
Submitted:
21 June 2023
Posted:
27 June 2023
You are already at the latest version
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a fatal disease due to early metastatic spread, late diagnosis and the lack of efficient therapies. A major driver of cancer progression and hurdle to successful treatment is transforming growth factor (TGF)-β. Recent data from pancreatic cancer mouse models have shown that transcriptionally active p73 (TAp73), a p53 family member, inhibits tumor progression through promoting tumor suppressive canonical TGF-β/Smad signaling, while preventing non-canonical TGF-β signaling through extracellular signal-regulated kinases (ERK)1/2. Here, we have studied whether this mechanism also operates in human PDAC. Using the PDAC-derived tumor cell lines PANC-1 and HPAFII, we show that TAp73 induces the expression of the epithelial marker and invasion suppressor E-cadherin and the common-mediator Smad, SMAD4, while at the same time suppressing expression of the EMT master regulator SNAIL and basal and TGF-β1-induced activation of ERK1 and ERK2. Using dominant-negative and RNA interference-based inhibition of SMAD4 function we went on to show that inhibition of ERK activation by TAp73 is mediated through SMAD4. Intriguingly, both SMAD4 and the α isoform of TAp73 - but not the isoform - interfered with cell migration as shown by xCELLigence technology. Our findings highlight the role of TAp73-SMAD4 signaling in tumor suppression of human PDAC and identify direct inhibition of basal and TGF-β-stimulated pro-invasive ERK activation as an underlying mechanism.
Keywords:
TAp73
; PDAC
; SMAD4
; transforming growth factor-β
; epithelial-mesenchymal transition
; cell migration
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC), the most common form of pancreatic cancer, has a very poor prognosis due to early metastatic spread, late diagnosis and the lack of efficient therapies [1,2,3]. This type of cancer is predicted in 2030 to become the second leading cause of cancer-related deaths worldwide, which highlights the urgent need to better understand PDAC development and progression. This knowledge is crucial to identify vulnerabilities that can be targeted therapeutically to improve the patient’s fate and quality of life. Recent advances in PDAC biology have focused on the intratumor microenvironment, which comprises up to 90% of the tumor mass [4,5]. The tumor stroma is mainly composed of cancer-associated fibroblasts, immune cells and excess extracellular matrix (ECM) [5], a phenomenon termed desmoplastic reaction. There is growing interest in targeting this non-malignant but nevertheless transformed compartment and its consequent impact on tumor development [4,6] in order to reduce tumor aggressiveness. The formation and composition of the ECM is orchestrated primarily by transforming growth factor-β (TGF-β) along with specific ECM components, the expression of which is induced by this growth factor. In PDAC, TGF-β can act as both a tumor suppressor and tumor promoter depending on the cellular context and the stage of disease progression [8]. TGF-β signals primarily via the Sma and Mad-related (Smad) proteins SMAD2, 3 and 4 [9] but also through Smad independent pathways, i.e., PI3K/AKT, JNK/p38 or the extracellular signal-regulated kinase (ERK) pathway [9,10,11,12]. Alterations in the TGF-β/SMAD4 signaling pathway, particularly in DPC4 (the gene encoding SMAD4) are critical events in PDAC progression [4].
Recently, it was revealed in two relevant and specific engineered pancreatic cancer mouse models made homozygous-null for transcriptionally active p73 (TAp73) that this p53 family member is a critical regulator of the TGF-β pathway. Mechanistically, TAp73 induces biglycan, a small proteoglycan and TGF-β inhibitor, via intermittent activation of Smad signaling [13]. The absence of TAp73, and as a consequence loss of expression of Smad4 and biglycan, led to activation of TGF-β signaling through Smad independent pathway(s), i.e., ERK1/2, favoring oncogenic TGF-β effects and epithelial-mesenchymal transition (EMT) in the tumor cells of TAp73−/− mice. The enhanced EMT phenotype of TAp73−/− as compared to TAp73 wildtype (WT) cells was associated with greater invasive abilities and a reduced sensitivity to gemcitabine treatment. The higher aggressiveness of PDAC from TAp73−/− mice was most convincingly demonstrated by an increase of stromal compartment with enhanced deposition of ECM, and reduced survival of the TAp73−/− mice [13]. These findings in the murine PDAC models suggest that TAp73 functions as a potent barrier to PDAC progression and implicates deletion of the TP73 locus in PDAC initiation or progression. Although in human cancers mutations in TP73 are less frequent than those in TP53, genetic aberrations of TP73 were nevertheless reported in PDAC and previous studies had already shown that loss of TAp73 induced spontaneous tumor development because of enhanced genomic instability [14]. In human pancreatic cancer cells, TP73 monoallelic expression was also observed [15] and correlated with patient outcome [13]. With respect to functional activities TAp73 has so far been implicated in the regulation of cell growth/death, neoangiogenesis and cellular metabolism/energy production [13,16,17].
In the PDAC mouse model carrying a loss of TP73, Thakur and colleagues have shown that TAp73 functions as an inhibitor of the EMT process and potent barrier to PDAC progression through modulation of TGF-β signaling. In their study, the authors focused on the murine system and on TAp73-dependent regulation of the TGF-β pathway via intermittent secretion of biglycan [13]. However, the role of SMAD4 as a downstream target of TAp73 and possible upstream repressor of the ERK pathway has not been functionally dissected. Hence it remains open whether the cellular effects of TAp73 in murine cells, particularly the induction of DPC4 and the impact of SMAD4 on TGF-β-induced ERK activation, also operate in human PDAC. The goal of this study, therefore, was to reveal if the regulatory interactions between TAp73 and SMAD4 dependent and independent signaling are of biological significance in human pancreatic cancer cells and whether these affect cancer relevant functions such as spontaneous and TGF-β1 dependent cell migration. Based on the data presented here, we conclude that TAp73 and SMAD4 signaling independently block basal and TGF-β1-induced ERK activation and cell migration in human PDAC-derived tumor cells.
2. Materials and Methods
2.1. Reagents
The following primary antibodies were used: anti-p73 Rabbit mAB (D3G10, #14620), anti-phospho-ERK1/2 (#4370), anti-GAPDH (14C10, #2118), and anti-Snail (#3895), Cell Signaling Technology (Frankfurt am Main, Germany); anti-HSP90 (F-8, #sc-13119), Santa Cruz Biotechnology (Heidelberg, Germany). HRP-linked anti-rabbit (#7074), and anti-mouse (#7076) secondary antibodies were from Cell Signaling Technology. Recombinant human TGF-β1 (#300-023) was provided by ReliaTech (Wolfenbüttel, Germany). An siRNA to TAp73 (5’-cgg auu cca gca ugg acg uTT-3’ and 5’-acg ucc aug cug gaa ucc gTT-3’) and a scrambled control siRNA were commercially synthesized by Metabion Int. AG (Planegg, Germany). An siRNA to SMAD4 (#1027415) was purchased from Qiagen (Hilden, Germany). The TAp73α and TAp73β vectors were a kind gift from Drs. Bertrand Joseph and Pinelopi Engskog Vlachos (Stockholm, Sweden).
2.2. Cells
PANC-1 human PDAC cells were obtained from the ATCC (Manassas, VA), while HPAFII cells were a kind gift of Dr. U. F. Wellner. Both cell lines were maintained in RPMI 1640 supplemented with fetal bovine serum (FBS), 1% Penicillin-Streptomycin-Glutamine (Life Technologies, Darmstadt, Germany) and 1% sodium pyruvate (Merck Millipore). PANC-1 cells stably expressing a Flag-tagged mutant version of SMAD4 (DPC4-1-514) were described and characterized in detail earlier [18,19].
2.3. Transfection of SiRNA and Reporter Gene Assays
On d 1, PANC-1 or HPAFII cells were seeded into NunclonTM Delta Surface plates (Nunc, Roskilde, Denmark), and transfected twice, on d 2 and 3, serum-free with 50 nM of prevalidated small interfering RNAs (siRNAs) specific for p73 or SMAD4, or scrambled siRNAs as control for 4 h using Lipofectamine 2000 (PANC-1) or RNAiMAX (HPAFII) (both from Life Technologies/Thermo Fisher Scientific) according to the manufacturer‘s instructions. Transfected cells were subjected to immunoblot analyses, reporter gene assays, or cell migration assays. For reporter gene assays, PANC-1 cells were seeded in 96-well plates on d 1 and co-transfected on d 2 serum-free with various siRNAs and Lipofectamine 2000. Twenty-four h later (d 3) cells again received Lipofectamine 2000 with the same siRNAs together with p(CAGA)12 MLP-Luc and the Renilla luciferase encoding vector pRL-TK-Luc. On d 4, cells were treated with TGF-β1 for 24 h and luciferase activities were determined with the Dual Luciferase Assay System (Promega, Mannheim, Germany). In all reporter gene assays, the data were derived from 6 wells processed in parallel and normalized with Renilla luciferase activity as described in detail elsewhere [7].
2.4. RT-PCR Analysis
Following transfection, PANC-1 and HPAFII cells were lysed in RL buffer and total RNA purified by affinity chromatography (Qiagen). For each sample, 2.5 μg RNA were subjected to reverse transcription for 1 h at 37 °C using 200 U M-MLV Reverse Transcriptase and 2.5 μM random hexamers (Thermo Fisher Scientific) in a total volume of 20 μl. Relative mRNA expression of target genes was quantified by quantitative RT-PCR on an I-Cycler (BioRad, Munich, Germany) using Maxima SYBR Green Mastermix (Thermo Fisher Scientific). Data were normalized to the expression of the gene for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). All PCR primer sequences have been given in previous publications [7,18,20].
