
Submitted:
23 June 2023
Posted:
26 June 2023
You are already at the latest version
Abstract
Despite being an abundant marine organism in Indonesia, black sea cucumber is still underutilised due to its slightly bitter taste. Previous studies have hinted at the potential of black sea cucumber as an anti-cancer agent. However, specific identification of bioactive compounds that can interact with cancer proteins is still lacking. In the same place, cancer ranks third as Indonesia's leading cause of death. Therefore, this study aims to identify potential anti-cancer compounds from black sea cucumbers using a comprehensive in silico drug discovery approach. This research uses machine learning, molecular docking, and ADMET analysis to identify bioactive compounds that specifically interact with cancer proteins. A combination of the Cascade Deep Forest algorithm and ECFP-AAIndex1 feature combination proved to be the most effective in predicting these interactions. Through molecular docking validation, four bioactive compounds with strong binding affinity were identified: Afimoxifene, Danazol, Taxifolin, and Terfenadine. ADMET analysis highlighted Taxifolin as the most promising candidate, as it passed most ADMET parameters. Further wet laboratory studies are required to confirm the effects and potential of these compounds as anti-cancer agents. This study builds a foundation for future investigations into alternative cancer treatments using abundant natural resources.
Keywords:
bioactive compounds
; black sea cucumbers
; cancer
; drug-target interactions
; machine learning
; molecular docking
1. Introduction
The black sea cucumber (Holothuria atra), a marine organism, has been traditionally used in medicine for various purposes with its potential medicinal properties [1]. Recent in vitro and in vivo studies have suggested that extracts from black sea cucumbers exhibit anti-cancer properties [2,3]. However, it is crucial to conduct additional research to identify bioactive compounds in black sea cucumber extracts that specifically target cancer-related proteins.
The burden of cancer in Indonesia is considerable, with cervical cancer now holding the distressing position of being the second-leading cause of death [4]. Between 2014 and 2018, the Indonesian government allocated Rp3.5 trillion to cancer-related expenditures. The top five cancers in terms of prevalence during this period were cervical, breast, lung, colorectal, and liver cancer [5]. Therefore, it is crucial to explore innovative strategies beyond conventional medicines to improve patient outcomes, overcome drug resistance, and address unmet medical needs associated with this devastating disease.
The process of identifying new drugs has been transformed by introducing cutting-edge technologies, including machine learning, molecular docking, and ADMET prediction (Absorption, Distribution, Metabolism, Excretion, and Toxicity). These cutting-edge techniques have greatly improved the discovery and development of prospective medicinal medicines [6,7,8].
The bioactive compounds present in an organism, including black sea cucumbers, that may interact with cancer-related proteins can be identified using drug-target interactions (DTI) [9]. Machine learning has recently become a potent and revolutionary tool in the field of drug discovery, particularly for understanding and predicting drug-target interactions [10]. Machine learning algorithms can uncover complex patterns and correlations that control these interactions by examining large datasets that include molecular and protein structures [11].
Molecular docking is one of the most utilized approaches to validate machine learning predictions in DTI. It is sophisticated in silico method essential for bridging the gap between computational predictions and experimental reality [12]. This method provides insights into the feasibility and strength of projected drug-target interactions [13]. It enables the determination of binding affinity and elucidation of interaction details between a drug and its target protein.
Incorporating ADMET analysis is crucial for exploring the potential of the selected compounds from molecular docking [14]. ADMET analysis provides insights into a compound’s pharmacokinetic properties and potential safety risks, aiding in identifying compounds with favorable ADMET profiles [15]. By considering the ADMET factors, researchers can prioritize compounds with a higher likelihood of successful translation into safe and effective therapies, ultimately enhancing the overall success rate of the drug discovery process.
In recent years, in silico drug discovery encompassing drug-target interactions, molecular docking, and ADMET analysis has gained significant importance in developing innovative cancer therapies. For instance, a study focusing on breast cancer utilized molecular docking and in vitro techniques to expedite drug discovery, demonstrating a strong consistency between the two approaches [8]. Another research investigated the in silico drug design of anti-breast cancer agents, encompassing molecular docking studies, MD simulations, and ADMET prediction [16]. Another study has utilized molecular docking studies and ADMET screening, which has led to the synthesis of novel pharmaceuticals as highly effective anti-hepatic cancer medicines [17].
In predicting DTI, numerous in silico approaches leverage the capabilities of machine learning algorithms. For example, [18] conducted a DTI study employing a newly developed algorithm based on chemogenomics feature space. In another study, [19] demonstrated that a general-purpose novel algorithm called Cascade Deep Forest (CDF) outperformed other state-of-the-art DTI algorithms. Other DTI studies also incorporated machine learning algorithmic options [20,21,22,23]. However, none have been found to continuously perform the three in silico approaches mentioned before to identify black sea cucumber’s bioactive compounds as a potential alternative cancer medicine.
This study aims to identify bioactive compounds from black sea cucumbers with anti-cancer properties through an integrated approach of DTI predictions, molecular docking, and ADMET analysis. The research’s significance lies in its ability to discover novel compounds, understand their interactions with target proteins, and evaluate their pharmacokinetic properties and toxicity. The findings can potentially contribute to developing effective anti-cancer therapies, expanding treatment options in cancer research.
This research integrated DTI predictions, molecular docking, and ADMET analysis. Initially, DTI predictions were conducted to identify target proteins involved in cancer and its potential inhibitors. Subsequently, molecular docking was employed to evaluate the binding affinities and interaction details between the bioactive compounds and target proteins. Furthermore, ADMET analysis was performed to assess the compounds’ pharmacokinetic properties and potential toxicity.
This paper is structured into five sections. The introduction provides background information on the significance of identifying bioactive compounds from black sea cucumbers for anti-cancer purposes. The results section presents the findings, including the identified compounds, their interactions with target proteins, and their ADMET profiles. The discussion section analyzes and interprets the results, discussing their implications and potential applications. The materials and methods section outlines the approach, including DTI predictions, molecular docking, and ADMET analysis. Finally, the conclusions section summarizes the essential findings and highlights the implications for developing anti-cancer therapies derived from black sea cucumbers.
2. Results
2.1. Data Acquisitions and Preprocessing
A total of 550 unique cancer-related proteins were acquired from three databases: The Cancer Genome Atlas (TCGA) [24], The Human Protein Atlas (THPA) [25], and Ijah Analytics (http://ijah.apps.cs.ipb.ac.id/). These proteins are presented in Table S1 of supplementary materials. Querying the interactions of these 550 proteins in the BindingDB database resulted in 139,881 interaction data. Meanwhile, 86 unique bioactive compounds were obtained from previous research. These bioactive compounds are presented in Table S2 of supplementary materials. Negative interaction samples were generated with a 1:1 ratio, resulting in 279,762 interaction data. The concatenated interaction data (positive and negative) is provided in CSV format in Spreadsheet S1 of supplementary materials.
2.2. Feature Engineering and Data Sampling
Five chemical features and four protein characteristics were combined to provide 20 feature space combinations. Two hundred datasets were produced by sampling each feature space combination ten times. Calculations revealed that the minimal sample size was 9,521. However, for convenience, that number was rounded to 10,000.
2.3. Machine Learning Modelling and Evaluation
A total of 1,400 models were trained using seven machine learning algorithms, with each algorithm trained on 20 different feature combinations. Each feature combination was sampled into ten different datasets, resulting in 1,400 trained models. A separate test dataset was used to evaluate the performance of these machine learning models. The evaluation results, measured in terms of performance metrics, were recorded for each model. Since each feature combination had ten evaluation results, the average value of each performance metric was calculated. This comprehensive approach allows for a thorough assessment of the trained models and provides an overall understanding of their performances.
The performance metrics indicated that CDF was outperformed in almost every metric, except for the recall value, which was surpassed by k-Nearest Neighbours (KNN). The highest performance was achieved by CDF, with an accuracy of 82.7%, an F1-score of 86.5%, an AUC score of 93.7%, a precision of 91.8%, and Cohen’s Kappa of 74.9%. On the other hand, the highest recall value was obtained by KNN (87%), followed by LightGBM (86.2%) and CDF (86.1%). The recall value difference was not appreciably siginifcant, so CDF was still preferred. The selection of the machine learning algorithm for the prediction stage was based on comparing the AUC score, as it was deemed to represent the overall effectiveness of an algorithm [26]. The AUC scores are calculated from ROC curves, which are presented in Figure 1.
The highest AUC score of 93.7% was achieved by CDF using the ECFP-AAIndex1 feature combination. CDF demonstrated this exceptional performance, further justifying the feature combination selection and the prediction stage’s algorithm. By leveraging the ECFP-AAIndex1 feature space and employing CDF, reliable and robust results can be anticipated.
2.4. Interaction Predictions
The prediction was carried out using ten trained CDF models on a black sea cucumbers’ bioactive compounds dataset comprising the ECFP-AAIndex1 feature combination. Subsequently, ten sets of prediction outcomes were obtained, necessitating the identification of common elements across these sets to determine the consensus among the models. This process identified seven compound-protein pairs as the intersection of all ten prediction sets. In Table 1, these seven pairs are presented along with the average confidence score values, which reflect the degree of agreement among the models regarding the interaction of each pair.
2.5. Molecular Docking
Seven compound-protein pairs that were previously obtained consisting of three types of proteins: Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit beta isoform (PIK3CB), NEDD8-conjugating enzyme (UBE2F), and Cysteinyl leukotriene receptor 2 (CYSLTR2). The 3D protein structures based on X-ray crystallography available in the Protein Data Bank (PDB) were only accessible for the UBE2F protein. However, for PIK3CB and CYSLTR2, only predicted structures obtained from AlphaFold were available. AlphaFold provides reliable predictions of 3D protein structures, which can be utilized in the molecular docking process [27]. Additionally, the 3D structures of the seven compounds were obtained from PubChem and converted to the appropriate format using Open Babel [28]. Autodock Vina [29] simulated the docking of each pair in five poses, and the best binding affinities for each pair are presented in Table 2. The visualization of the bindings with the best binding affinity is shown in Figure 3, while Figure 4 provides a 2D visualization.
