
Submitted:
28 June 2023
Posted:
28 June 2023
You are already at the latest version
Abstract
The emergence of CDK4/6 inhibitors, such as palbociclib, ribociclib, and abemaciclib, has revolutionized the treatment landscape for hormone receptor-positive breast cancer. These agents have demonstrated significant clinical benefits in terms of both progression-free survival and overall survival. However, resistance to CDK4/6 inhibitors remains a challenge, limiting their long-term efficacy. Understanding the complex mechanisms driving resistance is crucial for the development of novel therapeutic strategies and improvement of patient outcomes. Translational research efforts, such as preclinical models and biomarker studies, offer valuable insight into resistance mechanisms and may guide the identification of novel combination therapies. This review paper aims to outline the reported mechanisms underlying CDK4/6 inhibitor resistance, drawing insights from both clinical data and translational research in order to help direct the future of treatment for hormone receptor positive metastatic breast cancer.
Keywords:
breast cancer
; CDK4/6 inhibitors
; estrogen receptor
; therapeutic resistance
1. Introduction
Hormone receptor (HR)-positive metastatic breast cancer (MBC) is characterized by the expression of HR (estrogen receptor and/or progesterone receptor) and the absence of human epidermal growth factor receptor-2 (HER2) overexpression. Although endocrine based therapy has improved the prognosis of this subtype, HR-positive MBC (HR+ MBC) continues to present a significant clinical challenge and is responsible for a substantial number of breast cancer deaths[1]. However, the emergence of cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitors, including palbociclib, ribociclib, and abemaciclib, has transformed the treatment landscape for HR+ MBC. When combined with endocrine therapy, these agents have demonstrated remarkable efficacy, leading to improved progression-free survival (PFS) and overall survival (OS) rates as shown in Table 1. Building on the success of CDK4/6 inhibitors in the metastatic setting, further studies have been conducted to assess their efficacy in early breast cancer. For instance, abemaciclib was recently approved for use in the adjuvant setting based on the results of the MonarchE trial, which revealed sustained benefits in invasive disease-free survival (iDFS) even after discontinuation of the drug[2]. Furthermore, ribociclib has also demonstrated improved iDFS in patients with stage II or III high risk HR+ breast cancer when used for 3 years in the adjuvant setting[3].
CDK4/6 inhibitors exert their function through the targeted inhibition of kinases CDK4/6, which play crucial roles in regulating cell cycle progression and proliferation[17], particularly the transition from the G1 to S phase. Upon binding with cyclin D, CDK 4/6 form an active complex that triggers DNA synthesis and entry into the S phase via phosphorylation of tumor suppressor protein retinoblastoma (Rb). This phosphorylation event releases the repression of the E2F family, facilitating hyperphosphorylation of Rb via the activity of cyclin E1 and E2. This cascade further promotes the expression of E2F target genes that are crucial for cell progression into the S phase (Figure 1). By inhibiting these kinases, CDK 4/6 inhibitors induce cell cycle arrest at the G1 phase, thereby preventing entry into the S phase and subsequent DNA synthesis. This mechanism of action effectively disrupts tumor growth and slows down disease progression. Of note, CDK4/6 have also been demonstrated to phosphorylate proteins involved in cell differentiation, apoptosis, and mitochondrial function. This may partially explain the broad anti-tumor effects of CKD 4/6 inhibitors[18]. All three CDK4/6 inhibitors used in the clinical setting demonstrate inhibition of both CDK 4 and CDK 6. However, abemaciclib and ribociclib specifically exhibit greater potency against CDK4 compared to CDK6. Additionally, abemaciclib inhibits multiple other kinases, including CDK1 and CDK2[19]. With the wide range of effects of kinases on the progression of the cell cycle, these drugs may have multiple mechanisms through which they contribute to cancer cell death.
Despite the initial clinical success of CDK 4/6 inhibitors, the development of resistance remains a critical challenge in the long-term management of HR+ MBC. Unfortunately, many patients will continue to experience disease progression despite receiving continuous treatment with these agents. Additionally, the recent approval of abemaciclib in the adjuvant setting will certainly present new clinical challenges in determining treatment strategies for patients experiencing relapse while on or following the use of CDK 4/6 inhibitors. Understanding the underlying mechanisms of resistance is essential in developing strategies to overcome this limitation and improve patient outcomes. Further research and exploration of novel combinations are needed to address these challenges and advance treatment options for resistant disease.
This review aims to comprehensively explore the mechanisms of CDK 4/6 inhibitor resistance, drawing insights from both clinical data and translational research. By examining the available evidence, we seek to identify the molecular alterations, signaling pathways, and cellular processes that contribute to resistance development. Furthermore, we will highlight the implications of these resistance mechanisms on clinical practice and discuss potential strategies to overcome these resistance patterns in order to enhance treatment efficacy.
2. Potential mechanisms of CDK 4/6 Inhibitor Resistance in preclinical studies
2.1. Alterations in Cell Cycle Pathway Components
The CDK 4/6 pathway plays a crucial role in cell cycle progression and is the primary target of CDK 4/6 inhibitors. Alterations in various components of this pathway can contribute to CDK 4/6 inhibitor resistance. One potential mechanism involves alterations to cyclin D1, the primary activator of CDK 4/6. For example, cyclin D1 amplification or overexpression has been observed in a subset of patients who develop resistance to CDK 4/6 inhibitors[20]. In contrast, another study demonstrated that mutations activating cyclin D could potentially increase the sensitivity to CDK4/6 inhibitors. Meanwhile deficiency in cyclin D, which may be attributed to the oncogene addiction property in some cancer cells, is associated with resistance to CKD4/6 inhibitors[21,22]. Mutations or alterations in kinases CDK4 and 6 themselves have also been implicated in causing resistance[23]. These alterations can impair the binding of CDK 4/6 inhibitors to their targets, reducing the inhibitory effect and diminishing the response to therapy. For example, mutations in the ATP-binding pocket of CDK4/6 may interfere with the binding of CDK 4/6 inhibitors, thereby confer resistance to treatment[24].
The relationships between tumor cell cycle mediators are highly heterogenous, influenced by tumor origin and genetic alterations. These overlapping pathways allow for the development of CDK4/6 resistance via utilization of bypass mechanisms. Knudsen et al. demonstrated diverse patterns of dependency of CDK4/6 in breast cancer cells for cell cycle progression[25]. While some cells conform to a conventional CDK4/6-dependent cycle, others can bypass this dependency through mechanisms involving cyclin E activation or cyclin D1. Tumor cells with Rb loss display heterogeneous CDK and cyclin requirements, with some relying on cyclin E for progression. Moreover, Palafox et al. showed p16 overexpression was associated with reduced response to CDK4/6 inhibitors in patient derived xenografts and breast cancer cell lines, while heterozygous RB1 loss served as a biomarker for acquired resistance and poor clinical outcomes[26]. Cyclin E2 (CCNE2) overexpression was also proposed as a potential mechanism of resistance to both endocrine therapy and CDK4 inhibition from preclinical studies[27], and CCNE1 overexpression was associated with lower response to palbociclib in the PALOMA-3 trial[28]. Taken together, these findings highlight the complex nature of cancer cell cycles, including multiple mechanisms for G1/S progression and differential dependencies on CDKs and cyclins. Modifications or mutations across various elements of these pathways can lead to resistance against CDK4/6 inhibitors. It will be important to adopt precision therapeutic strategies that are tailored based on specific gene expressions and susceptibilities in order to overcome these resistance mechanisms in a clinically relevant way.
