
Submitted:
27 June 2023
Posted:
27 June 2023
You are already at the latest version
Abstract
In the past decade, targeted therapies for solid tumors, including non-small cell lung cancer (NSCLC), have advanced significantly, offering tailored treatment options for patients. However, individuals without targetable mutations pose a clinical challenge, as they may not respond to standard treatments like immune-checkpoint inhibitors (ICIs) and novel targeted therapies. While the mechanism of action of ICIs seems promising, the lack of a robust response limits their widespread use. Although the expression levels of programmed death ligand 1 (PD-L1) on tumor cells are used to predict ICI response, identifying new biomarkers, particularly those associated with the tumor microenvironment (TME), is crucial to address this unmet need. Recently, inflammatory cytokines such as interleukin-1 beta (IL-1β) have emerged as a key area of focus and hold significant potential implications for future clinical practice. Combinatorial approaches of IL-1β inhibitors and ICIs may provide a potential therapeutic modality for NSCLC patients without targetable mutations. In this review, we discuss the role of IL-1β in NSCLC, its involvement in inflammatory pathways, and explore its potential role in the treatment of NSCLC.
Keywords:
interleukin-1 beta (IL-1β)
; immune-checkpoint inhibitors (ICIs)
; non-small cell lung cancer (NSCLC)
; programmed death ligand 1 (PD-L1)
; therapeutic resistance
Introduction
Lung cancer remains the leading cause of cancer-related mortality and the second most common malignancy worldwide [1]. Currently, in NSCLC, initial management begins with searching for targetable drivers such as EGFR, ALK, ROS-1, NTRK, RET, NRG or BRAF, MET; however, for patients without such mutations, treatment options are limited [2]. Recent studies have demonstrated that the TME of NSCLC involves a proinflammatory state that can be targeted [3]. Dysregulated inflammatory conditions contribute to lung carcinogenesis by causing protumor inflammation, tumor suppression, and activation of oncogenes [1,4]. Recently, the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial incidentally demonstrated that inhibiting IL-1β is associated with significantly reduced incidence and mortality of lung cancer [5]. Therefore, proposing IL-1β, a key inflammatory cytokine, is a targetable area that directly affects the proinflammatory nature of NSCLC development and progression [5]. TME is becoming recognized as a key factor in carcinogenesis and is a topic of great interest for seeking management options for NSCLC without targetable mutations. In the TME, overexpression of the PD-L1 on tumor cells and engagement with its receptor programmed cell death protein 1 (PD-1) causes T-cell exhaustion and results in decreased anti-tumor activity [6,7]. Immunomodulators targeting the PD-1/PD-L1 axis have had promising results, but therapeutic resistance is common and challenging for clinicians. Here, the role of IL-1β inhibitors is an area of great interest as preclinical and clinical studies have demonstrated that increased levels of IL-1β are associated with elevated expression of PD-L1 and decreased response to anti-PD-L1 therapy [8,9].
Recent advances in understanding the IL-1β/PD-1/PD-L1 pathway have prompted attempts to utilize combinatorial strategies that involve IL-1β inhibitors together with ICIs. To identify biomarkers that predict ICI response in tumors without actionable mutations and provide a solution for patients with actionable mutations who develop resistance to currently approved targeted therapies, understanding the crosstalk that occurs within the IL-1β/PD-1/PD-L1 pathway and the TME alterations that arise when ICIs are combined with IL-1β inhibitors is crucial. Therefore, in this review, we aim to explore the pathophysiological mechanism of the inflammatory process of carcinogenesis in NSCLC and the dynamic relationship within the IL-1β/PD-1/PD-L1 pathway. We hope to shed light on the use of combination therapies for NSCLC without targetable mutations and provide a potential treatment option for NSCLC patients.
Il-1β signaling in NSCLC
The TME is altered by many immune factors and recent studies have shown that the TME of lung cancer is proinflammatory [10]. Due to the relationship between chronic inflammatory states and cancer, serum levels of IL-1β, IL-6, IL-8 [11] have been studied as potential biomarkers of malignancy. For example, patients with breast, lung, cervical, hepatocellular, and gastric cancer tumor cells revealed increased levels of IL-1β alleles [12]. In general, expression of IL-1β is induced by the presence of stressful stimuli such as hypoxia, inflammation, infection, which induces toll-like receptors (TLRs) to stimulate tumor necrosis factor (TNF). Inactive pro-IL1β is activated by intracellular protein complexes, known as inflammasomes, and then cleaved by caspase-1 (also known as IL-1β-converting enzyme (ICE) into its active form. Immune cells involved in tumor suppression, T cells, dendritic cells (DCs), epithelial cells, neutrophils, macrophages, and other antigen-presenting cells (APCs) express IL-1 receptors (IL-1R and IL-1R2) which interact with IL-1β (Figure 1). IL-1β signaling recruits myeloid differentiation primary response-88 (MyD88) and IL-1R associated kinases (IRAKs), which directly interact with TNF receptor-associated factor 6 (TRAF6). This activates the mitogen activated protein kinase (MAPK) pathway and (nuclear factor kappa B) NF-κB, thus activating downstream inflammatory pathways and promoting unchecked growth of malignant cells and inhibiting apoptosis [8,12,13].
Chronic inflammation has been linked to an increased risk of cancer by providing survival signals, suppressing T-cells’ effector functions, inducing angiogenesis, and promoting invasion and metastasis [3,14]. The active form of IL-1β induces protumor inflammation, tumorigenesis, immune invasion, and metastasis (Figure 2) [3,4,13]. IL-1β may also have a positive feedback loop, perpetuating the course of the cancer [12]. Within the TME, tumor cells are the source of chronic inflammation, and IL-1β becomes an upstream regulator, altering immunologic responses, hormonal signaling, neovascularization, and enhancing metastatic potential through downstream pathways such as NF-κB/MAPK/AKT/Wnt/β-catenin [13].
The role of IL-1β in cancer is complex, and its ability to directly alter the TME has been studied in the preclinical setting. For example, increased expression of IL-1β has been shown to promote the accumulation of myeloid-derived suppressor cells (MDSCs), decrease the number of natural killer (NK) cells, and increase tumor size [4,15]. The levels of MDSCs, our body’s major suppressors of immunological responses to tumors, are significantly increased in the TME, which leads to the maintenance and acceleration of tumor growth and metastasis [4]. Increased accumulation of MDSCs results in the expansion of CD4+ CD25+ Foxp3+ regulatory T cells (Tregs), leading to downregulation of the antitumor capability of NK and cytotoxic T cells. IL-1β also functions as a chemoattractant to recruit tumor-associated macrophages (TAMs) by attaching monocyte chemoattractant protein (MCP-1) on tumor cells [16]. This cascade further potentiates IL-1β production via TAMs and triggers inflammasomes to promote further tumor growth [15].
Furthermore, IL-1β has been implicated in the promotion of angiogenesis in several types of malignancies, such as melanoma and fibrosarcoma, potentiating their metastatic potential [12,17,18]. This is due to its ability to induce the expression of pro-angiogenic factors such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF), transforming growth factor (TGF-αβ), platelet-derived endothelial growth factor (PDGF), interleukin-8 (IL-8, also known as CXCL8), and fibroblast growth factors (FGF). They mediate the migration and proliferation of endothelial cells to expand the existing vascular system, which is necessary for the delivery of nutrients and oxygen to rapidly growing tumor cells [17]. IL-1β also increases the expression of matrix metalloproteinases (MMPs), which degrade extracellular matrix (ECM) proteins, facilitating the migration and invasion of tumor cells into surrounding tissues [3,17]. MMPs further promote angiogenesis by releasing sequestered pro-angiogenic factors from the ECM, making them available to surrounding cells. In addition, in lung cancer, exposure to IL-1β decreases phosphatase and tensin homolog (PTEN) expression, PI3K/AKT signaling activation, and the induction of epithelial-mesenchymal transition (EMT), conferring primary lung carcinoma cells with the ability to mobilize, invade, and damage distant sites, leading to angiogenesis [9,19,20].
Specifically, for NSCLC, IL-1β has a multifaceted impact on the development and progression of the disease. Patients with NSCLC have been found to have elevated levels of IL-1β in plasma, and IL-1β mRNA expression, and serum IL-1β has been associated with worse prognosis [14,21,22]. In NSCLC, IL-1β potentiates accelerated neoplastic progression by repressing miR-101 expression through the cyclooxygenase 2 (COX2)/HIF1α pathway [21]. Tumor-derived IL-1β activates γδT-cells, which in turn secrete IL-17. Secretion of IL-17 causes neutrophils to increase inducible nitric oxide synthase (iNOS) and suppress antitumor CD8+ T cells, leading to an increase in metastatic potential [23]. In lung cancer, it has also been shown that commensal bacteria promote IL-1β production from local macrophages, inducing tumor cell proliferation and inflammation [24].
