
Submitted:
27 June 2023
Posted:
28 June 2023
You are already at the latest version
Abstract
Thalassemia is a heterogeneous congenital hemoglobinopathy common in the Mediterranean region, Middle East, Indian subcontinent, and Southeast Asia with increasing incidence in Northern Europe and North America due to immigration. Iron overloading is one of the major long-term complications in patients with thalassemia and can lead to organ damage and carcin-ogenesis. Hepatocellular carcinoma (HCC) is one of the most common malignancies in both transfusion-dependent thalassemia (TDT) and non-transfusion-dependent thalassemia (NTDT). The incidence of HCC in patients with thalassemia has increased over time, as better chelation therapy confers a sufficiently long lifespan for the development of HCC. The mechanisms of iron overloading-associated HCC development include the increased reactive oxygen species (ROS), inflammation cytokines, dysregulated hepcidin, and ferroportin metabolism. The treat-ment of HCC in patients with thalassemia was basically similar to those in general population. However, due to the younger age of HCC onset in thalassemia, regular surveillance for HCC development is mandatory in TDT and NTDT. Other supplemental therapy and experience of novel treatment for HCC in thalassemia population were also reviewed in this article.
Keywords:
Hepatocellular carcinoma (HCC)
; Thalassemia
; Iron
; Transfusion-Dependent Thalassemia (TDT)
; Non-Transfusion-Dependent Thalassemia (NTDT)
; Reactive Oxygen Species (ROS)
1. Introduction
Thalassemia is a heterogeneous congenital hemoglobinopathy common in the Mediterranean region, Middle East, Indian subcontinent, and Southeast Asia [1,2,3,4]. Immigration has led to the gradual increase in the incidence of thalassemia in Northern Europe and North America [5,6]. Thalassemia can be classified based on defects in the globin chain, α-globin chain, or β-globin chain as α-thalassemia or β-thalassemia, respectively. Thalassemia is the most common monogenic disease worldwide, with approximately 1.5% of the population carrying the β-thalassemia allele and 5% carrying the α-thalassemia allele [5,7]. Clinical manifestations include anemia, jaundice, and iron overload, and disease severity ranges from near-normal without complications to requiring lifelong transfusion support [3,6]. Patients with thalassemia requiring transfusion support are classified into transfusion-dependent thalassemia (TDT) and non-transfusion-dependent thalassemia (NTDT) based on the frequency and regularity required. The lifespan will be less than 10–15 years without transfusions, and with transfusion therapy, it can be extended to approximately 50 years depending on the management of iron overloading resulting from repeated red cell transfusions, which damage organ functions, especially heart, liver, lung, pancreas, and pituitary glands [3,8,9]. Besides transfusions, increased dietary iron absorption is one of the major causes of liver iron overloading, typically in patients with NTDT [6,8]. Liver iron deposit lead to liver fibrosis, cirrhosis, and HCC [8]. The relationship between iron overload and HCC development is clear in that the incidence of HCC is much higher in patients with hereditary iron overload than in the general population [10]. A prospective study found that the incidence of HCC in patients with a beta-thalassemia major was approximately the same as the risk for HCC in the general population but with a significantly younger age at HCC diagnosis [11]. The incidence of HCC in patients with thalassemia has increased over time as better chelation therapy confers a sufficiently long lifespan for HCC development [12]. According to an analysis based on the Taiwan Health Insurance Longitudinal Database (1998–2010), patients with thalassemia had a significantly higher risk for abdominal cancer (aHR = 1.96, 95% CI 1.22–3.15) compared with the comparison group. Patients with thalassemia who received blood transfusions were 9.12 times more likely to develop abdominal cancer than those who did not receive blood transfusions [13]. Iron accumulation can also lead to various endocrinopathies, such as hypogonadism, impaired pancreatic excrete function, hypothyroidism [14]. This review discusses the pathogenesis, molecular mechanisms, management, and perspectives of iron overloading associated with HCC in patients with thalassemia.
2. Risk factors for HCC
2.1. Risk factors for HCC in the general population- focus on hepatitis virus, fatty liver disease
HCC is the sixth common cancer worldwide and the third leading cause of cancer death. The incidence in general population varies widely in different world regions, with the highest in Mongolia (85.6 per 10,0000) and the lowest in Sri Lanka (1.2 per 10,0000) [15]. Before the advent of vaccine against hepatitis B virus (HBV), hepatitis B and C viruses (HCV) were the main risk factors for HCC in the past 40 years. The risk of HCC development in HBV carriers were ranged from 9.6% in Hepatitis B surface antigen (HBsAg) positive patients to 60% in double positive (HBsAg + Hepatitis B e antigen (HBeAg)) patients [16]. In HCV-related cirrhosis patients, the risk of progression to HCC is estimated to be 2%–6% each year [17].
Nowadays, with modern high-fat/sugar diet, sedentary lifestyle and increasing alcohol consumption, alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD) are gradually recognized as risk factors for HCC nowadays. Alcohol-related cirrhosis accounted for about 15–30% of HCC cases in report [18]. The pathogenesis from ALD to HCC is complex, liver injury from the insult of reactive oxygen species (ROS) and acetaldehyde are contributing factors [16]. In chronic viral hepatitis patients, alcohol intake is associated with higher all-cause mortality and increases HCC development risk then non-alcoholic intake group [19,20]. The global prevalence of NAFLD is estimated about 25% [21] and it is recognized now as the most common cause of cirrhosis and a growing risk factor for HCC [22]. Oxidative stress, chronic inflammation, immune cell infiltration of fatty liver are all factors to HCC formation [23].
2.2. Risk factors for HCC in the thalassemia group: focus on hepatitis virus, fatty liver disease
Because of life-long, frequent blood transfusions, thalassemic patients had higher incidence of acquiring transfusion-transmitted viral infection, especially HBV and HCV. HBsAg positive was estimated in 0.3~5.7% of thalassemic patients [24]. Anti-HCV Ab was estimated 4.4%~85.4% [25]. The actual incidence may varied widely because of different reginal HBV/HCV prevalence.