2.5. Immunoblotting
Cell lysis and immunoblotting was essentially performed as described previously [7,18]. In brief, cells were washed once with ice-cold PBS and lysed with 1 x PhosphoSafe lysis buffer (Merck Millipore). Following clearance of the lysates by centrifugation, their total protein concentrations were determined with the DC Protein Assay (BioRad). Equal amounts of proteins were fractionated by polyacrylamide gel electrophoresis on mini-PROTEAN TGX any-kD precast gels (BioRad) and blotted to PVDF membranes. Membranes were blocked with nonfat dry milk or bovine serum albumin and incubated with primary antibodies either for 2 h at RT or overnight at 4°C. After washing and incubation with HRP-linked secondary antibodies, chemoluminescent detection of proteins was done on a ChemiDoc XRS imaging system (BioRad) with Amersham ECL Prime Detection Reagent (GE Healthcare). The signals for the proteins of interest were normalized to those for GAPDH or HSP90.
2.6. Real-Time Cell Migration Assays
We employed the xCELLigence® DP system (ACEA Biosciences, San Diego, USA) to measure random/spontaneous cell migration in a chemokinesis setup according to previous descriptions [18,20]. To enhance the adhesion of cells to the lower side of transwell membrane equipped with the gold electrodes, it was coated with a 1:1 mixture of collagens I and IV. Following assembly of the CIM plate-16 and a 1-h equilibration in an incubator each well of the CIM plates-16 received 60,000-80,000 cells in standard growth medium supplemented with 1% (rather than 10%) FBS to minimize proliferation. Data acquisition was done at intervals of 15 min and the assays were run for various lengths of time and analyzed with RTCA software (version 1.2, ACEA Biosciences).
2.7. Statistical Analysis
Statistical significance was calculated using the unpaired two-tailed Student’s t test or the Wilcoxon test. Results were considered significant at p < 0.05 and denoted in the graphs by an asterisk (*) or a rhombus (♦).
3. Results
3.1. TAp73 Up-Regulates ECAD and SMAD4 in in Human PDAC Cells
Thakur and colleagues revealed in murine cells that TAp73 inhibits EMT by inducing the expression of CDH1 and other epithelial genes, while suppressing that of mesenchymal genes as well as EMT-associated functions like cell migration [13]. We therefore set out to study the impact of TAp73 on EMT marker expression in human cells employing primarily the human PDAC cell lines, PANC-1 and HPAFII.
We first transfected PANC-1 cells with a p73-specific siRNA and monitored the transfectants by immunoblotting for successful downregulation of TAp73α protein levels (Figure S1). We detected only one band of low intensity that displayed the same electrophoretic mobility as TAp73α ectopically expressed in PANC-1 cells and run side-by-side as a control (Figure S1). The absence of a second band of higher mobility strongly suggested that TAp73α is the only - or at least the most abundant - p73 isoform expressed in these cells. The low endogenous protein levels of TAp73α in PANC-1 cells might be due to low stability of the WT TAp73 protein similar to WT p53 protein. Indeed, an increase in the half-life of the p73 protein and its subsequent accumulation has been observed in HCT116 cells upon treatment with cisplatin [21]. Next, we analysed the p73 siRNA-transfected PANC-1 cells by qPCR analysis for expression of epithelial marker genes. Interestingly, knockdown of p73 reduced the mRNA abundance of ECAD, Grainyhead-like 2 (GRHL2), an epithelial-specific transcription factor, and SMAD4 (Figure 1A). Regulation of ECAD and SMAD4 by p73 was also evident at the protein level (Figure 1B). Similar results with respect to ECAD and SMAD4 regulation by TAp73 were obtained for HPAFII cells (Figure S2).
The p73 siRNA employed here likely inhibits more than one isoform derived from TP73, i.e., not only TAp73α but also TAp73β. To obtain information as to whether both isoforms differ in their ability to stimulate expression of CDH1, GRHL2 and DPC4, we transfected PANC-1 cells with expression vectors for TAp73α or TAp73β and initially monitored the transfectants by immunoblotting for successful ectopic expression of the transgenes. Both isoforms were expressed with TAp73β exhibiting a higher electrophoretic mobility than TAp73 α due to lack of the SAM domain [22] (Figure 1C). In qPCR analysis of the transfectants we found increased mRNA levels for ECAD, GRHL2 and SMAD4 in response to transfection of TAp73α, while that of TAp73β did not exhibit a statistically significant effect on either gene (Figure 1D). We have, therefore, focused here on TAp73α since expression of this isoform is the major outcome of TP73 gene expression and the relevance and contribution of the other isoforms remain unclear [22].
3.2. Knockdown of TAp73 Interfered with TGF-β1-induced Luciferase Activity on a SMAD-Responsive Promoter and Regulation of TGF-β/SMAD Target Genes in Human PDAC Cells
Above we have shown that TAp73 sustains the protein levels of SMAD4, suggesting that TAp73 may impact on Smad-mediated transcriptional activation of TGF-β1 target genes. To demonstrate this more directly, PANC-1 cells silenced for TAp73 were transiently transfected with a TGF-β/Smad-responsive reporter gene and monitored for their sensitivity to TGF-β1 stimulation. We employed here the p(CAGA)12 MLP-luc plasmid rather than p3TPlux used by Thakur and colleagues [13] since unlike p3TPlux, which is also sensitive to TGF-β dependent non-Smad-mediated transcriptional activation [21], p(CAGA)12 MLP-luc carries twelve Smad binding elements (SBEs) in tandem and hence only responds to SBE binding of SMAD3/4 [23]. With this reporter TGF-β1-induced firefly luciferase activity was dramatically reduced in cells that received either p73 siRNA, or SMAD4 siRNA as positive control, compared to scrambled siRNA-transfected cells (Figure 2A). This suggests that in pancreatic tumor cells of human origin, too, TAp73 promotes TGF-β signaling through activation of a SMAD4 dependent pathway.
The failure pf TGF-β1 to induce luciferase activity in a TAp73 deficient context suggested that TAp73 promotes TGF-β signaling through activation of Smad signaling. If so, this should also impact the response of TGF-β target genes in human cells that are regulated in a SMAD dependent manner such as CDH1, TGFB1 and SNAI1. To this end, in TAp73-silenced PANC-1 cells the inhibitory effect of TGF-β1 treatment on CDH1 was reduced from 80% to 30% after 24 h of treatment (Figure 2B), suggesting that TAp73 facilitated its TGF-β1-induced down-regulation. The stimulatory effect of TGF-β1 on TGFB1 (a gene partially dependent on SMADs and ERK) dropped from 27-fold to 10-fold (Figure 2B). Moreover, basal protein levels of ECAD or SNAIL, a transcriptional regulator of CDH1, were decreased or increased, respectively, in TAp73-silenced PANC-1 cells (consistent with the anti-EMT function of TAp73 and previously observed also in TAp73 deficient PDAC cells from mice [13]). However, down- or upregulation of the ECAD or SNAIL proteins, respectively, by exogenous TGF-β1 in the TAp73-silenced cells was alleviated (Figure 2C, lanes 3 and 4). We conclude from these data that in PDAC cells TAp73 is required for a full-blown response of SMAD dependent TGF-β target genes to TGF-β1 treatment, most likely by its ability to promote SMAD4 expression.
3.3. TAp73 Inhibits Basal and TGF-β1-induced ERK Activation
Thakur and coworkers have shown that TAp73 knockout in mice caused downregulation of Smad4 in vivo as demonstrated by immunohistochemistry in PDAC tissues and in cell lines derived from TAp73 deficient mice compared with control mice [13], and that non-Smad, i.e., ERK signaling, is enhanced [13]. We therefore asked whether in human PDAC cells, too, TAp73 suppresses ERK activation. Knockdown of p73 in PANC-1 cells by siRNA enhanced both the basal (Figure 3A, left-hand graph) and TGF-β1-induced levels (Figure 3A, right-hand graph) of phospho-ERK1/2 (pERK1/2). The inducive effect of TGF-β1 was maximal at 0.25 h and subsequently declined when monitored over a period of 2 h, and was generally greater for ERK2 than for ERK1. Very similar effects were observed in HPAFII cells (Figure 3B). Our results therefore suggest that ERK activation is suppressed by TAp73 in pancreatic cancer cells.
3.4. The Inhibitory Effect of TAp73 on ERK Activation is Mediated via SMAD4
Given the induction of SMAD4 by TAp73 (Figure 1B), the question arose as to whether TAp73-mediated inhibition of ERK activation is mediated by SMAD4, or in other words, whether SMAD4 can mimic the effects of TAp73 on ERK activation. To explore this in more detail, we employed previously characterized PANC-1 cells, in which WT SMAD4 function is inhibited by a stable ectopic expression of C-terminally truncated SMAD4 protein, SMAD4-(1-514), that acts in a dominant-negative fashion [18,19]. When these cells were monitored for the activation status of ERK1/2, we noted an increase in the levels of pERK1/2 under basal conditions (Figure 4A) and after challenge with exogenous TGF-β1, peaking at 0.25 h of TGF-β1 stimulation (Figure 4B). To confirm this observation, we knocked down SMAD4 in PANC-1 (Figure 4C) and HPAFII (Figure 4D) cells by siRNA followed by ERK phosphoimmunoblotting. The results obtained were consistent with those of the dominant-negative inhibition approach, namely that pERK1/2 activation was enhanced in p73 knockdown cells with the greatest abundance of pERK1 and 2 seen at the 0.25 h time point. Taken together, these data show that SMAD4 inhibition phenocopied the stimulatory effect of the p73 knockdown on ERK1/2 activation.
