
Submitted:
26 June 2023
Posted:
27 June 2023
You are already at the latest version
Abstract
Peroxiredoxins play central roles in the detoxification of reactive oxygen species and have been modelled across multiple organisms using a variety of kinetic methods. However, the peroxiredoxin dimer-to-decamer transition has been underappreciated in these studies despite the 100-fold difference in activity between these forms. This is due to the lack of available kinetics and theoretical framework for modelling this process. Using published isothermal titration calorimetry data, we obtained association and dissociation rate constants of 93.0 µM-4·s-1 and 102 s-1, respectively, for the dimer-decamer transition of human PRDX1. We developed an approach that greatly reduces the number of reactions and species needed to model the peroxiredoxin decamer oxidation cycle. Using these data, we simulated horse radish peroxidase competition and NADPH-oxidation linked assays and found that the dimer-decamer transition had an inhibition-like effect on peroxidase activity. Further, we incorporated this dimer-decamer topology and kinetics into a published and validated in vivo model of PRDX2 in the erythrocyte and found that it almost perfectly reconciled experimental and simulated responses of PRDX2 oxidation to hydrogen peroxide insult. By accounting for the dimer-decamer transition of peroxiredoxins, we were able to resolve several discrepancies between experimental data and available kinetic models.
Keywords:
Enzyme kinetics
; Hydrogen peroxide
; Isothermal titration calorimetry
; Oligomerisation
; Parameter estimation
; Peroxiredoxin
; Quantitative redox biology
; Systems biology
1. Introduction
Peroxiredoxins (Prxs) are ubiquitous and highly abundant antioxidant proteins, which play crucial roles in reactive oxygen species detoxification, signalling, and heat stress response. The first member of the Prx family to be discovered was named “torin” after the toroid shape formed when Prx dimers oligomerise [1]; later, the peroxidase activity of Prxs was discovered and the family has been referred to as “peroxiredoxins” [2]. In addition to this oligomeric configuration, reduced and sulfenic Prx dimers are at equilibrium between the “fully folded” (FF) and “locally unfolded” (LU) conformations. In the FF conformation, peroxide substrates are able to bind in the active site and are thereby exposed to nucleophilic attack by the peroxidatic cysteine. By contrast, in the LU conformation, the peroxidatic cysteine is not available to substrate and is instead brought closer to the resolving cysteine, thereby facilitating disulphide formation with the resolving cysteine [3,4]. Formation of the disulphide bond locks the Prx into the LU formation which destabilizes the decamer [5]. The connection between Prx’s quaternary structure and peroxidase activity was established when it was demonstrated that abrogating decamer formation reduced the rate of hydrogen peroxide reduction by 100-fold [6]. However, this relationship has yet to be explored in any dynamic sense, likely owing to the lack of available kinetics or a theoretical framework.
The thermodynamic relationship between decameric and dimeric Prx has been described with values in the range of 1–2 µM4 or alternatively, since the interpretation of a cannot be directly linked to a concentration value for a fifth-order reaction, by a critical transition threshold (CTC) of approximately 0.8 µM, which defines the concentration above which all of the dimers would aggregate to form decamers [7], or by a C0.5 of 1.36 µM, which defines the concentration at which half of the total Prx protein on a subunit molar basis would be present in decameric form [8]. Curiously, a fully cooperative association of five dimers into a decamer cannot describe the “switch-like” relationship between total Prx and decameric Prx, where zero decamers are found below the CTC but above it they can be calculated as . Instead, the equilibrium relationship has been described using mass-action kinetics with Prx dimers raised to the power of 130 (instead of 5 which would be expected for a reaction with five reactants) and a dissociation constant of µM129 [7]. At present, there is no mechanistic interpretation for this phenomenological description.
Given that both Prx decamers and dimers are present at equilibrium at concentrations relevant to in vitro assays, we can expect that these assays have measured the combined activities of both oligomeric forms. The / of Prx during reduction of hydrogen peroxide has been determined at 4–5 µM-1·s-1 with an NADPH linked enzyme assay [9] and at M-1·s-1 using horse radish peroxidase (HRP) competition assays [10,11]. The rate constants of obligate dimer and obligate decamer Prx mutants have been directly compared [6], but not the relative ratio of the rate constants of the different oligomeric forms of the wild-type. Indeed, even at 0.4 µM PRDX2, below the CTC where no Prx decamers are expected, a rate constant of M-1·s-1 in a HRP assay has been reported [11]. In the present study, we used in silico analyses to disentangle the effects of Prx dimers and decamers on in vitro peroxidase assays and explore how Prx decamer activity might be observed at Prx concentrations below the CTC.
Most evidence suggests that the decameric form of Prx is dominant under reduced conditions [12,13] and thus, Prx oligomerisation has been excluded from computational models on the implicit assumption that Prx is always in the decameric form [14,15]. However, studying redox stress inherently requires consideration of oxidized conditions. Simulations with a computational model of the Prx system in the red blood cell (RBC) using well established kinetics [14] (termed “Model A” by the authors) showed Prx fully oxidised at hydrogen peroxide levels below 5 µM, which conflicts with in vivo studies showing only partial oxidation of Prx at this level of H2O2 [12]. In the model [14], this discrepancy was resolved by introducing an inhibited form of Prx, yielding “Model B”; however, this addition lacks a mechanistic explanation. Here, we investigated whether this discrepancy could, instead, be resolved by accounting for low-activity Prx dimers.
The question we aimed to address with this study was: can the Prx oligomerisation cycle be sufficiently described by reaction kinetics and incorporated into a kinetic model and, if so, what effect does this have on peroxidase activity? Incorporating the Prx decamerisation into a computational model required both the kinetics of the Prx dimer-decamer transition and a theoretical framework for modelling the peroxidase reactions of the 10-site Prx decamer. We were able to obtain the kinetics for the association and dissociation reactions by developing a model for isothermal titration calorimetry (ITC) and using it to analyse published ITC data [7]. We developed a new approach to modelling the activity of the Prx decamer that greatly reduced the complexity of the system, and show how best to incorporate these data into a kinetic model. Finally, we explore with kinetic modelling the influence of dynamically cycling Prx dimers and decamers on the in vitro peroxidase activity and the oxidation state of Prx in vivo. The cycling of Prx between the highly peroxidatically active decamers and less active dimers under hydrogen peroxide load has not been investigated before. Our results show that this process can be modelled and that it is crucial to understanding the role of Prxs in the cellular context as well as the relationship between Prx and redox homeostasis more broadly.
2. Materials and Methods
Modelling and computational analyses were performed in a Jupyter notebook [16,17] using the Python programming language [18] with the PySCeS [19], NumPy [20], SciPy [21], pandas [22], lmfit [23], sklearn [24], and Matplotlib [25] packages. The models, code and data for the simulations are available from https://github.com/Rohwer-Lab/Barry2023; detailed instructions are provided in the repository’s README file and the required versions of the packages are specified in the requirements files. The models are described in detail in Supplementary Material Section 1 and are available in both PySCeS input file (*.psc) format, as well as in the standard SBML (*.xml) format [26] from the GitHub repository. All analyses are available as Jupyter notebooks.
2.1. Topology of decamerisation
Prx proteins undergo reversible concatenation from dimers to decamers (red square of Figure 1). Both dimers and decamers are able to undergo a redox reaction which converts H2O2 to H2O, although at different rates [6]. These oxidised Prxs undergo a condensation reaction by forming a disulphide bridge between the peroxidatic and resolving cysteines [27]. This disulphide-bridge formation causes decamers to dissociate into dimers [28]. Additionally, Prx dimers follow the typical reaction network for Prxs, which includes sulfinilation [29] and subsequent reduction by sulfiredoxin [30], as well as reduction of disulphide Prx by the thioredoxin (Trx) system [31]. In this study, Prx dimers were modelled with two reaction sites that have oxidation states of: reduced, SH; sulfenilated, SOH; disulphide-bridge form, SS; or sulfinilated, SOOH. To unambiguously denote the different Prx dimer species, they were written such that the left-hand site followed the priority of (see also subsection 3.5).
2.2. Model construction
Kinetic models were drafted as a collection of stoichiometric and rate equations, parameters, and initial species concentrations, using the PySCeS input file syntax [19]. The models were formalized into sets of ordinary differential equations using the pysces.model() function and solved by the builtin PySCeS interface to CVODE [32]. See Supplementary Material Section 1 for a summary and descriptions of models used in this study.
2.3. Isothermal titration calorimetry simulations
ITC experiment simulations were set up to mimic the experimental protocol followed by Barranco-Medina and co-workers [7]. Repeated injections were simulated with an interval matching the experimental protocol. In the model, these injection events were effectuated by an increase in the quantity of the various Prx species (depending on the model and ) and an increase in the reaction vessel volume.
The first injection of an ITC experiment is commonly ignored during data analysis [33] as diffusion between the contents of the reaction cell and the injection syringe causes reagents to partially react prior to the injection. However, the reagent still enters the reaction vessel and thereby influences subsequent injections. In our simulations, this was compensated for by initiating the system with a quantity of already dissociated Prx equal to that of a single injection.
The delay between the heat released by chemical dissociation and the instrument reading was accounted for using a Laplace transform [34]. Further details of the data processing procedure can be found in Supplementary Material Section 2.
ITC data [7] were digitized using the online data digitizer, WebPlotDigitizer [35]. For simulating the PRDX1 experiments the parameters used were: injection interval, 200 s; injection volume, 1.6 µl; total Prx dimers, 51 µM; PRDX1 dissociation enthalpy, 156 kcal/mol; and initial cell volume, 1400 µl.
2.4. Parameter fitting
Parameters were estimated by non-linear least-squares regression of the digitized data to a model output of species concentrations from a simulation. The fitting was performed using the minimize() function of an lmfit.Minimizer() object with the “least-squares” method and an “epsfcn” parameter of 0.0001.
2.5. Simulations of the Prx in the Red blood cell
SBML code for the model of the Prx cycle in the human RBC was obtained from the authors of the original publication [14]. “Model B” was converted to PySCeS (*.psc) input file format and modified as described below. To replicate the published simulations, a reaction for diffusion of hydrogen peroxide across the cell membrane, a process which was described in the original publication, was added to the model. To replicate “Model A” (the Prx cycle without inhibition [14]), all reactions involving the inhibited form of Prx, i.e. containing the species “iPrx_R_R_ox”, “iPrx_R_O_oox”, “iPrx_S_O2_disulf_form”, “iPrx_R_S_red_Trx1SH_Trx2SH”, “iPrx_R_S_red_Trx1SH_Trx2SOH”, and “iPrx_R_O2_srx”, were disabled.
The state of the Prx dimer population was described by several measures as calculated below.
Fraction of Prx dimers without a disulphide bridge:
Fraction of Prx dimers with a single disulphide bridge only:
Fraction of Prx dimers with two disulphide bridges:
Fraction of non-disulphide bridge Prx dimers with a sulfinic site:
Fraction of disulphide bridge Prx dimers with a sulfinic site:
2.6. Horse radish peroxidase competition assay simulations
The HRP competition assay measures the activity of Prx using competition with HRP for a substrate, in this case hydrogen peroxide. The course of the reaction is tracked by measuring the change in absorbance at 398 nm () originating from the absorbance of compound I, the product of the reaction of HRP with the substrate. Prx activity consumes hydrogen peroxide, which results in less compound I and a smaller . The rate constant of Prx is calculated by comparing the in a reaction with Prx to one without Prx (refer to [10] for a detailed description).
