
Submitted:
28 June 2023
Posted:
28 June 2023
You are already at the latest version
Abstract
Optic nerve disorders encompass a wide spectrum of conditions characterized by the loss of retinal ganglion cells (RGCs) and subsequent degeneration of the optic nerve. The etiology of these disorders can vary significantly, but emerging research highlights the crucial role of oxidative stress, an imbalance in the redox status characterized by an excess of reactive oxygen species (ROS), in driving cell death through apoptosis, autophagy, and inflammation. This review provides an overview of ROS-related processes underlying four extensively studied optic nerve diseases: glaucoma, Leber's hereditary optic neuropathy (LHON), anterior ischemic optic neuropathy (AION), and optic neuritis (ON). Furthermore, we present preclinical findings on antioxidants, with the objective of evaluating the potential therapeutic benefits of targeting oxidative stress in the treatment of optic neuropathies.
Keywords:
oxidative stress
; optic nerve
; retinal ganglion cell
; glaucoma
; Leber’s hereditary optic neuropathy
; ischemic optic neuropathy
; optic neuritis
1. Introduction
Optic nerve diseases encompass a wide range of disorders characterized by optic nerve atrophy, resulting from the loss of retinal ganglion cells (RGCs) and leading to sight-threatening conditions [1,2,3]. These pathologies include
- Anterior ischemic optic neuropathies (AION) This category includes arteritic forms like giant cell arteritis (GCA), which has a pooled prevalence of approximately 51.74 in 100,000 for individuals over the age of 50 [15]. Non-arteritic forms have a reported prevalence of approximately 102.87 in 100,000 in the general population over the age of 40 in South Korea [16].
- Infiltrative optic neuropathies, such as leukemic optic neuropathy, which presents in approximately 16% and 18% of all chronic and acute leukemia cases, respectively [28].
Glaucoma is the most prevalent optic nerve disease worldwide (Figure 1) [6,7]. LHON has a low estimated prevalence for complete penetrance cases [8,9,10,11,12], while the prevalence is much higher for carriers of mutation variants in the general population [13,14]. The common underlying feature in all optic nerve diseases is the damage and loss of RGCs and their axons, which gradually leads to optic nerve degeneration [3,33,34]. RGCs have high energy requirements and are particularly susceptible to alterations in their energy supply, mainly generated in the mitochondria through the electron transport chain (ETC) [35]. Oxidative stress plays a pivotal role in the pathophysiology of optic nerve diseases such as glaucoma, LHON, and AION. Imbalances between reactive oxygen species (ROS) as well as reactive nitrogen species (RNS) generation and antioxidant systems lead to reactive species overproduction, adenosintriphosphat (ATP) insufficiency, irreversible cellular injuries, and ultimately RGC loss [3,36,37,38,39,40,41,42,43,44,45].
Considering the high global prevalence of glaucoma and the limited treatment options available for most optic neuropathies [33], it becomes essential to explore new research avenues and investigate novel therapeutic approaches. This review aims to shed light on the role of oxidative damage in the pathophysiology of optic nerve diseases. Specifically, we provide an overview of the current understanding of oxidative stress as a key pathological factor and explore its potential as a viable therapeutic target. We will especially focus on four of the most prevalent and clinically challenging optic nerve diseases, glaucoma, LHON, AION and ON. Ultimately, our goal is to identify novel curative strategies that may pave the way for improved treatment options in these conditions.
2. Anatomy and Perfusion of the Visual Pathway
The optic nerve, also known as the second cranial nerve [46], is composed of thin (0.1 µm) and lengthy (~50 mm) RGC axons that extend from the retina to the lateral geniculate nucleus, resulting the soma/axon ratio of approximatively 1:10,000 [47]. Within the retinal layers, these axons merge to form the retinal nerve fiber layer [48], which runs parallel to the superficial blood vessels. The inner retina, including the outer plexiform layer through the nerve fiber layer, is supplied by the central retinal artery. On the other hand, the avascular outer retina, consisting of the outer nuclear layer and photoreceptors, receives its blood supply through diffusion from the choriocapillaris. The choriocapillaris is nourished by short posterior ciliary arteries, which branch from the ophthalmic artery [49,50]. Notably, retinal oxygenation exhibits variability depending on light or dark conditions due to the different oxygen demand of rods and cones. In humans, rods, which are responsible for vision in low light, are present in approximately 120 million, while cones number around 6 million [51]. Consequently, retinal oxygen consumption is reduced by half in the presence of light, attributed to the decreased activity of rods compared to cones [51].
Optic nerve axons account for approximately 38% of all axons within the central nervous system [52]. Around 1.2 million RGC axons converge to form the optic nerve head (ONH), also referred to as the optic papilla or optic disc. The ONH exhibits a brighter central depression known as the optic cup [52,53,54,55]. Blood supply to the ONH in humans is provided by the arterial circle of Zinn-Haller [49].
The optic nerve can be divided into four compartments: the intraocular segment (1-2 mm), which includes the retinal nerve fiber layer (RNFL) and extends from the ONH to the lamina cribrosa; the intraorbital segment (25-30 mm), which spans from the retrobulbar tract to the optic canal; the intracanalicular segment (5-9 mm); and the intracranial segment (9-10 mm), which extends from the optic canal to the optic chiasm [52]. Four distinct regions can be identified within the optic nerve head: the nerve fiber layer, the prelaminar region, the lamina cribrosa, and the retrolaminar region [48]. The lamina cribrosa serves as a supportive structure for the RGC axons within the ONH [56,57], and consists of approximately 200-300 porous apertures through which the optic nerve passes from the sclera into the retrobulbar cavity [52]. Deformations of the lamina cribrosa may indicate RGC loss and can signify the initial stages of glaucomatous optic neuropathy [58]. Once beyond the lamina cribrosa, the optic nerve becomes myelinated by oligodendrocytes, increasing its diameter from 1-2 mm to 3-4 mm [52]. The intraorbital, intracanalicular, and intracranial segments receive their blood supply from the posterior ciliary arteries as well as the circle of Willis [59].
The two optic nerves converge at the optic chiasm, where nerve fibers originating from the nasal retina of each eye cross over to join the temporal fibers of the contralateral eye [48,60,61]. The blood supply to the chiasm is provided by the circle of Willis [48,53]. From the chiasm, the RGC axons continue their course into the optic tract, which receives perfusion from the posterior communicating and internal carotid artery [53]. Within the optic tract, the nerve fibers undergo rearrangement to align with their corresponding positions in the lateral geniculate nucleus [60]. Fibers carrying visual information from the right visual field project to the left cerebral hemisphere and vice versa [60]. In the lateral geniculate nucleus, the RGC-axons synapse with the second-order neurons of the visual pathway, organized in six layers consisting mainly of large and small neurons [48]. Some fibers from the optic tract also synapse with the olivary pretectal nucleus, regulating the pupillary light reflex [53,62]. Additionally, RGC axons containing melanopsin terminate in the suprachiasmatic nucleus, a crucial center for controlling the circadian rhythms [48,62].
The large and small axons of the lateral geniculate nucleus form the optic radiation, which initially projects anteriorly and then turns posteriorly, terminating in the occipital lobe where the visual cortex (Brodmann area 17) is located [48,60]. These regions receive blood supply from branches of the internal carotid artery (lateral geniculate nucleus, optic radiation) and the posterior cerebral artery (visual cortex) [53]. In Figure 2, the perfusion and the anatomy of the visual pathway are illustrated.
An interesting aspect of the optic nerve anatomy is the presence of an unmyelinated portion in the RGCs [47]. In the unmyelinated compartment, specifically in the intraocular segment of the optic nerve, potential signals cannot be transmitted through saltatory conduction due to the absence of myelin [63]. To compensate for this limitation and enable rapid transmission, RGCs generate higher quantities of ATP in their axons to repolarize the plasma membrane [64]. Mitochondrial bi-directional transport (antero- and retrograde) along the axons plays a crucial role in this process. These organelles move toward regions with high energy demands, such as the unmyelinated portions, and ATP gradients are believed to guide this transport [47]. This mechanism may explain the specific vulnerability of RGCs to mitochondrial dysfunction, leading to the triggering of ROS production in a vicious cycle [44,64].
3. General Mechanisms of Nitro-Oxidative Stress in the Optic Nerve
3.1. Generation of Reactive Oxygen and Nitrogen Species
Mitochondria are vital intracellular organelles responsible for essential chemical reactions that produce energy substrates [65,66]. They possess two membranes (outer and inner mitochondrial membrane), their own DNA (mtDNA), ribosomes and RNAs [67]. In addition to their various cellular functions such as modulating intracellular calcium levels, synthesizing nucleotides, lipids, and amino acids, and regulating apoptosis, mitochondria also generate ROS [65,68,69,70]. ROS, at basal levels, serve as critical mediators of signaling pathways, including hypoxic and inflammatory pathways [44,65,71,72]. The fundamental function of mitochondria is to regulate oxygen metabolism and produce energy in the form of ATP. This process involves the reduction of oxygen to water through oxidative phosphorylation, with the simultaneous generation of ATP as the major energy molecule for the cell [65,66,73]. The electron transport chain (ETC) within the inner mitochondrial membrane plays a central role in this process. The ETC consists of five membrane protein complexes (I, II, III, IV, V) and two electron carriers (ubiquinone (COQ) and cytochrome c (Cyt c)). Electrons are introduced into the chain by Complex I and subsequently transferred from one complex to the next via electron carriers until they reach Complex IV. This electron transfer is accompanied by the generation of a proton gradient between the mitochondrial matrix and the intermembrane space [67]. Complex V, also known as ATP synthase, utilizes this proton gradient to convert adenosine diphosphate (ADP) to ATP [67]. Despite the efficiency of oxidative phosphorylation electron leaks can occur, leading to the direct interaction of electron carriers with molecular oxygen (O₂) in the mitochondrial matrix. This interaction results in the donation of electrons and the generation of superoxide (O2•−) [66]. These events are known as electron leaks and correspond to the sites of ROS-generation in the mitochondrial matrix [74]. Under physiological conditions, approximately 0.2-2% of electrons “leak out” of the ETC and directly interact with O₂, producing ROS [66,73,74]. Researchers have identified eleven sites of ROS generation in the context of the ETC [75]. While mitochondria are recognized as the main source of ROS in the cell, other significant sources include the enzymatic activities of nitric oxide synthase (NOS) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) [3,74,76,77]. NOS generates nitric oxide (NO), while NOX (comprising seven isoforms: NOX1, -2, -3, -4, -5, DUOX1, -2) transfers electrons from cytosolic NADPH to molecular O₂, generating O2•− [78]. Superoxide serves as the precursor of most other ROS [73], which can be categorized into “free radicals” such as hydroxyl (•OH), alkoxyl (RO•), peroxyl (ROO•), hydroperoxyl (HOO•) radical [36], characterized by an unpaired electron at the outer orbital level [79]. Additionally, there are “non-radical oxidizing chemical species” such as hydrogen peroxide (H₂O₂), hypochlorous acid (HOCl), ozone (O3), and singlet oxygen (1△g) [36], which have electron pairs in covalent binding [79].
