
Submitted:
26 June 2023
Posted:
27 June 2023
You are already at the latest version
Abstract
Fisetin has been shown to be beneficial for brain injury and age-related brain disease via different mechanisms. The purpose of this study was to determine the presence of senescent cells and the effects of fisetin on cellular senescence in the brain and other organs in old sheep, a more translational model. Approximately 6-7 years old female sheep (N=6) were treated with 100mg/kg fisetin or vehicle two consecutive days a week for 8 weeks. All organs were harvested at the time of sacrifice. Histology, immunofluorescent staining, as well as Q-PCR was performed on different regions of brain tissues and organs. Our results indicated that Fisetin treatment at the current regimen did not affect general morphology of the brain. The presence of senescent cells in both cerebral brain cortex and cerebellum was detected by SA-β-Gal staining. More senescent cells were observed in the gray matter when compared to the white matter of the cerebral brain cortex. These senescent cells are mainly neurons in both gray and white matter of either cerebral brain cortex or cerebellum. Fisetin treatment showed a trend of decrease SA-β-Gal cells in gray matter of both cerebral brain cortex and cerebellum and significantly decreased in brain cortex white matter. Furthermore, fisetin treatment significantly decreased P16+ cells in brain cortex NEUN+ neurons, GFAP+ astrocytes, IBA+ microglia cells in both gray and white matter of cerebral brain cortex. Fisetin treatment also significantly decreased P16+ cells in microglia cells and a trend of decrease of P16+ cells in astrocytes in the non- (Cornu Ammonis) CA area of hippocampus. However, fisetin treatment did not change P16+ cells in the NEUN+ neurons in the CA1-4 area of the hippocampus. At the mRNA level, fisetin showed a decreased in GLB1 in heart ventricle muscle tissue and spleen tissues but not in other organs tested. Fisetin treatment also showed a decreased antioxidant gene SOD1 and increased CAT in spleen and bone marrow. Fisetin treatment showed variable effects in SASP and inflammasome genes in different organs. In conclusion, we found senescent cells are widely present in the cerebral brain cortex and cerebellum of old sheep. In addition, fisetin treatment decreased senescent cells as well as P16+ cells in the neurons in both gray and white matter of cerebral brain cortex and in astrocytes and microglia cells of cerebral brain cortex and non-CA area of hippocampus. Fisetin treatment represents a promising therapeutic strategy for age-related brain disease.
Keywords:
Fisetin
; cell senescence
; sheep
; cerebral brain cortex
; cerebellum
; hippocampus
; neurons
; astrocytes
; microglia
1. Introduction
Fisetin (3, 7, 3', 4' tetrahydroxyflavone) is a natural flavonoid that exists in strawberries, cucumber, apples, onions and so on [1](36558979). Early study by Pamela Maher et al showed that fisetin activated ERK and induced cAMP response element-binding protein (CREB) phosphorylation in rat hippocampal slices, facilitated long-term potentiation in rat hippocampal slices, and enhanced object recognition in mice[2]. Fisetin also possesses neurotrophic effects by promoting rat cortical neurons cell survival and generating long neurites via promoting proteasome activity not through activation of pERK or increase glutathione[3,4]. Cumulative evidence of this research team indicate fisetin is a novel neuroprotective and cognition-enhancing molecule. Fisetin not only has direct antioxidant activity, but also can increase the intracellular levels of glutathione (GSH), the major intracellular antioxidant. Fisetin maintains mitochondrial function in the presence of oxidative stress. In addition, it has anti-inflammatory activity against microglial cells and inhibits the activity of 5-lipoxygenase, and consequently reducing the production of lipid peroxides and their pro-inflammatory by-products. These wide ranges of actions suggest that fisetin has potential to reduce the age-related decline in brain function[5]. Fisetin can increase GSH level mainly by activating transcription factor 4 (ATF4) under basal conditions while activating both ATF 4 and NF-E2-related factor 2 (NRF2) under oxidative stress[6]. In animal model, oral administration of fisetin to APPswe/PS1dE9 double transgenic Alzheimer’s disease (AD) mice from 3 to 12 months of age prevented the development of learning and memory deficits. This correlated with an increase in ERK phosphorylation and a decrease in protein carbonylation, a marker of oxidative stress. In addition, fisetin decreased the levels of p25, the cyclin-dependent kinase 5 (Cdk5) activator p35 cleavage product, in both control and AD brains. It is known elevated levels of p25 relative to p35 cause dysregulation of Cdk5 activity leading to neuroinflammation and neurodegeneration. These effects were also mediated by its anti-inflammatory effects, including changes in eicosanoid synthesis, and the maintenance of markers of synaptic function in the AD mice [7]. Fisetin was also found to reduce cognitive deficits in old senescence-accelerated prone 8 (SAMP8) mice, a model for sporadic AD and dementia, while restoring multiple markers associated with impaired synaptic function, stress, and inflammation [8]. Fisetin and other flavonoids also function as mitochondrial uncouplers to mitigate neurodegeneration in aged C. elegans, possibly via a PINK1/Parkin mitophagy process [9]. Further, fisetin boosted mental health in the aged animals. Supplementing fisetin (oral 20mg/kg/BW/day) for four weeks improved relative electroencephalograph α-power, β-power, and multi-unit activity (MUA) count in aged rats. Fisetin treated aged rats also showed significantly improved cognitive and behavioral performances than non-treated aged rats [10]. Further, fisetin can inhibit aggregation of the tau fragment, K18, and can disaggregate tau K18 filaments in vitro and prevent the formation of tau aggregates in cells [11].
Fisetin served as caloric restriction mimetics. Fisetin, administrated (15mg/kg b.w., orally) to young D-gal induced aged (D-gal 500mg/kg b.w subcutaneously) or naturally aged rats for 6 weeks significantly decreased the level of pro-oxidants and increased the level of antioxidants. Furthermore, fisetin also ameliorated mitochondrial membrane depolarization, apoptotic cell death and impairments in the activities of synaptosomal membrane-bound ion transporters in aged rat brain. Fisetin up-regulated the expression of autophagy genes (Atg-3 and Beclin-1), sirtuin-1 and neuronal markers (NSE and Ngb), and down-regulated the inflammatory (IL-1β and TNF-α) and Sirt-2 genes mRNA respectively in aged rat brain [11]. Same treatment regimen also suppresses aging-induced increases in the levels of reactive oxygen species, eryptosis, lipid peroxidation, and protein oxidation in rat erythrocytes [12]. Fisetin prevented the D-gal-mediated reactive oxygen species (ROS) accumulation, by regulating the endogenous antioxidant mechanisms, such as Sirt1/Nrf2 signaling, suppressed the activated p-JNK/NF-kB pathway, and its downstream targets, such as inflammatory cytokines[13]. Fisetin enhanced phosphorylation and nuclear translocation of Nrf2, which subsequently activated antioxidant enzyme heme oxygenase-1 (HO-1) in RPE cells, and therefore contributing to the amelioration of oxidative stress-induced ocular disorders [14]. Similar effect was observed in C2C12 myoblasts cells [15].
More recently, fisetin was found to be a potent senolytic agent. Acute or intermittent treatment of progeroid and old mice with fisetin reduced senescence markers in multiple tissues. Fisetin reduced senescence in a subset of cells in murine and human adipose tissue. Administration of fisetin to wild-type aged mice restored tissue homeostasis, reduced age-related pathology, and extended median and maximum lifespan [16]. Senescent cells accumulate with ageing and are resistant to apoptosis and have up-regulation of anti-apoptotic pathways which defend them against their own inflammatory senescence-associated secretory phenotype (SASP), allowing them to survive, but killing neighboring cells. Because senescent cells take weeks to reaccumulate, senolytics can be administered intermittently - a 'hit-and-run' approach [17]. Fisetin pretreatment or treatment of old mice after COVID19 viral infection significantly reduced mortality, cellular senescence, and inflammatory markers and increased antiviral antibodies [18]. There may be crosstalk between fisetin’s anti-cell senescent and neuroprotection[19]. Fisetin treatment also has been shown to restore muscle stem cells function in muscular dystrophy mice or progeria mice by targeting senescent cells in the diseased or aged muscle tissues [20,21]. Our recent work showed fisetin selectively attenuated markers of senescence in a dose-dependent manner while maintaining the differentiation potential of the expanded human adipose stem cells in vitro [22].
Fisetin also suppressed renal tubular senescence and attenuated renal fibrosis and improved tubular repair, as indicated by restoration of tubular regeneration and renal function[23].. Fisetin treatment reduced renal fibrosis by inhibiting the phosphorylation of SMAD3, oxidative damage, inflammation, apoptotic cell death, and accumulation of profibrotic M2 macrophages in the obstructed kidneys. In cultured human proximal tubular cells, fisetin treatment inhibited TGF-β1-induced phosphorylation of SMAD3 and SMAD2[24].
Fisetin also demonstrated the potent cardioprotective efficacy against Ang-II induced apoptosis in H9c2 cells and in spontaneous hypertension rat model. Fisetin treatment significantly reduced the apoptotic nuclei number and apoptotic proteins such as TNF- α, Fas L, FADD, Cleaved caspase-3 and Cleaved PARP and increased the cell survival and anti-apoptotic proteins like Bcl-2, Bcl-xL, p-IGF1R, p-PI3K and p-AKT in both in vitro and in vivo models[25]. Further, fisetin treatment reduced both the atherosclerosis plaque and the lipid accumulation in the aortic sinus in apoE-/- mice fed with a high-fat diet, and the expressions of PCSK9, LOX-1 and aging markers, including p53, p21 and p16 were downregulated [26]. In vascular smooth muscle cell (VSMC) cells, fisetin inhibited cellular senescence induced by the PTEN-PKCδ-NOX1-ROS signaling pathway after hydrogen peroxide (H2O2) treatment, and this anti-aging effect was attributed to reduced ROS production caused by suppressing NOX1 activation[27].
Taken together, fisetin has been shown broad beneficial effects on age related disease. However, majority of the previous studies used mice. Therefore, the goal of this study is to take advantage of large animal model’s translational value and investigate whether systemic fisetin treatment of aged sheep would have beneficial to multiple organs via targeting cell senescent cells.
2. Materials and Methods
2.1. Animal use ethics
This study was approved by Institutional animal care and use committee (IACUC) of Colorado State University (#3159). 8 years old Nordic sheep of both male and females were divided into two groups (N=6/group), fisetin group received fisetin at 100mg/kg in DMSO and 10 mM in ethanol by intravenous infusion two days every week for 8 weeks, the control group received vehicle intravenous infusion using same regimen. Sheep were sacrificed at 8 weeks following the protocol and several organ tissues were harvested and dissected for histology or Q-PCR analysis.
2.2. Histology
Brain tissues were harvested after 8 weeks, left hemisphere were dissected and fixed in neutral buffered formalin for 9 days and then midbrain was sliced into three different level and processed using 15% sucrose 30% sucrose in PBS until tissues sink, and embedded in NEG freezing medium. Cerebellum was also harvested and sliced in the middle and embedded in NEG freezing medium after 9 days of fixation. Furthermore, hippocampus was dissected (after 6 months fixation) and processed using 15% sucrose and 30% sucrose and embedding in freezing medium and snap frozen in liquid nitrogen. Cerebral brain cortex (mid brain level), cerebellum and hippocampus sections were cut into 8µm thickness using Leica Cryostat for histology and immunofluorescent staining. The right hemispheres were used to collect fresh cerebral brain cortex and frozen in -80oC for isolation RNA and real time quantitative polymerase chain reaction (Q-PCR). H&E staining were performed using AnaTech extra-strength Hematoxylin and eosin Y in 75% alcohol following the manufactures protocol.
