
Submitted:
25 June 2023
Posted:
26 June 2023
You are already at the latest version
Abstract
Respiratory syncytial virus (RSV) RNA synthesis takes place in cytoplasmic viral factories also called inclusion bodies (IBs), which are membrane-less organelles concentrating the viral RNA polymerase complex. IBs assembly is driven by liquid-liquid phase separation promoted by interactions between the viral nucleoprotein N and the phosphoprotein P. We recently demonstrated that cyclopamine (CPM) inhibits RSV multiplication by disorganizing and hardening IBs. Although a single mutation in the viral transcription factor M2-1 induced resistance to CPM, the mechanism of action of CPM still remains to be characterized. Here, using FRAP experiments on reconstituted pseudo-IBs both in cellula and in vitro, we first demonstrated that CPM activity depends on the presence of M2-1 together with N and P. We show that CPM impairs the competition between P and RNA binding to M2-1. As mutations on both P and M2-1 induced resistance against CPM activity, we suggest that CPM may affect the dynamics of the M2-1−P interaction, thereby affecting the relative mobility of proteins contained in RSV IBs. Overall, our results reveal that stabilizing viral protein-protein interactions is an attractive new antiviral approach. They pave the way for the rational chemical optimization of new specific anti-RSV molecules.
Keywords:
RSV
; cyclopamine
; M2-1−P interaction
; antiviral mechanism
; inclusion bodies
1. Introduction
Replication of many viruses depends on the formation upon infection of cellular biomolecular condensates through a liquid-liquid phase separation (LLPS) mechanism, which biogenesis is driven by scaffold proteins [1,2,3]. More specifically, viral RNA synthesis of human and bovine respiratory syncytial viruses (hRSV and bRSV, respectively) was shown to occur in cytoplasmic membrane-less viral factories, also termed inclusion bodies (IBs) [4,5]. RSV belongs to the Mononegavirales order (MNVs), which gathers single-stranded negative sense RNA viruses [6]. The genomic RNA is tightly encapsidated by the viral nucleoprotein N, forming helical ribonucleocapsids (NCs), which are the template for both viral transcription and replication. Similarly to other MNVs, RSV IBs concentrate all the elements required for viral RNA synthesis, i.e. the encapsidated genomic and antigenomic viral RNA, the viral polymerase L and its co-factor P, and the viral transcription factor M2-1. The N and P proteins are regarded as scaffold proteins for IBs’ formation, their expression being sufficient to induce LLPS and pseudo-IBs morphogenesis in cells and condensation in vitro [7] [8]. Furthermore, for RSV the identification of sub-compartments within IBs called IBAGs (IBs associated granules), where viral mRNAs and the viral transcription factor M2-1 specifically concentrate, recently revealed that IBs are complex and present a high degree of organization [4]. Given their critical role in the viral cycle, IBs represent targets of interest for the development of antivirals.
hRSV is the main cause of severe lower respiratory tract infection in children worldwide, and the leading cause of hospitalization of children under 6 months of age [9]. Re-infections occur throughout life, leading to common cold in healthy adults, but to severe lower respiratory infections in immunocompromised and elderly people, with a burden comparable to influenza virus [10].Despite the high impact of hRSV infections on public health, and although several vaccine candidates are currently in advanced stages of development [11], the first vaccine to prevent RSV severe disease was approved by the Food and Drug Administration only on May 2023 and is restricted to elderly population (GSK’s Arexvy®) [12]. The only specific treatment commercialized is a humanised monoclonal antibody directed against the fusion protein F responsible for viral entry (Palivizumab, Synagis). However, a poor benefit-cost ratio limits its use to prophylaxis of severe infection in pre-term and high-risk infants [13]. Of note, a new prophylactic antibody targeting the pre-fusion form of F (Nirsevimab, Beyfortus®) has been very recently approved in the European Union for the prevention of RSV lower respiratory tract disease in newborns and infants during their first RSV season [14]. Similarly, bRSV is a major cause of respiratory disease in young calves and responsible for large economic losses worldwide [15]. Although commercial attenuated and inactivated bRSV vaccines are available, they remain poorly efficient to protect animals [16,17].
We have previously shown that cyclopamine (CPM), a plant alkaloid known to target the Hedgehog pathway, inhibits hRSV replication both in cell culture and in vivo in a mouse model [18,19]. We have also recently demonstrated that CPM acts by hardening hRSV IBs. The R151K mutation in the M2-1 protein has been shown to induce resistance to CPM treatment. However, no direct interaction between M2-1 and CPM could be observed. Interestingly, we have also recently shown that CPM inhibits bRSV replication [20]. The sequence alignement of orthopneumovirus M2-1 proteins highlights the conservation of M2-1 R151 residue suggesting that CPM could also be active against ovine RSV [20]. Although CPM represents a new potential antiviral against RSV, its mechanism of action remains to be characterized to consider further improvements of the molecule.
RSV M2-1 is a 22 kDa protein that forms tetramers in solution [21,22]. It is composed of an N-terminal zinc-finger domain, a central oligomerisation domain, and a C-terminal globular domain (M2-1 core). The structure of hRSV M2-1 was obtained using X-ray crystallography [23]. It displays a compact disk-shaped tetramer, referred to as a closed conformation. Later, it has been suggested that M2-1 might display conformational plasticity, by analogy with the M2-1 protein of the closely related human metapneumovirus (HMPV). The crystal structures of HMPV M2-1 indeed revealed M2-1 tetramer with a partially open conformation, where the core domain of one M2-1 protomer is projected to the outside of the disk in an open conformation, while the three other protomers adopt the same closed conformation as in the structure of RSV M2-1 [24]. The hRSV M2-1 core domain interacts with both the P protein and viral mRNA, in a competitive manner [21]. The recruitment of M2-1 into IBs depends on its interaction with P. The M2-1−P interaction is moreover required for dephosphorylation of M2-1 by the cellular phosphatase PP1 by a mechanism in which P serves as a scaffold protein that binds both M2-1 and PP1 [25,26]. M2-1 contains two phosphorylation sites, on residues S58 and S61. Dephosphorylation of M2-1 induces a switch of interaction from P to mRNA, leading to the co-localization of M2-1 with viral mRNA in IBAGs. Binding surfaces of P and RNA on M2-1 core domain, determined by Nuclear Magnetic Resonance (NMR), partly overlap [27]. The binding surface of P was confirmed by the X-ray crystal structure of M2-1 in a closed conformation, in complex with a P peptide corresponding to the M2-1 binding motif (residues 95-110) of P [28]. Furthermore, crystallographic data of hRSV M2-1 in complex with short RNA disclosed a dual RNA-binding mode [29]. The M2-1 zinc finger domain contains an RNA binding site that recognizes an RNA base, that can be considered as specific. The M2-1 core domain binds to the RNA backbone, thus with no sequence specificity expected, although higher affinities were observed for some short RNAs, in particular A-rich sequences [27]. Moreover M2-1 displays annealing of longer RNA with secondary structures, resulting in cooperative binding to two sites with increased affinity [30]. These data highlight the complexity of M2-1 interactions with both P and RNA. Of note, M2-1 R151 residue, which induces resistance to CPM when mutated into a lysine, is critical for the interaction with RNA, and also involved in P-binding [23,27]. However, previous data suggested that CPM does not impair the interaction of M2-1 with RNA, nor that with P [18].
In the present study, we aimed at shedding light on the mechanism of action of CPM. Using fluorescence recovery after photobleaching (FRAP) and hypotonic shock experiments, we first showed that CPM induces hardening of pseudo-IBs formed in cells upon transient expression of hRSV N and P only when M2-1 is co-expressed. Similar results were obtained in vitro by co-incubation of recombinant N, P, and M2-1 proteins. Study of M2-1 interaction with P and RNA using in vitro band shift assay by native gel electrophoresis, together with pull-down assays, revealed that CPM impairs the displacement by RNA of the M2-1−P interaction. Based on the structure of the M2-1−P complex, we generated mutations of the residue Y102 of P and showed that the mutation Y102L of P induces resistance to CPM, similarly to the R151K mutation of M2-1. Together, our results suggest that CPM acts on the M2-1−P interaction, and freezes M2-1 and P dynamics and induces IBs hardening.
2. Results
2.1. CPM treatment induces hardening of pseudo-IBs formed in cells
We have previously shown that CPM induces IBs hardening in the context of infected cells ([19]. Furthermore, using a functional minigenome assay, CPM has been shown to impair RSV polymerase activity [18]. We here decided to study the impact of CPM treatment on pseudo-IBs formed upon co-expression of hRSV N and P proteins, which are the minimal elements required for LLPS involved in RSV IBs morphogenesis [8]. We first tested the impact of CPM treatment on the fluidity of pseudo-IBs in cells expressing N and P-BFP (P fused to Blue Fluorescent Protein), by performing fluorescence recovery after photobleaching (FRAP) experiments. Compared to the diffusion of P-BFP previously observed in IBs of infected cells [19], P-BFP fluorescence recovery remained below 50% after 1 min suggesting slower diffusion of P-BFP in N+P pseudo-IBs (Figure 1A). P-BFP fluorescence recovery in pseudo-IBs was not affected by the treatment with 10 µM CPM (Figure 1A). A similar analysis was then performed in the presence of wild type (WT) M2-1 or M2-1R151K mutant. Upon M2-1 expression, fluorescence recovery of P-BFP in pseudo-IBs was close to 70% after 1 min, similarly to what was observed in RSV-P-BFP infected cells (Figure 1B), suggesting that M2-1 could play a role in the mobility of P within IBs. Similar fluorescence recovery of P-BFP was obtained in the presence of M2-1R151K (Figure 1C), showing that this M2-1 mutation does not affect the fluidity of pseudo-IBs. Treatement by CPM strongly inhibited P-BFP diffusion in the context of WT M2-1 expression, with only 10% recovery after 1 min, while the treatment had no impact in the presence of M2-1R151K (Figure 1B,C). In parallel, we analysed the nature of pseudo-IBs by studying their sensivity to hypotonic shock, which dirupts LLPS biocondensates [31,32]. As previously observed for IBs in infected cells [19], in the absence of CPM, pseudo-IBs obtained in the context of co-expression of both N and P disassembled upon a hypotonic shock with or without M2-1 expression, whereas CPM treatment prevented pseudo-IBs disruption only in the presence of M2-1 (Figure 1D).
Altogether these results highlight the role of M2-1 in the fluidity of hRSV IBs. They also show that hardening of these IBs by CPM is dependent on the M2-1 protein and reveal that the mechanism of action of CPM is independent of the viral RNA polymerase activity.
2.2. Impact of CPM on the fluidity of pseudo-IBs formed in vitro
We have previously shown that pseudo-IBs form by LLPS upon mixing recombinant fluorescent hRSV N and P proteins in the presence of the crowding agent Ficoll [8,33]. Using this approach, we wanted to assess the impact of CPM on in vitro reconstituted pseudo-IBs in the absence or presence of M2-1. In order to observe M2-1 in these structures, we took advantage of previous data showing that fusion of mCherry at the C-terminus of M2-1 had no major impact on the RNA polymerase activity [25], and engineered a recombinant M2-1-mCherry fusion protein expressed in E.coli. Both WT and R151K mutated M2-1-mCherry were produced and purified as RNA-free proteins (Figure 2A). Analysis of pseudo-IBs formed in vitro in the presence of recombinant P-BFP and mCherry-N confirmed that addition of CPM had no major impact on pseudo-IBs morphogenesis (Figure 2B left panel, Figure S1). Addition of M2-1-mCherry to P-BFP and untagged N did not modify pseudo-IBs morphogenesis, and red fluorescence was observed in all the pseudo-IBs, suggesting that M2-1 is homogeneously incorporated into these structures (Figure 2B right panel, Figure S1).