2.2. Activation of Alternative Pathways
In addition to alterations of major cell cycle regulators, CDK4/6 inhibitor resistance in breast cancer also involves various genomic alterations, with the RAS/MAPK and PI3K/AKT/mTOR pathways playing key roles. The PI3K/AKT/mTOR pathway is a critical signaling pathway involved in cell survival and proliferation. Activation of this pathway, often through genetic alterations or aberrant signaling, can promote resistance to CDK 4/6 inhibitors[29], 17. Dysregulation of the PI3K/AKT/mTOR pathway can bypass the inhibitory effects of CDK 4/6 inhibitors and promote cell cycle progression, leading to disease progression despite treatment. O’Brien et al. demonstrated that treatment with a p110α-selective PI3K inhibitor, alpelisib (BYL719), completely blocked the progression of acquired CDK4/6 inhibitor-resistant xenografts, proposing a possible clinical solution to PI3K-driven resistant tumors[30]. Of note, a major cause of anti-cancer therapeutic resistance is the increase of multidrug resistance (MDR) genes, which are well-established targets of PI3K signaling[31]. A recent study found upregulation of MDR proteins in CDK4/6 inhibitor-resistant breast cancer cells[32]. Based on these data, it is postulated that the PI3K/MDR axis may play a critical role in dictating response to CDK4/6 inhibitors in MBC.
Loss or alterations of Neurofibromin 1(NF1), a gene that regulates RAS and cell proliferation, have been linked to endocrine therapy resistance and disease progression, but its relationship with CDK4/6 inhibitors is still not well understood. One study suggested that patients with pathogenic NF1 mutations may be more likely to develop resistance to CDK4/6 inhibitors. However the impact of other NF1 mutations and the potential for targeting the MAPK pathway in this context require further exploration[33].
The fibroblast growth factor receptor (FGFR) signaling pathway, which is known to be a critical regulator of breast cancer progression, has emerged as another potential mechanism of resistance to CDK 4/6 inhibitors[34,35]. Activation of FGFR signaling can drive cell proliferation and survival, independent of CDK 4/6 activity. The activation of this pathway may confer resistance to CDK 4/6 inhibitors and limit their effectiveness.
2.3. Epigenetic Regulation and Transcriptional Rewiring
Epigenetic changes, such as chromatin remodeling and histone modifications, can also contribute to the development of resistance to CDK 4/6 inhibitors[36,37]. Chromatin remodeling involves altering the physical structure of DNA, affecting its access to various regulatory proteins[38,39]. These changes in structure can affect the binding of transcription factors to their target genes, leading to dysregulated gene expression patterns. Histone modifications, including acetylation, methylation, and phosphorylation, can also influence chromatin structure and gene transcription, causing activation of resistance mechanisms. For example, histone deacetylases can increase CKD4/6 inhibition efficacy and mediate cell cycle arrest by upregulating p21 expression in CKD4/6 inhibitor resistant tumors[40]. These various epigenetic modifications can modulate the expression of genes involved in the CDK 4/6 pathway and other resistance-associated pathways, ultimately contributing to further drug resistance development
Transcription factors also play a crucial role in regulating gene expression. They function by binding to specific DNA sequences and modulating transcriptional activity. In the context of CDK 4/6 inhibitor resistance, altered expression or activity of transcription factors can impact the response to therapy[41]. Transcription factors, such as E2F, AP-1, and NF-κB, can drive the expression of genes involved in cell cycle regulation, DNA repair, and cell survival, thereby promoting resistance. Additionally, enhancer hijacking, a phenomenon where active enhancers are redirected to different genes, can lead to transcriptional rewiring and altered gene expression patterns[42]. This rewiring may result in the activation of pathways that bypass the inhibitory effects of CDK 4/6 inhibitors, conferring drug resistance.
3. Clinical trials and CDK 4/6 Inhibitor Resistance
Clinical manifestations of CDK 4/6 inhibitor resistance can vary greatly. A subset of patients with HR+ MBC demonstrates intrinsic or primary resistance to CDK 4/6 inhibitors, presenting as a lack of initial response or limited sustained benefit. Conversely, another subset of patients may initially exhibit a favorable response to CDK 4/6 inhibitor therapy, but then develop disease progression despite continuous treatment, suggestive of acquired resistance. Careful clinical observation and identification of resistance patterns can provide valuable insight into the essential characteristics of differing resistance patterns. Understanding the underlying factors that contribute to both intrinsic and acquired resistance is imperative to identify patients who may not derive significant benefits from CDK 4/6 inhibitors, thus necessitating exploration of alternative therapeutic strategies. Although extensive studies have focused on the identification of biomarkers that can predict poor responsiveness to CDK 4/6 inhibitors, the results have not been concordance. Further analysis of molecular alterations and genomic landscapes can help elucidate novel indicators that may predict a patient’s response to treatment.
Turner et al. performed an analysis utilizing formalin-fixed paraffin-embedded (FFPE) tissue samples obtained from patients in the PALOMA-3 study to investigate the significance of varying gene expression levels on response to CDK 4/6 inhibitors[28]. In this study, the efficacy of palbociclib was lower in patients with elevated expression of cyclin E1 (CCNE1) mRNA compared to those with lower expression levels. Notably, no significant interaction was identified between treatment efficacy and the expression levels of CDK4, CDK6, cyclin D1, and RB1. A separate investigation utilizing plasma samples from patients in the PALOMA-3 study revealed that the relative change in PIK3CA circulating tumor DNA (ctDNA) levels after 15 days of treatment of palbociclib and fulvestrant strongly correlated with PFS[43]. Furthermore, an additional investigation using paired baseline and end-of-treatment ctDNA sequencing from the PALOMA-3 study demonstrated that acquired mutations resulting from fulvestrant treatment, such as the ESR1 Y537S mutation, play a significant role in driving resistance to fulvestrant and palbociclib combination therapy[44].
Further biomarker studies have conducted in the different clinical trials. In the MONARCH-2 trial, ctDNA analysis showed that abemaciclib plus fulvestrant improved PFS and OS in patients with PIK3CA and ESR1 mutations, as well as in those without these mutations, indicating the effectiveness of this combination therapy across certain mutation subgroups[45]. Similarly, a pooled ctDNA analysis from the MONALEESA phase III trial with 1503 patients identified specific genomic alterations, such as FRS2 and PRKCA, that were associated with increased PFS from treatment with ribociclib, while alterations in genes such as CHD4, BCL11B, ATM, or CDKN2A/2B/2C provided limited benefit[46]. Another genomic study using human tissue samples identified RB and PTEN expression loss as key drivers of acquired resistance to ribociclib and letrozole[47].