Despite recent advances in targeted therapies for NSCLC patients with specific genetic mutations, the lack of robust response has been a challenge for clinicians [25]. IL-1β has been implicated in the development of therapeutic resistance. For example, it has been observed that varying levels of IL-1β are present in therapy-resistant patients [3,26]. Thus, the heterogeneity of serum IL-1β levels may explain the variability of response to treatment options in NSCLC [26,27]. Additionally, mutations found in lung cancer cells are associated with the IL-1β axis, making it a potential target for therapy [8]. BRAF(V600E) mutations are rare in NSCLC but can occur in up to 2% of cases [28]. Treatment of these neoplasms with BRAF inhibitors (BRAFi) has been shown to increase dendritic cell-mediated IL-1β production, worsening the inflammatory positive feedback loop and promoting resistance. Hajek et al demonstrated that the combination of dabrafenib with vemurafenib or trametinib, BRAFi’s used in melanoma and NSCLC patients with BRAFV600E mutation, strongly upregulated IL-1β production in myeloid mouse APCs, due to BRAFi-induced activation of the inflammasome leading to caspase-8 activation and pro-IL-1β processing. Alternative mechanisms explain BRAFi resistance by a cytokine-signaling network involving TAM-derived IL-1β, cancer-associated fibroblasts (CAFs)-derived CXCR2 ligands, and PTEN inactivation [29]. Still, the mechanism of resistance to BRAFi and BRAFi combinatorial regimens remains an unmet area of further study. Given that IL-1β production affects PTEN, it is reasonable to hypothesize that IL-1β inhibitors may aid in combating resistance to BRAFi. Therefore, further studies are warranted to explore the potential of IL-1β inhibitors in combination with BRAFi in NSCLC patients with BRAF mutations.
The role of IL-1β in the development of resistance to treatment in NSCLC is further implicated in current studies with bortezomib and EGFR inhibitors. Davies et al. showed that bortezomib, a proteasome inhibitor that partially inhibits NF-κB, is not effective as a single agent in the treatment of NSCLC [30]. Further investigation revealed that the inhibition of NF-κB signaling was associated with an increase in IL-1β production and enhanced tumorigenesis in the lungs. Interestingly, McLeod et al. demonstrated that dual blockade with an IL-1R antagonist (anakinra) and bortezomib resulted in increased therapeutic efficacy by reducing lung tumor burden [31]. These findings suggest that the inefficiency of monotherapy with NF-κB inhibitors in NSCLC may be due to the potentiation of neutrophil-dependent production of IL-1β, leading to enhanced pulmonary carcinogenesis. The study also highlights the role of IL-1β in resistance to currently available treatment modalities and suggests that combined NF-κB and IL-1β targeted treatments may lead to reduced tumor formation and growth [16,31].
In addition, Huang et al. found that IL-1β upregulates EH domain-containing protein 1 (EHD1) expression, which activates the PTEN/PI3K/AKT signaling pathway, leading to off-site EGFR-TKI resistance in NSCLC. Interestingly, the inhibition of the IL-1β/EHD1/TUBB3 axis has shown promising results in overcoming EGFR-TKI resistance [9]. These findings suggest that inhibition of the IL-1β/EHD1 signaling pathway may be a target for patients who develop EGFR-TKI resistance. Overall, the combinatorial, targeted strategies towards the IL-1β axis may be a solution for NSCLC patients with actionable mutations who have developed resistance to currently approved targeted therapies.
Among targeted therapies for NSCLC, tyrosine kinase inhibitors (TKIs) against specific driver-mutations have shown significant improvement in survival outcomes [32]. However, only a subset of patients with NSCLC carries single-driver mutations against EGFR, ALK, ROS-1, or BRAF, while the majority lack such targetable mutations [32,33]. This has been a major challenge for the management of NSCLC. While novel treatment options for NSCLC with driver-mutations are standard of care, some patients develop resistance to such treatments, requiring another line of therapy [33]. In patients with NSCLC without targetable driver mutations and those with resistance to TKI therapy, the use of ICIs targeting the PD-1/PD-L1 pathway has been a topic of focused studies [34].
Preclinical studies have shown that the PD-1/PD-L1 pathway is interconnected with the TME [35,36]. PD-1 is expressed on T cells, specifically on tumor-specific T, B, and NK cells [37]. PD-L1 is usually expressed by macrophages, DCs, epithelial cells, along with some activated T and B cells and upregulated in certain tumor cells [37]. The interaction of PD-1 on T-cells and PD-L1 on tumor cells causes phosphorylation of Lck, a tyrosine kinase, leading to T-cell deactivation and subsequently compromised anti-tumor activity of T cells [38]. PD-L1 expression is a critical component of the adaptive immune mechanism of tumor cells to escape antitumor cells [39,40]. The interaction of PD-1/PD-L1 proteins in the TME results in T cell exhaustion, suppressing the antitumor effects of cells such as APCs and tumor infiltrating lymphocytes (TILs), leading to a state of dysfunction characterized by decreased T cell effector activity [36]. An “active” TME consists of the presence of PD-1+ CD8+ activated T cells and the expression of PD-L1 [35], which is a favorable setting for ICIs.
ICIs like pembrolizumab (PD-1 inhibitor), atezolizumab (PD-L1 inhibitor), cemiplimab (PD-L1 inhibitor), and durvalumab (PD-L1 inhibitor) have shown promising results in improving overall survival (OS) and progression-free survival (PFS) in certain types of NSCLC patients (Supplemental Table S1). Prominent clinical trials involving ICIs include KEYNOTE-010 and KEYNOTE-024, which showed improved survival with pembrolizumab, and CheckMate 017 and CheckMate 057, which reported longer survival with nivolumab [26,41,42,43,44]. Numerous studies have explored the use of ICIs for the treatment of various settings in NSCLC. In EMPOWER-Lung 1, cemiplimab was used as a first-line monotherapy for advanced NSCLC with at least 50% PD-L1 expression, and showed significant improvement in OS and PFS compared to platinum-doublet chemotherapy [45]. Similarly, durvalumab after chemoradiotherapy in stage III NSCLC in PACIFIC showed significantly longer PFS compared to placebo [46]. However, MYSTIC demonstrated that durvalumab as a first-line treatment for metastatic NSCLC did not improve OS [47]. In other studies, the combination of atezolizumab with chemotherapy in stage IV NSCLC showed improved PFS in IMpower131 and significant improvements in OS and PFS in IMpower130 for non-squamous NSCLC [48,49]. Similarly, atezolizumab also showed superior OS as a first-line treatment for metastatic NSCLC with at least 1% PD-L1 expression in IMpower110, and added on to bevacizumab-carboplatin-paclitaxel for metastatic nonsquamous NSCLC in IMpower150 [50]. Additionally, the OAK trial revealed improved survival with atezolizumab in patients with previously treated NSCLC with low or undetectable PD-L1 expression [51].
Despite these promising results, resistance to ICIs is a significant challenge [4,52,53]. The use of ICIs, specifically PD-1/PD-L1 inhibitors, have shown variable clinical benefit with major pathological regression rates ranging widely from 18-83% [8]. Recent studies have shown that resistance to anti-PD therapy may be one of explanations for this widely varied response [4,54,55]. Primary and acquired resistance occur from immune-mediated mechanisms which derive from the TME and can occur at any step of the immunoregulatory process [4,54,56,57]. Primary resistance is the lack of initial response to ICIs, whereas acquired resistance is the lack of further response after initial improvement with ICIs [58]. Some of the mechanisms of resistance to anti-PD therapy include immunologic loss or lack of neoantigens, signaling defects in the interferon (IFN) pathway, the lack of PD-L1 receptors, and local immune dysfunction, including exclusion of T cells [54,59]. A key mechanism of resistance to ICIs, specifically anti-PD therapy, is the lack of PD-L1 receptors. Recently, the TME of malignancies such as NSCLC and melanoma have been classified into four categories based on the expression of PD-L1 and TILs [54,59]. Type 1 is TIL- and PD-L1-, type 2 TIL+ and PD-L1+, type 3 TIL+ and PD-L1- and type 4 TIL- and PD-L1+. Unfortunately, only about 17% of NSCLCs demonstrate type TME, which may explain the unpredictable response to ICIs [54]. T cell infiltration is closely related to PTEN, and the loss of PTEN expression is associated with resistance to anti-PD therapy [60,61,62]. Further, though not fully understood, the gut microbiome may play a role in altering the TME by stimulating DCs and affecting antigen-presentation [63,64]. Interestingly, sensitivity to anti-PD therapy has been associated with tumor mutational burden (TMB), with higher levels of mutations indicating high immunogenicity, which can be targets for anti-PD therapy [60,65]. However, studies have shown that some gene mutations can lead to mixed results to anti-PD therapy. For example, patients with positive EGFR mutation have been shown to have lower levels of PD-L1 expression with possible resistance to anti-PD therapy [66,67]. Such observations of resistance to ICIs may help guide their use in NSCLC and further studies are needed.
In a retrospective study, Sakai et al stratified NSCLC patients who had received ICIs therapy by morphological, immune, and genetic features to further address concerns about ICIs resistance in sub-groups [68]. The study revealed that the absence of definitive morphological features in non-squamous NSCLC was associated with better prognosis, whereas tumors diagnosed as morphologic adenocarcinoma were more associated with PD-L1 score < 1% and less likely to be associated with samples with PD-L1 expression > 50%. These results suggest the use of morphological features of NSCLC and levels of PD-L1 expression to predict response and resistance to ICIs [68]. IFN-γ has been shown to maintain levels of PD-L1, and the clinical significance of the IFN-γ pathway and other cytokines are areas requiring further evaluation [54,55]. Further, biomarkers such as microsatellite instability (MSI), high TMB along with CD8+ T cell infiltrates are noted as foremost predictive markers for ICIs response [69]. Understanding these mechanisms of resistance is crucial for improving the effectiveness of ICIs in NSCLC.