NO is a free radical that plays a pivotal role in various physiological functions [80]. It serves as a regulator of vascular tone [81,82,83]. Additionally, NO acts as a signaling molecule in neurotransmission and as a regulator of gene transcription [84,85,86,87,88,89]. The production of NO is facilitated by the activity of NOS, an enzyme that has three isoforms: neuronal NOS (nNOS or NOS I), which is predominantly abundant in the central nervous system; inducible NOS (iNOS or NOS II), primarily expressed in the immune system but also detected in the vascular endothelium as a response to inflammatory stimuli; and endothelial NOS (eNOS or NOS III), mainly expressed in vascular cells and perivascular fat tissue [80,90]. NO rapidly and spontaneously reacts with O2•− through a “diffusion-limited reaction” [91,92]. The reaction occurs so swiftly that NO can outcompete superoxide dismutase (SOD), an antioxidant enzyme responsible for converting superoxide into hydrogen peroxide [91]. As a result, a highly damaging RNS termed peroxynitrite (ONOO−) is generated [80,91,92]. Under physiological pH conditions, a substantial portion of ONOO− is protonated, forming peroxynitrous acid (ONOOH) [93]. ONOOH can further decompose, leading to the formation of nitric dioxide (•NO2) and hydroxyl radicals [93]. Peroxynitrite contributes to the pathogenesis of diverse retinal disorders, being also newly proposed as a critical factor in the pathogenesis of glaucoma [94,95,96].
3.2. Oxidative Damage and Antioxidant Defense Systems
ROS and RNS play a physiological role in cellular responses to hypoxia, cell proliferation, cell death, inflammation or infection [44,72]. Immune cells, such as phagocytes, produce ROS, which provide reactions necessary for an appropriate killing of pathogens [97,98]. Exogenous factors such as alcohol, smoking, radiation, air pollutants, and some drugs can also lead to generation of ROS and RNS [99]. Hence, due to endogenous or exogenous trigger factors, the balance between pro- and antioxidant systems can be critically undermined, resulting in nitro-oxidative stress. In this context, radicals begin to compete for paired electrons with intracellular substrates [100], creating oxidative damage. Oxidative injuries are recognized to be a crucial player in the pathogenesis of a variety of diseases, such as tumors, atherosclerosis, neurodegenerative diseases, ocular diseases, rheumatic and immunological diseases [36,101,102,103,104]. At the biomolecular level, three general forms of injuries caused by reactive species can be distinguished: DNA-lesions, for example represented by guanine oxidation, with subsequent formation of 8-oxo guanine [105,106], protein alterations primarily via tyrosine nitration and cysteine oxidation [107,108], and lipid peroxidation via oxidation of cell membrane phospholipids, of liposomes, and of lipoproteins [91,109]. Consequences of DNA-damage are modifications in expression of proteins and altered regulation of fundamental activities like oxidative phosphorylation, according to the vicious cycle theory [110,111], where a free radical imbalance triggers further ROS production [112]. Likewise, DNA lesions promote apoptosis, ferroptosis, autophagy and aging [100,112,113,114]. In this context, mitochondrial ROS also induce activation of the nod-like receptor family pyrin domain-containing 3 (Nlrp3) inflammasome, a key factor in pyroptotic cell death during inflammation [115]. As a result of tyrosine nitration, enzyme inactivation can occur, such as in case of the mitochondrial SOD2 (also termed manganese SOD, MnSOD) [116]. Lipid peroxidation can induce demyelination [117], and also cause disruption of the cellular membrane, inflammation, and apoptosis [118,119,120].
Antioxidant systems are responsible to defend cells and tissues from the damaging impact of reactive species, which are constantly produced as a “by-product” of the oxidative phosphorylation, but also serve, at basal levels, for physiological functions [72]. Antioxidant agents act to “prevent, block or repair” ROS injuries, and can be classified into enzymatic and nonenzymatic [100,121]. Enzymatic antioxidants comprise SOD, catalase (CAT), glutathione peroxidase (GPX), glutathione-S-transferase (GST), heme oxygenase (HO), peroxiredoxin, and thioredoxin [100,122]. SOD is a major antioxidant enzyme [79], reducing O2•− into H2O2, while CAT und GPX provide the conversion of H2O2 into H2O [123]. GST acts to reduce peroxidized lipids via selenium-independent glutathione peroxidase activity [124]. HO catalyzes the degradation of heme to biliverdin/bilirubin, carbon monoxide, and free iron [125]. Finally, peroxiredoxin reduces oxidized substrates through a conserved cysteine residue, designated the “peroxidatic” Cys [126], while thioredoxin acts through disulfide reductase activity [127].
Nonenzymatic antioxidants can be classified into direct and indirect agents. Direct antioxidants react with ROS or RNS, “being sacrificed in the process of their antioxidant actions” [128]. These “scavenge” free radicals by donating electrons and thereby reducing reactive species, preventing their “attack” on biological intracellular substrates [129]. Free radical scavengers are for example glutathione (GSH) [130], carotenoids [131], vitamin C (ascorbic acid) [132], and vitamin E (α-tocopherol) [133]. Alternatively, indirect antioxidants are molecules, such as the same GSH or also vitamin C, that upregulate antioxidant proteins for example via nuclear factor erythroid-2-related factor 2 (Nrf-2) [128,132], or molecules like α-lipoic acid, which can bind metal ions, thereby preventing a metal-dependent inhibition of antioxidants [134]. Examples for antioxidant compounds adsorbed with the food are resveratrol and betulinic acid, which act in part through a direct antioxidant mechanism as free radical scavenger but also via an indirect mechanism, by inhibiting ROS-mediated proapoptotic pathways or by triggering antioxidant enzyme expression [94,135,136].
3.3. Oxidative Stress in Retinal Ganglion Cells
The retina belongs to the metabolically most active organs in the human body [137], and requires a relatively large amount of energy substrates [138], which makes it particularly vulnerable to energy insufficiency [139]. Oxygen supply is essential for retinal function [140], and its consume occurs very rapidly like in the brain [141,142,143]. Hence, conditions which can modify the supply of molecules such as O2, necessary for the production of energy substrates like ATP, may rapidly generate significant damage in RGCs, due to their susceptibility to oxygen deficiency (Figure 3). Thus, an appropriate blood supply by retrobulbar and retinal vessels is crucial for the proper function of RGCs. Studies in retrobulbar blood vessels reported that ROS blunted endothelial function partially by reducing the contribution of the NOS pathway to endothelium-dependent vasodilation [144]. Likewise, moderately elevated IOP induced endothelial dysfunction in retinal arterioles together with RGC loss [145,146]. Zadeh et al. found in apolipoprotein E (ApoE)-deficient mice that hypercholesterolemia caused oxidative stress and endothelial dysfunction in retinal arterioles, but did neither lead to increased ROS levels in the RGC layer nor to loss of RGCs, indicative of compensatory effects [147]. In contrast, a study in pigs reported that after only 12 minutes of ocular ischemia and 20 hours of reperfusion, endothelial dysfunction, retinal edema and RGC loss occurred [148]. ROS generation due to ischemia/reperfusion (I/R) injury is reported to be caused by diverse enzymes involved in regulation of oxidative metabolism, such as NOX2, xanthine oxidase (XO), uncoupled eNOS, and by ETC dysfunction [148,149,150,151]. Hyperglycemia was also described to be a cause of endothelial dysfunction and oxidative stress in the retina [152,153,154], via involvement of NOX2, due to activation of receptor of advanced glycation end product (RAGE)-, mitogen activated protein kinase (MAPK)-, Polyol-, protein kinase C (PKC)-, renin-angiotensin system (RAS)- signaling pathways [155,156,157,158,159,160].
4. Oxidative Stress in Individual Optic Nerve Diseases
4.1. Glaucoma
4.1.1. General Aspects
The term “glaucoma” encompasses a group of optic nerve diseases that share similar morphological characteristics but may have diverse origins. They represent the most common optic nerve disorders [34,161,162], characterized by alterations of the ONH, which include a decrease of the neuroretinal rim and an enlargement of the optic cup, leading to deformation of the lamina cribrosa [34,163]. As a result, in early stages, typical visual field defects, which follow the arcuate pattern corresponding to the arrangement of nerve fiber bundles, can be observed [164]. Elevated intraocular pressure (IOP) is described as the primary risk factor, which can result from pathological resistance to aqueous humor drainage through the trabecular meshwork (TM), causing mechanical stress and compression on the axonal fibers and the ONH [165,166,167]. Glaucomatous disorders are classified into primary forms, where there is no associated ocular disease, and secondary forms, where known coexisting pathological processes (such as uveitis, neovascularization, trauma, or lens-related conditions) lead to IOP elevation and thus to the development of glaucomatous damage [34,165,168]. Primary and secondary forms are further divided into open-angle glaucoma, which among others includes a subtype known as normal tension glaucoma (NTG), and angle-closure glaucoma, in relation to the angle between the iris and cornea in the anterior chamber [34,169,170]. Primary open angle glaucoma (POAG), the worldwide major form of glaucoma, describes a disorder where, under conditions of an open chamber angle, structural damages to the optic nerve emerge silently, gradually and chronically [165]. While high IOP values can be a contributing factor in this disorder, it is not always present in POAG. Elevated IOP is defined as a pressure value that exceeds the 97.5th percentile for the population under consideration and is commonly assumed to be a value higher than 21 mm Hg [34,168]. However, it is important to note that elevated IOP is recognized as a risk factor but not a diagnostic criterion for glaucoma [165,168]. In fact, approximately 30–90% of POAG cases have IOP values below the 21 mm Hg cutoff, which is considered NTG. The prevalence of NTG varies significantly geographically [171]. Additionally, the term “ocular hypertension” (OHT) refers to a condition where elevated IOP is detected without evidence of glaucomatous optic neuropathy [34]. Angle-closure glaucoma is considered an ocular emergency resulting from anatomical contact between the iris and lens. This contact can cause obstruction of the aqueous humor outflow leading to a condition known as pupillary block, present in approximately 90% of cases of primary angle closure glaucoma (PACG) [170,172]. In this context a sudden and significant increase in IOP occurs and can reach values as high as 60 to 80 mm Hg [169]. PACG is estimated to affect approximately 26% of the glaucoma population and is responsible for about half of the cases of glaucoma-related visual loss worldwide [173,174,175].