2.3. Senescent associated β-galactosidase (SA-β-Gal) staining
Brain cortex and cerebellum cryosections were used to perform SA-β-Gal staining using Senescence beta-galactosidase Staining kits (#9860, Cell Signaling Technology). The staining procedure followed the manufacture’s protocol. Blue staining indicated positive cells. Both intense blue staining and light blue that can be distinguished from surrounding cells were counted positive. Positive cells were counted using Image J. Positive cells number in 200X microscope field was used to compare between groups. Hippocampus tissues SA-β-Gal staining did not work due to long time fixation before dissecting for making cryosections.
2.4. Immunofluorescent staining.
To detect which cells become senescent in the brain tissues, immunofluorescent (IF) staining of P16ink4A (P16) and colocalized with astrocytes markers glial fibrillary acidic protein (GFAP), neuronal marker NeuN and microglia marker ionized calcium-binding adaptor molecule 1(IBA1) was performed for the brain cortex, and dissected hippocampus. IF was performed using the same principle and procedure as previously described[28]. Briefly, sections slides were dried at room temperature and refixed in neutral buffered formalin for 8 minutes to avoid section come off during antigen retrieval. Heat retrieval in pH=6.0 citrate buffer at 92oC water bath were performed and cool 10 minutes. Slides were washed with PBS for three times and blocking with 5% donkey serum at ambient temperature for 1 hour. Then primary antibodies diluted in 5% donkey serum were added on the sections and incubated at 40C overnight. The primary antibodies dilutions are rabbit anti-GFAP (ab68428, Abcam, 1:100), rabbit anti-IBA1(10904-1-AP, Proteintech 1:200 dilution), rabbit anti-NeuN (ab177487, 1:2000), mouse anti-P16 (SC1661, Santa Cruz Biotechnology,1:50). For dissected hippocampus staining, we used mouse anti-P16 from Thermofisher Scientific (MA5-17142) because the anti-P16 antibody from Santa-Cruz Biotechnology did not work for hippocampus sections. After primary antibody incubation, slides were washed with PBS for three times. Then sections were incubated with donkey anti-rabbit-594 (711-585-152, Jackson ImmunoReseach Laboratory Inc.) and donkey anti-mouse 488 (715-545-150, Jackson ImmunoResearch Laboratory Inc.) at 1:200 dilution and incubated at ambient temperature for 2 hrs. After secondary antibody incubation, slides were washed three times and nuclear were stained with 4′,6-diamidino-2-phenylindole (DAPI, Thermofisher Invitrogen) at 1ug/ml in PBS for 10 minutes. Slides were finally washed three times with PBS and three times of deionized water and mounted with VectaMount® AQ Aqueous Mounting Medium (H-5501-60, Vector Laboratories). Immunofluorescent images were captured using Nikon Ti microscope at 200X magnification and cell numbers were quantified with Image J software.
2.5. Brain cortex, heart, spleen tissue and bone marrow harvest
Brain cortex tissues from the same location were cut and snap frozen in liquid nitrogen and subsequently store in -80oC for later RNA isolation. Left ventricle muscle of the heart was cut at the same location and snap frozen in liquid nitrogen and stored in -80oC for isolation of RNA and Q-PCR analysis. Spleen tissues from the same location of each sheep were cut and frozen in -80oC for RNA extraction. Briefly, heart muscle or spleen tissues were rinsed with PBS and similar size around 0.5g of tissues were transferred to 2 ml tube and cut with scissors to around 1mm size, and then were homogenized with IKA-T10 tissue dispenser in 2ml TRIzol™ Reagent (15596026, Themofisher Scientific) until no visible tissue chunk left. Tissues were homogenized using 30 second homogenization and then 30 second on ice. After homogenization, tissues lysates were centrifuged at 12000g for 5 minutes to remove fat or tissue debris. Supernatant were transferred to a new tube for RNA isolation following the protocol by manufacturer. Furthermore, bone marrow was aspirated from sheep iliac crest and nucleated cells were isolated using gradient centrifugation using Percoll gradient reagent and nucleated cells were used for RNA extraction using Trizol reagent as indicated below.
2.6. RNA isolation, cDNA synthesis and semi-quantitative and Quantitative -PCR (Q-PCR).
All RNAs were isolated using the protocol provided by the manufacturer (#15596026,Thermofisher Scientific). Total RNA was air dried for 7 minutes and resuspended in DNase/RNase free water using appropriate volume based on the RNA pellet size and allowed RNA to dissolve for 10 minutes at room temperature. RNA concentrations were measured with Nanodrop plate equipped in Tecan Plate Reader (Switzerland). First strand cDNA synthesis was then conducted using iScript™ Reverse Transcription Supermix, 100 x 20 µl rxns, 400 µl (1708841, BioRad) in 20ul reaction using 25oC 5 minutes, 46oC 30 minutes and 95oC for 1 minute. After cDNA synthesis, cDNA was diluted using DNase/RNase free H2O to 10 ng/µl. Subsequently semi-quantitative or Q-PCR was performed. For the brain tissues, we performed both semi-quantitative-PCR and Q-PCR. Semi-quantitative PCR was performed using GoTaq® G2 Master Mixes(M7823). PCR products were electrophoresed using 1.5% agarose gel (BioRad) and gel images were captured using ChemiDoc MP System (BioRad). Housekeeping gene and target band density were quantified with Image Lab software (BioRad). Q-PCR was performed using SsoAdvanced Universal SYBR® Green Supermix, 1,000 x 20 µl rxns, 10 ml (10 x 1 ml) (1725272, BioRad) using 10µl reaction using 1µl diluted cDNA. Q-PCR was conducted on ABI system. The analyzed genes included Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), GFAP, lysomal β-galactosidase (GLB1), super oxide dismutase 1 (SOD1), catalase (CAT), neurofilament heavy chain (NEFH), neurofilament light chain (NEFL), interleukin 10 (IL10), interleukin 8 (IL8), P21, P53, NOD-, LRR- and pyrin domain-containing protein 3(NLRP3), Triggering Receptor Expressed On Myeloid Cells 2 (TREM2). Gene expression levels were expressed as fold change of control (2-Delta-Delra CT) methods. All primers were designed using primer 3 input free online software [29,30](22730293, 17379693). Primer information are shown on Table 1:
2.7. Statistical analysis: All data was analyzed with two group t test using Graphpad Prism 9. P<0.05 was considered statistically significant.
3. Results
3.1. Fisetin treatment did not significantly affect sheep brain general morphology.
Gross images showed no obvious differences on the cerebral brain cortex (midbrain) between fisetin treated sheep and vehicle treated sheep (Figure 1A). H&E staining did not reveal significant difference between fisetin treated and vehicle treated sheep brain for the cerebral cortex (Figure 1B), cerebellum (Figure 1C) and different regions of the hippocampus (Figure 1D).
3.2. Fisetin treatment decreased SA- β-Gal positive cells.
We found many SA-β-Gal positive cells in the gray matter of the control sheep’s cerebral brain cortex tissues which indicated the presence of senescent cells at this age of sheep (Blue staining, insets highlighted positive cells (Figure 2A). After Fisetin treatment, we still observed many positive cells. These positive cells are mainly large neurons which have loose large nuclear that not stained. Fisetin treatment showed trend of decreased SA-β-Gal positive cells compared to control group (P=0.1449) (Figure 2B). We also found many SA-β-Gal positive cells in the white matter of cerebral brain cortex, but relative fewer than gray matter. These SA-β-Gal positive cells were also mostly neurons. Fisetin treatment significantly decreased the number of SA-β-Gal positive cells in white matter compared to control (P=0.0306, Figure 2 C-D). Furthermore, we also found SA-β-Gal positive cells in the gray matter of the cerebellum, but not as many as in brain cortex region. These positive cells are also mainly large neurons (Figure 2E). Fisetin treatment showed trend of reduction of SA-β-Gal positive cells compared to control group (P=0.1804, Figure 2F). Further, SA-β-Gal positive cells were also found in the white matter of cerebellum and they are also mainly large neurons (Figure 2G). Fisetin treatment did not significantly decreased the number of SA-β-Gal positive cells in the white matter of cerebellum (Figure 2H).
3.3. Effects of fisetin treatment on the cellular senescence of neurons, astrocytes, and microglial cells of cerebral cortex.
Since we found many senescent cells in the aged sheep both in cerebral brain cortex and cerebellum, we want to identify which cells become cell senescent and if fisetin treatment decreased the senescent cells burden. We performed P16 staining, a hall marker of senescent cells and colocalized with different neuronal cells markers. We first tested P16 and colocalized with NEUN, a neuronal marker. We found many intense NEUN positive red stained cells in the gray matter while P16 staining localized in the nuclei of both neurons and non-neurons. P16 staining in the neurons are relative defused due to the loose nuclei structure of neurons (green channel). Colocalized cells appeared orange color (Figure 3A gray matter). Quantification indicated fisetin treatment significantly decreased P16+/NEUN+ cells/Total NEUN+ cells percentage in the NEUN positive neurons in the brain cortex gray matter (P=0.0079) (Figure 3B). Furthermore, in the white matter, very few NEUN+ cells are detected while many small P16+ green cells that are not colocalized with NEUN were detected (Figure 3A white matter). Therefore, we counted the NEUN-P16+cells/Total DAPI cells percentage. Fisetin treatment significantly decreased the NEUN-P16+cells/Total DAPI cells percentage in the white matter (P=0.0002) (Figure 3C). We further performed P16 staining and colocalized with astrocytes marker GFAP. GFAP+ astrocytes were stained red with many processes with small cytoplasm in the gray matter (Figure 3D gray matter). Fisetin treatment significantly reduced the P16+/GFAP+/Total GFAP+ cells percentage of astrocytes in the gray matter (P=0.0255) (Figure 3E). In the white matter, many more GFAP+ cells are detected (Figure 3D white matter). Fisetin treatment significantly decreased P16+/GFAP+/Total GFAP+ cells percentage of astrocytes in the white matter(P=0.0521) (Figure 3F). Additionally, we performed double staining of P16 and colocalized with microglia markers IBA1. IBA1+ cells were stained in red and are small cells with some process and some were colocalized with P16 (Figure 3G gray matter). Fisetin treatment significantly decreased P16+/IBA1+/Total IBA1+ cells percentage in the microglia cells (P=0.0407) (Figure 3H). Furthermore, in the white matter, many more IBA1+ (red) and P16+ (green) small cells with some colocalized were found (Figure.3G white matter). Fisetin treatment significantly reduced the P16+/IBA1+/Total IBA1+ percentage in microglia cells in the white matter (P=0.0004) (Figure 3I).