Complex networking in the IL-1β/PD-1/PD-L1 pathway
The relationship between IL-1β and PD-1/PD-L1 is complex and may provide insight into overcoming the limitations and variable response to current therapies and the role of combination therapies of ICIs and interleukin inhibitors (Figure 2) [70]. Research involving hepatocellular carcinoma (HCC) [71,72,73], gastric cancer [74], melanoma [75], multiple myeloma [76] and other malignancies [77] demonstrated a positive association between IL-1β levels and PD-L1 expression. In a murine renal cell carcinoma (RCC) model, Aggen et al showed that combinatorial inhibition of IL-1β and PD-1 led to increased anti-tumor activity [78]. In a study of Kaposi’s sarcoma, Chen et al. found that silencing IL-1β by RNA inhibitors (RNAi’s) reduced PD-L1 expression [79]. Further, targeting the IL-1β/PD-1/PD-L1 pathway can enhance the response to chemotherapy and radiation therapy, indicating its potential as a broad-spectrum cancer therapy [80].
Similarly, in NSCLC, studies have shown the fascinating interplay between IL-1β and PD-L1/ PD-1 [3,80]. According to a retrospective study by De Alencar et al., smoking history was associated with increased production of IL-1β in combination with higher expression of PD-L1 in NSCLC tumor cells [1]. The antitumor effects of co-inhibition of the IL-1β and PD-1/PD-L1 pathways may be due to dynamic changes in the TME. Li et al. demonstrated that acute IL-1β exposure led to chemoresistance and PD-L1 upregulation in the TME of NSCLC [80]. In a murine model of NSCLC, Kaplanov et al. demonstrated that combination therapy with both anti-IL-1β and anti-PD-1 treatment had a synergistic effect, leading to complete inhibition of tumor growth for 30 days [81]. Tumor volume and weight were significantly reduced compared to the control group and either of the single-agent groups. The TME of tumor cells treated with the combination of anti-IL-1β and anti-PD-1 therapies showed a statistically higher percentage of CD8+ T cells, indicating a reversal of the immunosuppressive effects of IL-1β [81]. Similar findings were shown by Jayaraman et al., who demonstrated that canakinumab treatment in humanized NSCLC cells slowed tumor growth by remodeling the TME [82].
Studies have suggested a potential amplifying effect between PD-1 signaling and the immunosuppressive activity of MDSC’s [83,84,85] and a positive correlation between IL-1β and expansion of MDSC’s [86]. The amplifying nature identified in the interaction between PD-1 and IL-1β with MDSC’s which serve as significant suppressors of immune activity suggest a deeper interplay between these two signaling molecules in creating an immunosuppressive, carcinogenic TME. While ICIs as a standalone treatment for NSCLC have limited efficacy, studies suggest that IL-1β and PD-1/PD-L1 pathways may work synergistically as targets for NSCLC treatment due to the notable correlations and intersection between IL-1β and PD-1 expression (Figure 3) [87]. Inhibition of IL-1β may lead to decreased release of dysregulated inflammatory cytokines, while simultaneous PD-1/PD-L1 inhibition targets T cells, overcoming their antitumor immunity [4]. IL-1β inhibitors have emerged as a promising treatment modality for NSCLC without targetable mutations. As discussed earlier, IL-1β promotes tumorigenesis by stimulating the production of inflammatory cytokines, promoting angiogenesis and invasion, and inhibiting apoptosis. Targeting IL-1β, therefore, has the potential to enhance the effectiveness of ICIs and other cancer treatments. The interconnectedness of the PD-1/PD-L1 and IL-1β pathways is not yet fully understood but elucidating their relationship may provide promising avenues for NSCLC treatment [4].
Recent and ongoing trials have targeted the IL-1β/PD-1/PD-L1 pathway. Though CANOPY-1 did not meet its primary end points, further trials to evaluate this combinatorial approach are ongoing (Table 1) [88]. Similarly, CANOPY-2, a trial investigating the role of canakinumab in patients with NSCLC previously treated with PD-1/ PD-L1 inhibitors and platinum-based chemotherapy, did not show significant improvement in PFS or OS in previously treated patients with advanced or metastatic NSCLC who received canakinumab with docetaxel compared to docetaxel alone [89]. Furthermore, there was a higher incidence of fatal infections, a concern seen in previous canakinumab trials. Although CANOPY-A, the latest trial of canakinumab, did not meet its primary endpoint of disease-free survival, its safety profile was comparable to previous trials [90]. Hence, while the relationship between IL-1β and PD-1/PD-L1 pathways in the TME of NSCLC is significant, further research is needed to elucidate the exact mechanisms and improve outcomes with IL-1β and PD-1/PD-L1 inhibitors.
There are ongoing clinical trials investigating the efficacy of anti-IL-1 strategies for NSCLC. These trials are examining the use of anti-IL-1 therapies either alone or in combination with chemotherapy or other immunotherapeutic agents (Table 2).
Biomarkers of response
Given the heterogeneity of response to ICIs and combination therapies with IL-1β and PD-1/PD-L1 inhibitors, there is an unmet need for predictive biomarkers of response and resistance. As stated above, PD-L1 expression is directly correlated with the function of IL-1β and the elevated serum levels of IL-1β may portend improved response to ICIs. Elevated serum IL-1β levels were correlated with decreased response to PD-L1 treatment and worsened survival outcomes, alluding to the potential role for serum IL-1β as a biomarker of response to combination therapies [3]. In another study, Wang et al noted the potential prognostic implication of tracking M-MDSCs-related genes (monocytic myeloid-derived suppressor cells) in the TME of lung adenocarcinoma (LUAD) [91]. Following the distinct genetic signatures of M-MDSCs-related genes may aid in our efforts to distinguish the effects of upregulated IL-1β and PD-1 expression in studying TME compositions that may have better responses to ICIs [91].
In the setting of the significance of the TME in the carcinogenesis and treatment of NSCLC along with the proinflammatory nature of NSCLC, studies have demonstrated the potential role of inflammatory cytokines and serum markers for predicting response to combination therapies. Levels of cytokines such as IL-6 and IL-8 may be relevant biomarkers for response to combined IL-1β and PD-1/PD-L1 inhibition [92,93]. Studies have shown a positive correlation between IL-6 and PD-1/PD-L1 expression, and in colorectal cancer, a strong correlation exists among IL-8, IL-β1, and MMP-2 [87,94]. In preclinical models of KRAS-mutant lung cancer, the use of IL-1β inhibitors has been shown to reduce tumor burden, increase infiltration of CD8+ T cells into the TME, elevate IFN-γ levels, decrease PD-1 expression, and reduce the number of exhausted PD-1+ T cells [95]. Furthermore, inhibiting IL-1β can increase the percentage of activated cytotoxic T-cells, leading to improved tumor suppression [96].
In addition, C-reactive protein (CRP), a clinical marker of inflammation associated with increased lung cancer risk [97] and progression [98], has been shown to have an interesting relationship with ICIs [97,98]. Given the association between chronic inflammatory states and lung cancer, and the role of IL-1β as an upstream activator of a wide range of inflammatory cytokines and tumorigenesis, CRP may serve as a potential predictive biomarker of response [26]. A study of advanced NSCLC found that elevated CRP levels before treatment with ICIs were predictive of worse PFS and OS, along with lower overall response rate (ORR) [99]. Moreover, high levels of pretreatment CRP were associated with worse PFS and OS, and more rapid increases in CRP levels during treatment were strong predictors of higher risk progressive disease [99]. In the multivariable analysis, there was an association between elevated baseline CRP and lower odds of response (adjusted odds ratio (OR) per doubling of baseline CRP = 0.66, 95% CI: 0.47–0.91, p = 0.011). When comparing the baseline and 8-week CRP levels, an early decline of CRP after the initiation of ICIs, defined as a change of 15.6% in the level of CRP, was associated with improved PFS [99]. Thus, validated serum biomarkers are needed to predict response to combination therapy with IL-1β and PD-1/PD-L1 inhibitors.
Despite these potential biomarkers of response to ICIs, there remains room for further identification of robust markers of response to ICIs. Recently, Assaf et al. demonstrated the clinical significance of circulating tumor DNA (ctDNA) in predicting OS and even response to therapy including ICIs [100]. This study utilized the NSCLC patients from the IMpower150 and the OAK trials and were able to risk stratify patients based on the level of ctDNA [100]. Interestingly, the role of ctDNA levels may be further applied to IL-1β inhibitors. Wong et al., in a molecular analysis of the patients from the CANTOS cohort who developed lung cancer, observed that detectable ctDNA levels were associated with earlier lung cancer diagnosis compared to patients without detectable ctDNA levels [101]. Thus, further studies on the role of ctDNA levels in guiding NSCLC, specifically in the setting of individual or combinatorial inhibition of the IL-1β/PD-1/PD-L1 pathway, are warranted.
Conclusions
Both preclinical and clinical studies suggest that the IL-1β/PD-1/PD-L1 pathway plays a crucial role in the carcinogenesis of NSCLC and interacts within the TME. The IL-1β/PD-1/PD-L1 pathway may be associated with resistance to therapy and represents a novel therapeutic target for NSCLC. However, further studies are warranted to fully elucidate the underlying mechanisms and determine the optimal combination regimens. As research into the IL-1β pathway continues to progress, it is hoped that the development of new therapeutic strategies will lead to standardized prediction models and biomarkers of response, and ultimately, improved outcomes for patients with NSCLC.
Supplementary Materials
Author Contributions
SM and HM designed the outline. DRC and JJW designed and drafted the manuscript. DP, BP, and CY designed the figures and tables. BJ, JHM, and JL contributed to the discussion. MER, KP, and EC offered professional suggestions for the manuscript. YLW and YQL provided the language editing. All authors read and approved the final manuscript.
Funding/Acknowledgment
This study was funded by the Loma Linda University Cancer Center and School of Medicine Dean's Office through the Grants to Promote Collaborative and Translational Research program (GCAT grant no. 2200460).
Informed Consent Statement
Not applicable.
Data Availability Statement
This original research has not been submitted elsewhere, is not under review by another journal, and has not been published previously.