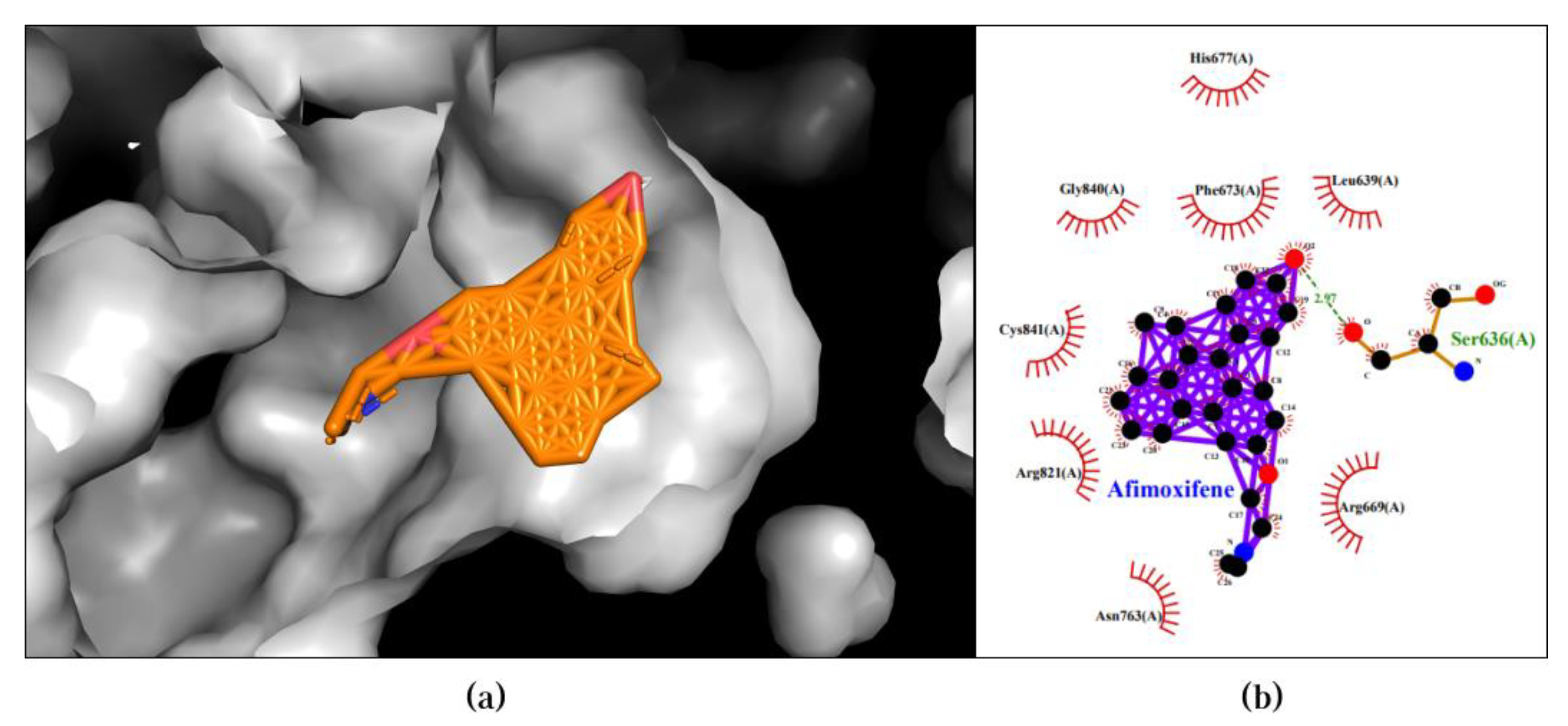
The Afimoxifene-PIK3CB pair exhibited the best binding affinity of -12.7 kcal/mol in the first pose. This pose revealed two types of bonds: hydrogen bonds and hydrophobic interactions. Hydrogen bonds formed between the compound and Ser636, while hydrophobic interactions were observed with Leu639, Phe673, Gly840, Cys841, Arg821, and Asn763. The 3D and 2D visualization of the best pose of the Afimoxifene-PIK3CB pair is presented in Figure 2.
Figure 2.
Best docking pose of Afimoxifene-PIK3CB resulted in binding affinity of -12.7 kcal/mol. Left and right panels are described as follow: (a) A 3D docking pose depicting the Afimoxifene-PIK3CB complex forming a ligand-protein interaction; (b) A 2D visualization highlighting the detailed interactions observed in the best pose.
Figure 2.
Best docking pose of Afimoxifene-PIK3CB resulted in binding affinity of -12.7 kcal/mol. Left and right panels are described as follow: (a) A 3D docking pose depicting the Afimoxifene-PIK3CB complex forming a ligand-protein interaction; (b) A 2D visualization highlighting the detailed interactions observed in the best pose.
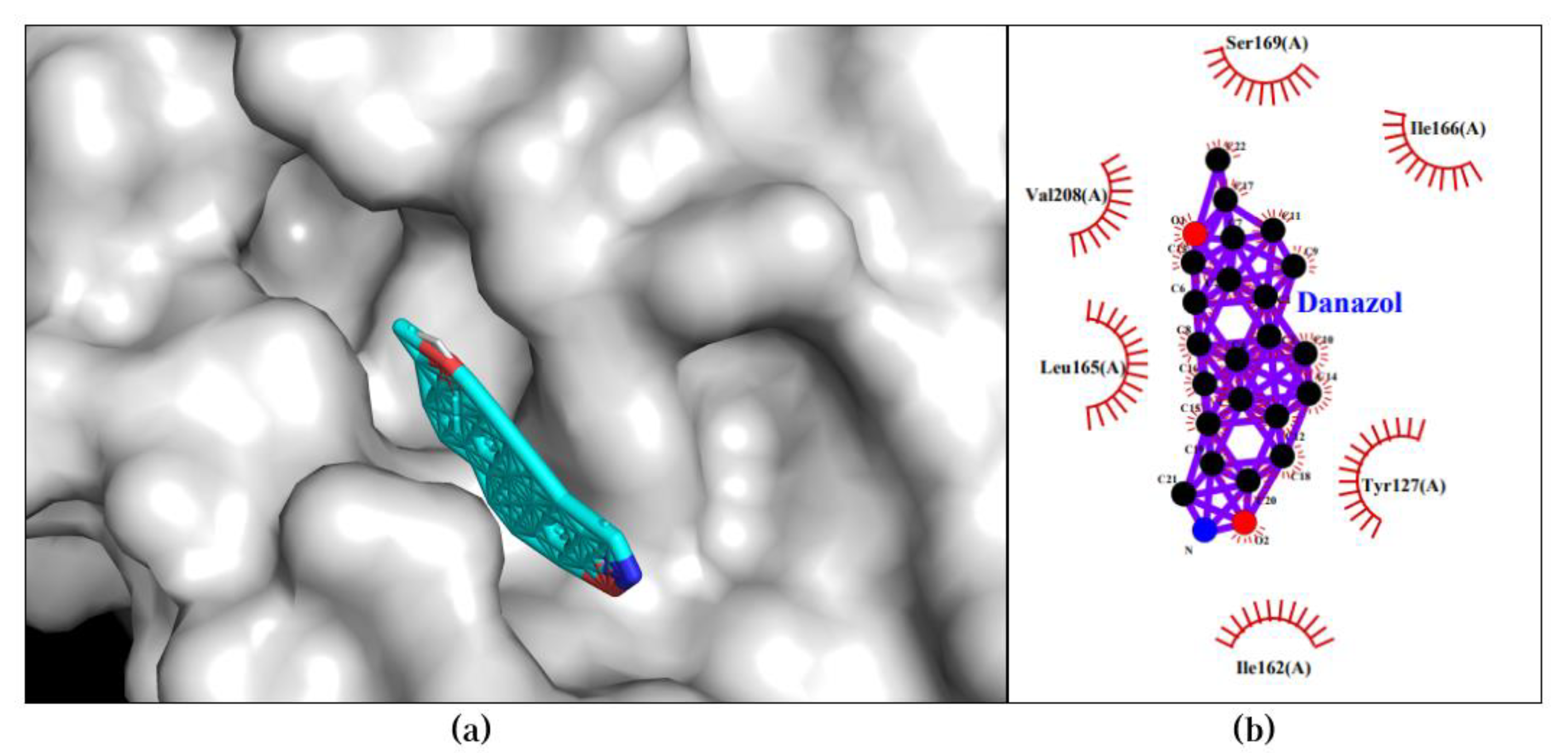
The Danazol-CYSLTR2 pair attained a binding affinity of -12.3 kcal/mol in its best pose. This pose displayed a single type of bond, specifically a hydrophobic interaction with Tyr127, His162, Leu165, His166, Val208, and Ser169. The 3D and 2D visualization of the best pose of the Danazol-CYSLTR2 pair is presented in Figure 2.
Figure 3.
Best docking pose of Danazol-CYSLTR2 resulted in binding affinity of -12.3 kcal/mol. Left and right panels are described as follow: (a) A 3D docking pose depicting the Danazol-CYSLTR2 complex forming a ligand-protein interaction; (b) A 2D visualization highlighting the detailed interactions observed in the best pose.
Figure 3.
Best docking pose of Danazol-CYSLTR2 resulted in binding affinity of -12.3 kcal/mol. Left and right panels are described as follow: (a) A 3D docking pose depicting the Danazol-CYSLTR2 complex forming a ligand-protein interaction; (b) A 2D visualization highlighting the detailed interactions observed in the best pose.
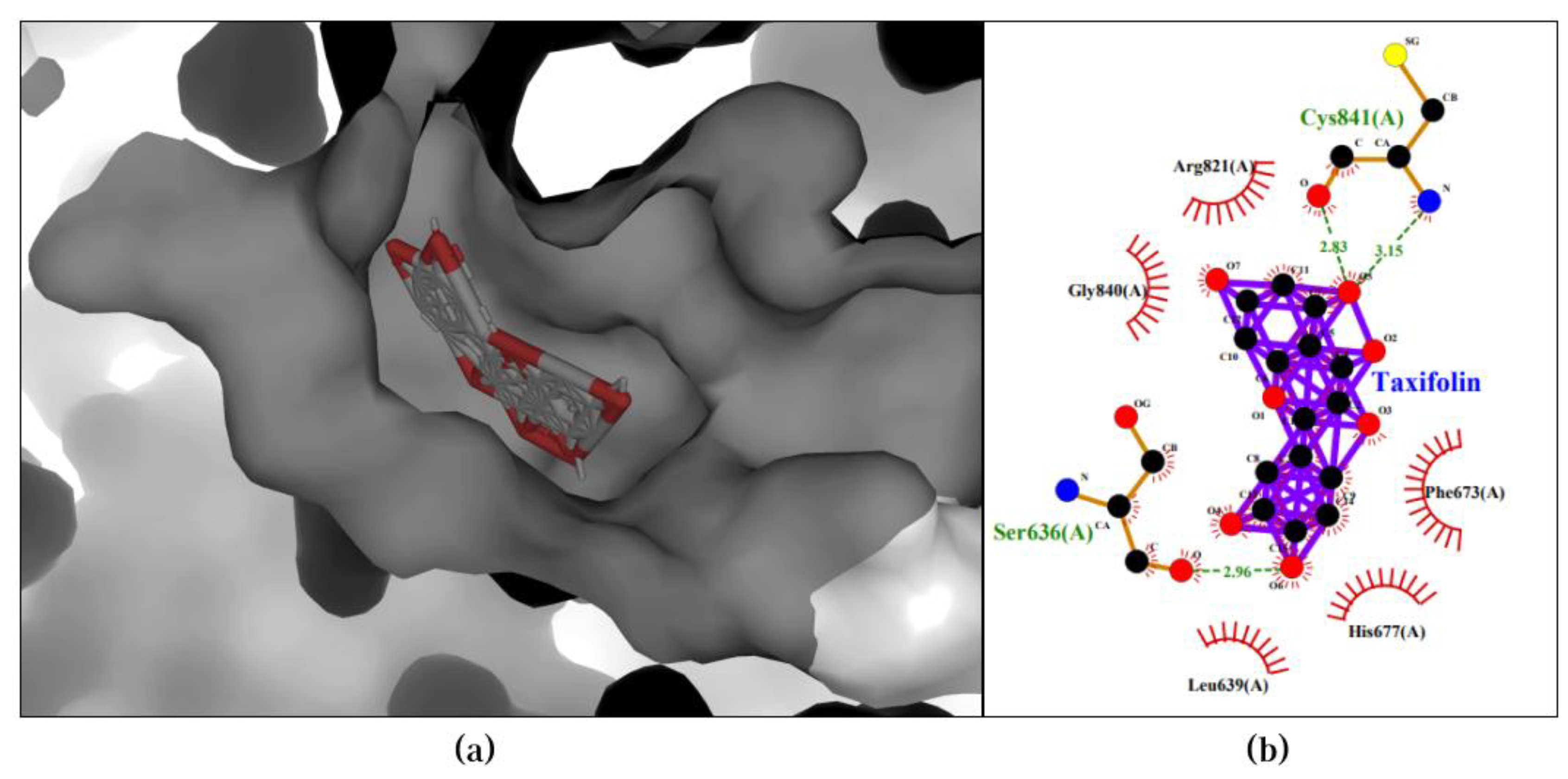
Moreover, the Taxifolin-PIK3CB pair achieved the highest binding affinity of -10.0 kcal/mol in the first pose. Two hydrogen bonds were formed between the compound and Cys841, and an additional hydrogen bond was observed with Ser636. Furthermore, hydrophobic interactions occurred with Phe673, His677, Leu639, Gly840, and Arg821. The 3D and 2D visualization of the best pose of the Taxifolin-PIK3CB pair is presented in Figure 4.