The main risk factors for POAG include elevated IOP, advanced age, black race, myopia, and a positive family history [168]. On the other hand, risk factors for PACG include hyperopia, shallow central anterior chamber depth, shallower limbal anterior chamber depth, anteriorly positioned lens, increased lens thickness, small corneal diameter, steep corneal curvature, as well as high IOP values, advanced age, Asian ethnicity and female gender [173,176,177]. Elevated IOP is the only identified modifiable risk factor [178], and reducing IOP is currently the only proven method to treat glaucoma effectively [176,179]. IOP has been considered a central factor in both the “mechanical” and “vascular” theories proposed to explain the etiopathogenesis of glaucoma. According to the mechanical theory, elevated IOP leads to compression and deformation of RGC axons and the optic nerve, ultimately resulting in cell death due to reduced or blocked axoplasmic flow and a deficit of cellular supply elements [180]. On the other hand, the vascular theory suggests that decreased perfusion to the optic nerve causes ischemia in RGCs, leading to neurodegenerative damage [166]. Reduced blood supply may be a consequence of IOP-related compression or functional deficits of blood or vessels that supply the laminar regions of the ONH [181]. Elevated IOP can be a result, for example, of pathological resistance in the aqueous humor drainage process through the TM [167].
The primary goal of antiglaucomatous drugs is to lower IOP to a target level, which is an acceptable range individually set to prevent disease progression [182]. Antiglaucomatous drugs are generally administered topically as eye drops and can be categorized into different classes based on their mechanism of action. Prostaglandin analogues, such as latanoprost, increase uveoscleral and trabecular outflow, while β-blockers (e.g., timolol), α2-adrenoceptor agonists (e.g., brimonidine), and carbonic anhydrase inhibitors (e.g., dorzolamide) reduce aqueous humor production [178]. Additionally, α2- adrenoceptor agonists like brimonidine, clonidine, and epinephrine can also increase trabecular outflow, similar to prostaglandins [178]. Miotic agents, such as pilocarpine, widen the chamber angle, and osmotically active drugs like mannitol increase water removal from the eye through the systemic circulation when administered intravenously (IV) [178]. Moreover, pilocarpine was reported to provide a neuroprotective effect through activation of muscarinic receptors [183]. In case of PACG, the primary treatment is peripheral iridotomy, a laser procedure that creates a full-thickness opening in the iris to relieve pupillary blockage [203]. Additionally, osmotic substances like mannitol or carbonic anhydrase inhibitors such as acetazolamide can be administered intravenously [178].
The economic implications of glaucoma have been extensively studied worldwide, highlighting its significance as a critical social problem. Cost-of-illness studies have shed light on the financial impact of glaucoma. For instance, a study conducted in the UK in 2002 reported that over £300 million was spent on glaucoma-related expenses [184]. Similarly, an Australian review analyzed published randomized trials and population-based studies since 1985, projecting that the total costs (including direct, indirect, and costs related to loss of well-being) for POAG in Australia would increase from AU$1.9 billion in 2005 to AU$4.3 billion in 2025 [185]. Examining the indirect costs of glaucoma, a study from the UK investigated whether glaucoma can be a risk factor for falls. The study found that between 2012 and 2018, 11.7% of hospital admissions for falls in a national health service hospital were in patients with a secondary diagnosis of glaucoma [186]. Furthermore, a recent retrospective cohort conducted in the US, extrapolating data from 2015 to 2017 and based on a database of patients with POAG and ocular hypertension (OHT), compared these two conditions. The study revealed that advanced-stage POAG was associated with a higher risk of falls compared to OHT. Additionally, it was found that the annual eye-related outpatient costs for POAG patients were higher (median: $516) than for OHT patients (median: $344). Among patients with POAG, those in advanced stages exhibited even higher annual eye-related costs (median: $639) compared to those in moderate (median: $546) and mild (median: $476) stages [187]. Similarly, a previous multicenter study conducted in Germany between 2009 and 2010, involving two university hospitals and 13 ophthalmological practices, reported that direct costs for therapies were higher for glaucoma compared to OHT and were directly associated with disease progression. The study highlighted that additional treatment changes necessitated by uncontrolled IOP were the major contributors to the increased costs associated with glaucoma. The study concluded that by effectively managing IOP over the long term and avoiding advanced disease stages, both disease progression and associated costs could be reduced [188]. In summary, publications on this topic consistently emphasize that early first-line therapies play a vital role in reducing the economic burden of glaucoma. By preventing disease progression to advanced stages, these early interventions can help minimize the need for more expensive and sometimes less effective treatments, ultimately alleviating the economic strain imposed by glaucoma [189].
4.1.2. Redox Parameters and Oxidative Stress Biomarkers in Glaucoma
Numerous studies have been conducted to gather evidence of altered redox status in glaucoma by examining oxidant or antioxidant levels in various samples, including blood, plasma, serum, aqueous humor, and TM. Common systemic parameters used to assess oxidative stress include total antioxidant status (TAS), total antioxidant capacity (TAC), biological antioxidant potential (BAP), total reactive antioxidant potential (TRAP), and total oxidant status (TOS). Biomarkers of oxidative stress, such as 8-hydroxy-2’-deoxyguanosine (8-OHdG), poly-(adenosindiphosphat-ribose)-polymerase (PARP1), oxoguanine-DNA-glycosylase (OGG1), and malondialdehyde (MDA), are indicative of DNA oxidative damage, base excision repair activity, and lipid peroxidation.
An Indian study examining redox-related DNA damage in POAG patients, reported elevated levels of 8-OHdG levels in both plasma and aqueous humor. They also observed a negative correlation between 8-OHdG levels and PARP1 and OGG1 levels, which were concurrently lower [190]. Increased levels of 8-OHdG were also found in the TM of patients with POAG, correlating with more severe visual field defects [191,192].
TAC levels were found to be lower in the serum and aqueous humor of glaucoma patients, while MDA level were increased [193]. Decreased levels of TAS [194] and TRAP [195] were also observed in the aqueous humor of glaucoma patients. Lower TAS levels were additionally found in plasma samples of individuals with POAG, further associated with an increasing cup-to-disc ratio, which indicates cup-disc enlargement and retinal ganglion cell loss [196]. Studies on serum samples of glaucoma patients reported lower BAP levels, correlating with lower RGC density in young males [197], more advanced visual field loss [198], or higher IOP [199].
A meta-analysis on redox parameters in glaucoma revealed a general increase in oxidative stress parameters in both serum and aqueous humor, with MDA being the most significant biomarker, suggesting its potential clinical utility [200]. Antioxidative markers were found to be lower in serum, while aqueous humor showed an increase of antioxidant defense, indicating a possible compensatory response to oxidative stress [200]. Similarly, a recent meta-analysis on oxidative stress markers in glaucoma highlighted lower TAS levels in blood and elevated levels of antioxidant enzymes such as SOD, CAT, and GPX in the aqueous humor [201]. This systematic review also compared patients with POAG to those with exfoliation glaucoma, the most common secondary form of POAG characterized by the deposition of fibrillar material in the anterior segment [202,203]. It reported no increase in antioxidant defense in the aqueous humor of exfoliation glaucoma patients, along with a decrease in TAS levels in the blood [201].
RNS, including NO, were found to be increased in the aqueous humor of patients with POAG and PACG [204]. A Spanish study examining both TM and aqueous humor of POAG patients, reported upregulation of iNOS and downregulation of eNOS in trabecular meshwork cells (TMCs). They also observed increased levels of MDA in the aqueous humor, corresponding to an increase in visual field defects [205]. The authors of this study suggested that the increased production of NO induced by iNOS may play a role in the process of RGC-death, potentially leading to an elevation of MDA levels in the aqueous humor [205].
4.1.3. Oxidative Stress in the Pathogenesis of Glaucoma
To gain a better understanding on the role of reactive species in glaucoma, it is important to discuss the anatomy and function of the TM and the detrimental impact of ROS on these structures. The TM is located in the sclerocorneal angle and consists of three layers: the uveal TM, the corneoscleral TM, and the juxtacanalicular TM, also known as the cribriform TM region. This region is adjacent to the Schlemm’s canal, which allows for the drainage of aqueous humor into the episcleral veins [206]. The TM plays a crucial role in regulating IOP by controlling the outflow of aqueous humor. It allows for the turnover of aqueous humor through drainage into superficial veins towards the conjunctiva [206]. Any alterations in TM permeability, such as changes in TM structure, can result in increased resistance to aqueous humor outflow, leading to elevated IOP. Studies focusing on TMCs have investigated how increased IOP may develop in glaucoma patients. These studies have shown that the oxidative balance in aqueous humor, which has been found to be altered in individuals with glaucoma [207,208], can impact the structure and function of TMCs, particularly the TM endothelial cells lining the Schlemm’s canal [209]. Elevated levels of ROS in the aqueous humor can trigger changes in the TM [167]. This can occur through fusion of TMCs, leading to trabecular thickening [210]. Additionally, ROS can induce TMC apoptosis, resulting in TM disruption [209]. Alterations in the extracellular matrix (ECM) can also occur, affecting the adhesion of endothelial TMCs to the ECM and causing TM to collapse [211]. In all of the cases, increased resistance to aqueous humor circulation occurs, leading to elevated IOP [209]. In Figure 4, an anatomical overview of the described organs, with focus on the anterior segment of the eye, is presented.