Conflicts of Interest
The authors above do not declare any conflict of interest. All the authors are aware of and approve the manuscript as submitted to this journal.
List of Abbreviations
| non-small cell lung cancer | NSCLC |
| immune-checkpoint inhibitor | ICI |
| programmed death ligand 1 | PD-L1 or B7-H1 |
| tumor microenvironment | TME |
| interleukin-1 beta | IL-1β |
| the Canakinumab Anti-inflammatory Thrombosis Outcome Study | CANTOS trial |
| trial programmed cell death protein 1 | PD-1 |
| interferon | IFN |
| microsatellite instability | MSI |
| high tumor mutational burden | TMB |
| toll-like receptor | TLR |
| tumor necrosis factor | TNF |
| IL-1β-converting enzyme | ICE |
| dendritic cell | DC |
| antigen-presenting cell | APC |
| IL-1 receptors | IL-1R and IL-1R2 |
| overall survival | OS |
| progression-free survival | PFS |
| myeloid differentiation primary response-88 | MyD88 |
| IL-1R associated kinase | IRAK |
| tumor necrosis factor receptor-associated factor 6 | TRAF6 |
| mitogen activated protein kinase | MAPK |
| Gasdermin D | GSDMD |
| Inhibitor of nuclear factor kappa B | IkB |
| Nuclear factor kappa B | NF-kB |
| Activator protein 1 | AP-1 |
| myeloid-derived suppressor cell | MDSC |
| regulatory T cell | Treg |
| tumor-associated macrophage | TAM |
| monocyte chemoattractant protein | MCP-1 |
| vascular endothelial growth factor | VEGF |
| basic fibroblast growth factor | bFGF |
| transforming growth factor | TGF-αβ |
| platelet-derived endothelial growth factor | PDGF |
| interleukin-8 | IL-8 |
| fibroblast growth factor | FGF |
| matrix metalloproteinase | MMP |
| extracellular matrix | ECM |
| phosphatase and tensin homolog | PTEN |
| epithelial-mesenchymal transition | EMT |
| cyclooxygenase 2 | COX2 |
| inducible nitric oxide synthase | iNOS |
| renal cell carcinoma | RCC model |
| BRAF inhibitors | BRAFi |
| cancer-associated fibroblast | CAF |
| tumor infiltrating lymphocyte | TIL |
| M-MDSC | monocytic myeloid-derived suppressor cell |
| lung adenocarcinoma | LUAD |
| C-reactive protein | CRP |
| overall response rate | ORR |
| odds ratio | OR |
| circulating tumor DNA | ctDNA |
| immune-related adverse event | irAE |
References
- de Alencar, V.T.L.; Figueiredo, A.B.; Corassa, M.; Gollob, K.J.; de Lima, V.C.C. Lung cancer in never smokers: Tumor immunology and challenges for immunotherapy. Front. Immunol. 2022, 13, 984349. [CrossRef]
- Gridelli C, Rossi A, Carbone DP, et al. Non-small-cell lung cancer. Nat Rev Dis Primers. 2015, 1, 1500.
- Zhang, J.; Veeramachaneni, N. Targeting interleukin-1β and inflammation in lung cancer. Biomark. Res. 2022, 10, 1–9. [CrossRef]
- Lee, J.M.; Tsuboi, M.; Kim, E.S.; Mok, T.S.; Garrido, P. Overcoming immunosuppression and pro-tumor inflammation in lung cancer with combined IL-1β and PD-1 inhibition. Futur. Oncol. 2022, 18, 3085–3100. [CrossRef]
- Ridker PM, MacFadyen JG, Thuren T, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017, 390, 1833-1842. [CrossRef]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 1–13. [CrossRef]
- Budimir N, Thomas GD, Dolina JS, Salek-Ardakani S. Reversing T-cell Exhaustion in Cancer: Lessons Learned from PD-1/PD-L1 Immune Checkpoint Blockade. Cancer Immunol Res. 2022, 10, 146-153. [CrossRef]
- Wang, S.; Yuan, P.; Mao, B.; Li, N.; Ying, J.; Tao, X.; Tang, W.; Zhang, L.; Geng, X.; Zhang, F.; et al. Genomic features and tumor immune microenvironment alteration in NSCLC treated with neoadjuvant PD-1 blockade. npj Precis. Oncol. 2022, 6, 1–8. [CrossRef]
- Huang J, Lan X, Wang T, et al. Targeting the IL-1β/EHD1/TUBB3 axis overcomes resistance to EGFR-TKI in NSCLC. Oncogene. 2020, 39, 1739-1754. [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [CrossRef]
- Yan X, Han L, Zhao R, Fatima S, Zhao L, Gao F. Prognosis value of IL-6, IL-8, and IL-1β in serum of patients with lung cancer: A fresh look at interleukins as a biomarker. Heliyon. 2022, 8, e09953. [CrossRef]
- Rébé, C.; Ghiringhelli, F. Interleukin-1β and Cancer. Cancers 2020, 12, 1791. [CrossRef]
- Qin, X.; Li, T.; Li, S.; Yang, H.; Wu, C.; Zheng, C.; You, F.; Liu, Y. The tumor biochemical and biophysical microenvironments synergistically contribute to cancer cell malignancy. Cell. Mol. Immunol. 2019, 17, 1186–1187. [CrossRef]
- Litmanovich, A.; Khazim, K.; Cohen, I. The Role of Interleukin-1 in the Pathogenesis of Cancer and its Potential as a Therapeutic Target in Clinical Practice. Oncol. Ther. 2018, 6, 109–127. [CrossRef]
- Elkabets, M.; Ribeiro, V.S.G.; Dinarello, C.A.; Ostrand-Rosenberg, S.; Di Santo, J.P.; Apte, R.N.; Vosshenrich, C.A.J. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur. J. Immunol. 2010, 40, 3347–3357. [CrossRef]
- Li, S.; Wang, W.; Zhang, N.; Ma, T.; Zhao, C. IL-1β mediates MCP-1 induction by Wnt5a in gastric cancer cells. BMC Cancer 2014, 14, 480–480. [CrossRef]
- Kolb, R.; Phan, L.; Borcherding, N.; Liu, Y.; Yuan, F.; Janowski, A.M.; Xie, Q.; Markan, K.R.; Li, W.; Potthoff, M.J.; et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 2016, 7, 13007. [CrossRef]
- Carmi Y, Dotan S, Rider P, et al. The role of IL-1β in the early tumor cell-induced angiogenic response. J Immunol. 2013, 190, 3500-3509. [CrossRef]
- Fahey, E.; Doyle, S.L. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1426. [CrossRef]
- Ping P.H., Bo T.F., Li L., Hui Y.N., Zhu H. IL-1β/NF-kb signaling promotes colorectal cancer cell growth through miR-181a/PTEN axis. Arch. Biochem. Biophys. 2016, 604, 20–26. [CrossRef]
- Wu C., Xu B., Zhou Y., Ji M., Zhang D., Jiang J., Wu C. Correlation between serum IL-1β and miR-144-3p as well as their prognostic values in LUAD and LUSC patients. Oncotarget. 2016, 7, 85876-85887. [CrossRef]
- Sanmamed, M.F.; Perez-Gracia, J.L.; Schalper, K.A.; Fusco, J.P.; Gonzalez, A.; Rodriguez-Ruiz, M.E.; Oñate, C.; Perez, G.; Alfaro, C.; Martín-Algarra, S.; et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988–1995. [CrossRef]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.-S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [CrossRef]
- Jin C, Lagoudas GK, Zhao C, et al. Commensal Microbiota Promote Lung Cancer Development via γδ T Cells. Cell. 2019, 176, 998-1013.e16.
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [CrossRef]
- Garon, E.B.; Yang, J.C.-H.; Dubinett, S.M. The Role of Interleukin 1β in the Pathogenesis of Lung Cancer. JTO Clin. Res. Rep. 2020, 1, 100001. [CrossRef]
- Gelfo, V.; Romaniello, D.; Mazzeschi, M.; Sgarzi, M.; Grilli, G.; Morselli, A.; Manzan, B.; Rihawi, K.; Lauriola, M. Roles of IL-1 in Cancer: From Tumor Progression to Resistance to Targeted Therapies. Int. J. Mol. Sci. 2020, 21, 6009. [CrossRef]
- Yan, N.; Guo, S.; Zhang, H.; Zhang, Z.; Shen, S.; Li, X. BRAF-Mutated Non-Small Cell Lung Cancer: Current Treatment Status and Future Perspective. Front. Oncol. 2022, 12, 863043. [CrossRef]
- Hajek, E.; Krebs, F.; Bent, R.; Haas, K.; Bast, A.; Steinmetz, I.; Tuettenberg, A.; Grabbe, S.; Bros, M. BRAF inhibitors stimulate inflammasome activation and interleukin 1 beta production in dendritic cells. Oncotarget 2018, 9, 28294–28308. [CrossRef]
- Davies, A.M.; Lara, P.N.; Mack, P.C.; Gumerlock, P.H.; Bold, R.J.; Gandara, D.R. Bortezomib-Based Combinations in the Treatment of Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2005, 7, S59–S63. [CrossRef]
- McLoed AG, Sherrill TP, Cheng DS, et al. Neutrophil-Derived IL-1β Impairs the Efficacy of NF-κB Inhibitors against Lung Cancer. Cell Rep. 2016, 16, 120-137. [CrossRef]
- Russano, M.; La Cava, G.; Cortellini, A.; Citarella, F.; Galletti, A.; Di Fazio, G.R.; Santo, V.; Brunetti, L.; Vendittelli, A.; Fioroni, I.; et al. Immunotherapy for Metastatic No n-Small Cell Lung Cancer: Therapeutic Advances and Biomarkers. Curr. Oncol. 2023, 30, 2366–2387. [CrossRef]
- Addeo, A.; Passaro, A.; Malapelle, U.; Banna, G.L.; Subbiah, V.; Friedlaender, A. Immunotherapy in non-small cell lung cancer harbouring driver mutations. Cancer Treat. Rev. 2021, 96, 102179. [CrossRef]
- Negrao, M.V.; Skoulidis, F.; Montesion, M.; Schulze, K.; Bara, I.; Shen, V.; Xu, H.; Hu, S.; Sui, D.; Elamin, Y.Y.; et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J. Immunother. Cancer 2021, 9, e002891. [CrossRef]
- Simon S, Labarriere N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? Oncoimmunology. 2017, 7, e1364828.
