Submitted:
26 June 2023
Posted:
26 June 2023
You are already at the latest version
Abstract
Rabies, caused by the rabies virus (RABV), remains a significant public health issue in the Philippines despite efforts to control it. To eliminate rabies by 2030, effective surveillance strategies are crucial. In this study, we examined RABV evolution and phylodynamics in the Davao Region using genome sequences from Davao City and nearby provinces. We adapted the RABV ARTIC Protocol for Oxford Nanopore High-Throughput Sequencing to optimize workflow efficiency under limited resources. Comparing new virus samples collected from June 2019 to June 2021 (n=38) with baseline samples from June 2018 to May 2019 (n=49), new sub-clades were observed in the phylogenetic tree, suggesting divergence from older variants that were previously undetected. Most of the new viruses belonged to the Asian SEA4_A1.1.1 lineage, but new (SEA4_B1 and SEA4_B1.1) and emerging (SEA4_B1.1_E1) lineages that have never been reported in the Philippines were also identified. The baseline study reported phylogeographic clustering of RABV isolates from the same areas. However, this pattern was disrupted in the current biosurveillance, with variants detected in areas outside the original cluster. Furthermore, our findings revealed significant transmission routes between Davao City and neighboring provinces, contrasting with the predominantly intra-city transmission observed in the baseline study. These results underscore the need for ongoing and timely genomic surveillance to monitor genetic diversity changes and the emergence of novel strains, as well as to track alterations in transmission pathways. Implementing cost-effective next generation sequencing workflows will facilitate the integration of genomic surveillance into rabies control programs, particularly in resource-limited settings. Collaborations between different sectors can empower local laboratories and experts in genomic technologies and analysis.
Keywords:
canine rabies virus
; whole genome sequencing
; nanopore technology
; biosurveillance
; Philippines
1. Introduction
Rabies is a life-threatening acute encephalomyelitis caused by the rabies virus (RABV), a neurotropic Lyssavirus that is primarily transmitted to humans through the bite of domestic dogs [1]. Rabies is an ancient disease, but still remains as one of the most neglected diseases, particularly in developing countries. It causes an estimated 60,000 human rabies cases deaths each year; 50% of deaths are in children under the age of 15, mostly in Asia and Africa [1,2]. The World Health Organization is supporting a campaign to eliminate canine-mediated rabies in humans by 2030 [1,3].
The Philippines is a major rabies endemic country in Southeast Asia, with approximately 200 to 300 human cases of dog-mediated rabies annually. To prevent and control human rabies, Republic Act No. 9482, or the Anti-Rabies Act of 2007, was enacted by the Department of Health in 2007 [4]. The Anti-Rabies Act mandates the local government units to implement the mass vaccination of dogs, control interventions such as impounding and neutering, field control and disposition of unregistered, stray, and unvaccinated dogs, and the conduct of rabies prevention and control information and education campaigns [6]. In Davao City - one of the largest cities in the country with approximately 163,000 dogs- mass vaccination of the canine population was enforced in 2006, while control interventions such as impounding and neutering of free-roaming dogs have been implemented since 2011 [6]. Despite the enhanced implementation of these control measures, the incidence of human rabies in the city has not significantly decreased in recent years [6].
A major challenge in the eradication of the rabies virus is the absence of reliable surveillance data for countries, such as the Philippines where the disease is prevalent. Surveillance is essential for constructing informed infectious disease management and control programs [8]. Genomic data is emerging as an effective surveillance tool through an understanding of pathogen populations, confirming suspected cases, discriminating between strains, identifying infectious agents, and elucidating the evolutionary history and transmission patterns of the virus [9]. Due to breakthroughs in high-throughput sequencing technologies over the last two decades, generating microbial genomes is no longer considered difficult or expensive. Thus, whole-genome sequencing (WGS) has been envisioned as the obvious and inevitable future of diagnostics [10].
In 2014, Oxford Nanopore Technologies (ONT) introduced a portable and affordable sequencing device called the MinION [11,12]. MinION has been used to sequence a wide range of microorganisms from human and other animal clinical samples, which include bacteria, viruses, and fungi [13]. Given its small, agile physical presence and long-read capabilities, the MinION has been used in a number of significant microbiological studies, including the detection of Ebola, Zika virus, and vancomycin resistant Enterococci [14]. A sequencing workflow for the genomic surveillance of the rabies virus was developed by Brunker et al. [11] which provided genomic data that revealed significant and distinctive insights into rabies spread in Kenya, Tanzania, and Philippines.
Bacus et al. [15] generated baseline genomic data on rabies cases in Davao City, Philippines and its neighboring localities between June 2018 to May 2019 using traditional whole genome sequencing via the Sanger method, highlighting the local dynamics of canine rabies. To further monitor the genomic evolution and phylodynamics of rabies virus in these areas, genomic surveillance was carried out from June 2019 to June 2021 utilizing the Oxford Nanopore MinION sequencing platform. The genetic diversity and evolutionary placement of the new viruses in reference to the baseline data and newly identified transmission routes provide deeper insights on the longer term impact of rabies interventions in the region, enabling the local government to evaluate and improve on existing strategies.
2. Materials and Methods
2.1. Sample Collection and Processing
A total of 81 dog brain tissue samples that tested positive for rabies virus in a Fluorescent Antibody Test (FAT) were obtained from the City Veterinarian’s Office (CVO) of Davao City, Philippines from June 2019 to June 2021. Epidemiological data such as location, gender, vaccination status, and the type of ownership of the dogs were recorded. All samples were stored at -80°C until used. Ethics clearance was obtained from the Institutional Animal Care and Use Committee (IACUC) at Davao Medical School Foundation Inc. (DMSF) prior to sample processing. Brain tissue (100-120mg) was homogenized in a Bead Mill 24 Homogenizer (FisherBrand). The collected supernatant was then used for viral RNA extraction using the Biospin Viral DNA/RNA Extraction Kit (Bioflux) following the manufacturer's protocol. The supernatant from the lysed samples and the RNA extracts were kept at -80°C until used. RNA extracts were subjected to RT-PCR of the RABV N gene to confirm the identity and the integrity of the RNA extracts [15]. Out of the 81 samples, only 66 were confirmed positive for RABV via RT-PCR.
2.2. RABV Genome Enrichment
Viral RNA of the RT-PCR positive samples was reverse transcribed to its complementary DNA (cDNA) using 5X LunaScript RT SuperMix (NEB). This was followed by viral DNA amplification via multiplex PCR using pooled primers [11] and Q5 High Fidelity Hot-Start DNA Polymerase (NEB). Multiplex PCR primers were prepared based on the RABV Southeast Asia (rabvSEasia) primer scheme, which was produced from Philippine reference sequences: KX148260, KX148263, N-gene 99% consensus sequence of [AB116581, AB116582, AB683592:AB683635], AB981664, KX148255, JN786878, KX148250, KX148254, EU293111, KX148248, and KX148266 [11]. The following PCR cycling conditions were initially applied: heat inactivation at 98°C for 30s, 30-40 cycles of denaturation at 98°C for 15s, and a combined extension and annealing step at 65°C for 5 minutes. For the next batches of samples, touchdown PCR was performed, wherein the temperature during the combined extension and annealing step was decreased from 65°C by 0.1°C every cycle for the first 25 cycles and then kept at 62.5°C. PCR products were purified using the NEBNext Sample Purification Bead (NEB), and concentrations were measured using a Qubit dsDNA High Sensitivity assay kit on a Qubit 3.0 fluorometer (ThermoFisher). A negative control included at the cDNA synthesis stage was used to check for contamination. If the concentration of the negative control was above 1ng/ul, sample preparation was repeated from RNA extraction.
2.3. Library Preparation
Library preparation for the MinION was conducted using Ligation Sequencing 1D and Native Barcoding kits (Oxford Nanopore Technologies) on FLO-MIN106 flow cells according to the manufacturer’s instructions. RABV DNA was first end-repaired and dA-tailed using an end-prep master mix followed by the ligation of barcodes from the ONT Native Barcoding kit. Samples were pooled into a single tube and purified with SPRI beads (NEB). Adapter ligation of the barcoded amplicon pool was performed using the NEBNext ARTIC Companion Kit according to the manufacturer’s instructions, and the sample was cleaned up using SPRI beads (NEB). The acceptable amount of genetic material in the final library for all runs was between 5-50 fmol [11] as measured by a Qubit dsDNA High Sensitivity assay kit (Thermofisher) on a Qubit 3.0 fluorometer (Thermofisher).
2.4. Nanopore Sequencing
Sequencing was carried out on a DELL Inspiron 3511 computer with a 1920 x 1080 Intel Core i7-1165G processor, 8GB DDR4 3200 MHz, and 512GB SSD storage using MinKnow version 20.10.6 with live basecalling turned off. The ligation sequencing and native barcoding kits used during library preparation were specified in the MinKnow interface. Run length varied between 16 to 18 hours depending on the amount of sequence data generated over time. All other settings were set to the default value. After sequencing, all liquid in the flow cell was removed from the waste channel through the waste port. About 400ul of wash mix containing wash mix and wash diluent (ONT EXP-WSH004) was loaded into the priming port. The mixture was then removed after 30 minutes. After that, storage buffer was slowly added through the priming port to allow the storage of the flow cell for subsequent library additions.
2.5. Bioinformatics Data Processing and Analysis
Consensus sequences were generated from the raw reads using the RABV version of the ARTIC network open-source bioinformatics pipeline [16]. The resulting sequences were then aligned with the Southeast Asia reference sequence (KX148260) using MAFFT version 7 [17]. The identity and coding region coverage of the sequences were determined using the online RABV-GLUE genotyping tool (http://rabv-glue.cvr.gla.ac.uk), while the whole genome coverage for each sample was manually calculated by dividing the total number of non-N bases by the total length of the sequence x 100%. SAMtools version 1.16.1 was then used to calculate the mean read depth and breadth of coverage of the sequences using the BAM files generated from the RABV ARTIC pipeline [18]. Sequences for phylogenetic analysis were selected based on the following criteria: whole genome coverage (where depth at each position >20X) is greater than 70% [19], and the breadth of coverage (where depth at each position >1X) obtained via SAMtools is greater than 90% [11]. Bammix version 1.0.0 was used to search for nucleotide mixtures among samples suspected to be contaminated during the sequencing run using filters on base quality (q>7), mapping quality (m>40), depth of coverage (>20X), base proportion (>25%), and read position (30-11620bp) [20].
Phylogenetic inference was performed using the discrete continuous-time Markov chain model (CTMC) via BEAST version 1.10.4. A total of 38 new viruses and 49 samples from the baseline study (Accession No.: OQ326736 to OQ326784) were included in the analysis. Genomic data of reference sequences from the Philippines (KM148260), Brazil (KM594034, KM594033, KM594041, and KM594028) and China (JN234411) was used as the outgroup in constructing the phylogenetic tree. All sequences were first aligned using MUSCLE in MEGA version 11 [21], where the resulting multiple sequence alignment was trimmed and manually cleaned by removing all gaps. The best fit model with the least Akaike Information Criterion (AIC) was also identified using MEGA. An executable xml file was prepared using BEAUTi version 1.10.4 with the GTR + G + I DNA substitution and site heterogeneity model, a length of 200 million, uncorrelated relaxed molecular clock, and coalescent constant size model. Ancestral state reconstruction was enabled using an asymmetric trait substitution model. The rest of the tree priors were set to the default value. The resulting log files from BEAST were then assessed using Tracer version 1.7.2 [22], ensuring that the estimated sampling size (ESS) values for most of the continuous parameters are sufficient (>200). TreeAnnotator version 1.10.4 was then used to construct a maximum clade credibility (MCC) tree, where the first 20 million states were specified as burn-in and the posterior probability limit was set to 0.0. FigTree version 1.4.4 was used to visualize the resulting phylogenetic tree.
NextStrain was also used to generate a TimeTree phylogeny of the sampled sequences [23], while RABV lineage assignment was performed using the tool Method for Assignment, Definition and Designation of Global Lineages (MADDOG) [24]. Phylogeographic analysis was conducted using the Bayesian stochastic search variable selection (BSSVS) approach in BEAST software with the following criteria: Strong support (30 > BF > 10), Very Strong support (100 > BF > 30), and Decisive (BF > 100). The results were then visualized through SPREAD3 version 0.9.6 [25].