In this study, HRP competition assays were simulated with a model containing the compound I, HRP, Prx, hydrogen peroxide and a model without Prx as the control. The A398 from the simulations was calculated as:
where the extinction coefficient of compound I, Mcm [36].
2.7. Time to reach equilibrium after dilution simulations
Dilution of concentrated Prx was simulated in a PySCeS model by calculating the equilibrium concentrations prior to dilution, updating the model with these concentrations divided by the dilution factor and then simulating the re-eqilibration of the species over time.
2.8. Whole system assay simulations
Peroxidase assays were simulated in a model system that linked hydrogen peroxide reduction by Prx to NADPH oxidation, measured as decay at 340 nm, via Trx regeneration by thioredoxin reductase (TRR). Simulation parameters were: Trx, 50 µM; TRR, 0.5 µM; , 10 s; hydrogen peroxide, 5 µM; NADPH, 150 µM.
3. Results
3.1. Modelling Prx with and without decamer
In this study, Prx peroxidase activity was modelled in two ways. First, in what will be referred to as the mixed-activity dimer-decamer model, Prx decamer and dimer species were modelled explicitly with the peroxidase activity matched to the species. Further, this model included a decamer association and dissociation reaction (red rectangle in Figure 1). Second, in what will be referred to as the full-activity dimer-only model, Prx species were modelled explicitly as dimers only, except with the higher peroxidase activity as if they were perpetually in the decamer form. This is analogous to the topology of the green rectangle in Figure 1 with the Prx kinetics of the blue rectangle.
3.2. Models of Prx decamerisation
Several approaches to modelling Prx oligomer association (the red rectangle of Figure 1) and peroxidase activity were considered and, ultimately, the transition was modelled as a single mass-action reaction where five dimers associate into a decamer, using the published value of 1.1 µM4 for [8].
We explored by simulation the distribution between dimers and decamers as a function of total Prx concentration (Figure 2). Dimers dominate at below the Prx CTC but decamers increasingly dominate above this which is generally in line with the literature. However, the transition was not as stark as the experimental data, which found virtually no decamers below the CTC, and above it, the dimer concentration remained constant at the CTC [7].
3.3. Isothermal titration calorimetry simulation
Determining kinetic constants requires time-based experimental data and, as far as we are aware, the only time-based (non-equilibrium) data available for Prx decamer dissociation are the ITC experiments of Barranco-Medina et al. [7]. Therefore, in order to characterise the association and dissociation constants of the Prx decamer, a kinetic model simulating the ITC experiments was developed. During development of the model, we used the exponent of 130 and a of 2.4 × 10-10 µM129 as published in the original study to validate our approach (as discussed below, we subsequently used an exponent of 5 and a of 1.1 µM4 when fitting the rate constants). With this model, ITC experiments with Prx were simulated allowing for the Prx species (Figure 3 a) to be tracked and the rates (Figure 3 b) of Prx decamer dissociation and association to be calculated over the time-course of the experiment. Notably, the system behaviour changed around the 45 min mark, which corresponded to the ITC experiments of Barranco-Medina et al. and the CTC. From the rates, the power (heat transferred per second) vs. time trace (Figure 3 c) was calculated by:
where P is the power and the known for Prx of 130–160 kcal/mol dimer was used in the simulations, depending on Prx species [7]. The is often unknown when conducting an ITC experiment but can be determined from the P trace by finding the maximum area under the curve of all injections in an ITC experiment (Figure 3 d).
Several of the experimental traces published by Barranco-Medina et al. showed a single peak of heat generation at the CTC (the other peaks were endothermic), which the authors attributed to an association reaction. In the case of a system with multiple reactions at differing rates, an injection can produce an endothermic peak followed by an exothermic peak or vice versa, however, when this is observed, it occurs for several injections. ITC experimental guidelines recommend using an injection period that allows the system to reach and rest at baseline between injections [37]. Failing to reach baseline produces a trace that superficially resembles baseline drift, another common issue associated with ITC experiments [38]. By considering the above, we were able to replicate the single peak following CTC observed by Barranco-Medina et al. as an artefact by applying a baseline correction to an ITC trace of a system with an injection period that was not sufficient to reach baseline and had a sharp dissociation transition (Figure 3 e with the area under the curve in Figure 3 f).
3.4. Fitting and
The Prx decamer association and dissociation rate constants where fitted using simulated ITC experiments and ITC time-course data of PRDX1 dissociation, digitised from Barranco-Medina et al. [7], and processed as detailed in Supplementary Material Section 2. The decamer dissociation and association reactions were described by the following mass-action rate equations:
During parameter estimation, the value of was linked to (the fitted parameter) using the ratio
and the published value of [8]. The best-fit values from this procedure were a of 102 ± 1.47 (S.E.) s-1 and a of 93.0 ± 1.34 (S.E.) µM-4·s-1 (Figure 4 a). Until the CTC was reached, each simulated injection produced a similar heat-release vs. time profile and the model consistently under-estimated the data except at approximately 20 s after the injection where the heat release was, instead, over-estimated (Figure 4 b).
3.5. Enumerating the molecular states of decameric Prx
With the Prx decamer association and dissociation rate constants estimated, the final task required before incorporating decamerisation into a kinetic model was to describe the peroxidase-activity-associated reactions of the Prx decamer (the blue rectangle in Figure 1). Fully enumerated, there are 410 possible configurations of decamer and in the order of several million reactions; fortunately, several orders of magnitude of degeneracy can be introduced by using four assumptions.
First, mirror symmetry: since the two active sites of a Prx dimer are equivalent, the orientation of a dimer within a decamer can be rearranged by mirror symmetry (Figure 5 a). Second, rotational symmetry: the choice of “first position” in a linear representation of a circular molecule is arbitrary and can be selected as is convenient (Figure 5 b). Third, planar symmetry: without planar distinctions the plane of viewing a molecule can be changed as is convenient. In these three cases care must be taken that the relative positions of the dimers within the decamer remain the same and that the rate constants of each reaction are scaled by the corresponding number of degenerate reactions. Fourth, single-step decamer dissociation upon disulphide-bridge formation: the intermediate Prx oligomer species constitute a small fraction of the overall Prx population and the overall effect of including them is likely to be negligible; therefore, oxidised-decamer dissociation was assumed to take place in a single step.
Using the above assumptions, the peroxidase activity-associated reactions of the Prx decamer were enumerated using a Python script either with the sulfinilation reactions or without the sulfinilation reactions. The logic of the reaction enumerating script is further described in Supplementary Material Section 3.
We found that, when including the sulfinilation reactions in a model, the number of reactions was computationally prohibitive. No kinetics are available for the difference between the sulfinilation rate constant of the dimer and that of the decamer. Considering that disulphide-bridge formation is likely the primary driver of decamer dissociation, the proportion of decamers that contain hyperoxidised sites during the catalytic cycle is likely to be low. Therefore, the Prx decamer reaction network without sulfinilation reactions was incorporated into a kinetic model of the Prx system.
3.6. Comparing the Prx red blood cell model (Benfeitas et al. 2014) with and without decamerisation
Neither the kinetics for PRDX2 decamer association and dissociation nor a validated in vivo model of PRDX1 was available at present; thus, we added the PRDX1 kinetics to a model of PRDX2 for the RBC. The two Prx isoforms bear 77% sequence identity [39], have similar sulfenilation rate constants [40], with the most relevant distinction being that the rate of disulfide formation for PRDX1 is that of PRDX2 [40], diverting Prx away from the sulfinilation reaction. Considering that sulfinilation had a minor influence on simulations (Figure 6), we concluded that PRDX1 was a suitable stand-in for PRDX2 in this instance.
The effect of Prx decamer association and dissociation on the molecular state of the Prx population (as might be observed in SDS-PAGE and Western blot studies) was evaluated by adding the Prx decamer association and dissociation reactions and the decamer peroxidase reactions to a previously published model of PRDX2 peroxidase activity. Model A [14] contains the Prx cycle based solely on the available kinetics and predicts that Prx consumes the overwhelming majority of hydrogen peroxide at low hydrogen peroxide concentrations. In that work, the authors added an inhibited form of Prx to replicate experimental evidence, which shows equivalent contributions of catalase and Prx to hydrogen peroxide consumption at low hydrogen peroxide concentrations [41,42,43,44]; this was termed Model B [14]. Here, we added the Prx decamer association and dissociation reactions with kinetics as determined (Figure 4) to Model A (termed Model A with decamerisation). The steady-state response of the fraction of Prx dimers to changes in hydrogen peroxide concentration of Model A with decamerisation (Figure 6 a) was able to reproduce the results of Model B (Figure 6 e) without the addition of an inhibited form of Prx. This behaviour was quite different from the original Model A (Figure 6 c). A similar result was found for the time-dependent response to a hydrogen peroxide bolus (Figure 6 b, d, and f). By introducing decamerisation into Model A, we were therefore able to approximate experimental data without the further introduction of an inhibited form of Prx for which there is no direct evidence (see Discussion). Further comparisons between Model A and Model A with decamerisation regarding hydrogen peroxide metabolism and other redox factors are provided in Supplementary Material Section 4 as reproductions of the analyses found in Benfeitas et al. [14].
3.7. Incorporating Prx decamers lowers activity
To determine the extent to which Prx decamer dissociation influences Prx activity, we simulated assays comparing the full-activity dimer-only model to the mixed-activity dimer-decamer model (see subsection 3.1 for definitions of these models). Comparing the same-colour traces of the simulated HRP competition assay in Figure 7 a shows that including dimers with lower peroxidase activity and the dimer-decamer equilibrium resulted in an overall decrease in the peroxidase activity of Prx, indicated by a larger (a larger indicates that less hydrogen peroxide was consumed by Prx). Analysing the fractional inhibition of the simulated traces (Figure 7 b) yielded rate constants of 175 µM-1·s-1 and 106 µM-1·s-1 for the full-activity dimer-only model and the mixed-activity dimer-decamer model, respectively.
Next, to evaluate this decrease in activity in a dynamic system where Prx is regenerated after oxidation, we simulated an NAPDH oxidation assay with Prx, Trx, and TRR (Figure 7 c). Similar to the simulated HRP assay, including low-activity dimers in the model lowered the Prx peroxidase activity, indicated by a shallower initial slope (a smaller NADPH indicates that less NADPH has been oxidised and therefore less hydrogen peroxide has been consumed by Prx). The rate constants for the full-activity dimer-only model and the mixed-activity dimer-decamer model, were determined by initial rate kinetics to be 2.71 s-1 and 2.09 s-1, respectively (Figure 7 d). From these simulations, it is clear that models that partition the reduced Prx population into high-activity decameric Prx and low-activity dimeric Prx, exhibit lower overall Prx activity than models that only consider high-activity Prx. This effect is inversely correlated to the Prx concentration since, although both Prx dimers and decamers increase with total Prx pool size, dimers occupy a relatively smaller proportion of the total Prx pool as the Prx concentration increases. Although this inverse relationship is not as marked as when modelling the Prx dimer-decamer equilibrium stringently according to the CTC observed by Barranco-Medina et al. [7], it provides general support for such a dimer-decamer transition.