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [CrossRef]
- Sari MI, Ilyas S. The Expression Levels and Concentrations of PD-1 and PD-L1 Proteins in Septic Patients: A Systematic Review. Diagnostics (Basel). 2022, 12, 2004.
- Xu, X.; Hou, B.; Fulzele, A.; Masubuchi, T.; Zhao, Y.; Wu, Z.; Hu, Y.; Jiang, Y.; Ma, Y.; Wang, H.; et al. PD-1 and BTLA regulate T cell signaling differentially and only partially through SHP1 and SHP2. J. Cell Biol. 2020, 219. [CrossRef]
- Lei Q, Wang D, Sun K, Wang L, Zhang Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front Cell Dev Biol. 2020, 8, 672. [CrossRef]
- Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020, 10, 727-742.
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1–Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Five-Year Outcomes With Pembrolizumab Versus Chemotherapy for Metastatic Non–Small-Cell Lung Cancer With PD-L1 Tumor Proportion Score ≥ 50%. J. Clin. Oncol. 2021, 39, 2339–2349. [CrossRef]
- Herbst, R.S.; Garon, E.B.; Kim, D.-W.; Cho, B.C.; Gervais, R.; Perez-Gracia, J.L.; Han, J.-Y.; Majem, M.; Forster, M.D.; Monnet, I.; et al. Five Year Survival Update From KEYNOTE-010: Pembrolizumab Versus Docetaxel for Previously Treated, Programmed Death-Ligand 1–Positive Advanced NSCLC. J. Thorac. Oncol. 2021, 16, 1718–1732. [CrossRef]
- Horn L, Spigel DR, Vokes EE, et al. Nivolumab Versus Docetaxel in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer: Two-Year Outcomes From Two Randomized, Open-Label, Phase III Trials (CheckMate 017 and CheckMate 057). J Clin Oncol. 2017, 35, 3924-393. [CrossRef]
- Sezer, A.; Kilickap, S.; Gümüş, M.; Bondarenko, I.; Özgüroğlu, M.; Gogishvili, M.; Turk, H.M.; Cicin, I.; Bentsion, D.; Gladkov, O.; et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet 2021, 397, 592–604. [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; De Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [CrossRef]
- Rizvi NA, Cho BC, Reinmuth N, et al. Durvalumab With or Without Tremelimumab vs Standard Chemotherapy in First-line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial [published correction appears in JAMA Oncol. 2020 Nov 1, 6, 1815]. JAMA Oncol. 2020, 6, 661-674. [CrossRef]
- Jotte, R.; Cappuzzo, F.; Vynnychenko, I.; Stroyakovskiy, D.; Rodríguez-Abreu, D.; Hussein, M.; Soo, R.; Conter, H.J.; Kozuki, T.; Huang, K.-C.; et al. Atezolizumab in Combination With Carboplatin and Nab-Paclitaxel in Advanced Squamous NSCLC (IMpower131): Results From a Randomized Phase III Trial. J. Thorac. Oncol. 2020, 15, 1351–1360. [CrossRef]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.-G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 924–937. [CrossRef]
- Socinski, M.A.; Nishio, M.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; et al. IMpower150 Final Overall Survival Analyses for Atezolizumab Plus Bevacizumab and Chemotherapy in First-Line Metastatic Nonsquamous NSCLC. J. Thorac. Oncol. 2021, 16, 1909–1924. [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265; Erratum in Lancet 2017, 389, e5. [CrossRef]
- Pathak R, Pharaon RR, Mohanty A, Villaflor VM, Salgia R, Massarelli E. Acquired Resistance to PD-1/PD-L1 Blockade in Lung Cancer: Mechanisms and Patterns of Failure. Cancers (Basel). 2020, 12, 3851. [CrossRef]
- Passaro, A.; Brahmer, J.; Antonia, S.; Mok, T.; Peters, S. Managing Resistance to Immune Checkpoint Inhibitors in Lung Cancer: Treatment and Novel Strategies. J. Clin. Oncol. 2022, 40, 598–610. [CrossRef]
- Vesely, M.D.; Zhang, T.; Chen, L. Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu. Rev. Immunol. 2022, 40, 45–74. [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4–328rv4. [CrossRef]
- Cheng C, Zhuge L, Xiao X, Luan S, Yuan Y. Overcoming resistance to PD-1/PD-L1 inhibitors in esophageal cancer. Front Oncol. 2022, 12, 955163. [CrossRef]
- Walsh, R.J.; Soo, R.A. Resistance to immune checkpoint inhibitors in non-small cell lung cancer: biomarkers and therapeutic strategies. Ther. Adv. Med Oncol. 2020, 12. [CrossRef]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 223–249. [CrossRef]
- Zhang Y, Chen L. Classification of Advanced Human Cancers Based on Tumor Immunity in the MicroEnvironment (TIME) for Cancer Immunotherapy [published correction appears in JAMA Oncol. 2016 Nov 1, 2, 1511]. JAMA Oncol. 2016, 2, 1403-1404.
- Zhuang Y, Liu C, Liu J, Li G. Resistance Mechanism of PD-1/PD-L1 Blockade in the Cancer-Immunity Cycle. Onco Targets Ther. 2020, 13, 83-94. [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell–Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [CrossRef]
- Ngiow, S.F.; Young, A.; Jacquelot, N.; Yamazaki, T.; Enot, D.; Zitvogel, L.; Smyth, M.J. A threshold level of intratumor CD8 + T-cell PD1 expression dictates therapeutic response to anti-PD1. Cancer Res. 2015, 75, 3800–3811. [CrossRef]
- Lu, Y.; Yuan, X.; Wang, M.; He, Z.; Li, H.; Wang, J.; Li, Q. Gut microbiota influence immunotherapy responses: mechanisms and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 1–20. [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [CrossRef]
- Dong, Z.-Y.; Zhang, J.-T.; Liu, S.-Y.; Su, J.; Zhang, C.; Xie, Z.; Zhou, Q.; Tu, H.-Y.; Xu, C.-R.; Yan, L.-X.; et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. OncoImmunology 2017, 6, e1356145. [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non–Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [CrossRef]
- Sakai, T.; Udagawa, H.; Matsumoto, S.; Yoh, K.; Nosaki, K.; Ikeda, T.; Zenke, Y.; Kirita, K.; Niho, S.; Akimoto, T.; et al. Morphological, immune and genetic features in biopsy sample associated with the efficacy of pembrolizumab in patients with non-squamous non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2020, 147, 1227–1237. [CrossRef]
- Giustini, N.; Bazhenova, L. Recognizing Prognostic and Predictive Biomarkers in the Treatment of Non-Small Cell Lung Cancer (NSCLC) with Immune Checkpoint Inhibitors (ICIs). Lung Cancer: Targets Ther. 2021, ume 12, 21–34. [CrossRef]
- Mortezaee, K.; Majidpoor, J. Checkpoint inhibitor/interleukin-based combination therapy of cancer. Cancer Med. 2022, 11, 2934–2943. [CrossRef]
- Zong, Z.; Zou, J.; Mao, R.; Ma, C.; Li, N.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; Shi, Y. M1 Macrophages Induce PD-L1 Expression in Hepatocellular Carcinoma Cells Through IL-1β Signaling. Front. Immunol. 2019, 10, 1643. [CrossRef]
- Perrichet A, Ghiringhelli F, Rébé C. Understanding Inflammasomes and PD-1/PD-L1 Crosstalk to Improve Cancer Treatment Efficiency. Cancers (Basel). 2020, 12, 3550. [CrossRef]
- Numata, Y.; Akutsu, N.; Ishigami, K.; Koide, H.; Wagatsuma, K.; Motoya, M.; Sasaki, S.; Nakase, H. Synergistic effect of IFN-γ and IL-1β on PD-L1 expression in hepatocellular carcinoma. Biochem. Biophys. Rep. 2022, 30, 101270. [CrossRef]
- Lu X, Li Y, Yang W, Tao M, Dai Y, Xu J, Xu Q. Inhibition of NF-κB is required for oleanolic acid to downregulate PD-L1 by promoting DNA demethylation in gastric cancer cells. J Biochem Mol Toxicol. 2021, 35, e22621. [CrossRef]
- Khalili, J.S.; Liu, S.; Rodríguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) Promotes Stromal Cell-Mediated Immunosuppression Via Induction of Interleukin-1 in Melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [CrossRef]
- Bar, N.; Costa, F.; Das, R.; Duffy, A.; Samur, M.; McCachren, S.; Gettinger, S.N.; Neparidze, N.; Parker, T.L.; Bailur, J.K.; et al. Differential effects of PD-L1 versus PD-1 blockade on myeloid inflammation in human cancer. J. Clin. Investig. 2020, 5. [CrossRef]
- Takahashi, R.; Macchini, M.; Sunagawa, M.; Jiang, Z.; Tanaka, T.; Valenti, G.; Renz, B.W.; A White, R.; Hayakawa, Y.; Westphalen, C.B.; et al. Interleukin-1β-induced pancreatitis promotes pancreatic ductal adenocarcinoma via B lymphocyte–mediated immune suppression. Gut 2020, 70, 330–341. [CrossRef]