Wander et al. explored the genomic landscape of resistance to CDK4/6 inhibitors via whole-exome sequencing (WES) of metastatic tumor tissues from patients who underwent treatment with CDK4/6 inhibitors[48]. This study revealed the heterogeneous genomic landscape of resistance mechanisms, identifying multiple potential mediators such as RB1 disruption, AKT1 activation, and alterations in AURKA, CCNE2, RAS, ERBB2, and FGFR2. Lastly, our group conducted a comprehensive analysis of broad-panel next-generation sequencing (NGS) data obtained from patients receiving any CDK4/6 inhibitors in a real-world clinical setting[49]. This retrospective study analyzed genomic biomarkers of CDK4/6 inhibitor resistance in patients with HR-positive MBC treated with palbociclib, ribociclib, or abemaciclib. Genomic and RNA sequencing data were analyzed in relation to PFS, and associations were identified between specific genes. FGFR1 amplification, PTEN loss, and DNA repair pathway gene mutations showed significant associations with shorter PFS in patients receiving treatment with CDK4/6 inhibitor therapy. These findings highlight the role potential genomic biomarkers could play in helping to predict response to CDK4/6 inhibitors in the clinical practice.
In conclusion, clinical studies investigating treatment resistance to CDK4/6 inhibitors have provided valuable insights into this phenomenon. The emergence of resistance is characterized by clinical observations and patterns, with molecular alterations and genomic landscape playing a crucial role. The findings from various studies investigating biomarkers have provided valuable insights, but the results have not always been consistent, leading to controversy and the need for further research.
4. Biomarkers for Predicting and Monitoring Resistance
Identifying biomarkers that can predict and monitor resistance to CDK4/6 inhibitors is crucial for optimizing treatment strategies and patient outcomes. Various approaches, such as genomic and transcriptomic profiling, ctDNA analysis, and imaging techniques, have been studied to identify and characterize biomarkers associated with CDK4/6 inhibitor resistance.
4.1. Tumor tissue profiling
Genomic and transcriptomic profiling of tumor samples has provided valuable insight into the molecular alterations associated with CDK 4/6 inhibitor resistance. For example, studies have identified alterations in genes such as CCNE1, RB1, ERBB1, and ESR1 that contribute to resistance[28,47,49,50]. Genomic profiling using techniques like NGS has revealed the presence of specific mutations, amplifications, or deletions in these genes that can influence treatment response. Transcriptomic profiling has also uncovered gene expression signatures associated with CDK4/6 inhibitor resistance, providing potential predictive biomarkers. Recently, advanced single-cell sequencing has been used for genomic profiling. Griffiths et al. investigated resistance pathways of CDK4/6 inhibitor and endocrine therapy using sing single-cell RNA sequencing (scRNAseq)[51]. The findings showed that combination therapy led to the emergence of resistance through a shift from estrogen signaling to up-regulation of JNK signaling proliferation through growth factor receptors. These types of analyses help to further dissect the mechanisms enabling resistance and introduce potential new targets to mitigate tumor growth.
4.2. Circulating tumor DNA (ctDNA) analysis
ctDNA analysis has emerged as a non-invasive approach for monitoring treatment response and detecting emerging drug resistance. Studies have demonstrated the utility of ctDNA analysis in identifying specific genetic alterations associated with CDK4/6 inhibitor resistance. For example, high ctDNA fraction and specific genomic alterations, such as TP53 mutations and FGFR1 amplification, had worse PFS despite receiving CDK4/6 inhibition[52]. Additionally, changes in ctDNA over time, such as an increase in mutant allele fraction, have been associated with acquired resistance. One study identified detectable acquired RB1 mutations in ctDNA after exposure to CDK4/6 inhibitor[53]. These findings highlight the emergence of somatic RB1 mutations as a potential mechanism of resistance to CDK4/6 inhibitors. ctDNA analysis holds promise as a real-time monitoring tool for detecting resistance-associated alterations and guiding treatment decisions.
4.3. Imaging techniques for assessing resistance
Positron emission tomography (PET) has been explored as a means to assess resistance to CDK4/6 inhibitors. For example, PET imaging using radiotracers that target cell proliferation markers, such as 18F-fluorothymidine (FLT), has shown promise in monitoring treatment response and detecting early signs of resistance. Elmi et al. investigated the use of two PET-proliferation tracers, [18F]FLT and [18F]ISO-1, for evaluating the response to a combination of palbociclib and fulvestrant in breast cancer cell lines and MCF7 tumor-bearing mice[54]. The findings showed that [18F]FLT is more sensitive to immediate changes in S-phase proliferation, while [18F]ISO-1 can assess delayed changes associated with cell-cycle arrest and transition to G0 quiescence. These findings highlight the possible utility of imaging studies as a biomarker to indicate response and resistance to treatment.
The identification and validation of biomarkers associated with CDK4/6 inhibitor resistance are crucial for personalized treatment strategies. Multi-omics, ctDNA analysis, imaging techniques, and particularly a combination of multiple approaches, can provide valuable tools for predicting and monitoring resistance. However, it is important to note that further research and validation are required to establish the clinical utility of these biomarkers.
5. Rational Combinations and Novel Therapeutic Strategies
Overcoming resistance to CDK4/6 inhibitors requires the development of novel therapeutic strategies that target resistance pathways, which can be identified through translational research and clinical trials. One such example is the use of immunotherapy in CDK4/6 inhibitor-resistant breast cancer, which holds promise for patients with limited treatment options.
5.1. Overcoming resistance through combination therapies
Combination therapies that target multiple pathways involved in CDK4/6 inhibitor resistance have shown promise in preclinical and clinical studies (Table 2). For example, the combination of CDK4/6 inhibitors with PI3K/AKT/mTOR pathway inhibitors such as alpelisib in breast cancer cell lines and patient-derived models has demonstrated that alpelisib can overcome resistance mediated by PI3K pathway activation[30]. A phase Ib trial explored the combination of palbociclib, fulvestrant, and the PI3K inhibitor taselisib in 25 patients with PIK3CA-mutant HR-positive MBC. The triplet therapy demonstrated a 37.5% response rate[55].
HER-2 overexpression has also been linked to CDK4/6 inhibitor resistance in ER+ MBC. The combination of CDK 4/6 inhibitors with HER2-targeted therapies has shown synergistic effects in overcoming resistance in HER2-positive breast cancer models[56,57]. These novel combinations aim to simultaneously inhibit multiple pathways involved in resistance, thereby enhancing their therapeutic efficacy. The monarcHER trial compared the efficacy of abemaciclib plus trastuzumab with or without fulvestrant to standard-of-care chemotherapy plus trastuzumab in advanced HER2-positive breast cancer in patients who had received at least two previous therapies[58]. The combination of abemaciclib, fulvestrant, and trastuzumab showed a significant improvement in PFS compared to chemotherapy plus trastuzumab, indicating that this chemotherapy-free regimen could be a potential alternative treatment option for patients with HR-positive, HER2-positive advanced breast cancer.