3.4. Effects of Fisetin treatments on cellular senescent of neurons, astrocytes, and microglial cells of hippocampus.
To further determine if fisetin treatment affects important brain area hippocampus, we dissected hippocampus from the cerebral brain cortex after fixation and processed and cryosection were cut for double immunofluorescent staining. First, we performed NEUN and P16 (MA5 17142, Thermofisher Scientific) double immunofluorescent staining. We choose Thermofisher P16 antibody for all hippocampus staining because SC1661 P16 antibody from Santa-Cruz (SC1661) did not work for hippocampus likely because we dissected the hippocampus after 6 months fixation in NBF after failure to locate hippocampus using whole cerebral brain section. NEUN/P16 double staining showed strong NEUN positive staining in very organized manner in Cornu Ammonis (CA) while large NEUN+ cells were scattered in non-CA area of hippocampus (Figure 4A merged images). P16 was stained in green clearly in the nuclei (Figure 4A green channel). Large number of NEUN+/P16+cells (orange or yellow colocalized cells) were detected in CA area in both control and fisetin group (Figure 4A merged channel). Fisetin treatment did not significantly change P16+NEUN+/Total NEUN+ cells percentage in CA area (Figure 4B). We also performed double staining of P16 and GFAP to detect senescent cells in the astrocytes in hippocampus. GFAP+ cells are stained in red color with many red processes and located in non-CA area with some processes penetrat CA area (Figure 4C). Therefore, we count P16+/GFAP+ cells in the non-CA area. Fisetin treatment showed trend of reduction of P16+/GFAP+/Total GFAP+ cells percentage in the non-CA area of hippocampus (P=0.1076) (Figure 4D). Furthermore, we performed double immunoflurescent staining of IBA1 and P16 to detect microglia cells senescent in the hippocampus. IBA1 was stained in red in small cells with few processes. P16 was stained green in the nuclei. We counted P16+/IBA1+ cells in the non-CA area because IBA1+ cells are not expressed by neurons in CA area (Figure 4E). Fisetin treatment significantly decreased P16+IBA1+/Total IBA1+ cells percentage in the microglia cells in the non-CA area of hippocampus (P=0.0102) (Figure 4F).
3.5. Effects of fisetin treatment on gene expression of sheep brain cortex.
Semi-quantitative-PCR indicated fisetin treatment did not significantly change gene expression of antioxidant genes SOD1 and CAT, astrocytes marker (GFAP), neuronal markers (NEFH, NEFL), SASP markers interleukin 10 and 8 (IL10, IL8) and senescent marker gene galactosidase beta 1 (GLB1) (Figure 5 A-B). We further performed Q-PCR analysis. We found fisetin treatment did not significantly affect SOD1 and CAT expression (Figure 5C). Fisetin treatment did not affect neuronal marker NEFL and NEFH and astrocytes marker GFAP (Figure 5D). Fisetin treatment did not significantly changed GLB1 gene expression as well as P53 expression, but showed a trend of decrease P21 expression (P=0.127). Fisetin treatment did not significantly change SASP gene IL10 and IL8 expression (Figure 5F). Furthermore, fisetin treatment did not significantly affect inflammasome gene NLRP3, but showed a trend of decreasing TREM2, another inflammasome gene (Figure 5G).
3.6. Effect of fisetin treatment on gene expression of heart tissues.
To further investigate if fisetin treatment affects gene expression of other tissues, we harvest ventricle muscle of heart tissues and performed Q-PCR analysis. We found fisetin treatment did not significantly change gene expression of SOD1, CAT in the sheep’s heart ventricle muscle tissues (Figure 6A). Among the senescent related genes, we found fisetin showed a trend of down-regulating GLB1 (P=0.192), but not affecting P21 and P53 expression (Figure 6B). Furthermore, fisetin treatment demonstrated a trend of increase of IL10 (P=0.0695) and IL8 (P=0.0691(Figure 6C). Finally, fisetin treatment showed a trend of up-regulating inflammasome gene NLRP3, with no effect on TREM2 expression of heart tissues (Figure 6D).
3.7. Effects of Fisetin treatment on sheep spleen tissues gene expressions
We also harvested spleen tissues, an important immune organ and extract total RNA for gene expression analysis. We found SOD1 was significantly decreased (P=0.017) and CAT was significantly increased (P=0.053) by fisetin treatment (Figure 7A). We also found fisetin treatment showed a trend of decrease for senescent hall marker gene GLB1 (P=0.083) (Figure 7B). No significant changes were found for P21 and P53 mRNA expression in the spleen tissues (Figure 7B). We did not find significant changes for SASP genes IL10 and IL8 (Figure 7C). Fisetin treatment did not significant change the expression of NLRP3, but demonstrated a trend of increase for TREM2 (P=0.098) (Figure 7D).
3.8. Effects of fiestin treatment on gene expression of bone marrow nucleated cells.
Furthermore, we isolated bone marrow nucleated cells from iliac crest and performed Q-PCR to investigate if fisetin treatment affects bone marrow cell senescent. Fisetin treatment showed a trend of decreasing SOD1, but not affecting CAT expression (Figure 8A). We did not observe significant differences between fisetin treatment and control for GLB1 and P21 expression (Figure 8B). However, we found fisetin treatment significantly increased P53 expression in the bone marrow (P=0.046) (Figure 8C). No statistical significance was found for IL10 and IL8 expression between fisetin treatment and control in bone marrow (Figure 8D). Additionally, fisetin treatment did not change inflammasome gene expression level of NLRP3, but showed a trend of increase in TREM2(P=0.073) (Figure 8E).
Discussion
The goal of this study was to determine the presence of senescent cells and the effects of fisetin on cells senescent in the brain and other organs in aged sheep, a more translational model. Our results revealed wide presence of senescent cells in the 8 years old sheep (equivalent to 60-65 years old human) in both cerebral brain cortex and cerebellum as demonstrated by SA-β-Gal staining which was not reported before. Cerebral cortex gray matter has relative more senescent cells than white matter. The senescent cells are mainly large neurons in both gray and white matter of either cerebral brain cortex or cerebellum. Fisetin treatment at the current regimen (100mg/kg, two consecutive days a week for 8 weeks) did not affect general morphology of the brain. But fisetin treatment showed a trend of decrease SA-β-Gal cells in gray matter of both cerebral brain cortex and cerebellum and significantly decrease in brain cortex white matter compared to control. Furthermore, Fisetin treatment significant decreased P16+ cells in NEUN+ neurons, GFAP+ astrocytes, IBA+ microglia cells in both gray and white matter of cerebral brain cortex. Fisetin treatment also significantly decreased P16+ cells in microglia cells and a trend of decrease of P16+ cells in astrocytes in the non-CA area of hippocampus. But fisetin treatment did not change P16+ cells in the NEUN+ neurons in the CA1-4 area of hippocampus. At the mRNA level, fisetin showed a trend of decrease GLB1 in heart ventricle muscle tissue and spleen tissues but not brain and bone marrow. Fisetin treatment also showed significant or trend of decreased antioxidant gene SOD1 and increased CAT. Fisetin treatment showed variable effects in SASP and inflammasome genes in different organs.
Cell senescence was recently discovered as fundamental mechanism of aging and age-related diseases [31-34]. Targeting cell senescent using senolytic drugs have been shown to improve SASP and frailty and enhanced metabolic function in aged mice [35,36]. Treatment with different senotlytic drugs such as JAK1 inhibitor Ruxolitinib or senolytic cocktail, dasatinib plus quercetin (D+Q) systemically for aged mice (20-22 months) prevented age related bone loss [37]. Senolytic drug treatment also improved overall physical function of aged mice and extend lifespan of old mice by decreasing senescent cells [38]. A human clinical trial showed D+Q treatment decreased senescent cells burden in humans as evidenced by reduced adipose tissue senescent cell burden with decreases in p16-and p21-expressing cells, SA-β-Gal+ cells, and adipocyte progenitors with limited replicative potential within 11 days [39]. Most of previous studies on cell senescence used rodent animals. However, there is lacking of study to investigate the senescent burden in the brain of large animals. Our SA-β-Gal staining results indicated that senescent cells were widely present in both gray matter and white matter of 8 years old sheep brain. More senescent cells are present in the gray matter than in white matter. Further, from the morphology of the SA-β-Gal, they are mainly large neurons in these brain regions. These results indicated senescent burden in the brain is significant even in the not advanced aged sheep (equivalent to 60-65 years old human).
Alzheimer's disease (AD) is the most common form of dementia with the numbers expected to increase dramatically as populations ages. No treatments are available to cure, prevent, or delay the progression of the disease. Multiple changes associated with brain aging, including neuroinflammation and oxidative stress contributed to disease development and progression [40]. Many studies have shown the beneficial effects fisetin on the different neurological disorders via its effects on multiple pathways. These include its anti-inflammatory and antioxidant effects as well as regulating cell death xytosis/ferroptosis pathway, the gut microbiome [41,42]. Fisetin was deemed as one of the promising nutraceuticals for treatment of Alzheimer’s disease. A combination of nutraceutical substances and other preventive measures could have significant clinical impact in a multi-layered therapy approach to treat AD[43]. Fisetin treatment has also been shown to decrease senescence and SASP in aged or progeria mice and extend lifespan [16]. But no study has investigated the effect of fisetin on brain cells senescent in large animal models. In this study, we showed that fisetin treatment in our current treatment regimen decreased SA-β-Gal positive cells in gray matter and white matter of cerebral brain cortex and gray matter in the cerebellum. However, fisetin treatment did not kill all senescent cells in the brain because many SA-β-Gal positive cells are still present in fisetin treated sheep brain regions. Furthermore, to determine which cells become senescent in the brain, we performed colocalization of P16 with neuronal marker NEUN, astrocytes marker GFAP and microglia markers IBA1. We found Fisetin treatment significantly decrease P16+NEUN+/Total NEUN+, P16+GFAP+/Total GFAP+ and P16+IBA1+/Total IBA1+ cells percentages in the cerebral brain cortex in both gray and white matters. We revealed fisetin treatment significantly decreased P16+/IBA1+/Total IBA1+ cells percentage in the non-CA area and a trend of decreased P16+GFAP+/Total GFAP+ cells percentage in non-CA-area of hippocampus. But fisetin treatment has no effect on the P16+NEUN+/Total NEUN+ cells percentage in the CA1-4 area of hippocampus. These results are consistent with SA-β-Gal staining results and collectively showed fisetin decreased senescent cells burden in the sheep brain for the first time. However, it is recognized that not all P16+ cells are senescent cells although all senescent cells expressed P16 [44]. We have attempted to use GLB1 antibody that worked for mouse and human to colocalize with neuronal marker, astrocyte marker and microglia marker, however, our antibody did not work for sheep and no GLB1 antibody was developed for sheep. Therefore, we could not determine if the P16+ cells are all senescent cells. This is the limitation of this study. On the other hand, Fisetin may have only eliminated P16+ senescent cells not P16+ non-senescent cells. The reason why we see a decrease of P16+/NEUN+ cells /Total NEUN+ cells percentage in cerebral brain cortex but not in the hippocampus may be due to the P16+ cells in hippocampus are not senescent cells because nearly all CA1-4 neurons (NEUN+) are P16+ (Figure 4A). It might be that we used different P16 antibodies for cerebral brain cortex and hippocampus because SC1661 antibody from Santa Cruz Biotechnology did not work for hippocampus even with antigen retrieval likely due to long time formalin fixation before dissecting hippocampus. Whether other senolytic drug are more effective needs further study. Indeed, a recent clinical trial data using D+Q treatment to prevent Alzheimer’s disease showed promising results in decreasing senescent related cytokines and chemokines [45].