We then assessed the impact of CPM on P and M2-1 mobility in these in vitro pseudo-IBs by following fluorescence recovery after photobleaching. We here used a recombinant P-mCherry protein instead of P-BFP, which was not suited to FRAP experiment in our conditions. As previously published [8], we first observed that in the presence of only P-mCherry and N, fluorescence recovery of P-mCherry was fast, with 70 % of recovery after 1 min. In the presence of CPM, P-mCherry was still able to diffuse, although fluorescence recovery was close to 40% after 1 min, suggesting a slight decrease in P mobility in presence of CPM (Figure 2C). The presence of M2-1 did not affect the mobility of P-mCherry in pseudo-IBs, but incubation with CPM resulted in strong reduction of P-mCherry mobility, with only 15% recovery after 1 min (Figure 2D). In parallel, we investigated M2-1 diffusion in the context of co-incubation of recombinant N, P, and M2-1-mCherry (Figure 2E). Similarly to what we previously observed in infected cells and on reconstituted pseudo-IBs in transfected cells, M2-1 was mobile within in vitro reconstituted pseudo-IBs with quick recovery of 50% of fluorescence of M2-1-mCherry after 1 min, and addition of CPM strongly impaired M2-1 mobility (Figure 2E).
These data thus show that CPM induces hardening of in vitro formed pseudo-IBs, revealing that this phenomenon is independent of cellular partners, including RNA. Similarly to what was observed in cells for pseudo-IBs, the presence of M2-1 is critical for CPM activity. However, our results also revealed that CPM addition did alter P mobility in the context of N and P co-incubation.
2.3. CPM interferes with the competitive binding of P and RNA to M2-1
Based on these results, we hypothesized that CPM may affect the M2-1−P interaction. We have previously shown that the P and the RNA binding sites on the M2-1 core domain partly overlap [27], and that the M2-1−P interaction can be disrupted by tRNA in vitro [21]. We thus studied the impact of CPM on M2-1−P and M2-1−RNA interactions. The capacity of tRNA to compete with P for M2-1-mCherry binding was analyzed using native gel electrophoresis with M2-1-mCherry migration revealed by UV exposition. As shown on Figure 3A, M2-1-mCherry mobility was strongly modified upon addition of tRNA or P (lanes 1, 2 and 3), confirming the formation of the corresponding complexes. Of note, the M2-1-mCherry−P complex migrated slightly faster than the M2-1-mCherry−tRNA complex. The addition of CPM did not modify the migration profiles of these complexes (lanes 5, 6 and 7). According to the migration profile, tRNA addition displaced P from the M2-1-mCherry-P complex and led to the formation of the M2-1−tRNA complex (Figure 3A, lanes 3 versus 4). Since M2-1 contains two RNA-binding sites, this indicates that tRNA binds at least to the non specific site that overlaps with the P-binding site. In the presence of CPM, tRNA addition to the M2-1-mCherry−P complex resulted in the formation of a complex that migrated slightly faster than the M2-1-mCherry−P complex (Figure 3A, lanes 7 versus 8). These results suggest that CPM could interact with the M2-1−P complex and impair the switch to the M2-1−tRNA complex. In contrast, similar experiments performed in the presence of M2-1R151K-mCherry mutant revealed that tRNA was able to displace P from M2-1R151K-mCherry in the presence of CPM (Figure 3A, lanes 11-12 and 15-16), validating that M2-1 R151 residue is critical for CPM activity. To confirm these results and to further analyse the specificity of the CPM effect, we then studied the impact of CPM on the interaction between P and M2-1 core domain using a pulldown assay. Beads containing GST-M2-1core were incubated with P in the presence or absence of CPM, before tRNA addition. After washes, the presence of pulled-down P was analyzed by SDS-PAGE. As shown on Figure 3B, addition of tRNA induced release of P in the absence of CPM, but not in the presence of the drug.
Based on this last result, we wondered if we could evidence a direct effect of CPM on the M2-1−P interaction by using NMR. We used the 15N-labeled M2-1 core domain (~100 µM) and measured 2D 1H-15N correlation spectra of 15N-M2-1core. CPM alone did not induce significant spectral changes in the spectrum of 15N-M2-1core (Figure S2), excluding that CPM directly binds to M2-1core. We then probed binding of a synthetic FITC-P peptide derived from the P-binding motif in M2-1 (FITC-P95-112). We followed titration of ~100 µM 15N-M2-1core by FITC-P95-112 by measuring 2D 1H-15N correlation spectra. FITC-P95-112 induced chemical shift as well as intensity perturbations (Figure 4A). Saturation was reached with 1-1.5 molar equivalents of peptide. A control spectrum measured with 1.6 molar equivalents of fluorescein showed that FITC-P95-112 binds to 15N-M2-1core via its peptide moiety. Most signals (e.g. residue S108 in Figure 4A) displayed an intermediate exchange regime, as indicated by complete broadening at a peptide:protein molar ratio of 0.5:1. Some signals with small chemical shift variation amplitudes were in fast chemical exchange. Affinity in the form of dissociation constants (Kd) can be easily derived from this kind of data using simple binding models. However the concentration range used for these experiments was too high to determine Kds with high accuracy. We can only estimate an order of magnitude for Kd in the µM range. Strikingly, many signals in intermediate exchange remain broadened beyond detection at the titration endpoint (e.g. residues L152 and V156 in Figure 4A). These residues delineate the P-binding pocket (Figure 4B). Signals of residues that are further apart (e.g. residues S108 and D155 in Figure 4A) reappear. This behavior reports on motions within the M2-1core−FITC-P95-112 complex on a µs-ms time scale. Addition of 1 molar equivalent of CPM to WT 15N-M2-1core incubated with 0.25 equivalent of FITC-P95-112 did not result in any significant change (Figure S3). Under these conditions, there is no indication that CPM might change the equilibrium or the kinetics of the complex formation. A control experiment was performed by NMR to assess if CPM interacts with the peptide (Figure S4). 1D 1H NMR spectra were acquired for FITC-P95-112 alone, CPM alone and an equimolar mixture of both, under the same buffer conditions. Some subtle changes are observed in several CPM and FITC-P95-112 signals in the mixture, as compared to the molecules in isolation, hinting at intermolecular contacts.
In order to decipher if CPM could modify the M2-1−P interaction, we also investigated the affinity of FITC-P95-112 to the M2-1core domain, in the absence and the presence of CPM using microscale thermophoresis (MST). The FITC-P95-112 at 100 nM was incubated in the presence of increasing concentrations of recombinant M2-1core (from 0 to 33.5 µM) alone or in the presence of 1 or 10 µM of CPM. In our conditions the estimated Kd is around 20 µM in the absence and presence of CPM (Figure S5). Although the affinity is lower than estimated by NMR, these results corroborate those obtained by NMR in suggesting that CPM does not affect the affinity between P peptide and M2-1 core domain.
In a last part we assessed if the M2-1 R151K mutation could affect P binding. The R151K mutation induced only minor spectral perturbations in the 2D 1H-15N correlation spectrum compared to the WT M2-1core (Figure S6), showing that this mutation does not alter the fold of the protein. 1H-15N amide chemical shifts of the M2-1core R151K mutant were easily obtained by transferring the assignments of the WT. During titration of 15N-M2-1core R151K mutant by FITC-P95-112, 2D 1H-15N correlation spectra displayed similar chemical shift and intensity perturbations to those observed with the WT due to FITC-P95-112 binding. In particular the exchange regime was mainly intermediate, and broadening beyond detection was also observed at a molar ratio of 1.5, where saturation is reached (Figure 4C). This indicates that complex formation is not significantly impacted by the mutation. However, broadening seems to be more severe for the mutant (e.g. by comparing residues S108 and D155 in Figure 4A,C), suggesting that the R151K mutation could have an influence on conformational fluctuations within the complex.
Taken together, our observations suggest that CPM interferes with the displacement of P from M2-1 by RNA. In the absence of detection of CPM binding on M2-1core domain−P peptide or of increase of affinity of this interaction, possible explanations of these results are that CPM i/ induces a change in the dynamics of the M2-1−P complex, ii/ impairs RNA binding to M2-1 when it interacts with P, or iii/ contributes to the formation of a ternary complex between M2-1, P and RNA. Of note, the results obtained with the M2-1R151K-mCherry mutant suggest that the R151 residue must be involved in the competition between P and tRNA for M2-1 binding.
2.4. Residue Y102 of P is involved in CPM activity
Based on the results presented above, and on the fact that the M2-1R151K substitution induces RSV resistance to CPM [18,19], we wanted to further test the role of P for the activity of CPM. A close inspection of the crystal-structure of the M2-1−P complex [28] shows that the M2-1 R151 residue is positioned closely to the P Y102 residue, both residues being surface exposed. Their orientations are less well defined than those of residues that are buried in the interface, M2-1 R151K and P Y102 interacting only in one protomer via a non-optimal cation-pi interaction (Figure 5A). We thus wondered if the P Y102 residue could play a role in CPM activity. This residue has previously been shown to be critical for P binding to M2-1, as the Y102A substitution completely abrogated hRSV polymerase activity and P binding to M2-1 [25].
We first assessed the impact on the polymerase activity of the two P mutations Y102F and Y102L, where tyrosine was replaced by another aromatic and a long hydrophobic residue, respectively, using a functional minigenome assay. As shown on Figure 5B, the P Y102F substitution had no impact on the polymerase activity (p>0.05 using nested t test), whereas Y102L substitution induced a decrease of approximatively 70% of the polymerase activity (p<0.001 using nested t test). Similar experiments performed in the presence of increased concentrations of CPM revealed a dose dependent inhibition of the polymerase activity, with similar efficiency when expressing WT P or the Y102F mutant. On the contrary, CPM had no impact on the polymerase activity in the presence of the Y102L P mutant. This result suggests that, although affecting the polymerase activity, this substitution conferred resistance to CPM (Figure 5B). Importantly, as assessed by Western blot, no difference in P expression was observed between WT and P mutants, and CPM did not affect P expression (Figure 5B). Of note, we have previously shown that the M2-1-binding region of the P protein (P residues 97-109) is mainly disordered with low α-helical propensity in its unbound form in solution [34], but folds into an α-helix when the M2-1−P complex is formed. Mutations inside this region may change the α-helical propensity, and thus the affinity for M2-1. We therefore used the PEP-FOLD 3 server (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/) to compare the predicted structures of WT and Y102L mutant P95-110 peptides. The predicted α-helical propensity was higher (60 %) than experimentally observed, but the two peptides did not display a significant difference (Figure S7). This indicates that the mutation would not alter P folding upon M2-1 binding.
We then analysed the impact of CPM on the capacity of tRNA to compete with the M2-1−P interaction in the presence of CPM, by analyzing the migration of M2-1-mCherry on native agarose gels. Migration of M2-1 complexes with P Y102F and Y102L mutants was similar to that of the WT M2-1−P complex (Figure 5C lanes 3 and 6, compared with Figure 3A), and slighly faster than the M2-1−tRNA complex (Figure 5C, lanes 4 and 7). We thus assessed if tRNA could compete with the M2-1−P interaction in the presence of CPM. As observed for WT P, the presence of CPM impaired the switch from the M2-1−P to the M2-1−tRNA complex for the Y102F mutant of P after incubation of tRNA. A complex was formed, which migrated faster than M2-1−tRNA and slowlier than M2-1−P, suggesting that CPM interferes with the competition between tRNA and P for M2-1 binding. On the contrary, in the same conditions, tRNA was able to displace the Y102L mutant of P from M2-1, as a shift corresponding to the formation of M2-1−tRNA complex was observed. Overall, our results show that similarly to the residue R151 of M2-1, the residue Y102 of P is critical for CPM activity. Thus, although we did not manage to detect a signicant impact of CPM on M2-1−P affinity these data suggest that CPM may act on the M2-1−P complex.