As mentioned in introduction, Thalassemia major patients had impaired pancreatic function because of iron overload. The prevalence of diabetes mellitus (DM), mostly type 1, and impaired fasting sugar in Thalassemic patients were about 6.54% and 17.21% respectively [26]. NAFLD was deemed as a result of insulin resistance and metabolic syndrome in the past but now a high prevalence in type 1 DM patients is noted. Altered dynamic insulin pulsatile, altered insulin clearance, hyperglucagonemia and hepatic glucagon resistance are parts of NAFLD pathogenesis in type 1 DM. These are slightly different from type 2 DM and metabolic syndrome patients [27]. Although there are no formal statistics of NAFLD prevalence in Thalassemic patients. We inferred that owing to type 1 DM and iron overload (discussed next), the prevalence of NAFLD is high in thalassemic patients.
2.3. Risk factors for HCC in the thalassemia group: focus on iron overload
Besides the abovementioned risk factors, a special concern regarding HCC in the thalassemia group is iron overload resulting from lifelong blood transfusion and ineffective hematopoiesis. The NTDT group also suffers from iron overload to a lesser degree because of excessive hemolysis and chronic anemia. Iron overload was mainly due to blood transfusion and destroyed red blood cells (RBCs) in excess. To make things worse, patients with thalassemia generally had low hepcidin levels because of chronic anemia so that their enterocytes still absorb iron greedily under an iron overload status [28].
Iron, an essential element for most species to live, is the most abundant metal in our planet [29]. Iron can easily gain or lose electrons by switching between the iron form (Fe2+) and the iron form (Fe3+). This characteristic enables iron to participate in the respiratory chain reaction, electron transfer, and signal transduction in cells and functions as an oxygen carrier in hemoglobin and myoglobin. The human body absorbs iron from food through enterocytes. They mostly go to the bone marrow for hemoglobin formation. RBCs are the biggest iron reservoir in our body. After meeting the need for basic cellular function, surplus iron is mainly stored in the liver. Iron is such a precious metal for humans that we did not evolve the ability to excrete iron out of our body actively. Instead, we decrease our iron store reluctantly (passively) from blood loss. Menstruation is the only non-traumatic and natural way of losing blood [30,31]. To sum up, it is easy to accumulate iron in the body and almost impossible to excrete them, except menstruation in women.
From the cellular level, various iron importers exist on the cell membrane, such as transferrin receptor protein 1 (TfR1), transferrin receptor 2 (TfR2), L-type calcium channel (LTCC), divalent metal transporter 1 (DMT1), zinc transporter 8 (ZIP8), and transient Receptor Potential Cation Channel Subfamily C Member 6 (TRPC6); however, ferroportin is the only exporter [30,31]. When thalassemia patients are forced to receive iron (ex. blood transfusion), there is no way for them to excrete excessive iron. In the long-run, more and more iron participated in the body, main in liver, and cause organ damage and affect cell function profoundly.
Iron overload is a risk factor for HCC. As seem in hereditary hemochromatosis patients, they were more likely to have liver cancer comparing to general population [32,33]. It is estimated that iron-overloaded patients have 10.6 times risk to have HCC and even got HCC before cirrhosis development [34]. Furthermore, hepatitis virus and iron can interact with each other and aggravate liver cirrhosis process and HCC formation [35].
3. Pathogenesis of iron overload and HCC
3.1. Iron and inflammation
When intrahepatic iron overflowed, hepatocytes were also stimulated to differentiate into a proinflammatory state through the excessive intracellular ferritin by enhancing the NF-κB expression pathway [36]. Prolonged activated hepatocytes can differentiate into myofibroblasts and lead to neovascularization and extracellular matrix formation. Excessive iron molecules are not only deposited in but also injure hepatocytes. They also affect immune system function. Liver, as the largest reticuloendothelial system in the body, contains the largest population of macrophages (Kupffer cells). Macrophages, as scavengers in the body, phagocytize not only external harmful agents, such as bacteria, but also old, dysfunctional cells, such as senescent RBCs and destroyed hepatocytes. Thus, they are prone to contain excessive amounts of iron, especially in patients with thalassemia. Excessive amounts of iron induces the macrophage to secrete proinflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin-12 (IL-12) [37]. Together, with the proinflammatory status of hepatocytes, complicated chronic inflammatory interaction ensues. Hepatic stellate cells, endothelial cells, and lymphocytes inside the liver are all participated, and cirrhosis begins [38]. Cirrhosis provides an oncogenic environment for HCC formation [39,40,41]. A study of human liver tissue revealed that hepatocytes were polarized to myofibroblasts when the liver iron concentration (LIC) was >60 μmol/g. Cirrhosis ensues when the LIC is >250 μmol/g [42]. Fibrosis developed when the LIC is >400 μmol/g [43].