Figure 4.
Best docking pose of Taxifolin-PIK3CB resulted in binding affinity of -10.0 kcal/mol. Left and right panels are described as follow: (a) A 3D docking pose depicting the Taxifolin-PIK3CB complex forming a ligand-protein interaction; (b) A 2D visualization highlighting the detailed interactions observed in the best pose.
Figure 4.
Best docking pose of Taxifolin-PIK3CB resulted in binding affinity of -10.0 kcal/mol. Left and right panels are described as follow: (a) A 3D docking pose depicting the Taxifolin-PIK3CB complex forming a ligand-protein interaction; (b) A 2D visualization highlighting the detailed interactions observed in the best pose.
Lastly, the Terfenadine-UBE2F pair obtained a binding affinity of -6.6 kcal/mol in the first pose, where four hydrophobic interactions were observed between the compound and Lys9, Ala5, Leu4, and Leu9. The 3D and 2D visualization of the best pose of the Terfenadine-UBE2F pair is presented in Figure 5. The Phencyclidine-UBE2F, Meclizine-UBE2F, and Selegiline-UBE2F pairs exhibited binding affinities above -6.0 kcal/mol, hence they will not be further discussed or included in the subsequent analysis.
2.6. ADMET Analysis
Four compound-protein pairs considered to have good binding during the docking process underwent ADMET analysis. The results indicated that all four compounds (Afimoxifene, Danazol, Taxifolin, and Terfenadine) passed the Lipinski rule-of-five test. Except for Taxifolin, all compounds passed the identification of PAINS, with one warning. Absorption profiles showed that all compounds passed the MDCK permeability and intestinal absorption parameters. However, only Afimoxifene and Danazol passed the Caco-2 permeability parameter. Distribution profiles revealed that only Terfenadine passed the PPB parameter, while all compounds passed the VD parameter. Metabolism profiles indicated that Afimoxifene and Danazol acted as CYP1A2 inhibitors, whereas Taxifolin and Terfenadine did not. Excretion profiles showed that all compounds passed the CL parameter and had favorable T1/2 values.
Lastly, toxicity profiles indicated that only Taxifolin was not an hERG blocker. All compounds passed the H-HT parameter except for Danazol. The AMES toxicity parameter revealed suboptimal results only for Taxifolin. Danazol showed indications of carcinogenic properties, while the others did not. None of the compounds exhibited corrosive properties towards the eyes, but only Taxifolin was considered safe for the respiratory system. The full results of the analysis are presented in Table 2.

Table 3.
Complete results of ADMET analysis from ADMETLab 2.0 presented in the following order: compound name, molecular formula, druglikeliness, toxicology profiles, absorption profiles, distribution profiles, metabolism profiles, and excretion profiles.
Table 3.
Complete results of ADMET analysis from ADMETLab 2.0 presented in the following order: compound name, molecular formula, druglikeliness, toxicology profiles, absorption profiles, distribution profiles, metabolism profiles, and excretion profiles.
| Compound (PubChem ID) | Afimoxifene (449459) | Danazol (28417) | Taxifolin (439533) | Terfenadine (5405) |
| Molecular Formula | C26H29NO2 | C22H27NO2 | C15H12O7 | C32H41NO2 |
| Lipinski rule-of-five | Passed | Passed | Passed | Passed |
| hERG Blockers | ++ | +++ | --- | +++ |
| H-HT | - | +++ | --- | -- |
| AMES Toxicity | --- | --- | + | --- |
| Rat Oral Acute Toxicity | + | + | -- | --- |
| Carcinogencity | -- | +++ | --- | --- |
| Eye Corrossion | --- | --- | --- | --- |
| Respiratory Toxicity | ++ | +++ | --- | +++ |
| CaCO2 permeability | -4,46 | -4,88 | -6,06 | -5,274 |
| MDCK permeability | ||||
| Intestinal Absorption | --- | --- | --- | --- |
| PPB | 95,63% | 98,46% | 93,23% | 74,04% |
| VD | 1,745 | 3,01 | 0,56 | 2,28 |
| CYP1A2 inhibitor | ++ | +++ | - | --- |
| CL | 10,08 | 5,265 | 12,29 | 5,11 |
| T1/2 | 0,108 | 0,12 | 0,76 | 0,005 |
Legend: (---): suitable with a score of 0 - 0.3, (+)/(-): less suitable with a score of 0.3 - 0.7, (++)/(+++): not suitable with a score of 0.7 - 1.0.
3. Discussion
3.1. Machine Learning-Based DTI Analysis Reveals the Superiority of CDF Using ECFP-Aaindex1 as a Feature Combination
This study employed seven machine learning algorithms, including CDF, random forest (RF), XGBoost, LightGBM, KNN, multi-layer perceptron neural networks (MLPNN), and logistic regression (LR). The selection of CDF was based on previous research findings that demonstrated its superior performance compared to other state-of-the-art algorithms commonly used for DTI prediction [19]. In this study, CDF exhibited the best performance among the evaluated algorithms.
CDF outperformed the other algorithms in most metrics, except for the recall value, where KNN had the highest ranking. However, the difference in recall values between CDF, KNN, and LightGBM in the first and second positions was insignificant (0.9% and 0.1% difference, respectively). Recall is a metric that measures the ratio of true positives (correct positive predictions) to all positive instances in the data [30]. A high recall value indicates that a model effectively identifies positive instances [31]. However, recall alone is not sufficient to fully describe the overall performance of a model, so comprehensive measures like the F1-Score, which is the harmonic mean of precision and recall, are commonly used. Regarding the F1-Score, CDF achieved the highest score (86.49%) compared to the other algorithms.
The selection of the best algorithm for prediction was determined by comparing the AUC scores of each algorithm. The AUC score calculates the area under the ROC curve, which ranges from 0 to 1, and indicates the model’s reliability in distinguishing between positive and negative classes in binary classification [32]. An AUC value of 1 indicates that the model can ideally separate the positive and negative classes, while an AUC value of 0.5 indicates that the model is no better than random guessing [33]. CDF outperformed the other algorithms with an AUC score of 93.75%, indicating that it is the most effective algorithm compared to the others. This finding aligns with the study conducted by [19].
The CDF algorithm was developed by [34] to address various limitations of neural network-based algorithms. This algorithm leverages the properties of neural networks, such as layer-by-layer learning, simultaneous feature transformation, and complex structure, to achieve comparable performance. With similar reliability, this algorithm aims to overcome the dependence of neural networks on hyperparameter tuning, which is often done through trial and error and is inefficient. CDF is built using a layered structure like neural networks, but each node is replaced with ensemble learning techniques, such as RF. This design choice reduces the number of hyperparameters required for CDF.
The complexity of the CDF structure can adapt to the complexity of the training data [34]. Unlike neural network, whose complexity is determined upfront, the number of layers in CDF depends on the data. The addition of layers in CDF is based on the evaluation of the previous layers, and the process will be stopped if there is no significant improvement in performance. Additionally, unlike neural networks, CDF does not require backpropagation, which means it does not rely on training with a large amount of labelled data to achieve good performance. Considering the characteristics of this algorithm, the superior performance of CDF in this study is not surprising.
This study utilized five types of molecular fingerprints (ECFP, Morgan, PubChem, MACCS, and Klekota-Roth) and four types of protein descriptors (AAC, AAIndex1, PAAC, and ATC), resulting in twenty feature combinations. The combination of ECFP-AAIndex1 features with CDF showed consistent performance, where CDF obtained the highest AUC and F1-Score using this feature combination. This consistency further strengthens the rationale for using the CDF algorithm and ECFP-AAIndex1 combination in the interaction prediction stage.
3.2. Molecular Docking Validates Predictions by CDF and Enhances Understanding of Compound-Protein Interaction Mechanisms
In this study, molecular docking was used to validate the predicted interactions. The docking method employed was blind docking, which considers the entire surface area of the protein without specifying a specific binding site. The advantage of blind docking is that it can identify potential binding sites and predict the binding poses of compounds within those sites [35]. In the context of drug discovery, blind docking is essential as it does not require prior knowledge about the specific binding sites of the target protein [36,37].
The docking results revealed four compound-protein pairs with good binding affinities (below -6.0 kcal/mol). The pair with the lowest binding affinity was Afimoxifene-PIK3CB (-12.7 kcal/mol). According to information from clinicaltrials.gov, Afimoxifene is being investigated for its potential as an anti-breast cancer agent (ID: NCT03063619). The study has been ongoing since 2017 and is currently in Phase 2 clinical trials [38]. A similar case can be observed for the pair with the second-best binding affinity (Danazol-CYSLTR2), where Danazol has been found to effectively overcome multi-drug resistance in cancer patients by inhibiting the STAT3 pathway [39].