3. Discussion
The formation of cytoplasmic membrane-less organelles is a major characteristic of MNVs infections. These structures, which have initially been considered as aggregates of dead-end products of viral infection, were recently shown to be viral factories, where replication and transcription of the viral genomes take place [4]. Similarly to other cellular organelles such as stress granules or P bodies, the morphogenesis of these structures depends on a LLPS mechanism, driven by low-affinity interactions of multivalent proteins and/or proteins containing intrinsically disordered regions (IDRs), as well as RNA-protein interactions [35]. In the case of hRSV, we have previously shown that the N protein associated to RNA and the P protein are both required for pseudo-IBs reconstitution in cells and in vitro [8,33]. The tetrameric P protein with N- and C-terminal IDRs [26,34] has also a central role in the recruitment of the L polymerase, the transcription factor M2-1, as well as the cellular phosphatase PP1 to IBs [25,36].
IBs, in which viral RNA synthesis occurs, as well the protein-protein interactions sustaining their activity represent original targets for the development of new antiviral strategies. We have recently shown that CPM inhibits RSV replication by hardening IBs [19]. However, although the R151K M2-1 mutation has been shown to induce resistance to CPM treatment, the exact molecular mechanism of CPM action remains unknown. Here, by studying pseudo-IBs properties in cells or in vitro using FRAP, we first demonstrated that the activity of CPM on IBs relies on the presence of M2-1, and may also impact P mobility in vitro, independently of the polymerase activity or the presence of cellular proteins. Our results also revealed that within pseudo-IBs formed in cells, the presence of M2-1 increases P mobility compared to the condition where only N and P are expressed. On the contrary, in a minimal system of in vitro reconstituted pseudo-IBs, the addition of M2-1 did not affect P mobility. These observations suggest that transient interactions between P and its partners could be a key factor of IBs fluidity and dynamics.
Using in vitro approaches, we then showed that CPM impairs formation of an M2-1−tRNA complex, when M2-1 is already in complex with P. This observation could explain previous results showing that M2-1 presents a diffuse localisation within IBs upon CPM treatment of infected cells, contrasting with its concentration into IBAGs together with viral mRNAs, as observed in untreated cells [19]. Based on these results, we hypothesized that CPM could bind to M2-1−P complex. We thus thought to identify P residues that could interact with CPM. Analysis of the crystal structure of M2-1-P complex revealed that within the heterotetramer, the interaction between M2-1 and P slightly differs, in particular the orientation of the lateral chains of the residues R151 of M2-1 and Y102 of P. By investigating the impact of Y102F and Y102L substitutions on P, we observed that the P Y102L substitution induced a strong decrease in the polymerase activity but also viral resistance to CPM treatment. This result reveals the implication of this P residue in CPM activity. However, we were not able to validate the direct interaction of CPM with the M2-1−P complex using MST or NMR approaches with M2-1core domain and P peptide. We hypothesized that it could be explained by the poor water solubility of CPM, and a potential low affinity of CPM for the complex.
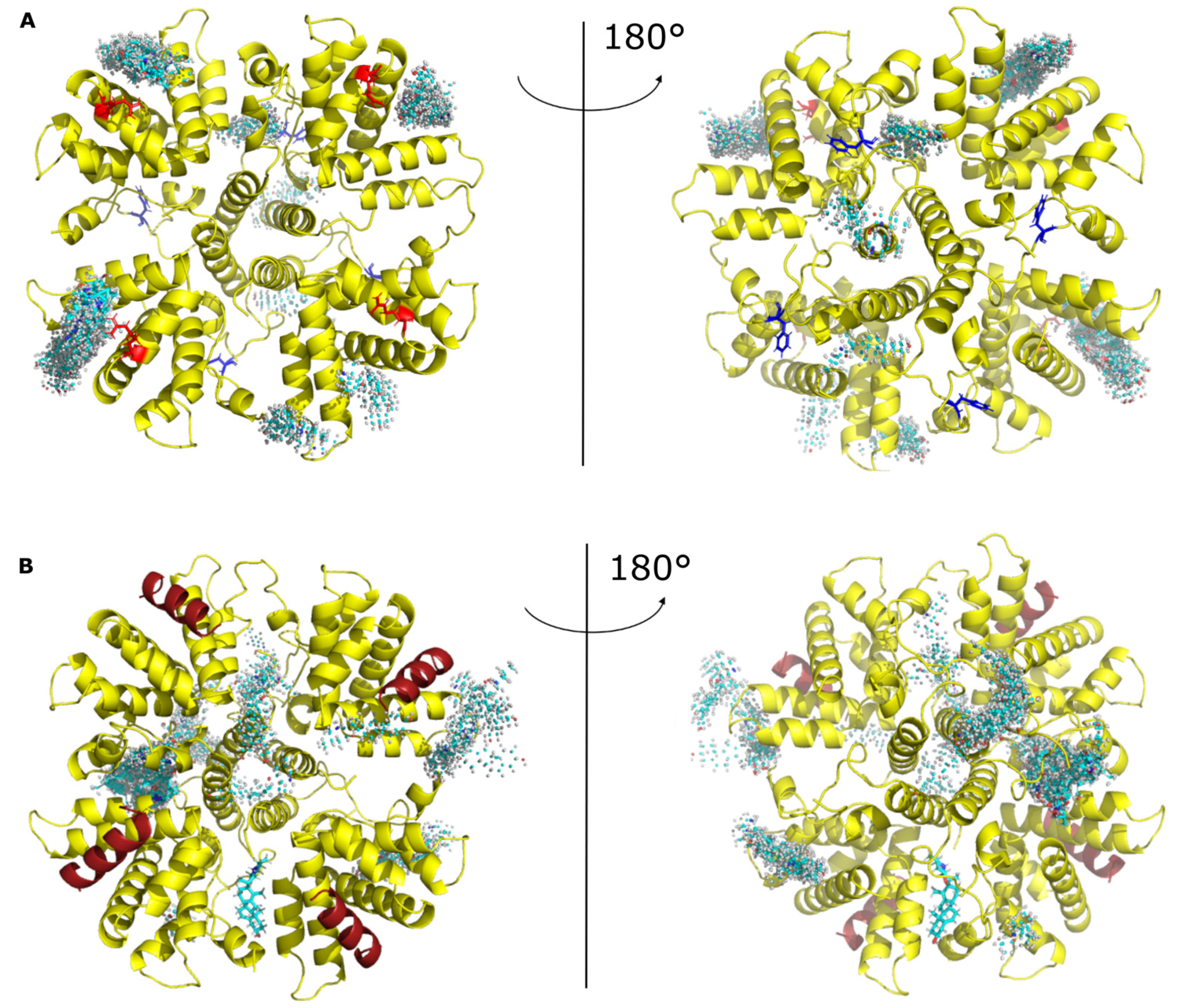
Our results also suggest that resistance to CPM does not depend on conformational changes of M2-1 and P binding sites. The direct implication of residues R151 of M2-1 and Y102 of P in a ternary complex with CPM thus remains to be determined. Interestingly, we recently showed that CPM also inhibited bRSV replication [20]. Although the structure of bRSV M2-1-P complex is not available, the sequence alignement of the domains of M2-1 and P involved in the interaction shows that residues R151 of M2-1 and Y102 of P are conserved among orthopneumoviruses. During the course of this study, we probed the binding of CPM on the M2-1−P complex by crystallography. However, we did not succeed to observe CPM in crystals of M2-1 in complex with P peptide. To get additional insight, we thus performed docking experiments using the M2-1 tetramer, alone or in complex with a P90-110 peptide (PDB accession code 6g0y). Many poses with M2-1 alone show CPM docking in the P-binding groove of M2-1, in the vicinity of R151 (Figure 6A). Some pauses show CPM on the other side of the disk, close to where RNA was shown to bind in the crystal structure of the complex [29]. Others show binding on the edge of the disk. With the M2-1−P heterotetramer, CPM predominantly docked next to the “specific RNA binding site” and on the edges of the disk formed by the tetramer (Figure 6B). Overall this indicates that CPM might explore different binding sites, including binding sites of P and RNA, and therefore interfere with M2-1−RNA and M2-1−P complex formation.
Nevertheless, structural characterization of the mechanism of CPM action on the RSV M2-1−P complex would be necessary to allow a potential rational optimisation of this antiviral compound. Finally, this study clearly suggests that stabilisation of protein-protein interaction is a potent new approach to specifically modify LLPS dynamics and function. Given the central role of viral factories formed by LLPS upon MNVs infections and the numerous transient protein-protein interactions involved in their functionning, our data open new perspective to develop specific inhibitors against all these viruses.
4. Materials and methods
4.1. Plasmid constructs
All the viral sequences were derived from the hRSV strain Long, ATCC VR-26 (Genbank accession n° AY911262.1). Expression plasmids pCI-N, pCI-P and pCI-M2-1 were obtained by cloning mammalian codon optimized coding sequences of N, P and M2-1 into pCI (Genbank accession n° U47119). The coding sequences were amplified by PCR (Phusion High-Fidelity DNA Polymerase, Thermofisher) using specific primers (sequences available upon request) and cloned into pCI by standart molecular biology procedures using NheI and EcoRI for N, MluI and XbaI for P, and MluI and NotI for M2-1. The BFP coding sequence was inserted into mammalian codon optimized coding sequence of P between residues 76 and 77 as described in (ref risso ballester 2021) to obtain the pCI-P-BFP expression vector. Plasmids for minigenome assay expressing the hRSV N, P, M2-1, and L proteins designated pN, pP, pM2-1 and pL, as well as the pM/Luc subgenomic minigenome expressing the firefly luciferase (Luc) reporter gene under the control of the M/SH gene start sequence were described previously [21].
For bacterial expression of recombinant mCherry-N, N, P-BFP, P and M2-1 core proteins, previously described pET-mCherry-N, pET-N, pGEX-P-BFP, pGEX-P and pGEX-M2-1core plasmids were used [8,27,37]. For expression of recombinant M2-1-mCherry fusion protein, the mCherry gene was amplified by PCR from the pmCherry vector (Clontech) and subcloned at the 3’ end of the M2-1 gene at SmaI-XhoI sites in pGEX-M2-1 plasmid [27]. Point mutations were introduced in pP, pGEX-P, pM2-1 and pGEX-M2-1-mCherry by site-directed mutagenesis, using the Quikchange site-directed mutagenesis kit (Stratagene). Sequence analysis was carried out to check the integrity of all the constructs.
4.2. Cells
BSRT7/5 cells (BHK-21 cells that constitutively express the T7 RNA polymerase2) [38] and HEp-2 cells (ATCC: CCL-23) were maintained respectively in DMEM and MEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), glutamine penicillin–streptomycin solution. Cells were grown in an incubator at 37 °C in 5% CO2. Transfection were performed with 2.5µL of Lipofectamine 2000 (Thermofisher) per 1 µg of DNA according to the manufacturer instructions.
4.3. Time-lapse microscopy and photobleaching experiments on pseudo-IBs
Live-cell imaging and FRAP experiments were realized using HEp-2 cells seeded in 15 or 4-wells Ibidi µ-Slide dishes with a polymer coverslip bottom and transfected with 0.22 µg pCI-P-BFP, 0.22 µg of pCI-N and 0.056 µg of pCI-M2-1 (WT or R151K mutant) for 24h. For FRAP experiments, imaging was performed using Leica SP8 inverted scanning confocal microscope with a 63× oil-immersion objective and a ×8 numerical zoom. Cells were maintained in a climate-controlled chamber (37 °C, 5% CO2) during imaging. FRAP acquisition was performed 1 h after addition of 10 μM CPM, or 0.5% DMSO. FRAP experiments were realized using the following settings: 8 s pre-bleach, 1 ms bleach and 60 s post-bleach at a frame rate of 1 image every 126 ms. Photobleaching of BFP was performed in a circular region at 100% laser intensity. Post-photobleaching fluorescence signals of the bleached region were quantified using the “ROI Intensity Evolution” tool of the Icy software [39], in parallel of regions with identical dimensions in i) a non-photobleached pseudo-IBs located in the vicinity of the targeted pseudo-IB (controls for potential loss of fluorescence in the imaging field studied), ii) a background region. Normalization and averaging of the recovery curves were then performed using the easyFRAP web-based tool [40]. For each experimental condition, two individual experiments were performed (n=2), during which 10 to 12 pseudo-IBs were analysed. Images from one in cellulo FRAP replicate are shown in Figure 1. In vitro pseudo-IBs sizes were quantified using the “Spot detector” tool of the Icy software, incremented with a filter for >90% sphericity to discard artefactual values generated by the detection of two adjacent pseudo-IBs as a single entity.