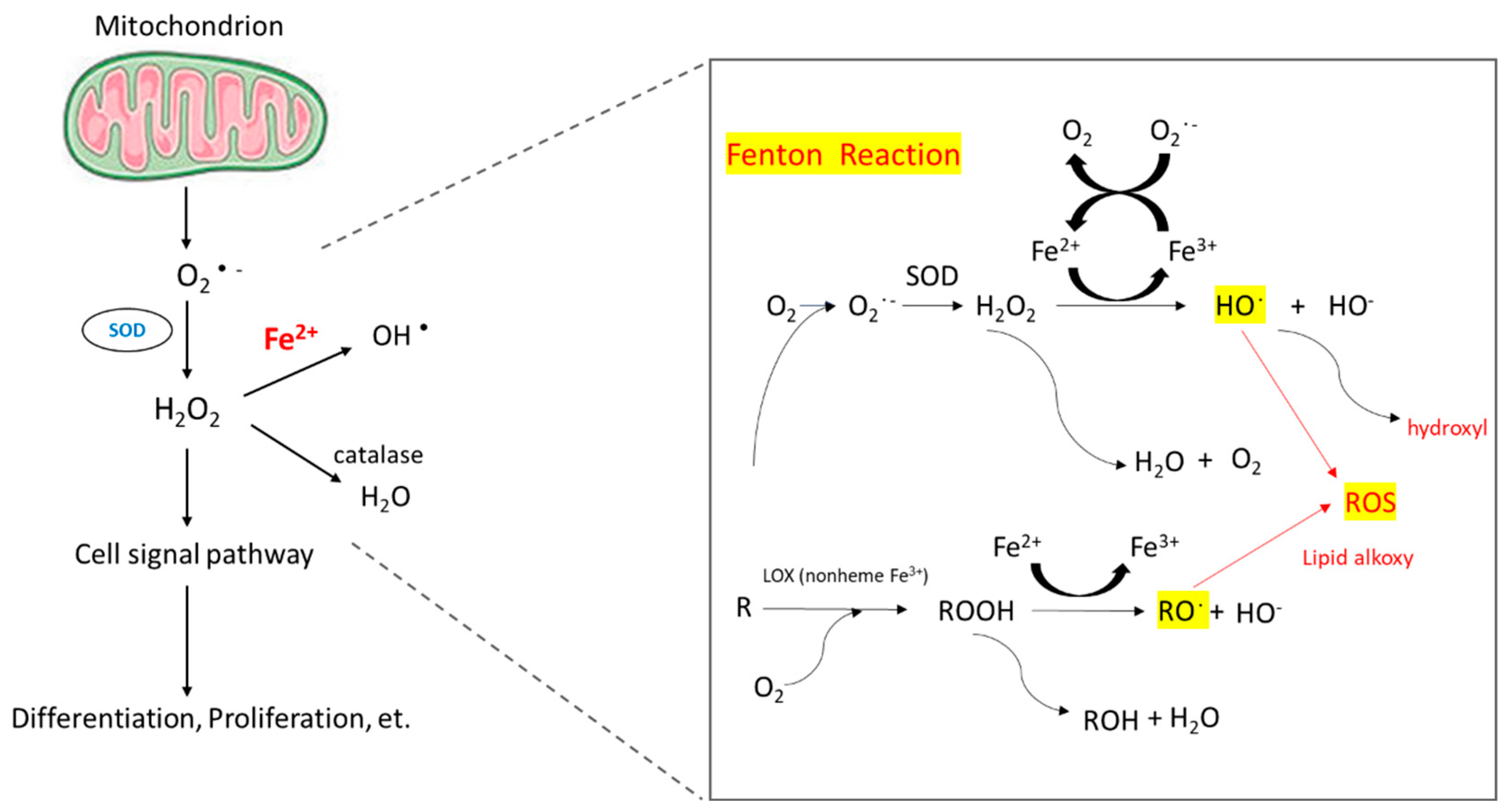
The reaction of peroxides with Fe2+ to yield soluble hydroxyl (HO•) or lipid alkoxy (RO•) radicals is referred to as the Fenton reaction.
3.2. Iron and reactive oxygen species (ROS)
When intracellular iron is excessive, these iron molecules participate in the Fenton reaction (Figure 1) and cause the accumulation of ROS [44]. Healthy cells can detoxicify ROS through non-enzymatic and enzymatic antioxidants, such as glutathione, flavonoids, superoxide reductase, catalase, and glutathione peroxidase. If ROS formation exceeds the detoxicifying ability of cells, excess ROS can cause genomic instability and reprogram cell metabolism toward carcinogenesis [45]. ROS induce hypoxia-inducible factor-1α expression and related angiogenic gene expression and thus promote angiogenesis [46]. ROS can also promote ERK/NF-kB pathway activation and facilitate tumor cell growth, migration and deposition of extracellular matrix [47].
3.3. Hepcidin and ferriportin
Hepcidin is a polypeptide hormone, isolated from plasma and urine samples with antimicrobial activities by two research groups in 2000 [48,49]. It showed that hepcidin is mainly produced by liver, followed by heart, brain and lung [48]. Almost at the same time, Pigeon et al tested the hepatic gene expressions in the iron overload mouse models and showed that hepcidin (HEPC), the gene encoding hepcidin, overexpressed in iron overload hepatic cells. In the report, they also mapped the gene to the human chromosome 19 close to the upstream stimulatory factor-2 (USF2) gene [50]. Lipopolysaccharide, a main component of pathogenic molecules, act on macrophage, including hepatic Kupffer cells, to induce interleukin-6 (IL-6) production, which in turn induces the overexpression of hepcidin mRNA in hepatocytes [51,52]. In summary, iron overloading, infection, and inflammation can induce hepatocyte hepcidin secretion, which leads to hypoferremia and the anemia of chronic inflammation. Anemia and hypoxia suppressed hepcidin expression and results in tissue iron overload. Hepcidin regulates the absorption of dietary iron from the intestine, the release of recycled hemoglobin iron by macrophages, and the movement of stored iron from hepatocytes by blocking iron transport in the intestinal epithelium, the placenta and macrophage [51]. Ferriportin, the only ion exporter in human cells, play a critical role in hepcidin-regulated iron metabolism that ferriportin-hepcidin binding leads to internalization and degradation of ferriportin and due to decreased amounts of ferripotin, iron is trapped inhepatocytes, macrophages, and absorptive enterocytes. In consequences, cytoplasmic iron cumulates and reduce cell uptake of iron. After plasma iron become depleted, mainly through hemoglobin synthesis by red cell precursors in the bone marrow, the hepcidin expression would be suppressed and restore the iron hemostasis system [53].