FRFG alteration is another potential resistance mechanism that has been studied for combined therapeutic approaches. One study suggests that breast cancer cells with FGFR1 alterations that exhibit resistance to CDK4/6 inhibitors and fulvestrant can be overcome by combining FGFR tyrosine kinase inhibitor (TKI) with CDK4/6 inhibitors and ER antagonists.[35]. Additionally, A phase Ib trial evaluated the combination of the FGFR inhibitor erdafitinib with fulvestrant and palbociclib in patients with FGFR-amplified HR-positive MBC[59]. The treatment demonstrated clinical activity, particularly in patients with high levels of FGFR amplification, but erdafitinib-related side effects led to treatment discontinuation in several patients. These studies highlight the potential for combination therapies targeting the FGFR pathway as a mechanism to overcome CDK4/6 inhibitor resistance.
5.2. Targeting alternative pathways
Insights gained from translational research have identified specific alternative pathways that can be targeted to overcome CDK4/6 inhibitor resistance (Figure 2). For example, Chand et al. Demonstrated cell growth inhibition and anti-tumor effect in CCNE1 over-expressed breast cancer cells using INCB123667, a potent and selective small molecular inhibitor of CDK2 currently in clinical development[60].
Targeting WEE1, a crucial regulator of the G2 checkpoint in endocrine and CDK4/6 inhibitor resistant breast cancer cells, effectively reduces cell proliferation, induces G2/M arrest, apoptosis, and DNA double-stranded breaks, offering a promising therapeutic strategy for treatment resistant HR+ breast cancer[61]. A phase II study evaluated the efficacy of adavosertib, a WEE1 inhibitor, in patients with CCNE1-amplified, advanced refractory solid tumors. Adavosertib demonstrated promising clinical activity with an objective response rate of 27% and a manageable toxicity profile, suggesting further investigation of WEE1 inhibitors as a potential treatment option for CCNE1-amplified tumors, alone or in combination with other therapies[62], is warranted.
5.3. Immunotherapy approaches combined with CDK4/6 inhibitor
Immunotherapy has revolutionized cancer treatment. For instance, immune checkpoint inhibitors (ICIs), such as pembrolizumab, have shown clinical benefit in triple negative breast cancer. However, single agent pembrolizumab demonstrated only modest responses in HR+ MBC[63]. Recent research has highlighted the immune modulatory effects of CDK4/6 inhibitors and their combined efficacy when used with standard immunotherapy. Several studies have demonstrated that CDK4/6 inhibitors can enhance antitumor immunity by promoting interferon production, improving tumor antigen presentation, suppressing regulatory T cell proliferation, and increasing tumor infiltration and activation of effector T cells, thus potentially enhancing the response to ICIs (Figure 2) [64,65]. A phase I/II trial investigated the safety and efficacy of combining palbociclib, pembrolizumab, and letrozole in women with HR+ MBC[66]. The combination therapy demonstrated good tolerability, with a complete response rate of 31% observed in patients receiving this regimen as a first-line treatment. Further trials are needed to explore the immune-priming effects of CDK4/6 inhibitors in HR+ MBC.
Rational combinations and novel therapeutic strategies offer potential new avenues to overcome CDK4/6 inhibitor resistance. The development of these approaches requires a comprehensive understanding of the underlying resistance mechanisms and their targeted modulation. Preclinical studies and ongoing clinical trials provide valuable insights into the efficacy and safety of these strategies in overcoming drug resistance and improving patient outcomes.
6. Future Directions and conclusion
Despite significant progress, several questions regarding CDK4/6 inhibitor resistance remain unanswered. Further research is needed to elucidate the precise mechanisms underlying resistance development, including the interplay between different resistance pathways and the role of tumor heterogeneity in driving resistance. Additionally, the impact of the tumor microenvironment, including immune cells and stromal components, on CDK4/6 inhibitor response and resistance requires further exploration. Moreover, understanding the dynamics of resistance acquisition and the potential for reversibility or persistence of resistance is crucial. Ongoing efforts in preclinical and clinical research aimed to address these questions will provide further insights into novel therapeutic targets and strategies.
Author Contributions
Conceptualization, J.L; original draft preparation, J.L.; review and editing, H.H., Y.Y. and X.C.; visualization, J.L., H.H.; All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Xiaojiang Cui is supported by National Institutes of Health (2R01CA151610), Department of Defense (W81XWH-18-1-0067), and Samuel Oschin Cancer Institute Research Development Fund.
Conflicts of Interest
Y.Y has contracted research sponsored by Agenus, Merck, Genentech, and Pfizer; is a consultant for Pfizer and Gilead; and is on the Speakers Bureau for AstraZeneca, Daiichi Sankyo, Merck, and Gilead. J.L is a consultant for Novartis. Other authors declare that there is no competing interest.