3. Results
3.1. Sample Characteristics
A total of 81 reported samples that tested positive for rabies virus in a Fluorescent Antibody Test (FAT) conducted by the Davao CVO were processed in this study. Out of 81, 30 samples were obtained from June to December 2019, 35 in the year 2020, and 16 from January to June 2021 (Figure 1A). The Davao Region is located in Southern Mindanao of the Philippines and is composed of one major city, Davao City, and five provinces Davao de Oro, Davao del Norte, Davao del Sur, Davao Oriental and Davao Occidental. The samples were collected from reported cases of rabies in various locations of Davao City (76.54%) and the neighboring provinces of Davao del Sur (4.94%), Davao del Norte (14.81%), and Davao de Oro (3.70%) (Figure 1B–D).
Out of the 81 FAT-positive brain samples, 66 (81.5%) were confirmed by RABV N RT-PCR and subjected to RABV whole genome enrichment via multiplex PCR. Modifications of the RABV MinION sequencing protocol established by Brunker et al. [11] were adopted in this study (Table 1). The RNA extraction kit and native barcoding kits were changed to accommodate available resources. Other modifications were introduced to improve the sequencing workflow, which was first tested out in a trial run. For the multiplex PCR, nucleic acid concentrations of the initial batch of purified amplicons were too low to be detected. Touchdown PCR was subsequently employed to increase primer specificity, in which the reaction temperature was decreased from 65°C by 0.1°C until it reached 62.5°C, resulting in purified amplicons with concentrations ranging from 1.39 to 108 ng/ul (data not shown). The oldest samples from 2019 (n=9) were excluded from library preparation due to undetectable nucleic acid concentrations post PCR cleanup.
About 5ng of input DNA was initially used for end-prep and barcode ligation reactions during the trial run of the library preparation (Table 1). The barcoded samples were then pooled together, totaling to 30ng of genetic material for adapter ligation. Prior to adapter ligation, the pooled amplicons were purified, resulting in a concentration of only 0.7 ng/ul (data not shown). In subsequent runs, the input DNA was increased from 5ng to 50ng. To minimize DNA loss during cleanup, Short Fragment Buffer (SFB) and 80% ethanol were used for the bead washings. Precise incubation temperatures were also applied by using a thermocycler instead of a heat block. With these modifications, the nucleic acid concentration of the barcoded amplicon pool in four batches was recorded to be 0.84 to 3.78 ng/ul (Table 2). A total of 57 samples (86.4% of sample input) were subjected to nanopore sequencing.
During sequencing, the run was extended to 16 hours or more with the basecalling mode off instead of six hours with the basecalling mode on to improve the read number (Table 1). To maximize resources, flow cells were recycled for a total of two uses. In batches sequenced consecutively using the same flow cell, traces of contamination from the previous run were observed despite the flow cell wash step. For example, thousands of reads were recorded from the negative control of the second batch, enough to create a consensus sequence with >70% coverage despite having a nucleic acid concentration <1 ng/ul prior to pooling during library preparation. The negative control in question shares the same native barcode with a RABV-positive sample from the previous run. To investigate possible contamination in this batch, the binary alignment map (BAM) files of the samples ran in the reused flow cell were further examined for nucleotide mixtures using bammix. Samples (n=11) that overlapped barcodes with the previous batch were flagged to have possible nucleotide mixtures in multiple positions (data not shown). Thus, these sequences were discarded and not used in downstream analysis. It is important to note, however, that nucleotide mixtures were also observed in clean samples albeit only a few hundred reads. Given that unique barcodes and flow cell were used when running them, clean sequences were still included in the analysis.
3.2. RABV Whole Genome Sequences
Consensus sequences were generated for 46 rabies viruses (80.7% of sequenced samples). One sample was not confirmed to be RABV through RABV-GLUE due to very low sequence read depth, while the remaining 45 samples were identified to belong to the Asian SEA4 clade (Table 3). However, only 38 out of the 45 viruses had acceptable whole genome coverage and breadth of coverage based on the set threshold values. A few samples (n=7) that dated back to 2019 had very low coverage and were not used for downstream phylogenetic and phylodynamic analysis (Table 3).
Lineage designation for the sampled viruses, including those from the baseline study in 2018-2019 [15], was based on the global reference set in the MADDOG database (Table A1). The nomenclature system by Rambaut et al. [26] was applied. Majority of the sequences from the Davao Region were assigned to the lineage Asian SEA4_A1.1.1 (69.4% of the old sequences and 63.2% of the new sequences), which emerged from the SEA4_A1.1 parent lineage (Figure 2). Some of the remaining sequences were suspected to belong to two new lineages descending from the A1.1.1 lineage: SEA4_B1 (30.6% of the old sequences and 7.9% of the new sequences) and SEA4_B1.1 (26.3% of the new sequences), while one (2.6%) of the new sequences was assigned to the SEA4_A1.1.2 lineage. Note that the A1.1.2 and B1.1 lineages were identified only among the new samples. Interestingly, one potentially emerging but currently undersampled lineage (Asian SEA4_B1.1_E1) was also identified by MADDOG among the new viruses.
3.3. Genetic Diversity and Phylogeographic Distribution of RABV
Phylogenetic analysis was performed to assess the genetic relationships of the rabies viral strains circulating within the different districts in Davao City and its neighboring provinces in Davao Region. Samples from the baseline study in 2018-2019 [15] were included to monitor changes in virus clades and transmission. A phylogenetic tree was produced through Bayesian inference, mapping a total of 87 reported samples from June 2018 to June 2021 (Figure 3). Three major clades were observed, with each composed of samples from both baseline (highlighted in red) and new sequence data (highlighted in green). In all three clades, the newly sampled viruses from 2019-2021 formed a divergent sub-clade with respect to the baseline sequences from 2018-2019 with ancestral placement in the phylogenetic tree. The most recent common ancestor for each clade dated back to 2014, 2013, and 2010 for Clade 1, 2, and 3, respectively. Bayesian posterior probabilities (≥ 0.95) are indicated on the branch labels.
To visualize the geographic distribution of the samples among the provinces in Davao Region, a time-resolved phylogenetic tree was inferred. Similar to the Bayesian inferred phylogeny, three major clades were evident. Clade 1 was composed of 40 samples from Davao City: Toril (n=6), Tugbok (n=8), Talomo (n=20), Calinan (n=1), Baguio (n=1), Buhangin (n=2), Sasa (n=1), and Poblacion (n=1); four samples from Davao del Norte: Tagum (n=1), Panabo (n=1), and Samal (n=2); three samples from Davao de Oro: New Bataan (n=1) and Compostela Valley (n=2); and one sample from Digos, Davao del Sur (Figure 4). The inferred date of divergence of this clade from its parent clade was 2014-09-30, indicating that the most recent common ancestor for these samples dated back to 2014. The time resolution of the samples was consistent with the sampling dates.
Majority of the samples mapped in Clade 2 were from the various districts of Davao City: Buhangin (n=8), Poblacion (n=4), Tugbok (n=7), Talomo (n=8), and Calinan (n=1) (Figure 5). Newly sampled viruses from neighboring provinces such as Sulop (n=1) of Davao del Sur, Samal (n=3) of Davao del Norte, and New Bataan (n=1) of Davao de Oro were also found in this clade. The most recent common ancestor for all samples in Clade 2 dated back to 2014, with the time resolution of the samples consistent with the sampling dates.
Clade 3 on the other hand was mainly composed of samples from Davao del Sur in the southern part of Davao Region (n=4), although samples from the northern Davao del Norte (Samal, n=1) and outside Davao Region (Cotabato City, n=1) were also mapped (Figure 6). The inferred date of divergence of Davao Region samples from the Cotabato strain was 2010-05-25, indicating that the most common ancestor for these samples dated back to 2010. The time resolution of the samples was also consistent with the sampling dates.
3.4. Phylodynamics of RABV in Davao Region
The ancestral state of the phylogeny from the BEAST analysis was reconstructed to shed light on the transmission routes of rabies in Davao Region. A total of 16 routes were identified with strong to decisive Bayes Factor support (BF > 10) as shown in Table 4 and Figure 7. Out of the 16 routes, two were recorded as intra-city transmission within Davao City, nine inter-city transmissions between Davao City and neighboring provinces, one intra-province transmission within a Davao Region province, and four inter-province transmissions. The reconstructed transmission routes are specific only for the RABV isolates captured in this study and from Bacus et al. [15] and do not cover other potentially circulating variants during the study period.
4. Discussion
4.1. Emerging Trends on RABV Diversity and Transmission Patterns
We obtained the whole genome sequence of 38 RABV isolates from Davao City, Philippines collected between June 2019 and June 2021 using NGS by Oxford Nanopore Technology. Integrating our new sequences to the baseline data of 49 RABV isolates in Davao City collected in 2018 to 2019 [15] showed that the more recent viruses collected between 2019 to 2021 diverged from the 2018 to 2019 older variants, suggesting emergence of new sub-clades. This hypothesis is supported by the lineage analysis via MADDOG, where 28.9% of the 2019-2021 sequences belonged to new sub-lineages dubbed as SEA4_A1.1.2 and SEA4_B1.1; these were previously not detected among isolates in the baseline study for Davao Region. Instead, the isolates in the baseline study were all composed of the SEA4 B1 lineage. The A1.1.2 lineage was first detected in Luzon in 2013 [11], while the B1 and B1.1 lineages from the Davao Region detected in 2018-2021 have never been reported elsewhere in the country. Note that the A1.1.2 and B1.1 lineages were detected among the new but not the baseline samples, suggesting they emerged in Davao Region sometime after 2019. This finding is consistent with our TreeTime phylogenetic data. However, it is also possible that these variants were just not captured in the baseline study, hence the prior unavailability of sequences in these lineages.
We also report an emerging but currently undersampled lineage, SEA4_B1.1_E1. New lineages are assigned only when at least ten samples are identified under it within a five-year period [24]. Only five samples were associated with the B1.1_E1 lineage, all of which were isolated in Davao Region, Philippines between 2019-2021. Hence, continuous monitoring is recommended. To our knowledge, we are the first to report the B1.1_E1 lineage in the country, although it is difficult to say at this point whether this is emerging only in Davao Region or more broadly in the Philippines.
Mapping our data with the baseline sequences also revealed that the recent samples formed new sub-clades which diverged from much older ancestral strains, instead of branching out from the posterior lineages. This implies the possible existence of at least one undetected, unidentified, older variant circulating around Davao City. New and previously undetected variants could have advanced characteristics on transmissibility, virulence, and vaccine resistance, among others. Thus, the epidemiological fitness of these new sub-clades and whether they would outcompete the other clades in Davao Region will be of great interest.
Aside from the emergence of new lineages and clades, notable changes in transmission pathways were also observed. In the baseline surveillance from 2018 to 2019, RABV variants clustered according to geographic locations [15]. Our genomic surveillance in the subsequent years from 2019-2021 revealed that variants previously associated with a geographic area have been introduced to other locations. For example, Clade 1 was mainly composed of samples from Davao City and neighboring areas in the northern parts of Davao Region [15]. However, our analysis showed that these variants have now reached Digos City in Davao del Sur. Clade 2 on the other hand was initially exclusively found in Davao City, but is now populated with new viruses from other provinces, such as from Davao del Norte, Davao de Oro, and Davao del Sur.
These observations were validated by the phylogeographic spread analysis where transmission across districts and provinces were evident. Compared to the predominantly inter-city transmission in the baseline study [15], most of the movements in the current study were between Davao City and its neighboring provinces, suggesting heightened long-distance transmissions. Davao City is a major urban hub located at the center of Davao Region in Southern Mindanao [27]. The interconnecting factors between human socioeconomic and population density might determine the transmission dynamics of RABV [28]. Thus, frequent rural-to-urban viral transmission within Davao Region is highly possible. However, the noted changes may also be due to a widened range of sample submission from areas outside Davao City (approximately 25% of total sample collection in the current study as opposed to 16% in the baseline study of Bacus et al, 2021).