As a key regulator of iron metabolism, hepcidin levels are influenced in the pathogenesis of anemia. Inefficient erythropoietic anemias, such as thalassemia, whose clinical manifestation is iron overload anemia, iron deficiency anemia, and congenital erythropoiesis, often have low or abnormal hepcidin relative to the anemia [28]. Kijima et al. We examined hepcidin expression in cancerous and noncancerous liver tissues from 40 HCC patients and found that hepcidin mRNA expression was significantly suppressed in cancerous tissues compared to noncancerous liver tissues. It is non-cancerous in HCC patients, regardless of disease state such as tumor differentiation. or time to relapse. There was no significant difference in the mRNA expression of ferripotin and ransferrin receptor 2 (TfR2) between cancerous and non-cancer tissues [54]. On the other hand, Tan et al. found that hepcidin, TfR2, transferrin (Tf), ceruloplasmin (Cp), and iron regulatory protein 1 (IRP1) isolated in 24 HCC patients with chronic HBV infection were downregulated compared to adjacent tumor-free liver tissue and normal liver controls [55]. Similarly, Tseng et al. found that hepcidin, ceruloplasmin, transferrin and transferrin-phosphorus receptor 2 were downregulated, transferrin receptor 1 was upregulated, and hepcidin levels consistently correlated with hepatic iron stores in 50 HCC patients [56]. Kessler et al. showed that incirrhotic tissues, hepcidin expression was lower compared to healthy liver samples in an HBV-related cohort as well as in HCV-infected patients and hepcidin expression was even lower in HCC samples, and in their mouse models, they showed that hepcidin expression is decreased in early hepatocarcinogenesis as well as in a later stage of murine tumorigenesis [57]. Hepcidin also ameliorates liver fibrosis by inhibiting hepatic stellate cells (HSCs) activation which facilitates liver fibrosis via degradation of ferroportin in HSCs, increasing Akt phosphorylationand finally prohibiting TGFb1-inducible Smad3 phosphorylation [58].
The underlying mechanisms of suppressed hepcidin in HCC included the suppression of HAMP, TfR2, HJV, ALK2 and/or circular RNA circ_0004913, upregulations of matriptase-2 and/or GDF15, and inactivation of RUNX3 and/or mutations in TP53 [59]. Hepcidin induced ferriportin dysregulation has been reported associated with increased risk of cancer, including HCC. FPN overexpression has been reported association with a significant reduction in clonogenic ability, tumor regression, and liver metastasis in breast cancer cells [60,61]. Effects of hepcidin downregulation involved increased cancer proliferation via activation of CDK1/STAT3 pathway and JAK/STAT pathway-dependent manner [59,62].
4. Management
4.1. Management of iron overload
Once iron overloaded, patients with thalassemia should receive iron-chelation therapy. There are three iron-chelating drugs commercially available, deferoxamine (DFO) in subcutaneous or intravenous form, deferiprone (DFP) in oral form, and deferasirox (DFX) in oral form. All three drugs are useful in TDT patients, while DFX remains the only approved drug in NTDT patients. Selection of iron chelator should be on the basis of patient age, iron overload profile, presence or absence of comorbidities, patient preference and adherence, and side effects. For TDT patients with serum ferritin ≥1000 ng/mL or after transfusion of 10 units of packed red blood cells, iron-chelation therapy is recommended. For NTDT patients, iron-chelation therapy starts when serum ferritin ≥800 ng/mL or LIC ≥5 mg/g [9,63].
Iron overload and increased non-transferrin-bound iron lead to ROS production and subsequent oxidative damage. It is reasonable that antioxidant therapy will be beneficial to iron overloaded thalassemia patients. Encouraging results from several randomized controlled trials have shown that natural antioxidants such as green tea and curcumin may be helpful; however, studies with longer duration are needed to assess long-term effects [64,65,66,67]. In 2021 Thalassemia International Federation guidelines for the management of TDT, a diet rich in foods high in vitamin E, such as plant oils, eggs, nuts, and cereals, is recommended. To support prolonged use of vitamin E supplements, further research is still needed [68].
4.2. Management of HCC
Because of limited data, the treatment of HCC in patients with thalassemia is primarily based on experience in non-thalassemic patients. Current treatment recommendations for HCC in the general population follow the Barcelona Clinic Liver Cancer (BCLC) staging system, in which patients with HCC are classified into five stages (stage 0: very early HCC, stage A: early HCC, stage B: intermediate HCC, stage C: advanced HCC, and stage D: terminal HCC) according to several prognostic variables, including tumor status, liver function (defined by Child-Pugh class), and performance status. In principle, patients with stage 0 and stage A should undergo surgical resection if they are candidates or local ablation. Liver transplantation is also a potentially curative treatment option for unresectable disease if eligible. For patients with stage B, embolization is the preferred treatment. For patients with advanced disease, systemic therapies will be applied first [69,70,71].
The treatment modalities have been reported and shown to be safe for HCC in thalassemia patients include surgical resection, radiofrequency ablation, microwave ablation, percutaneous ethanol injection, chemoembolization, radioembolization, sorafenib, and liver transplantation [11,72,73,74,75,76,77,78].
For a long time, liver transplantation has been refused to thalassemia patients mainly due to cardiac comorbidities. Based on the success stories reported in a series of cases, thalassemia per se should no longer be considered a contraindication to liver transplantation [73,78]. Liver transplantation is a potential treatment option for thalassemia patients once there is no significant comorbidities, such as severe pulmonary hypertension and overt heart disease [12,63,79,80].
In 2007, sorafenib became the first approved systemic therapy for HCC. Currently there are many approved therapies for advanced HCC, such as lenvatinib, regorafenib, carbozantinib, ramucirumab, nivolumab, pembrolizumab, combination of atezolizumab and bevacizumab, combination of nivolumab and ipilimumab, and combination of tremelimumab and durvalumab [69,70,71,81,82,83]. Published data are lacking on use of these agents in patients with thalassemia and HCC. However, treatment of HCC in thalassemia patients should follow the same recommendations as in non-thalassemic population. Novel immunotherapies, combination therapies, and chimeric antigen receptor (CAR)-T cell therapies are being tested for HCC, and thalassemia should not be regarded as a contraindication.