3.8. Diluting Prx can influence peroxidase activity
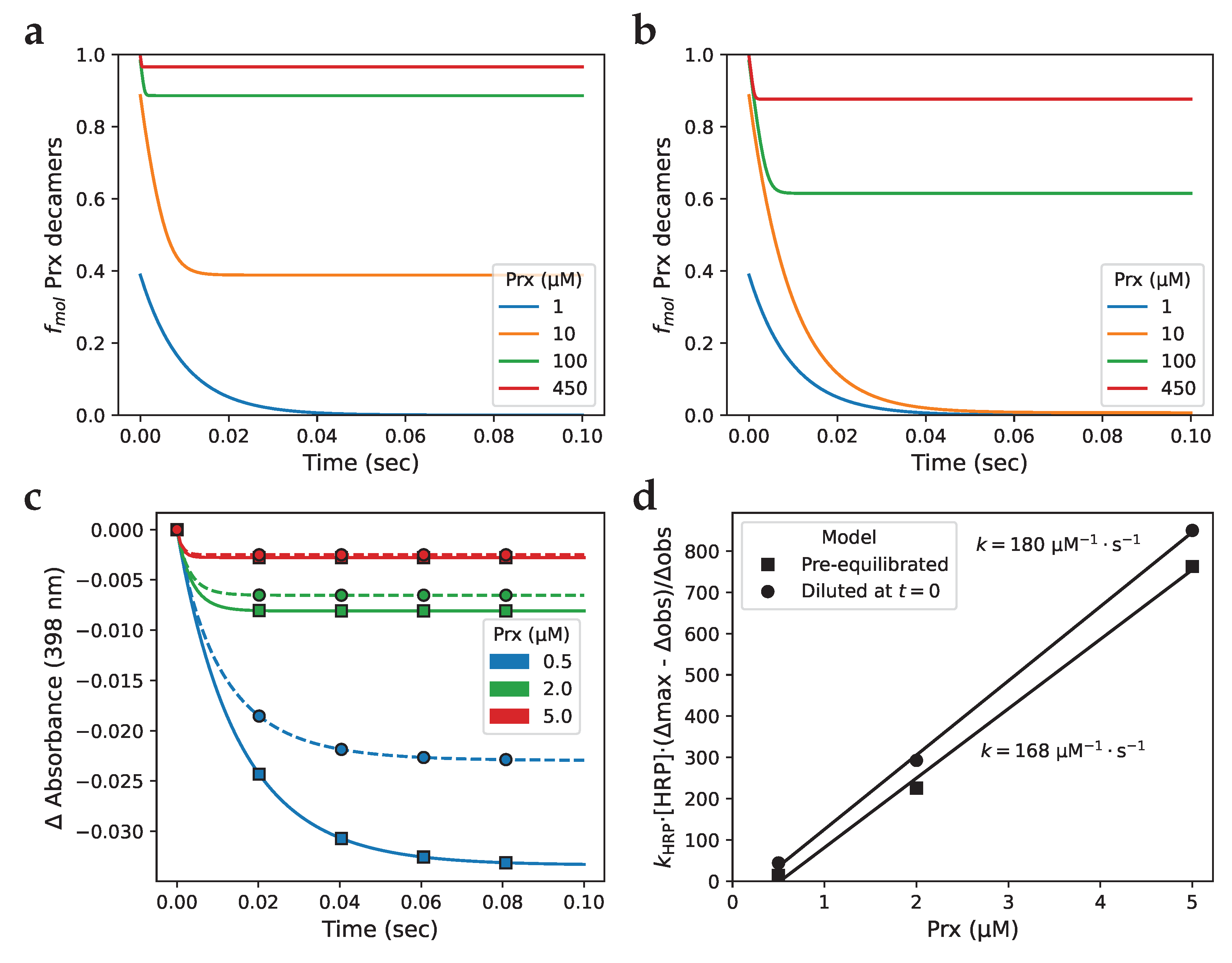
We were curious whether decamer dissociation following dilution of a Prx solution whose concentration is above the CTC needs to be considered by researchers handling Prx in the laboratory. Specifically, we considered dilution of frozen protein solutions where, commonly, purified Prx solutions are stored at approximately 10 mg/ml [45,46,47], which translates to ± 450 µM (using an Mr of 21.892 kDa for PRDX2 [48] via Uniprot), as well as dilutions of 10–100 µM Prx as would occur during HRP competition assays. Our simulations show that following a 10× (Figure 8 a) or 50× (Figure 8 b) dilution, solutions ranging in concentration between 1 and 450 µM Prx equilibrated over a time-span of ±45 ms to as fast as <1 ms. Comparing Figure 8 a to Figure 8 b, we find that increasing the magnitude of the dilution resulted in a longer the time to equilibrium for all Prx concentrations except for the 1 µM solution, which took the same time to reach equilibrium in following a 10× or a 50× dilution. Additional examples of solution dilution simulations can be found in Supplementary Material Section 5.
This equilibration is fast enough that a Prx solution would reach equilibrium after dilution in most laboratory applications, with the exception of HRP competition assays of the peroxidase activity of Prx, which can run to completion in approximately 100 ms [10,11,45]. To evaluate the extent that this affects assays of Prx peroxidase activity, we simulated a HRP competition assay (Figure 8 c) using the mixed-activity dimer-decamer model and compared the hypothetical scenario of Prx dimers and decamers equilibrating instantly to the scenario of Prx dimers and decamers equilibrating as per their mass action kinetics following injection by a stopped-flow apparatus, i.e. dilution at . These traces show that an instantly equilibrated system is expected to have lower peroxidase activity than one that follows the dissociation kinetics, which is confirmed by the rate constants of 168 µM-1·s-1 and 180 µM-1·s-1 (Figure 8 d), obtained from simulations with pre-equilibrated Prx and from those following the dissociation kinetics after dilution, respectively. Together, these results show that the equilibration of Prx following dilution can affect HRP competition assays when the time between dilution and assay is sufficiently short that the system cannot reach equilibrium, but should not affect most other assays where there is a longer time between dilution and data acquisition.
4. Discussion
The capacity of Prx to form reduced decamers and then to dissociate into dimers upon oxidation is well established [49]; however, to date, no studies have incorporated this process into kinetic models. In part, this is due to the difficulty measuring oligomerisation kinetics and researchers have circumvented the issue by modelling Prx activity with dimer topology and decamer kinetics [14,15], i.e. with the assumption that Prx is always in the high-activity decamer form. Evidence supports this assumption under basal conditions and low hydrogen peroxide [12,13]; however, it is desirable to model the response of cellular redox factors and hydrogen peroxide protection mechanisms under hydrogen peroxide load to understand how these systems respond to oxidative stress. We endeavoured to model the dimer-decamer transition of Prx in a dynamically responding system of Prx activity.
We were able to, for the first time for any Prx, determine the association and dissociation rate constants for the dimer-decamer transition as 93.0 µM-4·s-1 and 102 s-1, respectively. Using the tools of computational biology we were able to extract additional value from data that have been publicly available for over a decade [7] by performing a relatively complex analysis to derive these novel kinetic parameters. By measuring heat release directly, ITC allows for measurement of oligomerisation reactions without fluorogenic prosthetic groups and has been used extensively to study physical and chemical binding equilibria and to determine thermodynamic parameters in molecular biology [50]. Although the application of ITC to enzyme kinetics has been long established [51], it has not been as widely adopted as other kinetic assay techniques, owing to several factors that complicate data analysis. An upper limit of 2 s-1 has been proposed for determining kinetic parameters from ITC [52], which is below the values determined in this study; however, this limit was based on simulations of Ca2+ binding to EDTA, a system where the ITC peaks were dominated by the instrument. The dissociation of PRDX1 in the original ITC experiments [7] released 13 µcal of heat per injection and heat was detected over ±170 s, compared to the 11 µcal heat release and 40 s detection range of the above-mentioned experiments. Therefore, the experimental data used in this study should be able to measure kinetics on a faster time-scale than the Ca2+–EDTA system, which justifies a relaxation of the aforementioned upper limit. Moreover, while it is preferable to estimate the true instrument response time during data analysis as the value varies between machines and experimental setups [53], we had to rely on the published machine specifications. Considering these factors with the 45 ms dilution times (Figure 8), the data generated by an ITC apparatus with a ±20 s reading delay may not have sufficient resolution to accurately determine kinetic constants of the order of M-1·s-1. In light of this, the kinetics presented here should be considered preliminary and, as with any first time reporting, confidence in the accuracy of the Prx oligomerisation kinetics would be greatly improved by additional studies, in particular ones with additional experimental data. Nevertheless, our work highlights the power of ITC as an aid for the construction of systems-biology models, as time-dependent rate constants are crucial to simulating dynamic behaviour.
With these novel kinetic parameters, we were able to model the reduced Prx pool as a dynamic population of decamers and less peroxidatically active dimers. Comparing this to a model of the Prx pool using the topology of dimers with the kinetics of decamers (Figure 7), we found that our expansion of the model resulted in less peroxidase activity, an effect that arose from three factors: first, in a system at rest, the equilibrium between Prx dimers and decamers requires that a portion of the reduced Prx pool is dimeric, roughly corresponding to the CTC of 0.8 µM [7]. Second, since dimers contain two active sites, a reduced Prx site will regularly be paired with a site that is either sulfenic, disulphide, sulfinic, or—although not modelled in this study—sulfonic during Prx oxidation and regeneration. These hetero-oxidized dimers are unable to form decamers and therefore the reduced sites contained in them will have the corresponding low peroxidase activity of dimeric Prx. Third, when the sum of the rates of Prx regeneration and decamer dissociation is greater than the rate of decamer formation, the pool of reduced dimers will accumulate until the net rate is zero. In this way, while a system is under hydrogen peroxide stress, the proportion of the reduced Prx pool that is dimeric will be greater than that of the same system at rest.
We adopted a systems biology approach and evaluated the effect of the Prx dimer-decamer transition on the Prx redox cycle in silico by adding the Prx decamer association and dissociation reactions, with the associated kinetics determined in this study, to an existing model of PRDX2 activity [14]. Simulations of Prx species oxidation vs. hydrogen peroxide supply (Figure 6) show good agreement with the SDS-PAGE results of the same system [12]. Considering that the parameters used to model the Prx dimer-decamer transition are derived entirely independently, we view this as strong validation that the Prx dimer-decamer transition is essential to modelling the dynamics of the Prx redox cycle. Furthermore, the RBC contains far above the average level of Prx, which across various tissues is in the low dozen micro-molar range as derived from proteomics data (Barry et al., unpublished). Hence, the Prx dimer should have a proportionately larger effect on Prx activity and oxidised species profile in other cell types compared to the RBC.