For osmotic shock experiments, imaging was performed using an Olympus IX73 inverted microscope with a 63× oil-immersion objective and a ×2 numerical zoom. Hypotonic shocks were performed by incubating the cells in 10% MEM diluted in water (v/v) for 5 min. One image was acquired before hypotonic shock and the same cells were imaged every 1 min for 5 min during the shock. For each experimental data point, the hypotonic shock was applied to an entire well of a 15-well Ibidi µ-Slide dish, and one position was studied in order to keep the cell into focus. 7 to 10 acquisitions from 2 independent experiments were performed. Imaging fields of interest comprised 2 to 4 cells, and IBs present before and 5 min following the hypotonic shock were counted manually.
4.4. Photobleaching experiments on in vitro reconstituted pseudo-IBs.
As previously described [8,33], in vitro pseudo-IBs were reconstituted in 10-20 µL droplets using mCherry-N and P-BFP proteins (3 and 12 µM, respectively), or N, P-BFP and mCherry-M2-1 (3, 12, and 12 µM, respectively) recombinant proteins in a 20 mM Tris/HCl pH 8.5, 150 mM NaCl, 10% molecular-crowding agent Ficoll buffer, supplemented with 150 µM CPM or 0.5% DMSO. Drops were incubated on 8-well Ibidi µ-Slide dishes for 30 min before imaging at room temperature, and imaged using a SP8 Leica scanning confocal microscope with a 63× oil-immersion objective and a ×4 numerical zoom.
FRAP experiments were realized using the following settings: 2 s pre-bleach, 1 ms bleach and 60 s post-bleach at a frame rate of 1 image every 1 or 1.476 s for N/P and N/P/M2-1 experiments, respectively (increase of time-interval was applied to overcome additional photobleaching during the post-bleach step of the experiments). Photobleaching of mCherry was performed in a circular region at 100% laser intensity. In vitro FRAP data processing was identical to the in vitro FRAP data processing described above. Images from one in cellulo FRAP replicate are shown in Figure 1.
4.5. Minigenome assay
BSRT7/5 cells at 90% confluence in 96-well dishes were transfected with a plasmid mixture containing 125 ng of pM/Luc, 125 ng of pN, 125 ng of pP, 62.5 ng of pL, and 31 ng of pM2-1 as well as 31 ng of pRSV-β-Gal (Promega) to normalize transfection efficiencies [21]. Transfections were done in triplicate, and each independent transfection was performed three times. Cells were harvested 24 h post-transfection, then lyzed in luciferase lysis buffer (30 mM Tris pH 7.9, 10 mM MgCl2, 1 mM DTT, 1% Triton X-100, and 15% glycerol). The luciferase activities were determined for each cell lysate with an Infinite 200 Pro (Tecan, Männedorf, Switzerland) and normalized based on β-galactosidase (β-Gal) expression.
4.6. Expression and purification of the recombinant proteins
The Escherichia coli BL21 (DE3) bacteria strain (Novagen, Madison, WI) was transformed with the plasmids. Cultures were grown at 37 °C in 2xYT medium containing either 100 µg/ml of ampiciline (pGEX vectors) or 50 μg/ml kanamycin (pET vectors). After 8 h, an equal volume of 2xYT medium containing antibiotic was added to the cultures, and protein expression was induced by the addition of 80 μg/ml isopropyl β-d-1-thiogalactopyranoside (IPTG) overnight at 28 °C. Bacteria were then harvested by centrifugation. Purification of recombinant N, P, and M2-1core proteins has already been described ([8,27,37].
For M2-1-mCherry (WT and R151K mutant) purification, pellets were resuspended in lysis buffer (20 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.1% Triton X-100, 1 mg/ml lysozyme, and complete protease inhibitor cocktail (Roche)). After incubation on ice for 1h, the lysates were sonicated and benzonase (Novagen) (final concentration 5 U/ml) was added to the lysate, followed by incubation for 30 min at room temperature, before addition of NaCl up to a concentration of 1 M. After centrifugation at 10,000 g for 30 min at 4 °C, the lysates were incubated with Glutathione-Sepharose 4B beads (GE Healthcare) for 1 h at room temperature. Beads were washed three times with washing high-salt buffer (20 mM Tris–HCl, pH 7.4, 1 M NaCl) and three times with washing low-salt buffer (20 mM Tris–HCl, pH 7.4, 150 mM NaCl). To isolate GST-free M2-1-mCherry, beads were incubated with thrombin (Novagen). Purified proteins were then loaded on a Hi-Load 16/600 Superdex 200 column (Cytiva) and eluted in 20 mM Tris–HCl, pH 7.4, 150 mM NaCl. Finally, proteins were concentrated using a centrifugal concentrator with a MWCO of 100 kDa (Vivaspin turbo 4, Sartorius).
4.7. Band shift on native agarose gels.
Recombinant M2-1-mCherry (3 µM) and P proteins (30 µM) in 20 mM TrisHCl, pH 7.4,150 mM NaCl buffer were co-incubated for 1h at room temperature. CPM (675 µM) or equivalent volume of DMSO was then added to the samples before incubation 30 min at room temperature. tRNAs (10 µM) were then added, and samples incubated for 1h at room temperature. 50% sucrose loading buffer was added to the samples before loading on native 0.9% agarose gel stained with SYBR Safe (Invitrogen). The migration was performed in Tris–Glycine buffer during 6h at 80 V, before gel staining with amido black 10B.
4.8. Pulldown assay
GST-M2-1core proteins fixed on bead were incubated in the presence of purified recombinant P protein in PBS, in a final volume of 300 µl, in the presence of 400 µM CPM or DMSO (control condition). After 1 hour of incubation under agitation at 4°C beads were rinsed 2 times in 500 µl PBS and resuspended in the presence of 160 µg/ml of tRNA for 30 min, then washed 3 times in 500 µl PBS. 40 µl of Laemmli were added to beads, before boiling 5 min at 95°C for analysis by SDS-PAGE and Coomassie blue staining.
4.9. Docking of CPM onto M2-1
The structure of the CPM molecule (CAS 4449-51-8) was retrieved from PUBCHEM and converted into MOL2 format. Docking was performed on the Swiss-dock server (http://www.swissdock.ch/docking#). Docking was performed on the M2-1 tetramer and on the M2-1−P protomer.
4.10. P peptide fold prediction
Structure predictions were run on the PEP-FOLD 3 server (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/) [41], using peptides spanning P residues 95-110. This stretch comprises the M2-1 binding region.
4.11. Microscale thermophoresis
Microscale thermophoresis is a technology that uses the motion of fluorescent molecules along a microscale temperature gradient to detect any changes in their hydration shell, which can be induced by the binding to a partner [42,43,44,45]}. A fixed concentration of FITC-P95-112 (100 nM) was incubated with increasing amounts of M2-1 core domain at room temperature for 15 min in PBS and 1% DMSO containing either 0 or 1 or 10 μM of CPM. The measurements were performed for 30 s using a Monolith NT.115 (NanoTemper Technologies GmbH, Munich, Germany) at 20°C (blue LED power at 80% and infrared laser power at medium). The data from two independent measurements were averaged and analysed using the temperature-jump phase and the standard fitting mode (derived from law of mass action) of the NTAnalysis software (Nanotemper technologies).
4.12. NMR
Preparation of 15N-labeled M2-1 core domain was reported before ([27]. The same protocol was applied to wild-type and the R151K mutant. M2-1 core domain was in PBS pH 6.4 buffer supplemented with 1 mM DTT and 7.5% D2O to lock the NMR spectrometer frequency. Measurements were carried out on a Bruker 700 MHz spectrometer equipped with a TXO cryoprobe. The temperature was set to 298 K. 1H-15N correlation spectra were acquired using the BTROSY sequence. NMR data were processed with TopSpin 4.0 software (Bruker) and analysed with CcpNmr Analysis Assign 3.1 software [46]. 1H chemical shifts were referenced to DSS. Chemical shift assignment of the wild-type M2-1 core domain was reported previously [47].
FITC-P95-112 peptide was purchased from Proteogenix. A stock solution was prepared by dissolving peptide in pure water, and by adjusting the pH to neutral by addition of 1 M NaOH. The final concentration was 1 mM and determined by UV-VIS, using ε(500nM) = 80.000 cm-1.M-1. Cyclopamine was dissolved in DMSO-d6 (Eurisotop) at 5 mg/mL, equivalent to 12 mM. Interaction experiments with FITC-P95-112 were performed using a constant protein concentration of 105 and 115 µM for WT and R151K mutant, respectively, and by adding small volumes of concentrated peptides, so that protein dilution remained negligible. Titration points were made with 0, 0.1, 0.25, 0.5, 1.0 and 1.5 molar equivalents of peptide.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Droplets size is not altered by M2-1 or CPM addition; Figure S2: CPM does not bind to M2-1core with high affinity, Figure S3: CPM does not perturb the complex between 15N-labeled M2-1core domain and the FITC-P95-112 peptide; Figure S4: Analysis of the interaction between FITC-P95-112 and CPM by NMR; Figure S5: CPM does not affect the affinity of the complex between M2-1 core domain and P90-112 peptide; Figure S6: Impact of the R151K mutation on the structure of M2-1, Figure S7: Impact of the Y102L mutation on the conformation of the M2-1 binding motif in RSV P.
Author Contributions
Conceptualization, M.G., M.-A. R.-W., C. S., D. M., and J.-F.E; methodology, M.G., M.-A. R.-W., C. S., D. M., J.R.-B. and C.D.; validation, M.G., M.-A. R.-W., C. S., D. M.; formal analysis, C.D., J.R.-B., D.M., J.F., C.-A. R.., C.S and M.G..; investigation, C.D., J.R.-B., D.M., J.F., C.-A. R.., C.S and M.G..; writing—original draft preparation, M.-A R.-W. and M.G.; writing—review and editing, M.G., M.-A. R.-W., D.M., C. S., and J.-F.E.; supervision, M.-A. R.-W. and M.G.; project administration, M.G.; funding acquisition, M.G., M.-A. R.-W., and C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the French Agence Nationale de la Recherche, specific programs ANR Antibronchio (ANR-19-CE18-0012-01) and ANR RSVFact (ANR-21-CE15-0030-02), and the ATIP-AVENIR INSERM program and a grant from the Fondation Del Duca - Institut de France. Protein purification benefited from the purchase of a gel filtration system funded by the Région Ile de France (DIM- OneHealth 2018).
Acknowledgments
We thank Ralph Altmeyer (Medusa Therapeutics) for extensive discussions, and Thomas Edwards and John Barr (University of Leeds) for crystallography trials. We are grateful to Drs. Paola Llinas and Julie Ménétrey (I2BC, UMR9198, Gif-sur-Yvette) for advices and assistance for MST experiments which were performed with an equipment from the Macromolecular interaction measurements Platform of I2BC supported by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01. We thank the Cymages platform for access to the Leica SP8 microscope, which was supported by grants from the region Ile-de-France.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sagan, S. M.; Weber, S. C., Let's phase it: viruses are master architects of biomolecular condensates. Trends Biochem Sci 2023, 48, (3), 229-243.
- Li, H.; Ernst, C.; Kolonko-Adamska, M.; Greb-Markiewicz, B.; Man, J.; Parissi, V.; Ng, B. W., Phase separation in viral infections. Trends Microbiol 2022, 30, (12), 1217-1231.
- Wei, W.; Bai, L.; Yan, B.; Meng, W.; Wang, H.; Zhai, J.; Si, F.; Zheng, C., When liquid-liquid phase separation meets viral infections. Front Immunol 2022, 13, 985622.