3.5. TAp73α and SMAD4 Mediate Inhibition of Cell Migration in Human PDAC Cells
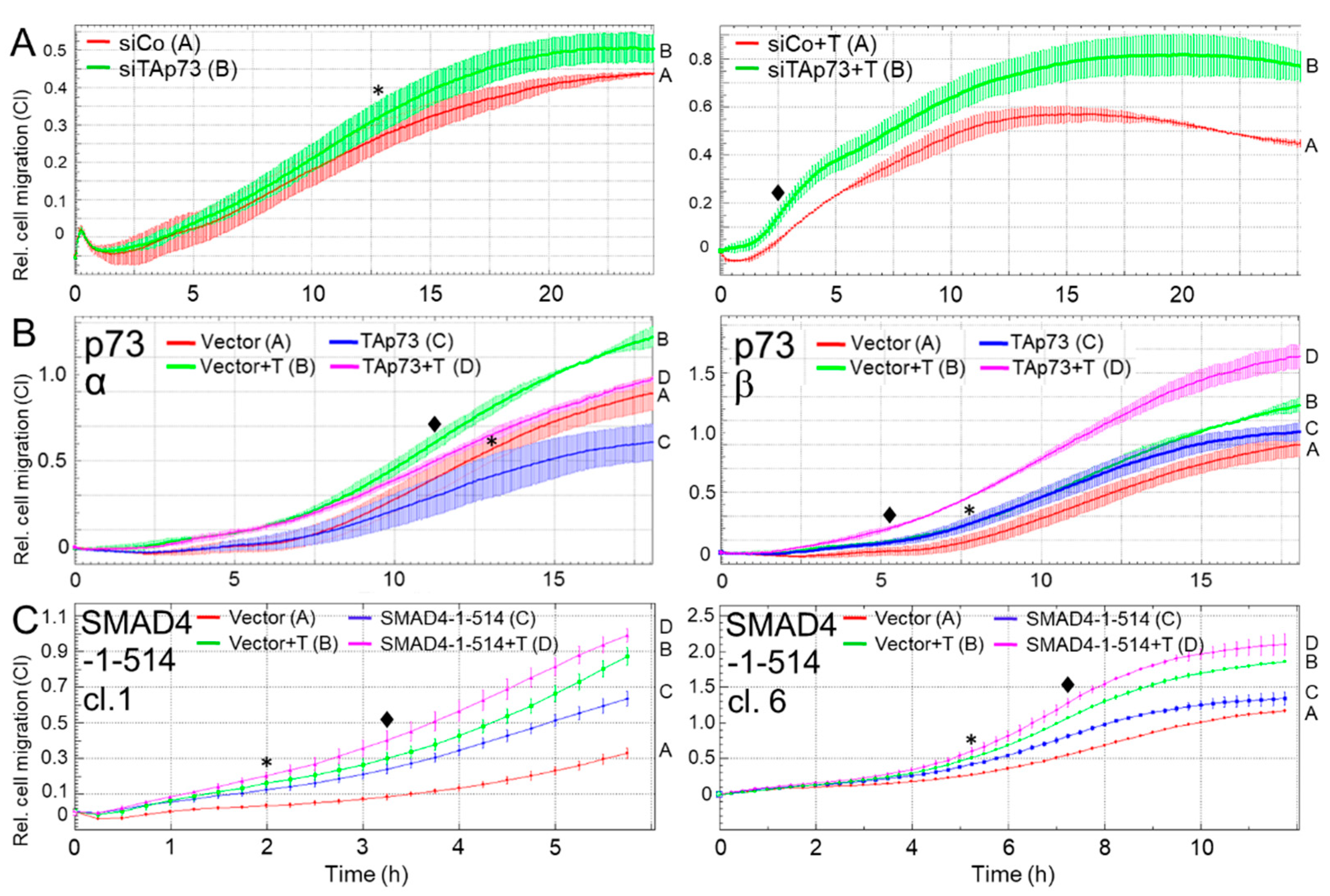
Thakur and colleagues [13] observed that TAp73 deficient murine tumor cells harbor an EMT phenotype associated with enhanced invasive properties. Above, we have shown that TAp73 promoted expression of ECAD, an epithelial marker and a well-known invasion inhibitor, but inhibited that of SNAIL, a mesenchymal marker and driver of invasion. To reveal whether enhanced migration or invasion is also a consequence of silencing TAp73 in human PDAC cells, we transfected PANC-1 or HPAFII cells with the TAp73-specific siRNA and subjected them to real-time measurement of random cell migration on an xCELLigence platform. Of note, knockdown of TAp73 in PANC-1 cells resulted in a moderate promigratory effect under basal conditions (Figure 5A, left-hand graph) and a more pronounced one under TGF-β1 stimulation (Figure 5A, right-hand graph). A similar migration profile was seen with HPAFII cells silenced for p73 (Figure S3). Conversely, ectopic overexpression of TAp73α (verified by p73 immunoblotting, Figure 1C) inhibited both basal and TGF-β1-induced migration in PANC-1 cells (Figure 5B, left-hand graph), while that of TAp73β (lacking the SAM domain) had the opposite effect (Figure 5B, right-hand graph). We therefore conclude that TAp73α, but not TAp73β, inhibits cell migration in human PDAC-derived tumor cells.
Above, we have shown that TAp73 induces SMAD4 and that both TAp73 and SMAD4 inhibit basal and TGF-β1-induced ERK activation. This suggested the possibility that migration inhibition by TAp73, too, is mediated through SMAD4. To test this directly, we again utilized PANC-1 cells stably expressing dominant-negative SMAD4-(1-514) and subjected these cells to xCELLigence migration assay. Intriguingly, impairing WT SMAD4 function enhanced both basal and TGF-β1-induced migratory activity in two individual clones (Figure 5C), and thus mimicked the effect of the TAp73 knockdown on cell migration.
4. Discussion
Despite recent advances in chemotherapeutic treatments, the prognosis for PDAC is still poor and urgently requires a deeper understanding of the molecular events and critical signaling pathways that drive tumor development and evolution. Previous studies reported that the p53 homolog, TAp73, is involved in cancer development through cell growth and death regulatory mechanisms. However, the significance of its altered expression in various cancers, including PDAC, has not yet been clearly defined. Using endogenous mouse models of PDAC, a pioneering study by Thakur and coworkers investigated the role of TAp73 in pancreatic carcinogenesis and showed that TAp73 deficient PDAC exhibited characteristics of EMT and enhanced desmoplasia, suggesting enhanced activity of TGF-β [13]. Interestingly, the increased amount of free TGF-β, which is suspected to be associated with a higher risk of pancreatic cancer [24] and resistance to anticancer treatment [25] resulted from the inability to trigger activation of the Smad dependent pathway (primarily due to downregulation of Smad4) and to induce expression of the Smad4 target and TGF-β inhibitor biglycan. As a consequence of the increased levels of free TGF-β, the tumor cells can display high levels of Smad independent pathway activation, such as that involving ERK derepressed under conditions of TAp73 deficiency [13]. These favor the expression of EMT-related transcription factors such as Snail and Zeb and promote EMT, enhanced migratory capacity and invasiveness as well as resistance to chemotherapeutic agents in TAp73 deficient murine PDAC cells. The data from murine cells suggest that both cell-intrinsic and paracrine effects brought about by TAp73 deficiency force a switch in function of TGF-β in carcinogenesis from tumor suppressive to tumor promoting [13].
While plentiful data from mice have been presented in the Thakur study, any conclusions as to whether this TAp73 driven network also operates in human PDAC has remained open. A clue that TAp73 is involved in EMT regulation in human cells, however, came from the observation that knockdown of TAp73 induced MCF10A mammary epithelial cells to undergo EMT via downregulation of CDH1 and upregulation of SNAI1, and an increase in cell migration [26]. By suppressing EMT, TAp73 physiologically achieves maintenance of normal cell polarity in these cells [26]. This is consistent with the role of TAp73 in cellular differentiation and suppression of the mesenchymal phenotype [27,28,29].
In our study, we initially found evidence for a tumor suppressive role of TAp73 in PDAC cells of human origin by observing that TAp73 upregulated the basal expression of ECAD and SMAD4, while downregulating that of SNAIL. By employing luciferase assays with a strictly SMAD-responsive reporter plasmid we were, in addition, able to reveal that TAp73 promotes TGF-β signaling through activation of a SMAD4 dependent pathway. In line with this, TAp73 was required for TGF-β1 regulatory effects on TGF-β/SMAD dependent target genes, like CDH1 (downregulation) and TGFB1 (upregulation). Besides its role as a promoter of epithelial gene expression, TAp73 was identified in murine cells as an inhibitor of mesenchymal gene expression, i.e., Snail, and mesenchymal non-Smad signaling, i.e., Erk1/2 [13], two functions that we have demonstrated here to operate also in human PDAC cells. The coordinated induction of epithelial genes and concurrent suppression of mesenchymal genes and pathways suggests that TAp73 is also a crucial antagonist of EMT and cell motility in human PDAC cells.
Regarding Smad independent signaling we observed in both PANC-1 and HPAFII cells constitutive ERK pathway activation that was enhanced as a result of TAp73 silencing. Since we had shown previously that exogenous TGF-β1 was able to rapidly stimulate ERK1/2 activation in PANC-1 and other PDAC cell lines that have retained sensitivity to this growth factor [18], we asked whether another tumor suppressive function of TAp73 could be repression of TGF-β1-driven ERK activation. To this end, in TAp73 depleted cells short-term ERK activation was significantly enhanced. Moreover, using dominant-negative and RNA interference with WT SMAD4 function in two different PDAC lines, we were able to show that higher ERK activation was a direct consequence of SMAD4 inhibition.