During construction of their erythrocyte PRDX2 model [14] (termed “Model A”), Benfeitas and co-workers found that there was a large discrepancy between their simulations and the aforementioned SDS-PAGE experimental evidence [12]. To reconcile these discrepancies, they augmented Model A with a hypothetical inhibition of Prx to create Model B while granting that there was no evidence for this inhibitor. Based on the evidence that that Prx and catalase contribute comparably to peroxidase activity at low hydrogen peroxide [41,42,44], the authors parameterised the inhibitor such that it had near equilibrium binding and disabled the peroxidatic activity of > 99 % of Prx. This sequestration-based inhibition bears similarity to our approach in that reduced Prx sites can be “sequestered” into a low-activity dimer until they can reform into a decamer but differs in that the entire Prx population is immediately available in the event of a surge in hydrogen peroxide. Without using kinetics derived in any part from their Model B, our simulation of Model A with the Prx dimer-decamer transition added was able to replicate Model B almost perfectly in both steady-state and hydrogen peroxide bolus simulations. Therefore, we propose that the Prx dimer-decamer transition with lower peroxidase activity of the Prx dimer is, in fact, the “inhibition mechanism” proposed by Benfeitas et al. [14].
In our approach we modelled the process of Prx decamer association and dissociation as a single reaction instead of a series of concatenating or splitting steps. Implicitly, this assumes perfect cooperativity and negligible presence of the intermediate oligomers, which is congruent with several studies [7,8]. However, it could be argued that while these assumptions are valid given the current limits of available techniques, this may not hold up to methods with greater resolution. Examples which tenuously hint that relevant amounts of intermediate Prx oligomers may be present in low concentrations of reduced Prx include the gradual increase in for the initial injections of several of Barranco-Medina’s ITC experiments [7] as well as the slight non-linearity visible in Villar’s phasor plots [8]. Therefore, the validity of assumption of perfect cooperativity will need to be reevaluated when more evidence becomes available.
If Prx dimers have lower Prx activity than Prx decamers, should we not consistently find that the Prx activity drops off around the CTC of the Prx decamer in peroxidase assays? Indeed, we believe that evidence for this can be found in several studies but, to our knowledge, has thus far been overlooked. The studies of Prx activity by HRP competition assays [10,54,55,56,57] show that a linear fit of fractional inhibition vs. Prx has an x-axis intercept in the µM range, suggesting that there is µM of peroxidatically inhibited Prx. As a counter-example, Manta and co-workers [11] report a peroxidase rate constant of M-1·s-1 for 0.4 µM PRDX2, and while this is lower than the rate constant they reported for higher Prx concentrations (, , and M-1·s-1 for , , and µM Prx, respectively), it is not congruent with a CTC of 0.8 µM [7] and a 100-fold lower Prx activity for dimers than for decamers [6] (see also discussion below).
As a caveat, the rate constants derived by analysing simulations of HRP assays reported here should only be considered in comparison to each other and not as kinetics for PRDX1. The established fractional inhibition method for determining rate constants from HRP competitive assays systemically and increasingly miss-estimates the target rate constant, the more this target rate constant differs from (Barry et al., in preparation).
The observed rate of hydrogen peroxide consumption during peroxidase activity assay with Prxs is a combination of the activity of the dimeric and the decameric Prx. This raises the question of how published Prx rate constants should be interpreted. Considering that, typically, Prx is incubated with DTT prior to activity assays to ensure that it is fully reduced [10] and, canonically, reduced Prx is decameric, this has lead to the equating of reported Prx rate constants to decameric Prx rate constants, which has carried through implicitly to modelling studies [14]. Since the Prx population is divided between dimers and decamers, decameric Prx is likely overestimated in these assays. Considering that catalytic rate constants are determined by dividing activity by enzyme concentration, this leads us to the conclusion that reported rate constants for decameric Prx are somewhat under-estimated, which is supported by the finding that obligate decamer mutants of Prx have higher activity than wild-type [6]. Confoundingly, this effect has been masked in millisecond assays by the fact that Prx is unable to fully dissociate prior to oxidation, since these assays are initiated by diluting the enzyme solution by mixing with the substrate solution (Figure 8). This may explain why Manta and co-workers [11] were able to detect significant peroxidase activity at concentrations of PRDX2 where only low-activity dimers should have been present. Protein concentrations prior to substrate addition and injection volumes or assay-to-stock dilution ratios are rarely reported, making it difficult to evaluate the extent of these two effects on published Prx kinetics, an issue that could be alleviated by improved assay reporting standards [58,59,60].
5. Conclusions
In this work, we have developed a model of the Prx decamer association and dissociation cycle. This model was parameterised by fitting the relevant rate constants to digitized ITC data for Prx dilution experiments. We developed a script that enumerates the reactions of the Prx decamer oxidation cycle. Together, these reactions were incorporated into an established model of hydrogen peroxide neutralisation by PRDX2 in the RBC. With this, we demonstrated that incorporating decamer dissociation causes an inhibition-like effect on peroxidase activity. This allowed us to mechanistically resolve a discrepancy between experimental data and kinetic simulations by showing that reduced Prx sites can be sequestered in a less active dimeric form. Additionally, we have demonstrated that Prx decamer dissociation occurs within a time-frame relevant to peroxidase assays and other oxidation experiments and needs to be considered when working with Prx in a laboratory. Our findings strongly emphasise the the link between the quaternary structure of Prxs and their peroxidase activity, a connection that has been underappreciated so far.
In conclusion, Prx kinetics have been studied using a range of methods such as steady-state and competition kinetic assays, ITC as well as newer methods such as phasor analysis. Computational modelling offers a platform to combine and organise different experimental data into a single framework to better understand these important antioxidant proteins.
Supplementary Materials
The following supporting information can be downloaded at: the website of this paper posted on Preprints.org., the website of this paper posted on Preprints.org., Document S1: Supplementary Material.
Author Contributions
Conceptualization, J.M.R., C.J.B. and C.S.P.; methodology, J.M.R. and C.J.B.; software, C.J.B.; validation, C.J.B. and J.M.R.; formal analysis, C.J.B. and J.M.R.; investigation, C.J.B.; resources, J.M.R.; data curation, C.J.B. and J.M.R.; writing—original draft preparation, C.J.B.; writing—review and editing, J.M.R. and C.S.P.; visualization, C.J.B.; supervision, J.M.R. and C.S.P.; project administration, J.M.R.; funding acquisition, J.M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Research Foundation (South Africa) grant numbers 93670, 114748, 117864, and 138096. Any opinion, finding and conclusion or recommendation expressed in this material is that of the author(s) and the NRF does not accept any liability in this regard. The APC was funded in part by Stellenbosch University.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data and code to reproduce the results presented in this manuscript are available from GitHub at https://github.com/Rohwer-Lab/Barry2023.
Acknowledgments
The assistance of Dr Carl Christensen in converting the Benfeitas model description into PySCeS format is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| CTC | critical transition threshold |
| FF | fully folded |
| HRP | horse radish peroxidase |
| ITC | isothermal titration calorimetry |
| LU | locally unfolded |
| Prx | peroxiredoxin |
| RBC | red blood cell |
| SH | reduced peroxiredoxin site |
| SOH | sulfenilated peroxiredoxin site |
| SOOH | sulfinilated peroxiredoxin site |
| SS | disulfide-bridge peroxiredoxin site |
| TRR | thioredoxin reductase |
| Trx | thioredoxin |
References
- Harris, J. Release of a macromolecular protein component from human erythrocyte ghosts. BBA - Biomembranes 1968, 150, 534–537. [Google Scholar] [CrossRef]
- Chae, H.; Rhee, S. A thiol-specific antioxidant and sequence homology to various proteins of unknown function. BioFactors 1994, 4, 177–180. [Google Scholar]
- Schröder, E.; Littlechild, J.; Lebedev, A.; Errington, N.; Vagin, A.; Isupov, M. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure 2000, 8, 605–615. [Google Scholar] [CrossRef]
- Alphey, M.; Bond, C.; Tetaud, E.; Fairlamb, A.; Hunter, W. The structure of reduced tryparedoxin peroxidase reveals a decamer and insight into reactivity of 2Cys-peroxiredoxins. Journal of Molecular Biology 2000, 300, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.; Poole, L.; Hantgan, R.; Karplus, P. Dimers to doughnuts: Redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry 2002, 41, 5493–5504. [Google Scholar] [CrossRef]
- Parsonage, D.; Youngblood, D.; Sarma, G.; Wood, Z.; Karplus, P.; Poole, L. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry 2005, 44, 10583–10592. [Google Scholar] [CrossRef] [PubMed]
- Barranco-Medina, S.; Kakorin, S.; Lázaro, J.; Dietz, K.J. Thermodynamics of the dimer-decamer transition of reduced human and plant 2-cys peroxiredoxin. Biochemistry 2008, 47, 7196–7204. [Google Scholar] [CrossRef] [PubMed]
- Villar, S.; Dalla-Rizza, J.; Möller, M.; Ferrer-Sueta, G.; Malacrida, L.; Jameson, D.; Denicola, A. Fluorescence lifetime phasor analysis of the decamer–dimer equilibrium of human peroxiredoxin 1. International Journal of Molecular Sciences 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, D.; Netto, L. Cytosolic thioredoxin peroxidase I and II are important defenses of yeast against organic hydroperoxide insult: Catalases and peroxiredoxins cooperate in the decomposition of H2O2 by yeast. Journal of Biological Chemistry 2004, 279, 35219–35227. [Google Scholar] [CrossRef]
- Ogusucu, R.; Rettori, D.; Munhoz, D.; Soares Netto, L.; Augusto, O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: Rate constants by competitive kinetics. Free Radical Biology and Medicine 2007, 42, 326–334. [Google Scholar] [CrossRef]
- Manta, B.; Hugo, M.; Ortiz, C.; Ferrer-Sueta, G.; Trujillo, M.; Denicola, A. The peroxidase and peroxynitrite reductase activity of human erythrocyte peroxiredoxin 2. Archives of Biochemistry and Biophysics 2009, 484, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Low, F.; Hampton, M.; Peskin, A.; Winterbourn, C. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood 2007, 109, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Bayer, S.; Maghzal, G.; Stocker, R.; Hampton, M.; Winterbourn, C. Neutrophil-mediated oxidation of erythrocyte peroxiredoxin 2 as a potential marker of oxidative stress in inflammation. FASEB Journal 2013, 27, 3315–3322. [Google Scholar] [CrossRef] [PubMed]
- Benfeitas, R.; Selvaggio, G.; Antunes, F.; Coelho, P.; Salvador, A. Hydrogen peroxide metabolism and sensing in human erythrocytes: A validated kinetic model and reappraisal of the role of peroxiredoxin II. Free Radical Biology and Medicine 2014, 74, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Tomalin, L.; Day, A.; Underwood, Z.; Smith, G.; Dalle Pezze, P.; Rallis, C.; Patel, W.; Dickinson, B.; Bähler, J.; Brewer, T.; et al. Increasing extracellular H2O2 produces a bi-phasic response in intracellular H2O2, with peroxiredoxin hyperoxidation only triggered once the cellular H2O2-buffering capacity is overwhelmed. Free Radical Biology and Medicine 2016, 95, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Pérez, F.; Granger, B.E. IPython: A system for interactive scientific computing. Computing in Science & Engineering 2007, 9, 21–29. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.E.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.B.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks – a publishing format for reproducible computational workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas. Proceedings of the 20th International Conference on Electronic Publishing; pp. 201687–90. [CrossRef]
- Van Rossum, G.; Drake, F.L. Python 3 Reference Manual; 2009.