- Aggen, D.H.; Ager, C.R.; Obradovic, A.Z.; Chowdhury, N.; Ghasemzadeh, A.; Mao, W.; Chaimowitz, M.G.; Lopez-Bujanda, Z.A.; Spina, C.S.; Hawley, J.E.; et al. Blocking IL1 Beta Promotes Tumor Regression and Remodeling of the Myeloid Compartment in a Renal Cell Carcinoma Model: Multidimensional Analyses. Clin. Cancer Res. 2021, 27, 608–621. [CrossRef]
- Chen J, Del Valle L, Lin HY, et al. Expression of PD-1 and PD-Ls in Kaposi's sarcoma and regulation by oncogenic herpesvirus lytic reactivation. Virology. 2019, 536, 16-19. [CrossRef]
- Li R, Ong SL, Tran LM, et al. Chronic IL-1β-induced inflammation regulates epithelial-to-mesenchymal transition memory phenotypes via epigenetic modifications in non-small cell lung cancer [published correction appears in Sci Rep. 2020, 10, 4386. Sci Rep. 2020, 10, 377. [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti–PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [CrossRef]
- Jayaraman, P.; Millholland, J.; O’brien, N.; Wong, C.; Diwanji, R.; Wang, M.; Choi, E.; Linnartz, R.; Rose, K.; Rodrik-Outmezguine, V.; et al. Abstract C103: Targeting IL-1β pathway for cancer immunotherapy. Mol. Cancer Ther. 2019, 18, C103–C103. [CrossRef]
- Loeuillard, E.; Yang, J.; Buckarma, E.; Wang, J.; Liu, Y.; Conboy, C.B.; Pavelko, K.D.; Li, Y.; O’brien, D.; Wang, C.; et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [CrossRef]
- Kim, W.; Chu, T.H.; Nienhüser, H.; Jiang, Z.; Del Portillo, A.; Remotti, H.E.; White, R.A.; Hayakawa, Y.; Tomita, H.; Fox, J.G.; et al. PD-1 Signaling Promotes Tumor-Infiltrating Myeloid-Derived Suppressor Cells and Gastric Tumorigenesis in Mice. Gastroenterology 2021, 160, 781–796. [CrossRef]
- Long H, Jia Q, Wang L, Fang W, Wang Z, Jiang T, Zhou F, Jin Z, Huang J, Zhou L, Hu C, Wang X, Zhang J, Ba Y, Gong Y, Zeng X, Zeng D, Su X, Alexander PB, Wang L, Wang L, Wan YY, Wang XF, Zhang L, Li QJ, Zhu B. Tumor-induced erythroid precursor-differentiated myeloid cells mediate immunosuppression and curtail anti-PD-1/PD-L1 treatment efficacy. Cancer Cell. 2022, 40, 674-693.e7. [CrossRef]
- Tengesdal IW, Dinarello A, Powers NE, Burchill MA, Joosten LAB, Marchetti C, Dinarello CA. Tumor NLRP3-Derived IL-1β Drives the IL-6/STAT3 Axis Resulting in Sustained MDSC-Mediated Immunosuppression. Front Immunol. 2021, 12, 661323. [CrossRef]
- Ding, X.; Zhang, J.; Shi, M.; Liu, D.; Zhang, L.; Zhang, R.; Su, B.; Ai, K. High expression level of interleukin-1β is correlated with poor prognosis and PD-1 expression in patients with lung adenocarcinoma. Clin. Transl. Oncol. 2020, 23, 35–42. [CrossRef]
- Pretre, V.; Papadopoulos, D.; Regard, J.; Pelletier, M.; Woo, J. Interleukin-1 (IL-1) and the inflammasome in cancer. Cytokine 2022, 153, 155850. [CrossRef]
- Paz-Ares L, Goto Y, Lim WDT, Halmos B, Cho BC, Dols MC, et al. 1194MO Canakinumab (CAN) + docetaxel (DTX) for the second- or third-line (2/3L) treatment of advanced non-small cell lung cancer (NSCLC): CANOPY-2 phase III results. Ann Oncol. 2021, 32, S953–4. [CrossRef]
- AG NP. Novartis provides update on Phase III CANOPY-A study evaluating canakinumab as adjuvant treatment in non-small cell lung cancer. GlobeNewswire News Room. 2022. Available online: https://www.globenewswire.com/en/news-release/2022/08/15/2497914/0/en/Novartis-provides-update-on-Phase-III-CANOPY-A-study-evaluating-canakinumab-as-adjuvant-treatment-in-non-small-cell-lung-cancer.html (accessed on 28 April 2023).
- Wang, G.-C.; Zhou, M.; Zhang, Y.; Cai, H.-M.; Chiang, S.-T.; Chen, Q.; Han, T.-Z.; Li, R.-X. Screening and identifying a novel M-MDSCs-related gene signature for predicting prognostic risk and immunotherapeutic responses in patients with lung adenocarcinoma. Front. Genet. 2023, 13, 989141. [CrossRef]
- Mizuno T, Katsuya Y, Sato J, Koyama T, Shimizu T, Yamamoto N. Emerging PD-1/PD-L1 targeting immunotherapy in non-small cell lung cancer: Current status and future perspective in Japan, US, EU, and China. Front Oncol. 2022, 12, 925938. [CrossRef]
- Keegan, A.; Ricciuti, B.; Garden, P.; Cohen, L.; Nishihara, R.; Adeni, A.; Paweletz, C.; Supplee, J.; A Jänne, P.; Severgnini, M.; et al. Plasma IL-6 changes correlate to PD-1 inhibitor responses in NSCLC. J. Immunother. Cancer 2020, 8, e000678. [CrossRef]
- Calu, V.; Ionescu, A.; Stanca, L.; Geicu, O.I.; Iordache, F.; Pisoschi, A.M.; Serban, A.I.; Bilteanu, L. Key biomarkers within the colorectal cancer related inflammatory microenvironment. Sci. Rep. 2021, 11, 1–14. [CrossRef]
- Yuan, B.; Clowers, M.J.; Velasco, W.V.; Peng, S.; Peng, Q.; Shi, Y.; Ramos-Castaneda, M.; Zarghooni, M.; Yang, S.; Babcock, R.L.; et al. Targeting IL-1β as an immunopreventive and therapeutic modality for K-ras–mutant lung cancer. J. Clin. Investig. 2022, 7. [CrossRef]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor Cell–Derived IL1β Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res 2020, 80, 1088–1101. [CrossRef]
- Chaturvedi, A.K.; Caporaso, N.E.; Katki, H.A.; Wong, H.-L.; Chatterjee, N.; Pine, S.R.; Chanock, S.J.; Goedert, J.J.; Engels, E.A. C-Reactive Protein and Risk of Lung Cancer. J. Clin. Oncol. 2010, 28, 2719–2726. [CrossRef]
- Kanoh, Y.; Abe, T.; Masuda, N.; Akahoshi, T. Progression of non-small cell lung cancer: Diagnostic and prognostic utility of matrix metalloproteinase-2, C-reactive protein and serum amyloid A. Oncol. Rep. 2012, 29, 469–473. [CrossRef]
- Riedl, J.M.; Barth, D.A.; Brueckl, W.M.; Zeitler, G.; Foris, V.; Mollnar, S.; Stotz, M.; Rossmann, C.H.; Terbuch, A.; Balic, M.; et al. C-Reactive Protein (CRP) Levels in Immune Checkpoint Inhibitor Response and Progression in Advanced Non-Small Cell Lung Cancer: A Bi-Center Study. Cancers 2020, 12, 2319. [CrossRef]
- Assaf, Z.J.F.; Zou, W.; Fine, A.D.; Socinski, M.A.; Young, A.; Lipson, D.; Freidin, J.F.; Kennedy, M.; Polisecki, E.; Nishio, M.; et al. A longitudinal circulating tumor DNA-based model associated with survival in metastatic non-small-cell lung cancer. Nat. Med. 2023, 29, 859–868. [CrossRef]
- Wong CC, Baum J, Silvestro A, Beste MT, Bharani-Dharan B, Xu S, et al. Inhibition of IL1β by Canakinumab May Be Effective against Diverse Molecular Subtypes of Lung Cancer: An Exploratory Analysis of the CANTOS Trial. Cancer Research. 2020, 80, 5597-605. [CrossRef]
- Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB. Current Diagnosis and Management of Immune-Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitor Therapy [published correction appears in Front Pharmacol. 2017, 8, 311]. Front Pharmacol. 2017, 8, 49. [CrossRef]
- Gogishvili, M.; Melkadze, T.; Makharadze, T.; Giorgadze, D.; Dvorkin, M.; Penkov, K.; Laktionov, K.; Nemsadze, G.; Nechaeva, M.; Rozhkova, I.; et al. Cemiplimab plus chemotherapy versus chemotherapy alone in non-small cell lung cancer: a randomized, controlled, double-blind phase 3 trial. Nat. Med. 2022, 28, 2374–2380. [CrossRef]
- Kelly K, Infante JR, Taylor MH, et al. Safety profile of avelumab in patients with advanced solid tumors: A pooled analysis of data from the phase 1 JAVELIN solid tumor and phase 2 JAVELIN Merkel 200 clinical trials. Cancer. 2018, 124, 2010-2017. [CrossRef]
- Song, P.; Zhang, D.; Cui, X.; Zhang, L. Meta-analysis of immune-related adverse events of immune checkpoint inhibitor therapy in cancer patients. Thorac. Cancer 2020, 11, 2406–2430. [CrossRef]
- Gross, N.D.; Miller, D.M.; Khushalani, N.I.; Divi, V.; Ruiz, E.S.; Lipson, E.J.; Meier, F.; Su, Y.B.; Swiecicki, P.L.; Atlas, J.; et al. Neoadjuvant Cemiplimab for Stage II to IV Cutaneous Squamous-Cell Carcinoma. New Engl. J. Med. 2022, 387, 1557–1568. [CrossRef]
- Vaddepally R, Doddamani R, Sodavarapu S, et al. Review of Immune-Related Adverse Events (irAEs) in Non-Small-Cell Lung Cancer (NSCLC)-Their Incidence, Management, Multiorgan irAEs, and Rechallenge. Biomedicines. 2022, 10, 790. [CrossRef]
Figure 1.