Next, the pair with the third-best binding affinity was Taxifolin-PIK3CB (-10.0 kcal/mol). Taxifolin has been associated with various uses, including as an additive substance [40], an antioxidant, an enzyme inhibitor in diseases such as diabetes, Alzheimer’s, and liver disorders [41]. One study demonstrated a strong correlation between Taxifolin and its antiproliferative effects on MCF-7 cells (breast cancer cells) [42].
Lastly, the pair Terfenadine-UBE2F achieved a binding affinity of -6.6 kcal/mol. Several studies have indicated that Terfenadine has inhibitory effects on various enzymes and pathways, such as CYP3A4, CaMKIID/CREB1, and CYP2J2, which are involved in drug metabolism and cancer cell resistance (Racha et al., 2003; Wu et al., 2020; Huang et al., 2022) [43,44,45].
3.3. Enrichment Analysis of Validated Genes/Proteins Reveals Potential Biological Processes and Pathways in Cancer
The protein/gene targets obtained from the validation results using docking present an exciting opportunity for further enrichment analysis since no previous study has linked the highlighted pairs in this study.. In this discussion, enrichment analysis was conducted on three proteins/genes: Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit beta isoform (PIK3CB), NEDD8-conjugating enzyme (UBE2F), and Cysteinyl leukotriene receptor 2 (CYSLTR2). The analysis was performed using Enrichr (https://maayanlab.cloud/Enrichr/), with Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases as the resources. The results were visualized using Cytoscape v3.9.1 [46]. Tables corresponding to the visualizations are presented in Tables S3 and S4 of supplementary materials. Enrichment analysis explored biological processes and KEGG pathways associated with these proteins/genes.
Enrichment analysis revealed that only two of the three proteins (PIK3CB and UBE2F) yielded significant results. It is important to emphasize that the absence of enrichment does not diminish the potential usefulness or significance of a gene for biological purposes. It may simply indicate that the available information and resources are currently insufficient to determine its functional role or involvement in specific pathways [47]. Regarding the obtained results, a total of 20 cellular biological processes and 7 KEGG pathways were enriched by these two genes. The results are presented in Figure 6.
The most enriched biological process identified was Positive Regulation Of the Myeloid Cell Apoptotic Process (GO:0033034, p-value = 0.001050), which involves enhancing anti-apoptotic mechanisms in myeloid cells and maintaining the balance between cell proliferation and apoptosis for normal tissue homeostasis [48,49]. Apoptosis serves as a protective mechanism against cancer development [48]. The most enriched pathway, Aldosterone-regulated sodium reabsorption, controls salt-water homeostasis through interaction with the mineralocorticoid receptor (MR) in renal epithelial cells [50]. Recent research has suggested the potential involvement of the aldosterone-regulated epithelial sodium channel (ENaC) in cancer [51]. Although not enriched, CYSLTR2 has been implicated in specific cancer types [52,53,54,55]. This enrichment analysis provides valuable insights into the biological processes and pathways associated with the selected genes, serving as a basis for further experimental studies to elucidate the underlying mechanisms between the identified anti-cancer candidates and cancer pathways in the human body.
3.4. ADMET Analysis Reveals Insights into Druglikeliness and Bioavailability of Selected Bioactive Compounds
The bioactive compounds validated through docking were further subjected to ADMET analysis. The analysis results indicated that all the compounds met the criteria of the Lipinski rule-of-five. This result suggests that the identified compounds possess favourable pharmacokinetic properties for drug development and can be administered orally [56]. The Lipinski rule-of-five consists of five criteria, including a molecular weight of fewer than five hundred Daltons, no more than five hydrogen bond donors, no more than ten hydrogen bond acceptors, and a partition coefficient (LogP) of less than five [57]. Bioactive compounds that satisfy these criteria have a higher potential for further drug development [58].
The ADMET analysis extended to the perspective of toxicology and revealed exciting findings. Among the compounds analyzed, only Taxifolin did not act as an hERG blocker, which is significant because compounds that inhibit the human-Ether-a-go-go-Related Gene (hERG) can potentially increase the risk of heart attacks [59]. However, it is important to note that not all hERG blockers exhibit the same level of toxicity. Factors such as the potency and selectivity of the inhibitor, dosage and treatment duration, and the patient’s health condition play a role in determining the risk [60].
The results of the ADMET analysis also shed light on the compounds’ hepatotoxicity, mutagenicity, and acute toxicity. Only Danazol did not meet the H-HT (human hepatotoxicity) parameter criteria, suggesting that the other three compounds undergo hepatic metabolism and can circulate throughout the body via the bloodstream [61]. In terms of mutagenicity, all compounds passed the AMES toxicity parameter except Taxifolin, which indicates that it may potentially cause mutations in bacteria [62]. Furthermore, Danazol and Afimoxifene received low scores (+: 0.3-0.7) in the Rat Oral Acute Toxicity parameter, which measures the potential of a compound to cause various effects, including death, in rats when administered at high oral doses [63].
Regarding specific toxicological properties, the carcinogenicity parameter indicated that only Danazol potentially possesses such properties, while the respiratory toxicity parameter suggested that only Taxifolin is not potentially harmful to the respiratory system. None of the compounds exhibited corrosive properties to the eyes, as per the eye corrosion parameter results. These findings from the ADMET analysis provide valuable insights into the potential safety and toxicity profiles of the compounds, highlighting the need for further research and evaluation in wet lab settings to confirm and expand upon these predictions.
As previously described, the various parameters in the toxicological profile of the four selected compounds indicate that none passed all parameters. However, this does not automatically rule out the potential for these compounds to be developed as drugs, as the results are still predictions and further extensive studies are needed to confirm these findings. Nonetheless, Taxifolin demonstrates the safest toxicological profile among the compounds, as it only did not pass the AMES toxicity parameter but with a low score (+: 0.3-0.7). This information can serve as a basis for further research in the future.
The next step in the ADMET analysis involves assessing the absorption profiles. The absorption profiles of the four compounds were evaluated, yielding interesting findings. Among these compounds, only Afimoxifene and Danazol passed the Caco-2 permeability test, indicating their potential for absorption through the intestinal epithelium. However, contrasting results were obtained when testing the compounds using the MDCK permeability and HIA (human intestinal absorption) methods. In these tests, all compounds demonstrated favourable outcomes, suggesting their potential for good absorption within the human body. The inconsistency between the Caco-2 permeability test and the MDCK permeability and HIA results emphasizes the need for further investigation into the permeability characteristics of these compounds. Nevertheless, the consistent results obtained from the MDCK permeability and HIA testing indicate that all compounds are promising for oral drug development, as they have the potential for effective absorption by the body [64].
Continuing the analysis, the distribution profiles reveal that only Terfenadine does not bind to plasma proteins according to the PPB (plasma protein binding) test. However, all compounds exhibit good volume distribution based on the VD (volume distribution) test. This suggests that all compounds can readily distribute throughout the body but may tend to bind to proteins in the blood, potentially reducing their therapeutic effects [65,66].
The following analysis is focused on the metabolic profiles of the selected compounds. Notably, Afimoxifene and Danazol are identified as inhibitors of the CYP1A2 enzyme, while Taxifolin and Terfenadine do not exhibit inhibitory effects. The CYP1A2 enzyme plays a crucial role in metabolising various drugs and toxins within the liver [67]. Compounds that act as inhibitors of this enzyme can significantly impact its activity, leading to potential alterations in the metabolism of other drugs typically metabolized by CYP1A2. This property, in turn, may result in reduced therapeutic efficacy or even drug-drug interactions [68]. Understanding these compounds’ metabolic profiles and potential enzyme interactions becomes essential for further evaluation and development.
In conclusion, this study employed a comprehensive in silico approach to identify several bioactive compounds from sea cucumbers with potential anti-cancer properties. Combining machine learning predictions, molecular docking, and ADMET analysis provided valuable insights into these compounds’ binding affinities, interaction with potential protein targets, pharmacokinetic properties, toxicity profiles, and metabolic characteristics. While each compound exhibited unique ADMET characteristics, Taxifolin emerged as an up-and-coming candidate. However, further in vitro and in vivo studies are warranted to validate and explore specific aspects of these compounds’ activities. The findings of this research lay the foundation for future extensive investigations, which have the potential to lead to the development of novel anti-cancer therapeutics derived from sea cucumber bioactive compounds.
4. Materials and Methods
4.1. Data Acquisition
This study utilized cancer protein data from three databases: The Cancer Genome Atlas (TCGA) [24], Ijah Analytics (http://ijah.apps.cs.ipb.ac.id/), and The Human Protein Atlas (THPA) [25]. Queries were conducted using keywords representing the five most prevalent types of cancer in Indonesia, and the resulting data were downloaded. Additionally, bioactive compound data of black sea cucumbers were obtained from a previous study [69]. Interactions between various cancer proteins and compounds were extracted from the BindingDB database leveraging API as a query tool and amino acid sequences as query parameters [70]. This interaction data was utilized for machine learning model training.
4.2. Data Preprocessing
The interaction data used in this study were obtained from BindingDB, which consists solely of positive interactions. To obtain the negative interaction data, random sampling was performed using compounds and proteins from the positive interaction dataset. This resulted in the creation of compound-protein pairs that were absent from the original data and were then labeled as negative interactions. To maintain a balanced dataset, the positive and negative labeled data were carefully balanced to achieve a 1:1 ratio.
Regarding cancer protein data, information was gathered from the three mentioned sources (THPA, TCGA, and Ijah Analytics) and subsequently merged. Redundant entries that occurred after the merging process were eliminated, ensuring a consolidated and nonrepetitive dataset for further analysis.
4.3. Feature Engineering
The chemogenomics feature space will be utilized for training the machine learning model. This feature space will be extracted from the amino acid sequence data of proteins and the chemical structure of compounds in FASTA and SMILES formats. The protein feature space will be extracted using protein descriptors. Similarly, the compound feature space will be extracted using molecular fingerprints. The protein descriptors to be used will include amino acid compositions (AAC) [71], a physicochemical feature named AAIndex1 [72], pseudo-amino acid compositions [73], and atomic and bond compositions (ATC) [74]. Additionally, the molecular fingerprints to be employed will consist of Extended-Connectivity Fingerprints (ECFP) [75], Klekota-Roth Fingerprints [76], Molecular Access System (MACCS), Morgan Fingerprints [77], and PubChem Fingerprints [78].
4.4. Data Sampling
The data size of compound-protein interactions is typically huge. This huge data size is further compounded by the generation of negative samples. The extensive data size can significantly increase the computational resource requirements. To prevent potential failures during the machine learning training process, researchers can anticipate by conducting sampling from the interaction data. Ten samples are drawn for each feature combination, resulting in two hundred data samples. The minimum sample size for each sample was determined using an online calculator accessible at the following address: https://www.calculator.net/sample-size-calculator.html. The calculator was employed with a margin of error of 0.01 and a confidence interval of 95%.
