
Submitted:
16 June 2023
Posted:
26 June 2023
You are already at the latest version
Abstract
Rabies is a fatal zoonosis that is considered a re-emerging infectious disease. Although rabies remains endemic in canines throughout much of the world, vaccination programs have essentially eliminated dog rabies in the Americas and much of Europe. However, despite the goal of eradicating dog rabies in the European Union by 2020, sporadic cases of dog rabies still occur in Eastern Europe, including Georgia. To assess the genetic diversity of strains recently circulating in Georgia, we sequenced 78 RABV-positive samples from brain tissues of rabid dogs and jackals using Illumina short-read sequencing of total RNA shotgun libraries. Seventy-seven RABV genomes were successfully assembled and annotated, 74 of them to the coding complete status. Phylogenetic analyses of the nucleoprotein (N) and attachment glycoprotein (G) genes placed all the assembled genomes into the Cosmopolitan clade, consistent with the Georgian origin of the samples. Amino acid alignment of the G glycoprotein ectodomain identified twelve different sequences for this domain among the samples. Only one of the ectodomain groups contained a residue change in an antigenic site, an R264H change in the G5 antigenic site. Three isolates were cultured, and these were found to be efficiently neutralized by human monoclonal antibody A6. Overall, our data show that recently circulating RABV isolates from Georgian canines are predominantly closely related phylogroup I viruses of the Cosmopolitan clade. Current rabies vaccines should offer protection against infection by Georgian canine RABVs. The genomes have been deposited in GenBank (accessions: OQ603609-OQ603685).
Keywords:
Rabies
; lyssaviruses
; RABV
; dog
; jackal
; canine
; neutralization
; genomics
; next-generation sequencing
; phylogeny
1. Introduction
Rabies virus (RABV), the prototypical lyssavirus, and rabies-related viruses are neurotropic pathogens that cause rabies, a uniformly fatal encephalitic disease of both humans and a diversity of mammals. Despite the existence of highly effective post-exposure prophylaxis (PEP) for known or potential exposures, RABV remains a neglected tropical disease and is estimated to cause approximately 60,000 human fatalities per year, the majority occurring in children across Africa and Asia. These deaths are attributable to a combination of poor access to PEP, the absence of effective treatments for symptomatic rabies, and inadequate domestic dog vaccination coverage[1]. Notably, domestic dogs are responsible for 99% of transmission events to humans and subsequent rabies fatalities [2]. Highly successful domestic dog vaccination programs in resource-rich countries have largely eliminated dog-mediated rabies in the Americas and Western Europe (although persistence occurs in wildlife sources, including bats, raccoons, and foxes). However, bat-mediated rabies is becoming a larger source of rabies disease in the Americas[3,4], since the effective interruption of dog-mediated transmission. The goal of Zero by 30, a program promoted by the United Nations, the World Health Organization and several other international health organizations, is to eliminate dog-mediated rabies deaths by 2030 [5]. Although a goal of the European Union (EU) was to eliminate dog rabies, by 2020, this goal has not been met, as evidenced by the fact that sporadic cases of dog-mediated rabies still occur in Eastern Europe[6]. Thus, detailed information regarding currently circulating rabies strains in Eastern European countries is of high relevance.
Lyssaviruses are negative strand RNA viruses of the family Rhabdoviridae. The majority of lyssavirus species are classified as belonging to one of two phylogroups (phylogroups I and II), with a few divergent species remaining ungrouped. RABV and most other lyssavirus species reported to infect humans are members of phylogroup I. Various species of canines are the primary animal hosts of RABV, whereas, specific species of bats are the primary hosts for the other lyssaviruses [7]. Transmission of RABV to humans occurs following a bite or scratch from a rabid animal. RABV generally replicates in muscle cells before transiting into peripheral nerves at the neuromuscular junction. RABV then uses these afferent nerves to enter the spinal cord, where another round of viral replication occurs, followed by ascension to the brain; death of the host is via encephalitis. Following further replication in the brain, RABV spreads to multiple peripheral tissues via efferent nerves. Spread to and replication within salivary glands enables transmission to subsequent hosts [7,8].
Lyssavirus genomes encode five genes, N (nucleoprotein), P (phosphoprotein), M (matrix), G (glycoprotein) and L (polymerase). Among these genes, N and G have been most characterized at the level of nucleic acid sequence [9]. Because lyssaviruses, like other RNA viruses, are replicated by an error-prone RNA-directed RNA polymerase, mutations accumulate over relatively short periods of time [10]. However, there is also selective pressure against many non-synonymous mutations, and the evolution of RABV over time is thus slow [11]. Canine RABV is dispersed across most of the world, with genomic sequences segregated into six major phylogenetic clades. The Cosmopolitan clade is globally distributed, whereas other clades are more regional. The Africa-2 clade is found in west Africa; the Africa-3 clade predominates in east Africa; the Asian clade is primarily in central and eastern Asia; the Arctic cluster is found mostly in regions bordering the arctic circle and in western Asia; and the Indian Subcontinent clade is present in Sri Lanka and southern India [11,12,13].
To date, the majority of phylogenetic studies of RABV isolates from infected hosts have focused either on limited sequencing of a large number of isolates (e.g., N only) or on full genome sequencing of small numbers of isolates. Although a few studies have provided complete sequences of a large number of isolates [11,12,14,15], we are aware of no such studies that have reported complete sequences of large numbers of samples collected in Eastern Europe. In this work, brain tissues from rabid dogs and jackals from Georgia, Caucasus region, were collected during the years 2018-2021, and RABV PCR-positive samples were processed for genome sequencing. RNA was extracted from 78 selected tissue samples for sequencing and genomic analysis. These analyses resulted in the production of 74 complete plus 3 quasi-complete RABV sequences from Georgian canines. Herein, we provide complete RABV sequences recovered from approximately 80 canines in Georgia, including eight sequences from jackals. We relate the distribution of identified amino acid variations to known antigenic sites in the G ectodomain, and we test the ability of a potent human anti-G monoclonal antibody (mAb) to neutralize three cultured isolates.
2. Materials and Methods
2.1. Collection of brain tissue from suspected rabid dogs and jackals in Georgia
As part of an ongoing rabies surveillance program in Georgia, brain tissue was collected from dogs and jackals suspected of being rabid, based on observation of disease signs consistent with rabies. Brain tissue was harvested from these canines as soon as possible, post-mortem. Brain tissue was divided into portions of approximately 1 g, which were placed into labeled cryovials. Brain tissue was then frozen and stored on dry ice or in a -80 C freezer until the time of RNA extraction or viral culture. Each sample was given an identifier, RABies Virus-GEOrgia-x (RABV-GEO-x), in which x is a unique number between 1 and 100.
2.2. RNA purification and degenerate PCR amplification and sequencing of the lyssavirus N gene
Total RNA was prepared by placing approximately 100 mg of frozen brain tissue into a 5 mL Eppendorf Safe-Lock tube with TRIzol (product number 15596026, Invitrogen, Waltham, MA) and zirconium oxide lysis beads (Next Advance, ZROB20-RNA). The brain tissue was homogenized using a BulletBlender (model BB5E-AU, Next Advance, Troy, NY) within a biosafety cabinet. The homogenate was then transferred to 1.5 mL microcentrifuge tubes to produce total RNA, following the TRIzol RNA purification protocol, as per the manufacturer (Invitrogen, Waltham, MA). To degrade any contaminating DNA, RNA samples were treated with RQ1 DNAse (Promega, Madison, WI), following the manufacturer’s protocol. After phenol extraction and ethanol precipitation, total RNA was resuspended and quantified using a NanoDrop One spectrophotometer (Thermo Scientific, Waltham, MA).