- Olivier, B.G.; Rohwer, J.M.; Hofmeyr, J.H.S. Modelling cellular systems with PySCeS. Bioinformatics 2005, 21, 560–561. [Google Scholar] [CrossRef]
- van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy array: A structure for efficient numerical computation. Computing in Science & Engineering 2011, 13, 22–30. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, T.; Peterson, P.; et al. SciPy: Open source scientific tools for Python, 2001.
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the Proceedings of the 9th Python in Science Conference; van der Walt, S.; Millman, J., Eds., 2010, pp. 56–61. [CrossRef]
- Newville, M.; Otten, R.; Nelson, A.; Stensitzki, T.; Ingargiola, A.; Allan, D.; Fox, A.; Carter, F. ; Michał. ; Osborn, R.; et al. lmfit/lmfit-py: 1.1.0 2022. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. Journal of Machine Learning Research 2011, 12, 2825–2830. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Computing in Science & Engineering 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Keating, S.M.; Waltemath, D.; König, M.; Zhang, F.; Dräger, A.; Chaouiya, C.; Bergmann, F.T.; Finney, A.; Gillespie, C.S.; Helikar, T.; et al. SBML Level 3: an extensible format for the exchange and reuse of biological models. Mol Syst. Biol. 2020, 16, e9110. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.; Schröder, E.; Harris, J.; Poole, L. Structure, mechanism and regulation of peroxiredoxins. Trends in Biochemical Sciences 2003, 28, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.; Poole, L.; Hantgan, R.; Karplus, P. Dimers to doughnuts: Redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry 2002, 41, 5493–5504. [Google Scholar] [CrossRef]
- Yang, K.S.; Kang, S.; Woo, H.; Hwang, S.; Chae, H.; Kim, K.; Rhee, S. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. Journal of Biological Chemistry 2002, 277, 38029–38036. [Google Scholar] [CrossRef]
- Chang, T.S.; Jeong, W.; Woo, H.; Sun, M.; Park, S.; Sue, G. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. Journal of Biological Chemistry 2004, 279, 50994–51001. [Google Scholar] [CrossRef]
- Poole, L.; Michael Reynolds, C.; Wood, Z.; Andrew Karplus, P.; Ellis, H.; Li Calzi, M. AhpF and other NADH:peroxiredoxin oxidoreductases, homologues of low M(r) thioredoxin reductase. European Journal of Biochemistry 2000, 267, 6126–6133. [Google Scholar] [CrossRef]
- Cohen, S.D.; Hindmarsh, A.C. CVODE, a stiff/nonstiff ODE solver in C. Computers in Physics 1996, 10, 138–143. [Google Scholar] [CrossRef]
- Sgarlata, C.; Zito, V.; Arena, G. Conditions for calibration of an isothermal titration calorimeter using chemical reactions. Analytical and Bioanalytical Chemistry 2013, 405, 1085–1094. [Google Scholar] [CrossRef]
- Di Trani, J.M. Measuring Enzyme Kinetics Using Isothermal Titration Calorimetry. PhD thesis, McGill University, 2018.
- Rohatgi, A. Webplotdigitizer: Version 4.6, 2022. https://automeris.io/WebPlotDigitizer.
- Hayashi, Y.; Yamazaki, I. The oxidation-reduction potentials of compound I/compound II and compound II/ferric couples of horseradish peroxidases A2 and C. Journal of Biological Chemistry 1979, 254, 9101–9106. [Google Scholar] [CrossRef]
- VP-ITC MicroCalorimeter User’s Manual.
- Kantonen, S.; Henriksen, N.; Gilson, M. Evaluation and minimization of uncertainty in ITC binding measurements: Heat error, concentration error, saturation, and stoichiometry. Biochimica et Biophysica Acta - General Subjects 2017, 1861, 485–498. [Google Scholar] [CrossRef]
- Dalla Rizza, J.; Randall, L.; Santos, J.; Ferrer-Sueta, G.; Denicola, A. Differential parameters between cytosolic 2-Cys peroxiredoxins, PRDX1 and PRDX2. Protein Science 2019, 28, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Portillo-Ledesma, S.; Randall, L.; Parsonage, D.; Rizza, J.; Andrew Karplus, P.; Poole, L.; Denicola, A.; Ferrer-Sueta, G. Differential kinetics of two-cysteine peroxiredoxin disulfide formation reveal a novel model for peroxide sensing. Biochemistry 2018, 57, 3416–3424. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Goyette Jr., G.; Ravindranath, Y.; Ho, Y.S. Hemoglobin autoxidation and regulation of endogenous H2O2 levels in erythrocytes. Free Radical Biology and Medicine 2005, 39, 1407–1417. [Google Scholar] [CrossRef]
- Johnson, R.M.; Ho, Y.S.; Yu, D.Y.; Kuypers, F.A.; Ravindranath, Y.; Goyette, G.W. The effects of disruption of genes for peroxiredoxin-2, glutathione peroxidase-1, and catalase on erythrocyte oxidative metabolism. Free Radical Biology and Medicine 2010, 48, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Jacob, H.; Ingbar, S.; Jandl, J. Oxidative hemolysis and erythrocyte metabolism in hereditary acatalasia. The Journal of Clinical Investigation 1965, 44, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S.; Kato, G.J.; Yang, S.H.; Bae, S.W.; Lee, J.S.; Gladwin, M.T.; Rhee, S.G. Hydroxyurea-induced expression of glutathione peroxidase 1 in red blood cells of individuals with sickle cell anemia. Antioxidants and Redox Signaling 2010, 13, 1–11. [Google Scholar] [CrossRef]
- Nelson, K.; Parsonage, D. Measurement of peroxiredoxin activity. Current Protocols in Toxicology 2011. [Google Scholar] [CrossRef]
- Reeves, S.; Parsonage, D.; Nelson, K.; Poole, L. Kinetic and thermodynamic features reveal that Escherichia coli BCP is an unusually versatile peroxiredoxin. Biochemistry 2011, 50, 8970–8981. [Google Scholar] [CrossRef]
- Perkins, A.; Parsonage, D.; Nelson, K.; Ogba, O.; Cheong, P.Y.; Poole, L.; Karplus, P. Peroxiredoxin catalysis at atomic resolution. Structure 2016, 24, 1668–1678. [Google Scholar] [CrossRef]
- Pahl, P.; Berger, R.; Hart, I.; Chae, H.; Rhee, S.; Patterson, D. Localization of TDPX1, a human homologue of the yeast thioredoxin-dependent peroxide reductase gene (TPX), to chromosome 13q12. Genomics 1995, 26, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Barranco-Medina, S.; Lázaro, J.J.; Dietz, K.J. The oligomeric conformation of peroxiredoxins links redox state to function. FEBS Letters 2009, 583, 1809–1816. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y. Applications of isothermal titration calorimetry in protein science. Acta Biochimica et Biophysica Sinica 2008, 40, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Todd, M.; Gomez, J. Enzyme kinetics determined using calorimetry: A general assay for enzyme activity? Analytical Biochemistry 2001, 296, 179–187. [Google Scholar] [CrossRef]
- Di Trani, J.M.; Moitessier, N.; Mittermaier, A.K. Measuring rapid time-scale reaction kinetics using isothermal titration calorimetry. Analytical Chemistry 2017, 89, 7022–7030. [Google Scholar] [CrossRef] [PubMed]
- Transtrum, M.; Hansen, L.; Quinn, C. Enzyme kinetics determined by single-injection isothermal titration calorimetry. Methods 2015, 76, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.; Peskin, A.; Paton, L.; Winterbourn, C.; Hampton, M. Redox potential and peroxide reactivity of human peroxiredoxin 3. Biochemistry 2009, 48, 6495–6501. [Google Scholar] [CrossRef]
- Toledo Jr., J.; Audi, R.; Ogusucu, R.; Monteiro, G.; Netto, L.; Augusto, O. Horseradish peroxidase compound i as a tool to investigate reactive protein-cysteine residues: From quantification to kinetics. Free Radical Biology and Medicine 2011, 50, 1032–1038. [Google Scholar] [CrossRef]
- Nelson, K.; Knutson, S.; Soito, L.; Klomsiri, C.; Poole, L.; Fetrow, J. Analysis of the peroxiredoxin family: Using active-site structure and sequence information for global classification and residue analysis. Proteins: Structure, Function and Bioinformatics 2011, 79, 947–964. [Google Scholar] [CrossRef]
- Peskin, A.; Pace, P.; Winterbourn, C. Enhanced hyperoxidation of peroxiredoxin 2 and peroxiredoxin 3 in the presence of bicarbonate/CO2. Free Radical Biology and Medicine 2019, 145, 1–7. [Google Scholar] [CrossRef]
- Tipton, K.F.; Armstrong, R.N.; Bakker, B.M.; Bairoch, A.; Cornish-Bowden, A.; Halling, P.J.; Hofmeyr, J.H.; Leyh, T.S.; Kettner, C.; Raushel, F.M.; et al. Standards for Reporting Enzyme Data: The STRENDA Consortium: What it aims to do and why it should be helpful. Perspectives in Science 2014, 1, 131–137. [Google Scholar] [CrossRef]
- Swainston, N.; Baici, A.; Bakker, B.M.; Cornish-Bowden, A.; Fitzpatrick, P.F.; Halling, P.; Leyh, T.S.; O’Donovan, C.; Raushel, F.M.; Reschel, U.; et al. STRENDA DB: enabling the validation and sharing of enzyme kinetics data. FEBS J. 2018, 285, 2193–2204. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, S.; Dienhart, H.; Range, J.; Malzacher, S.; Spöring, J.D.; Rother, D.; Pinto, M.F.; Martins, P.; Lagerman, C.E.; Bommarius, A.S.; et al. EnzymeML: seamless data flow and modeling of enzymatic data. Nature Methods 2023, 20, 400–402. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Peroxiredoxin (Prx) decamer association and dissociation pathways and their relationship to 2-Cys Prx activity and regeneration. Decamerisation can be modelled as a reaction where five dimers associate into a decamer in a single step. Prx sites are denoted as: SH, reduced; SOH, sulfenilated; or SS, disulphide-bridge. SS sites are regenerated by thioredoxin (Trx), which is, in turn, regenerated by thioredoxin reductase (TRR) using NADPH. Red rectangle: the reactions involved in decamer association and dissociation. Blue rectangle: the reactions of Prx decamer peroxidase activity and disulphide-bridge formation. Sulfinilation and reduction by sulfiredoxin are omitted for brevity. Green rectangle: the reactions of Prx dimer peroxidase activity, disulphide-bridge formation, and Prx regeneration. Dashed arrows represent reactions that can occur recursively. Prxdim. and Prxdec. are placeholders for several Prx dimer species and the multitude of Prx decamer species, respectively, with the number of chemical residues of each oxidation state listed below. * One Prx dimer species is formed per reaction.