A similar scenario was observed in our analysis for the Island Garden City of Samal in Davao del Norte. All three clades contained samples from Samal, suggesting that various areas of Davao Region are involved in the transmission events for this island. Although bodies of water can be a strong barrier of rabies transmission [29], Samal island is a popular tourist destination in Southern Mindanao and is only a few kilometers away from the mainland. The high level of human movement from several parts of Davao Region to Samal island therefore makes the island highly susceptible to human-mediated inter-island transmission. Human-mediated inter-island transmission has been previously reported in other regions in the Philippines [29] and in other countries such as in Thailand [28] and North Africa [30].
The long-distance spread of RABV is likely due to human-mediated displacement of dogs infected with rabies, a significant driver in repeated and successful reintroduction of RABV in rabies-free areas [30]. Multiple studies have reported that frequent reintroductions counteract local rabies elimination after vaccination campaigns, allowing rabies to continually spread in endemic areas like the Philippines [31,32,33]. Long-distance transmission has also been observed among recent RABV variants in Ghana, which belonged to both Africa 1a and Africa 1b sub-lineages, contrary to earlier reports that a single lineage (Africa 2) circulates in West Africa [34]. Africa 1a has been previously known to dominate in northern and eastern Africa, while Africa 1b has been detected in eastern, central, and southern Africa, suggesting probable repeated introduction of RABV from other parts of Africa into Ghana [30,35]. With the long-distance and inter-island transmission events observed for Davao City and Samal, local government units (LGU) should explore interventions that will strictly regulate such movements to limit transmission and prevent introduction or re-introduction of the virus. For example, Samal was about to be declared as rabies free in 2011 (personal communication, Davao City Veterinary Office), but our current findings suggest that weak border control may have forestalled this.
Our analysis also showed that the Talomo District in Davao City had the highest number of reported cases, making it the city's RABV hotspot. This supports findings from the baseline study [15], in which this district has been identified as both donor and recipient in the RABV transmission chain within Davao City. Population structure is a driving force for rabies maintenance, particularly in urban areas where human and dog population density are high [31]. As a result, the emergence and recurrence of rabies cases in large urban areas are caused by an increasing human and dog population [36,37].
4.2. Genomic Surveillance in a Low Resource Setting
Our new findings on formerly undetected and emerging lineages and new transmission pathways highlight the importance of continued rabies monitoring through genomic surveillance. Unlike traditional clinical reporting, genomic surveillance can provide deeper insights on virus characteristics and behavior that could be crucial in guiding rabies endgame [9]. Expanding surveillance including samples outside the target locality could capture long distance transmission pathways that would answer key questions about viral circulation, potential host shifts, and provide insights in persisting lineages [38].
In many third-world countries where rabies remains to be endemic, funding opportunities for next-generation sequencing are limited [39]. Hence, available resources have to be maximized and an efficient workflow for genomic surveillance is very important. Relative to the laborious and time-consuming conventional sequencing which was utilized in the baseline study [15], the MinION workflow provided a speedy yet affordable platform for WGS. In our study, 86% of genome enriched samples made it to sequencing. Whole genome data was successfully generated for 67% of these samples after MinION workflow optimization, where output was improved by increasing input DNA, utilizing alternative techniques such as touchdown PCR, and optimizing clean-up steps.
Use of reagents other than the prescribed kits in the sequencing workflow developed by Brunker et al. [11] may have led to variable efficiency. This could explain why only 81% of the reported RABV-positive samples tested positive in our targeted RT-PCR confirmatory step after viral RNA extraction. The age of the samples and storage conditions have to be considered as well. In total, 15 samples repeatedly tested negative during targeted RT-PCR, most of which (73%) were shelved for more than two years prior to the conduct of the study. Among those that were sequenced, the whole genome coverage for older samples (14%) was recorded to be significantly lower than the threshold value of 90% [11]. This suggests that the quality of the tissue samples and the viral RNA itself might have been compromised during storage. Loss of genetic material during long periods of storage or during transport could be reduced by keeping samples in an appropriate preservation buffer [40]. Alternatively, sequencing samples immediately after collection might increase percent yield.
Reusing flow cells should be done cautiously. We found that the ONT storage buffer can clog up waste ports when not properly and completely removed from the waste channel as residual buffer can later crystalize inside the flow cell. Moreover, we found that reusing flow cells can result in a low read number (i.e. fewer Fast5 files after a 16-hour runtime) when the remaining pore number after the first run is less than 400. We also note contamination in the reused flow cell, where native barcodes from the preceding run appeared during the demultiplexing step, even when flow cell was incubated with the ONT wash reagents for 30 minutes after the first use. This was supported by our analysis using the bammix tool, which revealed traces of nucleotide mixtures in multiple positions of the samples in question. Surprisingly, nucleotide mixtures were also detected even in clean samples run in unique flow cells (<1% of total genome). This may indicate possible lapses in the reading efficiency and accuracy of the nanopores in the R9.4.1 flow cell or may be due to errors during basecalling given that “fast” mode was selected in this study rather than the “high accuracy” option. It is important to consider, however, that the bammix tool counts possible nucleotide mixtures using BAM files as input. The medaka tool (https://github.com/nanoporetech/medaka) in the RABV ARTIC pipeline was used in this study for generating the consensus sequences from the same BAM files, which guarantees that the true variant is called regardless of any basecalling or nanopore errors, so long as there is enough coverage at each position. Hence, clean samples (i.e. samples processed in unique R9.4.1 flow cells and using unique barcodes) were processed for subsequent phylogenetic analysis given that they pass all criteria set in this study and that the negative control processed along with the samples had concentration <1ng/ul prior to library preparation. We recommend extending incubation time with the DNase-containing wash buffer for an hour or more to fully eliminate contaminating samples. We also suggest using different sets of barcodes for consecutive runs in the same flow cell, if possible, to avoid barcode overlapping.
The introduction of efficient and less expensive NGS workflows will be a strong impetus for inclusion of genomic surveillance in rabies control programs, especially in low resource settings. This comes with the need to also empower local laboratories that will be capable of implementing the technology with expertise on genomic analysis. This is achievable through the collaboration of academe and public health and veterinary agencies.
5. Conclusions
Our study reports a follow-up on the genomic surveillance of rabies virus in the southern Philippines. Here we shifted from conventional sequencing to next-generation sequencing using the Oxford Nanopore High-Throughput Sequencing Technology, which allowed us to efficiently and economically process samples in bulk and in a shorter period of time. The modifications introduced in our sequencing workflow validated the utility of the technology in low-resource settings and may be adopted in other laboratories. Our findings revealed emerging trends on the genetic diversity and transmission patterns of RABV in Davao Region, highlighting the value of consistent monitoring through genomic surveillance. The inclusion of genomic surveillance in rabies control programs may be facilitated by efficient and less expensive next generation sequencing workflows, especially in low resource settings. This would necessitate capacity building of local laboratories and assistance from genomic experts, which can be realized through inter-sectoral collaborations.
Author Contributions
Conceptualization, L.A.E.M; methodology, L.A.E.M. and J.B.G.C; software, J.B.G.C and A.J.C.S.; validation, L.A.E.M, Y.Y; formal analysis, J.B.G.C and A.J.C.S; investigation, J.B.G.C and A.J.C.S; resources, L.A.E.M, M.N.J.E, A.L., and M.C.B.S; data curation, J.B.G.C; writing—original draft preparation, J.B.G.C and A.J.C.S; writing—review and editing, L.A.E.M, J.B.G.C, and A.J.C.S.; visualization, J.B.G.C. and A.J.C.S; supervision, L.A.E.M; project administration, L.A.E.M and M.C.B.S; funding acquisition, L.A.E.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Department of Science and Technology- Philippine Council for Health Research and Development through the project entitled “RabCast: Proof-of-Concept for a Forecasting Tool for Rabies Spread in Davao City Through Combined Genomic and Mathematical Approaches“, which involves the University of the Philippines – Mindanao and the Davao City Veterinary Office.
Institutional Review Board Statement
The animal study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Davao Medical School Foundation, Inc.
Data Availability Statement
Data is contained within the article or supplementary material. All other data that support the findings of this study are available from the corresponding authors upon reasonable request.
Acknowledgments
The authors express their gratitude and recognition to Dr. Kirstyn Brunker and Kathryn Campbell from the University of Glasgow for their valuable assistance during investigation and sequence data analysis.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A

Table A1.
MADDOG Reference Sequence.
| Sample ID | Country | Year | Lineage |
|---|---|---|---|
| AB573762 | Japan | 2006 | Asian SEA4 |
| AB573763 | Japan | 2006 | Asian SEA4 |
| EU086201 | Philippines | 1994 | Asian SEA4 |
| EU086202 | Philippines | 1994 | Asian SEA4 |
| EU086203 | Philippines | 2000 | Asian SEA4 |
| EU086204 | Philippines | 2001 | Asian SEA4 |
| KF620487 | Taiwan | 2012 | Asian SEA5 |
| KF620488 | Taiwan | 2012 | Asian SEA5 |
| KF620489 | Taiwan | 2013 | Asian SEA5 |
| KP860149 | Taiwan | 2013 | Asian SEA5 |
| KP860153 | Taiwan | 2013 | Asian SEA5 |
| KX148259 | Philippines | 1994 | Asian SEA4 |
| KX148260 | Philippines | 2004 | Asian SEA4 |
| KX148261 | Philippines | 1994 | Asian SEA4 |
| KX148262 | Philippines | 1994 | Asian SEA4 |
| KX148263 | Philippines | 1994 | Asian SEA4 |
| LC571945 | Japan | 2006 | Asian SEA4 |
| LC571946 | Japan | 2020 | Asian SEA4 |
| MN726836 | Philippines | 2013 | Asian SEA4 |
| MN726837 | Philippines | 2013 | Asian SEA4 |
| MN726838 | Philippines | 2013 | Asian SEA4 |
| MN726839 | Philippines | 2015 | Asian SEA4 |
| MN726840 | Philippines | 2015 | Asian SEA4 |
| MN726841 | Philippines | 2014 | Asian SEA4 |
| MN726843 | Philippines | 2012 | Asian SEA4 |
| MN726846 | Philippines | 2015 | Asian SEA4 |
| MN726847 | Philippines | 2017 | Asian SEA4 |
| MN726848 | Philippines | 2016 | Asian SEA4 |
| MN726851 | Philippines | 2014 | Asian SEA4 |
| MN726853 | Philippines | 2012 | Asian SEA4 |
| MN726854 | Philippines | 2015 | Asian SEA4 |
| MN726855 | Philippines | 2016 | Asian SEA4 |
| MN726856 | Philippines | 2012 | Asian SEA4 |
| MN726858 | Philippines | 2016 | Asian SEA4 |
| MN726861 | Philippines | 2015 | Asian SEA4 |
| MN726862 | Philippines | 2013 | Asian SEA4 |
| MN726865 | Philippines | 2014 | Asian SEA4 |
| MN726866 | Philippines | 2014 | Asian SEA4 |
| MN726867 | Philippines | 2012 | Asian SEA4 |
| MN726868 | Philippines | 2016 | Asian SEA4 |
| MN726869 | Philippines | 2013 | Asian SEA4 |
| MN726870 | Philippines | 2014 | Asian SEA4 |
| MN726871 | Philippines | 2013 | Asian SEA4 |
| MN726872 | Philippines | 2017 | Asian SEA4 |
| MN726873 | Philippines | 2014 | Asian SEA4 |
| MN726874 | Philippines | 2012 | Asian SEA4 |
| MN726879 | Philippines | 2012 | Asian SEA4 |
| MN726881 | Philippines | 2015 | Asian SEA4 |
| MN726882 | Philippines | 2019 | Asian SEA4 |
| MN857169 | Philippines | 2019 | Asian SEA4 |
Table A2.