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J. F.; Rameix-Welti, M. A., Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat Commun 2017, 8, (1), 563.
- Jobe, F.; Simpson, J.; Hawes, P.; Guzman, E.; Bailey, D., Respiratory Syncytial Virus Sequesters NF-kappaB Subunit p65 to Cytoplasmic Inclusion Bodies To Inhibit Innate Immune Signaling. J Virol 2020, 94, (22).
- Afonso, C. L.; Amarasinghe, G. K.; Banyai, K.; Bao, Y.; Basler, C. F.; Bavari, S.; Bejerman, N.; Blasdell, K. R.; Briand, F. X.; Briese, T.; Bukreyev, A.; Calisher, C. H.; Chandran, K.; Cheng, J.; Clawson, A. N.; Collins, P. L.; Dietzgen, R. G.; Dolnik, O.; Domier, L. L.; Durrwald, R.; Dye, J. M.; Easton, A. J.; Ebihara, H.; Farkas, S. L.; Freitas-Astua, J.; Formenty, P.; Fouchier, R. A.; Fu, Y.; Ghedin, E.; Goodin, M. M.; Hewson, R.; Horie, M.; Hyndman, T. H.; Jiang, D.; Kitajima, E. W.; Kobinger, G. P.; Kondo, H.; Kurath, G.; Lamb, R. A.; Lenardon, S.; Leroy, E. M.; Li, C. X.; Lin, X. D.; Liu, L.; Longdon, B.; Marton, S.; Maisner, A.; Muhlberger, E.; Netesov, S. V.; Nowotny, N.; Patterson, J. L.; Payne, S. L.; Paweska, J. T.; Randall, R. E.; Rima, B. K.; Rota, P.; Rubbenstroth, D.; Schwemmle, M.; Shi, M.; Smither, S. J.; Stenglein, M. D.; Stone, D. M.; Takada, A.; Terregino, C.; Tesh, R. B.; Tian, J. H.; Tomonaga, K.; Tordo, N.; Towner, J. S.; Vasilakis, N.; Verbeek, M.; Volchkov, V. E.; Wahl-Jensen, V.; Walsh, J. A.; Walker, P. J.; Wang, D.; Wang, L. F.; Wetzel, T.; Whitfield, A. E.; Xie, J. T.; Yuen, K. Y.; Zhang, Y. Z.; Kuhn, J. H., Taxonomy of the order Mononegavirales: update 2016. Arch Virol 2016, 161, (8), 2351-60.
- Garcia, J.; Garcia-Barreno, B.; Vivo, A.; Melero, J. A., Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 1993, 195, (1), 243-7.
- Galloux, M.; Risso-Ballester, J.; Richard, C. A.; Fix, J.; Rameix-Welti, M. A.; Eleouet, J. F., Minimal Elements Required for the Formation of Respiratory Syncytial Virus Cytoplasmic Inclusion Bodies In Vivo and In Vitro. mBio 2020, 11, (5).
- Shi, T.; McAllister, D. A.; O'Brien, K. L.; Simoes, E. A. F.; Madhi, S. A.; Gessner, B. D.; Polack, F. P.; Balsells, E.; Acacio, S.; Aguayo, C.; Alassani, I.; Ali, A.; Antonio, M.; Awasthi, S.; Awori, J. O.; Azziz-Baumgartner, E.; Baggett, H. C.; Baillie, V. L.; Balmaseda, A.; Barahona, A.; Basnet, S.; Bassat, Q.; Basualdo, W.; Bigogo, G.; Bont, L.; Breiman, R. F.; Brooks, W. A.; Broor, S.; Bruce, N.; Bruden, D.; Buchy, P.; Campbell, S.; Carosone-Link, P.; Chadha, M.; Chipeta, J.; Chou, M.; Clara, W.; Cohen, C.; de Cuellar, E.; Dang, D. A.; Dash-Yandag, B.; Deloria-Knoll, M.; Dherani, M.; Eap, T.; Ebruke, B. E.; Echavarria, M.; de Freitas Lazaro Emediato, C. C.; Fasce, R. A.; Feikin, D. R.; Feng, L.; Gentile, A.; Gordon, A.; Goswami, D.; Goyet, S.; Groome, M.; Halasa, N.; Hirve, S.; Homaira, N.; Howie, S. R. C.; Jara, J.; Jroundi, I.; Kartasasmita, C. B.; Khuri-Bulos, N.; Kotloff, K. L.; Krishnan, A.; Libster, R.; Lopez, O.; Lucero, M. G.; Lucion, F.; Lupisan, S. P.; Marcone, D. N.; McCracken, J. P.; Mejia, M.; Moisi, J. C.; Montgomery, J. M.; Moore, D. P.; Moraleda, C.; Moyes, J.; Munywoki, P.; Mutyara, K.; Nicol, M. P.; Nokes, D. J.; Nymadawa, P.; da Costa Oliveira, M. T.; Oshitani, H.; Pandey, N.; Paranhos-Baccala, G.; Phillips, L. N.; Picot, V. S.; Rahman, M.; Rakoto-Andrianarivelo, M.; Rasmussen, Z. A.; Rath, B. A.; Robinson, A.; Romero, C.; Russomando, G.; Salimi, V.; Sawatwong, P.; Scheltema, N.; Schweiger, B.; Scott, J. A. G.; Seidenberg, P.; Shen, K.; Singleton, R.; Sotomayor, V.; Strand, T. A.; Sutanto, A.; Sylla, M.; Tapia, M. D.; Thamthitiwat, S.; Thomas, E. D.; Tokarz, R.; Turner, C.; Venter, M.; Waicharoen, S.; Wang, J.; Watthanaworawit, W.; Yoshida, L. M.; Yu, H.; Zar, H. J.; Campbell, H.; Nair, H.; Network, R. S. V. G. E., Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 2017, 390, (10098), 946-958.
- Busack, B.; Shorr, A. F., Going Viral-RSV as the Neglected Adult Respiratory Virus. Pathogens 2022, 11, (11).
- Mazur, N. I.; Terstappen, J.; Baral, R.; Bardaji, A.; Beutels, P.; Buchholz, U. J.; Cohen, C.; Crowe, J. E., Jr.; Cutland, C. L.; Eckert, L.; Feikin, D.; Fitzpatrick, T.; Fong, Y.; Graham, B. S.; Heikkinen, T.; Higgins, D.; Hirve, S.; Klugman, K. P.; Kragten-Tabatabaie, L.; Lemey, P.; Libster, R.; Lowensteyn, Y.; Mejias, A.; Munoz, F. M.; Munywoki, P. K.; Mwananyanda, L.; Nair, H.; Nunes, M. C.; Ramilo, O.; Richmond, P.; Ruckwardt, T. J.; Sande, C.; Srikantiah, P.; Thacker, N.; Waldstein, K. A.; Weinberger, D.; Wildenbeest, J.; Wiseman, D.; Zar, H. J.; Zambon, M.; Bont, L., Respiratory syncytial virus prevention within reach: the vaccine and monoclonal antibody landscape. Lancet Infect Dis 2023, 23, (1), e2-e21.
- Vidal Valero, M., 'A good day': FDA approves world's first RSV vaccine. Nature 2023, 617, (7960), 234-235.
- Pinquier, D.; Crepey, P.; Tissieres, P.; Vabret, A.; Roze, J. C.; Dubos, F.; Cahn-Sellem, F.; Javouhey, E.; Cohen, R.; Weil-Olivier, C., Preventing Respiratory Syncytial Virus in Children in France: A Narrative Review of the Importance of a Reinforced Partnership Between Parents, Healthcare Professionals, and Public Health Authorities. Infect Dis Ther 2023, 12, (2), 317-332.
- Hammitt, L. L.; Dagan, R.; Yuan, Y.; Baca Cots, M.; Bosheva, M.; Madhi, S. A.; Muller, W. J.; Zar, H. J.; Brooks, D.; Grenham, A.; Wahlby Hamren, U.; Mankad, V. S.; Ren, P.; Takas, T.; Abram, M. E.; Leach, A.; Griffin, M. P.; Villafana, T.; Group, M. S., Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants. N Engl J Med 2022, 386, (9), 837-846.
- Valarcher, J. F.; Taylor, G., Bovine respiratory syncytial virus infection. Veterinary research 2007, 38, (2), 153-80.
- Kimman, T. G.; Westenbrink, F.; Straver, P. J., Priming for local and systemic antibody memory responses to bovine respiratory syncytial virus: effect of amount of virus, virus replication, route of administration and maternal antibodies. Vet Immunol Immunopathol 1989, 22, (2), 145-60.
- Ellis, J. A., How efficacious are vaccines against bovine respiratory syncytial virus in cattle? Vet Microbiol 2017, 206, 59-68.
- Bailly, B.; Richard, C. A.; Sharma, G.; Wang, L.; Johansen, L.; Cao, J.; Pendharkar, V.; Sharma, D. C.; Galloux, M.; Wang, Y.; Cui, R.; Zou, G.; Guillon, P.; von Itzstein, M.; Eleouet, J. F.; Altmeyer, R., Targeting human respiratory syncytial virus transcription anti-termination factor M2-1 to inhibit in vivo viral replication. Sci Rep 2016, 6, 25806.
- Risso-Ballester, J.; Galloux, M.; Cao, J.; Le Goffic, R.; Hontonnou, F.; Jobart-Malfait, A.; Desquesnes, A.; Sake, S. M.; Haid, S.; Du, M.; Zhang, X.; Zhang, H.; Wang, Z.; Rincheval, V.; Zhang, Y.; Pietschmann, T.; Eleouet, J. F.; Rameix-Welti, M. A.; Altmeyer, R., A condensate-hardening drug blocks RSV replication in vivo. Nature 2021, 595, (7868), 596-599.
- Fix, J.; Descamps, D.; Galloux, M.; Ferret, C.; Bouguyon, E.; Zohari, S.; Naslund, K.; Hagglund, S.; Altmeyer, R.; Valarcher, J. F.; Riffault, S.; Eleouet, J. F., Screening antivirals with a mCherry-expressing recombinant bovine respiratory syncytial virus: a proof of concept using cyclopamine. Vet Res 2023, 54, (1), 36.
- Tran, T. L.; Castagne, N.; Dubosclard, V.; Noinville, S.; Koch, E.; Moudjou, M.; Henry, C.; Bernard, J.; Yeo, R. P.; Eleouet, J. F., The respiratory syncytial virus M2-1 protein forms tetramers and interacts with RNA and P in a competitive manner. J Virol 2009, 83, (13), 6363-74.
- Esperante, S. A.; Paris, G.; de Prat-Gay, G., Modular unfolding and dissociation of the human respiratory syncytial virus phosphoprotein p and its interaction with the m(2-1) antiterminator: a singular tetramer-tetramer interface arrangement. Biochemistry 2012, 51, (41), 8100-10.
- Tanner, S. J.; Ariza, A.; Richard, C. A.; Kyle, H. F.; Dods, R. L.; Blondot, M. L.; Wu, W.; Trincao, J.; Trinh, C. H.; Hiscox, J. A.; Carroll, M. W.; Silman, N. J.; Eleouet, J. F.; Edwards, T. A.; Barr, J. N., Crystal structure of the essential transcription antiterminator M2-1 protein of human respiratory syncytial virus and implications of its phosphorylation. Proc Natl Acad Sci U S A 2014, 111, (4), 1580-5.
- Leyrat, C.; Renner, M.; Harlos, K.; Huiskonen, J. T.; Grimes, J. M., Drastic changes in conformational dynamics of the antiterminator M2-1 regulate transcription efficiency in Pneumovirinae. Elife 2014, 3, e02674.