Prompted by suppression of the EMT phenotype we reasoned that TAp73 should also interfere with cell migration/invasion. Therefore, in another set of experiments we applied the real-time xCELLigence technology to monitor the migratory potential of PANC-1 and HPAFII cells after siRNA-mediated knockdown of TAp73 or after ectopic expression of plasmids encoding either TAp73α (the most abundant p73 isoform [22]) or TAp73β. These assays showed strong de-repression of cell migration in the p73 knockdown cells and inhibition after ectopic expression of TAp73α but not TAp73β. Finally, to verify that the anti-migratory effect of TAp73 was due to induction of SMAD4 expression, we performed migration assays with PANC-1 cells harboring defective SMAD4 function as a result of ectopic expression of dominant-negatively acting SMAD4 mutant. In accordance with the p73 siRNA data, cells with defective SMAD4 function also exhibited an increased migratory activity. This is consistent with studies in other PDAC-derived tumor cell lines, in which SMAD4 acted as an inhibitor of migration or invasion [30,31] confirming the tumor suppressing function of SMAD4 in PDAC cells. The observation that both TAp73 and SMAD4 target ERK1/2 activation for inhibition are in perfect agreement with earlier findings from us [32] and others [12] on the crucial role of this pathway in driving EMT and migration/invasion in PDAC. Moreover, suppression of ERK activation by (endothelial) Smad4 has been demonstrated to restrain the transition to hematopoietic progenitors [33].
While the primary goal of this study was to demonstrate that the original findings on TAp73 in murine PDAC cells also operate in their human orthologues, we provided additional data that were not contained in the Thakur study. Specifically, we revealed that only the α but not the β isoform was able to stimulate ECAD and SMAD4 expression (Figure 1), and to inhibit cell migration, while the latter response was even promoted by the β isoform (Figure 5). TAp73β differs from TAp73α by the lack of the SAM domain, a potential protein–protein interaction domain that might contribute to the control of TAp73 transcriptional activity [22]. Mechanistically, TAp73 may act via transactivation of the DPC4 gene promoter, which harbors a p53 response element [13].
Figure 6.
Cartoon illustrating the role of TAp73 in negative regulation of TGF-β dependent EMT and cell migration (CM) in human PDAC cells. Left-hand side, in WT TAp73 epithelial tumor cells TAp73α induces SMAD4 expression and following stimulation of cells with TGF-β1 (blue ovals) and activation of its receptors (blue rectangles) SMAD4 inhibits the basal and TGF-β1-induced formation of pERK and an ERK-mediated increase in EMT and CM. Right-hand side, following siRNA-mediated knockdown (KD) of TAp73α the subsequent decrease in SMAD4 expression and a failure to activate a SMAD dependent (dep.) pathway, reinforced TGF-β signaling switching to SMAD4 independent (indep.) pathways, e.g., MEK-ERK1/2, by removing the inhibitory effect of SMAD4 on pERK1/2 formation. In the course of this study, we have also carved out that only the α isoform of TAp73 (TAp73α) is able to promote SMAD4 and ECAD expression and to inhibit CM. The green arrows indicate activation, while the red lines indicate suppression. Grey-shaded arrows and lines indicate the inactive state. The stippled lines and arrows denote the possibility that these effects are indirect. For details see text.
Figure 6.
Cartoon illustrating the role of TAp73 in negative regulation of TGF-β dependent EMT and cell migration (CM) in human PDAC cells. Left-hand side, in WT TAp73 epithelial tumor cells TAp73α induces SMAD4 expression and following stimulation of cells with TGF-β1 (blue ovals) and activation of its receptors (blue rectangles) SMAD4 inhibits the basal and TGF-β1-induced formation of pERK and an ERK-mediated increase in EMT and CM. Right-hand side, following siRNA-mediated knockdown (KD) of TAp73α the subsequent decrease in SMAD4 expression and a failure to activate a SMAD dependent (dep.) pathway, reinforced TGF-β signaling switching to SMAD4 independent (indep.) pathways, e.g., MEK-ERK1/2, by removing the inhibitory effect of SMAD4 on pERK1/2 formation. In the course of this study, we have also carved out that only the α isoform of TAp73 (TAp73α) is able to promote SMAD4 and ECAD expression and to inhibit CM. The green arrows indicate activation, while the red lines indicate suppression. Grey-shaded arrows and lines indicate the inactive state. The stippled lines and arrows denote the possibility that these effects are indirect. For details see text.
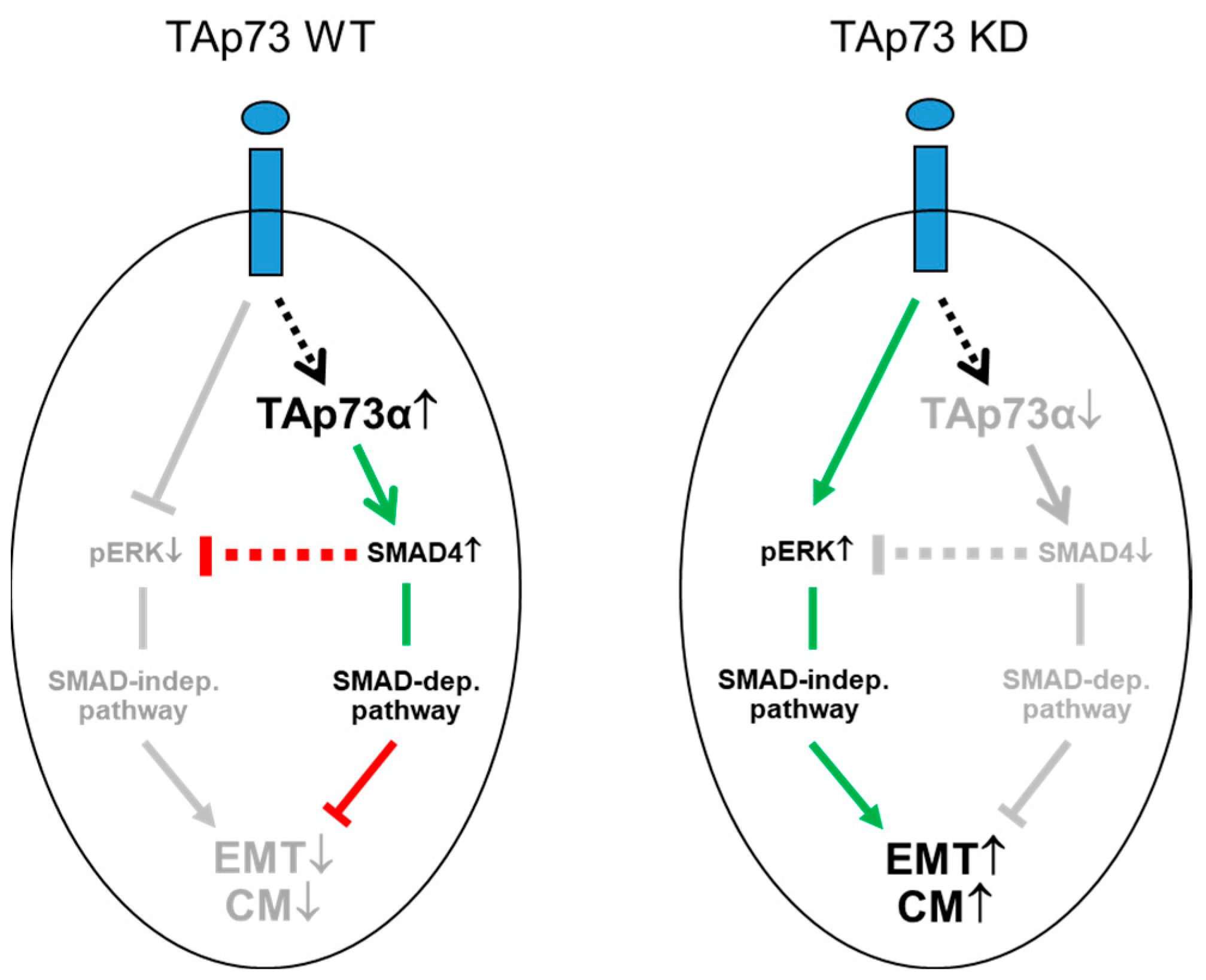
In the present study, we provide evidence that TAp73 through induction of DPC4 suppresses basal and TGF-β1 dependent activity of the MEK-ERK signaling pathway. According to the model proposed by Thakur et al., de-repression of ERK activation and an ensuing increase in EMT and migration/invasion - rather than being executed directly by SMAD4 - is believed to be an indirect effect, ultimately resulting from increased levels of free TGF-β due to the absence of TGF-β trapping by biglycan that, in turn, is induced via TAp73 and SMAD4 [7,13]. The issue of whether in human PDAC, too, Biglycan and endogenous TGF-β are involved here - although being highly relevant - was beyond the scope of the present study.