References
- Gong, Y.; Liu, Y.R.; Ji, P.; Hu, X.; Shao, Z.M. "Impact of molecular subtypes on metastatic breast cancer patients: a SEER population-based study. Sci Rep 2017, 7, 45411. [Google Scholar] [CrossRef]
- Johnston, S.R.D.; Toi, M.; O'Shaughnessy, J.; Rastogi, P.; Campone, M.; Neven, P.; Huang, C.S.; Huober, J.; Jaliffe, G.G.; Cicin, I.; et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol 2023, 24, 77–90. [Google Scholar] [CrossRef]
- Stroyakovskiy, D.; Yardley, D.A.; Huang, C.-S.; Fasching, P.A.; Crown, J.; Bardia, A.; Chia, S.; Im, S.-A.; Martin, M.; Loi, S.; et al. Ribociclib and endocrine therapy as adjuvant treatment in patients with HR+/HER2- early breast cancer: Primary results from the phase III NATALEE trial. Journal of Clinical Oncology 2023, 41, LBA500–LBA500. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 2016, 375, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Rugo, H.S.; Dieras, V.C.; Harbeck, N.; Im, S.-A.; Gelmon, K.A.; Walshe, J.M.; Martin, M.; Gregor, M.C.M.; Bananis, E.; et al. Overall survival (OS) with first-line palbociclib plus letrozole (PAL+LET) versus placebo plus letrozole (PBO+LET) in women with estrogen receptor–positive/human epidermal growth factor receptor 2–negative advanced breast cancer (ER+/HER2− ABC): Analyses from PALOMA-2. Journal of Clinical Oncology 2022, 40, LBA1003–LBA1003. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; et al. Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. N Engl J Med 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann Oncol 2018, 29, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.; Martin, M.; Di Leo, A.; Im, S.A.; Awada, A.; Forrester, T.; Frenzel, M.; Hardebeck, M.C.; Cox, J.; Barriga, S.; et al. MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 2019, 5, 5. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 2016, 17, 425–439. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N Engl J Med 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- George, W. Sledge, J.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. Journal of Clinical Oncology 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor–Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy—MONARCH 2: A Randomized Clinical Trial. JAMA Oncology 2020, 6, 116–124. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martin, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N Engl J Med 2020, 382, 514–524. [Google Scholar] [CrossRef]
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J Clin Oncol 2018, 36, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Im, S.A.; Lu, Y.S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N Engl J Med 2019, 381, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol 2018, 19, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Klein, M.E.; Kovatcheva, M.; Davis, L.E.; Tap, W.D.; Koff, A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 2018, 34, 9–20. [Google Scholar] [CrossRef]
- George, M.A.; Qureshi, S.; Omene, C.; Toppmeyer, D.L.; Ganesan, S. Clinical and Pharmacologic Differences of CDK4/6 Inhibitors in Breast Cancer. Front Oncol 2021, 11, 693104. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, J.; Li, Y.; Shi, Q.; Jin, L.; Li, S.; Zhu, M.; Wang, Q.; Wong, L.L.; Yang, W.; et al. Overexpressed Cyclin D1 and CDK4 proteins are responsible for the resistance to CDK4/6 inhibitor in breast cancer that can be reversed by PI3K/mTOR inhibitors. Science China Life Sciences 2023, 66, 94–109. [Google Scholar] [CrossRef]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T.; Shan, J.; Gu, W. Targeting the degradation of cyclin D1 will help to eliminate oncogene addiction. Cell Cycle 2010, 9, 857–858. [Google Scholar] [CrossRef]
- Yang, C.; Li, Z.; Bhatt, T.; Dickler, M.; Giri, D.; Scaltriti, M.; Baselga, J.; Rosen, N.; Chandarlapaty, S. Acquired CDK6 amplification promotes breast cancer resistance to CDK4/6 inhibitors and loss of ER signaling and dependence. Oncogene 2017, 36, 2255–2264. [Google Scholar] [CrossRef]
- Islam, R.; Rahaman, M.; Hoque, H.; Hasan, N.; Prodhan, S.H.; Ruhama, A.; Jewel, N.A. Computational and structural based approach to identify malignant nonsynonymous single nucleotide polymorphisms associated with CDK4 gene. PLoS One 2021, 16, e0259691. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, E.S.; Kumarasamy, V.; Nambiar, R.; Pearson, J.D.; Vail, P.; Rosenheck, H.; Wang, J.; Eng, K.; Bremner, R.; Schramek, D.; et al. CDK/cyclin dependencies define extreme cancer cell-cycle heterogeneity and collateral vulnerabilities. Cell Rep 2022, 38, 110448. [Google Scholar] [CrossRef] [PubMed]
- Palafox, M.; Monserrat, L.; Bellet, M.; Villacampa, G.; Gonzalez-Perez, A.; Oliveira, M.; Brasó-Maristany, F.; Ibrahimi, N.; Kannan, S.; Mina, L.; et al. High p16 expression and heterozygous RB1 loss are biomarkers for CDK4/6 inhibitor resistance in ER+ breast cancer. Nature Communications 2022, 13, 5258. [Google Scholar] [CrossRef]
- Caldon, C.E.; Sergio, C.M.; Kang, J.; Muthukaruppan, A.; Boersma, M.N.; Stone, A.; Barraclough, J.; Lee, C.S.; Black, M.A.; Miller, L.D.; et al. Cyclin E2 overexpression is associated with endocrine resistance but not insensitivity to CDK2 inhibition in human breast cancer cells. Mol Cancer Ther 2012, 11, 1488–1499. [Google Scholar] [CrossRef]