Lineage designation of new and baseline samples.
| Sample ID | New/Baseline | Length | Year | Lineage |
|---|---|---|---|---|
| 21-002_Sulop_d2021-01 | New | 10370 | 2021 | Asian SEA4_A1.1.2 |
| 21-008_Tugbok_d2021-01 | New | 10551 | 2021 | Asian SEA4_A1.1.1 |
| 21-010_Tugbok_d2021-02 | New | 11331 | 2021 | Asian SEA4_B1.1 |
| 20-047_Buhangin_d2020-07 | New | 9965 | 2020 | Asian SEA4_A1.1.1 |
| 20-053_Samal_d2020-08 | New | 9481 | 2020 | Asian SEA4_A1.1.1 |
| 20-054_Talomo_d2020-08 | New | 10851 | 2020 | Asian SEA4_A1.1.1 |
| 20-055_Talomo_d2020-08 | New | 11035 | 2020 | Asian SEA4_A1.1.1 |
| 20-056_Talomo_d2020-08 | New | 11195 | 2020 | Asian SEA4_A1.1.1 |
| 20-057_Tugbok_d2020-08 | New | 10219 | 2020 | Asian SEA4_A1.1.1 |
| 20-059_Tugbok_d2020-09 | New | 11039 | 2020 | Asian SEA4_A1.1.1 |
| 20-060_Buhangin_d2020-09 | New | 10347 | 2020 | Asian SEA4_A1.1.1 |
| 20-066_Samal_d2020-10 | New | 10737 | 2020 | Asian SEA4_A1.1.1 |
| 20-067_Samal_d2020-10 | New | 11626 | 2020 | Asian SEA4_A1.1.1 |
| 20-068_Talomo_d2020-10 | New | 11397 | 2020 | Asian SEA4_A1.1.1 |
| 20-069_Samal_d2020-10 | New | 11013 | 2020 | Asian SEA4_B1 |
| 20-082_NewBataan_d2020-12 | New | 10550 | 2020 | Asian SEA4_A1.1.1 |
| 20-083_Sasa_d2020-12 | New | 10473 | 2020 | Asian SEA4_B1.1 |
| 21-027_Digos_d2021-04 | New | 11221 | 2021 | Asian SEA4_A1.1.1 |
| 21-029_Talomo_d2021-04 | New | 11422 | 2021 | Asian SEA4_A1.1.1 |
| 21-033_Talomo_d2021-05 | New | 11619 | 2021 | Asian SEA4_B1.1 |
| 21-035_Tugbok_d2021-05 | New | 10958 | 2021 | Asian SEA4_A1.1.1 |
| 21-037_Digos_d2021-05 | New | 11570 | 2021 | Asian SEA4_A1.1.1 |
| 21-042_Buhangin_d2021-06 | New | 11587 | 2021 | Asian SEA4_B1.1 |
| 18-092_Talomo_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-094_Talomo_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-096_Buhangin_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-099_Talomo_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-101_Talomo_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-102_Tugbok_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-103_Talomo_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-106_Talomo_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-107_ComVal_d2018-06 | New | 11800 | 2018 | Asian SEA4_B1 |
| 18-114_Talomo_d2018-06 | New | 11800 | 2018 | Asian SEA4_B1 |
| 18-116_Tugbok_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-118_DigosCogon_d2018-06 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-120_Tugbok_d2018-07 | New | 11800 | 2018 | Asian SEA4_B1 |
| 18-121_Tugbok_d2018-07 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-130_Poblacion_d2018-07 | New | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-133_Tagum_d2018-07 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-135_Talomo_d2018-07 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-138_Talomo_d2018-07 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-142_Nabunturan_d2018-07 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-144_Buhangin_d2018-07 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-147_Buhangin_d2018-08 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-151_Buhangin_d2018-08 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-153_Tugbok_d2018-08 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-162_Samal_d2018-08 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-163_Toril_d2018-08 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-164_Digos_d2018-08 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-172_Talomo_d2018-09 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-174_Toril_d2018-09 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-179_Cotabato_d2018-09 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-181_Talomo_d2018-09 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-183_Tugbok_d2018-09 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-188_Talomo_d2018-10 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-191_Talomo_d2018-10 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-194_Calinan_d2018-10 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-196_Talomo_d2018-10 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-199_Tugbok_d2018-10 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-209_Toril_d2018-11 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-214_Talomo_d2018-11 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-216_Poblacion_d2018-11 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-221_Toril_d2018-11 | Baseline | 11800 | 2018 | Asian SEA4_B1 |
| 18-223_Buhangin_d2018-12 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-224_Poblacion_d2018-12 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 18-225_Talomo_d2018-12 | Baseline | 11800 | 2018 | Asian SEA4_A1.1.1 |
| 19-004_Talomo_d2019-01 | Baseline | 11800 | 2019 | Asian SEA4_A1.1.1 |
| 19-011_Talomo_d2019-01 | Baseline | 11800 | 2019 | Asian SEA4_B1 |
| 19-015_Talomo_d2019-02 | Baseline | 11800 | 2019 | Asian SEA4_B1 |
| 19-022_Buhangin_d2019-02 | Baseline | 11800 | 2019 | Asian SEA4_A1.1.1 |
| 19-045_Poblacion_d2019-04 | Baseline | 11800 | 2019 | Asian SEA4_A1.1.1 |
| 19-050_Panabo_d2019-04 | Baseline | 11800 | 2019 | Asian SEA4_B1 |
| 19-102_Samal_d2019-09 | Baseline | 10122 | 2019 | Asian SEA4_A1.1.1 |
| 19-131_Calinan_d2019-11 | Baseline | 9804 | 2019 | Asian SEA4_B1 |
| 19-136_Buhangin_d2019-12 | Baseline | 10697 | 2019 | Asian SEA4_B1.1 |
| 20-001_Toril_d2020-01 | Baseline | 10531 | 2020 | Asian SEA4_A1.1.1 |
| 20-004_Tugbok_d2020-01 | Baseline | 10958 | 2020 | Asian SEA4_A1.1.1 |
| 20-007_Toril_d2020-01 | Baseline | 11284 | 2020 | Asian SEA4_A1.1.1 |
| 20-011_Talomo_d2020-02 | Baseline | 9553 | 2020 | Asian SEA4_A1.1.1 |
| 20-014_Talomo_d2020-02 | Baseline | 10535 | 2020 | Asian SEA4_A1.1.1 |
| 20-015_Talomo_d2020-02 | Baseline | 11378 | 2020 | Asian SEA4_B1.1 |
| 21-014_Poblacion_d2021-02 | Baseline | 11547 | 2021 | Asian SEA4_B1.1 |
| 21-015_Tugbok_d2021-02 | Baseline | 11182 | 2021 | Asian SEA4_B1.1 |
| 21-019_Tugbok_d2021-02 | Baseline | 11404 | 2021 | Asian SEA4_B1.1 |
| 21-021_NewBataan_d2021-02 | Baseline | 11411 | 2021 | Asian SEA4_B1 |
| 21-025_Digos_d2021-04 | Baseline | 11243 | 2021 | Asian SEA4_B1.1 |
| 21-026_Baguio_d2021-04 | Baseline | 11163 | 2021 | Asian SEA4_A1.1.1 |
References
- World Health Organization. Zero by 30: the global strategic plan to end human deaths from dog-mediated rabies by 2030: United Against Rabies Collaboration: first annual progress report: Global strategic plan to end human deaths from dog-mediated rabies by 2030. No. WHO/CDS/NTD/NZD/2019.04. World Health Organization, 2019.
- Tarantola, Arnaud. "Four thousand years of concepts relating to rabies in animals and humans, its prevention and its cure." Tropical medicine and infectious disease 2, no. 2 (2017): 5.
- de Melo, Guilherme Dias, Florian Sonthonnax, Gabriel Lepousez, Grégory Jouvion, Andrea Minola, Fabrizia Zatta, Florence Larrous et al. "A combination of two human monoclonal antibodies cures symptomatic rabies." EMBO molecular medicine 12, no. 11 (2020): e12628. [CrossRef]
- Cantaert, Tineke, Laurence Borand, Lauriane Kergoat, Chanthy Leng, Sivlin Ung, Sotheary In, Yiksing Peng et al. "A 1-week intradermal dose-sparing regimen for rabies post-exposure prophylaxis (RESIST-2): an observational cohort study." The Lancet Infectious Diseases 19, no. 12 (2019): 1355-1362. [CrossRef]
- Department of Health. "Anti-Rabies Act (RA 9482) and Other Issuances on Rabies Control and Prevention Programs." Anti-Rabies Act (RA 9482) and Other Issuances on Rabies Control and Prevention Programs, (2007). Accessed December 16, 2022. https://rabies.doh.gov.ph/images/PDF/IRR-INSIDE-REV2_new.pdf.
- Department of Health. "National Rabies Prevention and Control Program." Strategic Plan 2020-2025, (2020). Accessed December 16, 2022. https://rr-asia.woah.org/wp-content/uploads/2020/03/final-mtp-rabies_philippines.pdf.
- Lachica, Zython Paul T., Sherelyn A. Evangelio, Eliezer O. Diamante, Abigail J. Clemente, Johanna Marie Peralta, Lyre Anni E. Murao, May Anne E. Mata, and Pedro A. Alviola IV. "Trends of canine rabies lyssavirus and impact of the intensified rabies control program in Davao city, Philippines: 2006–2017." Philipp J Sci 148 (2019): 751-763.
- Klepac, Petra, C. Jessica E. Metcalf, Angela R. McLean, and Katie Hampson. "Towards the endgame and beyond: complexities and challenges for the elimination of infectious diseases." Philosophical Transactions of the Royal Society B: Biological Sciences 368, no. 1623 (2013): 20120137. [CrossRef]
- Cotton, James A., Matthew Berriman, Love Dalén, and Ian Barnes. "Eradication genomics—lessons for parasite control." Science 361, no. 6398 (2018): 130-131.
- Balloux, Francois, Ola Brønstad Brynildsrud, Lucy Van Dorp, Liam P. Shaw, Hongbin Chen, Kathryn A. Harris, Hui Wang, and Vegard Eldholm. "From theory to practice: translating whole-genome sequencing (WGS) into the clinic." Trends in microbiology 26, no. 12 (2018): 1035-1048. [CrossRef]
- Brunker, Kirstyn, Gurdeep Jaswant, S. M. Thumbi, Kennedy Lushasi, Ahmed Lugelo, Anna M. Czupryna, Fred Ade et al. "Rapid in-country sequencing of whole virus genomes to inform rabies elimination programmes." Wellcome Open Research 5 (2020). [CrossRef]
- Suzuki, Yutaka. "Advent of a new sequencing era: long-read and on-site sequencing." Journal of Human Genetics 65, no. 1 (2020): 1-1. [CrossRef]
- Mikheyev, Alexander S., and Mandy MY Tin. "A first look at the Oxford Nanopore MinION sequencer." Molecular ecology resources 14, no. 6 (2014): 1097-1102. [CrossRef]
- Tyler, Andrea D., Laura Mataseje, Chantel J. Urfano, Lisa Schmidt, Kym S. Antonation, Michael R. Mulvey, and Cindi R. Corbett. "Evaluation of Oxford Nanopore’s MinION sequencing device for microbial whole genome sequencing applications." Scientific reports 8, no. 1 (2018): 1-12. [CrossRef]
- Bacus, Michael G., Sheryl Grace C. Buenaventura, Allan Michael C. Mamites, Hannah G. Elizagaque, Christian C. Labrador, Frederick C. Delfin, Ma Noreen J. Eng, Arlene P. Lagare, Gloria N. Marquez, and Lyre Anni E. Murao. "Genome-based local dynamics of canine rabies virus epidemiology, transmission, and evolution in Davao City, Philippines, 2018–2019." Infection, Genetics and Evolution 92 (2021): 104868. [CrossRef]
- Quick, Joshua, Nathan D. Grubaugh, Steven T. Pullan, Ingra M. Claro, Andrew D. Smith, Karthik Gangavarapu, Glenn Oliveira et al. "Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples." Nature protocols 12, no. 6 (2017): 1261-1276. [CrossRef]
- Katoh, Kazutaka, and Hiroyuki Toh. "Parallelization of the MAFFT multiple sequence alignment program." Bioinformatics 26, no. 15 (2010): 1899-1900. [CrossRef]
- Penir, Sarah. 2017. Calculating Mapping Statistics from a SAM/BAM File Using SAMtools and Awk. Retrieved from https://sarahpenir.github.io/bioinformatics/awk/calculating-mapping-stats-from-a-bam-file-using-samtools-and-awk/.