For initial screening of samples, we used a modified degenerate nested reverse-transcriptase PCR protocol to amplify a region of the N gene, a region that codes for approximately the N-terminal half of the protein. This protocol was chosen because it can successfully amplify a wide array of lyssavirus N genes, including those outside of phylogroup I [16]. The JW12 primer (ATGTAACACCYCTACAATTG) was used for first-strand cDNA synthesis, followed by primary PCR, using JW12 and an equimolar mix of primers JW6(DPL), JW6(E) and JW6(M) (CAATTCGCACACATTTTGTG; CAGTTGGCACACATCTTGTG; and CAGTTAGCGCACATCTTATG, respectively). The secondary PCR was performed with primers modified by the addition of binding sites for sequencing primers M13R49 and M13F43. In this manner, secondary PCR was performed with primer M13R49-JW12 (GCTGAGCGGATAACAATTTCACACAGGATGTAACACCYCTACAATTG), and an equimolar mix of M13F43-JW10(DLE2), M13F43-JW10(ME1) and M13F43-JW10(P) (GCTAGGGTTTTCCCAGTCACGACGTTGTCATCAAAGTGTGRTGCTC; GCTAGGGTTTTCCCAGTCACGACGTTGTCATCAATGTGTGRTGTTC; and GCTAGGGTTTTCCCAGTCACGACGTTGTCATTAGAGTATGGTGTTC, respectively). Samples yielding visible PCR products of the predicted size (~635 bp) were sequenced from both ends using primers M13R49 (GAGCGGATAACAATTTCACACAGG) and M13F43 (AGGGTTTTCCCAGTCACGACGTT) via Sanger sequencing (Eurofins Genomics, Louisville, KY). Primer sequences were removed, and the remaining sequence of each isolate (approximately 542 bp) was subjected to phylogenetic relatedness analysis using Geneious Prime software (Dotmatics, Boston, MA). Briefly, after removing primer sequences from each read, forward and reverse sequences were aligned to create a consensus sequence for each clone. The RABV-GEO N gene PCR product sequences were then compared to N gene sequences from a variety of known GenBank clones using the Geneious Tree Builder function. A Global alignment was performed and the Tamura-Nei model with neighbor-joining and no outgroup was used to produce the phylogenetic tree.
2.3. Shotgun sequencing of total RNA samples
Total RNA was used to prepare shotgun libraries for sequencing using the NEBNext Ultra II RNA Library Prep for Illumina (New England Biolabs; Ipswich, MA) following the manufacturer’s instructions. Briefly, 5 μl of RNA at ~50 ng/μl was first fragmented and reverse transcribed into ssDNA. The ssDNA was then used as template for the synthesis of the complementary DNA strand to obtain dsDNA. The dsDNA was end-repaired and 3’-end extended to add dA overhangs. Hairpin sequencing adaptors, containing 5’-dT overhangs and a U ribonucleotide in the hairpin loop, were added by DNA ligation, and subsequently cleaved at the U sites. The libraries were then amplified and indexed by PCR using NEBNext Unique Dual Indexes. Prior to sequencing, the libraries were evaluated for quality using Agilent D1000 kit (Agilent Technologies; Santa Clara, CA). The libraries that passed QC were then quantified using Qubit dsDNA BR assay (ThermoFisher Scientific; Waltham, MA) and pooled for sequencing. An initial set of 4 samples (RABV-GEO-6, -9, -13 and -20) were sequenced using a MiSeq Reagent Kit v3 600 cycle and a MiSeq sequencer (Illumina; San Diego, CA). The remaining libraries were sequenced using a NovaSeq6000 S4 Reagent Kit v1.5 300 cycles sequencing kit and a NovaSeq6000 sequencer (Illumina; San Diego, CA).
2.4. Sequencing-data processing and genome assembly
Raw reads were trimmed and filtered prior to assembly. Briefly, bbtools v39.01 suite [17] was used to trim raw reads using bbduk and to map quality-controlled reads >Q20 to Lyssavirus taxonid 11286, downloaded from NCBI using taxonkit [18]. Positively filtered lyssavirus reads were subsequently assembled using metaSPAdes v3.15.3 [19]. Assembly quality and query cover were evaluated using Bandage v0.8.1 [20]. Contigs that had sparse coverage of the genome were abandoned. For samples that did not produce complete RABV genomes using metaSPAdes, reads were further subsampled to 3,000-25,000 paired-end reads using bbtools v39.01 and assembled using Unicycler v0.5.0 [21]. When appropriate, manual genome closure was performed with evidence supported by contigs assembled using both metaSPAdes and Unicycler. All genomes were manually reviewed for quality and annotated using ORF finder in CLC Genomics Workbench v23 (QIAGEN).
2.5. Analysis of heterozygous single nucleotide variants (SNVs)
To evaluate the possibility of coinfection with multiple genotypes or lineages of rabies virus, trimmed reads were mapped back to assemblies requiring at least half the read to map with a minimum of 80% identity and evaluated for heterozygous SNVs using CLC Genomics Workbench v23 (QIAGEN, Hilden, Germany) with a minimum frequency of 35% and a minimum coverage of 10.
2.6. Phylogenetic analysis
Alignments of the open reading frames predicted for N and G genes each were generated using CLC Genomics Workbench v23 (QIAGEN, Hilden, Germany). To generate maximum likelihood trees, iqtree v2.0.3 [22,23] was used with 1000 bootstraps [24] and visualized using FigTree v1.4.4 [25]. In both cases, the best-fit model was TVMe+G4.
2.7. Sequence analysis of the G glycoprotein ectodomain
Amino acid sequences of the G genes were obtained by direct translation of the genomic assemblies using ORFfinder [26]. Predicted G glycoprotein sequences were aligned with Clustal Omega [27] and trimmed to eliminate the N-terminal signal peptide, the C-proximal transmembrane domain and the C-terminal cytoplasmic domain [28,29]. The resulting 439 aa ectodomains were aligned using Clustal Omega and resulting alignments and phylograms used to group samples with identical G glycoprotein ectodomain sequences and to obtain the consensus sequence.
2.8. Principal components analysis (PCA) of ectodomain group and geographic origin of samples
2.9. Clustering of RABV genomes and RABV protein sequences
Full length genomes of RABV, as well as amino acid sequences for each gene product, were downloaded from NCBI (taxID: 11292, Lyssavirus rabies; accessed March 2023) [32] and combined with the RABV-GEO sequences. Amino acid sequences were subsequently filtered to include only full or nearly full-length gene products. Genome and polypeptide sequences were then clustered using MMseqs2 v13.45111 [33] specifying a minimum of 0.95 and 0.8 sequence identity respectively. Target clustering mode was utilized so that partial sequences were heavily weighted against being representative of a cluster.
2.10. Alignment of clustered RABV amino acid sequences
Representative amino acid sequences were extracted for each cluster and aligned using CLC Workbench v23 (QIAGEN; Hilden, Germany) using default parameters (gap open cost = 10; gap extension cost = 1). RABV-GEO-97, selected automatically by MMseqs2 as the representative for P cluster 8, was also included as an additional representative in N, M, G and L alignments. BlastP [32] was used to identify conserved domains from the Pfam database [34], and the relevant positions were subsequently evaluated for the degree of conservation among representative sequences.
2.11. Analysis of predicted epitopes for RABV-GEO P, M, L proteins
2.12. RABV isolation and neutralization with mAb A6
To isolate RABV, frozen brain tissue was homogenized into a 20% [w/v] suspension in 1 mL of sterile 1✕ PBS using a Bullet Blender (Next Advance, model BB5E-AU). This homogenate was then clarified by centrifugation. Next, 0.5 mL of clarified supernatant was transferred to a 50 mL disposable centrifuge tube containing 106 N2a cells (a mouse neuroblast cell line; ATCC CCL-131), followed by 30 min incubation in a 37°C, 5% CO2, with occasional agitation. Complete DMEM (10 mL) was then added, and tubes were centrifuged for 15 min at 700 × g. The growth media was replaced with 15 mL of fresh DMEM, cells were resuspended and aliquoted to one well of a 96-well plate (75 μL), with the remainder transferred to a T75 flask. After 48 h, the cells in the 96-well plate were stained with FITC anti-Rabies G (Cat #800-092, Fujirebio, Malvern, PA) at a dilution of 1:100 overnight at 4°C to identify foci of RABV-infected cells. At approximately 72 h post-infection low-titer viral supernatant and half of the cells from the T75 flask were cultured with fresh N2a cells (5 × 105), again distributing between one well of a 96-well plate and a T75 flask. Viral supernatant from this second passage was concentrated by ultracentrifugation, followed by titering on N2a cells.