Oxidative damage contributes to axonal injuries, leading to the death of RGCs [37]. RGC loss may occur as a result of a process involving ROS, elevated IOP, mechanical compression on RGC axons, or vascular compression of optic nerve-perfusing vessels [212]. Our previous research in mice has demonstrated that already moderately elevated IOP can impair vascular autoregulation and cause endothelial dysfunction in the retina, which is associated with upregulation of NOX2 [145]. Additionally, high hydrostatic pressure and ischemia have been found to stimulate the release of tumor necrosis factor α (TNF-α) from glial cells, activating apoptotic signaling pathways in RGCs [213]. TNF-α released by active astrocytes, microglia, and Müller glial cells, is one of the most significant pro-inflammatory cytokines in glial neuroinflammation, mitochondrial dysfunction, and oxidative stress [214]. Moreover, TNF-α can induce apoptosis through caspase-8 activation [214]. Mitochondrial dysfunction also plays a pivotal role in glaucomatous optic disorders. Increased IOP has been shown to damage mitochondria, leading to mitochondrial fission and alterations in the expression of the OPA1 gene [215,216]. Via ROS-induced activation of the transcription factor nuclear factor ’kappa-light-chain-enhancer’ of activated B-cells (NF-kB), mitochondria indirectly contribute to the initiation or amplification of glial neuroinflammation processes [215], resulting in the production of inflammatory cytokines. Furthermore, oxidized mitochondrial DNA and mitochondrial fragments released from microglia can activate inflammasomes [215]. Oxidative stress also disrupts the glutamate/glutamine metabolism, leading to the neurotoxic extracellular accumulation of glutamate [212]. Glutamate is released from dying RGCs and activated glia, and the dysfunctional glial cells are unable to adequately buffer excessive extracellular glutamate [212,217,218]. Additionally, oxidized substrates, such as advanced glycation end products (AGEs) and oxidized low density lipoproteins (oxLDLs) can act as “antigenic stimuli”, inducing ROS production, triggering NF-kB activation, glial activation, neuroinflammation and apoptosis [212,215]. In this context, the overproduction of ROS activates the apoptosis signal-regulating kinase 1 (ASK-1) [219,220], which, in turn, activates the p38/ c-Jun N-terminal kinase (JNK)/ extracellular-signal-regulated kinase (ERK) axis. This axis triggers the mitochondrial apoptotic pathway, characterized by the release of the Bcl-2 associated X-protein (Bax) and cytochrome c into the cytosol, followed by apoptosome formation (cytochrome c/apoptotic protease-activating factor 1(Apaf-1)/caspase-9), ultimately leading to caspase-3 activation [219,221,222]. Moreover, hydrogen peroxide has been described to activate the phosphoinositide 3-kinase (PI3K)/ Ak strain transforming (Akt) axis and reduce the intracellular concentration of the mammalian target of rapamycin (mTor), promoting NF-kB activation [223].
Apart from elevated IOP, other potential risk factors have been investigated in the context of NTG. The “Collaborative Normal-Tension Glaucoma Study” (1998) examined the effectiveness of IOP-lowering therapy in patients with NTG and concluded that although reducing IOP can have a positive impact, it cannot prevent disease progression [224,225]. Recent reviews on NTG have focused on the multifactorial nature of the disease, considering various risk factors implicated in its pathogenesis, including translaminar cribrosa pressure difference, fractal dimensions, neurovascular coupling, vascular dysregulation, endothelial dysfunction, ocular perfusion pressure, and oxidative stress [171,225]. Regarding vascular pathological aspects, Leung and Tham have described NTG as part of a larger group of disorders known as small vessel diseases [171]. They focused on the correlation between NTG and cerebral silent infarcts detected in patients with NTG [226,227,228]. CSI can lead to hypoperfusion of the optic nerve head, hypoperfusion in the optic nerve head, increasing the risk of NTG [171]. Additionally, arterial hypotonia, especially during night time when blood pressure is reduced, may result in lower ocular perfusion pressure and decreased blood supply to the ONH [225]. In this context, it is relevant to consider another potential primary injury to RGCs independent of elevated IOP: tissue hypoxia, which can be associated with pathogenic mechanisms in glaucoma [37]. During hypoxia, hypoxia-inducible factor 1-alpha (HIF-1α) induces the transcription of various genes, including those encoding vascular endothelial growth factor (VEGF), heme oxygenase-1 (HO-1), and inducible nitric oxide synthase (iNOS), aiming to increase oxygen supply to the affected tissue [229,230]. However, HIF-1α also upregulates NOX, which generates ROS, creating a vicious cycle of HIF-1α overexpression and ROS production [231,232]. The presence of increased HIF-1α expression regions in the retina and optic nerve of glaucoma patients has been confirmed by Tezel and Wax, further supporting the pathogenic role of hypoxia in glaucoma [233]. Hypoxic conditions primarily cause energy depletion and disrupt ionic homeostasis, leading to increased ROS generation and inflammation [37,234], mediated by overproduction of ROS via NOX2 [235], and other sources such as XO and cyclooxygenase (COX) [236]. Additionally, activated glia release TNF-α, which, among other effects, activates NF-kB, intensifying glial activation, neuroinflammation, and apoptosis through caspase-mediated or caspase-independent signaling pathways [214,237].
Reviewing the literature on the pathogenesis of glaucoma (Figure 5), it becomes evident that the pathways leading to RGC loss are redundant, intersect, and overlap, making it challenging to identify a single initial cause-and-effect factor [212]. Ultimately, the outcome appears to be a combination of “vicious cycles”, in which inflammation and ROS interconnect and amplify each other.
4.1.4. Interplay Between Mitochondrial and ER Stress in TMCs and RGCs
Mechanical and vascular stress in glaucoma contribute to mitochondrial dysfunction [217], which leads to calcium imbalance and interconnects with endoplasmic reticulum (ER) stress, resulting in energy impairment and subsequent ROS generation. Numerous studies have explored the relationship between the endoplasmic reticulum and mitochondria in the pathogenesis of glaucoma. Understanding the main processes of this connection can help trace intracellular pathways that contribute to TM dysfunction and RGC death.
The endoplasmic reticulum is an intracellular organelle with multiple functions, including lipid synthesis, calcium storage, and protein processing. It regulates protein folding to ensure proper functionality, facilitates protein transportation, and detects misfolded proteins, which are then retained in the ER for degradation [238,239]. Various conditions such as hypoxia, oxidative stress, viral infections, nutrient depletion, protein mutations, impaired glycosylation, or disrupted disulfide bond formation can interfere with the ER’s physiological functions. These disturbances can lead to ER saturation and the accumulation of misfolded proteins in the ER lumen, resulting in endoplasmic reticulum stress [240,241,242]. ER stress activates multiple signaling pathways aimed at restoring cellular homeostasis [238,241]. The unfolded protein response (UPR) is triggered in response to ER stress and encompasses a range of signaling mechanisms designed to reduce protein synthesis, enhance protein folding, and increase protein degradation [243]. Additionally, the ER-associated degradation (ERAD) system is responsible for retro-transporting misfolded proteins from the ER lumen to the cytosol for clearance through the ubiquitin-proteasome system [241,244]. UPR and ERAD are two independent quality-control mechanisms that can interact to eliminate misfolded proteins and maintain protein folding homeostasis [244]. UPR can lead to either cell survival or apoptosis [242]. Prolonged activation of UPR pathways can shift the balance toward cell death [241]. Three main UPR regulators have been extensively described: activating transcription factor (ATF)-6, inositol-requiring protein 1 (IRE1), and protein kinase RNA-like endoplasmic reticulum kinase (PERK). These transmembrane proteins with ER lumen domains serve as sensors of ER stress [242]. ATF-6 activates cascades to enhance ER folding capacity, promote clearance of misfolded proteins through ERAD, and may also have a proapoptotic effect [238,242,245]. IRE1 initiates a pathway involving spliced X-box binding protein-1 (sXBP1), which promotes ERAD components, ER folding proteins, and autophagy [238]. Furthermore, IRE1 activates JNK through tumor necrosis factor receptor 2 (TNRF2) and ASK1, leading to apoptosis [242], as well as inflammation via NF-kB induction [246]. PERK activates eukaryotic initiation factor 2α (eIF2α), which downregulates overall protein translation, indirectly protecting the cell from protein misfolding [242]. PERK also upregulates activating transcription factor 4 (ATF-4), which triggers CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP), exerting a proapoptotic function [268]. In this regard, Han et al. demonstrated that a sustained activation of ATF-4 and CHOP increases protein synthesis and leads to cell death through oxidative stress and ATP depletion [243]. These findings align with previous research showing that CHOP deletion in multiple mouse models of diabetes reduces oxidative stress [247]. A noteworthy aspect regarding the connection between ER stress and ROS generation is the ER overload response, which is activated when there is a high concentration of misfolded proteins in the ER lumen [241]. During the ER overload response, a significant amount of calcium ions (Ca²⁺) may be released from the ER, possibly through Ca²⁺ release channels such as inositol 1,4,5-trisphosphate receptor (IP3R) or ryanodine receptor (RyR) [238,241]. This process can result in increased Ca²⁺ uptake from the ER to mitochondria, leading to abnormal production of H₂O₂ and disruption of the ETC, ultimately causing mitochondrial dysfunction [238,248]. Moreover, UPR-related signaling can activate the endoplasmic reticulum-oxidoreductin 1 (ERO1) and NOX, which are involved in oxidative protein folding under normal physiological conditions. However, in the context of ER stress, their activation can contribute to ROS production in the stressed ER [238,249]. Lastly, ROS generation resulting from both ER and mitochondrial dysfunction can activate NF-kB, a key transcription factor involved in inflammation and cell proliferation [250]. The main transductions in the interconnection between ER and mitochondria are summarized in Figure 6.
ER-related oxidative stress has been reported in both TMCs and RGCs in the context of glaucomatous optic neuropathy. Studies on TMCs have demonstrated the involvement of the PERK-eIF2α-ATF4-CHOP cascade in glaucomatous TM, both in human and murine cells, highlighting the activation of this pathway in glaucoma [251,252,253,254]. Similarly, various studies have described the implication of the PERK-eIF2α-ATF4-CHOP pathway in RGC loss [255,256,257,258]. Recent literature has highlighted the potential of targeting the PERK-eIF2α-ATF4-CHOP pathway as a therapeutic approach to prevent CHOP-related oxidative stress and apoptosis, thereby mitigating TM structure and function loss and potentially reducing IOP elevation. Notably, in a recent study conducted by Gao et al., the protective effect of valdecoxib, a selective COX-2 inhibitor commonly used in the treatment of conditions such as osteoarthritis and rheumatoid arthritis, was assessed against apoptosis induced by ER stress. The study demonstrated that valdecoxib inhibits the ATF4-CHOP pathway in “I/R-induced glaucoma-like” damaged cells, providing potential insights into its therapeutic efficacy in glaucoma management [259].
4.2. Therapeutic Perspectives in Glaucoma
4.2.1. Therapeutic Potential of Natural Compounds in Glaucoma
Naturally occurring antioxidant compounds offer a diverse range of potential therapeutic options. One such compound is resveratrol, a polyphenol found in peanuts, berries, grapes, and red wine, known for its anti-inflammatory and antioxidant properties in conditions like cancer, neurodegeneration, and aging [260]. In human glaucomatous TMCs, resveratrol has been reported to reduce the expression of proinflammatory molecules such as IL-1α, and iNOS, while increasing the production of NO by elevating eNOS expression. These effects contribute to its beneficial antioxidant effects in the TM [261]. Resveratrol is also an activator of suirtin1 (SIRT1), a nuclear NAD⁺-dependent deacetylase, highly relevant for regulation of several antioxidant genes, performed by triggering the Nrf2/ARE (antioxidant response elements) pathway [262,263]. In an experimental rat model of glaucoma, resveratrol has been shown to delay the loss of RGCs [264]. Additionally, studies by Ye and Meng have demonstrated that resveratrol protects RGCs from H₂O₂-induced apoptosis by inhibiting proteins involved in MAPK cascades (p38, JNK, ERK) and activating antioxidant enzymes such as SOD, CAT, and GSH [265].