5. Prognosis
Worldwide, liver cancer is the sixth most common cancer and the third leading cause of cancer-related death. HCC accounts for 75%–85% of liver cancer cases [15]. Thanks to advances in early diagnosis and treatment, survival rates for HCC have increased over the past three decades, with Surveillance Epidemiological Final Results reporting 1-year survival rates, with an overall increase from 18.2% to 51.2% [84]. The recently reported median survival of HCC in the general population was approximately 6.1–10.3 months. Survival differences were noted across etiologies of HCC due to differential screening and treatment practices, with more favorable survival in HBV-related HCC [80]. More studies have focused on HCC in patients with thalassemia; however, survival data for this population are still limited. In 2004, Borgna-Pignatti et al. from Italy reported that based on data collected from 52 centers, the median survival after HCC diagnosis was 3.5 months in patients with TDT and HCC [72]. Ten years later, in their update report, the median survival rose to 11.5 months [73]. In a previous study, the prognostic factor of survival in patients with TDT and HCC was investigated, and the only significant factor was Barcelona Clinic Liver Cancer staging [74].
6. Surveillance
In patients with thalassemia, iron overload and liver fibrosis must be monitored, and early-onset HCC detected. For the measurement of LIC, magnetic resonance imaging (MRI) T2* or R2* is a validated tool and has replaced invasive liver biopsy in current clinical practice. The current surveillance program suggests that the frequency of MRI should be based on the baseline LIC. If the baseline LIC is <3 mg/g, MRI is encouraged every 2 years; if 3–15 mg/g, MRI is recommended annually; and if >15 mg/g or the LIC or serum ferritin is rapidly increasing, MRI is advised every 6 months [85].Transient elastography (e.g., Fibroscan®) is a reliable and accurate noninvasive test to assess liver fibrosis. According to current practice, transient elastography should be performed every 6–12 months if available [85,86].
Surveillance programs for HCC aimed to detect cancer at early stages in patients at a high risk, for which potentially curative treatment options are indicated. The proposed mean doubling time of an HCC lesion is 117 days[87]. Current guidelines from the American Association for the Study of Liver Disease recommend biennial follow-ups with abdominal ultrasonography, with or without PFA. Considering cost-effectiveness and safety, multiphasic computed tomography (CT) and multiphasic MRI are reserved for diagnostic evaluation despite their high diagnostic efficiency [71]. In a previous meta-analysis, ultrasonography combined with Alpha-Fetoprotein (AFP) provides 63% sensitivity, higher than the 45% sensitivity with ultrasonography alone [88]. To improve the sensitivity for the early detection of HCC, further studies may clarify the role of advanced imaging as a primary modality in selected cases.
Besides AFP, several new biomarkers and models such as the GALAD score are being evaluated [89,90,91]. Novel biomarkers assays, such as cell-free DNA and microRNA, are also promising [90,91,92,93,94]. However, the optimistic results come from early-phase biomarker studies. New biomarkers need to be evaluated and validated in further studies before they can be incorporated into routine clinical practice.
HCC surveillance in thalassemia using abdominal ultrasonography, with or without AFP, has been proposed in the literature. The frequency of the majority of surveillance programs is every 6 months. However, differences exist regarding the target population. Some authors suggest screening all patients with thalassemia, while other authors suggest screening those with risk factors for HCC, including serum ferritin ≥1000 ng/mL, LIC ≥5 mg/g in NTDT, LIC ≥7 mg/g in TDT, concurrent HBV and/or HCV infection, and advanced cirrhosis [12,63,80]. In one review, the authors recommend screening all patients with thalassemia aged >30 and younger patients if a risk factor is present, given that the youngest patient reported to have thalassemia with HCC was 33 years old [86].
7. Conclusions
HCC is one of the most common cancers worldwide and the third leading cause of cancer death. In patients with thalassemia, repeated transfusion and increased gastrointestinal absorption of iron lead to liver iron deposit, which in turn result in ROS accumulation and proinflammatory pathway activation. Under the chronic inflammation status, cirrhosis gradually develops and become an oncogenic circumstance of hepatocyte. Therefore, HCC is one of the major complications in patients with thalassemia and management for iron chelation, regular surveillance and other supplementary treatment, like antioxidant therapy were recommended. Treatment of HCC in patients with thalassemia was based on that in non-thalassemia patients except more consideration should be given to the cardiac and pulmonary conditions which are also influenced by iron overload. In the future, new treatment modalities, such as Luspatercept and gene therapy for thalassemia, can reduce the transfusion requirement and the iron deposit, and may be beneficial for preventing HCC development in patients with thalassemia.
Author Contributions
Conceptualization, Pei-Chin Lin,Shyh-Shin Chiouand Shih-Hsien Hsu.; Investigation, Pei-Chin Lin, Wan-Yi Hsuand Po-Yi Lee.; Writing—original draft preparation, Pei-Chin Lin, Wan-Yi Hsu, Po-Yi Lee.; Writing—review and editing, Shyh-Shin Chiouand Shih-Hsien Hsu.; Visualization, Shih-Hsien Hsu.; Supervision, Shyh-Shin Chiou and Shih-Hsien Hsu. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by a grant from the Kaohsiung Medical University Hospital and the Ministry of Science and Technology, Taiwan: KMUH108-8M42, DK-DK(A)112002, MOST110-2314-B-037-072-MY3 MOST111-2314-B-037-078.
References
- Steinberg, M. H., Forget, B. G., Higgs, D. R. & Weatherall, D. J. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. (Cambridge University Press, 2009).
- Weatherall, D. J. The inherited diseases of hemoglobin are an emerging global health burden. Blood, The Journal of the American Society of Hematology 115, 4331-4336 (2010). [CrossRef]
- Kattamis, A., Kwiatkowski, J. L. & Aydinok, Y. Thalassaemia. (2022).