It is worthy to emphasize that fisetin decreased P16+IBA1+/Total IBA1+ cells percentage in microglia in both cerebral brain cortex and hippocampus although we used different P16 antibody. Microglia is macrophage lineage of cells in the brain [46]. It is known both activated inflammatory microglia and senescent microglia secreted TNFα, IL1β and IL6 [47]. Fisetin treatment inhibited the upregulation IL1β induced by LPS/IFN-γ- or peptidoglycan-induced inflammatory mediator in microglia cells. Fisetin also induced an endogenous anti-oxidative enzyme HO (heme oxygenase)-1 expression through the PI-3 kinase/AKT and p38 signaling pathways in microglia. Further, fisetin also significantly attenuated inflammation-related microglial activation and coordination deficit in mice in vivo[48]. Fisetin treatment was also reported to alleviate intracerebral hemorrhage (ICH)-induced brain injury by downregulating proinflammatory cytokines and attenuating NF-κB signaling and preventing microglia activation [49]. Hence our results that fisetin decreased P16+ cells in the microglia added another beneficial effect of fisetin for the brain.
We also found Fisetin treatment affected the antioxidant gene SOD1 and CAT differently in different organs. We found fisetin treatment decreased SOD1 and increased CAT in both spleen and bone marrow but did not significantly change these two genes in the brain cortex and heart tissues. Fisetin is well known for its antioxidant effects by increasing glutathione (GSH) mainly through activating antioxidant transcription factor NRF2 [4,6]. In a vascular dementia model, fisetin treatment attenuated histological injury, malondialdehyde levels, inflammasome pathway activation, apoptosis, as well as increased brain derived neural growth factor (BDNF) expression, reduced astrocyte, microglial activation, and cognitive deficits [50]. However, it has been reported fisetin could only reduce cell death induced by iron and copper in response to treatments that lower GSH levels, it is much less effective when the metals are combined with other inducers of oxidative stress. These effects correlated with the ability of iron but not copper to block the induction of the antioxidant transcription factor, Nrf2, by fisetin [51]. Previous study using pressure overload induced cardiac hypertrophy model showed fisetin markedly reduced ROS by increasing expression of SOD1 and CAT and HO1[52]. However, ours results only showed Fisetin treatment increased CAT but decreased SOD1. We did not detect other antioxidant genes due to unavailability of mRNA sequence for sheep. This inconsistence between our results and previous study might be due to different regimen of drug administration.
Finally, we found variable effect of fisetin on the expression of GLB1 in different organs. We found fisetin treatment relatively decreased GLB1 expression in spleen and bone marrow but not heart and brain cortex tissues by Q-PCR analysis. We also found different changes of SASP and inflammasome genes. It is possible this was because different organ has different working threshold of effects of fisetin. Further we used a hit-and run” strategy specific of senolytic treatment instead of regular drug treatment [16,39]. This treatment regimen does not produce sustained level of effective fisetin or its metabolites in the systemic level like other persistent administration regimen that used daily administration. Fisetin was shown to downregulate the activation of the NLRP3 inflammasome induced by LPS and ATP (LPS/ATP) and the subsequent maturation of IL-1β. Fisetin also activated mitophagy and prevented the accumulation of damaged mitochondria and the excessive production of mitochondrial ROS. Treatment of LPS/ATP-stimulated zebrafish model with fisetin facilitated the recovery of the impaired heart rate, decreased the recruitment of macrophage to the brain, and gradually downregulated the expression of inflammasome-related genes in a p62-dependent manner [53]. But our study did not reveal consistent effects of fisetin on inflammasome gene NLRP3 and TREM2 at the current senolytic treatment regimen likely due to that we were not using induced inflammatory model in this study.
Conclusion
In summary, our results indicated that senescent cells are widely present in the gray and white matter of cerebral brain cortex and cerebellum in relative aged sheep and are mainly neurons. Fisetin treatment decreased senescent cells in gray matter of brain cortex and cerebellum. Further, Fisetin treatment significantly decreased P16+ neurons, astrocytes and microglia cells in both gray and white matter on cerebral brain cortex as well as reduced P16+ astrocytes and microglia in the non-CA area of hippocampus. Fisetin treatment also showed decrease in GLB1 expression in the heart and spleen tissues and decreased SOD1 and increased CAT in the spleen and bone marrows. Fisetin showed promise to be used as senolytic drug to treat age related brain diseases and warrantee clinical trial.
Author Contributions
Conceptualization, J.H. and X.G.; Methodology, C.A.H, X.G., M.D.H.,R.D.H, K.L; Software, C.A.H, X.G.; Validation, X.G., J.H.; Formal Analysis, C.A.H. X.G.; Investigation, C.A.H, X.G., M.D.H.,R.D.H, K.L.; Resources, J.H.; Data Curation, X.G.; Writing – Original Draft Preparation, C.A.H. X.G.; Writing – Review & Editing, J.H., P.J.M; Visualization, C.A.H, X.G.; Supervision, X.G, J.H.; Project Administration, J.H.; Funding Acquisition, J.H.
Funding
This project was funded by the Philanthropic gift from Borgen Family and the Linda and Mitch Hart Family.
Institutional Review Board Statement
This study was approved by IACUC of Colorado State University (Fort Collins campus, Protocol #3159).
Informed Consent Statement
N/A.
Data Availability Statement
Original data will be available from corresponding authors upon request.
Conflicts of Interest
J.H. received Royalties from Cook Myocyte. All other authors declared no conflict of interest.
References
- Park, S.; Kim, B.K.; Park, S.K. Effects of Fisetin, a Plant-Derived Flavonoid, on Response to Oxidative Stress, Aging, and Age-Related Diseases in Caenorhabditis elegans. Pharmaceuticals (Basel) 2022, 15. [Google Scholar] [CrossRef]
- Maher, P.; Akaishi, T.; Abe, K. Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc Natl Acad Sci U S A 2006, 103, 16568–16573. [Google Scholar] [CrossRef]
- Maher, P. A comparison of the neurotrophic activities of the flavonoid fisetin and some of its derivatives. Free Radic Res 2006, 40, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. The flavonoid fisetin promotes nerve cell survival from trophic factor withdrawal by enhancement of proteasome activity. Arch Biochem Biophys 2008, 476, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Maher, P. Modulation of multiple pathways involved in the maintenance of neuronal function during aging by fisetin. Genes Nutr 2009, 4, 297–307. [Google Scholar] [CrossRef]
- Ehren, J.L.; Maher, P. Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem Pharmacol 2013, 85, 1816–1826. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Quehenberger, O.; Maher, P. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer's disease transgenic mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef]
- Currais, A.; Farrokhi, C.; Dargusch, R.; Armando, A.; Quehenberger, O.; Schubert, D.; Maher, P. Fisetin Reduces the Impact of Aging on Behavior and Physiology in the Rapidly Aging SAMP8 Mouse. J Gerontol A Biol Sci Med Sci 2018, 73, 299–307. [Google Scholar] [CrossRef]
- Cho, I.; Song, H.O.; Cho, J.H. Flavonoids mitigate neurodegeneration in aged Caenorhabditis elegans by mitochondrial uncoupling. Food Sci Nutr 2020, 8, 6633–6642. [Google Scholar] [CrossRef]
- Das, J.; Singh, R.; Ladol, S.; Nayak, S.K.; Sharma, D. Fisetin prevents the aging-associated decline in relative spectral power of alpha, beta and linked MUA in the cortex and behavioral alterations. Exp Gerontol 2020, 138, 111006. [Google Scholar] [CrossRef]
- Xiao, S.; Lu, Y.; Wu, Q.; Yang, J.; Chen, J.; Zhong, S.; Eliezer, D.; Tan, Q.; Wu, C. Fisetin inhibits tau aggregation by interacting with the protein and preventing the formation of beta-strands. Int J Biol Macromol 2021, 178, 381–393. [Google Scholar] [CrossRef]
- Singh, S.; Garg, G.; Singh, A.K.; Bissoyi, A.; Rizvi, S.I. Fisetin, a potential caloric restriction mimetic, attenuates senescence biomarkers in rat erythrocytes. Biochem Cell Biol 2019, 97, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Khan, A.; Ali, W.; Jo, M.H.; Park, J.; Ikram, M.; Kim, M.O. Fisetin Rescues the Mice Brains Against D-Galactose-Induced Oxidative Stress, Neuroinflammation and Memory Impairment. Front Pharmacol 2021, 12, 612078. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Noh, J.S.; Jung, Y.; Leem, S.H.; Hyun, J.W.; Chang, Y.C.; Kwon, T.K.; Kim, G.Y.; Lee, H.; Choi, Y.H. Fisetin Attenuated Oxidative Stress-Induced Cellular Damage in ARPE-19 Human Retinal Pigment Epithelial Cells Through Nrf2-Mediated Activation of Heme Oxygenase-1. Front Pharmacol 2022, 13, 927898. [Google Scholar] [CrossRef]