- Turner, N.C.; Liu, Y.; Zhu, Z.; Loi, S.; Colleoni, M.; Loibl, S.; DeMichele, A.; Harbeck, N.; Andre, F.; Bayar, M.A.; et al. Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J Clin Oncol 2019, 37, 1169–1178. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front Oncol 2022, 12, 819128. [Google Scholar] [CrossRef]
- O’Brien, N.A.; McDermott, M.S.J.; Conklin, D.; Luo, T.; Ayala, R.; Salgar, S.; Chau, K.; DiTomaso, E.; Babbar, N.; Su, F.; et al. Targeting activated PI3K/mTOR signaling overcomes acquired resistance to CDK4/6-based therapies in preclinical models of hormone receptor-positive breast cancer. Breast Cancer Research 2020, 22, 89. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Hu, C.; Chen, Y.; Chen, Z.; Chen, Z.S.; Zhang, J.Y.; Fang, S. CDK6-PI3K signaling axis is an efficient target for attenuating ABCB1/P-gp mediated multi-drug resistance (MDR) in cancer cells. Mol Cancer 2022, 21, 103. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Prasad, P.; Singh, R.; Sahu, R.K.; Singh, A.; Magani, S.J.; Hedau, S. Role of identified proteins in the proteome profiles of CDK4/6 inhibitor-resistant breast cancer cell lines. Mol Omics 2023. [Google Scholar] [CrossRef]
- Lloyd, M.R.; Ryan, L.; Medford, A.J.; Keenan, J.C.; Spring, L.M.; Vidula, N.; Moy, B.; Juric, D.; Ellisen, L.; Bardia, A.; et al. Abstract P1-13-07: Investigating NF1 Mutations in Circulating Tumor DNA of Patients with Hormone-receptor Positive (HR+) Breast Tumors Resistant to CDK4/6 Inhibition (CDK4/6i): A Retrospective Clinical Analysis. Cancer Research 2023, 83, P1–13. [Google Scholar] [CrossRef]
- Mouron, S.; Manso, L.; Caleiras, E.; Rodriguez-Peralto, J.L.; Rueda, O.M.; Caldas, C.; Colomer, R.; Quintela-Fandino, M.; Bueno, M.J. FGFR1 amplification or overexpression and hormonal resistance in luminal breast cancer: rationale for a triple blockade of ER, CDK4/6, and FGFR1. Breast Cancer Research 2021, 23, 21. [Google Scholar] [CrossRef]
- Formisano, L.; Lu, Y.; Servetto, A.; Hanker, A.B.; Jansen, V.M.; Bauer, J.A.; Sudhan, D.R.; Guerrero-Zotano, A.L.; Croessmann, S.; Guo, Y.; et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun 2019, 10, 1373. [Google Scholar] [CrossRef]
- Garcia-Martinez, L.; Zhang, Y.; Nakata, Y.; Chan, H.L.; Morey, L. Epigenetic mechanisms in breast cancer therapy and resistance. Nature Communications 2021, 12, 1786. [Google Scholar] [CrossRef] [PubMed]
- Watt, A.C.; Cejas, P.; DeCristo, M.J.; Metzger-Filho, O.; Lam, E.Y.N.; Qiu, X.; BrinJones, H.; Kesten, N.; Coulson, R.; Font-Tello, A.; et al. CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nature Cancer 2021, 2, 34–48. [Google Scholar] [CrossRef]
- Luo, R.X.; Dean, D.C. Chromatin Remodeling and Transcriptional Regulation. JNCI: Journal of the National Cancer Institute 1999, 91, 1288–1294. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Li, Z.; Zou, W.; Zhang, J.; Zhang, Y.; Xu, Q.; Li, S.; Chen, C. Mechanisms of CDK4/6 Inhibitor Resistance in Luminal Breast Cancer. Front Pharmacol 2020, 11, 580251. [Google Scholar] [CrossRef]
- Pancholi, S.; Ribas, R.; Simigdala, N.; Schuster, E.; Nikitorowicz-Buniak, J.; Ressa, A.; Gao, Q.; Leal, M.F.; Bhamra, A.; Thornhill, A.; et al. Tumour kinome re-wiring governs resistance to palbociclib in oestrogen receptor positive breast cancers, highlighting new therapeutic modalities. Oncogene 2020, 39, 4781–4797. [Google Scholar] [CrossRef]
- Okabe, A.; Kaneda, A. Transcriptional dysregulation by aberrant enhancer activation and rewiring in cancer. Cancer Science 2021, 112, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Hrebien, S.; Morden, J.P.; Beaney, M.; Fribbens, C.; Huang, X.; Liu, Y.; Bartlett, C.H.; Koehler, M.; Cristofanilli, M.; et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nature Communications 2018, 9, 896. [Google Scholar] [CrossRef] [PubMed]
- O'Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; Andre, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov 2018, 8, 1390–1403. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Toi, M.; Neven, P.; Sohn, J.; Grischke, E.M.; Llombart-Cussac, A.; Soliman, H.; Wang, H.; Wijayawardana, S.; Jansen, V.M.; et al. Clinical Significance of PIK3CA and ESR1 Mutations in Circulating Tumor DNA: Analysis from the MONARCH 2 Study of Abemaciclib plus Fulvestrant. Clin Cancer Res 2022, 28, 1500–1506. [Google Scholar] [CrossRef]
- Andre, F.; Su, F.; Solovieff, N.; Arteaga, C.L.; Hortobagyi, G.N.; Chia, S.K.L.; Neven, P.; Bardia, A.; Tripathy, D.; Lu, Y.-S.; et al. Pooled ctDNA analysis of the MONALEESA (ML) phase III advanced breast cancer (ABC) trials. Journal of Clinical Oncology 2020, 38, 1009–1009. [Google Scholar] [CrossRef]