4.5. Machine Learning Modelling
The machine learning algorithms used in this study included CDF, extreme gradient boosting machine (XGBoost), light gradient boosting machine (LightGBM), logistic regression, multi-layer perceptron neural networks (MLPNN), random forest, and k-nearest neighbors (KNN). The DF21 (package source code available here: https://github.com/LAMDA-NJU/Deep-Forest) package was utilized to implement the CDF algorithm, while the Scikit-Learn [79], XGBoost [80], and LightGBM [81] packages were used for the other algorithms. All algorithms were executed with default hyperparameter configurations. Considering the sample size of two hundred samples, two hundred models were trained for each algorithm. The modeling process employed 77% of the training data and 33% of the testing data, partitioned from the generated samples in the preceding stage. The project source code can be accessed via following link: https://github.com/TropBRC-BioinfoLab/black_sea_cucumbers_for_anti_cancers.
4.6. Model Evaluations
The various trained machine learning models from the previous stage were then tested using the training data partition. The testing results were presented in the form of performance metrics for each algorithm. These performance metrics included accuracy, precision, recall, F1-score, and AUC score. The performance metrics of each model (two hundred models) were averaged based on the feature combinations, resulting in a single value for each mentioned performance metric. This process aimed to evaluate which feature combination yielded the best model performance. Finally, the algorithm performance was compared using AUC score.
4.7. Interaction Predictions
The best feature combination and algorithm from the previous stage were utilized for this stage. However, in this stage, the data from the black sea cucumber bioactive compounds were employed. These compounds were paired with each previously obtained cancer protein, forming the compound-protein feature space as part of the feature engineering process. The data was then used as input for the model to predict their interactions. Since there were ten different models corresponding to the best feature combination from the previous sampling and training process, there would be ten different sets of prediction results. The intersection of positive predictions from these ten sets of interaction predictions was extracted and used for the subsequent stage.
4.8. Molecular Docking
The compound-protein pairs, representing the intersection of the prediction sets obtained in the previous stage, underwent further analysis using molecular docking. The 3D structures of the compounds were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov/) in *.sdf format and converted to *.pdb format using OpenBabel (O’Boyle et al., 2011). Meanwhile, the 3D structures of the proteins were obtained from the Protein Data Bank (https://www.rcsb.org/). The preparation of the protein 3D structures involved removing water molecules, adding charges, adding non-polar hydrogens, removing heteroatoms, and defining the search space configuration. AutoDock Tools [82] was used to perform these preparations.
In this study, blind docking was conducted, considering the entire protein surface instead of specific binding sites. The compound structures were also prepared (converted from *.pdb to *.pdbqt) using AutoDock Tools. The subsequent docking process utilized the prepared *.pdbqt files of the compounds and proteins, along with the configuration *.txt file obtained from the previous search space configuration process. The docking was performed using the AutoDock Vina program [29].
4.9. Absorption, Distribution, Metabolism, dan Toxicity (ADMET) Analysis
The compound-protein pairs with binding affinity ≤ -6.0 kcal/mol underwent ADMET analysis. Such compound-protein interactions with binding affinity in this range are considered to be genuinely binding and stable (Liu et al., 2021). The ADMET analysis was conducted using ADMETLab 2.0, where the SMILES of the selected compounds were inputted (Xiong et al., 2021). The results of the ADMET analysis will help evaluate the feasibility of the compounds as drug candidates.
The ADMET analysis will focus on several parameters, including (a) Absorption: CaCO2 permeability > -5.15 log cm/s, MDCK permeability (Papp) > 20×10^(-6) cm/s, and intestinal absorption > 30%; (b) Distribution: plasma protein binding (PPB) ≤ 90% and volume distribution (VD) ranging from 0.04 to 20 L/kg; (c) Metabolism: acting as an inhibitor of CYP1A2 (increasing the compound concentration in plasma but may have negative implications in certain situations); (d) Excretion: drug clearance ≥ 5, compound half-life T1/2 ranging from 0 to 0.3; (e) Toxicology: including hERG blockers, human hepatotoxicity (H-HT), AMES Toxicity, Rat Oral Acute Toxicity, carcinogenicity, eye corrosion, and respiratory toxicity ranging from 0 to 0.3 [85]. Furthermore, drug-likeness parameters such as Lipinski rule-of-five and Pan-Assay Interference Compounds (PAINS) represent the potential of the compounds to have therapeutic effects based on their physicochemical characteristics.
5. Conclusions
This research successfully utilized in silico approaches, including machine learning, molecular docking, and ADMET analyses, to identify bioactive compounds from the black sea cucumber with potential anti-cancer properties. The results highlighted the effectiveness of the CDF algorithm and the ECFP-AAIndex1 feature combination in determining drug-target interactions. Through molecular docking validation, four promising bioactive compounds were identified: Afimoxifene, Danazol, Taxifolin, and Terfenadine. Subsequent ADME analysis provided valuable insights into these compounds’ absorption, distribution, metabolism, excretion, and toxicity characteristics. Among them, Taxifolin exhibited the most favourable results, passing the highest number of ADME parameters. These findings underscore the significance of the black sea cucumber as a valuable source of bioactive compounds, with Taxifolin showing promise as a lead compound for further development of anti-cancer drugs. However, additional experimental validation is necessary to ascertain the efficacy and safety of these compounds, ultimately paving the way for potential therapeutic interventions against cancer.
Supplementary Materials
Cancer-related proteins acquired from THPA, TCGA, and Ijah Analytics are presented in Table S1; Black Sea Cucumbers’ bioactive compounds are presented in Table S2; Enrichment analysis results in term of GO Biological Process and KEGG Pathways are presented in Table S3 and Table S4 respectively; Complete interaction dataset is provided in Spreadsheet S1 in CSV format.
Author Contributions
Conceptualization, W.A.K. and M.F.R.; methodology, W.A.K. and M.F.R.; software, M.F.R.; validation, M.F.R.; formal analysis, M.F.R. and R.F.; resources, M.N.; data curation, M.N.; writing—original draft preparation, M.F.R.; writing—review and editing, W.A.K., A. and M.N.; visualization, M.F.R. and R.F.; supervision, W.A.K., A. and M.N.; project administration, W.A.K.; funding acquisition, W.A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the Directorate of Higher Education, Research and Technology. Ministry of Education, Culture, Research and Technology in accordance with the contract for the Implementation of the Research Programme Year 2023 No: 102/E5/PG.02.00.PL/2023 dated 19 June 2023.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article and supplementary materials.
Acknowledgments
The authors extend our sincere gratitude to the Department of Computer Sciences, IPB University, the Biopharmaceuticals Research Center, IPB University, and the Directorate General of Higher Education on behalf of the Indonesian Government for their invaluable support, financial supports, and guidance throughout this research project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- W.-T. Lee et al., “Black sea cucumber (Holothuria atra Jaeger, 1833) rescues Pseudomonas aeruginosa -infected Caenorhabditis elegans via reduction of pathogen virulence factors and enhancement of host immunity,” Food Funct., vol. 10, no. 9, pp. 5759–5767, 2019. [CrossRef]
- R. Pangestuti and Z. Arifin, “Medicinal and health benefit effects of functional sea cucumbers,” J. Tradit. Complement. Med., vol. 8, no. 3, pp. 341–351, Jul. 2018. [CrossRef]
- S. Bordbar, F. Anwar, and N. Saari, “High-Value Components and Bioactives from Sea Cucumbers for Functional Foods—A Review,” Mar. Drugs, vol. 9, no. 10, pp. 1761–1805, Oct. 2011. [CrossRef]
- S. A. Kristina, D. Endarti, and H. Aditama, “Prediction of Productivity Costs Related to Cervical Cancer Mortality in Indonesia 2018,” Malaysian J. Med. Sci., vol. 29, no. 1, pp. 138–144, Feb. 2022. Pusdatin Kemenkes RI, “Beban Kanker di Indonesia,” 2019. [CrossRef]
- A. S. Rifaioglu, H. Atas, M. J. Martin, et al., “Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases,” Briefings …, 2019, Available online: https://academic.oup.com/bib/article-abstract/20/5/1878/5062947.
- G. Bocci, E. Carosati, P. Vayer, A. Arrault, S. Lozano, and G. Cruciani, “ADME-Space: a new tool for medicinal chemists to explore ADME properties,” Sci. Rep., vol. 7, no. 1, p. 6359, Jul. 2017. [CrossRef]
- C. Cava and I. Castiglioni, “Integration of Molecular Docking and In Vitro Studies: A Powerful Approach for Drug Discovery in Breast Cancer,” Appl. Sci., vol. 10, no. 19, p. 6981, Oct. 2020. [CrossRef]
- M. T. F. Calangian and V. P. C. Magboo, “Predicting Drug-Target Interaction (DTI) based on Machine Learning with Lasso Dimensionality Reduction and SMOTE from Protein Sequence and Drug Fingerprint,” in 2022 International Conference on Electrical, Computer and Energy Technologies (ICECET), Jul. 2022, pp. 1–6. [CrossRef]
- R. Chen, X. Liu, S. Jin, J. Lin, and J. Liu, “Machine Learning for Drug-Target Interaction Prediction,” Molecules, vol. 23, no. 9, p. 2208, Aug. 2018. [CrossRef]
- A. Ezzat, M. Wu, X. Li, and C.-K. Kwoh, “Computational Prediction of Drug-Target Interactions via Ensemble Learning,” in Computational Methods for Drug Repurposing, 2019, pp. 239–254.
- B. Li, et al. “Machine Learning Models Combined with Virtual Screening and Molecular Docking to Predict Human Topoisomerase I Inhibitors,” Molecules, vol. 24, no. 11, p. 2107, Jun. 2019. [CrossRef]
- N. Srivastava, P. Garg, P. Srivastava, and P. K. Seth, “A molecular dynamics simulation study of the ACE2 receptor with screened natural inhibitors to identify novel drug candidate against COVID-19.,” PeerJ, vol. 9, p. e11171, Apr. 2021. [CrossRef]
- L. L. G. Ferreira and A. D. Andricopulo, “ADMET modeling approaches in drug discovery,” Drug Discov. Today, vol. 24, no. 5, pp. 1157–1165, May 2019. [CrossRef]
- K. E. Knoll, M. M. van der Walt, and D. T. Loots, “In Silico Drug Discovery Strategies Identified ADMET Properties of Decoquinate RMB041 and Its Potential Drug Targets against Mycobacterium tuberculosis,” Microbiol. Spectr., vol. 10, no. 2, Apr. 2022. [CrossRef]
- K. Rajagopal et al., “In Silico Drug Design of Anti-Breast Cancer Agents,” Molecules, vol. 28, no. 10, p. 4175, May 2023. [CrossRef]
- H. R. M. Rashdan, M. El-Naggar, and A. H. Abdelmonsef, “Synthesis, Molecular Docking Studies and In Silico ADMET Screening of New Heterocycles Linked Thiazole Conjugates as Potent Anti-Hepatic Cancer Agents,” Molecules, vol. 26, no. 6, p. 1705, Mar. 2021. [CrossRef]
- Y. Wang et al., “RoFDT: Identification of Drug–Target Interactions from Protein Sequence and Drug Molecular Structure Using Rotation Forest,” Biology (Basel)., vol. 11, no. 5, p. 741, May 2022. [CrossRef]
- Y. Chu et al., “DTI-CDF: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features,” Brief. Bioinform., vol. 22, no. 1, pp. 451–462, Jan. 2021. [CrossRef]
- K. Nasution, S. H. Wijaya, and W. A. Kusuma, “Prediction of Drug-Target Interaction on Jamu Formulas using Machine Learning Approaches,” in 2019 International Conference on Advanced Computer Science and information Systems (ICACSIS), Oct. 2019, pp. 169–174. [CrossRef]
- A. Fadli, A. Annisa, and W. A. Kusuma, “Prediction of Drug-Target Interaction Using Random Forest in Coronavirus Disease 2019 Case,” Bioinforma. Biomed. Res. J., vol. 4, no. 1, pp. 1–7, Nov. 2021. [CrossRef]
- Y. Ding, J. Tang, F. Guo, and Q. Zou, “Identification of drug–target interactions via multiple kernel-based triple collaborative matrix factorization,” Brief. Bioinform., vol. 23, no. 2, Mar. 2022. [CrossRef]
- Z. Cheng, C. Yan, F. X. Wu, and J. Wang, “Drug-target interaction prediction using multi-head self-attention and graph attention network,” IEEE/ACM Trans. …, 2021, Available online: https://ieeexplore.ieee.org/abstract/document/9425008/.