Another naturally occurring antioxidant molecule with promising characteristics is curcumin. In chronic high IOP rat models, curcumin has been shown to decrease ROS formation and prevent activation of apoptotic pathways by downregulating caspase-3, Bax, and cyt c [266]. Moreover, in an ex vivo mouse model of optic nerve cut, curcumin has been found to preserve RGC survival by preventing MAPK activation and inhibiting caspase-9 and caspase-3 activation [267].
Spermidine, a natural polyamine present in mushrooms and soybeans, possesses antioxidant properties and has demonstrated the ability to improve RGC loss and visual impairment in murine NTG models [268]. The same study also observed that spermidine treatment in murine optic nerve injury models promoted RGC survival by suppressing the ASK-1/p38/MAPK apoptotic pathway and inhibiting iNOS, particularly in microglial cells [269].
Flavonoids, known for their free radical scavenging properties, have been extensively studied. Extracts from the Gingko biloba L. plant, which contain over 70 different flavonoids, have shown interactions with apoptotic pathway proteins such as p53, Bax, Bcl-2, caspase-3, and caspase-9 [270]. These interactions suggest that Gingko biloba extracts may reduce RGC damage in glaucoma by inhibiting H₂O₂-related apoptosis through pathways involving p53, Bax/Bcl-2, and caspase-3/9 [271]. A dedicated clinical trial evaluated the use of oral antioxidants for glaucoma, comparing extracts of Gingko biloba with α-tocopherol for 3 months (NCT01544192), and completed phase III, reporting no clear clinical benefit for the use of Gingko biloba [272]. Another flavonoid, coenzyme Q₁₀, a natural lipophilic compound, has demonstrated in murine glaucoma models its ability to reduce glutamate excitotoxicity and oxidative stress, promoting RGC survival and preventing apoptotic cell death by decreasing Bax expression and increasing the Bcl-2 associated agonist of cell death (Bad) protein expression [273]. Currently, a clinical trial (NCT03611530) is underway to evaluate the effect of a medical solution containing coenzyme Q₁₀ and vitamin E on patients with primary open-angle glaucoma (POAG) [274].
The α-lipoic acid (ALA) is a naturally occurring antioxidant molecule found in various fruits, vegetables, as well as in the heart or liver of animals [275]. In murine glaucoma models (DBA/2J), Inman et al. demonstrated that ALA reduces oxidative stress and upregulates antioxidant agents such as HO-1 and NOS, possibly through the activation of Nrf-2 [275]. In a more recent prospective case-control study, an ALA-based solution (containing also taurine, vitamins C and E, lutein, zeaxanthin, zinc, copper and docosahexaenoic acid) was found to increase TAS and to decrease MDA, a marker of lipid peroxidation, in the plasma of patients with POAG [276].
Additionally, vitamin B3, also known as niacin, has been studied as a potential new treatment for glaucoma due to its antioxidant properties [277]. A study conducted in Korea revealed that patients with NTG have a lower intake of niacin compared to other nutrients, suggesting a possible association between vitamin B3 deficiency and NTG risk [278]. Williams et al. administrated nicotinamide (amide form of niacin) to DBA/2J mice and demonstrated both preventive and therapeutic effects against the development of glaucoma, preserving the age-dependent reduction of nicotinamide adenine dinucleotide (NAD), a crucial molecule for mitochondrial health [279]. In a recent randomized controlled trial involving 57 patients with glaucoma, nicotinamide supplementation was shown to improve inner retinal function [280].
Currently, there is an ongoing randomized controlled trial (NCT04784234) evaluating a mixture of curcumin, Ginkgo biloba extract, alpha-lipoic acid, coenzyme Q10, and other naturally occurring compounds in 100 patients with POAG. The expected completion date for this study is the end of 2023.
4.2.2. NOX-Inhibitors
A novel and promising class of antioxidant medications for glaucoma are NOX inhibitors. These compounds offer a new strategy to counteract glaucomatous damage by preserving RGCs from the detrimental effects of neuroinflammation and glial activation, potentially complementing traditional IOP lowering therapies [281]. Among these compounds, GKT137831, also known as setanaxib, is particularly noteworthy. It acts as a dual inhibitor of NOX1 and NOX4 and has demonstrated protective effects against retinal inflammation and ischemia by reducing hypoxia-induced ROS generation [282]. Another intriguing molecule in this class is GLX7013114, a specific NOX4 inhibitor. In a study involving rats with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-induced retinal excitotoxicity, intravitreal injections of GLX7013114 were found to attenuate glial activation [283]. Moreover, a recent publication reviewed the role of NOX and NOX inhibitors in glaucoma, highlighting the significant relationship between NOX4 and TGF-β in the fibrotic changes observed in glaucomatous TMCs, which contribute to functional impairment and elevated IOP [281,284]. These NOX inhibitors offer exciting prospects in antioxidant therapy for optic nerve diseases, as they target underlying mechanisms beyond IOP regulation, aiming to protect RGCs and to mitigate neuroinflammatory processes. Continued research in this field holds promise for the development of innovative treatments for glaucoma.
4.2.3. Exploring Nrf-2 Activation for Antioxidant Therapy in Glaucoma
Exciting new prospects in antioxidant therapy for glaucoma involve the use of Nrf-2 activators, which have the potential to provide benefits through their antioxidant and anti-inflammatory properties. One example is trimetazidine, an anti-ischemic medication that has been shown to activate the Nrf2-HO-1 pathway, leading to the inhibition of RGC- apoptosis [285]. Astaxanthin (AST), a potent natural antioxidant found in microalgae and seafood such as lobster, is another noteworthy compound in this context [286,287]. In a rat model of elevated IOP, AST has been demonstrated to reduce apoptotic pathways [288]. In mouse models of NTG, AST administration inhibited RGC degeneration and suppressed RGC loss [289]. Li et al. showed that AST activates Nrf2 and HO-1 in RGCs of mouse models, resulting in a decrease in RGC loss in glaucoma [290]. Sestrin2, a stress-induced protein with antioxidant properties, has also shown promise in activating Nrf2 by inhibiting kelch-like ECH-associated protein 1 (Keap-1), thus protecting against RGC apoptosis in mouse tissues [291]. Additionally, eye drops containing metformin, an antidiabetic drug, have been demonstrated to preserve against fibrosis following glaucoma filtration surgery in rodent models by activating the AMP-activated protein kinase (AMPK)/Nrf2 signaling pathway [292]. A recent study in mice demonstrated that sustained intraocular release of erythropoietin can counteract glaucomatous pathogenic processes by reducing superoxide levels in the retina, upregulating antioxidant agents, and activating the Nrf2/ARE pathway through MAPK signaling [293].
4.2.4. Rho Kinase Inhibitors in Glaucoma Treatment
The class of Rho-associated protein kinase (ROCK) inhibitors has shown promise in glaucoma treatment. One notable ROCK inhibitor is netarsudil, which has been demonstrated to reduce fibrosis in the TM, leading to improved aqueous humor outflow and lowered IOP [294]. It has received clinical approval for the use in the United States (2017) and Europe (2019) as a 0.02% ophthalmic solution for once-daily topical application [295]. Another ROCK inhibitor, Y-27632, has been shown to upregulate antioxidant enzymes such as CAT and to partially reduce ROS generation [296]. In addition to its beneficial effects on the TM, Y-27632 promotes phagocytosis in glaucomatous TM cells, leading to IOP reduction [297]. Ripasudil, also known as Rho kinase inhibitor K-115, has been evaluated in porcine retinal arterioles and shown to induce endothelium-independent relaxation and inhibit endothelin-1 activity. These findings highlight its potential as a potential antiglaucomatous medication [298]. The development of Rho kinase inhibitors offers a promising avenue for glaucoma treatment. By targeting the ROCK pathway, these inhibitors improve TM function, reduce fibrosis, and potentially lower IOP. Continued research and clinical investigations will further elucidate their efficacy and safety for glaucoma patients.
4.2.5. ER-Stress Antagonists: Potential Antioxidant Drugs
Another subgroup of potential antioxidant drug includes compounds that antagonize ER stress. Among these compounds, 4-phenylbutyric acid (4-PBA) has been the subject of several studies. Originally used in urea cycle disorders and later in the treatment of cystic fibrosis in the 1990s [299,300], phenylbutyrate has been reported to attenuate ROS production in activated microglia [301]. Additionally, 4-PBA has been found to reduce oxidative stress caused by a high-fat diet or acute ammonia challenge by counteracting ER stress [302]. Furthermore, 4-PBA has shown the ability to decrease ER stress and prevent disease phenotypes in murine glaucoma models [303]. Importantly, it has also been demonstrated to lower IOP by promoting ECM degradation through the activation of matrix metalloproteinase (MMP)-9 [304].
4.2.6. Other Antioxidant Molecules
In addition to the previously mentioned compounds, several other antioxidant molecules have shown potential therapeutic effects in glaucoma.
For example, edaravone, a medication primarily used against stroke and known for its free radical scavenging properties [305], was described to suppress pJNK/p38 proapoptotic pathways in glaucoma models [306], preserving RGCs from death [307,308].
Valproic acid (VPA), a widely used antiepileptic drug, has been shown to upregulate antioxidant enzymes such as SOD, CAT, and GPX while inhibiting apoptotic pathways in rodent models of retinal ischemia-reperfusion injuries [309]. In murine models of NTG, VPA has been found to decrease oxidative stress and enhance RGC survival through a pathway associated with ERK [310]. Additionally, a recent Swedish study using retina explant models suggested that VPA possesses anti-inflammatory properties by reducing the expression of pro-inflammatory cytokines and attenuating microglial changes, highlighting its potential as an anti-neuroinflammatory drug in retinal diseases [311]. A randomized controlled trial assessing the effectiveness of VPA in glaucoma patients reported an improvement of visual acuity among patients with advanced glaucoma [312].
N-acetylcysteine, a molecule with radical scavenger properties originally used as antidote in cases of paracetamol overdose and more recently as mucolytic agent in bronchopulmonary disorders [313], has shown promise in mitigating retinal oxidative stress caused by high IOP when combined with brimonidine therapy in rat models of OHT [314]. It has also been demonstrated to target the HIF-1α pathway via BNIP3 (Bcl2 interacting protein 3) and the PI3K/Akt/mTOR pathway, thereby preventing hypoxia-mimetic induced autophagy in RGCs [315]. In murine models of NTG, N-acetylcysteine suppressed oxidative stress and autophagy in RGCs and increased levels of glutathione [316].