Mechanism of the IL-1β pathway and its downstream effects. Legend: GSDMD- Gasdermin D; IRAK- IL-1 receptor-associated kinase; MYD88 - Myeloid differentiation primary response 88; TRAF6 – tumor necrosis factor receptor associated factor 6; IL-1R associated kinases MAPK – mitogen-activated protein kinase; IkB – inhibitor of nuclear factor kappa B; NF-kB (nuclear factor kappa B); AP-1 – Activator protein 1.
Figure 1.
Mechanism of the IL-1β pathway and its downstream effects. Legend: GSDMD- Gasdermin D; IRAK- IL-1 receptor-associated kinase; MYD88 - Myeloid differentiation primary response 88; TRAF6 – tumor necrosis factor receptor associated factor 6; IL-1R associated kinases MAPK – mitogen-activated protein kinase; IkB – inhibitor of nuclear factor kappa B; NF-kB (nuclear factor kappa B); AP-1 – Activator protein 1.
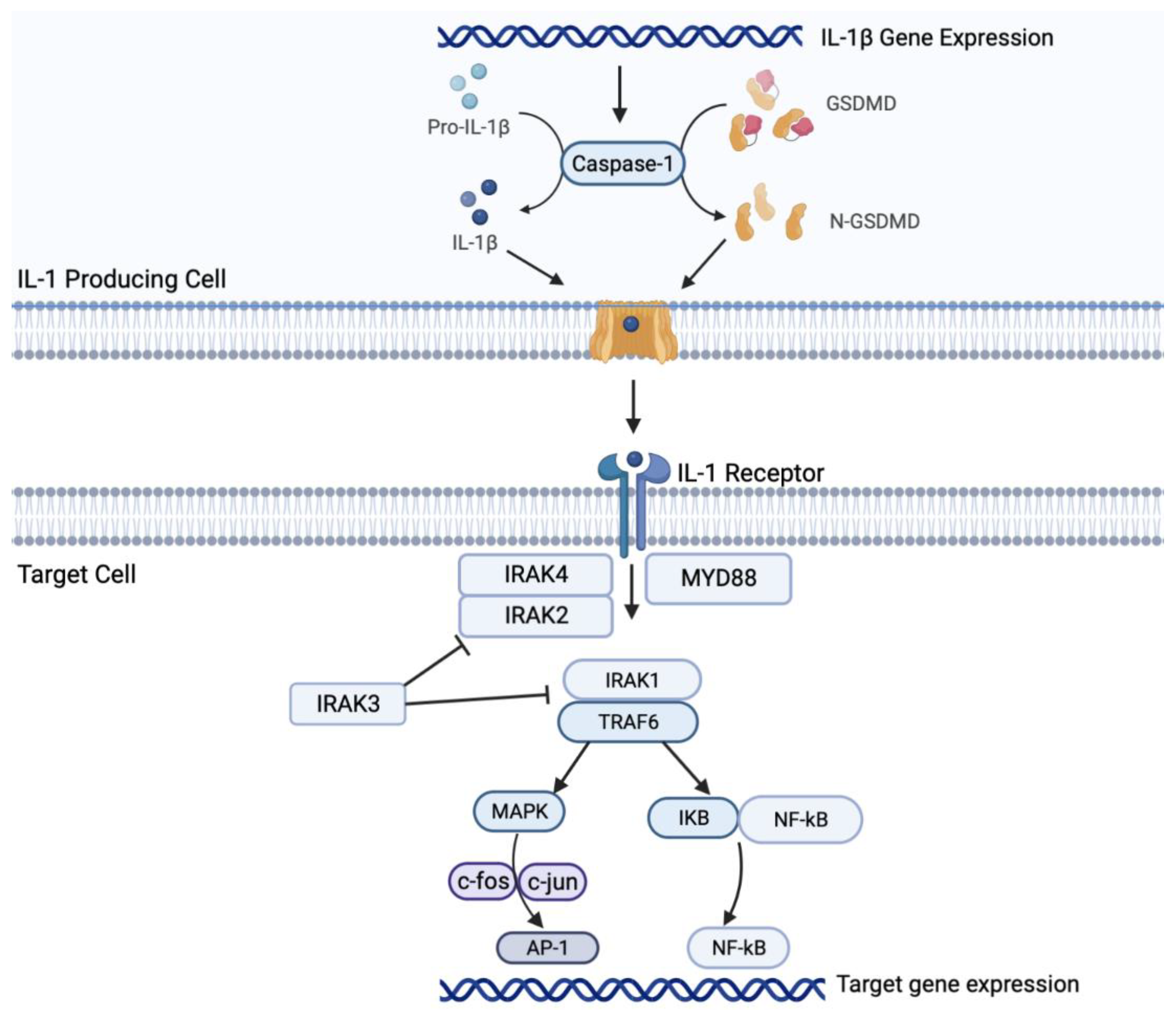
Figure 2.
The effects of IL-1β on the tumor microenvironment. The figure depicts immunoregulatory cells altering the TME via secretion of IL-1β, which promote pro-tumor changes including angiogenesis, immune suppression, and metastasis. Legend. TME- tumor microenvironment; APC – antigen presenting cell; IL-1β – interferon 1-beta.
Figure 2.
The effects of IL-1β on the tumor microenvironment. The figure depicts immunoregulatory cells altering the TME via secretion of IL-1β, which promote pro-tumor changes including angiogenesis, immune suppression, and metastasis. Legend. TME- tumor microenvironment; APC – antigen presenting cell; IL-1β – interferon 1-beta.
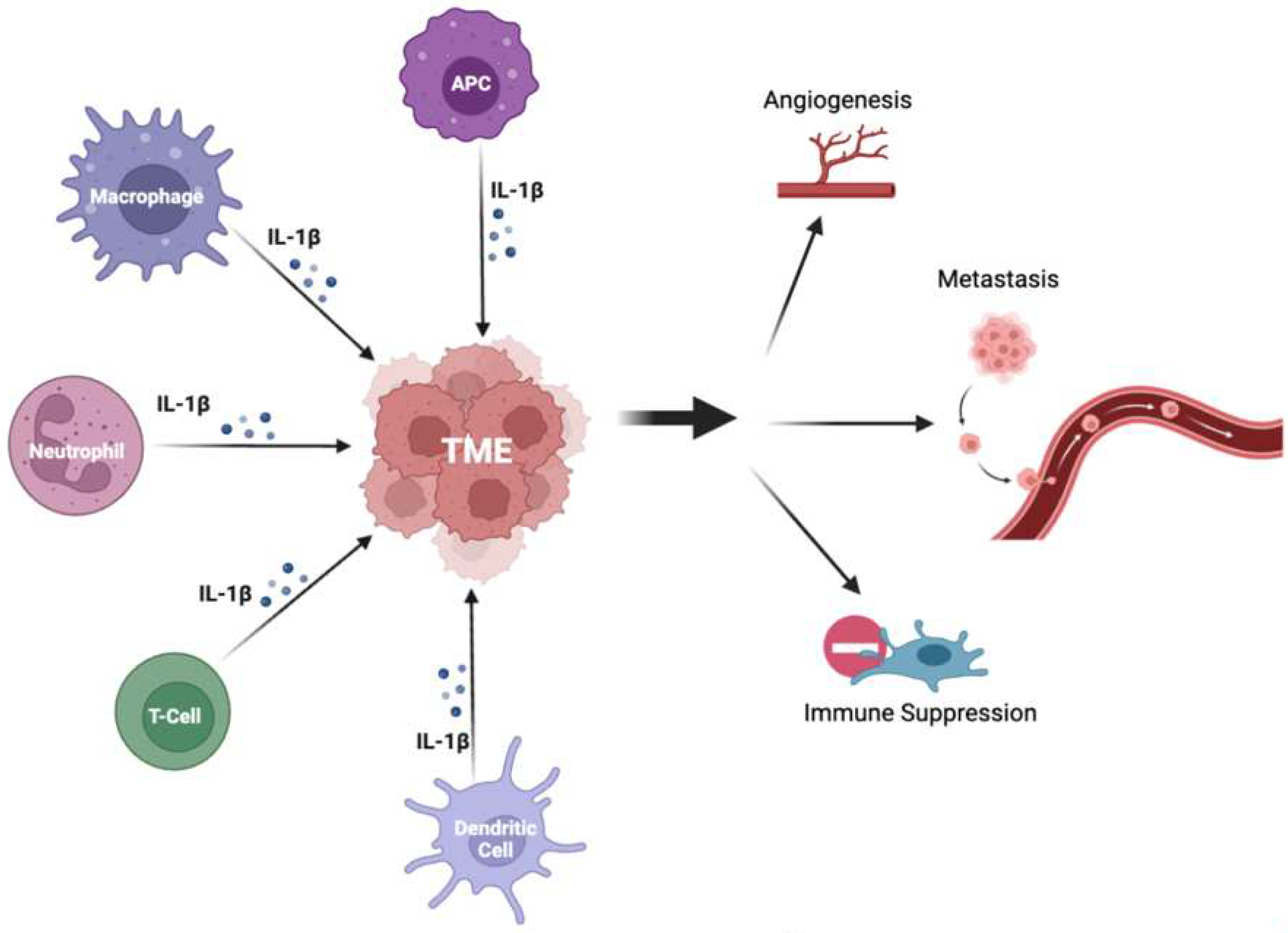
Figure 3.