Neutralization with anti-lyssavirus G human monoclonal antibody A6[37] was performed with the titered RABV-GEO viruses. Viral stocks at an MOI of 0.1 were incubated with 2-fold serial dilutions of A6 and incubated at 37°C in a 5% CO2 incubator for 90 min. The virus/A6 mix was transferred to corresponding wells with N2a cells (4 × 104 cells/well) in a 96-well plate. Plates were incubated at 37°C in a 5% CO2 incubator for 48 hr. Cells were fixed with 4% paraformaldehyde for 1 hr, followed by washing (1× PBS) and permeabilization (1× PBS, 0.2% Triton X-100, 0.1% sodium azide) for 10 min at ambient temperature. Following further washing, wells were blocked for 20 min at ambient temperature (DMEM with 10% fetal bovine serum). Staining was then performed with FITC anti-Rabies G (Cat #800-092, Fujirebio, Malvern, PA) at a dilution of 1:100, overnight at 4°C. Following washing, foci were counted using an ELISpot analyzer (model S6 Flex M2, Immunospot, Shaker heights, OH). All manipulations with RABV were performed in a biosafety cabinet by rabies-vaccinated personnel using appropriate BSL2 procedures for lyssaviruses as outlined in the CDC Biosafety in Microbiological and Biomedical Laboratories (BMBL) 6th Edition [38], following protocols approved by the USU Institutional Biosafety Committee.
3. Results
3.1. PCR screening and sequencing of N genes of Georgian canine brain tissue isolates
To gain further insight into the population of rabies viruses recently circulating in Eastern Europe, we analyzed a large group of canine samples collected in Georgia. Primary brain tissue isolates from suspected rabid dogs and jackals were processed to yield total RNA. Purified RNAs were then screened using nested degenerate PCR primers that amplify approximately the N-terminal half of the lyssavirus N gene [16]. The majority of isolates yielded a clear PCR product of the predicted molecular weight. Using the M13R43 and M13F49 sequencing primer binding sites that were incorporated into the N gene amplification primers, the PCR products were sequenced to provide an initial assessment of viral genotype. Phylogenetic analyses of these partial N gene sequences suggested the viruses were closely related to Eastern European isolates, consistent with their Georgian origin (Supplementary Figure S1). To better clarify the genetic relatedness to other strains, we proceeded to fully sequence these isolates.
3.2. Sequencing and assembly of seventy-seven complete Georgian RABV genomes
The majority of RABV sequences in publicly available databases such as NCBI are partial genome sequences, generally covering a portion or all of the N gene or G gene. To provide more complete information regarding the sequence diversity of RABVs circulating in Georgia, we sequenced, assembled, and annotated 77 total RNA samples (Supplementary Table S1). Based on the proportion of viral reads from the total RNA, the average load of RABV RNA in the brain tissue samples was approximately 0.2%. The depth of coverage ranged from 50 to 2000-fold for most of the samples. RABV-GEO-100 was positive for the presence of the RABV virus judging by the sequencing results but had low depth and sparse breath of coverage of the genome and therefore was not used in further analyses. RABV genomes were successfully assembled using metaSPAdes for seven samples (RABV-GEO-6, 9, 13, 20, 74, 78, and 97) while the assembly of the rest of the samples with Unicycler resulted in an additional set of 64 successfully assembled samples. An additional set of six samples (RABV-GEO-23, 30, 36, 54, 57, and 62) produced complete genomes when manually closed. From a total of 77 successfully assembled genomes, 74 samples were fully annotated for all five rabies virus genes (N, P, M, G and L) making them coding complete, while for the other three samples, RABV-GEO-19, -95 and -96, only partial genomes were obtained.
Heterozygous SNVs were observed in 24 samples, but only three samples were found to have more than three heterozygous SNVs across the genome: RABV-GEO-19, 62, and 95. They were found to have 27, 80, and 77 heterozygous SNVs, respectively. We interpret the relatively large number of heterozygous SNVs in these samples to indicate a potential mixed genotype in each, which may have contributed to the breakage (gaps) in the assemblies for these particular samples. A summary of each observed heterozygous SNV, including depth of coverage at each position and quality scores, can be found in Supplementary Table S2.
3.3. Phylogeny of the newly identified RABV genomes
To evaluate the diversity among the 77 RABV-GEO genomes successfully assembled, we first examined similarity by pairwise comparison of the N genes. This resulted in a range of 81.82-100% nucleotide identity. Then, to compare the new genomes with other rabies viruses from multiple geographical origins and clades, we created phylogenetic trees for the highly conserved N gene [39] as well as the more divergent G gene [40]. RABV-GEO-95 was excluded from the G tree due to incomplete sequence coverage. In both cases, we used as references sequences from selected samples previously described ([11,12,41]; Supplementary Table S3).
The N gene phylogenetic tree shows that the RABV-GEO samples clustered together with reference samples from the Cosmopolitan clade (Figure 1). As expected from Cosmopolitan samples, they were distant from the Bat, Asian and Indian Subcontinent clades, while reference samples from the Africa-2, Africa-3, and Arctic-related clades were located midway. Interestingly, while most of the RABV-GEO samples were tightly grouped with reference samples from Georgia and bordering countries, two of the samples, RABV-GEO-37 and -54, branched out with other Cosmopolitan samples from more diverse origins like Azerbaijan (LN879480), Tajikistan (KY765901), Russia (AY352481), Kazakhstan (AY352489), Hungary (U43025), Bosnia and Herzegovina (U42704; U42706) and Poland (U22840). Samples collected in the Georgian subregions of Samegrelo, Adjara, Shida Kartli, Imereti, Kvemo Kartli, and Kakheti mostly clustered together, with a few exceptions. Samples collected in Tbilisi and Guria did not cluster together clearly.
The G phylogenetic tree showed a similar pattern to the one observed in the N tree, grouping the RABV-GEO samples with the Cosmopolitan clade where most of the RABV-GEO samples were associated to reference samples for Georgia and bordering countries (Figure 2). In addition, RABV-GEO-37 and -54, branched out from the main cluster of RABV-GEO samples and associated with reference samples with more diverse geographical location like Azerbaijan (LN879480), Tajikistan (KY765901), Hungary (AF325462), and Poland (AF325464).
3.4. Clustering analysis of RABV full-length genomes
The N- and G-based phylogenetic trees support a close relatedness of the RABV-GEO viruses. Nevertheless, this analysis does not address the possibility of higher dissimilarity levels along the rest of their genomes, for example due to hypermutation or recombination events in loci outside the N and G genes. To further evaluate the relationship of the 77 RABV-GEO to previously described RABV isolates, we performed clustering analysis including 2,769 full genomes publicly available at NCBI. The complete set of RABV sequences grouped in 124 distinct clusters and the RABV-GEO samples were assigned to only two of these clusters. Interestingly, all but two samples, RABV-GEO-37 and -54, fell within one cluster. This result recapitulated what we observed in the phylogenetic analysis of N and G sequences and thus strengthens the notion that all RAVB-GEO viruses are close relatives to each other.
3.5. Amino acid sequence variability in the RABV G ectodomain
The RABV G gene encodes a transmembrane glycoprotein containing an ectodomain that protrudes from the viral membrane and is responsible for binding to cellular receptors, mediating viral entry into host cells. RABV G is also the target of both the rabies vaccine and the HRIG component of PEP [42]. Upon analysis of the amino acid composition of the G ectodomain of the RABV-GEO samples, the viral sequences were assigned to 12 groups, ECTO1 - ECTO12, based on sequence identity (i.e., members of each ECTO group are comprised of viruses having identical ectodomain sequences). The largest group was ECTO1 which included approximately 80% of the total sequenced viruses. Among the other 11 groups, ECTO2 contains about 7% of the sequenced viruses, while the remaining ECTO groups each represent <3% of the total number of samples (Supplementary Table S1). Using principal components analysis, we found no significant correlation among ectodomain group assignment and subregion from which samples were collected (Supplementary Figure S2). Figure 3A shows the sequence of the G ectodomain from viruses of ECTO1, with antigenic sites underlined and sequence variations across the different ECTO groups indicated in red. The specific amino acid changes characteristic of each individual ECTO group are shown in Figure 3B.