- Costa, C.; Wang, Y.; Ly, A.; Hosono, Y.; Murchie, E.; Walmsley, C.S.; Huynh, T.; Healy, C.; Peterson, R.; Yanase, S.; et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov 2020, 10, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Cohen, O.; Gong, X.; Johnson, G.N.; Buendia-Buendia, J.E.; Lloyd, M.R.; Kim, D.; Luo, F.; Mao, P.; Helvie, K.; et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov 2020, 10, 1174–1193. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yost, S.E.; Li, S.M.; Cui, Y.; Frankel, P.H.; Yuan, Y.C.; Schmolze, D.; Egelston, C.A.; Guo, W.; Murga, M.; et al. Genomic Markers of CDK 4/6 Inhibitor Resistance in Hormone Receptor Positive Metastatic Breast Cancer. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Zañudo, J.G.T.; Barroso-Sousa, R.; Jain, E.; Jin, Q.; Li, T.; Buendia-Buendia, J.E.; Pereslete, A.; Abravanel, D.L.; Ferreira, A.R.; Wrabel, E.; et al. Genomic and Transcriptomic Determinants of Resistance to CDK4/6 Inhibitors and Response to Combined Exemestane plus Everolimus and Palbociclib in Patients with Metastatic Hormone Receptor Positive Breast Cancer. medRxiv 2022. [Google Scholar] [CrossRef]
- Griffiths, J.I.; Chen, J.; Cosgrove, P.A.; O'Dea, A.; Sharma, P.; Ma, C.; Trivedi, M.; Kalinsky, K.; Wisinski, K.B.; O'Regan, R.; et al. Serial single-cell genomics reveals convergent subclonal evolution of resistance as early-stage breast cancer patients progress on endocrine plus CDK4/6 therapy. Nat Cancer 2021, 2, 658–671. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Cutts, R.J.; Huang, X.; Hrebien, S.; Liu, Y.; André, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; Cristofanilli, M.; et al. Circulating Tumor DNA Markers for Early Progression on Fulvestrant With or Without Palbociclib in ER+ Advanced Breast Cancer. JNCI: Journal of the National Cancer Institute 2020, 113, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, R.; Spring, L.; O'Shaughnessy, J.; Lacroix, L.; Bailleux, C.; Scott, V.; Dubois, J.; Nagy, R.J.; Lanman, R.B.; Iafrate, A.J.; et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol 2018, 29, 640–645. [Google Scholar] [CrossRef]
- Elmi, A.; Makvandi, M.; Weng, C.C.; Hou, C.; Clark, A.S.; Mach, R.H.; Mankoff, D.A. Cell-Proliferation Imaging for Monitoring Response to CDK4/6 Inhibition Combined with Endocrine-Therapy in Breast Cancer: Comparison of [(18)F]FLT and [(18)F]ISO-1 PET/CT. Clin Cancer Res 2019, 25, 3063–3073. [Google Scholar] [CrossRef]
- Pascual, J.; Lim, J.S.J.; Macpherson, I.R.; Armstrong, A.C.; Ring, A.; Okines, A.F.C.; Cutts, R.J.; Herrera-Abreu, M.T.; Garcia-Murillas, I.; Pearson, A.; et al. Triplet Therapy with Palbociclib, Taselisib, and Fulvestrant in PIK3CA-Mutant Breast Cancer and Doublet Palbociclib and Taselisib in Pathway-Mutant Solid Cancers. Cancer Discovery 2021, 11, 92–107. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009, 11, R77. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; Cox, D.; Knudsen, E.S. CDK4/6 inhibition provides a potent adjunct to Her2-targeted therapies in preclinical breast cancer models. Genes Cancer 2014, 5, 261–272. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Wardley, A.M.; Zambelli, S.; Hilton, J.F.; Troso-Sandoval, T.A.; Ricci, F.; Im, S.A.; Kim, S.B.; Johnston, S.R.; Chan, A.; et al. Abemaciclib plus trastuzumab with or without fulvestrant versus trastuzumab plus standard-of-care chemotherapy in women with hormone receptor-positive, HER2-positive advanced breast cancer (monarcHER): a randomised, open-label, phase 2 trial. Lancet Oncol 2020, 21, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Haley, B.B.; Abramson, V.G.; Brufsky, A.; Rexer, B.; Stringer-Reasor, E.; Jhaveri, K.L.; Sanders, M.; Ericsson-Gonzalez, P.I.; Ye, F.; et al. Abstract PD1-03: A phase Ib trial of fulvestrant + CDK4/6 inhibitor (CDK4/6i) palbociclib + pan-FGFR tyrosine kinase inhibitor (TKI) erdafitinib in FGFR-amplified/ ER+/ HER2-negative metastatic breast cancer (MBC). Cancer Research 2021, 81, PD1–03. [Google Scholar] [CrossRef]
- Chand, S.; Hansbury, M.; Lo, Y.; Feldman, P.; Carl, J.; Timmers, C.; Hummel, J.; Wee, S.; Kim, S. Abstract 1143: Development of a CDK2-selective small molecule inhibitor INCB123667 for the treatment of CCNE1hi breast cancers. Cancer Research 2023, 83, 1143–1143. [Google Scholar] [CrossRef]
- Fallah, Y.; Demas, D.M.; Jin, L.; He, W.; Shajahan-Haq, A.N. Targeting WEE1 Inhibits Growth of Breast Cancer Cells That Are Resistant to Endocrine Therapy and CDK4/6 Inhibitors. Front Oncol 2021, 11, 681530. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yao, S.; Yuan, Y.; Previs, R.A.; Elias, A.D.; Carvajal, R.D.; George, T.J.; Yuan, Y.; Yu, L.; Westin, S.N.; et al. Multicenter Phase II Trial of the WEE1 Inhibitor Adavosertib in Refractory Solid Tumors Harboring CCNE1 Amplification. Journal of Clinical Oncology 2023, 41, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Delord, J.P.; Im, S.A.; Ott, P.A.; Piha-Paul, S.A.; Bedard, P.L.; Sachdev, J.; Le Tourneau, C.; van Brummelen, E.M.J.; Varga, A.; et al. Safety and Antitumor Activity of Pembrolizumab in Patients with Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer. Clin Cancer Res 2018, 24, 2804–2811. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Lee, J.S.; Yost, S.E.; Frankel, P.H.; Ruel, C.; Egelston, C.A.; Guo, W.; Padam, S.; Tang, A.; Martinez, N.; et al. Phase I/II trial of palbociclib, pembrolizumab and letrozole in patients with hormone receptor-positive metastatic breast cancer. Eur J Cancer 2021, 154, 11–20. [Google Scholar] [CrossRef]
Figure 1.
Signaling pathway of CDK 4/6-Cycle D complex and mechanism of action of CDK 4/6 inhibitors.
Figure 1.
Signaling pathway of CDK 4/6-Cycle D complex and mechanism of action of CDK 4/6 inhibitors.
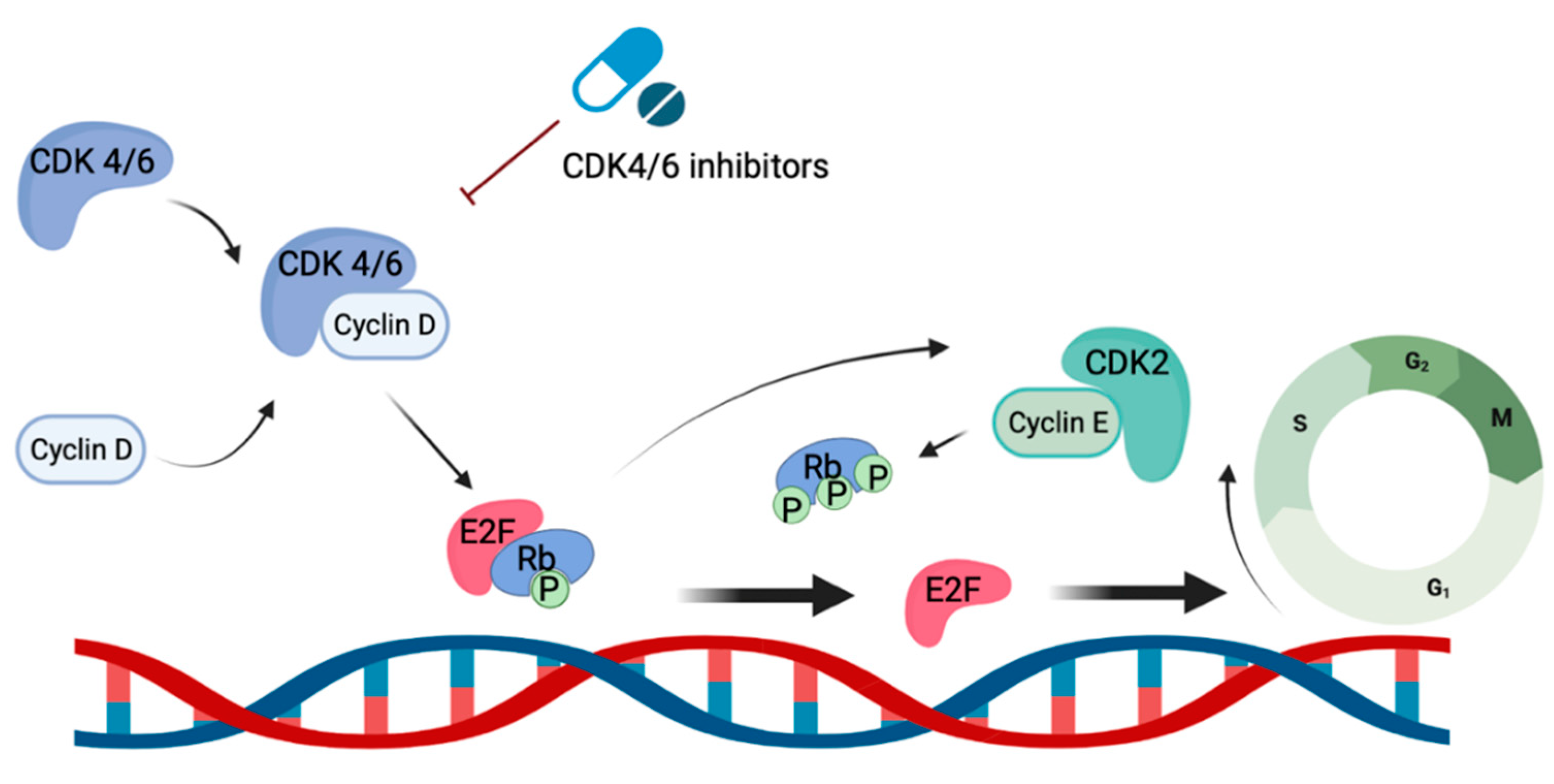
Figure 2.
Targeting alterative pathway.