- H. Lee, J. Palm, S. M. Grimes, and H. P. Ji, “The Cancer Genome Atlas Clinical Explorer: a web and mobile interface for identifying clinical–genomic driver associations,” Genome Med., vol. 7, no. 1, p. 112, Dec. 2015. [CrossRef]
- M. Uhlén et al., “A Human Protein Atlas for Normal and Cancer Tissues Based on Antibody Proteomics,” Mol. Cell. Proteomics, vol. 4, no. 12, pp. 1920–1932, Dec. 2005. [CrossRef]
- V. Premachandran and R. Kakarala, “Measuring the effectiveness of bad pixel detection algorithms using the ROC curve,” IEEE Trans. Consum. Electron., vol. 56, no. 4, pp. 2511–2519, Nov. 2010. [CrossRef]
- J. Jumper et al., “Highly accurate protein structure prediction with AlphaFold,” Nature, vol. 596, no. 7873, pp. 583–589, Aug. 2021. [CrossRef]
- N. M. O’Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch, and G. R. Hutchison, “Open Babel: An open chemical toolbox,” J. Cheminform., vol. 3, no. 1, p. 33, Dec. 2011. [CrossRef]
- O. Trott and A. J. Olson, “AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading,” J. Comput. Chem., p. NA-NA, 2009. [CrossRef]
- S. S. Islam, M. S. Haque, M. S. U. Miah, T. Bin Sarwar, and R. Nugraha, “Application of machine learning algorithms to predict the thyroid disease risk: an experimental comparative study,” PeerJ Comput. Sci., vol. 8, p. e898, Mar. 2022. [CrossRef]
- N. Nigar, M. Umar, M. K. Shahzad, S. Islam, and D. Abalo, “A Deep Learning Approach Based on Explainable Artificial Intelligence for Skin Lesion Classification,” IEEE Access, vol. 10, pp. 113715–113725, 2022. [CrossRef]
- V. Malathi, M. P. Gopinath, M. Kumar, S. Bhushan, and S. Jayaprakash, “Enhancing the Paddy Disease Classification by Using Cross-Validation Strategy for Artificial Neural Network over Baseline Classifiers,” J. Sensors, vol. 2023, pp. 1–13, Apr. 2023. [CrossRef]
- L. Nie, C. Li, F. Marzani, H. Wang, F. Thibouw, and A. B. Grayeli, “Classification of Wideband Tympanometry by Deep Transfer Learning With Data Augmentation for Automatic Diagnosis of Otosclerosis,” IEEE J. Biomed. Heal. Informatics, vol. 26, no. 2, pp. 888–897, Feb. 2022. [CrossRef]
- Z.-H. Zhou and J. Feng, “Deep forest,” Natl. Sci. Rev., vol. 6, no. 1, pp. 74–86, Jan. 2019. [CrossRef]
- Y. Liu, X. Yang, J. Gan, S. Chen, Z.-X. Xiao, and Y. Cao, “CB-Dock2: improved protein–ligand blind docking by integrating cavity detection, docking and homologous template fitting,” Nucleic Acids Res., vol. 50, no. W1, pp. W159–W164, Jul. 2022. [CrossRef]
- X.-Y. Meng, H.-X. Zhang, M. Mezei, and M. Cui, “Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery,” Curr. Comput. Aided-Drug Des., vol. 7, no. 2, pp. 146–157, Jun. 2011. [CrossRef]
- N. M. Hassan, A. A. Alhossary, Y. Mu, and C.-K. Kwoh, “Protein-Ligand Blind Docking Using QuickVina-W With Inter-Process Spatio-Temporal Integration,” Sci. Rep., vol. 7, no. 1, p. 15451, Nov. 2017. [CrossRef]
- M.D. Anderson Cancer Center, “Afimoxifene in Reducing the Risk of Breast Cancer in Women With Mammographically Dense Breast,” 2023. https://beta.clinicaltrials.gov/study/NCT03063619 (accessed May 29, 2023).
- Y.-T. Chang et al., “Danazol mediates collateral sensitivity via STAT3/Myc related pathway in multidrug-resistant cancer cells,” Sci. Rep., vol. 9, no. 1, p. 11628, Aug. 2019. [CrossRef]
- D. Turck et al., “Scientific Opinion on taxifolin-rich extract from Dahurian Larch (Larix gmelinii),” EFSA J., vol. 15, no. 2, Feb. 2017. [CrossRef]
- S. Begum, A. Banerjee, and B. De, “Antioxidant and Enzyme Inhibitory Properties of Mangifera indica leaf Extract,” Nat. Prod. J., vol. 10, no. 4, pp. 384–394, Aug. 2020. [CrossRef]
- V. S. Rogovskiĭ et al., “Antiproliferative and antioxidant activity of new dihydroquercetin derivatives,” Eksp. Klin. Farmakol., vol. 73, no. 9, pp. 39–42, Sep. 2010, Available online: http://www.ncbi.nlm.nih.gov/pubmed/21086652.
- J. K. Racha et al., “Substrate Dependent Inhibition Profiles of Fourteen Drugs on CYP3A4 Activity Measured by A High Throughput LCMS/MS Method with Four Probe Drugs, Midazolam, Testosterone, Nifedipine and Terfenadine,” Drug Metab. Pharmacokinet., vol. 18, no. 2, pp. 128–138, 2003. [CrossRef]
- Z. Wu, S.-N. Jang, S.-Y. Park, N. M. Phuc, and K.-H. Liu, “Inhibitory Potential of Bilobetin Against CYP2J2 Activities in Human Liver Microsomes,” Mass Spectrom. Lett., vol. 11, no. 4, pp. 113–117, 2020. UL - https://msletters/v.11/4/113/7217. [CrossRef]
- W. Huang et al., “Terfenadine resensitizes doxorubicin activity in drug-resistant ovarian cancer cells via an inhibition of CaMKII/CREB1 mediated ABCB1 expression,” Front. Oncol., vol. 12, Nov. 2022. [CrossRef]
- P. Shannon et al., “Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks,” Genome Res., vol. 13, no. 11, pp. 2498–2504, Nov. 2003. [CrossRef]
- Williams and, S. Halappanavar, “Application of biclustering of gene expression data and gene set enrichment analysis methods to identify potentially disease causing nanomaterials,” Beilstein J. Nanotechnol., vol. 6, pp. 2438–2448, Dec. 2015. [CrossRef]
- S. G. C. Mestrum et al., “Proliferative and anti-apoptotic fractions in maturing hematopoietic cell lineages and their role in homeostasis of normal bone marrow,” Cytom. Part A, vol. 101, no. 7, pp. 552–563, Jul. 2022. [CrossRef]
- X.-Q. Xu, C.-M. Huang, Y.-F. Zhang, L. Chen, H. Cheng, and J.-M. Wang, “S1PR1 mediates anti-apoptotic/pro-proliferative processes in human acute myeloid leukemia cells,” Mol. Med. Rep., vol. 14, no. 4, pp. 3369–3375, Oct. 2016. [CrossRef]
- A. Lother, F. Jaisser, and U. O. Wenzel, “Emerging fields for therapeutic targeting of the aldosterone–mineralocorticoid receptor signaling pathway,” Br. J. Pharmacol., vol. 179, no. 13, pp. 3099–3102, Jul. 2022. [CrossRef]
- W. Ware, J. J. Harris, T. L. Slatter, H. E. Cunliffe, and F. J. McDonald, “The epithelial sodium channel has a role in breast cancer cell proliferation,” Breast Cancer Res. Treat., vol. 187, no. 1, pp. 31–43, May 2021. [CrossRef]
- E. Ceraudo et al., “Direct evidence that the GPCR CysLTR2 mutant causative of uveal melanoma is constitutively active with highly biased signaling,” J. Biol. Chem., vol. 296, p. 100163, Jan. 2021. [CrossRef]
- D. F. Akin-Bali, “Bioinformatics analysis of GNAQ, GNA11, BAP1, SF3B1,SRSF2, EIF1AX, PLCB4, and CYSLTR2 genes and their role in the pathogenesis of Uveal Melanoma,” Ophthalmic Genet., vol. 42, no. 6, pp. 732–743, Nov. 2021. [CrossRef]
- M. Venerito et al., “Leukotriene receptor expression in esophageal squamous cell cancer and non-transformed esophageal epithelium: a matched case control study,” BMC Gastroenterol., vol. 16, no. 1, p. 85, Dec. 2016. [CrossRef]
- W. Liu, C. Zhang, X. Gong, W. Liao, J. Xu, and X. Zhang, “Prognostic value of immune-related genes in laryngeal squamous cell carcinoma,” Transl. Cancer Res., vol. 9, no. 10, pp. 6287–6302, Oct. 2020. [CrossRef]