3.6. Global sequence variability of RABV proteins
In order to have a global overview of variability of the full complement of the RABV proteins, we performed individual clustering analysis for amino acid sequences of N, P, M, G and L. We included previously reported sequences obtained from NCBI database, as well as the protein sequences for the viruses described in this work. As can be seen in Table 1, the number of sequences for N far exceeds the reported sequences for the other RABV proteins, followed by G, with P, M and L significantly behind. This disparity is likely the result of the wider interest of the scientific community in the sequence of N (as a conserved marker used for RABV phylogenetic analysis) and G (used both for phylogenetic analysis and for assessing likely responses to RABV vaccines and antibody therapy). As expected from its high degree of conservation, N grouped in only two clusters while P, M, G and L clustered in nine, four, five and five clusters respectively. Also, in agreement with the low stringency used for the protein clustering analysis (see Materials and Methods), RABV-GEO samples grouped in just one cluster for each of the proteins.
Representative sequences from the clusters in Table 2 were used for sequence alignment analysis to visualize regions of amino acid conservation all along the protein sequences (Supplementary Figures S3 and S4).
3.7. Epitopes in RABV-GEO P, M and L proteins
Antibodies that neutralize RABV bind to the surface G glycoprotein, preventing interaction with cellular receptors [42,43,44]. Because effectiveness of RABV vaccines and therapies depends on efficient neutralization, antibodies directed against G have been more intensely characterized than antibodies against other RABV proteins. However, monoclonal antibodies against P, M, and L have been isolated and characterized, mostly for the purpose of detection and diagnostics [45,46,47]. Additionally, understanding protein homology between distinct RABV isolates is important for evaluating direct-acting antivirals that have been investigated as possible routes for generation of novel RABV therapeutics. Such approaches include targeting specific matrix protein domains and preventing association between the phosphoprotein-polymerase complex and the N, to block formation of the fully assembled helical ribonucleoprotein complex [48]. To define the conservation of known epitopes located in P, M and L, we searched in BV-BRC for rabies epitopes, followed by comparison to the protein sequences in the RABV-GEO viruses (Table 2). Of the 19 epitopes identified in this analysis, two were not present in the selected sequences and six were conserved among the representative sequence for the cluster including RABV-GEO sequences. Each of the epitopes for M (2 epitopes) and L (4 epitopes) were conserved but 11/13 P epitopes were not conserved.
3.8. Neutralization of RABV isolates by mAb A6
Our previous studies have shown that mAb A6, a human IgG1 raised against the G of Australian bat lyssavirus (ABLV), is broadly neutralizing against a wide array of phylogroup I lyssaviruses [37]. To demonstrate that these phylogroup I Georgian RABV isolates could also be efficiently neutralized by A6, we cultured several distinct viral isolates from the harvested Georgian canine brain tissues. Specifically, we isolated and passaged RABV-GEO-6, -9 and -20. Viruses from these tissues reached a titer in the range of 105 – 106 focus-forming units (ffu) after the second passage, and these second-passage viral stocks were thus used for neutralization analysis. As shown in Figure 4, each of these cultured viruses was efficiently neutralized by mAb A6, with a potency similar to our previously reported analyses of other phylogroup I lyssaviruses [37].
4. Discussion
In this study, we sequenced and annotated 77 new RABV genomes isolated from rabid dogs and jackals in Georgia. The genomes have been deposited in GenBank (Supplementary Table S1). Phylogenetic analyses of the N and G RABV genes indicate that all the samples belong to the Cosmopolitan cluster, and they show a strong clustering with other RABV from samples isolated in Georgia and neighboring countries. This conclusion is further supported by cluster analysis of full genomes. These results recapitulate the finding that RABV generally cluster by geographic origin [11]. Given metadata on the precise location in the country of Georgia from which each sample was collected, we were further able to investigate the geospatial clustering patterns among our samples, as well as comparing them with the data from a similar study of N gene sequences from distinct Georgian samples [41]. Although regionally distinct isolates generally clustered together, two isolates from Tbilisi and Kvemo Kartli (RABV-GEO-37 and -54, respectively) were more diverse, perhaps reflecting human-facilitated movement of rabid animals from neighboring countries where these genotypes are more common.
Amino acid sequence analyses of the G protein ectodomain revealed a high degree of conservation in the antigenic sites. Indeed, among the 12 ECTO groups, we identified only one amino acid change within the RABV G antigenic sites: R264H in the ECTO2 group, involving the C-terminal residue within the antigenic site G5. This change has been previously reported in strains of Asian origin [49,50]. Notably, this amino acid is not critical for recognition by mAb AR16, and both His and Arg residues are common at position 264 in RABV G genes from a variety of sequenced isolates [27]. However, although R264H does not impair binding by mAb AR16, it does impede the binding of other anti-RABV G mAbs, such as 1D1 and 6-15C4 [28, 29], demonstrating that this amino acid substitution does impact the spectrum of antibodies capable of interacting with this site.
The remainder of residue changes among the ECTO groups do not alter putative glycosylation sites (N-X-S/T) or disulfide bonds and will require further analysis to evaluate putative effects on the tertiary structure of the G protein. In this regard, the I90T and S156G amino acids changes found in the ECTO6 group may be of particular interest, since this group contains RABV-GEO-37 and -54, the more phylogenetically distant samples identified in this study. It would be interesting to determine if these and other residue changes outside of the antigenic sites have significant effects on parameters such as viral titer or efficiency of cellular infection. Overall, sequence analysis of G genes from RABV-GEO isolates suggests that current vaccines should offer protection against recently circulating Georgian canine strains.
As expected, human mAb A6 neutralized two RABV-GEO isolates that we were able to culture successfully. These data support the conclusion that mAb A6 is highly efficient in the neutralization of lyssaviruses across phylogroup I [37], including currently circulating strains. Given the in vitro potency of this mAb, the utility of A6 for in vivo applications (e.g., as part of a PEP cocktail[51] or as a therapeutic[52]) should be explored.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, B.C.S.; design, conduct and analysis of experiments C.H., A.C.P., C.C., A.F., M.R., G.K.R., A.E.L., M.C.F., H.M.D., R.Z.C..; analyzed data, prepared original manuscript draft, edited and revised manuscript, C.H., A.C.P, C.C., F.M., M.A.W., E.D.L., K.A.B-L.; collected and provided samples, M.D., T.B., A.N., and C.E.H.; supervision, financial support, C.E.H., M.A.W., K.A.B-L., C.C.B., and B.C.S. All authors have read and agreed to the published version of the manuscript. Funding: Research support was provided by US Navy WUN A1417 at NMRC, NIH grant AI057168 to C.C.B. and Center for Global Health Engagement, Uniformed Services University grant HU00011920118 to B.C.S. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Complete viral genome sequences are available in GenBank, with IDs OQ603609 to OQ603685
Acknowledgments
The authors thank Dr. Wanda Markotter (University of Pretoria) for protocols and guidance regarding culture of lyssavirus from brain tissue isolates. The opinions and assertions expressed herein are those of the authors and do not reflect the official policy or position of the Uniformed Services University of the Health Sciences, Walter Reed Army Institute of Research, the U.S. Navy, the Department of Defense, nor the U.S. Government. Several of the authors are U.S. Government employees. This work was prepared as part of their official duties. Title 17 U.S.C. § 105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the global burden of endemic canine rabies. PLoS Negl Trop Dis 2015, 9, e0003709. [Google Scholar] [CrossRef]
- Knobel, D.L.; Jackson, A.C.; Bingham, J.; Ertl, H.C.J.; Gibson, A.D.; Hughes, D.; Joubert, K.; Mani, R.S.; Mohr, B.J.; Moore, S.M.; et al. A One Medicine Mission for an Effective Rabies Therapy. Front Vet Sci 2022, 9, 867382. [Google Scholar] [CrossRef] [PubMed]
- Benavides, J.A.; Valderrama, W.; Recuenco, S.; Uieda, W.; Suzan, G.; Avila-Flores, R.; Velasco-Villa, A.; Almeida, M.; Andrade, F.A.G.; Molina-Flores, B.; et al. Defining New Pathways to Manage the Ongoing Emergence of Bat Rabies in Latin America. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.P.; Chand, R.; Huettmann, F.; Ghimire, T.R. Rabies Elimination: Is It Feasible without Considering Wildlife? J Trop Med 2022, 2022, 5942693. [Google Scholar] [CrossRef] [PubMed]