Rapamycin, a macrolide antibiotic, with anti-neurodegenerative and neuroprotective properties described in Alzheimer’s and Parkinson’s diseases [317,318], has been found to enhance RGC survival in rodent glaucoma models. It achieves this by inhibiting the production of NO and TNF-α in microglia through modulation of NF-kB activity and by maintaining Akt phosphorylation to inhibit RGC apoptosis [319].
Geranylgeranylacetone (GGA), a molecule used in the therapy of gastric ulcers and known for its antioxidant features, has been shown to reduce oxidative stress in the retina by triggering thioredoxin and heat shock proteins (Hsp)-72, thereby protecting against apoptosis [320]. In murine models of NTG, GGA has been demonstrated to decrease RGC loss through upregulation of Hsp-70 and reduction of caspase-3 and -9 activities [321].
These findings highlight the potential of these antioxidant molecules in providing neuroprotection and preserving retinal health in glaucoma.
4.3. Leber’s hereditary optic neuropathy
4.3.1. General Aspects: Genetics, Clinical Presentation, and Current Therapeutic Options
Leber’s hereditary optic neuropathy (LHON) is a relatively rare disease compared to glaucoma, but it is considered the most frequent mitochondrial DNA (mtDNA) disorder [322]. It follows a maternal inheritance pattern and primarily affects young males, typically presenting between the ages of 15 and 35 [32]. The most prevalent mutations associated with LHON are m.3460G>A, m.11778G>A, and m.14484T>C, which account for approximately 90%-95% of all cases [13]. These mutations affect the protein subunits of complex I in the mitochondrial respiratory chain [323]. Among these mutations, m.11778G>A is most frequently observed in Northern Europe, Australia, and Japan [8,12,324,325], while m.14484T>C is more common among the French-Canadian population [326,327,328]. Studies examining the complete penetrance of LHON have reported varying prevalence estimates in Europe, ranging from 1 in 30,000 to 1 in 54,000, approximately 0.00002% of the population [8,9,10,11,12]. A comprehensive Australian review, which accounted for incomplete disease penetrance in variant carriers and used a well-characterized population-based control cohort to minimize sampling bias, estimated a prevalence of 1 in 800, or approximately 0.00125% [13]. Similarly, another study using a similar methodology concluded that LHON prevalence in the general population exceeds 1 in 1,000 [14]. These findings suggest that most LHON mutations remain silent until unknown triggers precipitate the conversion from asymptomatic mutation carriers to symptomatic individuals [329]. Indeed, only around 50% of males and 10% of females harboring a pathogenic mtDNA mutation effectively develop the disease [323]. LHON also exhibits a gender bias, with males being more likely to be affected than females [330]. This gender predisposition has been associated with X-linked susceptibility loci [331,332]. Other studies have explored potential factors that may explain the incomplete penetrance and have found that smoking and excessive alcohol consumption are more common in individuals with LHON compared to asymptomatic carriers [333]. Additionally, it has been suggested that vitamin B12 (cobalamin) deficiency could accelerate the symptomatology in LHON carriers [334], and that regular screening for vitamin B12 levels may be considered in LHON carriers and patients [322].
Clinically, LHON is highly disabling and leads to subacute bilateral irreversible vision loss [323]. A recent study examining the quality of life in 17 LHON patients from different countries (Germany, UK, France, and the US) described several daily life challenges in for example physical capabilities, interpersonal relationships, work and recreational activities, that significantly impact their well-being [335]. A cost-of-illness study focusing on inherited retinal disorders (IRD) estimated that in 2019, the overall costs associated with LHON amounted to $84–200 million (USD) and $10–42 million (CAD) in the US and Canada, respectively [336]. Moreover, healthcare costs, including therapy expenses, represented only 7% and 2% of the total costs in the US and Canada, respectively [336].
It is widely acknowledged that the treatment of LHON should ideally begin within one year from the onset of visual loss [329,337]. In relation to the disease progression, it is divided into three stages starting from the onset [338]:
- Subacute phase (<6 months): During this phase, patients commonly experience blurred vision and impaired color perception without pain, and their pupillary reflex remains unaffected [323]. Approximately 75% of cases initially experience visual loss in one eye, with the contralateral eye becoming affected within a few weeks [323,339]. Fundoscopy may reveal axonal loss in the papillomacular bundle and circumpapillary telangiectasias. Perimetry often demonstrates typical centrocaecal or central scotomas [323,339,340]. Optical coherence tomography (OCT) may show swelling of the peripapillary RNFL. Magnetic resonance imaging (MRI) is commonly performed for differential diagnosis [339].
- Dynamic phase (6–12 months): During this stage, fundoscopic signs such as telangiectasias and RNFL edema gradually regress [339].
- Chronic phase (>12 months): In the chronic phase, there is a further decline in visual acuity and visual field loss. Fundoscopic examinations may reveal optic nerve head atrophy, while OCT may indicate thinning of the RNFL [339].
An international consensus has established that idebenone (IDE), a synthetic analogue of coenzyme Q₁₀ (COQ₁₀), is the first disease-specific antioxidative drug authorized by the European Medicines Agency in 2015. It has been shown to provide benefits when administered during the subacute or dynamic stages of LHON at a dosage of 900 mg/day. However, it is not recommended for use during the chronic phase [337,338,341]. Extensive research has been conducted to discover medications that can restore the ETC, which is crucial for ATP synthesis and maintaining a normal redox status [329]. IDE functions as an electron carrier within the mitochondrial ETC, facilitating electron transfer from complex II to complex III and promoting ATP synthesis [342]. However, it is important to note that Jaber and Polster have described IDE as a pro-oxidative molecule that also inhibits complex I [342]. Additionally, Gueven et al. extensively reviewed the pro-oxidative properties of IDE and questioned the actual antioxidative activity of this drug [343]. They compared studies that detected low nanomolar concentrations of IDE in target tissues for only a short period of time with the majority of publications on IDE, which reported the need for micromolar concentrations to achieve an antioxidative effect [344,345,346]. One possible explanation for the apparent contradiction regarding the low bioavailability of IDE is the suggestion of an indirect antioxidant effect through the inhibition of NOX2, which normally produces damaging ROS such as superoxide [347]. IDE may also activate different signaling pathways that increase the activity of SOD, NAD(P)H quinone oxidoreductase 1 (NQO1), glutathione (GSH), and glutathione peroxidase (GPX), possibly through Nrf-2, a transcription factor that regulates the expression of SOD, NQO1, and GPX. Another proposed explanation for the rapid pharmacokinetics of IDE focuses on the possibility that some of its metabolites, such as 6-(9-carboxynonyl)-2,3-dimethoxy-5-methyl-1,4-benzoquinone (QS10), can also provide therapeutic effects [343]. In support of this hypothesis, a previous Italian study concluded that QS10, like IDE, can bypass the defect in complex I and, unlike IDE, can replace endogenous coenzyme Q₁₀ (CoQ₁₀), potentially exhibiting even greater activity than IDE in diseases caused by complex I defects or CoQ₁₀ deficiency [348]. In Figure 7, direct and indirect effects of idebenone are represented.
4.3.2. Oxidative Stress in the Pathogenesis of Leber’s hereditary optic neuropathy
Mitochondrial DNA mutations in LHON lead to a defective complex I, resulting in impaired ETC activity [349]. Studies on fibroblast and cybrid mitochondria affected by LHON mutations have shown varying levels of mitochondrial aerobic respiration alterations depending on the specific mutations [350]. Decreased respiration rates have been reported as approximately 20–28% for m.3460G>A, 30–36% for m.11778G>A, and 10-15% for m.14484T>C [350]. Despite the impaired oxidative phosphorylation and reduced ATP production associated with complex I dysfunction, it is proposed that LHON partially compensates for these deficiencies through glycolysis, an alternative energetic pathway observed in human tissues. This compensation is suggested to maintain the total cellular ATP concentration despite the severe decrease in complex I-related ATP synthesis [349,351]. Hence, additional processes need to be considered to explain the clinical manifestation of the disease. Carelli et al. proposed that decreased pH resulting from defective complex I affects redox sites during aerobic respiration, leading to overproduction of ROS [351]. Studies on cells carrying LHON mutations have reported increased levels of oxidative stress biomarkers such as 8-OHdG, reduced activity of antioxidant systems including glutathione reductase and Mn-SOD, and elevated levels of oxidized glutathione (GSSG) [352,353,354]. Lin et al. demonstrated in a murine model of mtDNA mutations that LHON mutations cause systemic elevation of ROS production, with chronic elevation of ROS generation observed in synaptosomes, optic nerve, and RGCs, suggesting a more significant damaging role of oxidative stress compared to ATP depletion [355]. Another study in mice aimed at establishing a murine model of LHON found that a mutation in the subunit ND4 of complex I led to mitochondrial structure disassembly, increased ROS levels, ONH swelling, and RGC apoptosis [356]. Additionally, an alternative hypothesis suggests that altered permeability transition pores, possibly due to Ca²⁺ and ROS-mediated depolarization of the mitochondrial membrane, may play a role in LHON. These opened permeability transition pores in cybrid cells carrying LHON mutations could facilitate the release of cytochrome c from mitochondria to the cytosol, activating the apoptotic pathway [357].
Dysfunctional complex I in LHON may lead to sensitization of permeability transition pores, primarily due to increased ROS generation and cytosolic Ca²⁺ overload. The reduced ATP supply to the Ca²⁺ pumps in the ER membranes may contribute to the intracellular Ca²⁺ overload by impairing Ca²⁺ uptake from the cytosol to the ER [357]. Consequently, the death of RGCs in LHON is likely influenced by altered redox status and intracellular Ca²⁺ overload, with an emphasis on the role of ROS in triggering proapoptotic pathways (Figure 8) [355,358,359,360,361,362]. Apoptosis in LHON has been described as mediated by caspases, including the Fas-induced pathway involving caspase-8 [363,364]. Activation of caspase-8 leads to the cleavage of protein Bid (BH3 interacting-domain death agonist), a component of the Bcl-2 family, indirectly causing the release of cytochrome c from mitochondria. Cytochrome c then binds to Apaf-1, along with caspase-9, forming a complex called the “apoptosome.” The apoptosome subsequently activates caspase-3, which is responsible for cellular disassembly and apoptosis. Additionally, caspase-independent pathways have been observed in LHON, involving the release of cytochrome c, AIF (apoptosis-inducing factor), and EndoG (endonuclease G) into the cytosol [365]. These apoptotic processes contribute to the demise of RGCs in LHON.