The proposed synergistic effect of co-inhibition of the IL-1β and PD-L1/PD-1 pathways. 1) Inhibition of IL-1β and PD-L1/PD-1 via monoclonal antibodies (inhibitors); 2) activation of cytotoxic T cells because of inhibition of IL-1β and PD-L1/PD-1 pathways; 3) anti-tumor immune response leading to decreased tumor metastasis and growth. Legend. TME- tumor microenvironment; PD-1 – programmed cell death 1; PD-L1 – programmed cell death ligand 1.
Figure 3.
The proposed synergistic effect of co-inhibition of the IL-1β and PD-L1/PD-1 pathways. 1) Inhibition of IL-1β and PD-L1/PD-1 via monoclonal antibodies (inhibitors); 2) activation of cytotoxic T cells because of inhibition of IL-1β and PD-L1/PD-1 pathways; 3) anti-tumor immune response leading to decreased tumor metastasis and growth. Legend. TME- tumor microenvironment; PD-1 – programmed cell death 1; PD-L1 – programmed cell death ligand 1.
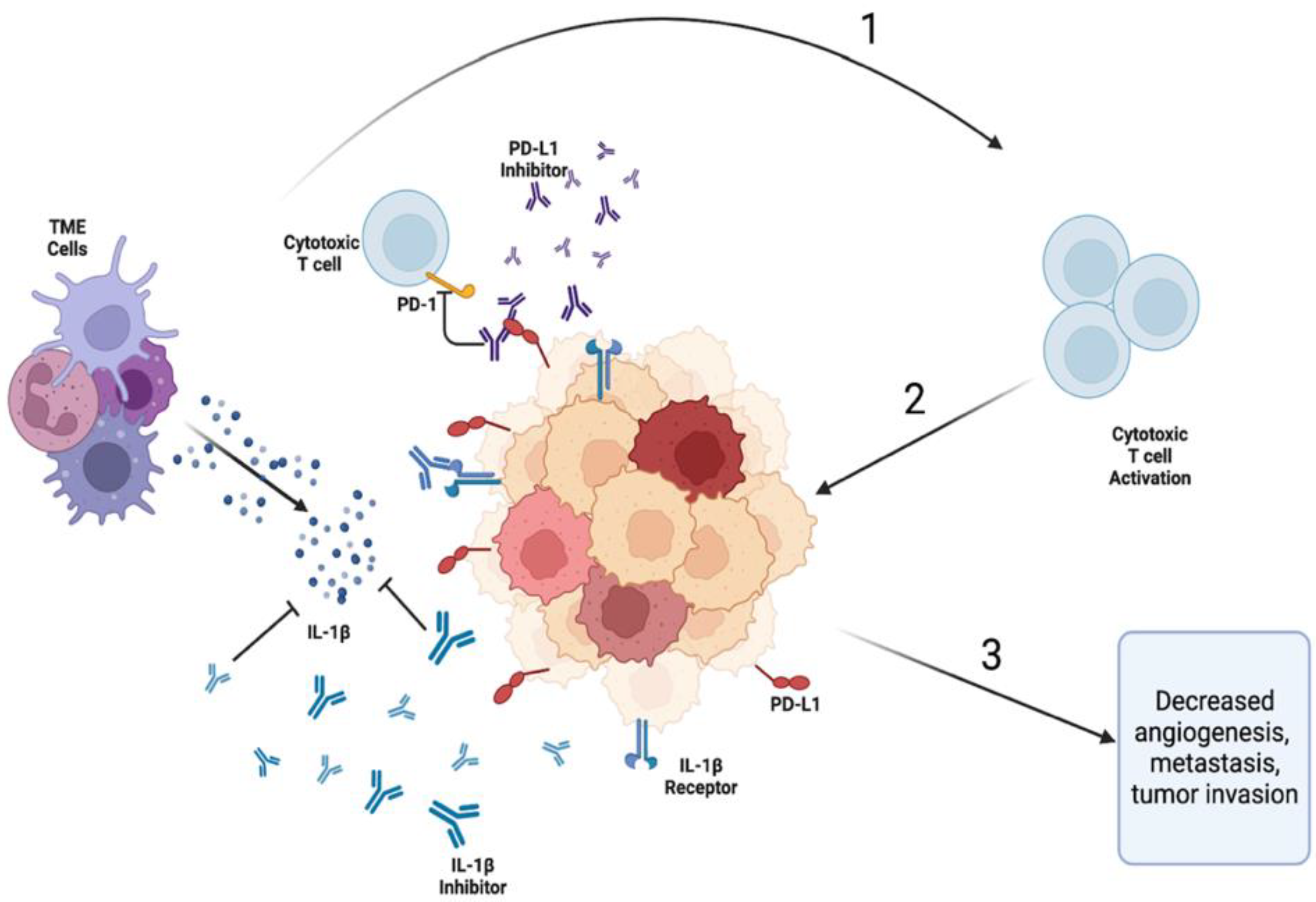

Table 1.
The summary of phase II/III clinical trials of IL-1β inhibitors in patients with NSCLC.
| IL-1β inhibitors | Type | Target | Trials | Phase, progress | Experimental arms | Primary outcome | Results of the primary outcome |
|---|---|---|---|---|---|---|---|
| Canakinumab | human IgGκ mAb | IL-1β | NCT03968419 (CANOPY-N) | Phase II, completed | canakinumab +/- pembrolizumab; pembrolizumab monotherapy | MPR | MPR: 2.9% (CAN)/ 17.1% (CAN+PEM)/11.1% (PEM), does not meet the primary endpoint |
| NCT03626545 (CANOPY-2) | Phase III, completed | docetaxel +/- canakinumab | OS and incidence of DLTs | Median OS: 10.5m (CAN) /11.3m (Placebo) (HR 1.06 95% CI, 0.76-1.48); does not meet the primary endpoint | |||
| NCT03631199 (CANOPY-1) | Phase III, active, not recruiting | pembrolizumab + platinum-based doublet chemotherapy +/- canakinumab | OS, PFS, and incidence DLTs | Median OS: 20.8m (CAN) /20.2 m (Placebo) (HR 0.87, 95% CI, 0.70-1.10; one-sided P=0.123); median PFS: 6.8m for both treatment arms (hazard ratio [HR], 0.85; 95% CI, 0.67-1.09; P =.1); does not meet the primary endpoint | |||
| NCT03447769 (CANOPY-A) | Phase III, terminated | Canakinumab (Canakinumab versus Placebo | DFS | Median DFS: 35m (CAN)/ 29.7m (Placebo) (HR 0.94; 95% CI 0.78–1.14; one-sided p=0.258); does not meet the primary endpoint | |||
| NCT04905316 (CHORUS) | Phase II, recruiting | Canakinumab + Chemoradiation + Durvalumab (Single-arm, prospective, phase I/II study) |
PFS | Recruiting, no result yet |
Legend: mAb- monoclonal antibody; MPR- major pathologic response; DLT- dose-limiting toxicity; DFS- disease free survival; PFS- progression free survival; OS- overall survival; m- months.
Table 2.
The summary of clinical trials of IL-1 receptor antagonists and anti-IL-1R-Antibodies related to lung cancer.
Table 2.
The summary of clinical trials of IL-1 receptor antagonists and anti-IL-1R-Antibodies related to lung cancer.
| IL-1 inhibitors | Type | Target | Trials | Phase, progress | Experimental arms | Primary outcome | Results of the primary outcome |
| Anakinra | IL-1 receptor mAb | IL-1 receptor | NCT01624766A888A | Phase I, completed | Everolimus (mTOR Inhibitor) + Anakinra/Denosumab | Incidence of AEs, MTD of everolimus | No result published yet |
| IL-1R inhibitors | Type | Target | Trials | Phase, progress | Experimental arms | Primary outcome | Results of the primary outcome |
| Nadunolimab (CAN04) | first-in-class fully humanized and ADCC enhanced mAb | IL1RAP | NCT03267316A888A(CANFOUR) | Phase I/II, Recruiting | CAN04 +/- standard of care treatment | Incidence of Treatment-Emergent AE (Safety and Tolerability) | infusion-related reactions (41%), fatigue (32%), constipation (27%), diarrhea (27%), decreased appetite (23%), nausea (23%), and vomiting (23%); ORR 53% |
| NCT05116891A888A(CESTAFOUR) | Phase I/II, active, not recruiting | CAN04 + chemotherapy (mFOLFOX or DTX or G/C) | ORR, frequency, duration, and severity of AEs | No result yet | |||
| NCT04452214 (CIRIFOUR) | Phase I, active, not recruiting | CAN04 +pembrolizumab +/- carboplatin and pemetrexed | TEAEs, DLEs, SAEs | No result yet |
Legend: MTD- maximum tolerated dose; AE - adverse event; TEAE- Treatment emergent adverse event; ORR- overall response rate.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