- Tidman, R.; Fahrion, A.S.; Thumbi, S.M.; Wallace, R.M.; De Balogh, K.; Iwar, V.; Yale, G.; Dieuzy-Labaye, I. United Against Rabies Forum: The first 2 years. Front Public Health 2023, 11, 1010071. [Google Scholar] [CrossRef] [PubMed]
- Lojkic, I.; Simic, I.; Bedekovic, T.; Kresic, N. Current Status of Rabies and Its Eradication in Eastern and Southeastern Europe. Pathogens 2021, 10. [Google Scholar] [CrossRef]
- Fisher, C.R.; Streicker, D.G.; Schnell, M.J. The spread and evolution of rabies virus: conquering new frontiers. Nat Rev Microbiol 2018, 16, 241–255. [Google Scholar] [CrossRef]
- Scott, T.P.; Nel, L.H. Lyssaviruses and the Fatal Encephalitic Disease Rabies. Front Immunol 2021, 12, 786953. [Google Scholar] [CrossRef]
- Brunker, K.; Nadin-Davis, S.; Biek, R. Genomic sequencing, evolution and molecular epidemiology of rabies virus. Rev Sci Tech 2018, 37, 401–408. [Google Scholar] [CrossRef]
- Holmes, E.C. The phylogeography of human viruses. Mol Ecol 2004, 13, 745–756. [Google Scholar] [CrossRef]
- Bourhy, H.; Reynes, J.M.; Dunham, E.J.; Dacheux, L.; Larrous, F.; Huong, V.T.Q.; Xu, G.; Yan, J.; Miranda, M.E.G.; Holmes, E.C. The origin and phylogeography of dog rabies virus. J Gen Virol 2008, 89, 2673–2681. [Google Scholar] [CrossRef]
- Troupin, C.; Dacheux, L.; Tanguy, M.; Sabeta, C.; Blanc, H.; Bouchier, C.; Vignuzzi, M.; Duchene, S.; Holmes, E.C.; Bourhy, H. Large-Scale Phylogenomic Analysis Reveals the Complex Evolutionary History of Rabies Virus in Multiple Carnivore Hosts. PLoS Pathog 2016, 12, e1006041. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Gifford, R.J.; Singer, J.; Hill, V.; O'Toole, A.; Rambaut, A.; Hampson, K.; Brunker, K. Making genomic surveillance deliver: A lineage classification and nomenclature system to inform rabies elimination. PLoS Pathog 2022, 18, e1010023. [Google Scholar] [CrossRef] [PubMed]
- Bacus, M.G.; Buenaventura, S.G.C.; Mamites, A.M.C.; Elizagaque, H.G.; Labrador, C.C.; Delfin, F.C.; Eng, M.N.J.; Lagare, A.P.; Marquez, G.N.; Murao, L.A.E. Genome-based local dynamics of canine rabies virus epidemiology, transmission, and evolution in Davao City, Philippines, 2018-2019. Infect Genet Evol 2021, 92, 104868. [Google Scholar] [CrossRef]
- Dellicour, S.; Troupin, C.; Jahanbakhsh, F.; Salama, A.; Massoudi, S.; Moghaddam, M.K.; Baele, G.; Lemey, P.; Gholami, A.; Bourhy, H. Using phylogeographic approaches to analyse the dispersal history, velocity and direction of viral lineages - Application to rabies virus spread in Iran. Mol Ecol 2019, 28, 4335–4350. [Google Scholar] [CrossRef]
- Heaton, P.R.; Johnstone, P.; McElhinney, L.M.; Cowley, R.; O'Sullivan, E.; Whitby, J.E. Heminested PCR assay for detection of six genotypes of rabies and rabies-related viruses. J Clin Microbiol 1997, 35, 2762–2766. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner. In Proceedings of the Conference: 9th Annual Genomics of Energy & Environment Meeting, Walnut Creek, CA, USA., 17-20 March 2014. [Google Scholar]
- Shen, W.; Ren, H. TaxonKit: A practical and efficient NCBI taxonomy toolkit. J Genet Genomics 2021, 48, 844–850. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: a new versatile metagenomic assembler. Genome Res 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol Biol Evol 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol Biol Evol 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on.
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 2008, 36, D13–21. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res 2022, 50, W276–279. [Google Scholar] [CrossRef]
- Lai, C.Y.; Dietzschold, B. Amino acid composition and terminal sequence analysis of the rabies virus glycoprotein: identification of the reading frame on the cDNA sequence. Biochem Biophys Res Commun 1981, 103, 536–542. [Google Scholar] [CrossRef]
- Yang, F.; Lin, S.; Ye, F.; Yang, J.; Qi, J.; Chen, Z.; Lin, X.; Wang, J.; Yue, D.; Cheng, Y. , et al. Structural Analysis of Rabies Virus Glycoprotein Reveals pH-Dependent Conformational Changes and Interactions with a Neutralizing Antibody. Cell Host Microbe 2020, 27, 441–453. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing. 2022. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York 2016.
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Rapp, B.A.; Wheeler, D.L. GenBank. Nucleic Acids Res 2000, 28, 15–18. [Google Scholar] [CrossRef]
- Steinegger, M.; Soding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J. , et al. Pfam: The protein families database in 2021. Nucleic Acids Res 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P. , et al. IEDB-AR: immune epitope database-analysis resource in 2019. Nucleic Acids Res 2019, 47, W502–W506. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W. , et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res 2023, 51, D678–D689. [Google Scholar] [CrossRef] [PubMed]
- Weir, D.L.; Coggins, S.A.; Vu, B.K.; Coertse, J.; Yan, L.; Smith, I.L.; Laing, E.D.; Markotter, W.; Broder, C.C.; Schaefer, B.C. Isolation and Characterization of Cross-Reactive Human Monoclonal Antibodies That Potently Neutralize Australian Bat Lyssavirus Variants and Other Phylogroup 1 Lyssaviruses. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Meechan, P.J.; Potts, J. Biosafety in microbiological and biomedical laboratories. 2020. 2020. [Google Scholar]
- Tordo, N.; Kouknetzoff, A. The rabies virus genome: an overview. Onderstepoort J Vet Res 1993, 60, 263–269. [Google Scholar] [PubMed]
- Tordo, N.; Badrane, H.; Bourhy, H.; Sacramento, D. Molecular epidemiology of lyssaviruses: focus on the glycoprotein and pseudogenes. Onderstepoort J Vet Res 1993, 60, 315–323. [Google Scholar]
- Tabatadze, L.; Gabashvili, E.; Kobakhidze, S.; Lomidze, G.; Loladze, J.; Tsitskishvili, L.; Kotetishvili, M. Evolutionary analysis of rabies virus isolates from Georgia. Arch Virol 2022, 167, 2293–2298. [Google Scholar] [CrossRef]
- Badrane, H.; Bahloul, C.; Perrin, P.; Tordo, N. Evidence of two Lyssavirus phylogroups with distinct pathogenicity and immunogenicity. J Virol 2001, 75, 3268–3276. [Google Scholar] [CrossRef]
- Wunner, W.H.; Larson, J.K.; Dietzschold, B.; Smith, C.L. The molecular biology of rabies viruses. Rev Infect Dis 1988, 10 Suppl 4, S771–784. [Google Scholar] [CrossRef]
- Dietzschold, B.; Tollis, M.; Lafon, M.; Wunner, W.H.; Koprowski, H. Mechanisms of rabies virus neutralization by glycoprotein-specific monoclonal antibodies. Virology 1987, 161, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhao, W.; He, W.; Wang, N.; Su, J.; Ji, S.; Chen, J.; Wang, D.; Zhou, J.; Su, S. Generation of Monoclonal Antibodies against Variable Epitopes of the M Protein of Rabies Virus. Viruses 2019, 11. [Google Scholar] [CrossRef]
- Zhao, W.; Su, J.; Zhao, N.; Liu, J.; Su, S. Development of Monoclonal Antibodies for Detection of Conserved and Variable Epitopes of Large Protein of Rabies Virus. Viruses 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Luo, Y.; Michel, F.; Hogan, R.J.; He, Y.; Fu, Z.F. Characterization of conformation-specific monoclonal antibodies against rabies virus nucleoprotein. Arch Virol 2010, 155, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Du Pont, V.; Plemper, R.K.; Schnell, M.J. Status of antiviral therapeutics against rabies virus and related emerging lyssaviruses. Curr Opin Virol 2019, 35, 1–13. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, S.; Zhang, S.; Liu, Y.; Zhang, J.; Zhang, F.; Mi, L.; Hu, R. Molecular characterization of a rabies virus isolate from a rabid dog in Hanzhong District, Shaanxi Province, China. Arch Virol 2014, 159, 1481–1486. [Google Scholar] [CrossRef]
- Tohma, K.; Saito, M.; Kamigaki, T.; Tuason, L.T.; Demetria, C.S.; Orbina, J.R.; Manalo, D.L.; Miranda, M.E.; Noguchi, A.; Inoue, S. , et al. Phylogeographic analysis of rabies viruses in the Philippines. Infect Genet Evol 2014, 23, 86–94. [Google Scholar] [CrossRef]
- de Melo, G.D.; Hellert, J.; Gupta, R.; Corti, D.; Bourhy, H. Monoclonal antibodies against rabies: current uses in prophylaxis and in therapy. Curr Opin Virol 2022, 53, 101204. [Google Scholar] [CrossRef]
- de Melo, G.D.; Sonthonnax, F.; Lepousez, G.; Jouvion, G.; Minola, A.; Zatta, F.; Larrous, F.; Kergoat, L.; Mazo, C.; Moigneu, C. , et al. A combination of two human monoclonal antibodies cures symptomatic rabies. EMBO Mol Med 2020, 12, e12628. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic tree of Georgian lyssavirus N gene sequences. A total of 140 sequences with 1,353 nucleotide sites were included and the log-likelihood of the consensus tree was -9,845. The tree is rooted at the midpoint. The labels of samples collected in Georgia with geographical metadata are colored by subregion: Kakheti (green), Kvemo Kartli (purple), Samgrelo (orange), Tbilisi (blue), Guria (pink), Adjara (red), Shida Kartli (teal). (A) Full phylogenetic tree (the area shadowed in blue is zoomed in the B panel). (B) Zoomed view of the branching area of the tree containing the RABV-GEO samples. (C) Map of the country of Georgia, indicating regions from which samples were collected. Colors coded to match samples in (A) and (B).