- Modell, B. & Darlison, M. J. B. o. t. W. H. O. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization 86, 480-487 (2008).
- Kattamis, A., Forni, G. L., Aydinok, Y. & Viprakasit, V. Changing patterns in the epidemiology of β-thalassemia. European Journal of Haematology 105, 692-703 (2020).
- Taher, A. T., Weatherall, D. J. & Cappellini, M. D. Thalassaemia. The Lancet 391, 155-167 (2018).
- Piel, F. B. & Weatherall, D. J. The α-thalassemias. New England Journal of Medicine 371, 1908-1916 (2014).
- Taher, A. T. & Saliba, A. N. J. H., the American Society of Hematology Education Program Book. Iron overload in thalassemia: different organs at different rates. Hematology , the American Society of Hematology Education Program Book 2017, 265-271 (2017). [CrossRef]
- Taher, A. T., Musallam, K. M. & Cappellini, M. D. J. N. E. J. o. M. β-Thalassemias. New England Journal of Medicine 384, 727-743 (2021).
- Kowdley, K. V. J. G. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 127, S79-S86 (2004).
- Mancuso, A., Sciarrino, E., Concetta Renda, M. & Maggio, A. A prospective study of hepatocellular carcinoma incidence in thalassemia. Hemoglobin 30, 119-124 (2006).
- Moukhadder, H. M., Halawi, R., Cappellini, M. D. & Taher, A. T. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: a comprehensive review. Cancer 123, 751-758 (2017). [CrossRef]
- Chung, W.-S., Lin, C.-L., Lin, C.-L. & Kao, C.-H. J. J. E. C. H. Thalassaemia and risk of cancer: a population-based cohort study. J Epidemiol Community Health 69, 1066-1070 (2015).
- Taher, A., Musallam, K. & Cappellini, M. D. Guidelines for the Management of Non-Transfusion Dependent Thalassaemia (NTDT). (2017).
- Sung, H. et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 71, 209-249 (2021). [CrossRef]
- Zhang, C. h., Cheng, Y., Zhang, S., Fan, J. & Gao, Q. Changing epidemiology of hepatocellular carcinoma in Asia. Liver International 42, 2029-2041 (2022). [CrossRef]
- Sangiovanni, A. et al. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterology 126, 1005-1014 (2004). [CrossRef]
- Jepsen, P., Ott, P., Andersen, P. K., Sørensen, H. T. & Vilstrup, H. Risk for hepatocellular carcinoma in patients with alcoholic cirrhosis: a Danish nationwide cohort study. Annals of internal medicine 156, 841-847 (2012).
- Lin, C.-W. et al. Heavy alcohol consumption increases the incidence of hepatocellular carcinoma in hepatitis B virus-related cirrhosis. Journal of hepatology 58, 730-735 (2013). [CrossRef]
- Sinn, D. H. et al. Alcohol intake and mortality in patients with chronic viral hepatitis: a nationwide cohort study. Official journal of the American College of Gastroenterology| ACG 116, 329-335 (2021). [CrossRef]
- Younossi, Z. et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nature reviews Gastroenterology & hepatology 15, 11-20 (2018).
- Estes, C., Razavi, H., Loomba, R., Younossi, Z. & Sanyal, A. J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123-133 (2018). [CrossRef]
- Josep M. Llovet, R. K. K., Augusto Villanueva, Amit G. Singal, Eli Pikarsky, Sasan Roayaie, Riccardo Lencioni, Kazuhiko Koike, Jessica Zucman-Rossi & Richard S. Finn Hepatocellular carcinoma. nature reviews disease primers (2021).
- Borgna-Pignatti, C. et al. Hepatocellular carcinoma in thalassaemia: an update of the Italian Registry. British journal of haematology 167, 121-126 (2014). [CrossRef]
- Wonke, B., Hoffbrand, A., Brown, D. & Dusheiko, G. Antibody to hepatitis C virus in multiply transfused patients with thalassaemia major. Journal of clinical pathology 43, 638-640 (1990). [CrossRef]
- He, L.-N. et al. Elevated prevalence of abnormal glucose metabolism and other endocrine disorders in patients with-thalassemia major: a meta-analysis. BioMed research international 2019 (2019).
- Memaj, P. & Jornayvaz, F. R. Non-alcoholic fatty liver disease in type 1 diabetes: Prevalence and pathophysiology. Frontiers in Endocrinology 13, 1031633 (2022). [CrossRef]
- Pagani, A., Nai, A., Silvestri, L. & Camaschella, C. J. F. i. p. Hepcidin and anemia: a tight relationship. Frontiers in physiology 10, 1294 (2019). [CrossRef]
- Frey, P. A. & Reed, G. H. (ACS Publications, 2012).
- Muckenthaler, M. U., Rivella, S., Hentze, M. W. & Galy, B. J. C. A red carpet for iron metabolism. Cell 168, 344-361 (2017). [CrossRef]
- Hentze, M. W., Muckenthaler, M. U., Galy, B. & Camaschella, C. J. C. Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24-38 (2010). [CrossRef]
- Grosse, S. D., Gurrin, L. C., Bertalli, N. A. & Allen, K. J. Clinical penetrance in hereditary hemochromatosis: estimates of the cumulative incidence of severe liver disease among HFE C282Y homozygotes. Genetics in Medicine 20, 383-389 (2018). [CrossRef]
- Atkins, J. L. et al. Association of hemochromatosis HFE p. C282Y homozygosity with hepatic malignancy. JAMA 324, 2048-2057 (2020). [CrossRef]
- Turlin, B. et al. Increased liver iron stores in patients with hepatocellular carcinoma developed on a noncirrhotic liver. Hepatology 22, 446-450 (1995).