- Richard, C. A.; Rincheval, V.; Lassoued, S.; Fix, J.; Cardone, C.; Esneau, C.; Nekhai, S.; Galloux, M.; Rameix-Welti, M. A.; Sizun, C.; Eleouet, J. F., RSV hijacks cellular protein phosphatase 1 to regulate M2-1 phosphorylation and viral transcription. PLoS Pathog 2018, 14, (3), e1006920.
- Cardone, C.; Caseau, C. M.; Pereira, N.; Sizun, C., Pneumoviral Phosphoprotein, a Multidomain Adaptor-Like Protein of Apparent Low Structural Complexity and High Conformational Versatility. Int J Mol Sci 2021, 22, (4).
- Blondot, M. L.; Dubosclard, V.; Fix, J.; Lassoued, S.; Aumont-Nicaise, M.; Bontems, F.; Eleouet, J. F.; Sizun, C., Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. PLoS pathogens 2012, 8, (5), e1002734.
- Selvaraj, M.; Yegambaram, K.; Todd, E.; Richard, C. A.; Dods, R. L.; Pangratiou, G. M.; Trinh, C. H.; Moul, S. L.; Murphy, J. C.; Mankouri, J.; Eleouet, J. F.; Barr, J. N.; Edwards, T. A., The Structure of the Human Respiratory Syncytial Virus M2-1 Protein Bound to the Interaction Domain of the Phosphoprotein P Defines the Orientation of the Complex. mBio 2018, 9, (6).
- Gao, Y.; Cao, D.; Pawnikar, S.; John, K. P.; Ahn, H. M.; Hill, S.; Ha, J. M.; Parikh, P.; Ogilvie, C.; Swain, A.; Yang, A.; Bell, A.; Salazar, A.; Miao, Y.; Liang, B., Structure of the Human Respiratory Syncytial Virus M2-1 Protein in Complex with a Short Positive-Sense Gene-End RNA. Structure 2020, 28, (9), 979-990 e4.
- Molina, I. G.; Esperante, S. A.; Marino-Buslje, C.; Chemes, L. B.; de Prat-Gay, G., Cooperative RNA Recognition by a Viral Transcription Antiterminator. J Mol Biol 2018, 430, (6), 777-792.
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V., Liquid-Liquid Phase Separation by Intrinsically Disordered Protein Regions of Viruses: Roles in Viral Life Cycle and Control of Virus-Host Interactions. Int J Mol Sci 2020, 21, (23).
- Lopez, N.; Camporeale, G.; Salgueiro, M.; Borkosky, S. S.; Visentin, A.; Peralta-Martinez, R.; Loureiro, M. E.; de Prat-Gay, G., Deconstructing virus condensation. PLoS Pathog 2021, 17, (10), e1009926.
- Gonnin, L.; Richard, C. A.; Gutsche, I.; Chevret, D.; Troussier, J.; Vasseur, J. J.; Debart, F.; Eleouet, J. F.; Galloux, M., Importance of RNA length for in vitro encapsidation by the nucleoprotein of human respiratory syncytial virus. J Biol Chem 2022, 298, (9), 102337.
- Pereira, N.; Cardone, C.; Lassoued, S.; Galloux, M.; Fix, J.; Assrir, N.; Lescop, E.; Bontems, F.; Eleouet, J. F.; Sizun, C., New Insights into Structural Disorder in Human Respiratory Syncytial Virus Phosphoprotein and Implications for Binding of Protein Partners. J Biol Chem 2017, 292, (6), 2120-2131.
- Banani, S. F.; Lee, H. O.; Hyman, A. A.; Rosen, M. K., Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 2017, 18, (5), 285-298.
- Sourimant, J.; Rameix-Welti, M. A.; Gaillard, A. L.; Chevret, D.; Galloux, M.; Gault, E.; Eleouet, J. F., Fine mapping and characterization of the L-polymerase-binding domain of the respiratory syncytial virus phosphoprotein. Journal of virology 2015, 89, (8), 4421-33.
- Galloux, M.; Gabiane, G.; Sourimant, J.; Richard, C. A.; England, P.; Moudjou, M.; Aumont-Nicaise, M.; Fix, J.; Rameix-Welti, M. A.; Eleouet, J. F., Identification and Characterization of the Binding Site of the Respiratory Syncytial Virus Phosphoprotein to RNA-Free Nucleoprotein. Journal of virology 2015, 89, (7), 3484-96.
- Buchholz, U. J.; Finke, S.; Conzelmann, K. K., Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. Journal of virology 1999, 73, (1), 251-9.
- de Chaumont, F.; Dallongeville, S.; Chenouard, N.; Herve, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; Lagache, T.; Dufour, A.; Olivo-Marin, J. C., Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods 2012, 9, (7), 690-6.
- Giakoumakis, N. N.; Rapsomaniki, M. A.; Lygerou, Z., Analysis of Protein Kinetics Using Fluorescence Recovery After Photobleaching (FRAP). Methods Mol Biol 2017, 1563, 243-267.
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P., PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res 2016, 44, (W1), W449-54.
- Baaske, P.; Wienken, C. J.; Reineck, P.; Duhr, S.; Braun, D., Optical thermophoresis for quantifying the buffer dependence of aptamer binding. Angew Chem Int Ed Engl 2010, 49, (12), 2238-41.
- Zillner, K.; Jerabek-Willemsen, M.; Duhr, S.; Braun, D.; Langst, G.; Baaske, P., Microscale thermophoresis as a sensitive method to quantify protein: nucleic acid interactions in solution. Methods Mol Biol 2012, 815, 241-52.
- Jerabek-Willemsen, M.; Wienken, C. J.; Braun, D.; Baaske, P.; Duhr, S., Molecular interaction studies using microscale thermophoresis. Assay Drug Dev Technol 2011, 9, (4), 342-53.
- Duhr, S.; Braun, D., Why molecules move along a temperature gradient. Proc Natl Acad Sci U S A 2006, 103, (52), 19678-82.
- Skinner, S. P.; Fogh, R. H.; Boucher, W.; Ragan, T. J.; Mureddu, L. G.; Vuister, G. W., CcpNmr AnalysisAssign: a flexible platform for integrated NMR analysis. J Biomol NMR 2016, 66, (2), 111-124.
- Dubosclard, V.; Blondot, M. L.; Eleouet, J. F.; Bontems, F.; Sizun, C., 1H, 13C, and 15N resonance assignment of the central domain of hRSV transcription antitermination factor M2-1. Biomol NMR Assign 2011, 5, (2), 237-9.
Figure 1.
Hardening of pseudo IBs by CPM in cells requires the presence of M2-1 . (A-F) P-BFP mobility in biomolecular condensates was analysed by FRAP in HEp-2 cells transiently expressing N, P-BFP, ± M2-1 (B) or M2-1(R151K) (C), and treated with 10 µM CPM (blue) or DMSO (red) for one hour. (A-C) Representative images of time-lapse microscopy from FRAP experiments (left panels). Scale bars: 2 µm. Quantification of spontaneous re-distribution of fluorescence after photobleaching, corrected for background and bleaching during post-bleach imaging, and normalized to the post-bleach signals (right panels). Data are represented as mean ± SD of ≥ 20 FRAP events out of 2 independent experiments. (D) Hypotonic shock was applied on HEp-2 cells transiently expressing N, P-BFP ± M2-1 and treated with 10 µM CPM (blue) or DMSO (red) for one hour. Representative images of the impact of hypotonic shock on IBs (left panel). Proportions of IBs resisting a 5 min hypotonic shock are represented as mean percentages ± SD in the right panel (8 to 9 acquisitions from two independent experiments). Significance was tested using an unpaired t test followed by Welch correction; ***p < 0.001, ns = not significant. Scale bars: 10 µm.
Figure 1.
Hardening of pseudo IBs by CPM in cells requires the presence of M2-1 . (A-F) P-BFP mobility in biomolecular condensates was analysed by FRAP in HEp-2 cells transiently expressing N, P-BFP, ± M2-1 (B) or M2-1(R151K) (C), and treated with 10 µM CPM (blue) or DMSO (red) for one hour. (A-C) Representative images of time-lapse microscopy from FRAP experiments (left panels). Scale bars: 2 µm. Quantification of spontaneous re-distribution of fluorescence after photobleaching, corrected for background and bleaching during post-bleach imaging, and normalized to the post-bleach signals (right panels). Data are represented as mean ± SD of ≥ 20 FRAP events out of 2 independent experiments. (D) Hypotonic shock was applied on HEp-2 cells transiently expressing N, P-BFP ± M2-1 and treated with 10 µM CPM (blue) or DMSO (red) for one hour. Representative images of the impact of hypotonic shock on IBs (left panel). Proportions of IBs resisting a 5 min hypotonic shock are represented as mean percentages ± SD in the right panel (8 to 9 acquisitions from two independent experiments). Significance was tested using an unpaired t test followed by Welch correction; ***p < 0.001, ns = not significant. Scale bars: 10 µm.
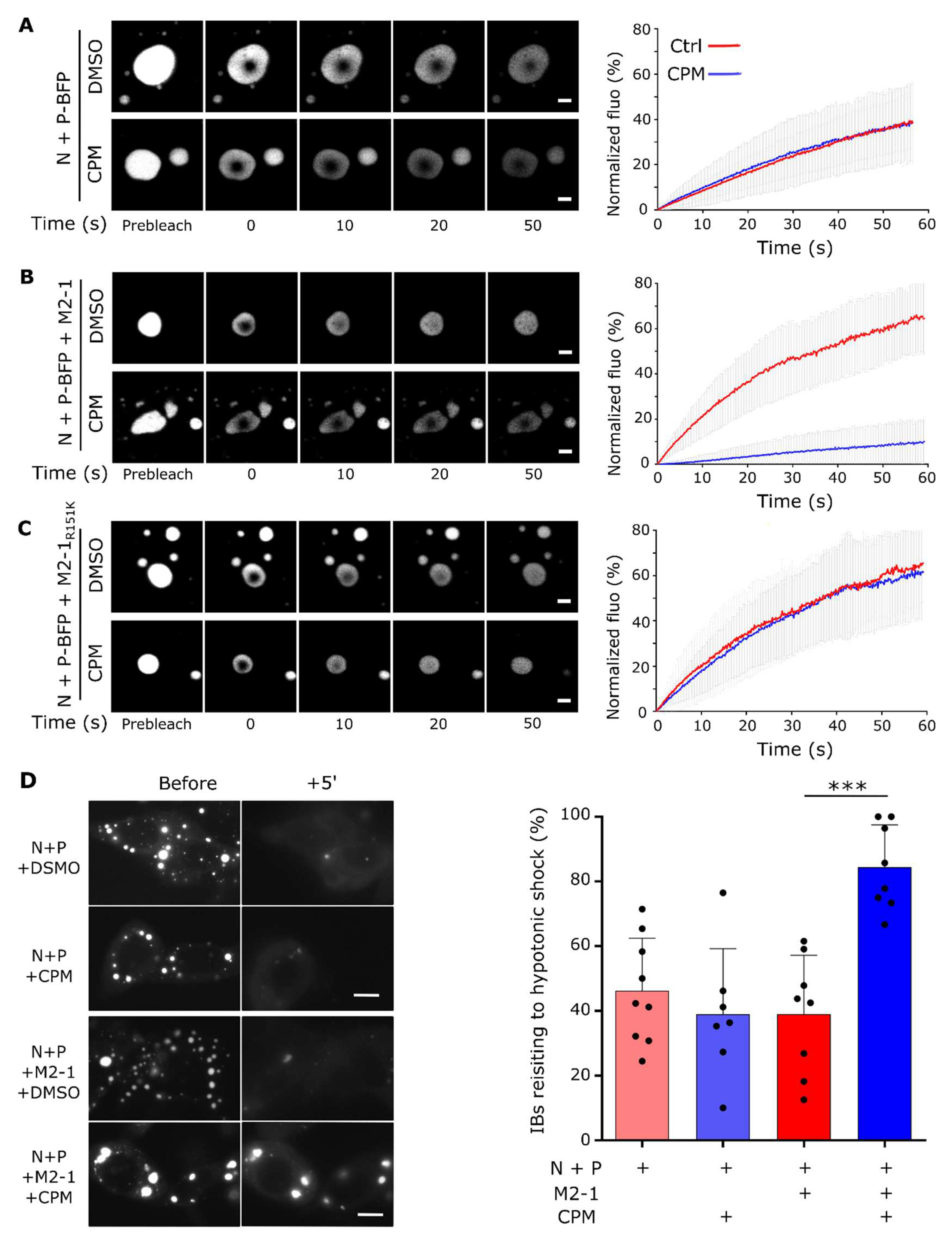
Figure 2.