- Morobe, John Mwita, Brigitte Pool, Lina Marie, Dwayne Didon, Arnold W. Lambisia, Timothy Makori, Khadija Said Mohammed et al. "Genomic Epidemiology of SARS-CoV-2 in Seychelles, 2020–2021." Viruses 14, no. 6 (2022): 1318. [CrossRef]
- Ruis, Chris. 2022. Bammix v1.0.0. Retrieved from https://github.com/chrisruis/bammix.
- Kumar, Sudhir, Glen Stecher, Michael Li, Christina Knyaz, and Koichiro Tamura. "MEGA X: molecular evolutionary genetics analysis across computing platforms." Molecular biology and evolution 35, no. 6 (2018): 1547. [CrossRef]
- Rambaut, Andrew, Alexei J. Drummond, Dong Xie, Guy Baele, and Marc A. Suchard. "Posterior summarization in Bayesian phylogenetics using Tracer 1.7." Systematic biology 67, no. 5 (2018): 901-904. [CrossRef]
- Hadfield, James, Colin Megill, Sidney M. Bell, John Huddleston, Barney Potter, Charlton Callender, Pavel Sagulenko, Trevor Bedford, and Richard A. Neher. "Nextstrain: real-time tracking of pathogen evolution." Bioinformatics 34, no. 23 (2018): 4121-4123. [CrossRef]
- Campbell, Kathryn, Robert J. Gifford, Joshua Singer, Verity Hill, Aine O’toole, Andrew Rambaut, Katie Hampson, and Kirstyn Brunker. "Making genomic surveillance deliver: A lineage classification and nomenclature system to inform rabies elimination." PLoS Pathogens 18, no. 5 (2022): e1010023. [CrossRef]
- Bielejec, Filip, Guy Baele, Bram Vrancken, Marc A. Suchard, Andrew Rambaut, and Philippe Lemey. "SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes." Molecular biology and evolution 33, no. 8 (2016): 2167-2169. [CrossRef]
- Rambaut, Andrew, Edward C. Holmes, Áine O’Toole, Verity Hill, John T. McCrone, Christopher Ruis, Louis du Plessis, and Oliver G. Pybus. "A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology." Nature microbiology 5, no. 11 (2020): 1403-1407. [CrossRef]
- Philippine Statistics Authority Gross Regional Domestic Product by Year Published. (2020). https://psa.gov.ph/grdp/ tables.
- Denduangboripant, Jessada, Supaporn Wacharapluesadee, Boonlert Lumlertdacha, Nipada Ruankaew, Wirongrong Hoonsuwan, Apirom Puanghat, and Thiravat Hemachudha. "Transmission dynamics of rabies virus in Thailand: implications for disease control." BMC Infectious Diseases 5, no. 1 (2005): 1-11. [CrossRef]
- Tohma, Kentaro, Mariko Saito, Taro Kamigaki, Laarni T. Tuason, Catalino S. Demetria, Jun Ryan C. Orbina, Daria L. Manalo et al. "Phylogeographic analysis of rabies viruses in the Philippines." Infection, Genetics and Evolution 23 (2014): 86-94. [CrossRef]
- Talbi, Chiraz, Philippe Lemey, Marc A. Suchard, Elbia Abdelatif, Mehdi Elharrak, Nourlil Jalal, Abdellah Faouzi et al. "Phylodynamics and human-mediated dispersal of a zoonotic virus." PLoS pathogens 6, no. 10 (2010): e1001166. [CrossRef]
- Layan, Maylis, Simon Dellicour, Guy Baele, Simon Cauchemez, and Hervé Bourhy. "Mathematical modelling and phylodynamics for the study of dog rabies dynamics and control: A scoping review." PLOS Neglected Tropical Diseases 15, no. 5 (2021): e0009449. [CrossRef]
- Zinsstag, Jakob, Monique Lechenne, Mirjam Laager, Rolande Mindekem, Service Naïssengar, Assandi Oussiguéré, Kebkiba Bidjeh et al. "Vaccination of dogs in an African city interrupts rabies transmission and reduces human exposure." Science translational medicine 9, no. 421 (2017): eaaf6984. [CrossRef]
- Bourhy, Hervé, Emmanuel Nakouné, Matthew Hall, Pierre Nouvellet, Anthony Lepelletier, Chiraz Talbi, Laurence Watier et al. "Revealing the micro-scale signature of endemic zoonotic disease transmission in an African urban setting." PLoS pathogens 12, no. 4 (2016): e1005525. [CrossRef]
- Hayman, David TS, Nicholas Johnson, Daniel L. Horton, Jessica Hedge, Philip R. Wakeley, Ashley C. Banyard, Shoufeng Zhang, Andy Alhassan, and Anthony R. Fooks. "Evolutionary history of rabies in Ghana." PLoS Neglected Tropical Diseases 5, no. 4 (2011): e1001. [CrossRef]
- De Benedictis, P., A. Sow, A. Fusaro, C. Veggiato, C. Talbi, A. Kaboré, W. G. Dundon, Hervé Bourhy, and I. Capua. "Phylogenetic analysis of rabies viruses from Burkina Faso, 2007." Zoonoses and public health 57, no. 7-8 (2010): e42-e46. [CrossRef]
- Salomao, Cristolde, Amílcar Nacima, Lutero Cuamba, Lorna Gujral, Olga Amiel, Cynthia Baltazar, Julie Cliff, and Eduardo Samo Gudo. "Epidemiology, clinical features and risk factors for human rabies and animal bites during an outbreak of rabies in Maputo and Matola cities, Mozambique, 2014: Implications for public health interventions for rabies control." PLOS Neglected Tropical Diseases 11, no. 7 (2017): e0005787. [CrossRef]
- Beran, George W. "Ecology of dogs in the Central Philippines in relation to rabies control efforts." Comparative immunology, microbiology and infectious diseases 5, no. 1-3 (1982): 265-270. [CrossRef]
- Rysava, Kristyna, Tamara Mancero, Eduardo Caldas, Mary Freire de Carvalho, André PB Castro, Veronica Gutiérrez, Daniel T. Haydon et al. "Towards the elimination of dog-mediated rabies: development and application of an evidence-based management tool." BMC infectious diseases 20, no. 1 (2020): 1-14. [CrossRef]
- Gigante, Crystal M., Gowri Yale, Rene Edgar Condori, Niceta Cunha Costa, Nguyen Van Long, Phan Quang Minh, Vo Dinh Chuong et al. "Portable rabies virus sequencing in canine rabies endemic countries using the Oxford Nanopore MinION." Viruses 12, no. 11 (2020): 1255. [CrossRef]
- Natarajan, Aravind, Alvin Han, Soumaya Zlitni, Erin F. Brooks, Summer E. Vance, Marlene Wolfe, Upinder Singh et al. "Standardized preservation, extraction and quantification techniques for detection of fecal SARS-CoV-2 RNA." Nature communications 12, no. 1 (2021): 5753. [CrossRef]
Figure 1.
Distribution of RABV-positive samples in Davao City and neighboring provinces in Davao Region, Philippines. (A) Collection period of samples from June 2019 to June 2021. (B) Municipalities and provinces of the collected samples. (C) Relative geographical location of provinces and corresponding number of samples collected during the study period. (D) Magnified view of Fig 1C for sampling locations within Davao City and Samal Island.
Figure 1.
Distribution of RABV-positive samples in Davao City and neighboring provinces in Davao Region, Philippines. (A) Collection period of samples from June 2019 to June 2021. (B) Municipalities and provinces of the collected samples. (C) Relative geographical location of provinces and corresponding number of samples collected during the study period. (D) Magnified view of Fig 1C for sampling locations within Davao City and Samal Island.
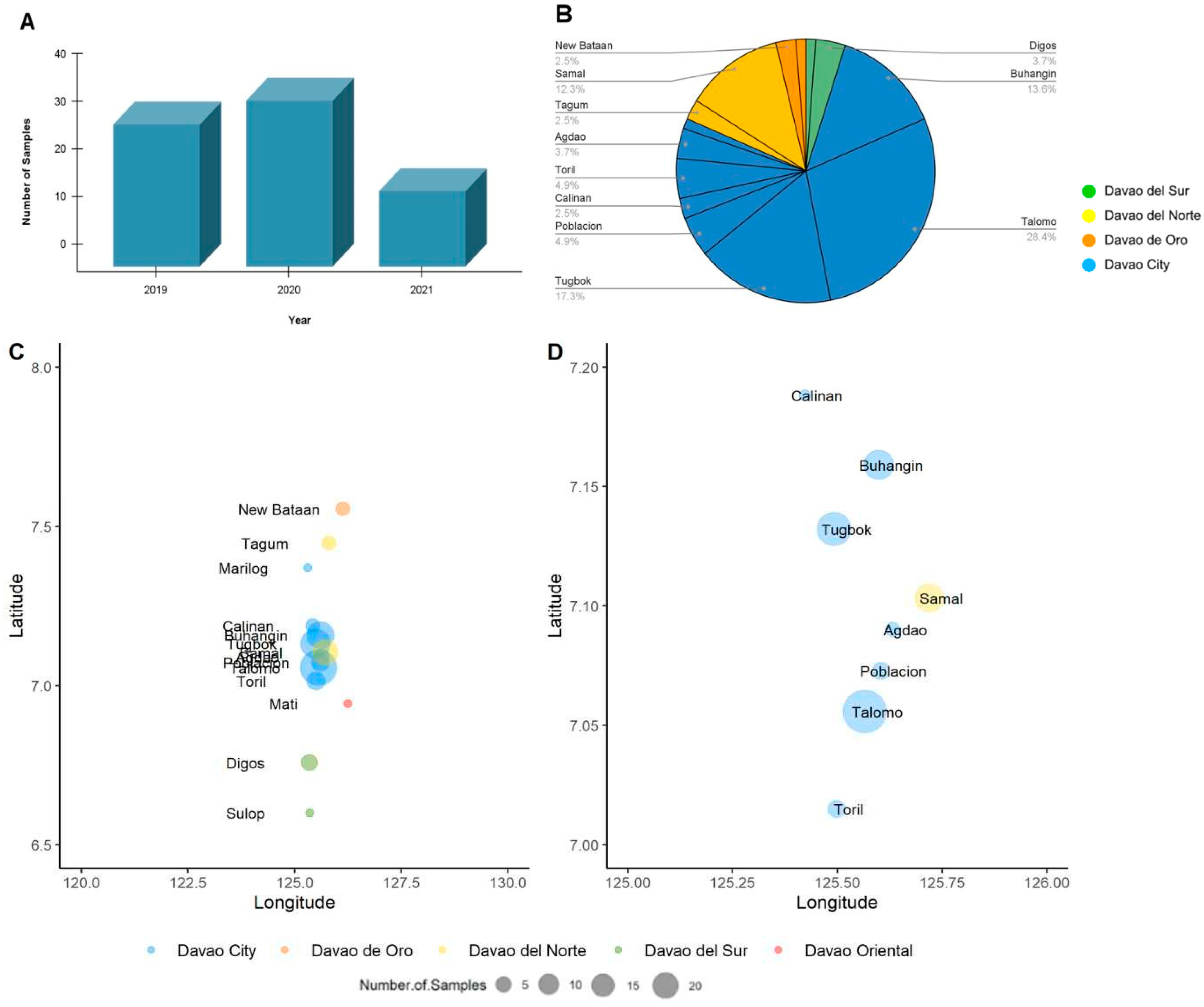
Figure 2.
Lineage Assignments of Rabies Whole Genome Sequences from Davao City, Philippines Against the MADDOG Global Reference Set, 2018-2021. Hierarchical relationships of new and existing lineages are represented in a sunburst plot in the leftmost panel, expanding outward from major (Asian_A1) to minor lineages (Asian SEA4_B1.1). The middle panel shows a phylogenetic tree of the input sequences (n=87) plus all sequences from relevant lineages, with tips colored according to the color scheme from the sunburst plot. Sequences from the baseline study (n=49) are indicated in the rightmost panel in red circles, new sequences from this study (n=38) are in green, while sequences from relevant existing lineages in the MADDOG repository are in gray (Table A2).