Altogether, our data on human PDAC clearly suggest that the absence of TAp73 impairs TGF-β signaling toward the tumor-suppressing SMAD4 dependent pathway. Hence, TAp73 in suppressing EMT and cell motility might have implications for other tumor suppressive functions, e.g., responsiveness to SMAD4 dependent cell death after TGF-β treatment [34]. It will be interesting to see if TAp73 deficiency can render PANC-1 or HPAFII less apoptosis-sensitive to TGF-β/SMAD4 dependent cell death. Moreover, in vivo data in mouse models will reveal if after intra-pancreatic injection of human TAp73 deficient cells (as observed for the murine counterparts [13]), the number of liver metastases that developed from these cells is higher than that with the TAp73 WT cells and if the loss of both tumor suppressive functions (anti-EMT/anti-invasion and apoptosis) contributes to the pro-metastatic effect.
5. Conclusions
Our findings, which highlight the complex role of TGF-β in pancreatic tumorigenesis, might have implications for therapeutic approaches targeting this growth factor for inhibition. Currently, several inhibitors of TGF-β receptors or ligands are being evaluated in clinical trials. For instance, galunisertib, the first small molecule TGF-β receptor inhibitor, plus gemcitabine resulted in the improvement of survival in patients with unresectable PDAC [35]. However, the lack of predictive biomarkers to identify patients likely to respond makes PDAC treatment with TGF-β inhibitors a challenging issue. Careful patient selection and timing of treatment with respect to activation of TGF-β signaling could help to improve this situation. Hence, measuring the levels of TAp73, SMAD4 and/or pERK could help to predict whether TGF-β preferentially uses an oncogenic or a tumor suppressive pathway in a given patient and at a specific time.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Verification of the RNA interference-mediated knockdown of TAp73; Figure S2: Effect of TAp73 knockdown on ECAD and SMAD4 expression in HPAFII cells. Figure S3: TAp73 inhibits cell migration in the human PDAC-derived cell line HPAFII.
Author Contributions
H.U. conceived and designed the experiments; H.U. and R.B. performed the experiments; H.U. analyzed the data; B.K. provided essential reagents; H.U. wrote the paper; B.K., U.F.W. H.L., and J.U.M. critically read and edited the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data reported are contained in the “Results” section and the supplementary material.
Acknowledgments
We are indebted to H. Albrecht for excellent technical assistance, and Bertrand Joseph and Pinelopi Engskog Vlachos (Stockholm, Sweden) for providing expression vectors for TAp73α and TAp73β, and Steven Dooley (Mannheim, Germany) for p(CAGA)12 MLP-luc.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kalthoff, H. How open is the therapeutic horizon for pancreatic cancer patients? Hepatobiliary Pancreat Dis Int 2022, 21, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int J Mol Sci 2017, 18, pii: E1338. [Google Scholar] [CrossRef]
- Chiang, K.C.; Yeh, C.N.; Ueng, S.H.; Hsu, J.T.; Yeh, T.S.; Jan, Y.Y.; Hwang, T.L.; Chen, M.F. Clinicodemographic aspect of resectable pancreatic cancer and prognostic factors for resectable cancer. World J Surg Oncol 2012, 10, 77–86. [Google Scholar] [CrossRef]
- Schneider, G.; Siveke, J.T.; Eckel, F.; Schmid, R.M. Pancreatic cancer: basic and clinical aspects. Gastroenterology 2005, 128, 1606–1625. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Pothula, S.P.; Wilson, J.S.; Apte, M.V. Pancreatic cancer and its stroma: a conspiracy theory. World J Gastroenterol 2014, 20, 11216–11229. [Google Scholar] [CrossRef] [PubMed]
- Hamada, S.; Masamune, A.; Shimosegawa, T. Novel therapeutic strategies targeting tumor stromal interactions in pancreatic cancer. Front Physiol 2013, 4, 331. [Google Scholar] [CrossRef]
- Chen, W.B.; Lenschow, W.; Tiede, K.; Fischer, J.W.; Kalthoff, H.; Ungefroren, H. Smad4/DPC4-dependent regulation of biglycan gene expression by transforming growth factor-beta in pancreatic tumor cells. J Biol Chem 2002, 277, 36118–3628. [Google Scholar] [CrossRef]
- Inman, G.J. Switching TGFbeta from a tumor suppressor to a tumor promoter. Curr Opin Genet Dev 2011, 21, 93–99. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, C.; Kong, Y.; Huang, H.; Wang, C.; Zhang, H. TGFbeta signaling in pancreatic ductal adenocarcinoma. Tumour Biol 2015, 36, 1613–1618. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Ellenrieder, V.; Hendler, S.F.; Boeck, W.; Seufferlein, T.; Menke, A.; Ruhland, C.; Adler, G.; Gress, T.M. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res 2001, 61, 4222–4228. [Google Scholar] [PubMed]
- Thakur, A.K.; Nigri, J.; Lac, S.; Leca, J.; Bressy, C.; Berthezene, P.; Bartholin, L.; Chan, P.; Calvo, E.; Iovanna, J.L.; et al. TAp73 loss favors Smad-independent TGF-β signaling that drives EMT in pancreatic ductal adenocarcinoma. Cell Death Differ 2016, 23, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, R.; Tsuchihara, K.; Wilhelm, M.; Fujitani, M.; Rufini, A.; Cheung, C.C.; Khan, F.; Itie-Youten, A.; Wakeham, A.; Tsao, M.S.; et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008, 22, 2677–2691. [Google Scholar] [CrossRef]
- Han, S.; Semba, S.; Abe, T.; Makino, N.; Furukawa, T.; Fukushige, S.; Takahashi, H.; Sakurada, A.; Sato, M.; Shiiba, K.; et al. Infrequent somatic mutations of the p73 gene in various human cancers. Eur J Surg Oncol 1999, 25, 194–198. [Google Scholar] [CrossRef]
- Shen, L.; Kim, S.H.; Chen, C.Y. Sensitization of human pancreatic cancer cells harboring mutated K-ras to apoptosis. PLoS One 2012, 7, e40435. [Google Scholar] [CrossRef]
- Azmi, A.S.; Ali, S.; Banerjee, S.; Bao, B.; Maitah, M.N.; Padhye, S.; Philip, P.A.; Mohammad, R.M.; Sarkar, F.H. Network modeling of CDF treated pancreatic cancer cells reveals a novel c-myc-p73 dependent apoptotic mechanism. Am J Transl Res 2011, 3, 374–382. [Google Scholar]
- Witte, D.; Otterbein, H.; Förster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-β1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci Rep 2017, 7, 17313. [Google Scholar] [CrossRef]
- Maurice, D.; Pierreux, C.E.; Howell, M.; Wilentz, R.E.; Owen, M.J.; Hill, C.S. Loss of Smad4 function in pancreatic tumors: C-terminal truncation leads to decreased stability. J Biol Chem. 2001, 16, 43175–43181. [Google Scholar] [CrossRef]
- Ungefroren, H.; Thürling, I.; Färber, B.; Kowalke, T.; Fischer, T.; De Assis, L.V.M.; Braun, R.; Castven, D.; Oster, H.; Konukiewitz, B.; et al. The Quasimesenchymal Pancreatic Ductal Epithelial Cell Line PANC-1-A Useful Model to Study Clonal Heterogeneity and EMT Subtype Shifting. Cancers 2022, 14, 2057. [Google Scholar] [CrossRef]
- Gong, J.G.; Costanzo, A.; Yang, H.Q.; Melino, G.; Kaelin, W.G., Jr.; Levrero, M.; Wang, J.Y. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999, 399, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Vikhreva, P.; Melino, G.; Amelio, I. p73 Alternative Splicing: Exploring a Biological Role for the C-Terminal Isoforms. J Mol Biol 2018, 430, 1829–1838. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Huet, S.; Gauthier, J.M. A short amino-acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene 1999, 18, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.J.; Newton, C.C.; Silverman, D.T.; Nogueira, L.M.; Albanes, D.; Männistö, S.; Pollak, M.; Stolzenberg-Solomon, R.Z. Serum transforming growth factor-β1 and risk of pancreatic cancer in three prospective cohort studies. Cancer Causes Control 2014, 25, 1083–1091. [Google Scholar] [CrossRef]
- Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; Arteaga, C.L. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest 2007, 117, 1305–1313. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, W.; Jung, Y.S.; Chen, X. Mammary epithelial cell polarity is regulated differentially by p73 isoforms via epithelial-to-mesenchymal transition. J Biol Chem 2012, 287, 17746–17753. [Google Scholar] [CrossRef]
- Nemajerova, A.; Moll, U.M. Tissue-specific roles of p73 in development and homeostasis. J Cell Sci 2019, 132, jcs233338. [Google Scholar] [CrossRef]
- Nemajerova, A.; Amelio, I.; Gebel, J.; Dötsch, V.; Melino, G.; Moll, U.M. Non-oncogenic roles of TAp73: from multiciliogenesis to metabolism. Cell Death Differ 2018, 25, 144–153. [Google Scholar] [CrossRef]
- De Laurenzi, V.; Raschella, G.; Barcaroli, D.; Annicchiarico-Petruzzelli, M.; Ranalli, M.; Catani, M.V.; Tanno, B.; Costanzo, A.; Levrero, M.; Melino, G. Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J Biol Chem 2000, 275, 15226–15231. [Google Scholar] [CrossRef]
- Fullerton, P.T., Jr.; Creighton, C.J.; Matzuk, M.M. Insights Into SMAD4 Loss in Pancreatic Cancer From Inducible Restoration of TGF-β Signaling. Mol Endocrinol 2015, 29, 1440–153. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, K.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Huang, C.; Zhao, Q.; Qiu, Z. MicroRNA-301a-3p promotes pancreatic cancer progression via negative regulation of SMAD4. Oncotarget 2015, 6, 21046–21063. [Google Scholar] [CrossRef]
- Ungefroren, H.; Witte, D.; Fiedler, C.; Gädeken, T.; Kaufmann, R.; Lehnert, H.; Gieseler, F.; Rauch, B.H. The Role of PAR2 in TGF-β1-Induced ERK Activation and Cell Motility. Int J Mol Sci 2017, 18, 2776. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; He, W.; Li, Z.; Wang, Y.; Wang, J.; Gao, J.; Wang, W.; Cheng, T.; Liu, B.; Yang, X. Endothelial Smad4 restrains the transition to hematopoietic progenitors via suppression of ERK activation. Blood 2014, 123, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Huang, Y.H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFβ receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother Pharmacol 2019, 83, 975–991. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
TAp73 induces the expression of epithelial genes and SMAD4. (A) PANC-1 cells were transiently transfected twice with 50 nM each of p73 siRNA or an irrelevant control (Co) siRNA and 48 h after transfection subjected to qPCR for ECAD, GRHL2, and SMAD4, as well as GAPDH as normalization control. Data represent the normalized mean ± SD of three assays. (B) As in (A), except that cells were subjected to immunoblotting for ECAD (left-hand panel) or SMAD4 (right-hand panel), and HSP90 as loading control. Successful knockdown of p73 was verified by immunoblot analysis (see Figure S1). (C) Immunoblot analysis show successful overexpression of TAp73α and TAp73β in PANC-1 cells transfected with either empty vector (V) or expression vectors for the two p73 isoforms and incubated with a p73 antibody. The bands of the endogenous p73 protein are not visible here due to the short exposure time of the blot. (D) PANC-1 cells were transiently transfected with empty vector (V) or expression vectors for TAp73α or TAp73β, and processed for qPCR analysis of ECAD, GRHL2 and SMAD4, and GAPDH as a control. Data represent the normalized mean ± SD of three experiments. The asterisks (∗) in (A) and (D) denote a significant difference relative to the control (p<0.05, Wilcoxon test).