- Park, C.; Cha, H.J.; Kim, D.H.; Kwon, C.Y.; Park, S.H.; Hong, S.H.; Bang, E.; Cheong, J.; Kim, G.Y.; Choi, Y.H. Fisetin Protects C2C12 Mouse Myoblasts from Oxidative Stress-Induced Cytotoxicity through Regulation of the Nrf2/HO-1 Signaling. J Microbiol Biotechnol 2023, 33, 591–599. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: from discovery to translation. J Intern Med 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Prata, L.; Huggins, M.A.; Pierson, M.; Zhang, L.; O'Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics reduce coronavirus-related mortality in old mice. Science 2021, 373. [Google Scholar] [CrossRef]
- Elsallabi, O.; Patruno, A.; Pesce, M.; Cataldi, A.; Carradori, S.; Gallorini, M. Fisetin as a Senotherapeutic Agent: Biopharmaceutical Properties and Crosstalk between Cell Senescence and Neuroprotection. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Liu, L.; Yue, X.; Sun, Z.; Hambright, W.S.; Feng, Q.; Cui, Y.; Huard, J.; Robbins, P.D.; Wang, Z.; Mu, X. Senolytic elimination of senescent macrophages restores muscle stem cell function in severely dystrophic muscle. Aging (Albany NY) 2022, 14, 7650–7661. [Google Scholar] [CrossRef]
- Liu, L.; Yue, X.; Sun, Z.; Hambright, W.S.; Wei, J.; Li, Y.; Matre, P.; Cui, Y.; Wang, Z.; Rodney, G.; et al. Reduction of senescent fibro-adipogenic progenitors in progeria-aged muscle by senolytics rescues the function of muscle stem cells. J Cachexia Sarcopenia Muscle 2022, 13, 3137–3148. [Google Scholar] [CrossRef] [PubMed]
- Mullen, M.; Nelson, A.L.; Goff, A.; Billings, J.; Kloser, H.; Huard, C.; Mitchell, J.; Hambright, W.S.; Ravuri, S.; Huard, J. Fisetin Attenuates Cellular Senescence Accumulation During Culture Expansion of Human Adipose-Derived Stem Cells. Stem Cells 2023. [Google Scholar] [CrossRef]
- Li, S.; Livingston, M.J.; Ma, Z.; Hu, X.; Wen, L.; Ding, H.F.; Zhou, D.; Dong, Z. Tubular cell senescence promotes maladaptive kidney repair and chronic kidney disease after cisplatin nephrotoxicity. JCI Insight 2023, 8. [Google Scholar] [CrossRef]
- Ju, H.Y.; Kim, J.; Han, S.J. The flavonoid fisetin ameliorates renal fibrosis by inhibiting SMAD3 phosphorylation, oxidative damage, and inflammation in ureteral obstructed kidney in mice. Kidney Res Clin Pract 2023. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Sivalingam, K.; Shibu, M.A.; Peramaiyan, R.; Day, C.H.; Shen, C.Y.; Lai, C.H.; Chen, R.J.; Viswanadha, V.P.; Chen, Y.F.; et al. Protective effect of Fisetin against angiotensin II-induced apoptosis by activation of IGF-IR-PI3K-Akt signaling in H9c2 cells and spontaneous hypertension rats. Phytomedicine 2019, 57, 1–8. [Google Scholar] [CrossRef]
- Yan, L.; Jia, Q.; Cao, H.; Chen, C.; Xing, S.; Huang, Y.; Shen, D. Fisetin ameliorates atherosclerosis by regulating PCSK9 and LOX-1 in apoE(-/-) mice. Exp Ther Med 2021, 21, 25. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Sung, J.Y.; Kang, Y.J.; Choi, H.C. Fisetin alleviates cellular senescence through PTEN mediated inhibition of PKCdelta-NOX1 pathway in vascular smooth muscle cells. Arch Gerontol Geriatr 2023, 108, 104927. [Google Scholar] [CrossRef]
- Gao, X.; Usas, A.; Proto, J.D.; Lu, A.; Cummins, J.H.; Proctor, A.; Chen, C.W.; Huard, J. Role of donor and host cells in muscle-derived stem cell-mediated bone repair: differentiation vs. paracrine effects. FASEB J 2014, 28, 3792–3809. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res 2012, 40, e115. [Google Scholar] [CrossRef]
- Koressaar, T.; Remm, M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007, 23, 1289–1291. [Google Scholar] [CrossRef]
- Wissler Gerdes, E.O.; Zhu, Y.; Weigand, B.M.; Tripathi, U.; Burns, T.C.; Tchkonia, T.; Kirkland, J.L. Cellular senescence in aging and age-related diseases: Implications for neurodegenerative diseases. Int Rev Neurobiol 2020, 155, 203–234. [Google Scholar] [CrossRef]
- Khosla, S.; Farr, J.N.; Tchkonia, T.; Kirkland, J.L. The role of cellular senescence in ageing and endocrine disease. Nat Rev Endocrinol 2020, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Tchkonia, T.; Jiang, J.; Kirkland, J.L.; Sun, Y. Targeting Senescent Cells for a Healthier Aging: Challenges and Opportunities. Adv Sci (Weinh) 2020, 7, 2002611. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol 2022, 18, 611–627. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A 2015, 112, E6301–E6310. [Google Scholar] [CrossRef]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 2015, 4, e12997. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Maher, P.A. Using Plants as a Source of Potential Therapeutics for the Treatment of Alzheimer's Disease. Yale J Biol Med 2020, 93, 365–373. [Google Scholar]
- Maher, P. Preventing and Treating Neurological Disorders with the Flavonol Fisetin. Brain Plast 2021, 6, 155–166. [Google Scholar] [CrossRef]
- Ravula, A.R.; Teegala, S.B.; Kalakotla, S.; Pasangulapati, J.P.; Perumal, V.; Boyina, H.K. Fisetin, potential flavonoid with multifarious targets for treating neurological disorders: An updated review. Eur J Pharmacol 2021, 910, 174492. [Google Scholar] [CrossRef]
- Gruendler, R.; Hippe, B.; Sendula Jengic, V.; Peterlin, B.; Haslberger, A.G. Nutraceutical Approaches of Autophagy and Neuroinflammation in Alzheimer's Disease: A Systematic Review. Molecules 2020, 25. [Google Scholar] [CrossRef]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.; Gonzales, M.; Garbarino, V.; Kautz, T.; Palavicini, J.; Lopez-Cruzan, M.; Dehkordi, S.K.; Mathews, J.; Zare, H.; Xu, P.; et al. Senolytic therapy to modulate the progression of Alzheimer's Disease (SToMP-AD) - Outcomes from the first clinical trial of senolytic therapy for Alzheimer's disease. Res Sq 2023. [Google Scholar] [CrossRef]
- Antignano, I.; Liu, Y.; Offermann, N.; Capasso, M. Aging microglia. Cell Mol Life Sci 2023, 80, 126. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.Y.; McNeely, T.L.; Baker, D.J. Untangling senescent and damage-associated microglia in the aging and diseased brain. FEBS J 2023, 290, 1326–1339. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.Y.; Chang, P.C.; Shen, Y.C.; Lin, C.; Tsai, C.F.; Chen, J.H.; Yeh, W.L.; Wu, L.H.; Lin, H.Y.; Liu, Y.S.; et al. Regulatory effects of fisetin on microglial activation. Molecules 2014, 19, 8820–8839. [Google Scholar] [CrossRef]
- Chen, C.; Yao, L.; Cui, J.; Liu, B. Fisetin Protects against Intracerebral Hemorrhage-Induced Neuroinflammation in Aged Mice. Cerebrovasc Dis 2018, 45, 154–161. [Google Scholar] [CrossRef]
- Cordaro, M.; D'Amico, R.; Fusco, R.; Peritore, A.F.; Genovese, T.; Interdonato, L.; Franco, G.; Arangia, A.; Gugliandolo, E.; Crupi, R.; et al. Discovering the Effects of Fisetin on NF-kappaB/NLRP-3/NRF-2 Molecular Pathways in a Mouse Model of Vascular Dementia Induced by Repeated Bilateral Carotid Occlusion. Biomedicines 2022, 10. [Google Scholar] [CrossRef]
- Maher, P. Modulation of the Neuroprotective and Anti-inflammatory Activities of the Flavonol Fisetin by the Transition Metals Iron and Copper. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Liu, C.; Xue, R.; Wang, Y.; Sun, Y.; Liang, Z.; Fan, W.; Jiang, J.; Zhao, J.; Su, Q.; et al. Fisetin inhibits cardiac hypertrophy by suppressing oxidative stress. J Nutr Biochem 2018, 62, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Molagoda, I.M.N.; Athapaththu, A.; Choi, Y.H.; Park, C.; Jin, C.Y.; Kang, C.H.; Lee, M.H.; Kim, G.Y. Fisetin Inhibits NLRP3 Inflammasome by Suppressing TLR4/MD2-Mediated Mitochondrial ROS Production. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Gross images H&E staining of brain tissues after Fisetin treatment. A. Cross sectional views of cerebellum, left hemisphere of cerebral cortex at different axial level. Red arrows indicated white matter, green arrows indicated gray matter, pink arrows pointed to hippocampus. B. H&E staining of cerebral cortex. C. H&E staining of cerebellum for gray matter and white matter showed different cellularity. D. H&E staining of hippocampus at CA1, CA3 and DG area. Fisetin treatment did not significantly affect general morphology of different regions.
Figure 1.
Gross images H&E staining of brain tissues after Fisetin treatment. A. Cross sectional views of cerebellum, left hemisphere of cerebral cortex at different axial level. Red arrows indicated white matter, green arrows indicated gray matter, pink arrows pointed to hippocampus. B. H&E staining of cerebral cortex. C. H&E staining of cerebellum for gray matter and white matter showed different cellularity. D. H&E staining of hippocampus at CA1, CA3 and DG area. Fisetin treatment did not significantly affect general morphology of different regions.
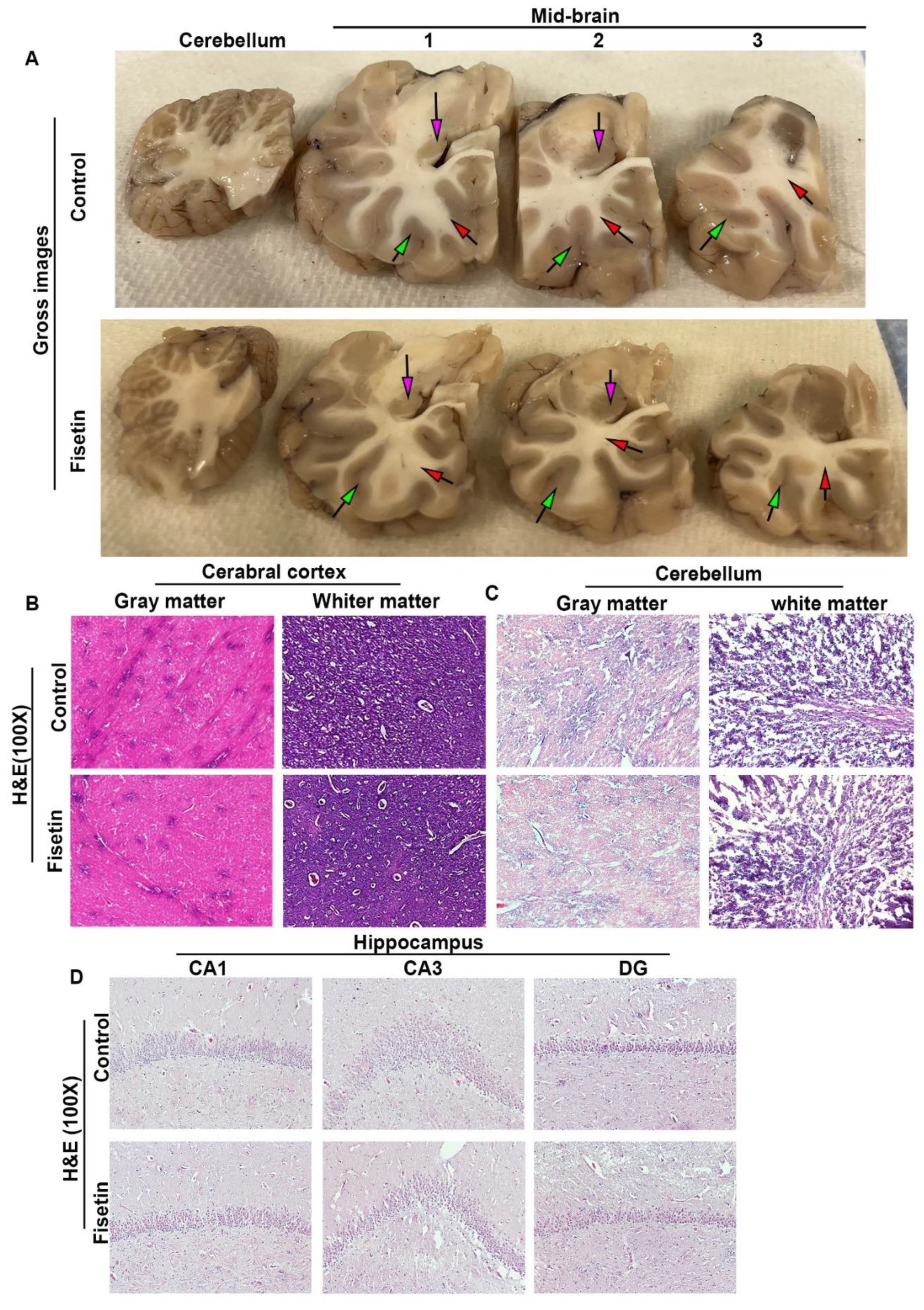
Figure 2.