Figure 1.
Peroxiredoxin (Prx) decamer association and dissociation pathways and their relationship to 2-Cys Prx activity and regeneration. Decamerisation can be modelled as a reaction where five dimers associate into a decamer in a single step. Prx sites are denoted as: SH, reduced; SOH, sulfenilated; or SS, disulphide-bridge. SS sites are regenerated by thioredoxin (Trx), which is, in turn, regenerated by thioredoxin reductase (TRR) using NADPH. Red rectangle: the reactions involved in decamer association and dissociation. Blue rectangle: the reactions of Prx decamer peroxidase activity and disulphide-bridge formation. Sulfinilation and reduction by sulfiredoxin are omitted for brevity. Green rectangle: the reactions of Prx dimer peroxidase activity, disulphide-bridge formation, and Prx regeneration. Dashed arrows represent reactions that can occur recursively. Prxdim. and Prxdec. are placeholders for several Prx dimer species and the multitude of Prx decamer species, respectively, with the number of chemical residues of each oxidation state listed below. * One Prx dimer species is formed per reaction.

Figure 2.
The proportion of reduced Prx oligomers that are in decameric or in dimeric form at equilibrium is related to the total reduced Prx concentration. The figure shows an equilibrium simulation of the distribution of dimeric and decameric Prx over a range of total Prx concentrations using a one-step mass-action model with exponent of 5. (—) Prx dimers, (—) Prx decamers, and (- -) the critical transition threshold or minimum concentration of total Prx where decamers are observed as described by Barranco-Medina et al. [7].
Figure 2.
The proportion of reduced Prx oligomers that are in decameric or in dimeric form at equilibrium is related to the total reduced Prx concentration. The figure shows an equilibrium simulation of the distribution of dimeric and decameric Prx over a range of total Prx concentrations using a one-step mass-action model with exponent of 5. (—) Prx dimers, (—) Prx decamers, and (- -) the critical transition threshold or minimum concentration of total Prx where decamers are observed as described by Barranco-Medina et al. [7].
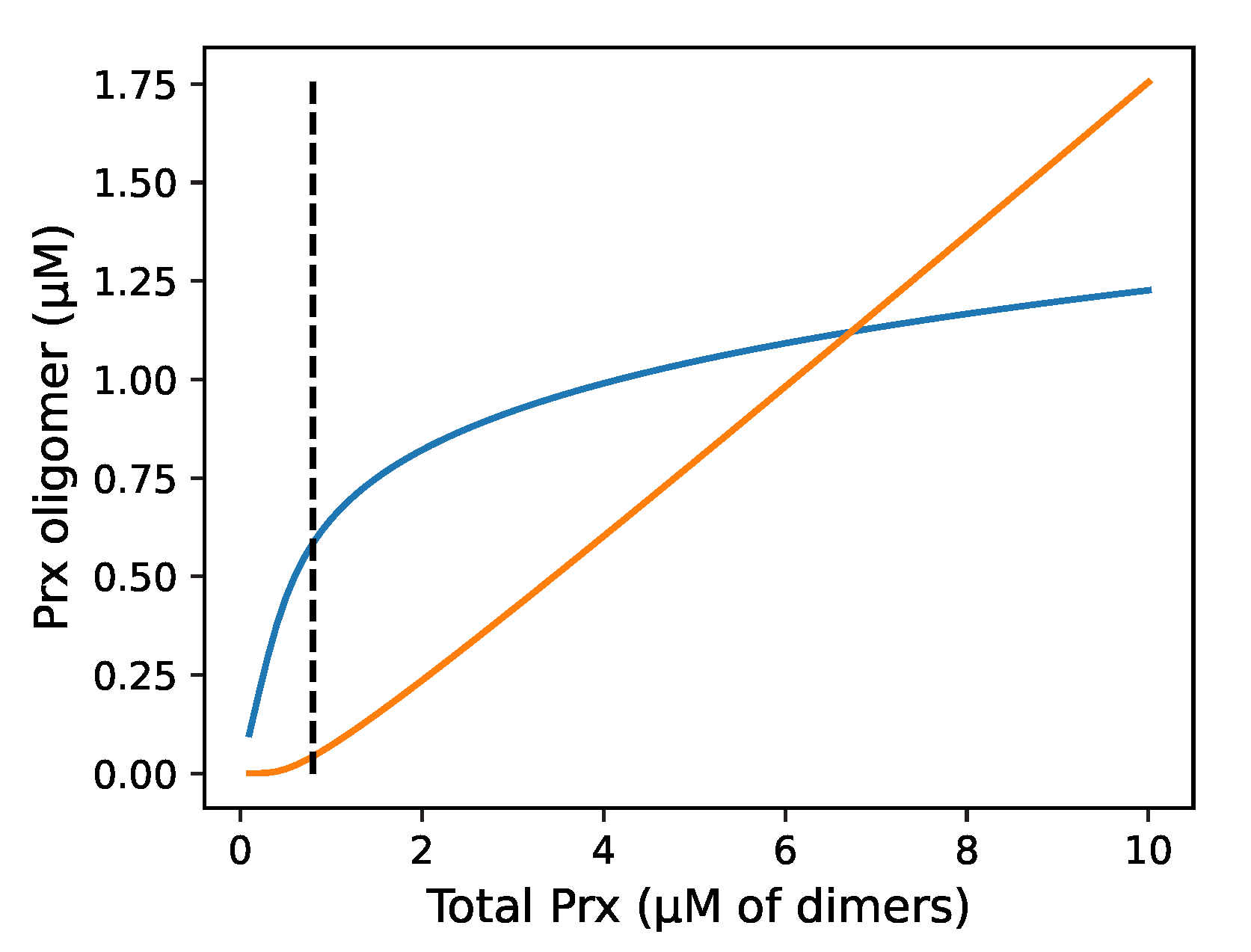
Figure 3.
Development of a model for simulating isothermal titration calorimetry (ITC) experiments. The rates and species concentrations, as well as heat generation, were calculated over time with model simulations. The injection parameters were set to match those in Fig. 1A of [7]. (a) Trace of species concentration over time; (b) rates over time; (c) heat generation over time calculated by the product of association enthalpy and the difference between dissociation and association rates; (d) heat released by each injection calculated as the area under the curve; (e) heat generation over time calculated as in (c) and with the baseline subtracted; (f) heat released by each injection calculated as the area under the curve after baseline correction. A full injection was assumed to occur before the beginning of the trace (the first injection of an ITC experiment does not produce a full heat curve but must be considered in the model simulation). Model parameters were: reaction exponent, 130; , 2.4 × 10-10 µM129; , 2.08 × 1010 µM-129·s-1; , 5.0 s-1; injection volume, 1.6 µl; syringe total Prx (in dimers), 87.5 µM; injection interval, 180 s; association enthalpy, 142 kcal/mol of dimer.
Figure 3.
Development of a model for simulating isothermal titration calorimetry (ITC) experiments. The rates and species concentrations, as well as heat generation, were calculated over time with model simulations. The injection parameters were set to match those in Fig. 1A of [7]. (a) Trace of species concentration over time; (b) rates over time; (c) heat generation over time calculated by the product of association enthalpy and the difference between dissociation and association rates; (d) heat released by each injection calculated as the area under the curve; (e) heat generation over time calculated as in (c) and with the baseline subtracted; (f) heat released by each injection calculated as the area under the curve after baseline correction. A full injection was assumed to occur before the beginning of the trace (the first injection of an ITC experiment does not produce a full heat curve but must be considered in the model simulation). Model parameters were: reaction exponent, 130; , 2.4 × 10-10 µM129; , 2.08 × 1010 µM-129·s-1; , 5.0 s-1; injection volume, 1.6 µl; syringe total Prx (in dimers), 87.5 µM; injection interval, 180 s; association enthalpy, 142 kcal/mol of dimer.
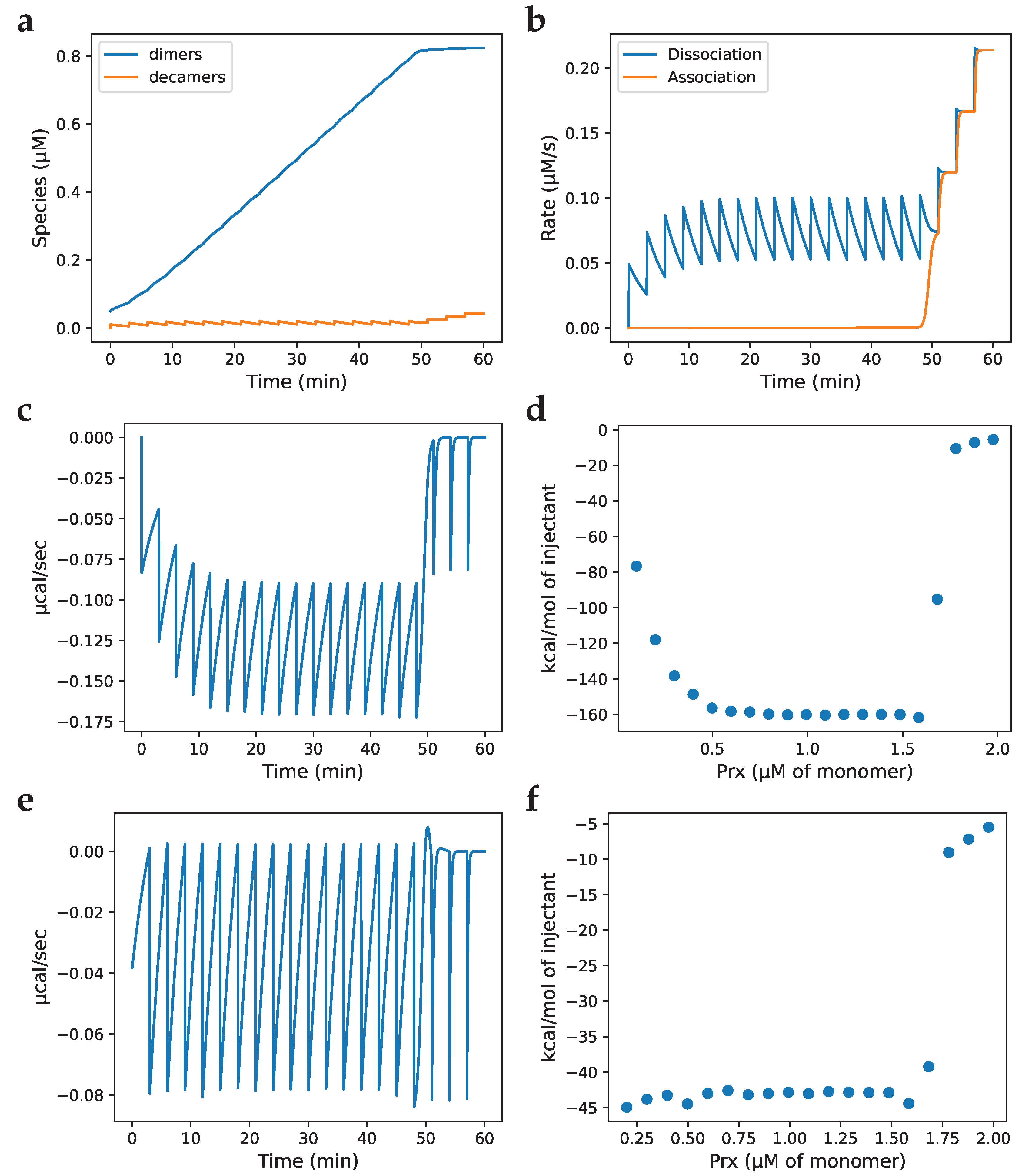
Figure 4.