Figure 1.
TAp73 induces the expression of epithelial genes and SMAD4. (A) PANC-1 cells were transiently transfected twice with 50 nM each of p73 siRNA or an irrelevant control (Co) siRNA and 48 h after transfection subjected to qPCR for ECAD, GRHL2, and SMAD4, as well as GAPDH as normalization control. Data represent the normalized mean ± SD of three assays. (B) As in (A), except that cells were subjected to immunoblotting for ECAD (left-hand panel) or SMAD4 (right-hand panel), and HSP90 as loading control. Successful knockdown of p73 was verified by immunoblot analysis (see Figure S1). (C) Immunoblot analysis show successful overexpression of TAp73α and TAp73β in PANC-1 cells transfected with either empty vector (V) or expression vectors for the two p73 isoforms and incubated with a p73 antibody. The bands of the endogenous p73 protein are not visible here due to the short exposure time of the blot. (D) PANC-1 cells were transiently transfected with empty vector (V) or expression vectors for TAp73α or TAp73β, and processed for qPCR analysis of ECAD, GRHL2 and SMAD4, and GAPDH as a control. Data represent the normalized mean ± SD of three experiments. The asterisks (∗) in (A) and (D) denote a significant difference relative to the control (p<0.05, Wilcoxon test).
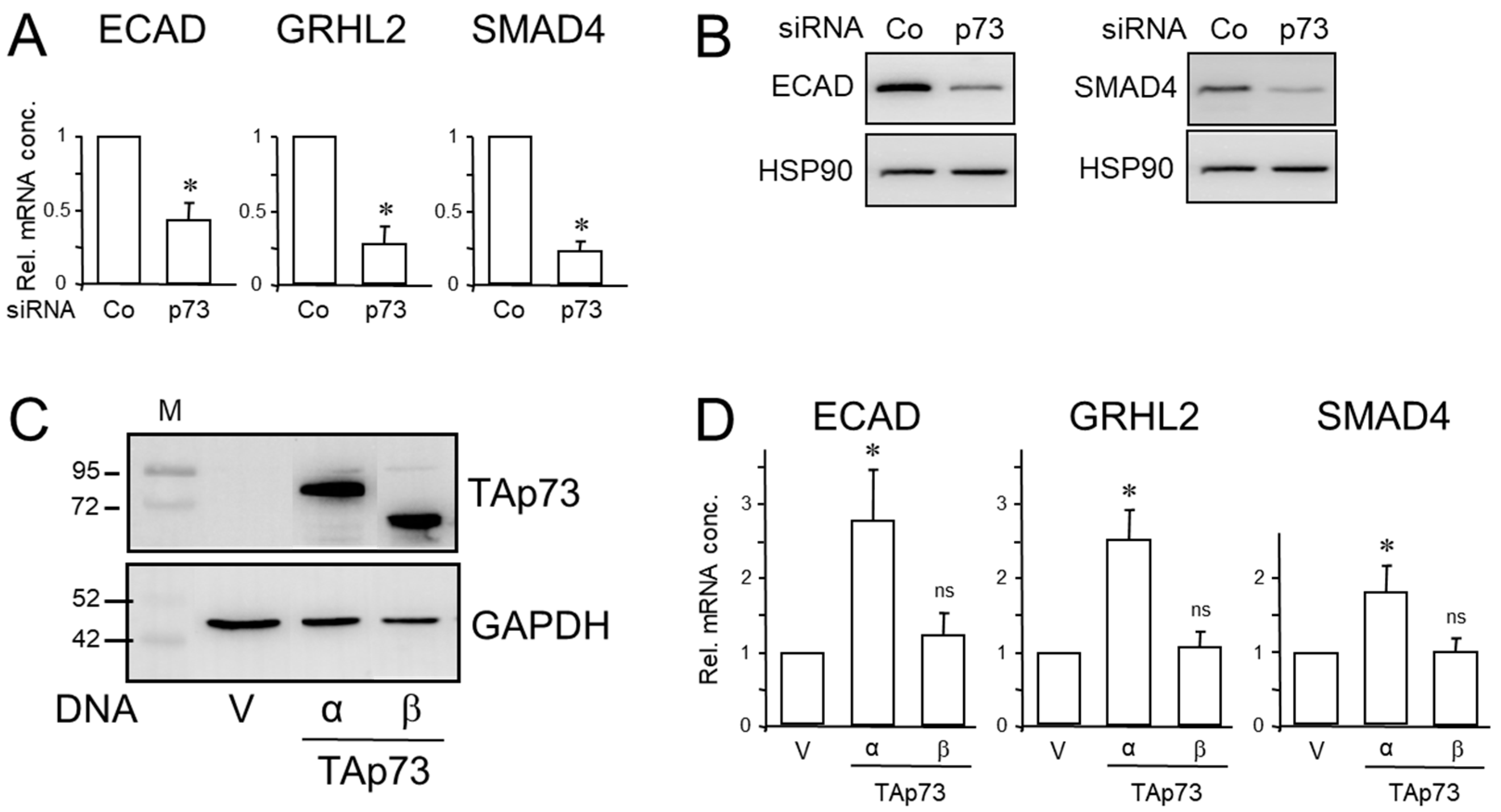
Figure 2.
Knockdown of TAp73 interfered with TGF-β/Smad-specific transcriptional activation and TGF-β1-induced regulation of TGF-β target genes in human pancreatic tumor cells. (A) PANC-1 cells (10,000) were seeded in 96 wells on d 1 and were transfected on d 2 with RNAiMAX along with 50 nM each of negative (scrambled) control (Co) siRNA, p73 siRNA, or SMAD4 siRNA. On d 3, cells received the same siRNAs along with 20 ng/well of p(CAGA)12 MLP-luc, and 5 ng/well of the Renilla luciferase encoding vector pRL-TK-Luc using Lipofectamine 2000. Forty-eight h after the start of the first transfection, cells were stimulated with TGF-β1 (5 ng/ml) for another 24 h. At the end of the stimulation period, cells were lysed in Glo lysis buffer and subjected to luciferase assay. Relative firefly luciferase activities were measured in PANC-1 cell extracts, normalized to those for Renilla luciferase. Three independent experiments were performed with similar results. Data shown are the mean ± SD of six wells processed in parallel. The asterisks (∗) indicate significance vs. TGF-β1-treated Co cells (unpaired two-tailed Student’s t-test; **p<0.01, ***p<0.001). (B) PANC-1 cells were treated for 24 h with recombinant human TGF-β1 and subjected to qPCR for ECAD or TGFB1 and p73 (from left to right) to verify successful knockdown. Data are the mean ± SD of three experiments (p<0.05, Wilcoxon test). (C) As in (B), except that cells were subjected to immunoblot analysis of ECAD, SNAIL, and HSP90 as a loading control. The graphs underneath the blots show densitometry-based signal quantification from three independent blots (mean ± SD, n = 3). The asterisks (∗) indicate significant differences relative to non-TGF-β1 treated controls (p<0.05, Wilcoxon test); ns, non-significant.
Figure 2.