Figure 2.
Lineage Assignments of Rabies Whole Genome Sequences from Davao City, Philippines Against the MADDOG Global Reference Set, 2018-2021. Hierarchical relationships of new and existing lineages are represented in a sunburst plot in the leftmost panel, expanding outward from major (Asian_A1) to minor lineages (Asian SEA4_B1.1). The middle panel shows a phylogenetic tree of the input sequences (n=87) plus all sequences from relevant lineages, with tips colored according to the color scheme from the sunburst plot. Sequences from the baseline study (n=49) are indicated in the rightmost panel in red circles, new sequences from this study (n=38) are in green, while sequences from relevant existing lineages in the MADDOG repository are in gray (Table A2).
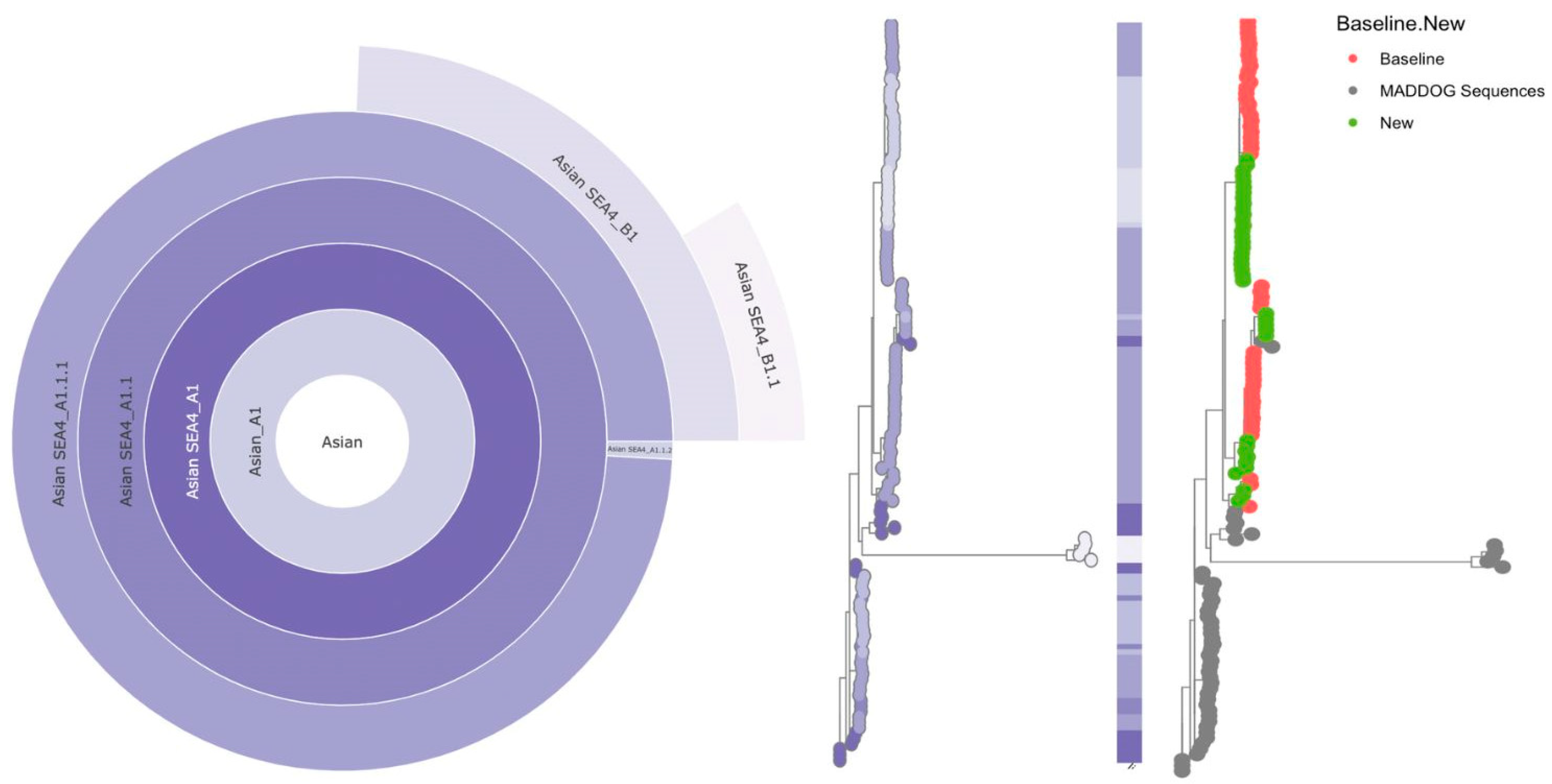
Figure 3.
Bayesian Phylogeny of 87 reported RABV Samples from Davao City and from neighboring provinces in Davao Region, Philippines. Samples are labeled according to the Sample ID_ District/Province_Barangay_Date of sampling. Genomic data of reference sequences from the Philippines (KM148260), Brazil (KM594034, KM594033, KM594041, and KM594028) and China (JN234411) was used as the outgroup in constructing the phylogenetic tree. Major clades are indicated in brackets, while old and new samples are in red and green texts, respectively.
Figure 3.
Bayesian Phylogeny of 87 reported RABV Samples from Davao City and from neighboring provinces in Davao Region, Philippines. Samples are labeled according to the Sample ID_ District/Province_Barangay_Date of sampling. Genomic data of reference sequences from the Philippines (KM148260), Brazil (KM594034, KM594033, KM594041, and KM594028) and China (JN234411) was used as the outgroup in constructing the phylogenetic tree. Major clades are indicated in brackets, while old and new samples are in red and green texts, respectively.
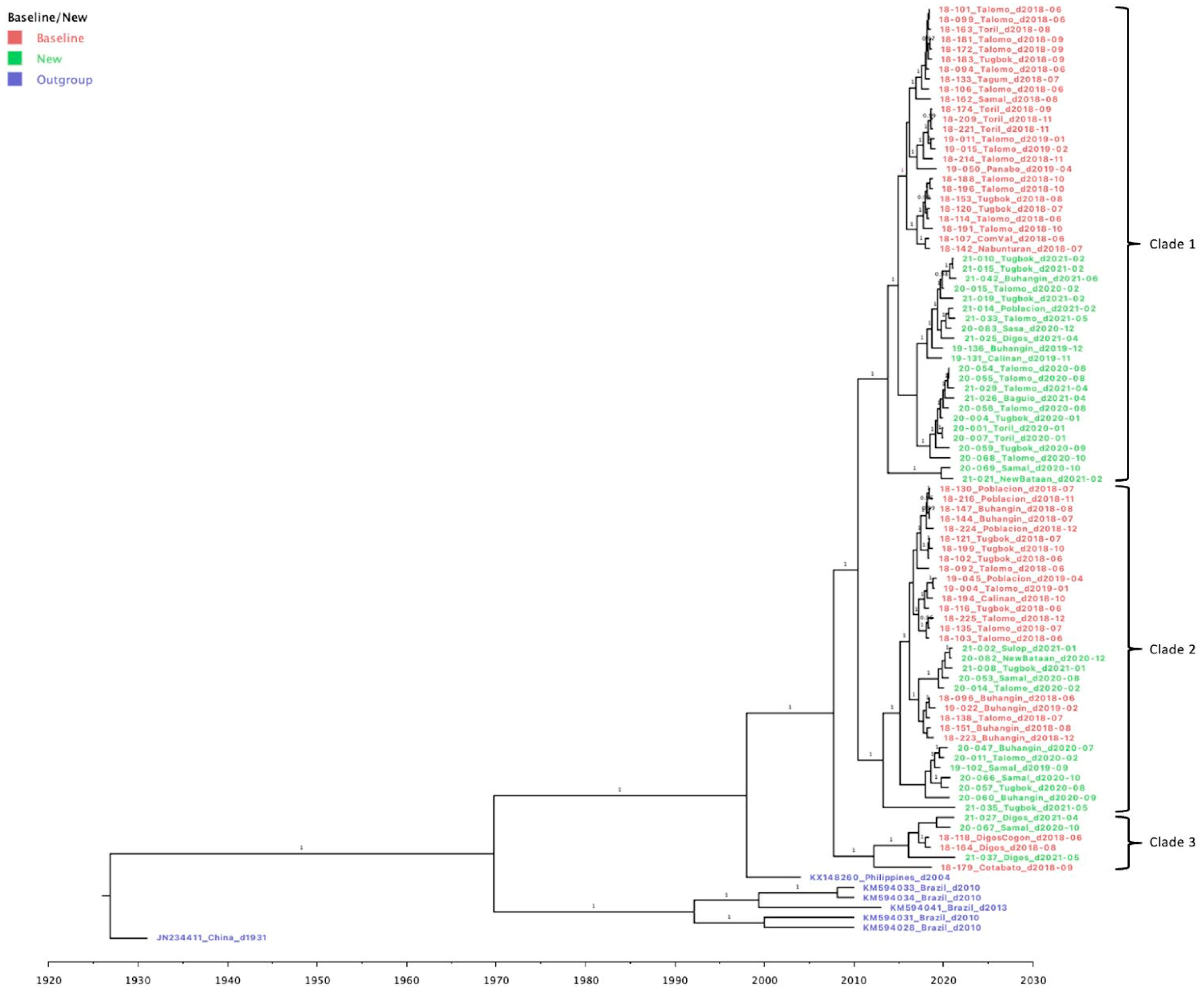
Figure 4.
TreeTime Phylogeny of reported samples from Davao City and neighboring provinces from Davao Region, Philippines (Clade 1). Samples are labeled according to the Sample ID_ District/Province_Barangay_Date of sampling and color-coded according to the city or province.
Figure 4.
TreeTime Phylogeny of reported samples from Davao City and neighboring provinces from Davao Region, Philippines (Clade 1). Samples are labeled according to the Sample ID_ District/Province_Barangay_Date of sampling and color-coded according to the city or province.
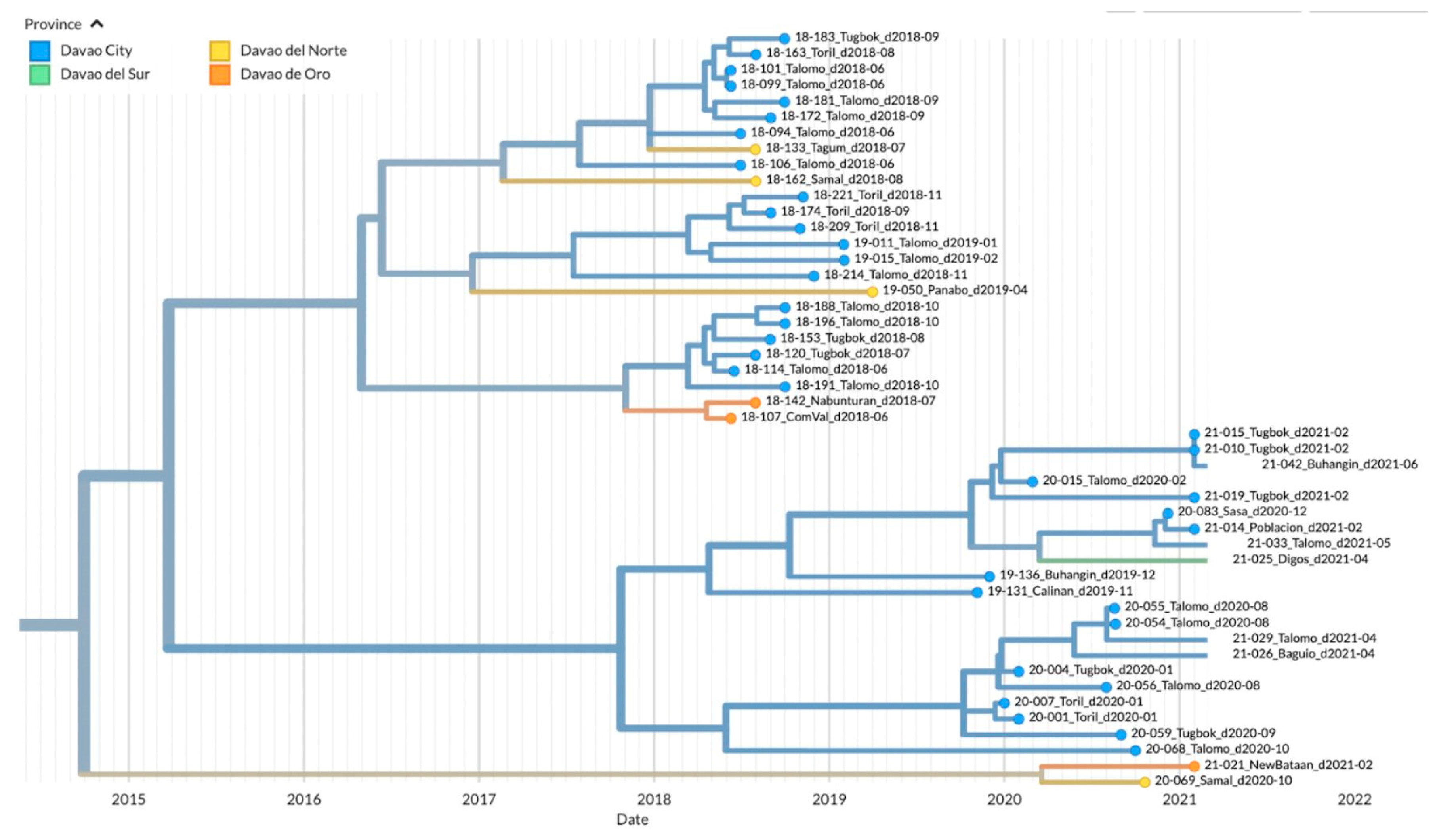
Figure 5.