Figure 1.
Phylogenetic tree of Georgian lyssavirus N gene sequences. A total of 140 sequences with 1,353 nucleotide sites were included and the log-likelihood of the consensus tree was -9,845. The tree is rooted at the midpoint. The labels of samples collected in Georgia with geographical metadata are colored by subregion: Kakheti (green), Kvemo Kartli (purple), Samgrelo (orange), Tbilisi (blue), Guria (pink), Adjara (red), Shida Kartli (teal). (A) Full phylogenetic tree (the area shadowed in blue is zoomed in the B panel). (B) Zoomed view of the branching area of the tree containing the RABV-GEO samples. (C) Map of the country of Georgia, indicating regions from which samples were collected. Colors coded to match samples in (A) and (B).
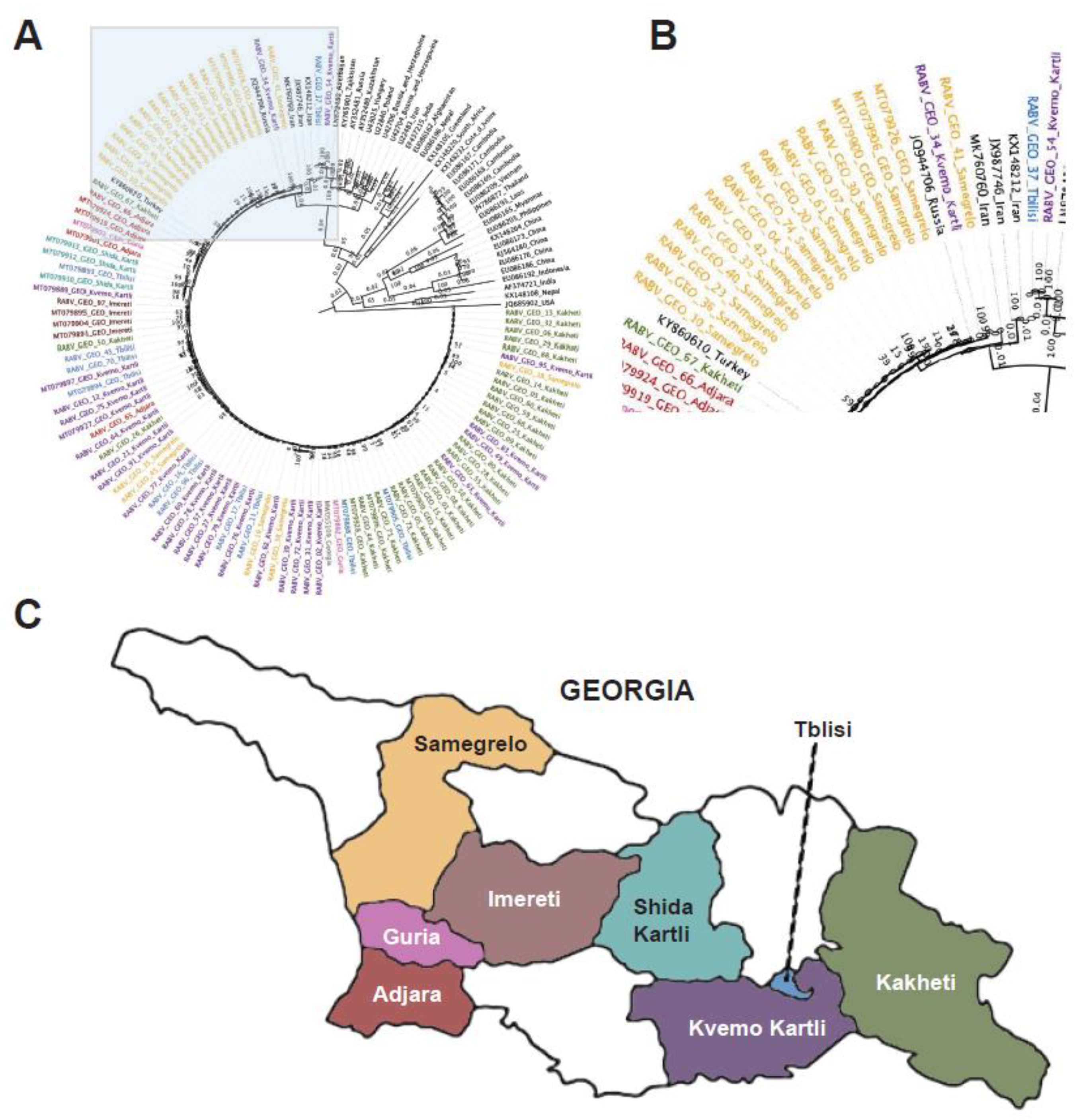
Figure 2.
Phylogenetic tree of Georgian lyssavirus G gene sequences. A total of 110 sequences with 1,578 nucleotide sites were included and the log-likelihood of the consensus tree was -12,159. The tree is rooted at the midpoint. The labels of samples collected in this study are colored green. (A) Full phylogenetic tree (the area shadowed in blue is zoomed in the B panel). (B) Zoomed view of the branching area of the tree containing the RABV-GEO samples.
Figure 2.
Phylogenetic tree of Georgian lyssavirus G gene sequences. A total of 110 sequences with 1,578 nucleotide sites were included and the log-likelihood of the consensus tree was -12,159. The tree is rooted at the midpoint. The labels of samples collected in this study are colored green. (A) Full phylogenetic tree (the area shadowed in blue is zoomed in the B panel). (B) Zoomed view of the branching area of the tree containing the RABV-GEO samples.