Estimation of PRDX1 dimer-decamer association and dissociation rate constants by fitting the ITC simulation model to experimental data [7], which were digitized digitized and processed as described in the text. The model injection interval (200 s) and total Prx in simulated injections were as per the original experiment. The for decamer dissociation was estimated by fitting the model to these data; the solid line shows simulation results with the best-fit parameters. (a) (—) Model simulation with best-fit and and values (see text), (- -) digitized ITC data; (b) close-up view of a single ITC injection.
Figure 4.
Estimation of PRDX1 dimer-decamer association and dissociation rate constants by fitting the ITC simulation model to experimental data [7], which were digitized digitized and processed as described in the text. The model injection interval (200 s) and total Prx in simulated injections were as per the original experiment. The for decamer dissociation was estimated by fitting the model to these data; the solid line shows simulation results with the best-fit parameters. (a) (—) Model simulation with best-fit and and values (see text), (- -) digitized ITC data; (b) close-up view of a single ITC injection.
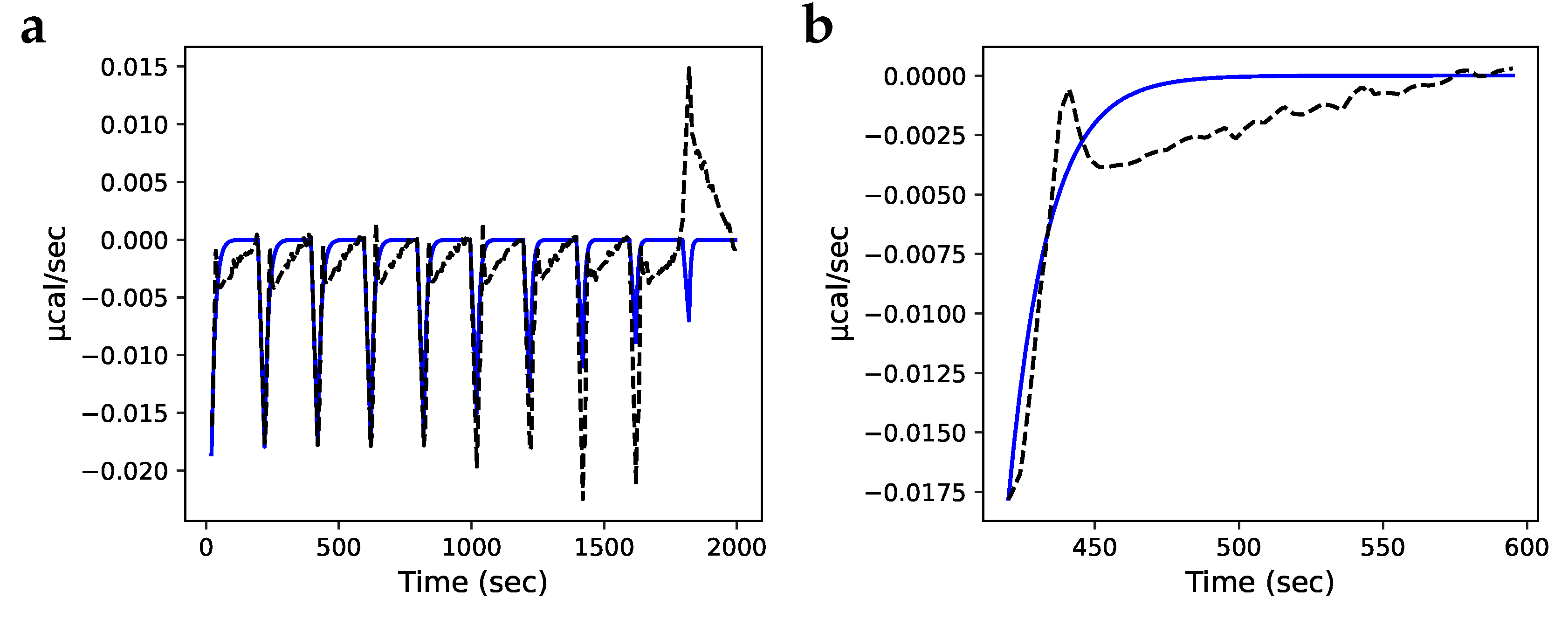
Figure 5.
Scheme for scaling of rate constants by the amount of degeneracy introduced from symmetry. (a) Degeneracy introduced by mirror symmetry within Prx dimers. Oxidation of one site in a reduced Prx dimer results in one of two products that are related by mirror symmetry. Therefore, the reactions can be collated into one reaction and the rate constant multiplied by a statistical factor of 2. This logic can be applied to the other reactions of Prx dimers. (b) Degeneracy introduced by rotational symmetry of Prx decamers. Taking mirror symmetry into account, oxidation of one site of a reduced Prx decamer results in 1 of 5 products that are related by rotational symmetry. Therefore, the reactions can be collated into one reaction with a statistical factor of 10. The same logic of symmetry can be applied to many of the Prx decamer species with other oxidation states, albeit resulting in less degeneracy.
Figure 5.
Scheme for scaling of rate constants by the amount of degeneracy introduced from symmetry. (a) Degeneracy introduced by mirror symmetry within Prx dimers. Oxidation of one site in a reduced Prx dimer results in one of two products that are related by mirror symmetry. Therefore, the reactions can be collated into one reaction and the rate constant multiplied by a statistical factor of 2. This logic can be applied to the other reactions of Prx dimers. (b) Degeneracy introduced by rotational symmetry of Prx decamers. Taking mirror symmetry into account, oxidation of one site of a reduced Prx decamer results in 1 of 5 products that are related by rotational symmetry. Therefore, the reactions can be collated into one reaction with a statistical factor of 10. The same logic of symmetry can be applied to many of the Prx decamer species with other oxidation states, albeit resulting in less degeneracy.
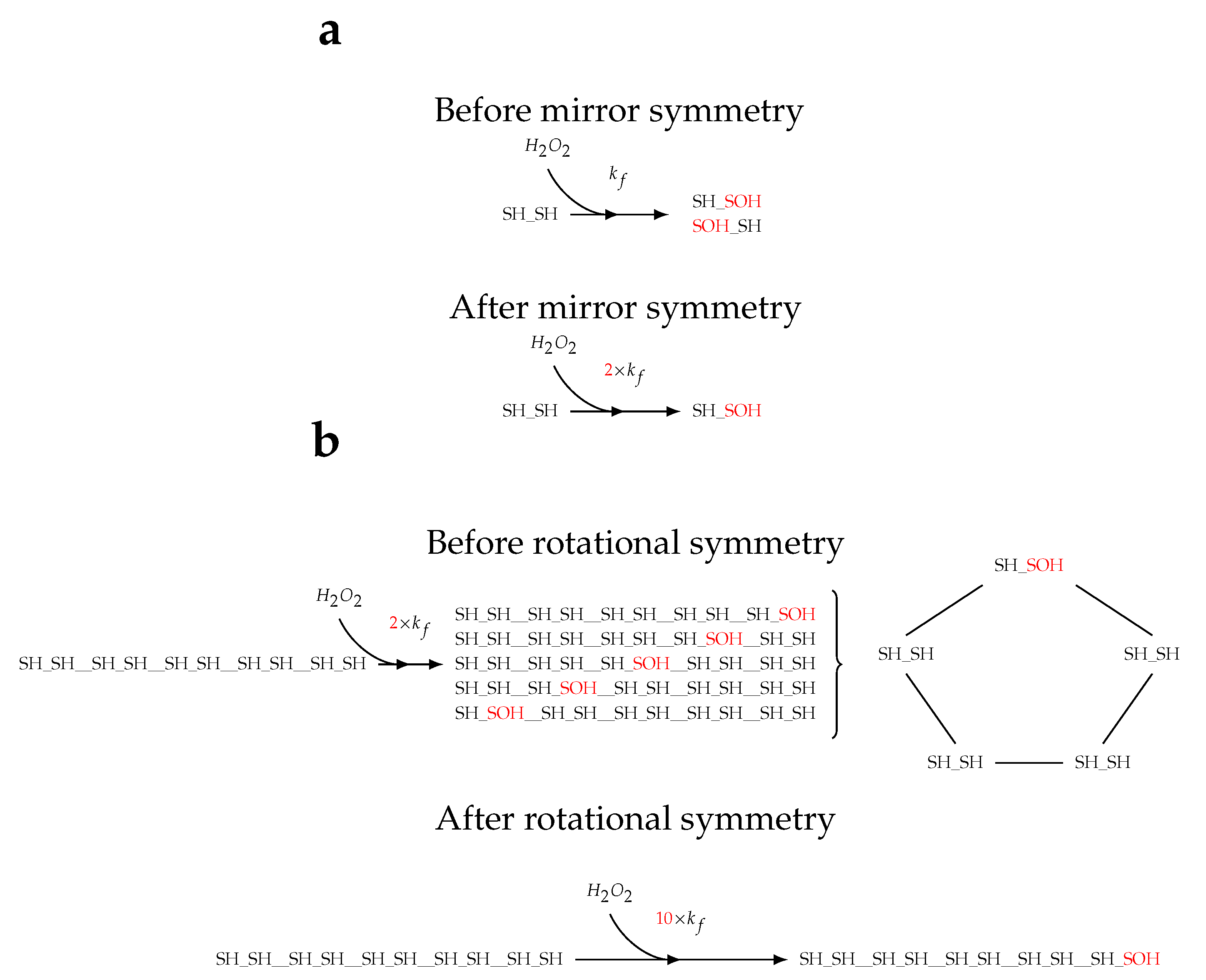
Figure 6.
Incorporating decamerisation solves the cryptic inhibition effect in a RBC PRXD2 model. (a) Steady-state and (b) time-based simulations of a RBC PRDX2 model based on Model A in [14] with decamerisation added. (c), (e) Steady-state and (d), (f) time-based simulations of the original RBC PRDX2 models by Benfeitas et al. [14]: (c) and (d) model A, (e) and (f) model B. (- -) Fraction of reduced and sulfenilated Prx sites; (—) fraction of Prx dimers with a single disulphide-bridge; ( ) fraction of Prx dimers with two disulphide-bridges; (—) fraction of Prx dimers with two sulfinic sites; (- -) fraction of Prx dimers with a disulphide-bridge and a sulfinic site.
) fraction of Prx dimers with two disulphide-bridges; (—) fraction of Prx dimers with two sulfinic sites; (- -) fraction of Prx dimers with a disulphide-bridge and a sulfinic site.