Effects of fisetin treatment on sheep brain senescent cells of different regions. A. Brain cortex SA-β-Gal staining of gray matter region showed many blue stained cells that are mainly large neurons. Nuclear did not stain. B. Quantification of SA-β-Gal positive cells. C. Brain cortex SA-β-Gal staining in the white matter region. D. Quantification of SA-β-Gal positive cells in the brain cortex white matter. E. Cerebellum gray matter SA-β-Gal staining. F. Quantification of SA-β-Gal positive cells in the cerebellum gray matter. G. Cerebellum white matter SA-β-Gal staining. H. Quantification of SA-β-Gal positive cells in the white matter of cerebellum. Insets in each image highlighted SA-β-Gal positive cells. Scale bars=100µm. Exact P values are indicated between group bars.
Figure 2.
Effects of fisetin treatment on sheep brain senescent cells of different regions. A. Brain cortex SA-β-Gal staining of gray matter region showed many blue stained cells that are mainly large neurons. Nuclear did not stain. B. Quantification of SA-β-Gal positive cells. C. Brain cortex SA-β-Gal staining in the white matter region. D. Quantification of SA-β-Gal positive cells in the brain cortex white matter. E. Cerebellum gray matter SA-β-Gal staining. F. Quantification of SA-β-Gal positive cells in the cerebellum gray matter. G. Cerebellum white matter SA-β-Gal staining. H. Quantification of SA-β-Gal positive cells in the white matter of cerebellum. Insets in each image highlighted SA-β-Gal positive cells. Scale bars=100µm. Exact P values are indicated between group bars.
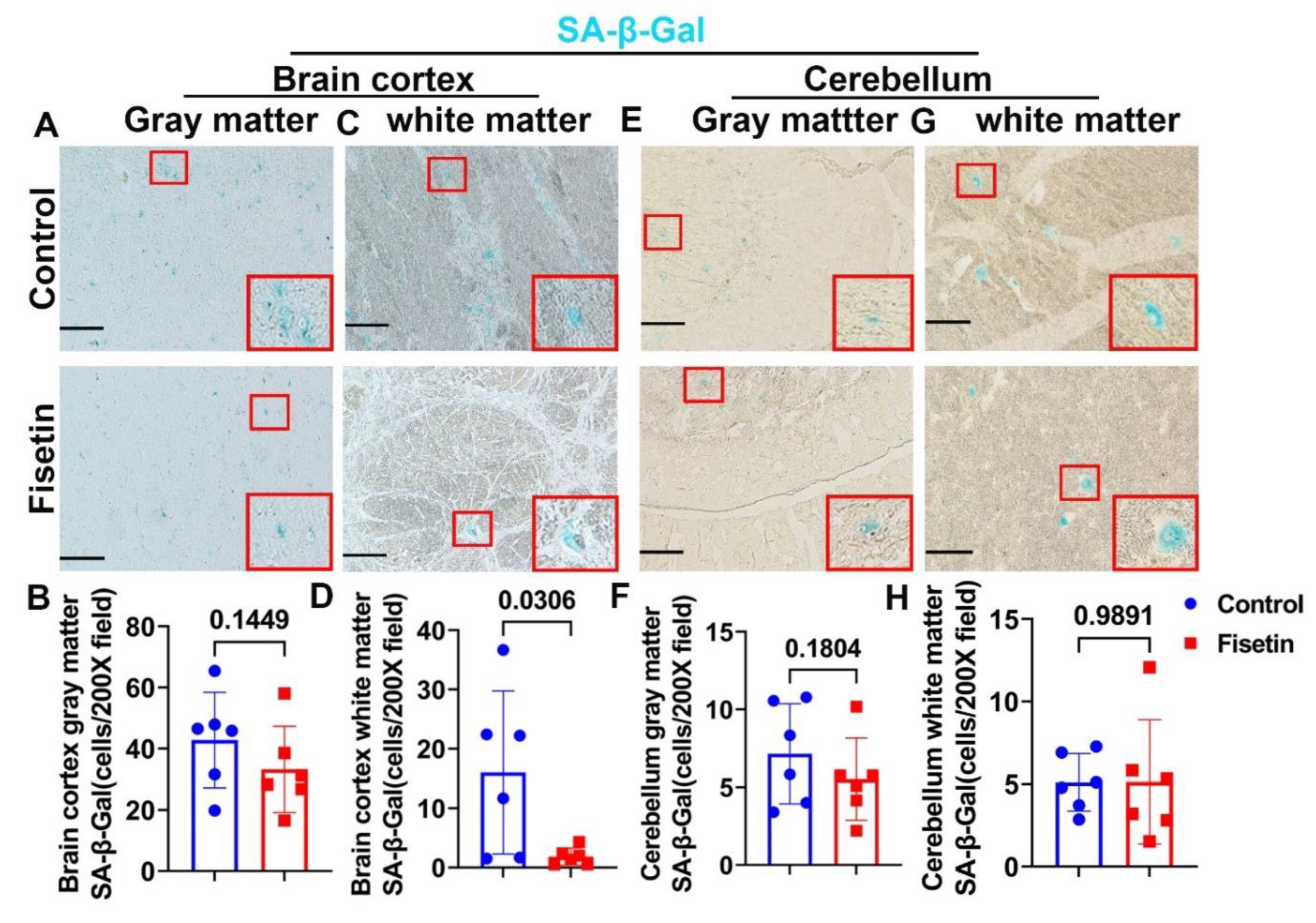
Figure 3.
Identification senescent cells of brain cortex. A. P16 and NEUN double immunofluorescent staining for neurons for brain cortex gray and white matter. P16 stained green in nuclei. NEUN labeled neurons as red color in the cytoplasm. Nuclei stained blue with DAPI. There are many red stained large neurons in the gray matter while very few in the white matter. Green channel showed P16+ cells in gray matter are bigger and diffused while small and strong signal in white matter. B. Quantification of P16+/NEUN+/Total NEUN+ cells percentage in the gray matter. C. Quantification of P16+/NEUN-/Total DAPI+ cells in the white matter. D. P16 and GFAP double immunofluorescent staining of astrocytes in brain cortex gray and white matter. P16 stained green in nuclei, GFAP stained red for astrocytes with many processes. E. Quantification of P16+/GFAP+/Total GFAP+ cells percentage in gray matter. F. Quantification of P16+/GFAP+/Total GFAP+ cells percentage in white matter. G. P16 and IBA1 double immunofluorescent staining for microglia cells. P16 stained nuclei in green. IBA1 stained microglia cells in red. H. Quantification of P16+/IBA1+/Total IBA1+cells percentage in gray matter. I.Quantification of P16+/IBA1+/Total IBA1+ cells percentage in white matter. Insets highlighted positive cells. Scale bars=100µm. Exact P values are between group bars.
Figure 3.
Identification senescent cells of brain cortex. A. P16 and NEUN double immunofluorescent staining for neurons for brain cortex gray and white matter. P16 stained green in nuclei. NEUN labeled neurons as red color in the cytoplasm. Nuclei stained blue with DAPI. There are many red stained large neurons in the gray matter while very few in the white matter. Green channel showed P16+ cells in gray matter are bigger and diffused while small and strong signal in white matter. B. Quantification of P16+/NEUN+/Total NEUN+ cells percentage in the gray matter. C. Quantification of P16+/NEUN-/Total DAPI+ cells in the white matter. D. P16 and GFAP double immunofluorescent staining of astrocytes in brain cortex gray and white matter. P16 stained green in nuclei, GFAP stained red for astrocytes with many processes. E. Quantification of P16+/GFAP+/Total GFAP+ cells percentage in gray matter. F. Quantification of P16+/GFAP+/Total GFAP+ cells percentage in white matter. G. P16 and IBA1 double immunofluorescent staining for microglia cells. P16 stained nuclei in green. IBA1 stained microglia cells in red. H. Quantification of P16+/IBA1+/Total IBA1+cells percentage in gray matter. I.Quantification of P16+/IBA1+/Total IBA1+ cells percentage in white matter. Insets highlighted positive cells. Scale bars=100µm. Exact P values are between group bars.
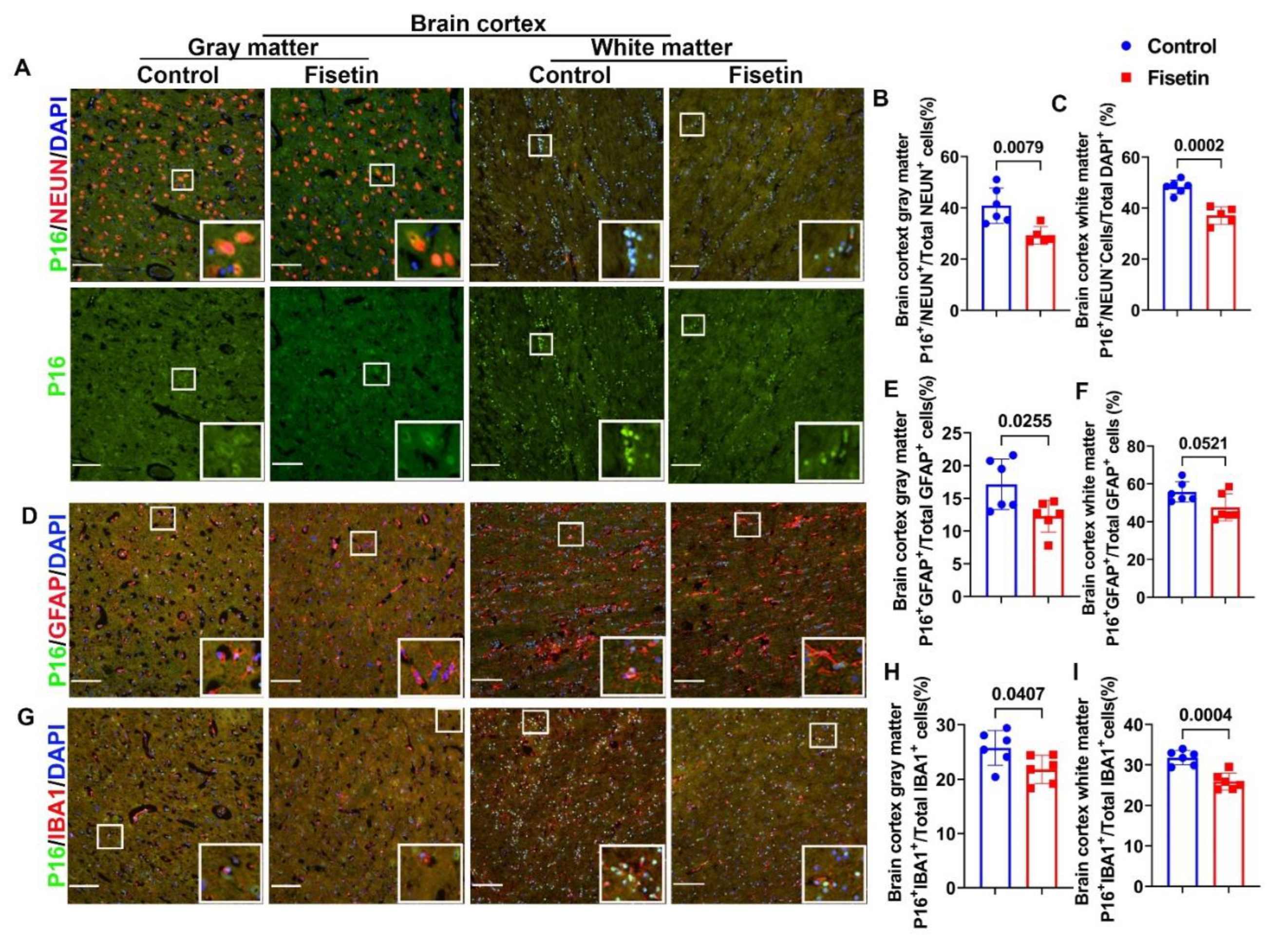
Figure 4.