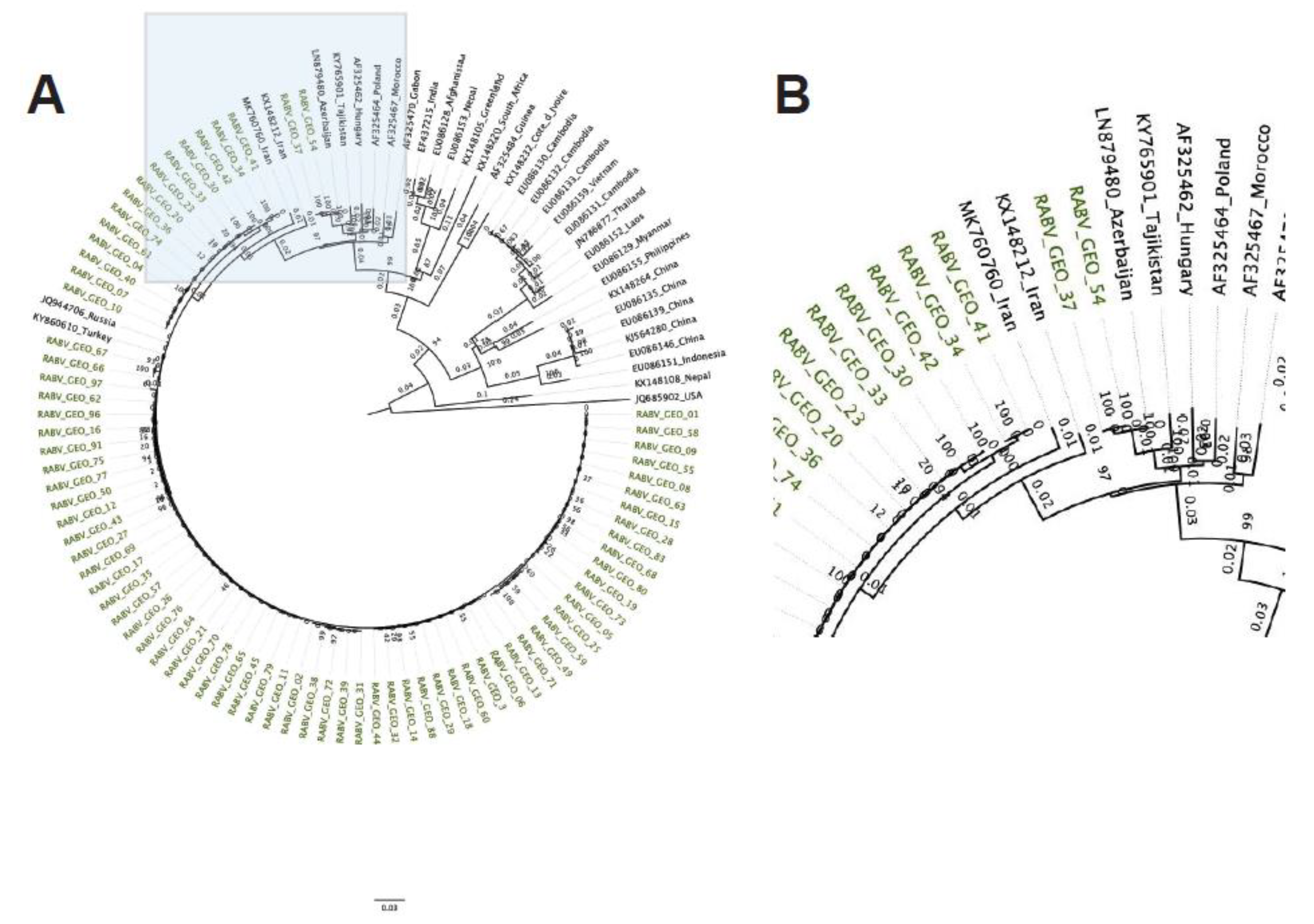
Figure 3.
Amino acid sequence of the G protein ectodomain of the Georgian ECTO1 group. (A) The amino acid sequence of the RABV G ectodomain is shown, using the sequence derived from the viruses of the ECTO1 group. The antigenic sites are underlined. Amino acids that vary between different ECTO groups are indicated in red. (B) Amino acid changes in different ECTO groups respect to the reference (ECTO1). The positions of the amino acids in the ectodomain are indicated in superscript.
Figure 3.
Amino acid sequence of the G protein ectodomain of the Georgian ECTO1 group. (A) The amino acid sequence of the RABV G ectodomain is shown, using the sequence derived from the viruses of the ECTO1 group. The antigenic sites are underlined. Amino acids that vary between different ECTO groups are indicated in red. (B) Amino acid changes in different ECTO groups respect to the reference (ECTO1). The positions of the amino acids in the ectodomain are indicated in superscript.
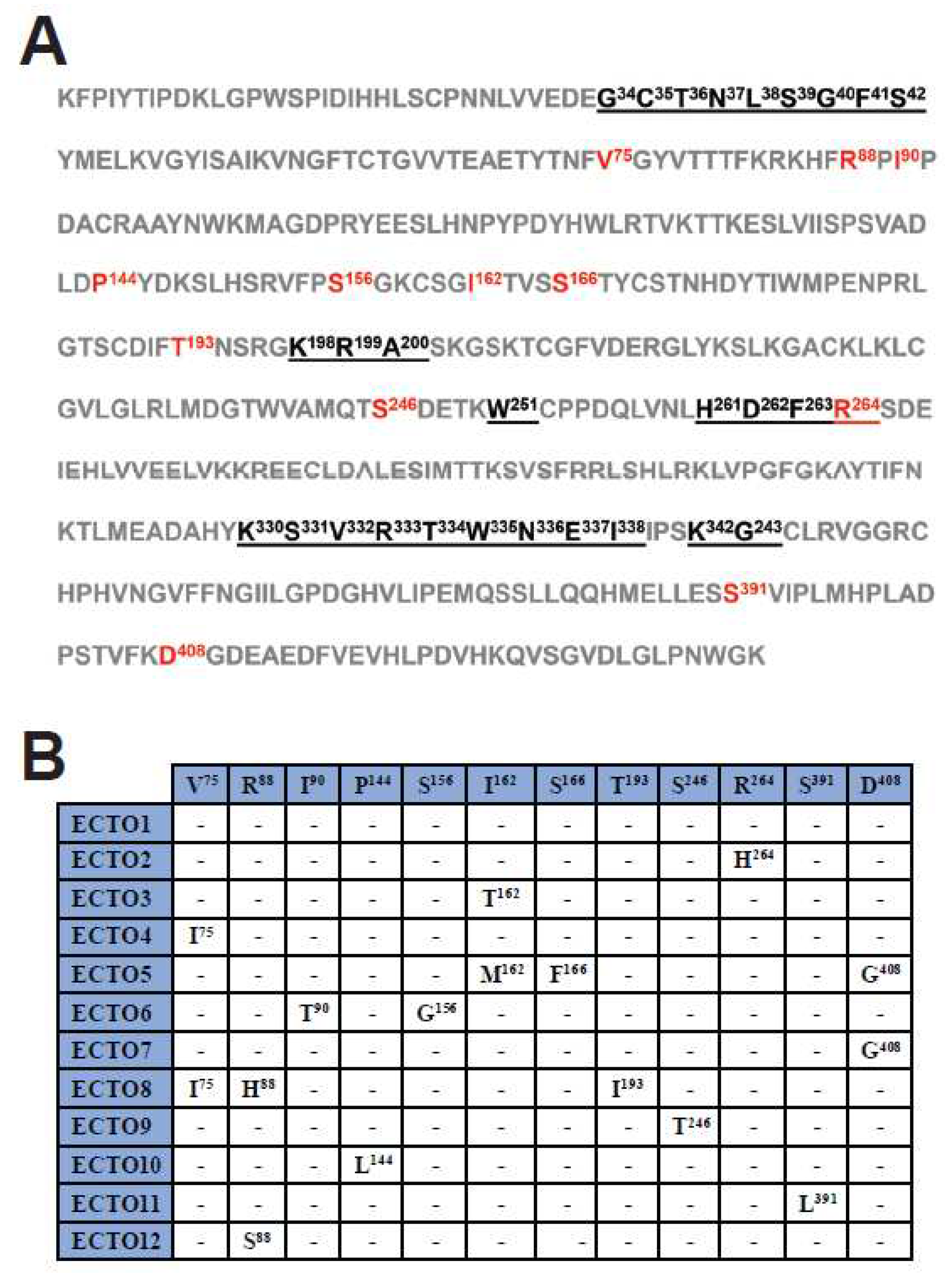
Figure 4.
Neutralizing activity of anti-ABLV G human mAb A6 against RABV-GEO isolates. RABV-GEO viruses 6, 9 and 20 were isolated from primary canine brain tissue, followed by one round of in vitro passaging. Virus neutralization was measured by incubation of viral supernatants with human mAb A6, followed by infection of N2a cells. Viral foci were identified by staining with anti-Rabies G. Graph shows percent virus neutralization over a series of 5-fold dilutions of mAb A6.
