTRL2, and LB correspond to the end of the different experimental conditions. * p < 0.05 (A). Hepatic Genes Expression in Liver Organoids (B). H&E staining of 3D organoids cultured in dynamic conditions for one week (C). Albumin immunolocalization (D). Immunolocalization of the three cell types using antibodies to CD31 (red), CD90 (brown) and AFP (blue) (E).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