- P. M. Natarajan, V. R. Umapathy, A. Murali, and B. Swamikannu, “Computational simulations of identified marine-derived natural bioactive compounds as potential inhibitors of oral cancer,” Futur. Sci. OA, vol. 8, no. 3, Mar. 2022. [CrossRef]
- M. R. R. Rahardhian, Y. Susilawati, I. Musfiroh, R. M. Febriyanti, Muchtaridi, and S. A. Sumiwi, “In Silico Study of Bioactive Compounds from Sungkai (Peronema Canescens) As Immunomodulator,” Int. J. Appl. Pharm., vol. 14, no. 4, pp. 135–141, Nov. 2022. [CrossRef]
- N. S. Aini et al., “In Silico Screening of Bioactive Compounds from Garcinia mangostana L. Against SARS-CoV-2 via Tetra Inhibitors,” Pharmacogn. J., vol. 14, no. 5, pp. 575–579, Nov. 2022. [CrossRef]
- M. J. Windley, W. Lee, J. I. Vandenberg, and A. P. Hill, “The Temperature Dependence of Kinetics Associated with Drug Block of hERG Channels Is Compound-Specific and an Important Factor for Proarrhythmic Risk Prediction,” Mol. Pharmacol., vol. 94, no. 1, pp. 760–769, Jul. 2018. [CrossRef]
- J. M. Kratz, U. Grienke, O. Scheel, S. A. Mann, and J. M. Rollinger, “Natural products modulating the hERG channel: heartaches and hope,” Nat. Prod. Rep., vol. 34, no. 8, pp. 957–980, 2017. [CrossRef]
- H. Park, B. Kim, J.-H. Oh, Y. Kim, and Y.-J. Lee, “First-pass Metabolism of Decursin, a Bioactive Compound of Angelica gigas, in Rats,” Planta Med., vol. 78, no. 09, pp. 909–913, Jun. 2012. [CrossRef]
- J. Kim, M. Han, and W. K. Jeon, “Acute and Subacute Oral Toxicity of Mumefural, Bioactive Compound Derived from Processed Fruit of Prunus mume Sieb. et Zucc., in ICR Mice,” Nutrients, vol. 12, no. 5, p. 1328, May 2020. [CrossRef]
- J. Yu and S.-J. Choi, “Particle Size and Biological Fate of ZnO Do Not Cause Acute Toxicity, but Affect Toxicokinetics and Gene Expression Profiles in the Rat Livers after Oral Administration,” Int. J. Mol. Sci., vol. 22, no. 4, p. 1698, Feb. 2021. [CrossRef]
- Y. H. Bai et al., “Assessment of a Bioactive Compound for Its Potential Antiinflammatory Property by Tight Junction Permeability,” Phyther. Res., vol. 19, no. 12, pp. 1009–1012, Dec. 2005. [CrossRef]
- K. S. Roy et al., “Optimizing a High-Throughput Solid-Phase Microextraction System to Determine the Plasma Protein Binding of Drugs in Human Plasma,” Anal. Chem., vol. 93, no. 32, pp. 11061–11065, Aug. 2021. [CrossRef]
- C. Fărcaș et al., “An Update Regarding the Bioactive Compound of Cereal By-Products: Health Benefits and Potential Applications,” Nutrients, vol. 14, no. 17, p. 3470, Aug. 2022. [CrossRef]
- F. Bachmann et al., “Metamizole is a Moderate Cytochrome P450 Inducer Via the Constitutive Androstane Receptor and a Weak Inhibitor of CYP1A2,” Clin. Pharmacol. Ther., vol. 109, no. 6, pp. 1505–1516, Jun. 2021. [CrossRef]
- T. J. K. Kaartinen et al., “Effect of High-Dose Esomeprazole on CYP1A2, CYP2C19, and CYP3A4 Activities in Humans: Evidence for Substantial and Long-lasting Inhibition of CYP2C19,” Clin. Pharmacol. Ther., vol. 108, no. 6, pp. 1254–1264, Dec. 2020. [CrossRef]
- I. R. Fadhlia, M. Nurilmala, and T. Nurhayati, “Karakterisasi Konsentrat dan Hidrolisat Protein Teripang Keling (Holothuria atra) dan Potensinya sebagai Imunomodulator,” Institut Pertanian Bogor, 2017.
- T. Liu, Y. Lin, X. Wen, R. N. Jorissen, and M. K. Gilson, “BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities.,” Nucleic Acids Res., vol. 35, no. Database issue, pp. D198-201, Jan. 2007. [CrossRef]
- B. Zhao et al., “DescribePROT: database of amino acid-level protein structure and function predictions,” Nucleic Acids Res., vol. 49, no. D1, pp. D298–D308, Jan. 2021. [CrossRef]
- S. Kawashima, P. Pokarowski, M. Pokarowska, A. Kolinski, T. Katayama, and M. Kanehisa, “AAindex: amino acid index database, progress report 2008.,” Nucleic Acids Res., vol. 36, no. Database issue, pp. D202-5, Jan. 2008. [CrossRef]
- K.-C. Chou, “Prediction of protein cellular attributes using pseudo-amino acid composition,” Proteins Struct. Funct. Genet., vol. 43, no. 3, pp. 246–255, May 2001. [CrossRef]
- R. Kumar et al., “An in silico platform for predicting, screening and designing of antihypertensive peptides,” Sci. Rep., vol. 5, no. 1, p. 12512, Jul. 2015. [CrossRef]
- D. Rogers and M. Hahn, “Extended-Connectivity Fingerprints,” J. Chem. Inf. Model., vol. 50, no. 5, pp. 742–754, May 2010. [CrossRef]
- J. Klekota and F. P. Roth, “Chemical substructures that enrich for biological activity,” Bioinformatics, vol. 24, no. 21, pp. 2518–2525, Nov. 2008. [CrossRef]
- H. L. Morgan, “The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service.,” J. Chem. Doc., vol. 5, no. 2, pp. 107–113, May 1965. [CrossRef]
- Y. Wang, J. Xiao, T. O. Suzek, J. Zhang, J. Wang, and S. H. Bryant, “PubChem: a public information system for analyzing bioactivities of small molecules,” Nucleic Acids Res., vol. 37, no. Web Server, pp. W623–W633, Jul. 2009. [CrossRef]
- F. Pedregosa et al., “Scikit-learn: Machine Learning in Python,” J. Mach. Learn. Res., vol. 12, no. 85, pp. 2825–2830, 2011, Available online: http://jmlr.org/papers/v12/pedregosa11a.html.
- T. Chen and C. Guestrin, “XGBoost,” in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Aug. 2016, pp. 785–794. [CrossRef]
- G. Ke et al., “LightGBM: A Highly Efficient Gradient Boosting Decision Tree,” in Advances in Neural Information Processing Systems, 2017, vol. 30, Available online: https://proceedings.neurips.cc/paper_files/paper/2017/file/6449f44a102fde848669bdd9eb6b76fa-Paper.pdf.
- G. M. Morris et al., “AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility,” J. Comput. Chem., vol. 30, no. 16, pp. 2785–2791, Dec. 2009. [CrossRef]
- Schrödinger, LLC, “The {PyMOL} Molecular Graphics System, Version~1.8,” Nov. 2015.
- R. A. Laskowski and M. B. Swindells, “LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery,” J. Chem. Inf. Model., vol. 51, no. 10, pp. 2778–2786, Oct. 2011. [CrossRef]
- S. Kumari and P. Kumar, “Design and Computational Analysis of an MMP9 Inhibitor in Hypoxia-Induced Glioblastoma Multiforme.” ACS omega, vol. 8, no. 11, pp. 10565–10590, Mar. 2023. [CrossRef]
Figure 1.
ROC Curve of each feature combination in multiple algorithms. Algorithm abbreviations are as follow: CDF: Cascade Deep Forest; RF: Random Forest; XGB: Xtreme Gradient Boosting; LGBM: Light Gradient Boosting Machine; KNN: K-Nearest Neighbours; LR: Logistic Regression; NN: Neural Networks (MLPNN).
Figure 1.
ROC Curve of each feature combination in multiple algorithms. Algorithm abbreviations are as follow: CDF: Cascade Deep Forest; RF: Random Forest; XGB: Xtreme Gradient Boosting; LGBM: Light Gradient Boosting Machine; KNN: K-Nearest Neighbours; LR: Logistic Regression; NN: Neural Networks (MLPNN).
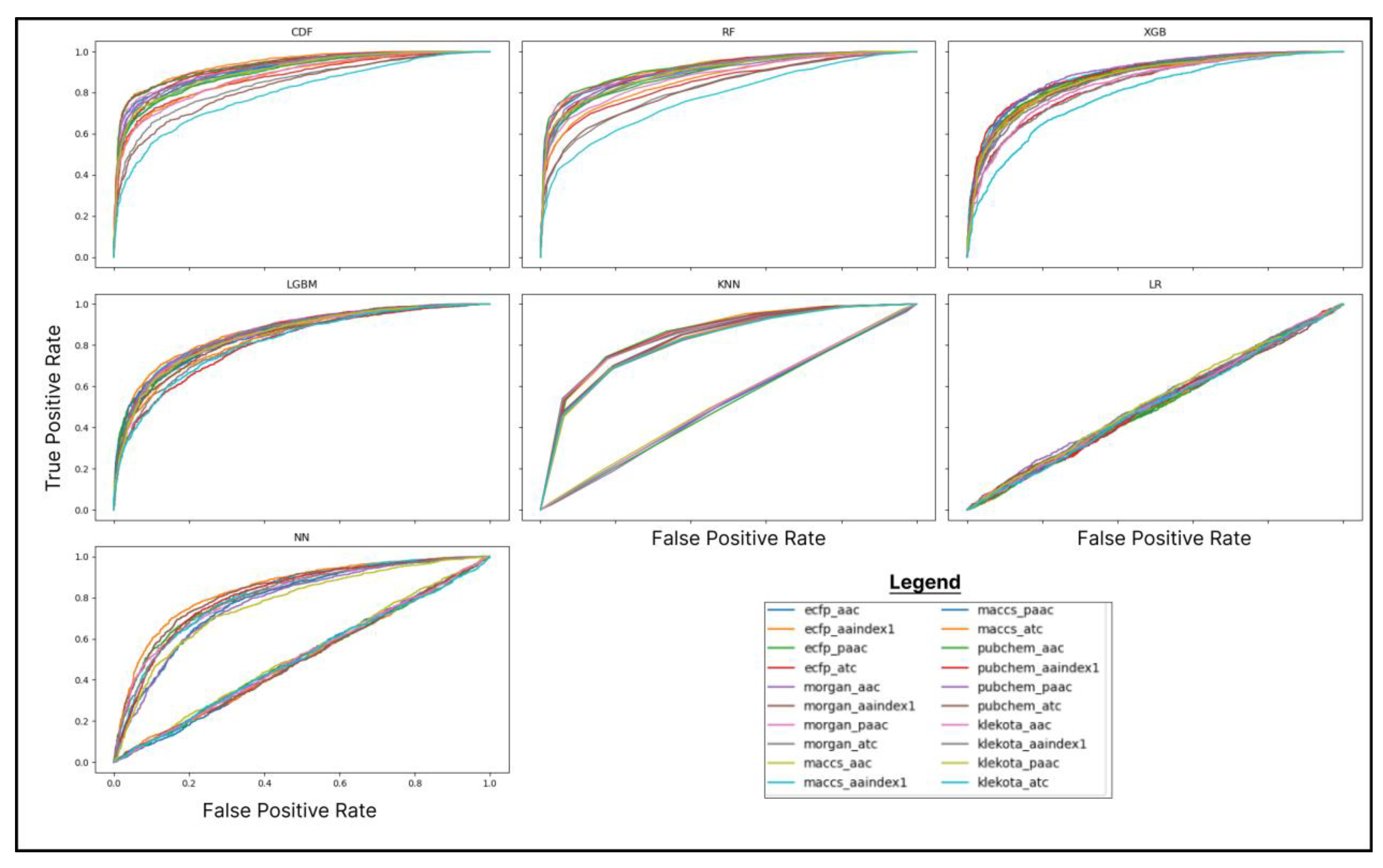
Figure 5.