4.4. Therapeutic Perspectives in Leber’s hereditary optic neuropathy
α-Tocotrienol quinone, also known as EPI-743, is a synthetic molecule classified as a para-benzoquinone. It targets NQO1 and leads to the replenishment of intracellular glutathione stores, thereby increasing antioxidant capabilities [366]. In vitro studies have shown that EPI-743 is over one thousand-fold more effective than IDE in protecting cells from oxidative stress [367]. An investigation into the effect of EPI-743 in LHON demonstrated disease progression arrest and reversal of vision loss in 4 out of 5 patients treated with EPI-743 within 4 months of the onset of visual loss [368]. Another study involving 12 patients with active-phase LHON concluded that EPI-743 stabilized or improved visual function in approximately 70% of the treated eyes [369]. However, larger trials in the LHON population are needed to further validate these findings. Currently, EPI-743 is in phase II of various clinical trials (NCT01370447; NCT04378075; NCT02352896) aimed at evaluating its effectiveness in primary mitochondrial disorders [370].
Elamipretide, also known as MTP-131, Bendavia, or SS-31, is a relatively new mitochondria-targeting peptide that has been shown to protect RGCs from oxidative stress-induced apoptosis [371]. Other interesting molecules that modulate mitochondrial redox status and enhance mitochondrial biogenesis include sonlicromanol (also known as KH176) and KL1333 [370]. KL1333 is an NAD⁺ modulator that improves the NAD+/NADH ratio and has been demonstrated to decrease lactate and ROS levels while increasing ATP synthesis in fibroblasts from patients with MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) [372].These emerging therapeutic options provide potential avenues for the treatment of LHON by targeting oxidative stress and mitochondrial dysfunction.
4.5. Anterior Ischemic Optic Neuropathy
4.5.1. General Aspects: Prevalence, Clinical Presentation, and Current Therapies
Among optic nerve disorders, anterior ischemic optic neuropathies stand out as one of the most complex and significant conditions. These conditions are classified into anterior and posterior optic neuropathies (AION and PION), distinguished by the presence or absence of disc edema, respectively [373]. They are further categorized into arteritic and nonarteritic diseases based on the presence or absence of vasculitis, which results in reduced perfusion to the optic nerve head [374]. Nonarteritic anterior optic neuropathy (NA-AION) is the most common acute optic nerve disease in patients older than 50 years [375]. Clinical presentations include sudden acute unilateral vision loss without pain. However, effective treatments for these conditions remain challenging [16,375,376]. Giant cell arteritis (GCA), is the most common subtype of arteritic anterior ischemic optic neuropathy (A-AION). High intravenous corticosteroid doses serve as the first-line therapy for GCA [377]. It is crucial to initiate rapid treatment once GCA is suspected [378]. A systematic review predicts that by 2050, over 3 million individuals in Europe, North America, and Oceania will suffer from giant cell arteritis, primarily due to aging [379]. Additionally, the estimated economic burden resulting from visual disability caused by GCA in the United States is expected to exceed $76 billion by 2050 [379]. Clinically, GCA manifests in individuals over 50 years old, presenting classic symptoms such as severe headache, jaw claudication, cutaneous allodynia, and, in the majority of patients, additional symptoms like fever, weight loss, depression, and night sweats [378]. Despite the administration of high steroid doses, including intravenous methylprednisolone 1000 mg/day for 3 days, followed by a maintenance dose of 1 mg/kg of prednisone [380], the chances of visual recovery in GCA-related visual loss remain very low [381]. Furthermore, long-term use of steroids is associated with various side effects and rebound syndrome, while GCA is prone to relapse [382]. Consequently, there is a significant research need for new therapeutic strategies, and immunomodulating therapies have emerged as promising avenues in the treatment of GCA. One example of an immunomodulating therapy is tolicizumab (TCZ), an interleukin-6 (IL-6) receptor antibody. TCZ has already received approval from the National Institute for Health and Care Excellence and NHS England for the treatment of refractory GCA [383]. However, the use of TCZ is restricted due to known side effects, including alterations in liver enzymes and cholesterol levels, gastrointestinal perforation, infections, headache, and arterial hypertension [384,385]. In the field of immunomodulation, several new immunoglobulins are currently undergoing clinical trials for the treatment of GCA [386].
Non-arteritic anterior ischemic optic neuropathies (NA-AION) occur as a result of events that lead to hypoperfusion of the optic nerve head. A recent comprehensive meta-analysis revealed several risk factors for NA-AION, including male gender, hypertension, hyperlipidemia, diabetes mellitus, coronary heart disease, sleep apnea, factor V Leiden heterozygosity, and a history of cardiovascular medication use [387]. Pathogenetically, Hayreh SS. described that NA-AION involves hypoperfusion of the optic nerve head, leading to hypoxia of retinal ganglion cell axons. This, in turn, results in stasis of axoplasmic flow and the generation of swollen axons [374]. Consequently, optic disc edema occurs, causing compression of capillaries supplying the optic nerve head, creating a vicious cycle. The primary trigger for the initial hypoperfusion of the optic nerve head is typically a transient drop in blood pressure, often occurring during sleep, such as nocturnal arterial hypotension or hypotension after sleeping during the day. Severe occlusion of the internal carotid or ophthalmic artery rarely causes ocular ischemia in NA-AION cases [374]. While embolic lesions can be an occasional cause of NA-AION, they result in more extensive and permanent damage to the optic nerve head compared to the hypotensive type [374]. The clinical onset of NA-AION is sudden and typically affects a single eye, resulting in vision loss without pain, often discovered by patients upon waking in the morning [388]. Fundoscopy and perimetry are essential diagnostic tools for NA-AION. The Goldmann perimeter, in particular, helps detect classic visual field defects in NA-AION. A study on 312 consecutive eyes reported central scotomas in nearly half of the cases, with the absolute inferior nasal defect being the most commonly detected [389].
Therapeutic options for NA-AION remain a challenging clinical issue. The effectiveness of steroids, whether administered orally or intravenously, is a subject of intensive debate [390]. A large nonrandomized cohort study in the United States involving 696 eyes, spanning from 1973 to 2000, suggested that systemic steroid treatment with oral prednisone (80 mg per day) during the acute phase may increase the probability of visual acuity and visual field improvement after six months [391]. However, due to the nonrandomized nature of this study and potential biases, the findings should be interpreted with caution [392]. Katz and Trobe extensively reviewed possible treatment strategies for NA-AION in both conservative and surgical fields and concluded that optic nerve fenestration surgery is ineffective and potentially harmful, while the efficacy of steroids remains uncertain [393]. In an Indian randomized controlled trial involving 38 patients with NA-AION, Saxena et al. found that oral prednisolone at 80 mg per day reduced the duration of disc edema and improved electrophysiological parameters of the optic nerve but did not result in a visual acuity benefit after six months [394]. One potential clinical approach is the use of α2-adrenergic agonists such as brimonidine in the acute phase of AION through topical application. A study investigating the neuroprotective effect of brimonidine on anterior optic neuropathy in rodents (rAION) demonstrated a decrease in RGC loss in mice treated with brimonidine in the acute phase, possibly through a reduction in VEGF-A and HIF-1α expression [395]. These findings align with previous studies that also demonstrated the effectiveness of brimonidine in the acute phase of rAION [396,397]. In summary, the significant lack of effective treatments for NA-AION and its relatively high prevalence underscore the urgent need for research in novel therapeutic approaches. Exploring the antioxidative branch of treatment holds promising potential to make significant contributions in this regard. By focusing on antioxidant strategies, new therapeutic avenues can be explored to address the challenges posed by NA-AION.
4.5.2. Oxidative Stress in the Pathogenesis of Anterior Ischemic Optic Neuropathy
GCA is an autoimmune disorder characterized by granulomatous infiltration involving T-cells and activated macrophages, including multinucleated giant cells, primarily observed in the vascular wall of the Aorta and its main branches [398,399,400,401]. Vascular aging is considered one of the critical risk factors for GCA, as oxidative stress is believed to play a significant role in its pathogenesis [399]. This is likely attributed to age-associated mitochondrial dysfunction leading to increased ROS generation in endothelial cells and vascular smooth muscle cells [400,402,403]. A study on GCA patients has demonstrated the presence of neutrophils in the vascular lumen and adventitia tissues surrounding the external lamina elastica of temporal arteries, producing high levels of extracellular ROS [404]. Additionally, systemic oxidative stress parameters such as TAC and MDA levels, along with intracellular leukocyte ROS levels, were found to be elevated in GCA patients [405]. findings support the hypothesis that reactive species contribute to the pathophysiology of GCA by inducing vascular stress in the large and medium-sized vessels.
In the pathogenesis of NA-AION, damage may arise as a consequence of hypoxia in the optic nerve head. Cellular hypoxia resulting from glucose and oxygen depletion, followed by reoxygenation during the ischemia/reperfusion (I/R) process, can lead to an overproduction of reactive species [406,407,408,409]. It has been proposed that systemic corticosteroids are effective in the acute phase of NA-AION, as they suppress NF-kB activation during inflammation [391]. NF-kB is a transcription factor that can be triggered by ROS-related cascades, as described in the pathogenesis of glaucoma [212,215,410]. ROS have been shown to cause immune-mediated neuronal injuries, disruption of the blood-optic nerve barrier, apoptosis, and autophagy through damages to DNA, proteins, and lipids [411,412]. Studies have revealed that I/R injuries in ischemic neurons lead to somatic autophagy of axonal mitochondria, resulting in increased retrograde movement and decreased anterograde movement of mitochondria, ultimately reducing functional mitochondria available for ATP synthesis [412]. Our laboratory’s study on short-term ischemia in porcine models demonstrated hypoxia-related alterations characterized by upregulation of HIF-1α, VEGF-A, NOX2, iNOS, and high levels of ROS [148]. Supporting these findings, a study on NOX2-deficient murine models (NOX2-/-) showed that these mice displayed neuroprotection in retinal I/R injury scenarios [235].
Interestingly, a Turkish study investigating plasma samples from 18 newly diagnosed NA-AION patients found no significant differences in systemic oxidative stress parameters, including TOS and TAS, when compared to healthy controls [411]. However, genetic investigations have revealed an association between NA-AION and a loss-of-function deletion in the gene GSTM1, which encodes one of the three isoforms of the antioxidative enzyme glutathione-S-transferase (GST) [413,414]. Additionally, another study reported a higher frequency of mitochondrial DNA-mutations in NA-AION patients compared to controls, suggesting that mitochondrial dysfunction may be a risk factor for NA-AION [415].
Based on the current literature and the pivotal role of hypoxia in NA-AION, some studies have provided evidence that conditions leading to increased ROS generation may contribute to the risk of developing NA-AION. These findings highlight the potential involvement of ROS in the pathogenesis of this disease.