Effects of fiestin treatment on the hippocampus. A. NEUN and P16 double immunofluorescent staining for hippocampus focus on CA area. NEUN+ cells were stained in red and many colocalized with P16 (Merged channel). P16+ cells were stained in green in the nuclei (Green channel). B. Quantification of P16+/NEUN+/Total NEUN+ cells in CA1-4 area. C. GFAP and P16 double immunofluorescent staining in hippocampus. GFAP+ cells were stained in red color with many processes and mainly located in the non-CA area. P16 positive cells were stained in green in the nuclei. D. Quantification of P16+GFAP+/Total GFAP+ cells in non-CA area of hippocampus. E. IBA1 and P16 double immunofluorescent staining in hippocampus. IBA1 positive cells were stained in red and mainly located in the non-CA area. P16+ cells were stained in green in nuclei. F. Quantification of P16+/IBA1+/Total IBA1+ cells in the non-CA area of hippocampus. Insets in each images highlighted positive cells of each staining. Scale bars=100µm. Exact P values are indicated between group bars.
Figure 4.
Effects of fiestin treatment on the hippocampus. A. NEUN and P16 double immunofluorescent staining for hippocampus focus on CA area. NEUN+ cells were stained in red and many colocalized with P16 (Merged channel). P16+ cells were stained in green in the nuclei (Green channel). B. Quantification of P16+/NEUN+/Total NEUN+ cells in CA1-4 area. C. GFAP and P16 double immunofluorescent staining in hippocampus. GFAP+ cells were stained in red color with many processes and mainly located in the non-CA area. P16 positive cells were stained in green in the nuclei. D. Quantification of P16+GFAP+/Total GFAP+ cells in non-CA area of hippocampus. E. IBA1 and P16 double immunofluorescent staining in hippocampus. IBA1 positive cells were stained in red and mainly located in the non-CA area. P16+ cells were stained in green in nuclei. F. Quantification of P16+/IBA1+/Total IBA1+ cells in the non-CA area of hippocampus. Insets in each images highlighted positive cells of each staining. Scale bars=100µm. Exact P values are indicated between group bars.
Figure 5.
Semi-quantitative and quantitative Q-PCR results in the brain cortex after Fisetin treatment. A. Electrophoresis images of different target genes. B. Band density quantification relative to GAPDH. C. Q-PCR analysis of antioxidant genes. D. Q-PCR analysis of NEFL and NEFH and GFAP. E. Q-PCR analysis of senescent related genes. F. Q-PCR analysis of SASP genes. G. Q-PCR analysis of inflammasome genes. Exact P values are shown between group bars.
Figure 5.
Semi-quantitative and quantitative Q-PCR results in the brain cortex after Fisetin treatment. A. Electrophoresis images of different target genes. B. Band density quantification relative to GAPDH. C. Q-PCR analysis of antioxidant genes. D. Q-PCR analysis of NEFL and NEFH and GFAP. E. Q-PCR analysis of senescent related genes. F. Q-PCR analysis of SASP genes. G. Q-PCR analysis of inflammasome genes. Exact P values are shown between group bars.
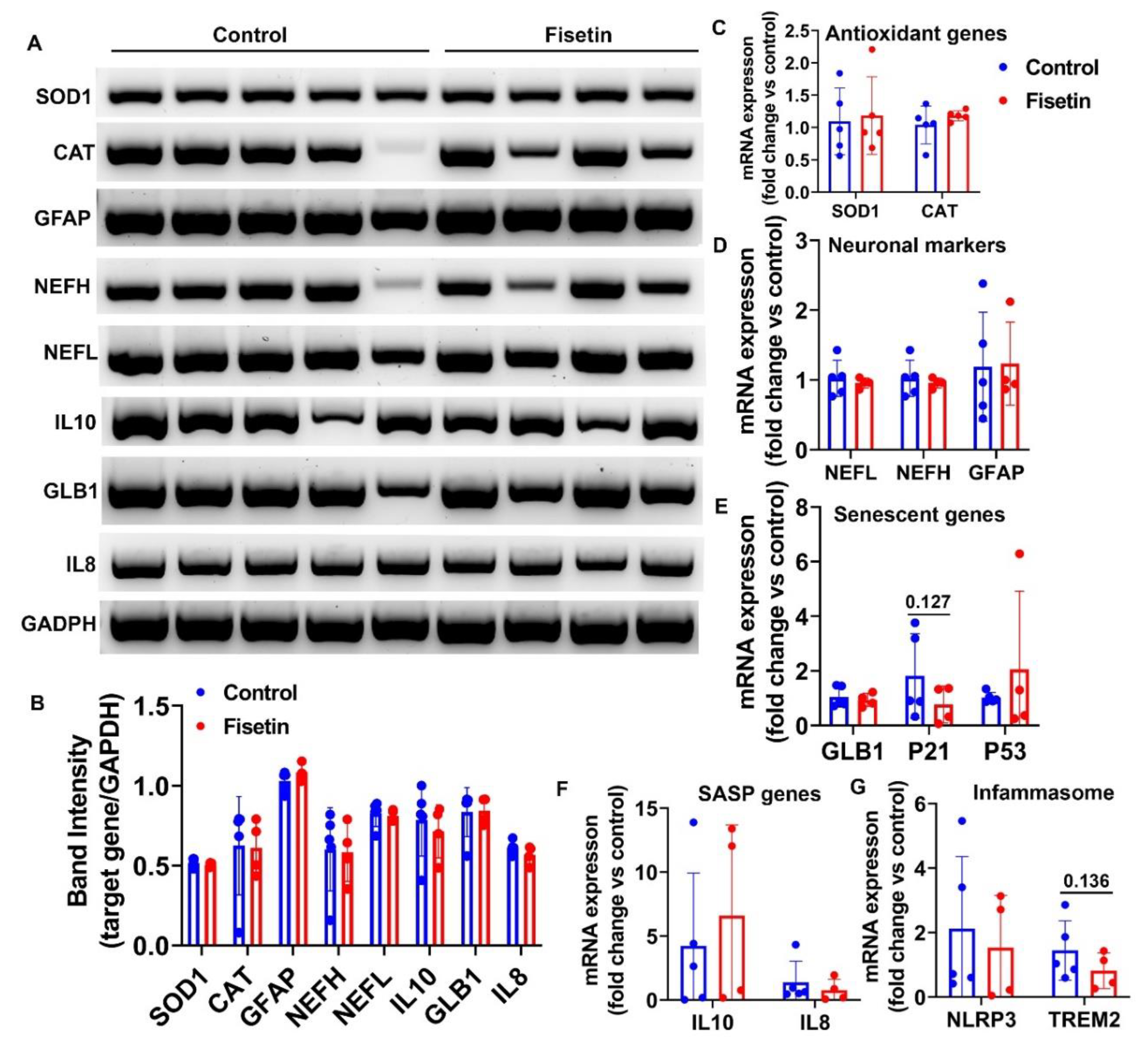
Figure 6.
Q-PCR analysis of gene expression of sheep heart tissues after fisetin treatment. A. Q-PCR results of antioxidant genes. B. Q-PCR analysis of senescent related genes. C. mRNA expression of SASP genes. D. mRNA expression of inflammasome genes. Exact P values are shown between group bars.
Figure 6.
Q-PCR analysis of gene expression of sheep heart tissues after fisetin treatment. A. Q-PCR results of antioxidant genes. B. Q-PCR analysis of senescent related genes. C. mRNA expression of SASP genes. D. mRNA expression of inflammasome genes. Exact P values are shown between group bars.
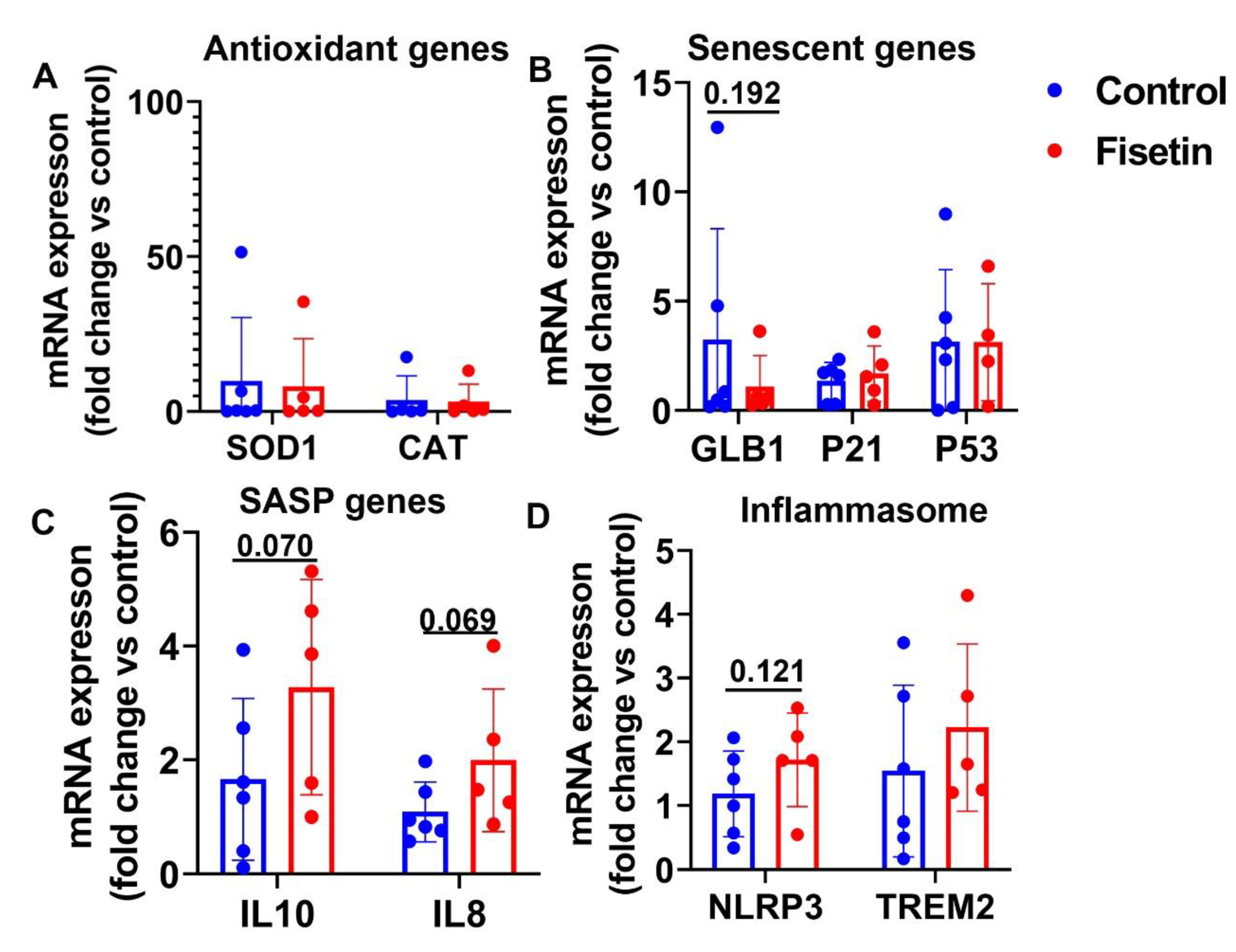
Figure 7.