Best docking pose of Terfenadine-UBE2F resulted in binding affinity of -6.6 kcal/mol. Left and right panels are described as follow: (a) A 3D docking pose depicting the Terfenadine-UBE2F complex forming a ligand-protein interaction; (b) A 2D visualization highlighting the detailed interactions observed in the best pose.
Figure 5.
Best docking pose of Terfenadine-UBE2F resulted in binding affinity of -6.6 kcal/mol. Left and right panels are described as follow: (a) A 3D docking pose depicting the Terfenadine-UBE2F complex forming a ligand-protein interaction; (b) A 2D visualization highlighting the detailed interactions observed in the best pose.
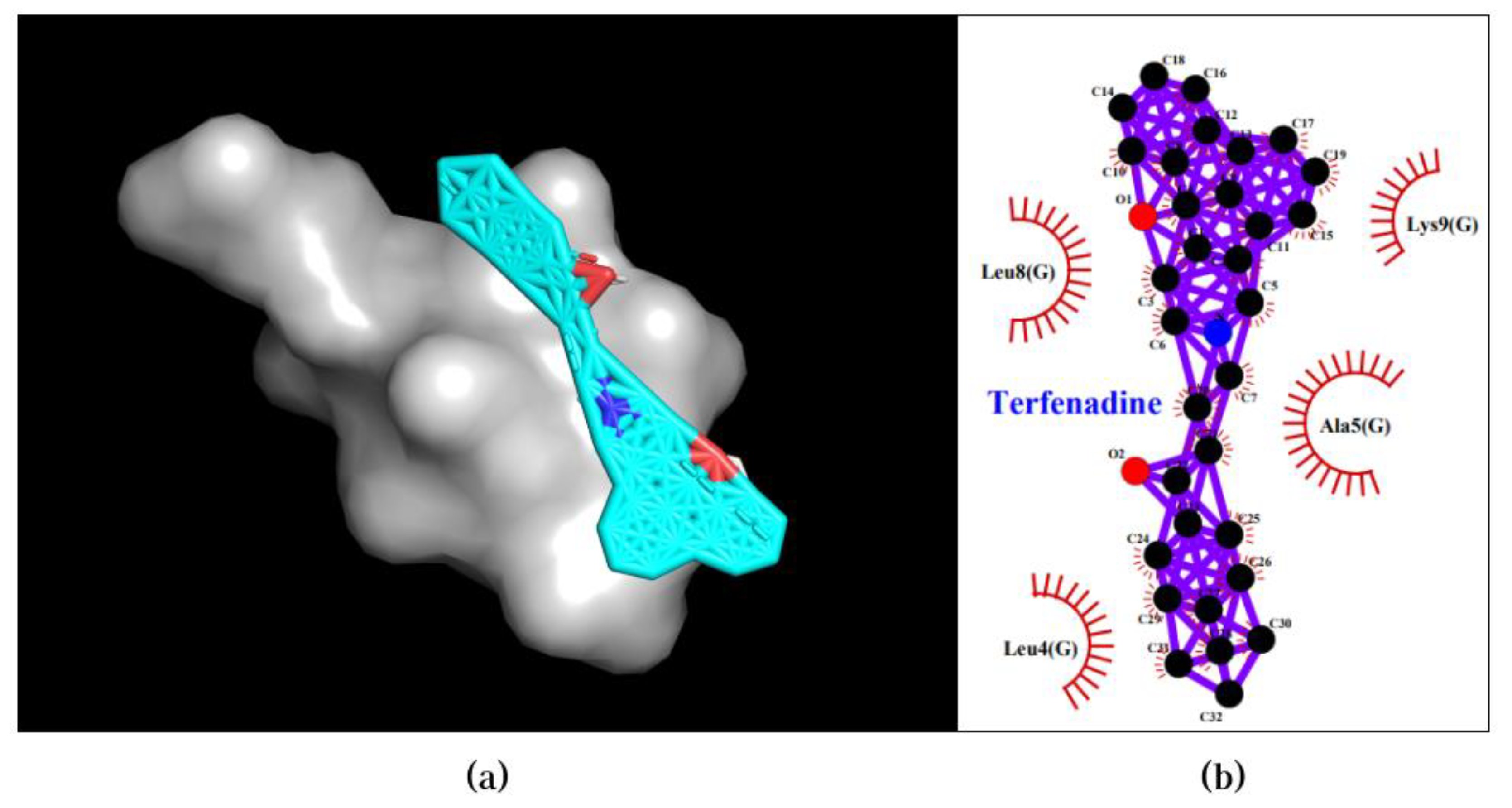
Figure 6.
The enrichment analysis results demonstrated that PIK3CB and UBE2F exhibited significant enrichment in terms of GO biological processes and KEGG Pathways. The results are presented as follows: (a) Topological representation of enriched biological processes; (b) Ranking of biological processes based on p-values (top 10); (c) Topological representation of enriched KEGG pathways; (d) Ranking of KEGG pathways based on p-values (top 10).
Figure 6.
The enrichment analysis results demonstrated that PIK3CB and UBE2F exhibited significant enrichment in terms of GO biological processes and KEGG Pathways. The results are presented as follows: (a) Topological representation of enriched biological processes; (b) Ranking of biological processes based on p-values (top 10); (c) Topological representation of enriched KEGG pathways; (d) Ranking of KEGG pathways based on p-values (top 10).
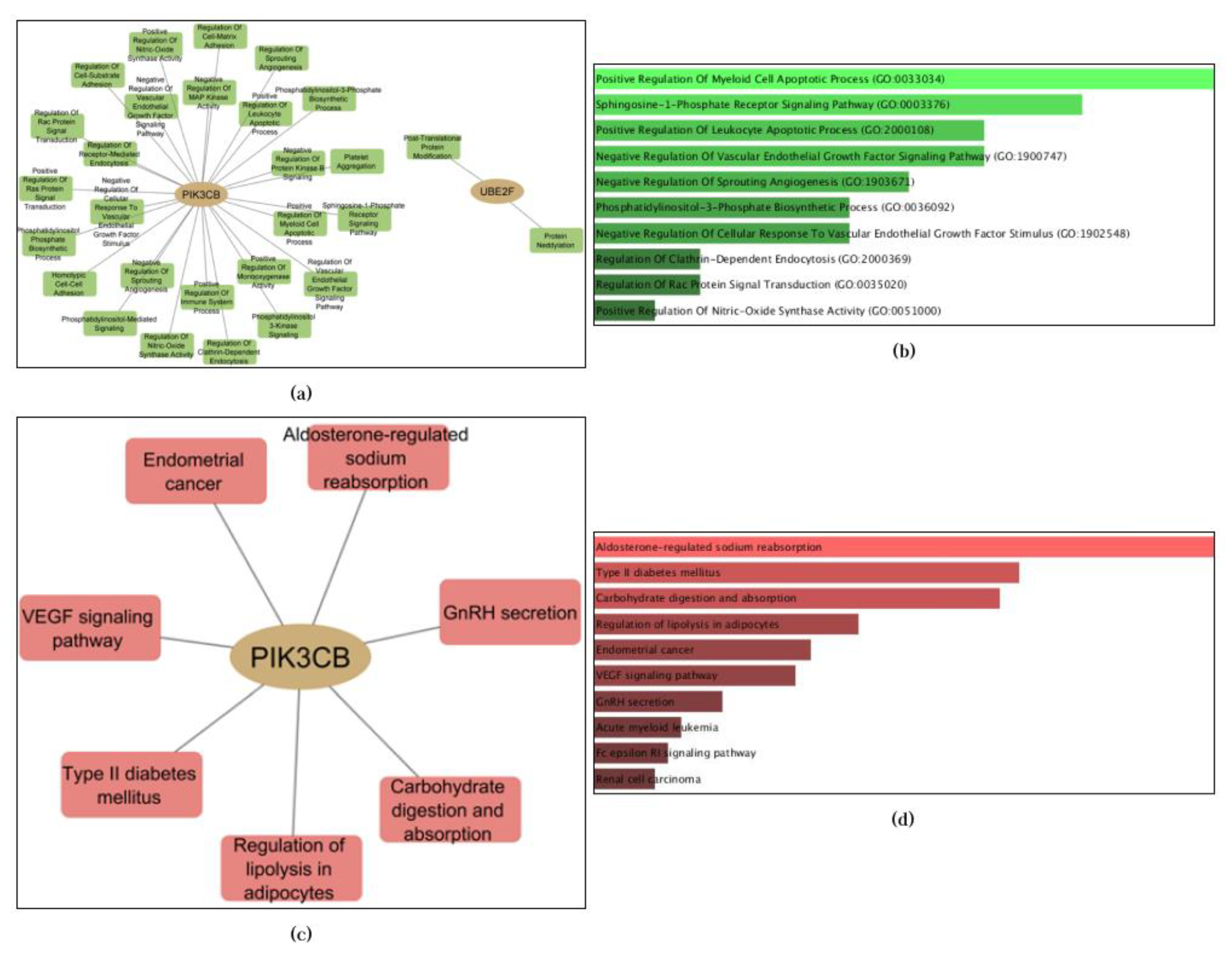
Table 1.
Common predictions from ten prediction sets yielded by ten trained CDF models.
| Compound Name (PubChem ID) |
Protein/Gene Name (Uniprot ID) |
Average Confidence Score |
|---|---|---|
| Meclizine (4034) | UBE2F (Q969M7) | 0.8445 |
| Taxifolin (439533) | PIK3CB (P42338) | 0.790625 |
| Terfenadine (5405) | UBE2F (Q969M7) | 0.856 |
| Afimoxifene (449459) | PIK3CB (P42338) | 0.892125 |
| Selegiline (26757) | UBE2F (Q969M7) | 0.87525 |
| Phencyclidine (6468) | UBE2F (Q969M7) | 0.89625 |
| Danazol (28417) | CYSLTR2 (Q9NS75) | 0.80275 |
Table 2.
Best binding affinities of each seven pairs resulted from molecular docking.
| Compound Name | Protein/Gene Name | Best Binding Affinity |
|---|---|---|
| Afimoxifene | PIK3CB | -12.7 |
| Danazol | CYSLTR2 | -12.3 |
| Taxifolin | PIK3CB | -10.0 |
| Terfenadine | UBE2F | -6.6 |
| Phencyclidine | UBE2F | -4.6 |
| Meclizine | UBE2F | -4.3 |
| Selegiline | UBE2F | -3.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