TreeTime Phylogeny of reported samples from Davao City and neighboring provinces from Davao Region, Philippines (Clade 2). Samples are labeled according to the Sample ID_ District/Province_Barangay_Date of sampling and color-coded according to the city or province.
Figure 5.
TreeTime Phylogeny of reported samples from Davao City and neighboring provinces from Davao Region, Philippines (Clade 2). Samples are labeled according to the Sample ID_ District/Province_Barangay_Date of sampling and color-coded according to the city or province.
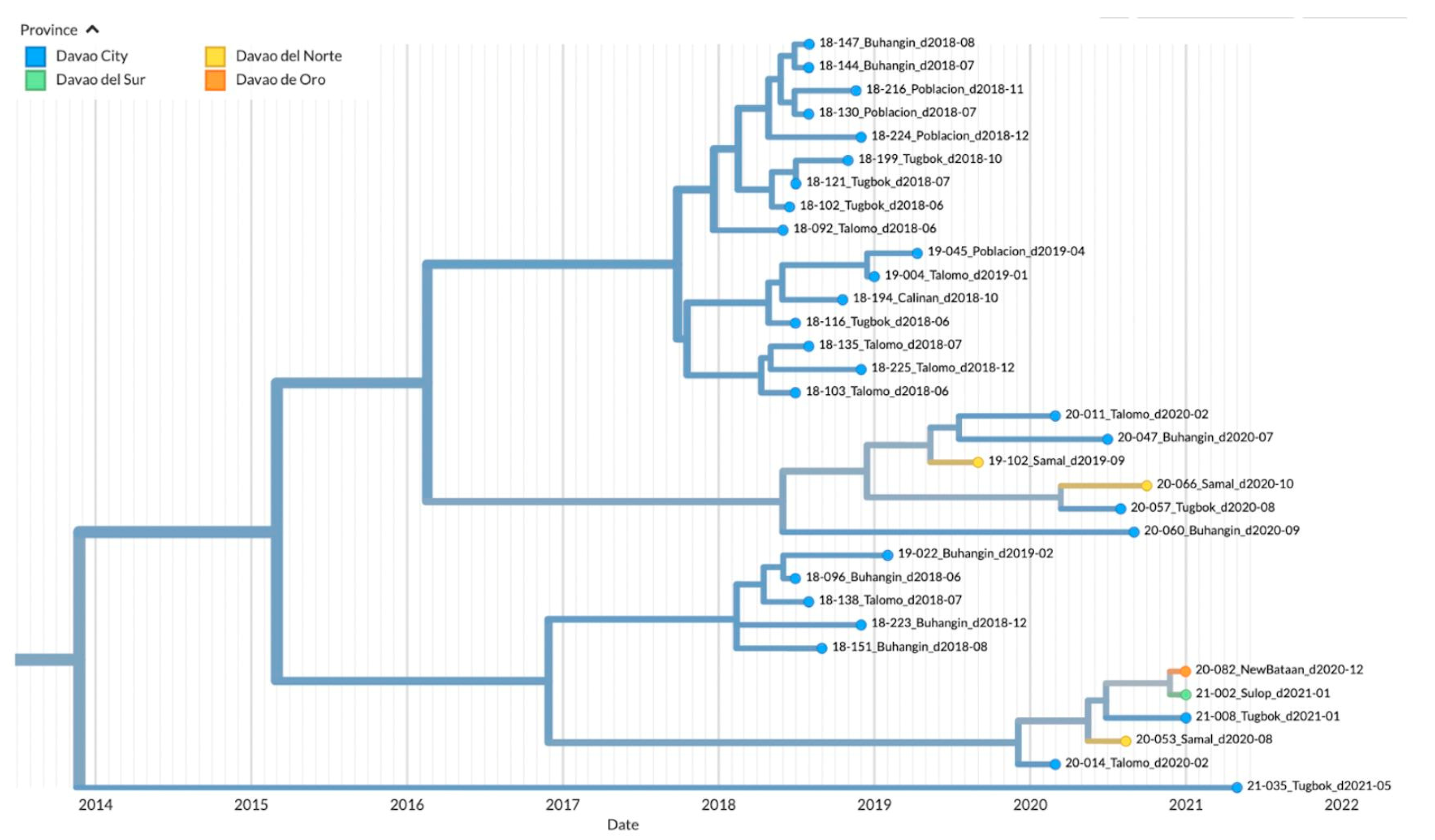
Figure 6.
TreeTime Phylogeny of reported samples from Davao City and neighboring provinces from Davao Region, Philippines (Clade 3). Samples are labeled according to the Sample ID_ District/Province_Barangay_Date of sampling and color-coded according to the city or province.
Figure 6.
TreeTime Phylogeny of reported samples from Davao City and neighboring provinces from Davao Region, Philippines (Clade 3). Samples are labeled according to the Sample ID_ District/Province_Barangay_Date of sampling and color-coded according to the city or province.
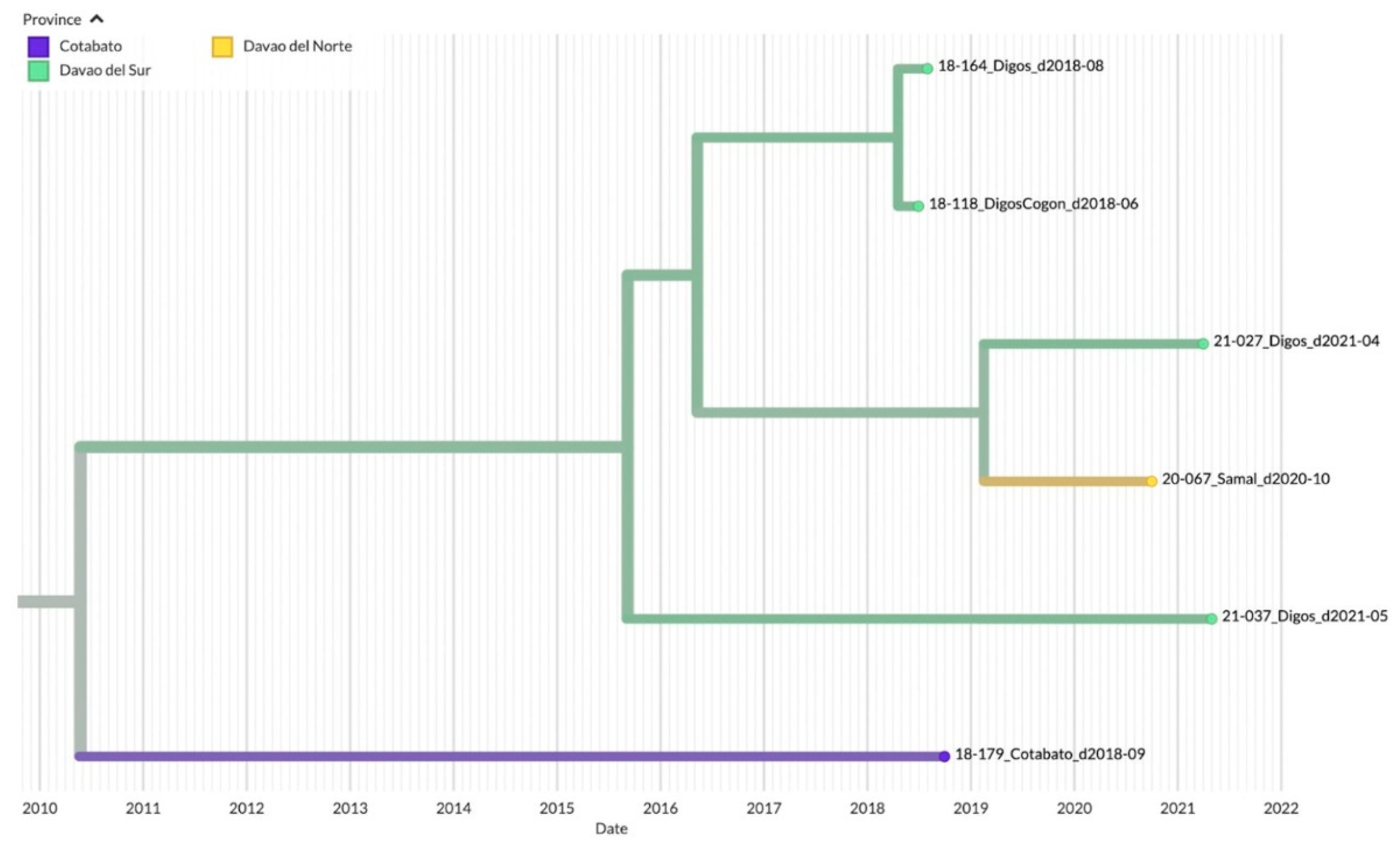
Figure 7.
Transmission routes of reported RABV in Davao Region. Sixteen significant transmission routes with Bayes Factor (BF) support > 10 were identified. Strong support: 30 > BF > 10; Very Strong support: 100 > BF > 30; Decisive: BF > 100. Map Source: QGIS version 3.22.10.
Figure 7.
Transmission routes of reported RABV in Davao Region. Sixteen significant transmission routes with Bayes Factor (BF) support > 10 were identified. Strong support: 30 > BF > 10; Very Strong support: 100 > BF > 30; Decisive: BF > 100. Map Source: QGIS version 3.22.10.