Figure 4.
Neutralizing activity of anti-ABLV G human mAb A6 against RABV-GEO isolates. RABV-GEO viruses 6, 9 and 20 were isolated from primary canine brain tissue, followed by one round of in vitro passaging. Virus neutralization was measured by incubation of viral supernatants with human mAb A6, followed by infection of N2a cells. Viral foci were identified by staining with anti-Rabies G. Graph shows percent virus neutralization over a series of 5-fold dilutions of mAb A6.
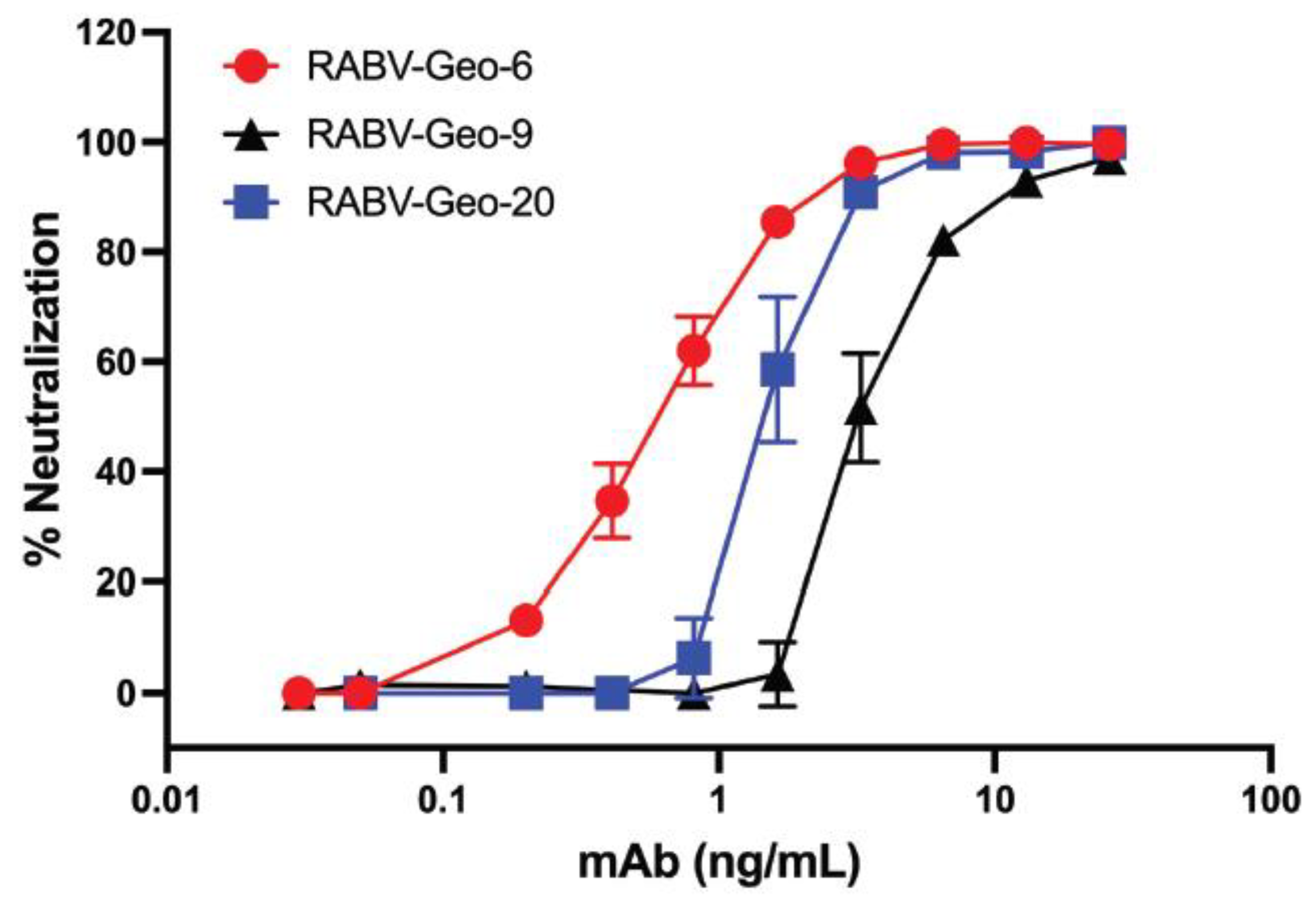

Table 1.
Cluster analysis of RABV-GEO N, P, M, G and L proteins.
| Protein | Number of sequences from NCBI | RABV-GEO sequences | Number of clusters | Number of clusters containing RABV-GEO samples |
|---|---|---|---|---|
| N | 7,489 | 77 | 2 | 1 |
| P | 2,579 | 77 | 9 | 1 |
| M | 2,178 | 77 | 4 | 1 |
| G | 5,915 | 77 | 5 | 1 |
| L | 2,948 | 77 | 5 | 1 |
Table 2.
Linear peptide epitopes within L, M, P among representative amino acid sequences. Differences are shown in bold red.
Table 2.
Linear peptide epitopes within L, M, P among representative amino acid sequences. Differences are shown in bold red.
| Epitope ID | Epitope Sequence | Difference in representative sequence (if relevant, colored red and bold) | Protein Name | Protein ID | Protein Accession | Start | End |
|---|---|---|---|---|---|---|---|
| 1336019 | EIFSIP | - | L | ADJ29912.1 | P11213 | 1479 | 1484 |
| 1336142 | RALSK | - | L | ADJ29912.1 | P11213 | 1659 | 1663 |
| 1336215 | VFNSL | - | L | ADJ29912.1 | P11213 | 1724 | 1728 |
| 93714 | RKLGWWLKL | not present in representative sequence set | L | SRC266014 | P11213 | ||
| 929568 | PPDDD | - | M | CEH11416.1 | P08671 | 42 | 46 |
| 929569 | PPYDDD | not present in representative sequence set | M | ADJ29910.1 | P08671 | 25 | 30 |
| 12638 | EKDDLSVEAEIAHQIA | EEDDLSVEAEIAHQIA | P | P69479.1 | P06747 | 191 | 206 |
| 1795 | AHLQGEPIEVDNLPEDMKRLQLDDKKPSGL | AHLQGEPIEVDNLPEDMRRLNLDDGKSPNL | P | AAK54996.1 | P06747 | 37 | 66 |
| 20735 | GKYREDFQMDEGDPS | GKYREDFQMDEGEDP | P | SRC279966 | P06747 | ||
| 23111 | GVQIVRQIRSGERFLKIWSQ | GVQIVRQMRSGERFLKIWSQ | P | NP_056794.1 | P06747 | 101 | 120 |
| 31389 | KIPLRCVLGWVALANSKKFQLLVEADKLSKIMQDDLNRYTSC | KLPLRCVLGWVALANSKKFQLLVEADKLSRIMQDDLNRYASS | P | AAZ07892.1 | P06747 | 256 | 297 |
| 31531 | KKETTSISSQRDSQSSKA | KKETTSTPSQRESQSSKA | P | AAK54996.1 | P06747 | 154 | 171 |
| 38005 | LMDEGEDPSLLFQSYLDNVGVQIVRQMRSGER | QMDEGEDPSLLFQSYLDNVGVQIVRQMRSGER | P | AAK54996.1 | P06747 | 82 | 113 |
| 451183 | VLGWV | - | P | AAK55085.1 | P06747 | 262 | 266 |
| 53736 | RFLKIWSQTVEEIISYVAVN | RFLKIWSQTVEEIISYVTVN | P | P69479.1 | P06747 | 113 | 132 |
| 59254 | SLLFQSYLDNVGVQIVRQIR | SLLFQSYLDNVGVQIVRQMR | P | NP_056794.1 | P06747 | 90 | 109 |
| 68113 | VEAEIAHQI | - | P | P15198.1 | P06747 | 197 | 205 |
| 93760 | RQMKSGGRF | RQMRSGERF | P | AAY23584.1 | P06747 | 68 | 76 |
| 94299 | YLDNVGVHI | YLDNVGVQI | P | AAK55014.1 | P06747 | 96 | 104 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