Knockdown of TAp73 interfered with TGF-β/Smad-specific transcriptional activation and TGF-β1-induced regulation of TGF-β target genes in human pancreatic tumor cells. (A) PANC-1 cells (10,000) were seeded in 96 wells on d 1 and were transfected on d 2 with RNAiMAX along with 50 nM each of negative (scrambled) control (Co) siRNA, p73 siRNA, or SMAD4 siRNA. On d 3, cells received the same siRNAs along with 20 ng/well of p(CAGA)12 MLP-luc, and 5 ng/well of the Renilla luciferase encoding vector pRL-TK-Luc using Lipofectamine 2000. Forty-eight h after the start of the first transfection, cells were stimulated with TGF-β1 (5 ng/ml) for another 24 h. At the end of the stimulation period, cells were lysed in Glo lysis buffer and subjected to luciferase assay. Relative firefly luciferase activities were measured in PANC-1 cell extracts, normalized to those for Renilla luciferase. Three independent experiments were performed with similar results. Data shown are the mean ± SD of six wells processed in parallel. The asterisks (∗) indicate significance vs. TGF-β1-treated Co cells (unpaired two-tailed Student’s t-test; **p<0.01, ***p<0.001). (B) PANC-1 cells were treated for 24 h with recombinant human TGF-β1 and subjected to qPCR for ECAD or TGFB1 and p73 (from left to right) to verify successful knockdown. Data are the mean ± SD of three experiments (p<0.05, Wilcoxon test). (C) As in (B), except that cells were subjected to immunoblot analysis of ECAD, SNAIL, and HSP90 as a loading control. The graphs underneath the blots show densitometry-based signal quantification from three independent blots (mean ± SD, n = 3). The asterisks (∗) indicate significant differences relative to non-TGF-β1 treated controls (p<0.05, Wilcoxon test); ns, non-significant.
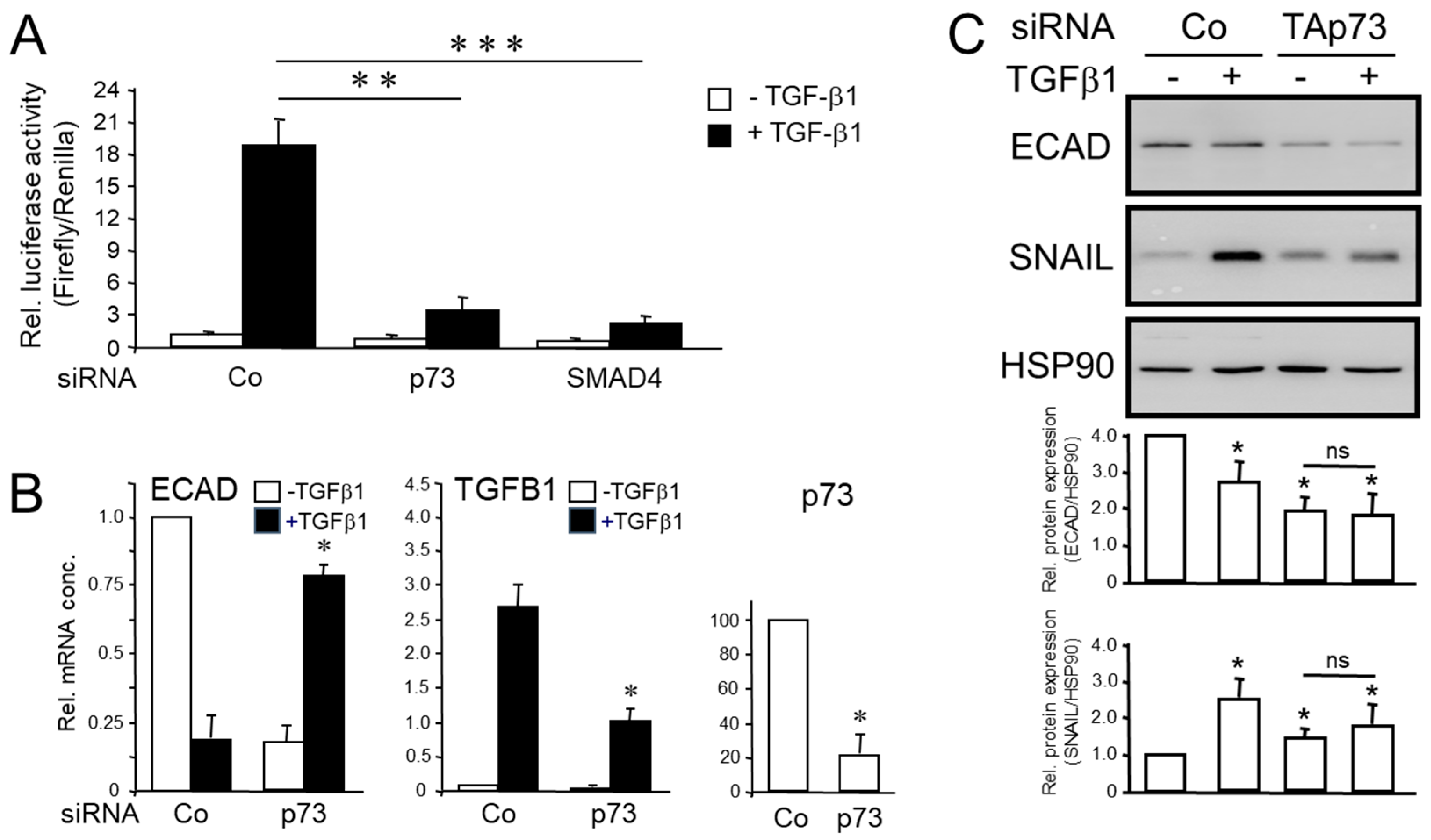
Figure 3.
TAp73 interferes with basal and TGF-β1-induced ERK activation. (A) PANC-1 cells were transiently transfected with 50 nM each of p73 siRNA or an irrelevant control (Co) siRNA and were either left untreated (left-hand blot) or were treated 48 h after transfection for various times (as indicated) with 5 ng/ml TGF-β1. Subsequently, cells were subjected to immunoblotting of pERK1/2 and either total ERK1/2 (left-hand blot) or GAPDH (right-hand blot) to control for equal loading. Since we observed that within the 2-h observation period neither GAPDH nor HSP90 expression was affected by modulation of TAp73 expression these housekeeping proteins rather than total ERK1/2 were used for normalization of pERK1 and pERK2 levels. (B) The same as in (A), except that HPAFII cells were used. Successful knockdown of TAp73 in (A) was verified by immunoblotting (Figure S1) and in (B) by qPCR (not shown). The graphs below the blots show results from densitometry-based signal quantification of three experiments (means ± SD, n = 3). The asterisks (∗) indicate significance (p<0.05, Wilcoxon test) relative to the respective Co siRNA sample.
Figure 3.
TAp73 interferes with basal and TGF-β1-induced ERK activation. (A) PANC-1 cells were transiently transfected with 50 nM each of p73 siRNA or an irrelevant control (Co) siRNA and were either left untreated (left-hand blot) or were treated 48 h after transfection for various times (as indicated) with 5 ng/ml TGF-β1. Subsequently, cells were subjected to immunoblotting of pERK1/2 and either total ERK1/2 (left-hand blot) or GAPDH (right-hand blot) to control for equal loading. Since we observed that within the 2-h observation period neither GAPDH nor HSP90 expression was affected by modulation of TAp73 expression these housekeeping proteins rather than total ERK1/2 were used for normalization of pERK1 and pERK2 levels. (B) The same as in (A), except that HPAFII cells were used. Successful knockdown of TAp73 in (A) was verified by immunoblotting (Figure S1) and in (B) by qPCR (not shown). The graphs below the blots show results from densitometry-based signal quantification of three experiments (means ± SD, n = 3). The asterisks (∗) indicate significance (p<0.05, Wilcoxon test) relative to the respective Co siRNA sample.
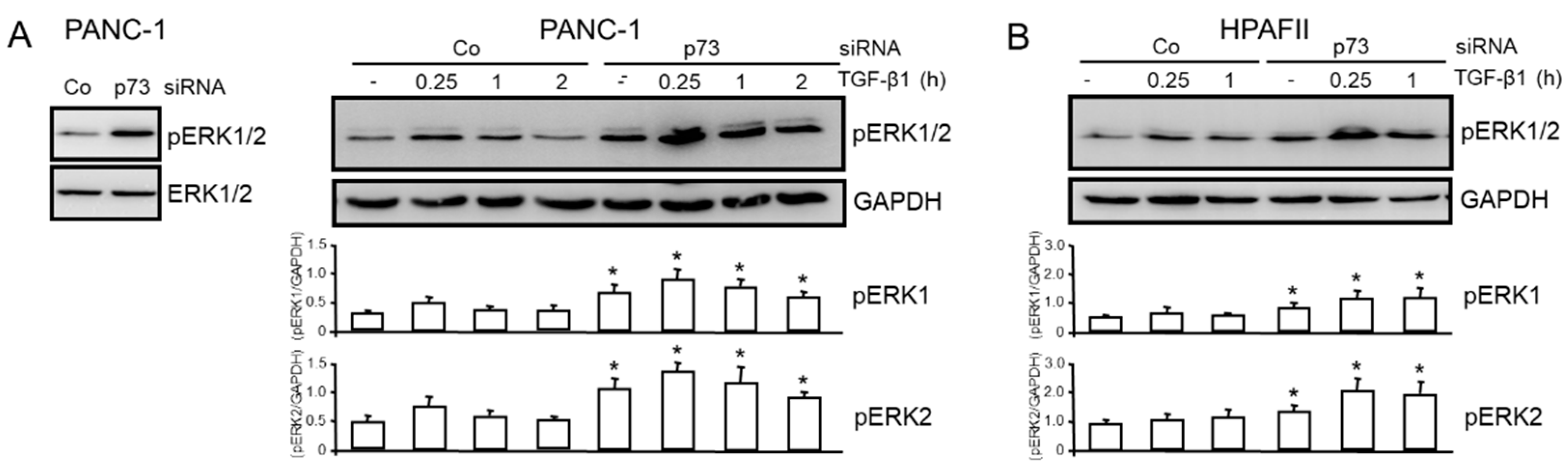
Figure 4.