Hardening of in vitro reconstituted pseudo-IBs by CPM depends on M2-1. (A) Recombinant GST-M2-1-mCherry proteins (WT and mutant R151K) were produced in E.coli. After GST cleavage, a gel filtration was performed to isolate M2-1-mCherry (left panel), (the arrow shows the peak of elution corresponding to M2-1-mCherry tetramer), and the purified proteins were analyzed by SDS-PAGE and Coomassie blue staining (right panel). (B-C) Recombinant mCherry-N and P-BFP (C) or N, P-BFP and M2-1-mCherry (D) proteins were co-incubated and phase separation was assessed using fluorescence microscopy. Scale bars: 10 µm. (D-F) mCherry-P (E, F, H, I) or M2-1-mCherry (G, J) mobility was analysed by FRAP in condensates following a 30 min incubation with 150 µM CPM (blue) or DMSO (red). Representative images of time-lapse microscopy from FRAP experiments on in vitro reconstituted pseudo-IBs (left panels). Scale bars 2 µm. Quantification of the spontaneous re-distribution of fluorescence of mCherry after photobleaching, corrected for background and bleaching during post-bleach imaging and normalized to the post-bleach signal (right panels). Data are represented as mean ± SD of ≥ 20 FRAP events out of 2 independent experiments.
Figure 2.
Hardening of in vitro reconstituted pseudo-IBs by CPM depends on M2-1. (A) Recombinant GST-M2-1-mCherry proteins (WT and mutant R151K) were produced in E.coli. After GST cleavage, a gel filtration was performed to isolate M2-1-mCherry (left panel), (the arrow shows the peak of elution corresponding to M2-1-mCherry tetramer), and the purified proteins were analyzed by SDS-PAGE and Coomassie blue staining (right panel). (B-C) Recombinant mCherry-N and P-BFP (C) or N, P-BFP and M2-1-mCherry (D) proteins were co-incubated and phase separation was assessed using fluorescence microscopy. Scale bars: 10 µm. (D-F) mCherry-P (E, F, H, I) or M2-1-mCherry (G, J) mobility was analysed by FRAP in condensates following a 30 min incubation with 150 µM CPM (blue) or DMSO (red). Representative images of time-lapse microscopy from FRAP experiments on in vitro reconstituted pseudo-IBs (left panels). Scale bars 2 µm. Quantification of the spontaneous re-distribution of fluorescence of mCherry after photobleaching, corrected for background and bleaching during post-bleach imaging and normalized to the post-bleach signal (right panels). Data are represented as mean ± SD of ≥ 20 FRAP events out of 2 independent experiments.
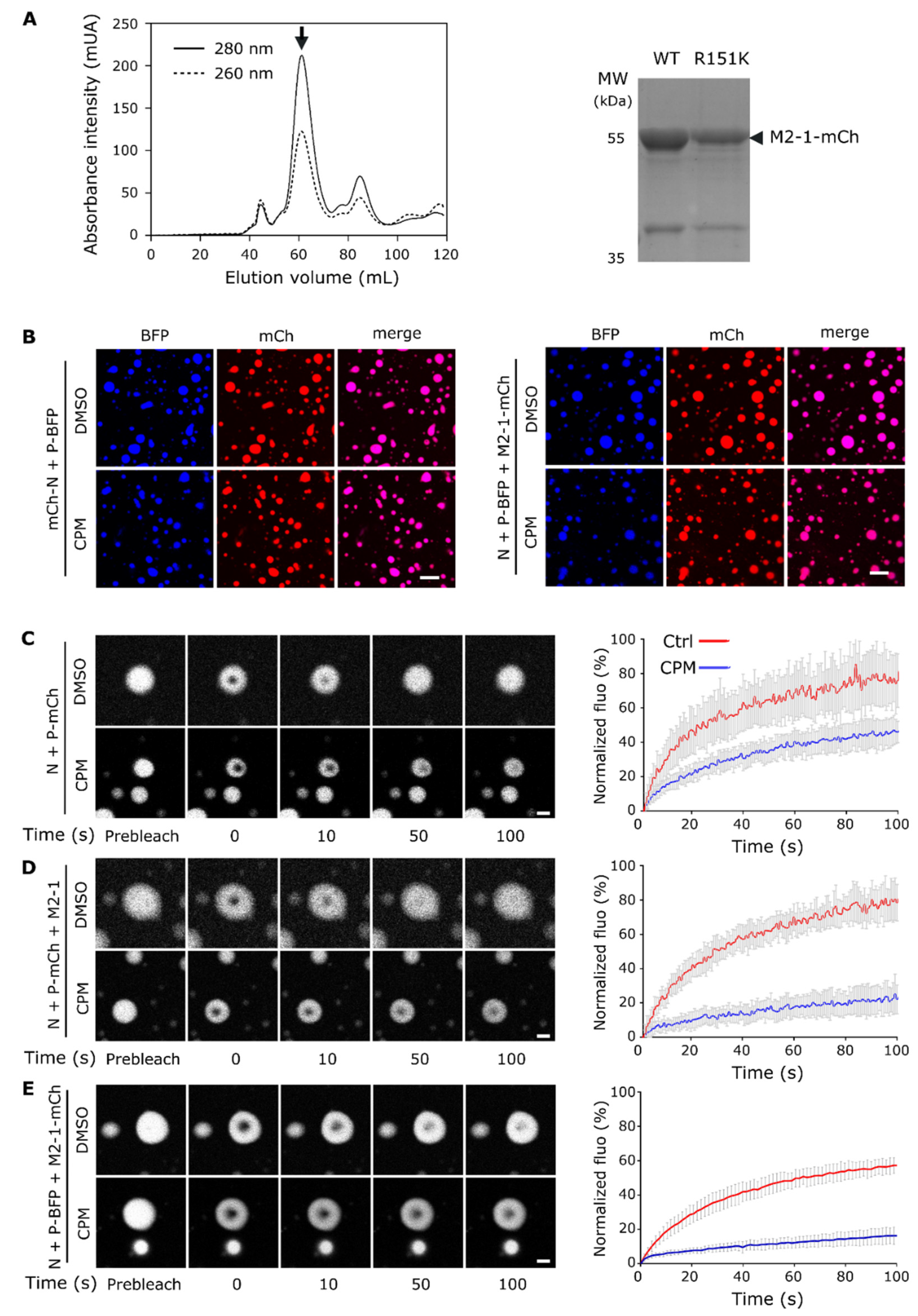
Figure 3.
CPM affects competition between P and tRNA for M2-1 binding in vitro. (A) Analysis of M2-1-mCherry migration alone or incubated in the presence of tRNA or P, in the absence or presence of CPM, by native polyacrylamide gel electrophoresis. M2-1-mCherry was observed using UV. (B) Pulldown of P by GST-M2-1core in the absence or the presence of CPM and/or tRNA; analyzed by SDS-PAGE and Coomassie blue staining.
Figure 3.
CPM affects competition between P and tRNA for M2-1 binding in vitro. (A) Analysis of M2-1-mCherry migration alone or incubated in the presence of tRNA or P, in the absence or presence of CPM, by native polyacrylamide gel electrophoresis. M2-1-mCherry was observed using UV. (B) Pulldown of P by GST-M2-1core in the absence or the presence of CPM and/or tRNA; analyzed by SDS-PAGE and Coomassie blue staining.
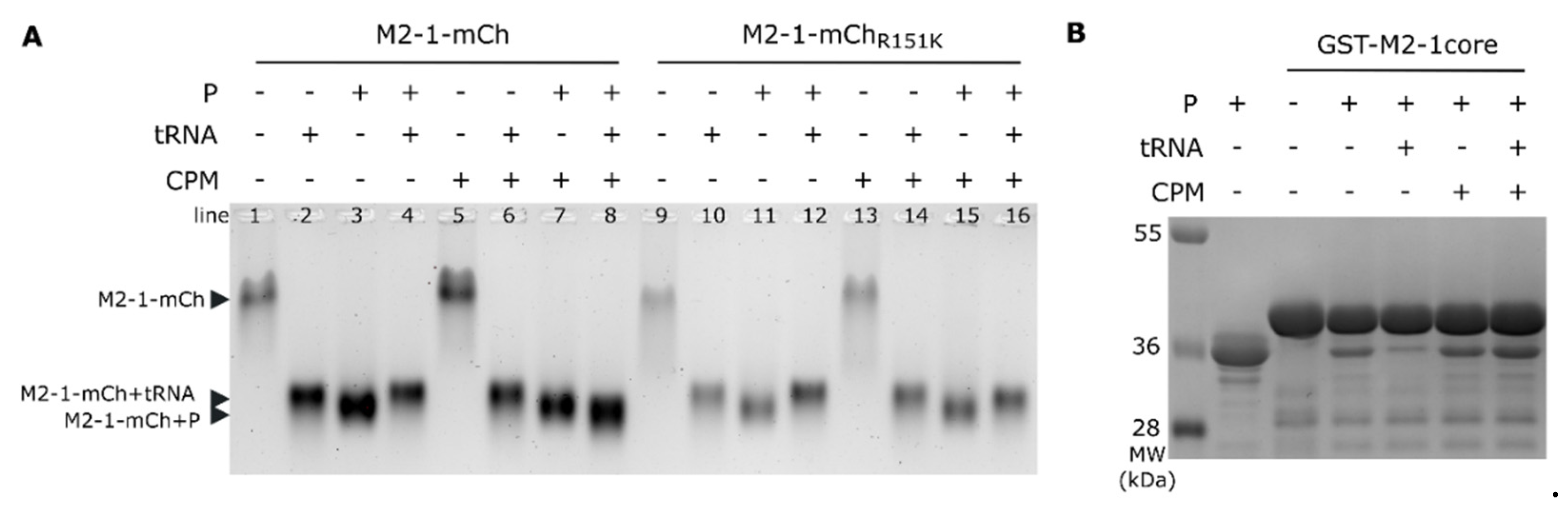
Figure 4.
The M2-1−P complex displays motions at the µs-ms time scale. (A) Binding of the FITC-P95-112 peptide to 15N-labeled M2-1core domain was followed by 2D NMR (1H frequency 700 MHz, temperature 298 K). Amide signals are annotated with the corresponding residue number. Increasing amounts (0.1 to 1.5 molar equivalents) of FITC-P95-112 peptide were added to 100 µM WT M2-1core and a 1H-15N BTROSY spectrum was acquired at each titrating point. Saturation was reached at a peptide:protein molar ratio of 1.5:1. (B) The signals of nearly all residues belonging to the P-binding site remain broad at the last titration point. They are mapped in red on the structure of M2-1core (PDB 2L9J). (C) Titration of 15N-labeled M2-1core R151K mutant by 0.1 to 1.5 molar equivalents FITC-P95-112 peptide was followed by measuring 1H-15N BTROSY spectra.
Figure 4.
The M2-1−P complex displays motions at the µs-ms time scale. (A) Binding of the FITC-P95-112 peptide to 15N-labeled M2-1core domain was followed by 2D NMR (1H frequency 700 MHz, temperature 298 K). Amide signals are annotated with the corresponding residue number. Increasing amounts (0.1 to 1.5 molar equivalents) of FITC-P95-112 peptide were added to 100 µM WT M2-1core and a 1H-15N BTROSY spectrum was acquired at each titrating point. Saturation was reached at a peptide:protein molar ratio of 1.5:1. (B) The signals of nearly all residues belonging to the P-binding site remain broad at the last titration point. They are mapped in red on the structure of M2-1core (PDB 2L9J). (C) Titration of 15N-labeled M2-1core R151K mutant by 0.1 to 1.5 molar equivalents FITC-P95-112 peptide was followed by measuring 1H-15N BTROSY spectra.