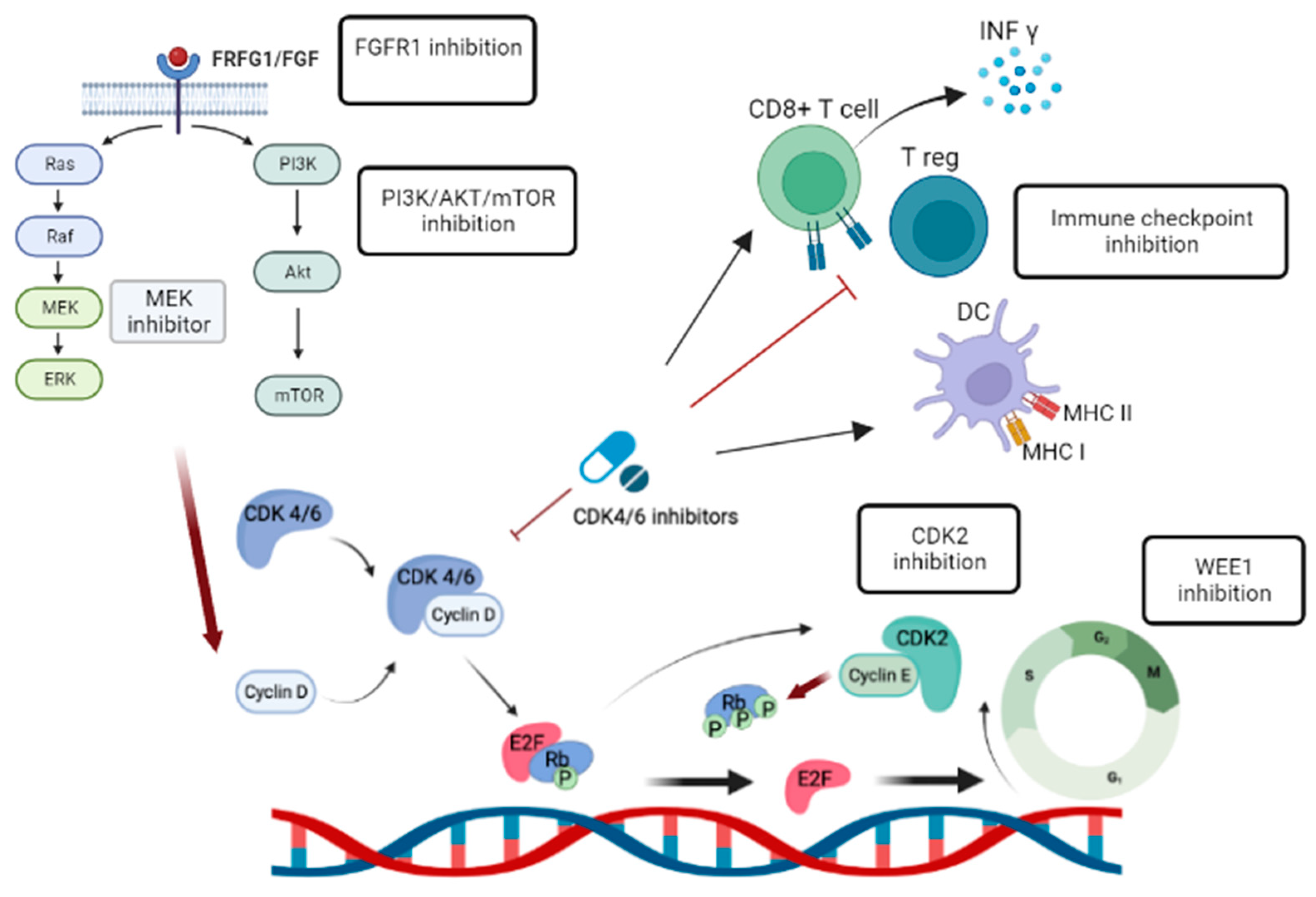

Table 1.
Clinical trials and survival benefits of CDK4/6 inhibitors.
| Endocrine sensitive MBC, postmenopausal | N (ratio) | mPFS | HR | mOS | HR | |
|---|---|---|---|---|---|---|
| PALOMA-2[4,5] | Palbociclib plus letrozole | 666 (2:1) | 24.8 | 0.58 | 53.9 | 0.956 |
| Placebo plus letrozole | 14.5 | 1 | 51.2 | 1 | ||
| MONALEESA-2[6,7] | Ribociclib plus letrozole | 668 (1:1) | 25.3 | 0.56 | 63.9 | 0.76 |
| Placebo plus letrozole | 16.0 | 1 | 51.3 | 1 | ||
| MONARCH-3[8] | Abemaciclib plus NSAI* | 493 (2:1) | 28.1 | 0.54 | pending | |
| Placebo plus NSAI* | 14.7 | 1 | ||||
| Endocrine resistant MBC, any menopausal status | ||||||
| PALOMA-3[9,10] | Palbociclib plus Fulvestrant | 521 (2:1) | 9.5 | 0.46 | 34.9 | 0.81 |
| Placebo plus Fulvestrant | 4.6 | 1 | 28.0 | 1 | ||
| MONARCH-2[11,12] | Abemaciclib plus Fulvestrant | 669 (2:1) | 16.3 | 0.55 | 46.7 | 0.76 |
| Placebo plus Fulvestrant | 9.3 | 1 | 37.3 | 1 | ||
| Endocrine sensitive or resistant MBC, postmenopausal | ||||||
| MONALEESA-3[13,14] | Ribociclib plus Fulvestrant | 726 (2:1) | 20.5 | 0.59 | NR | 0.72 |
| Placebo plus Fulvestrant | 12.8 | 1 | 40.0 | 1 | ||
| Endocrine sensitive MBC, pre/perimenopausal | ||||||
| MONALEESA-7[15,16] | Ribociclib plus NSAI/LHRH analog | 672 (2:1) | 23.8 | 0.55 | NR | 0.72 |
| Placebo plus NSAI/LHRH analog | 13.0 | 1 | 40.9 | 1 | ||
Table 2.
Clinical trials of combination therapy with CDK 4/6 inhibitors.
| Treatment Drug | Trial ID | Phase | Cancer Type |
|---|---|---|---|
| Ricociclib, everolimus, exemestane | NCT02732119 | 1 | HR+ HER2- locally advanced/metastatic breast cancer post progression on CDK 4/6 inhibitor |
| Palbociclib, bosutinib, fuvestrant | NCT03854903 | 1 | HR+ HER2- advanced breast cancer refractory to CDK 4/6 inhibitor |
| Palbociclib, letrozole, venetoclax | NCT03900884 | 1b | ER and BCL-2 positive breast cancer |
| Ribociclib, belinostat | NCT04315233 | 1/1b | Metastatic triple negative breast cancer |
| Palbociclib, avelumab | NCT04360941 | 1b | AR+ triple negative breast cancer |
| CDK4/6 inhibitor, fulvestrant, capivasertib | NCT04862663 | 3 | Metastatic HR+ HER2- breast cancer |
| CDK4/6 inhibitor, fulvestrant, ipatasertib | NCT04920708 | 2 | Metastatic HR+ HER2- breast cancer |
| Abemaciclib, tucidinostat | NCT05464173 | 1/2 | Previously treated with palbociclib in HR+ HER2- relapsed/metastatic breast cancer |
| Ribociclib, alpelisib | NCT05508906 | 1b | Metastatic HR+ HER2- breast cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