Gene expressions in the spleen tissues after fisetin treatment. A. Antioxidant genes mRNA expression. B. Senescent related genes mRNA expression. C. SASP gene mRNA expression. D. Inflammasome gene expression. Exact P values are shown between group bars.
Figure 7.
Gene expressions in the spleen tissues after fisetin treatment. A. Antioxidant genes mRNA expression. B. Senescent related genes mRNA expression. C. SASP gene mRNA expression. D. Inflammasome gene expression. Exact P values are shown between group bars.
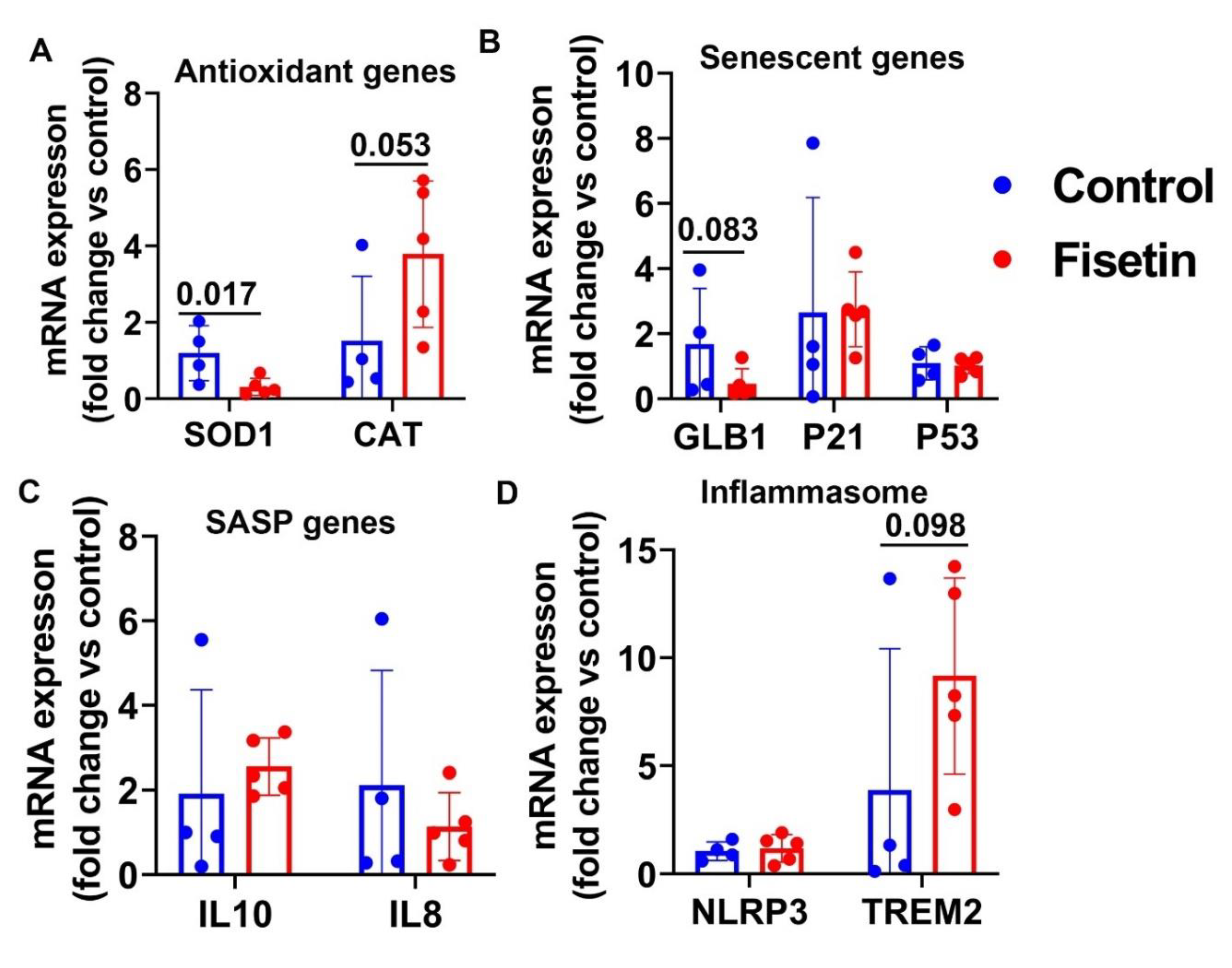
Figure 8.
mRNA Gene expression of sheep bone marrow after fisetin treatment. A. Antioxidant genes mRNA expression. B. Senescent related genes. C. mRNA expression of P53. D. SASP genes. E. Inflammasome genes. Exact P values are shown between group bars.
Figure 8.
mRNA Gene expression of sheep bone marrow after fisetin treatment. A. Antioxidant genes mRNA expression. B. Senescent related genes. C. mRNA expression of P53. D. SASP genes. E. Inflammasome genes. Exact P values are shown between group bars.
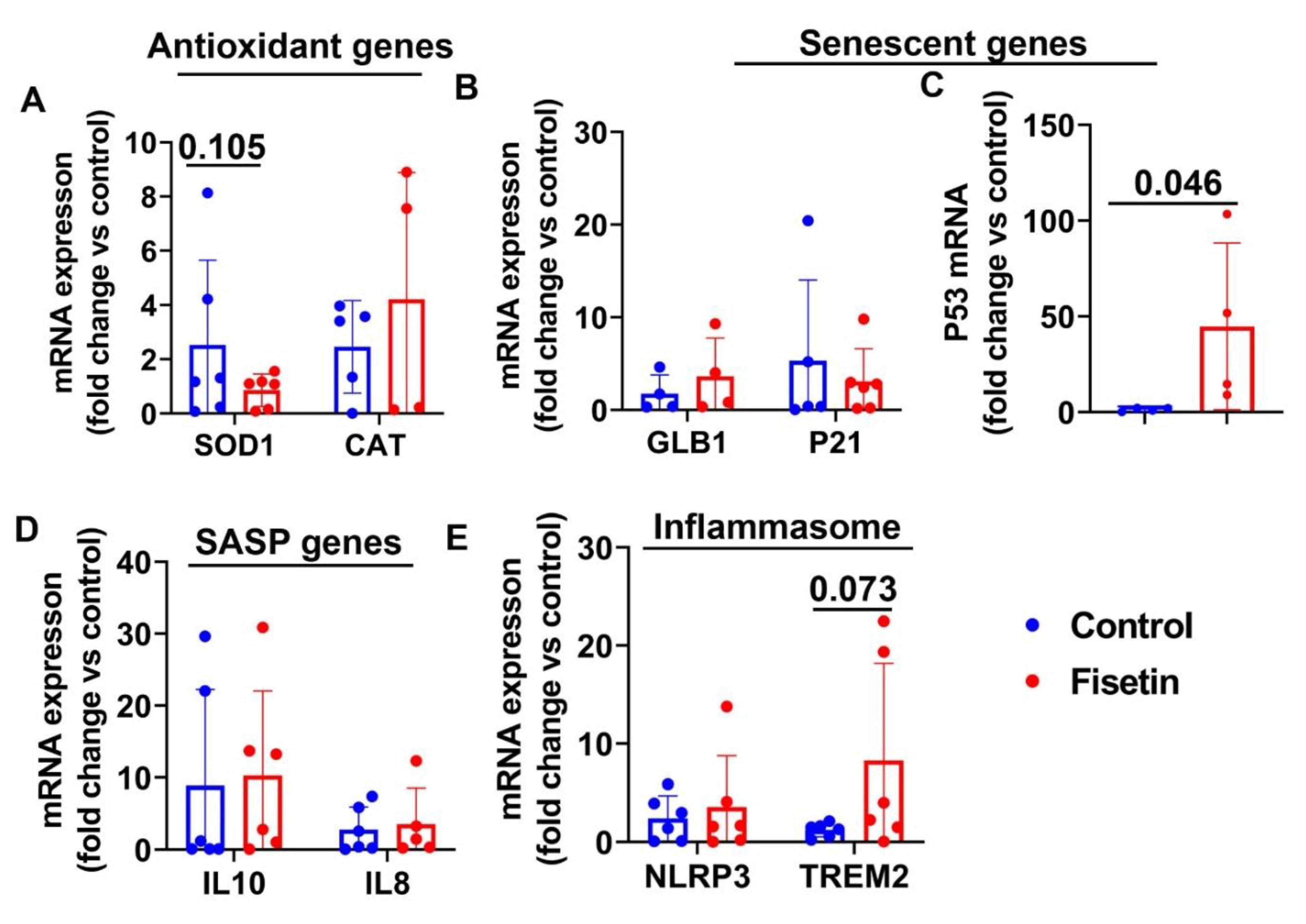

Table 1.
Primers information.
| Accession # | Gene ID | Forward primer(5’-3’) | Reverse primer(5’-3’) | Product size(bp) |
|---|---|---|---|---|
| XM_018065254.1 | Goat GFAP | caggatctgctcaacgtcaa | atctccacggtcttcaccac | 197 |
| XM_015102219.3 | Sheep GLB1 | agtccccctacctacgcact | ggtcgaagtcaccaggatgt | 213 |
| NM_001145185.2 | Sheep SOD1 | ggttccacgtccatcagttt | tttgtcagccttcacattgc | 143 |
| XM_005690077.3 | Goat CAT | ctcgtccaggatgtggtttt | gagcctgattctccagcaac | 215 |
| XM_042234806.1 | Sheep NEFH | acatcgcatcctaccaggag | cagcgatctcaatgtccaga | 135 |
| XM_015093090.3 | Sheep NEFL | aagcgcatagacagcctgat | ctctcggtcagcacagtgaa | 242 |
| HM043737.1 | Sheep GAPDH | acagtcaaggcagagaacgg | ggttcacgcccatcacaaac | 235 |
| X78306.1 | Sheep IL8 | tcgatgccaatgcataaaaa | ttggggtctaagcacacctc | 147 |
| NM_001009327.1 | Sheep IL10 | tgttgacccagtctctgctg | ttcacgtgctccttgatgtc | 136 |
| FJ943992.1 | Sheep P21 | gagagcgatggaacttcgac | agtggtcctcctgagacgtg | 186 |
| FJ855223.1 | Sheep P53 | cctgctcccgtactcagaag | ctggcagaacagcttgttga | 247 |
| XM_042250404.1 | Sheep NLRP3 | ctgtgcacacggtggtattc | ctctgagtcccaaggctcac | 157 |
| XM_004018807.5 | Sheep TREM2 | agcctttcggaagaggagag | agctggtaacctgggttgtg | 179 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