- Zanella, S., Garani, M. C. & Borgna-Pignatti, C. Malignancies and thalassemia: a review of the literature. Annals of the New York Academy of Sciences 1368, 140-148 (2016). [CrossRef]
- Ruddell, R. G. et al. Ferritin functions as a proinflammatory cytokine via iron-independent protein kinase C zeta/nuclear factor kappaB–regulated signaling in rat hepatic stellate cells. Hepatology 49, 887-900 (2009). [CrossRef]
- Sindrilaru, A. et al. An unrestrained proinflammatory M1 macrophage population induced by iron impairs wound healing in humans and mice. The Journal of clinical investigation 121, 985-997 (2011). [CrossRef]
- Ringelhan, M., Pfister, D., O’Connor, T., Pikarsky, E. & Heikenwalder, M. J. N. i. The immunology of hepatocellular carcinoma. Nature immunology 19, 222-232 (2018). [CrossRef]
- Hoshida, Y. et al. Prognostic gene expression signature for patients with hepatitis C–related early-stage cirrhosis. Gastroenterology 144, 1024-1030 (2013). [CrossRef]
- Budhu, A. et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer cell 10, 99-111 (2006). [CrossRef]
- Moeini, A. et al. An immune gene expression signature associated with development of human hepatocellular carcinoma identifies mice that respond to chemopreventive agents. Gastroenterology 157, 1383-1397. e1311 (2019). [CrossRef]
- Adams, P. C. J. T. A. j. o. g. Is there a threshold of hepatic iron concentration that leads to cirrhosis in C282Y hemochromatosis? The American journal of gastroenterology 96, 567-569 (2001).
- Angelucci, E. et al. Effects of iron overload and hepatitis C virus positivity in determining progression of liver fibrosis in thalassemia following bone marrow transplantation. Blood, The Journal of the American Society of Hematology 100, 17-21 (2002). [CrossRef]
- Dixon, S. J. & Stockwell, B. R. J. N. c. b. The role of iron and reactive oxygen species in cell death. Nature chemical biology 10, 9-17 (2014). [CrossRef]
- Moloney, J. N. & Cotter, T. G. in Seminars in cell & developmental biology. 50-64 (Elsevier).
- Choi, J.-E. et al. 15-Deoxy-Δ12, 14-prostaglandin J2 stabilizes hypoxia inducible factor-1α through induction of heme oxygenase-1 and direct modification ofprolyl-4-hydroxylase 2. Free radical research 50, 1140-1152 (2016).
- Cao, L., Liu, J., Zhang, L., Xiao, X. & Li, W. Curcumin inhibits H2O2-induced invasion and migration of human pancreatic cancer via suppression of the ERK/NF-κB pathway. Oncology reports 36, 2245-2251 (2016).
- Krause, A. et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 480, 147-150 (2000). [CrossRef]
- Park, C. H., Valore, E. V., Waring, A. J. & Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 276, 7806-7810 (2001). [CrossRef]
- Pigeon, C. et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 276, 7811-7819 (2001). [CrossRef]
- Ganz, T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 102, 783-788 (2003). [CrossRef]
- Nemeth, E. et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 101, 2461-2463 (2003). [CrossRef]
- Nemeth, E. et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306, 2090-2093 (2004). [CrossRef]
- Kijima, H., Sawada, T., Tomosugi, N. & Kubota, K. Expression of hepcidin mRNA is uniformly suppressed in hepatocellular carcinoma. BMC Cancer 8, 167 (2008). [CrossRef]
- Tan, M. G. K. et al. Modulation of iron-regulatory genes in human hepatocellular carcinoma and its physiological consequences. Experimental Biology and Medicine 234, 693-702 (2009). [CrossRef]
- Tseng, H. H. et al. Expression of hepcidin and other iron-regulatory genes in human hepatocellular carcinoma and its clinical implications. J Cancer Res Clin Oncol 135, 1413-1420 (2009). [CrossRef]
- Kessler, S. M., Barghash, A., Laggai, S., Helms, V. & Kiemer, A. K. Hepatic hepcidin expression is decreased in cirrhosis and HCC. J Hepatol 62, 977-979 (2015). [CrossRef]
- Han, C. Y. et al. Hepcidin inhibits Smad3 phosphorylation in hepatic stellate cells by impeding ferroportin-mediated regulation of Akt. Nat Commun 7, 13817 (2016). [CrossRef]
- Joachim, J. H. & Mehta, K. J. Hepcidin in hepatocellular carcinoma. Br J Cancer 127, 185-192 (2022). [CrossRef]
- Zhang, S. et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal 26, 2539-2550 (2014). [CrossRef]
- Guo, W. et al. An important role of the hepcidin-ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim Biophys Sin (Shanghai) 47, 703-715 (2015). [CrossRef]
- Rah, B., Farhat, N. M., Hamad, M. & Muhammad, J. S. JAK/STAT signaling and cellular iron metabolism in hepatocellular carcinoma: therapeutic implications. Clinical and Experimental Medicine (2023). [CrossRef]
- Finianos, A., Matar, C. F. & Taher, A. J. I. j. o. m. s. Hepatocellular carcinoma in β-thalassemia patients: review of the literature with molecular insight into liver carcinogenesis. International journal of molecular sciences 19, 4070 (2018). [CrossRef]
- Hatairaktham, S. et al. Curcuminoids supplementation ameliorates iron overload, oxidative stress, hypercoagulability, and inflammation in non-transfusion-dependent β-thalassemia/Hb E patients. Annals of Hematology 100, 891-901 (2021).
- Mohammadi, E. et al. An investigation of the effects of curcumin on iron overload, hepcidin level, and liver function in β-thalassemia major patients: A double-blind randomized controlled clinical trial. Phytotherapy Research 32, 1828-1835 (2018).