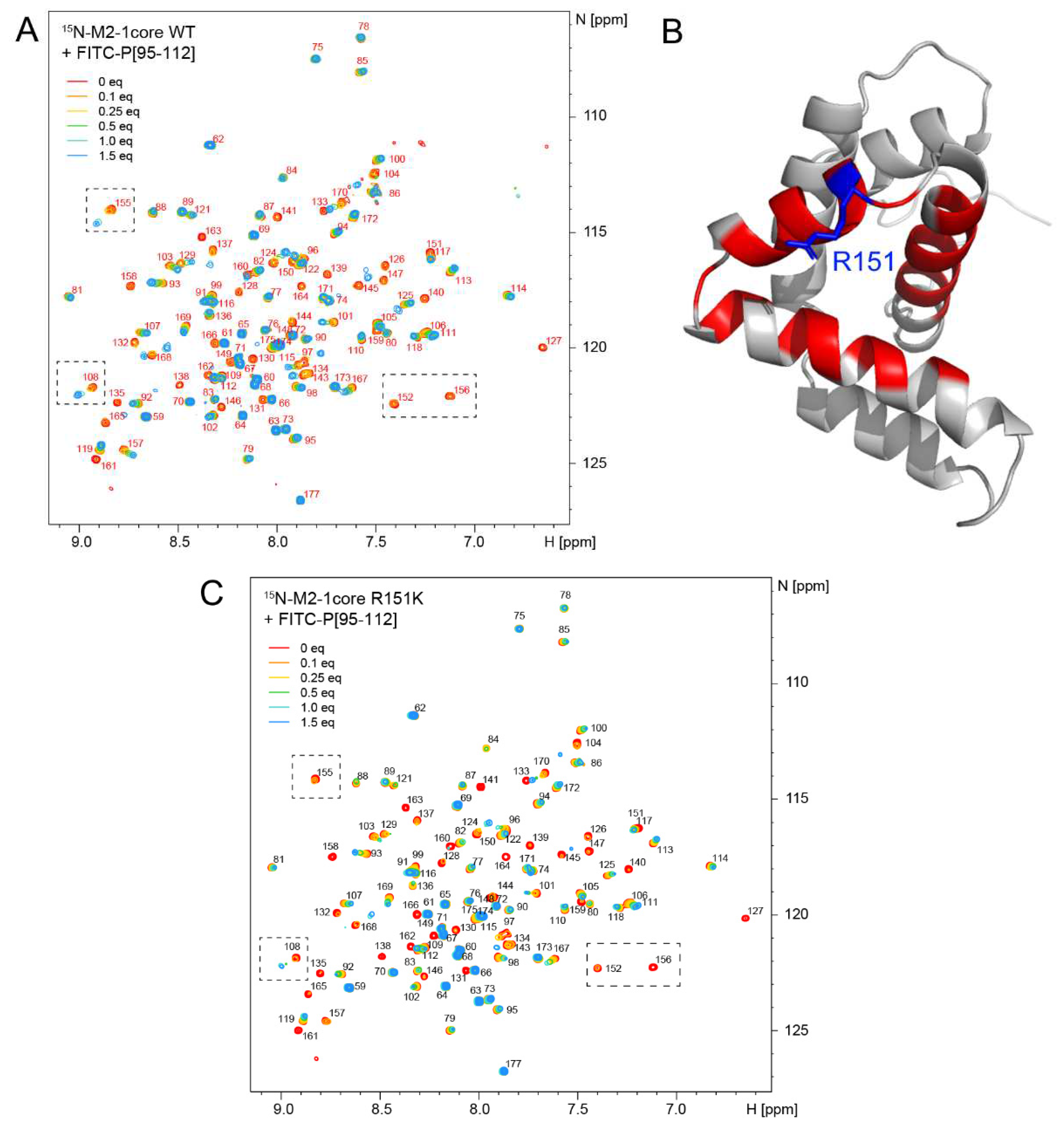
Figure 5.
Role of the residue Y102 of P protein in CPM antiviral activity. (A) Close-up view of the RSV M2-1−P binding site (PDB 6g0y). The four M2-1 protomers were structurally aligned. The M2-1 core domain is shown in gray ribbon, and the bound P90-110 peptide in green. The side chains of P and M2-1 residues that were previously shown to be critical for RSV replication in vitro by alanine scanning are shown in sticks. The cation-pi interaction between M2-1 R151 (blue) and P Y102 (yellow) is indicated with broken line. (B) Polymerase activity in the presence of mutated P and CPM. BSRT7/5 cells were transfected with plasmids encoding the N, P, L and M2-1 proteins and the M/Luc subgenomic minireplicon together with pCMV β-gal for transfection standardization. P mutants expressed instead of the corresponding wild type P are indicated below the histogram. Luciferase activity, reflecting viral RNA synthesis, was measured 24 h after transfection, normalized to the β-galactosidase activity, and expressed as percentage of the WT proteins activity. The mean value ± SD from 3 independent experiments performed in triplicate or quadruplicate are shown. Nested One way ANOVA followed by two-sided Dunn's multiple comparison tests against untreated group (ns= not significant, **** p< 0.0001; * p<0.05). Western blot analysis showing efficient expression of P mutant proteins in BSRT7/5 cells for one representative experiment is shown. (C) Analysis of M2-1-mCherry migration alone or incubated in the presence of tRNA or P mutants, in the absence or presence of CPM, by native polyacrylamide gel electrophoresis. M2-1-mCherry was observed using UV.
Figure 5.
Role of the residue Y102 of P protein in CPM antiviral activity. (A) Close-up view of the RSV M2-1−P binding site (PDB 6g0y). The four M2-1 protomers were structurally aligned. The M2-1 core domain is shown in gray ribbon, and the bound P90-110 peptide in green. The side chains of P and M2-1 residues that were previously shown to be critical for RSV replication in vitro by alanine scanning are shown in sticks. The cation-pi interaction between M2-1 R151 (blue) and P Y102 (yellow) is indicated with broken line. (B) Polymerase activity in the presence of mutated P and CPM. BSRT7/5 cells were transfected with plasmids encoding the N, P, L and M2-1 proteins and the M/Luc subgenomic minireplicon together with pCMV β-gal for transfection standardization. P mutants expressed instead of the corresponding wild type P are indicated below the histogram. Luciferase activity, reflecting viral RNA synthesis, was measured 24 h after transfection, normalized to the β-galactosidase activity, and expressed as percentage of the WT proteins activity. The mean value ± SD from 3 independent experiments performed in triplicate or quadruplicate are shown. Nested One way ANOVA followed by two-sided Dunn's multiple comparison tests against untreated group (ns= not significant, **** p< 0.0001; * p<0.05). Western blot analysis showing efficient expression of P mutant proteins in BSRT7/5 cells for one representative experiment is shown. (C) Analysis of M2-1-mCherry migration alone or incubated in the presence of tRNA or P mutants, in the absence or presence of CPM, by native polyacrylamide gel electrophoresis. M2-1-mCherry was observed using UV.
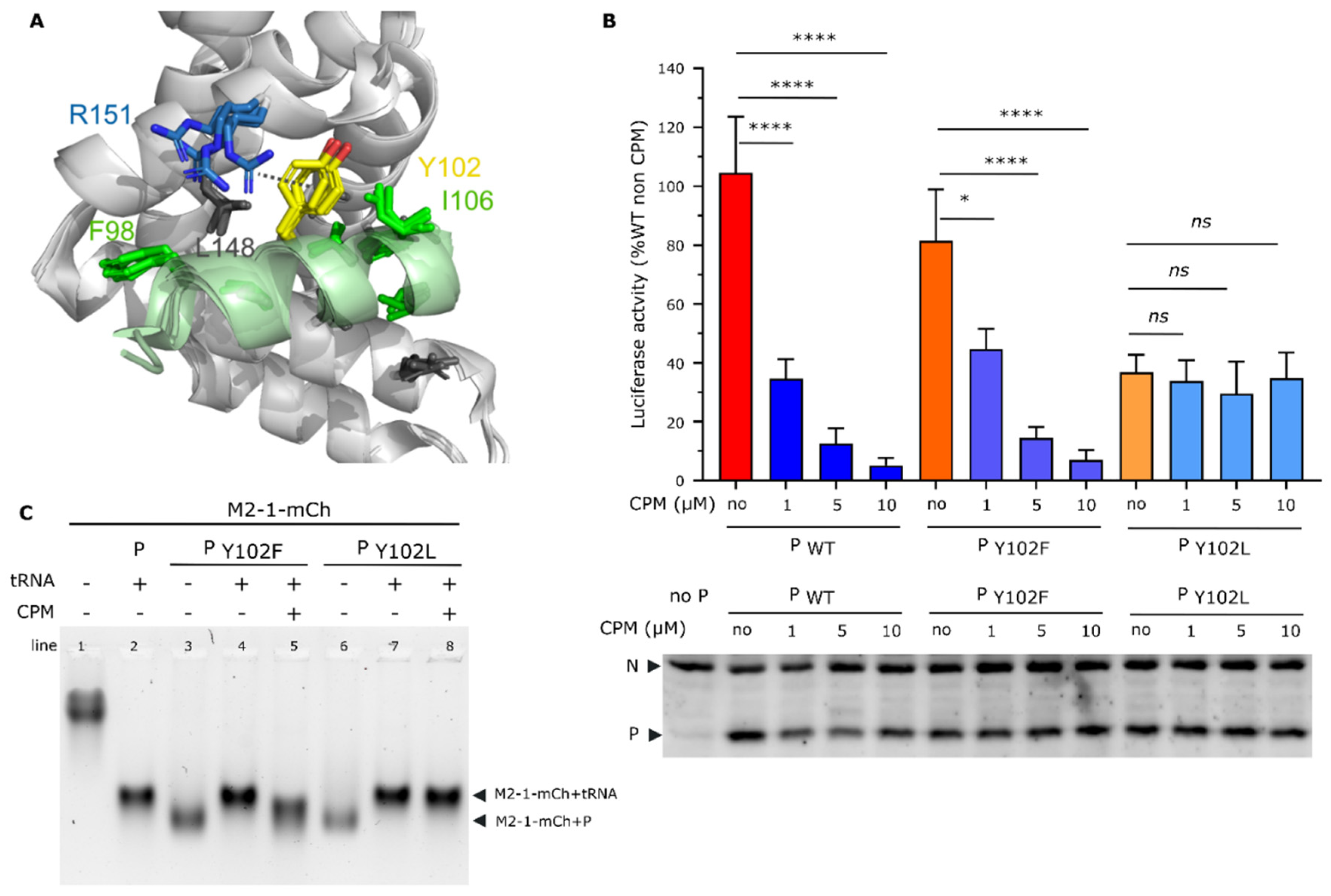
Figure 6.
CPM explores different binding sites on M2-1. Docking experiments were performed using (A) the M2-1 tetramer without peptide as well as (B) the M2-1 tetramer in complex with the P95-110 peptide (PDB 6g0y). Two views are shown for each, rotated by 180°. M2-1 is in yellow cartoon, and the R151 residue in red sticks. The P peptide is in brick red cartoon. CPM carbon atoms are in cyan sticks and spheres.
Figure 6.
CPM explores different binding sites on M2-1. Docking experiments were performed using (A) the M2-1 tetramer without peptide as well as (B) the M2-1 tetramer in complex with the P95-110 peptide (PDB 6g0y). Two views are shown for each, rotated by 180°. M2-1 is in yellow cartoon, and the R151 residue in red sticks. The P peptide is in brick red cartoon. CPM carbon atoms are in cyan sticks and spheres.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