) fraction of Prx dimers with two disulphide-bridges; (—) fraction of Prx dimers with two sulfinic sites; (- -) fraction of Prx dimers with a disulphide-bridge and a sulfinic site.
Figure 6.
Incorporating decamerisation solves the cryptic inhibition effect in a RBC PRXD2 model. (a) Steady-state and (b) time-based simulations of a RBC PRDX2 model based on Model A in [14] with decamerisation added. (c), (e) Steady-state and (d), (f) time-based simulations of the original RBC PRDX2 models by Benfeitas et al. [14]: (c) and (d) model A, (e) and (f) model B. (- -) Fraction of reduced and sulfenilated Prx sites; (—) fraction of Prx dimers with a single disulphide-bridge; () fraction of Prx dimers with two disulphide-bridges; (—) fraction of Prx dimers with two sulfinic sites; (- -) fraction of Prx dimers with a disulphide-bridge and a sulfinic site.
) fraction of Prx dimers with two disulphide-bridges; (—) fraction of Prx dimers with two sulfinic sites; (- -) fraction of Prx dimers with a disulphide-bridge and a sulfinic site.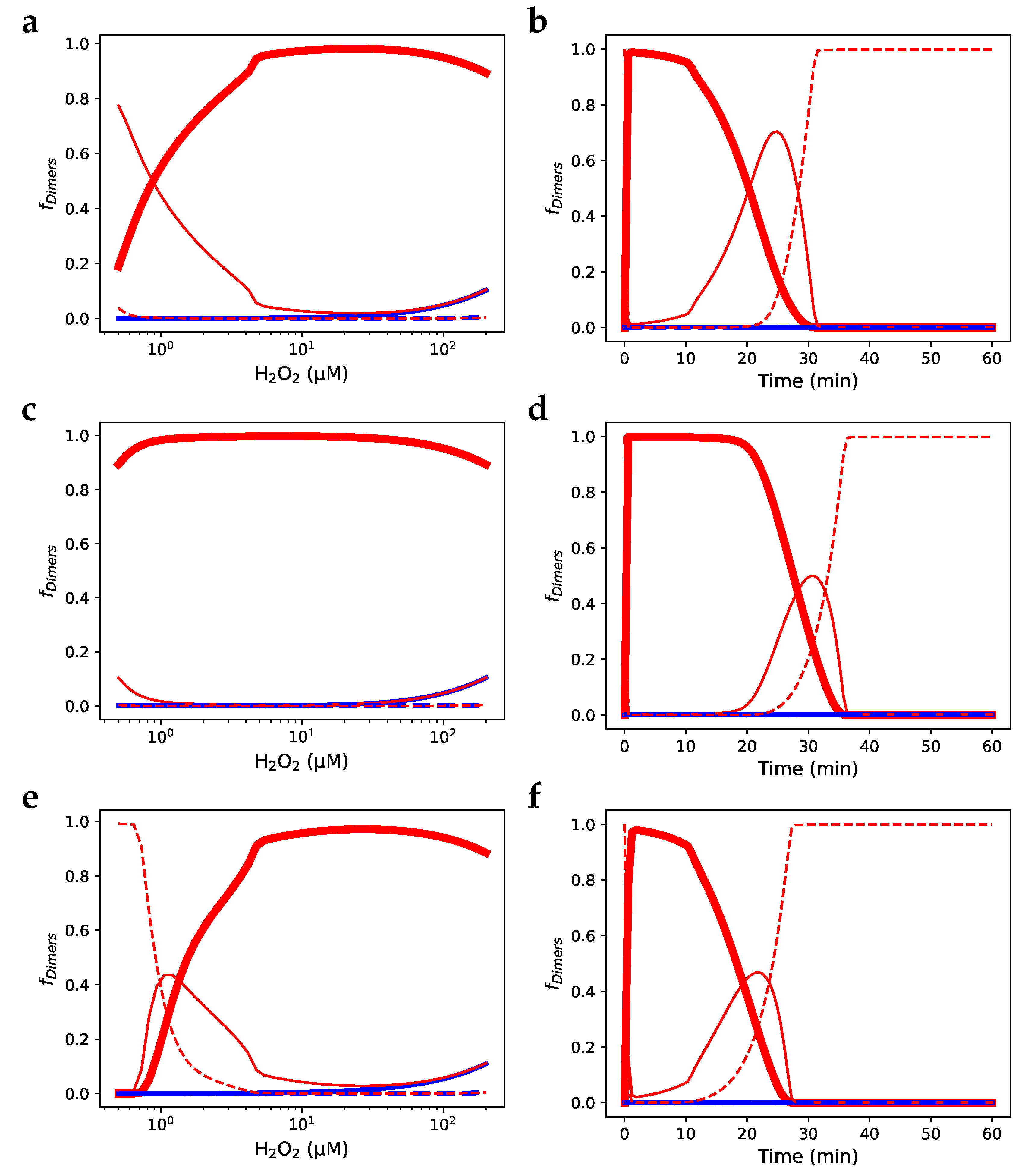
Figure 7.
Incorporating Prx dimer-decamer topology creates an inhibition-like effect during simulated in vitro activity assays. Simulations of peroxidase assays with () the full-activity dimer-only and (□) the mixed-activity dimer-decamer models of Prx. (a) Simulation of a horse radish peroxidase (HRP) competitive assay with parameters to replicate the traces from Fig. 2 in Manta et al. [11] and (b) the associated determination of the Prx rate constant by fractional inhibition analysis. Solid lines, simulations with the full-activity dimer-only model. Dashed lines, simulations with the mixed-activity dimer-decamer model. (c) Simulation of a NADPH reduction assay with the Prx system including Trx and TRR, and (d) the associated Prx rate constant determination by initial rate kinetics.
Figure 7.
Incorporating Prx dimer-decamer topology creates an inhibition-like effect during simulated in vitro activity assays. Simulations of peroxidase assays with () the full-activity dimer-only and (□) the mixed-activity dimer-decamer models of Prx. (a) Simulation of a horse radish peroxidase (HRP) competitive assay with parameters to replicate the traces from Fig. 2 in Manta et al. [11] and (b) the associated determination of the Prx rate constant by fractional inhibition analysis. Solid lines, simulations with the full-activity dimer-only model. Dashed lines, simulations with the mixed-activity dimer-decamer model. (c) Simulation of a NADPH reduction assay with the Prx system including Trx and TRR, and (d) the associated Prx rate constant determination by initial rate kinetics.
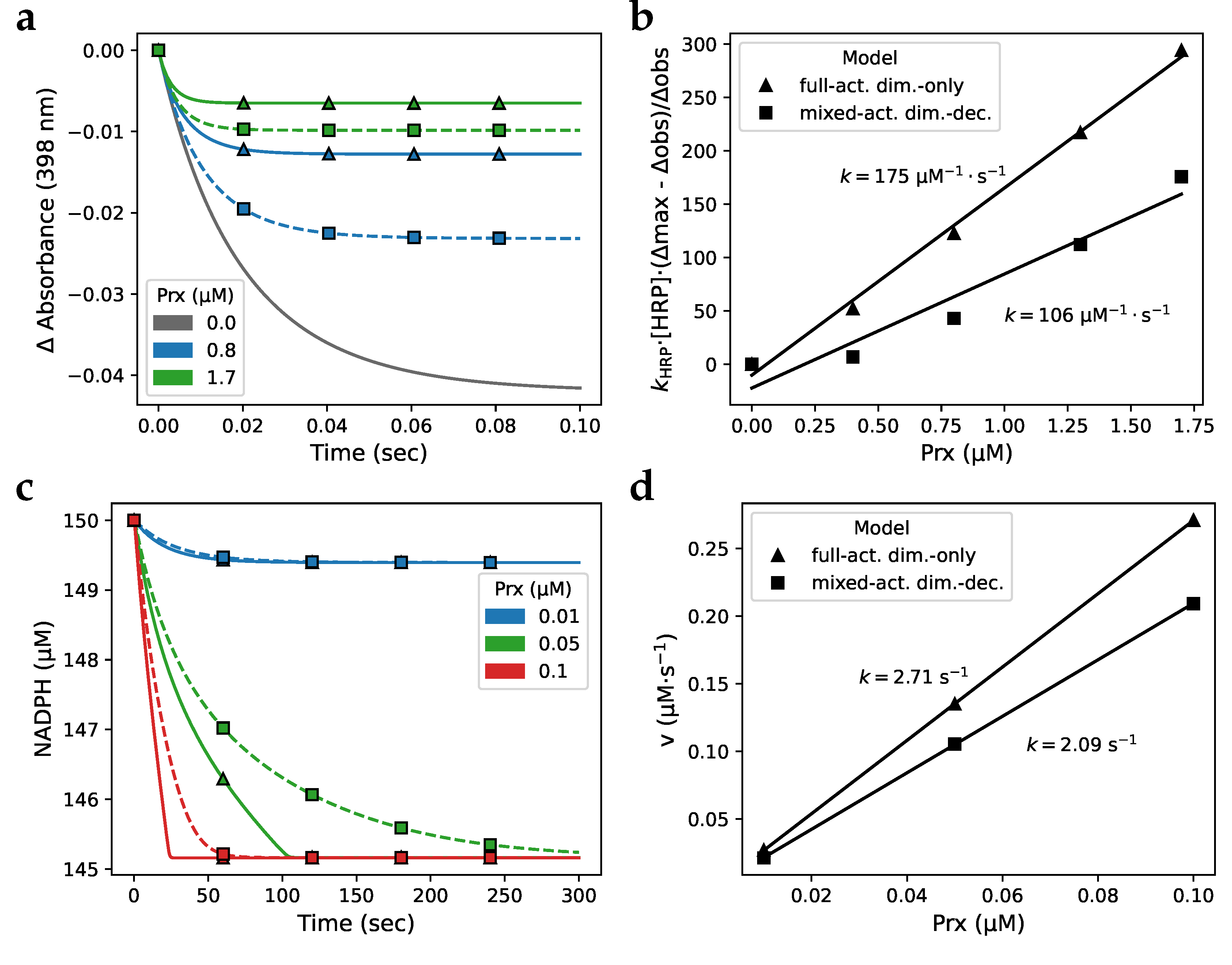
Figure 8.
The time for a Prx solution to re-equilibrate after dilution is relevant to fast kinetic experiments. Simulation of (a) 10× and (b) 50× dilution of Prx at equilibrium, starting at various initial concentrations. (c) Simulations of an HRP competition assay with the mixed-activity dimer-decamer model with (□) assay substrates pre-equilibrated and (∘) Prx diluted 10× at , and (d) the associated determination of the Prx rate constant by fractional inhibition analysis.
Figure 8.
The time for a Prx solution to re-equilibrate after dilution is relevant to fast kinetic experiments. Simulation of (a) 10× and (b) 50× dilution of Prx at equilibrium, starting at various initial concentrations. (c) Simulations of an HRP competition assay with the mixed-activity dimer-decamer model with (□) assay substrates pre-equilibrated and (∘) Prx diluted 10× at , and (d) the associated determination of the Prx rate constant by fractional inhibition analysis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