PANC-1 cells stably expressing (A) a C-terminally truncated form of SMAD4, SMAD4-(1-514) or empty vector controls (quadruplicate samples each) were either left untreated, or (B) were treated with TGF-β1 for the indicated times, and subjected to pERK1/2 immunoblotting. The data shown in (A) are representative of three experiments. The graphs in (A) represent the densitometric values (means ± SD) of four wells. The p values were calculated with the unpaired two-tailed Student’s t-test. (C) PANC-1 cells were transiently transfected with siRNAs to SMAD4 followed by immunoblotting of pERK1/2. (D) As in (C), except that HPAFII cells were employed. The quantitative data in B-D (graphs underneath the blots) represent densitometric data derived from three independent experiments (means ± SD, p<0.05, Wilcoxon test). The asterisks (∗) indicate significant differences relative to the respective Co siRNA transfected cells.
Figure 4.
PANC-1 cells stably expressing (A) a C-terminally truncated form of SMAD4, SMAD4-(1-514) or empty vector controls (quadruplicate samples each) were either left untreated, or (B) were treated with TGF-β1 for the indicated times, and subjected to pERK1/2 immunoblotting. The data shown in (A) are representative of three experiments. The graphs in (A) represent the densitometric values (means ± SD) of four wells. The p values were calculated with the unpaired two-tailed Student’s t-test. (C) PANC-1 cells were transiently transfected with siRNAs to SMAD4 followed by immunoblotting of pERK1/2. (D) As in (C), except that HPAFII cells were employed. The quantitative data in B-D (graphs underneath the blots) represent densitometric data derived from three independent experiments (means ± SD, p<0.05, Wilcoxon test). The asterisks (∗) indicate significant differences relative to the respective Co siRNA transfected cells.
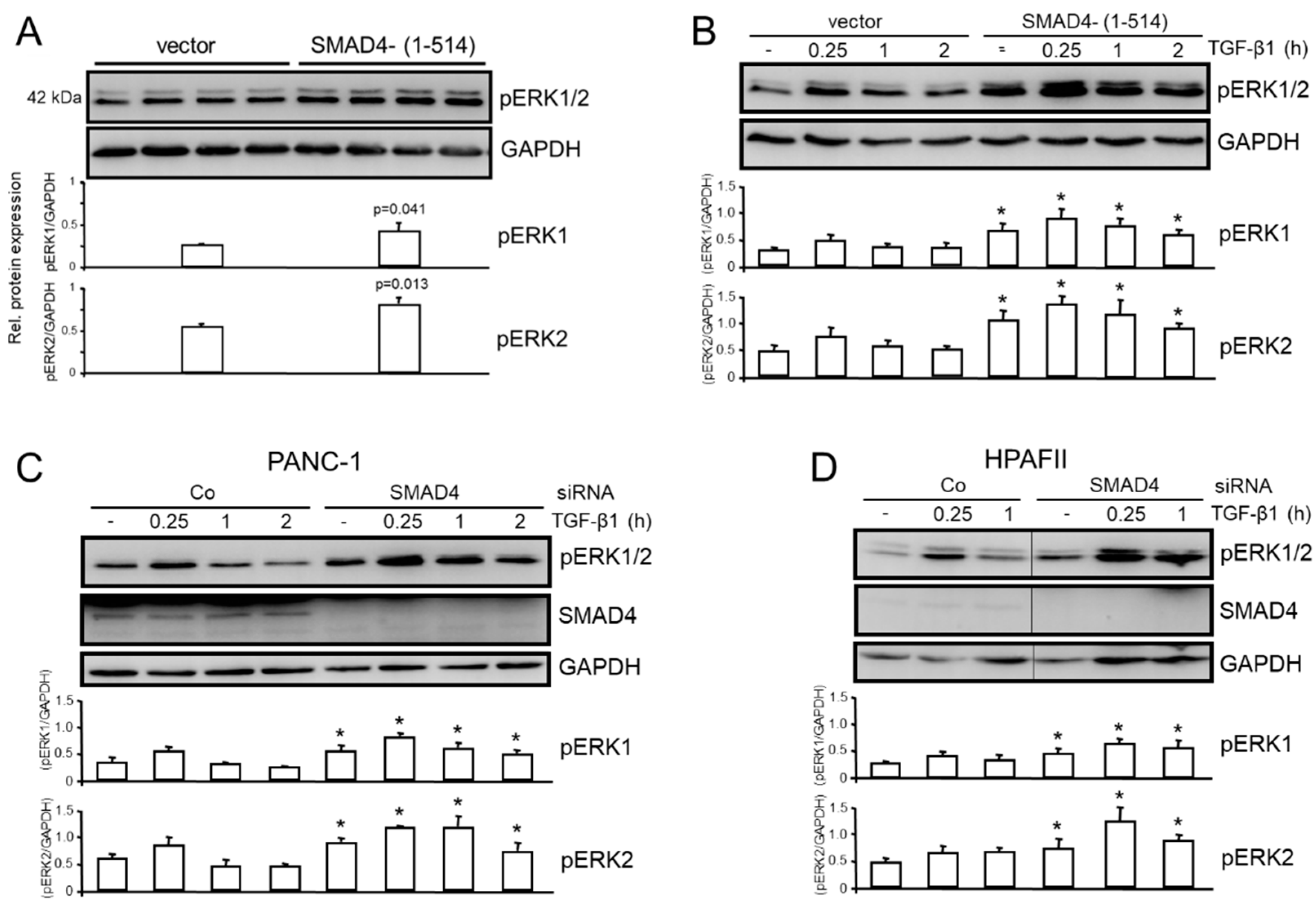
Figure 5.
TAp73 and SMAD4 inhibit cell migration in human PDAC-derived tumor cells as revealed by xCELLigence-based real-time assay. (A) PANC-1 cells were transiently transfected twice (on two consecutive days) with 50 nM of either control siRNA (siCo) or TAp73 siRNA (siTAp73) and subsequently subjected to cell migration assay with xCELLigence technology in the absence (left-hand graph) or presence (right-hand graph) of exogenous TGF-β1 (+T, 5 ng/ml). Measurements of migratory activity were taken every 15 min and graphically displayed as the dimensionless cell index (CI) plotted against assay time in h. Data are from a representative experiment out of three experiments performed in total (mean ± SD from quadruplicate wells). Successful knockdown of TAp73 was verified by immunoblotting and qPCR analysis (see Figures S1 and S2B). (B) PANC-1 cells were transfected with either TAp73α (p73α, left-hand graph) or TAp73β (p73β, right-hand graph), or empty vector as control, and 48 h later assayed as in (A) for migratory activity in the absence or presence (+T) of TGF-β1. In each panel, the assay shown is representative of three assays performed in total. Data are the means ± SD from 3-4 parallel wells. Successful overexpression of the TAp73α and TAp73β transgenes is shown in Figure 1C. (C) Two individual clones (cl. 1 and 6) of PANC-1-SMAD4-(1-514) cells were subjected to cell migration assay in the absence or presence of rec. human TGF-β1 (+T, 5 ng/ml). For each clone a representative experiment is shown out of three experiments performed in total (mean ± SD from 3-4 parallel wells). The asterisks (∗) indicate the first time point at which differences are significantly different between the untreated control cells (siRNA in panel A, vector in panels B and C) and the cells transfected with a TAp73-specific nucleic acid (siRNA in panel A and TAp73 encoding vector in panels B and C). The rhombuses (♦) indicate the first time point at which differences are significantly different between the corresponding TGF-β1-treated cell populations (p<0.05, unpaired two-tailed Student’s t-test).
Figure 5.
TAp73 and SMAD4 inhibit cell migration in human PDAC-derived tumor cells as revealed by xCELLigence-based real-time assay. (A) PANC-1 cells were transiently transfected twice (on two consecutive days) with 50 nM of either control siRNA (siCo) or TAp73 siRNA (siTAp73) and subsequently subjected to cell migration assay with xCELLigence technology in the absence (left-hand graph) or presence (right-hand graph) of exogenous TGF-β1 (+T, 5 ng/ml). Measurements of migratory activity were taken every 15 min and graphically displayed as the dimensionless cell index (CI) plotted against assay time in h. Data are from a representative experiment out of three experiments performed in total (mean ± SD from quadruplicate wells). Successful knockdown of TAp73 was verified by immunoblotting and qPCR analysis (see Figures S1 and S2B). (B) PANC-1 cells were transfected with either TAp73α (p73α, left-hand graph) or TAp73β (p73β, right-hand graph), or empty vector as control, and 48 h later assayed as in (A) for migratory activity in the absence or presence (+T) of TGF-β1. In each panel, the assay shown is representative of three assays performed in total. Data are the means ± SD from 3-4 parallel wells. Successful overexpression of the TAp73α and TAp73β transgenes is shown in Figure 1C. (C) Two individual clones (cl. 1 and 6) of PANC-1-SMAD4-(1-514) cells were subjected to cell migration assay in the absence or presence of rec. human TGF-β1 (+T, 5 ng/ml). For each clone a representative experiment is shown out of three experiments performed in total (mean ± SD from 3-4 parallel wells). The asterisks (∗) indicate the first time point at which differences are significantly different between the untreated control cells (siRNA in panel A, vector in panels B and C) and the cells transfected with a TAp73-specific nucleic acid (siRNA in panel A and TAp73 encoding vector in panels B and C). The rhombuses (♦) indicate the first time point at which differences are significantly different between the corresponding TGF-β1-treated cell populations (p<0.05, unpaired two-tailed Student’s t-test).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