- Al-Momen, H., Hussein, H. K., Al-Attar, Z. & Hussein, M. J. J. F. Green tea influence on iron overload in thalassemia intermedia patients: a randomized controlled trial. FResearch 9, 1136 (2020). [CrossRef]
- Saeidnia, M. et al. The Effect of Curcumin on Iron Overload in Patients with Beta-Thalassemia Intermedia. Clinical Laboratory 68 (2022). [CrossRef]
- Farmakis, D. et al. 2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-dependent Thalassemia. HemaSphere 6 (2022). [CrossRef]
- Galle, P. R. et al. EASL clinical practice guidelines: management of hepatocellular carcinoma. Journal of hepatology 69, 182-236 (2018). [CrossRef]
- Llovet, J., Kelley, R. & Villanueva, A. J. P. A. Hepatocellular carcinoma. Nat Rev Dis Primers. PubMed Article 7, 7 (2021). [CrossRef]
- Marrero, J. A. et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 68, 723-750 (2018).
- Borgna-Pignatti, C. et al. Hepatocellular carcinoma in the thalassaemia syndromes. British journal of haematology 124, 114-117 (2004). [CrossRef]
- Borgna-Pignatti, C. et al. Hepatocellular carcinoma in thalassaemia: an update of the Italian Registry. British journal of haematology 167, 121-126 (2014). [CrossRef]
- Papadopoulos, N. et al. Characteristics and prognosis of hepatocellular carcinoma in multi-transfused patients with β-thalassemia. Experience of a single tertiary center. Mediterranean Journal of Hematology 12 (2020).
- Filippiadis, D. et al. Percutaneous Microwave Ablation for the Management of Hepatocellular Carcinoma in Transfusion-Dependent Beta-Thalassemia Patients. CardioVascular 45, 709-711 (2022). [CrossRef]
- Ricchi, P. et al. Hepatocellular carcinoma in patients with thalassemia in the post-DAA era: not a disappearing entity. Annals of Hematology 100, 1907-1910 (2021). [CrossRef]
- Maakaron, J. E., Cappellini, M. D., Graziadei, G., Ayache, J. B. & Taher, A. T. J. A. o. h. Hepatocellular carcinoma in hepatitis-negative patients with thalassemia intermedia: a closer look at the role of siderosis. Annals of hepatology 12, 142-146 (2013). [CrossRef]
- Restivo Pantalone, G. et al. Hepatocellular carcinoma in patients with thalassaemia syndromes: clinical characteristics and outcome in a long term single centre experience. British journal of haematology 150, 245-247 (2010). [CrossRef]
- Mancuso, A. & Perricone, G. J. B. J. o. H. Time to define a new strategy for management of hepatocellular carcinoma in thalassaemia? British Journal of Haematology 168, 304-305 (2015).
- Mangia, A. et al. Hepatocellular carcinoma in adult thalassemia patients: an expert opinion based on current evidence. BMC gastroenterology 20, 1-14 (2020). [CrossRef]
- Girardi, D. M. et al. Systemic Therapy for Advanced Hepatocellular Carcinoma: Current Stand and Perspectives. Cancers 15, 1680 (2023). [CrossRef]
- Koulouris, A. et al. Hepatocellular carcinoma: an overview of the changing landscape of treatment options. Journal of hepatocellular carcinoma, 387-401 (2021). [CrossRef]
- Chan, S. L., Wong, N., Lam, W. J., Kuang, M. J. J. o. G. & Hepatology. Personalized treatment for hepatocellular carcinoma: Current status and Future perspectives. Journal of Gastroenterology 37, 1197-1206 (2022). [CrossRef]
- Ding, J. & Wen, Z. J. B. c. Survival improvement and prognosis for hepatocellular carcinoma: analysis of the SEER database. BMC cancer 21, 1-12 (2021). [CrossRef]
- Taher, A. T. & Cappellini, M. D. How I manage medical complications of β-thalassemia in adults. Blood, The Journal of the American Society of Hematology 132, 1781-1791 (2018). [CrossRef]
- De Sanctis, V. et al. A concise review on the frequency, major risk factors and surveillance of hepatocellular carcinoma (HCC) in β-thalassemias: past, present and future perspectives and the ICET-A experience. Mediterranean Journal of Hematology Infectious Diseases 12 (2020).
- Sheu, J.-C. et al. Growth rate of asymptomatic hepatocellular carcinoma and its clinical implications. Gastroenterology 89, 259-266 (1985). [CrossRef]
- Tzartzeva, K. et al. Surveillance imaging and alpha fetoprotein for early detection of hepatocellular carcinoma in patients with cirrhosis: a meta-analysis. Gastroenterology 154, 1706-1718. e1701 (2018). [CrossRef]
- Johnson, P. J. et al. The Detection of Hepatocellular Carcinoma Using a Prospectively Developed and Validated Model Based on Serological BiomarkersDetection of Hepatocellular Carcinoma. Cancer Epidemiology, Biomarkers 23, 144-153 (2014).
- Schlosser, S. et al. HCC biomarkers–state of the old and outlook to future promising biomarkers and their potential in everyday clinical practice. Frontiers in Oncology 12 (2022). [CrossRef]
- Shahini, E. et al. Updating the Clinical Application of Blood Biomarkers and Their Algorithms in the Diagnosis and Surveillance of Hepatocellular Carcinoma: A Critical Review. International Journal of Molecular Sciences 24, 4286 (2023). [CrossRef]
- Foda, Z. H. et al. Detecting liver cancer using cell-free DNA fragmentomes. Cancer discovery 13, 616-631 (2023). [CrossRef]
- Tran, N. H., Kisiel, J. & Roberts, L. R. Using cell-free DNA for HCC surveillance and prognosis. JHEP Reports 3, 100304 (2021). [CrossRef]
- Zhang, X. et al. Ultrasensitive and affordable assay for early detection of primary liver cancer using plasma cell-free DNA fragmentomics. Hepatology 76, 317-329 (2022). [CrossRef]
Figure 1.
Mechanism of ROS accumulation caused by Fenton reaction.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