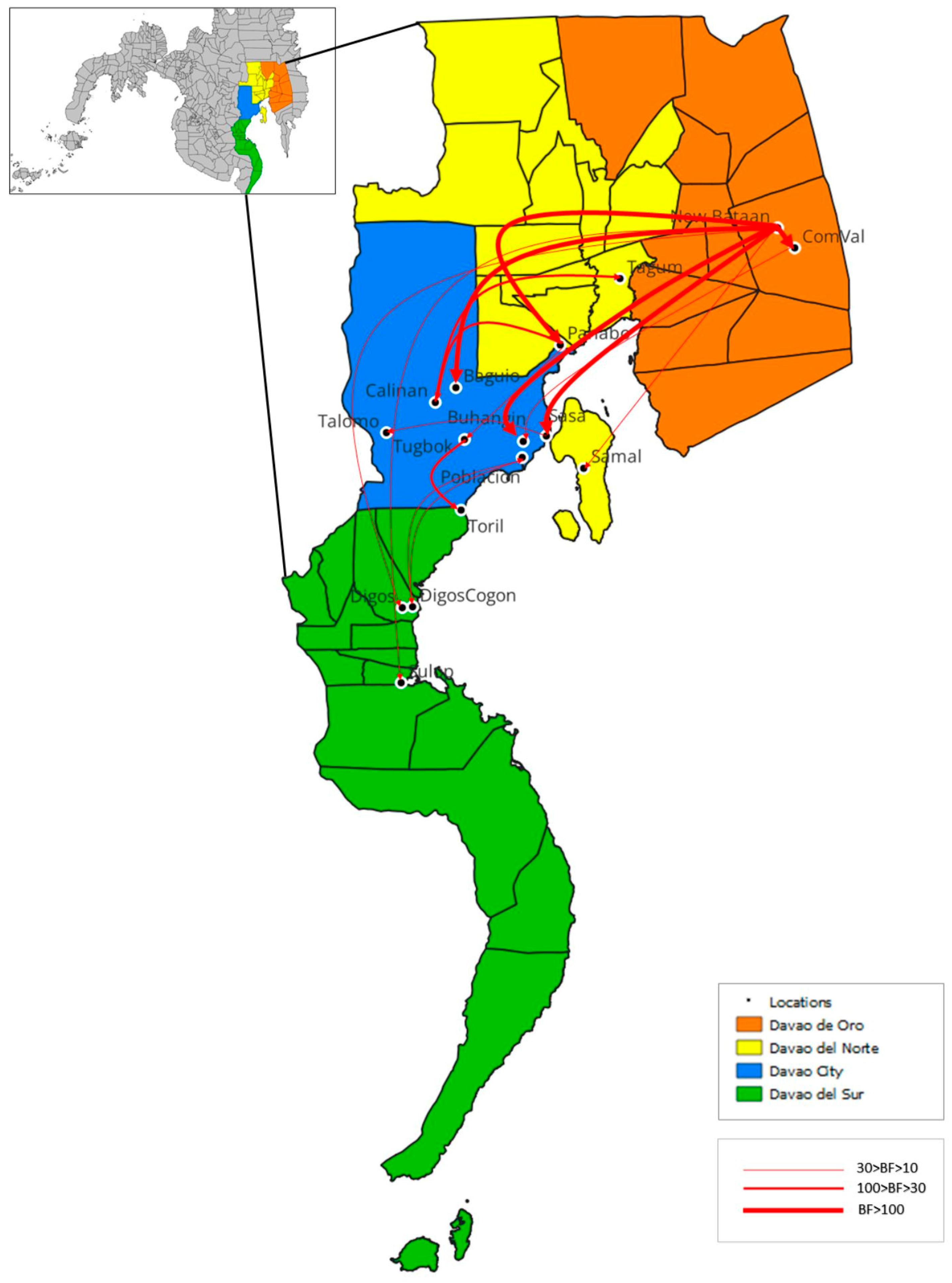
Table 1.
Modifications in the RABV MinION sequencing workflow of Brunker et al. (2020).
| Procedure | Challenges Encountered |
Original Protocol | Modification |
|---|---|---|---|
| RNA Extraction | Prescribed kit is unavailable | Zymo Research Quick-RNA miniprep kit, 2ml ceramic tubes (1.4mm), and Terralyzer | Biospin Viral DNA/RNA Extraction Kit, 2ml ceramic tubes (2.8mm), Bead Mill Homogenizer |
| Multiplex PCR | Low concentration of PCR products | Conventional multiplex PCR | Touchdown multiplex PCR |
| Library Preparation | Low concentration post barcode ligation | 5ng DNA input per sample; 70% ethanol for cleanup wash step |
50ng DNA input per sample; Freshly prepared 80% ethanol for cleanup wash step |
| Ultra II Ligation Module for Native Barcoding not available in purchased kit | Ultra II Ligation Master Mix and Ligation Enhancer | Blunt/TA Ligase Master Mix diluted with NFW | |
| Sequencing | Low read number Recycling of flow cells |
Run can finish in six hours, basecalling mode on for fast basecalling n/a |
Run extended to 16 hours or more, basecalling mode off Yes, but excluded sequences that overlapped barcodes with samples from previous run |
Table 2.
Total nucleic acid concentration of the pooled amplicons and libraries.
| Sequence Batch No. | No. of Samples a |
Concentration of Barcoded Amplicon Pool (ng/ul) |
Concentration of Final Library (ng/ul) |
|---|---|---|---|
| 1 | 12b | 3.78 | 0.956 |
| 2 | 17 | 1.39 | 1.28 |
| 3 | 17 | 1.18 | 1.21 |
| 4 | 17 | 0.84 | 0.14 |
aNegative control not included in count. b Samples were processed in duplicate.
Table 3.
Rabies Whole Genome Sequence Profile Using RABV-GLUE.
| Sequence Batch No. | Sample ID |
Identified as RABV? |
Closest full genome ref seq |
Whole genome coverage |
Breadth of coverage | Coding region coverage | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | P | M | G | L | ||||||
| 1 | 27-21 | Yes | KX149259 | 94% | 98% | 100% | 100% | 100% | 100% | 91.2% |
| 29-21 | Yes | KX149259 | 96% | 99% | 100% | 100% | 100% | 100% | 94.4% | |
| 33-21 | Yes | KX149259 | 97% | 99% | 100% | 100% | 100% | 100% | 97.5% | |
| 35-21 | Yes | KX149259 | 92% | 99% | 100% | 100% | 100% | 100% | 90.9% | |
| 37-21 | Yes | KX149259 | 97% | 99% | 100% | 100% | 100% | 100% | 97.5% | |
| 42-21 | Yes | KX149259 | 97% | 99% | 100% | 100% | 100% | 100% | 97.5% | |
| 2 | 47-20 | Yes | KX149259 | 84% | 92% | 100% | 100% | 100% | 100% | 80.2% |
| 53-20 | Yes | KX149259 | 79% | 94% | 100% | 100% | 100% | 100% | 80.2% | |
| 54-20 | Yes | KX149259 | 91% | 97% | 100% | 100% | 100% | 100% | 90.9% | |
| 55-20 | Yes | KX149259 | 92% | 96% | 100% | 100% | 80.1% | 100% | 90.9% | |
| 56-20 | Yes | KX149259 | 94% | 96% | 100% | 100% | 100% | 100% | 94.5% | |
| 57-20 | Yes | KX149259 | 86% | 91% | 100% | 100% | 100% | 100% | 83.8% | |
| 59-20 | Yes | KX149259 | 93% | 96% | 100% | 100% | 100% | 100% | 94.5% | |
| 60-20 | Yes | KX149259 | 87% | 94% | 100% | 100% | 100% | 100% | 80.3% | |
| 66-20 | Yes | KX149259 | 90% | 97% | 100% | 100% | 100% | 100% | 83.8% | |
| 67-20 | Yes | KX149259 | 97% | 98% | 100% | 100% | 100% | 100% | 97.5% | |
| 68-20 | Yes | KX149259 | 96% | 98% | 100% | 100% | 80.1% | 87.1% | 94.3% | |
| 69-20 | Yes | KX149259 | 92% | 98% | 100% | 100% | 100% | 100% | 94.5% | |
| 82-20 | Yes | KX149259 | 88% | 99% | 100% | 100% | 100% | 100% | 83.4% | |
| 83-20 | Yes | KX149259 | 94% | 94% | 100% | 100% | 100% | 100% | 87.4% | |
| 02-21 | Yes | KX149259 | 87% | 96% | 100% | 100% | 100% | 100% | 83.4% | |
| 08-21 | Yes | KX149259 | 88% | 98% | 100% | 100% | 100% | 100% | 83.4% | |
| 10-21 | Yes | KX149259 | 95% | 99% | 100% | 100% | 100% | 100% | 94.5% | |
| 3 | 14-21 | Yes | KX149259 | 97% | 99% | 100% | 99.9% | 100% | 99.9% | 97.5% |
| 15-21 | Yes | KX149259 | 94% | 98% | 100% | 99.9% | 100% | 99.9% | 94.5% | |
| 19-21 | Yes | KX149259 | 96% | 99% | 100% | 99.9% | 100% | 99.9% | 94.5% | |
| 21-21 | Yes | KX149259 | 96% | 99% | 99.9% | 99.9% | 100% | 99.9% | 94.5% | |
| 25-21 | Yes | KX149259 | 94% | 98% | 100% | 99.9% | 100% | 99.9% | 94.5% | |
| 26-21 | Yes | KX149259 | 94% | 97% | 100% | 99.9% | 100% | 99.9% | 90.9% | |
| 4 | 98-19 | Yesb | KX149259 | 38% | 50% | 15.6% | 75.7% | 63.1% | 41.3% | 35.5% |
| 101-19 | Yesb | KX149259 | 25% | 33% | 15.6% | 75.7% | 31.2% | 17.0% | 21.6% | |
| 105-19 | Yesb | KX149259 | 25% | 32% | 15.6% | 75.7% | 31.2% | 17.0% | 21.6% | |
| 118-19 | Yesb | KX149259 | 32% | 51% | 0.0% | 71.3% | 31.2% | 41.3% | 35.5% | |
| 122-19 | Yesb | KX149259 | 47% | 56% | 15.6% | 75.7% | 63.1% | 60.8% | 49.9% | |
| 131-19 | Yes | KX149259 | 82% | 94% | 78.3% | 100% | 80.1% | 100% | 80.6% | |
| 102-19 | Yes | KX149259 | 85% | 93% | 83.8% | 100% | 100% | 100% | 83.4% | |
| 107-19 | Yesb | KX149259 | 69% | 89% | 63.3% | 100% | 80.1% | 69.8% | 63.7% | |
| 135-19 | Yesb | KX149259 | 80% | 89% | 83.9% | 100% | 100% | 100% | 72.3% | |
| 136-19 | Yes | KX149259 | 90% | 96% | 83.2% | 100% | 100% | 100% | 90.9% | |
| 01-20 | Yes | KX149259 | 88% | 94% | 83.9% | 100% | 100% | 100% | 84.9% | |
| 04-20 | Yes | KX149259 | 92% | 96% | 84.3% | 100% | 100% | 100% | 91.0% | |
| 07-20 | Yes | KX149259 | 95% | 98% | 98.9% | 100% | 100% | 100% | 94.5% | |
| 08-20a | Noa | NAa | NAa | 2% | 0.0% | 0.0% | 0.0% | 0.0% | 0.0% | |
| 11-20 | Yes | KX149259 | 80% | 90% | 83.9% | 100% | 100% | 100% | 77.7% | |
| 14-20 | Yes | KX149259 | 88% | 93% | 100% | 100% | 100% | 100% | 83.4% | |
| 15-20 | Yes | KX149259 | 95% | 96% | 98.9% | 100% | 100% | 100% | 94.5% | |
a Very low sequence read depth, excluded from downstream analysis. b Whole genome coverage and breadth coverage below threshold, excluded from downstream analysis.
Table 4.
Bayes Factor-supported transmission routes of reported rabies viruses within Davao Region from 2018-2021.
Table 4.
Bayes Factor-supported transmission routes of reported rabies viruses within Davao Region from 2018-2021.
| From | To | Support |
|---|---|---|
| Intra-city transmission | ||
| Sasa1 | Talomo1 | strong |
| Tugbok1 | Toril1 | very strong |
| Inter-city transmission | ||
| Poblacion1 | DigosCogon2 | strong |
| ComVal4 | Buhangin1 | strong |
| DigosCogon2 | Poblacion1 | strong |
| Panabo3 | Tugbok1 | strong |
| Calinan1 | Tagum3 | very strong |
| Panabo3 | Calinan1 | very strong |
| New Bataan4 | Baguio1 | decisive |
| New Bataan4 | Sasa1 | decisive |
| New Bataan4 | Buhangin1 | decisive |
| Intra-province transmission | ||
| New Bataan4 | ComVal4 | decisive |
| Inter-province transmission | ||
| New Bataan4 | Digos2 | strong |
| New Bataan4 | Sulop2 | strong |
| New Bataan4 | Samal3 | strong |
| New Bataan4 | Panabo3 | decisive |
1 Sample collected from Davao City. 2Sample collected from Davao del Sur. 3Sample collected from Davao del Norte. 4Sample collected from Davao de Oro.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