4.6. Therapeutic Perspectives in Anterior Ischemic Optic Neuropathy
4.6.1. Giant Cell Arteritis
Nuclear sirtuins, such as the before mentioned SIRT1, are proteins that potentially have an antioxidative impact in GCA. These enzymes inhibit inflammation and oxidative stress by transcriptionally repressing various inflammation-related genes [416,417]. A recent study observed SIRT1 downregulation and subsequent ROS generation in individuals with GCA, suggesting a potential therapeutic effect of SIRT1 activators [405]. While immunomodulating molecules have been extensively investigated due to the autoimmune nature of GCA, there is growing scientific interest in exploring new antioxidant molecules such as nuclear suirtins.
4.6.2. Nonarteritic Anterior Ischemic Optic Neuropathy
An investigation using rAION mice, intravitreal injection of the rho-kinase inhibitor, E212, immediately after optic nerve infarction resulted in increased SOD activity, decreased ROS levels, reduced oxidative stress, and preservation of the blood-retinal barrier [418]. Our laboratory conducted a study on mice with I/R retinal injuries and found that betulinic acid, a natural compound found in plane bark, leaves, and fruit peel, preserved vascular function, attenuated ROS formation, and upregulated SOD and HO-1 [136]. Oroxylin A, a bioactive flavonoid extracted from Scutellariae baicalensis Georgi, showed potential protective effects in rAION by activating Nrf2, increasing HO-1 and NQO1 activity, promoting RGC survival, maintaining RNFL thickness, and exhibiting anti-inflammatory effects by reducing IL-6 and increasing TGF-β levels [419]. N-butylidenephthalide, another molecule, demonstrated a neuroprotective role in rAION by inhibiting the NF-κB signaling pathway [443]. Vitamin B3 (niacin) was found to provide neuroprotection from oxidative stress in rAION by activating Nrf2, increasing SOD expression, inhibiting mitochondrial apoptosis, and reducing the inflammatory response through NF-κB inhibition [376].
Geranylgeranylacetone (GGA), previously mentioned as a promising molecule for glaucoma therapies, was also tested in murine models of I/R retinal injury and demonstrated a decrease in RGC death by inhibiting the p38 MAPK apoptotic pathway [420]. In a more recent study, GGA was shown to induce an increase in Hsp70 levels, inhibiting glial activation, autophagy, and apoptosis [421]. Astaxanthin, previously identified as a potential antiglaucomatous drug, possesses antioxidant and anti-apoptotic properties. Lin et al. found that Astaxanthin preserved visual function, increased RGC survival, and inhibited apoptosis by blocking the Akt pathway in rAION [422]. Resveratrol, previously investigated for its potential in preventing RGC loss in glaucoma, has also been studied in I/R injuries. It was found to attenuate glial activation and RGC death by suppressing HIF-1α-VEGF-A upregulation, activating the downregulation of the PI3K/Akt pathway [423]. Resveratrol also protects RGCs from ischemia-related injuries by increasing Opa1 expression [424]. Our recent research demonstrated that resveratrol prevents vascular dysfunction and RGC death in I/R-induced murine models, possibly through the inhibition of I/R-related upregulation of NOX2 [94].These findings highlight potential therapeutic approaches for AION by targeting oxidative stress, inflammation, and mitochondrial dysfunction.
4.7. Optic Neuritis
4.7.1. General aspects
Optic neuritis (ON) is an idiopathic inflammatory demyelinating disease of the optic nerve [425,426], which can be associated with the neuromyelitis optica [427], or also can appear as initial manifestation of multiple sclerosis, a chronic demyelinating disorder of the central nervous system [428,429]. In addition, reported risk factors for ON are granulomatous diseases, infections, autoimmune pathologies [430]. In the pathogenesis of ON are involved activated T-cells, which releasing abnormal volume of inflammatory cytokines, elicit demyelination and disruption of the blood-optic nerve barrier, and guide to loss of retinal ganglion cells and ONH-atrophy [44,427,431,432,433]. ON is described as the major cause of acute optic nerve disorder in young patients [427], being commonly observed between 20 and 45 years, and predominantly in women with a ratio of 3:1 [17,428,434]. Its clinical presentation can occur through single or multiple episodes and usually consists of a sudden and unilateral loss of visual acuity, afferent pupillary impairment, orbital pain by eye movements and altered color perception, in presence or absence of disc edema [435]. Diagnostically, fundoscopy and OCT help to detect ONH-anomalies and RNFL-thinning, respectively. A study estimated that OCT can reveal thinning of the RNFL, with an average reduction of 33% compared to controls and an average reduction of 27% between impaired and unimpaired eyes of the same patient [436]. Furthermore, it has been reported that an RNFL thinning is visible in 85% of all patients suffering from ON within 3-6 months after the acute phase [437]. In addition, perimetry can evidence visual field loss limited to the nerve fiber bundle region, with paracentral, partial arcuate or arcuate defects [438]. Cranial MRI scan is performed for differential diagnosis and to evaluate the association with multiple sclerosis [428]. Current standard therapies of ON include high intravenous steroid doses (1000 mg IV methylprednisolone) during acute phases, which were reported to facilitate visual recovery, whereas oral prednisone alone is not recommended, since it failed to show comparable improvements [439]. However, a systematic review examining six clinical trials, globally including 750 patients affected by ON, did not evidence beneficial effects neither through intravenous nor via oral steroid therapy, comparing treated with placebo groups, in visual field, visual acuity and contrast sensitivity outcomes [440]. Alternatively, the use of intravenous immunoglobulins in acute ON, which may be considered in steroid-refractory cases [441], failed anyway to provide benefits for a generalized practice [442,443].
4.7.2. Oxidative Stress in the Pathogenesis of Optic Neuritis
Various investigations succeeded to provide evidence on an altered redox status in the context of ON, using experimental autoimmune encephalomyelitis (EAE) models [444,445,446,447], an animal model originally established for multiple sclerosis, and often employed in experiments aimed to study optic neuritis [435]. Moreover, studies on patients affected by ON showed significant ROS-related anomalies, such as elevated fraction of oxidized thiol [448], or reduced bilirubin serum levels [449], indicators of high ROS-generation and low antioxidant status, respectively. As mentioned before, similarly to NA-AION and LHON, also in ON loss-of-function deletions in the gene coding for the antioxidative enzyme glutathione-S-transferase were detected [414], therefore emphasizing a decreased antioxidant activity as a possible risk factor. As we previously highlighted in glaucoma, elevated ROS generation and active inflammatory grade are deeply interconnected and tend to trigger and intensify each other in a process where NF-kB and cytokines are fundamental pathomechanistic players [450]. In relation to ON, increased ROS are responsible for the myelin phagocytosis, stimulate changings in the permeability of the blood-brain barrier, favoring migration and infiltration of active T-cells, which in turn reflects in abnormal inflammation and augmented ROS-production [448]. Oxidative damage may possibly be an initiator for the demyelination and neurodegeneration [448], which finally drives to RGC-loss [425].
More large studies designed to interpret the multiple aspects in the pathogenesis of ON, including the role of oxidative stress, may deliver new knowledge for the design of more effective pharmacological drugs to treat this disease.
4.8. Therapeutic Perspectives in Optic Neuritis
While a prompt administration of anti-inflammatory drugs during the acute phase of optic neuritis (ON) may help preserve vision [451], the use of steroids alone has not been shown to protect the optic nerve from inflammatory demyelination or prevent degeneration of retinal ganglion cells (RGCs) [425,452]. Therefore, antioxidant approaches have been explored to support nerve fiber integrity by counteracting inflammation and demyelination, thus preventing RGC death [425,453]. In preclinical studies on ON models, several major antioxidant targets have been investigated, including the activation of Nrf2 and the suppression of ROS-related proapoptotic signaling, such as blocking ASK-1 and the pJNK/p38 pathway
Dimethyl fumarate, a Nrf2 activator that has been licensed for the treatment of relapsing-remitting multiple sclerosis in the US since 2013, has shown promising results in experimental autoimmune encephalomyelitis (EAE) models, reducing the severity and relapses of optic neuritis and preserving RGCs from cell death [453]. Gypenosides, an extract from Gynostemma pentaphyllum, have demonstrated beneficial antioxidant effects on the retina[454]. In a study on murine models of optic neuritis, gypenosides led to a decrease in iNOS and COX2 expression while activating Nrf2, resulting in the upregulation of HO and GPX, along with free radical scavenging and anti-inflammatory activities [455]. α-Lipoic acid, which potentially activates Nrf2 [275], has been investigated in EAE models, showing increased RGC survival and anti-inflammatory effects[456]. However, a dedicated trial on 31 patients with ON (NCT01294176), reported good tolerability after oral supplementation (600 mg twice a day) of α-lipoic acid [457], but did not show significant benefits in retinal nerve fiber layer thickness after 24 weeks compared to the placebo group [458]. Consequently, the trial ended in phase I due to insufficient recruitment [459]. As previously mentioned, SIRT1, known to activate Nrf2, has also been studied [425]. In an EAE study, an adeno-associated virus vector was utilized as a delivery vehicle to enhance SIRT1 expression, demonstrating that selective upregulation of SIRT1 promotes RGC survival and preserves axons from demyelination through intravitreal injection [425]. Another investigation explored the use of Matrine, an extract from the herb Radix Sophorae Flavescentis, in EAE models. Matrine induced overexpression of SIRT1, subsequent upregulation of Nrf2, and collectively resulted in mitochondrial biosynthesis, reduced ROS formation, and suppression of inflammation and demyelination [460].
Inhibition of ASK-1 also showed encouraging preclinical results in mitigating oxidative stress-induced RGC apoptosis. The ASK-1 inhibitor MSC2032964A has been demonstrated to alleviate neuroinflammation and diminish optic nerve demyelination in EAE murine models [461]. Spermidine, previously mentioned as a potential antioxidant for glaucoma, also suppresses the ASK-1/p38/pJNK pathway[269] and has been reported to reduce RGC apoptosis in EAE mice [462].
A recent publication investigated the effects of edaravone, previously mentioned, in cultures and mice with neuromyelitis optica spectrum disorder, and found that it promoted remyelination through activation of the mTOR complex I (mTORC1) signaling pathway[463]. Currently, there is an ongoing trial (NCT05540262) that aims to assess the impact of edaravone in patients with optic neuritis who are positive for aquaporin-4 antibodies. The trial is expected to be completed by the end of 2024.
5. Other Rare Optic Neuropathies
5.1. Traumatic optic neuropathies
5.1.1. General Characteristics
Craniofacial traumas can directly or indirectly insult the optic nerve, leading to so called traumatic optic neuropathies (TON) [464,465]. Direct traumas are caused by injuries to the optic nerve through intracranial fragmentation of bones, or also through contusion causing anatomical disruption [466]. Indirect traumas occur when compression and disruption of pial vessels cause a reduction of the vascular perfusion in the optic nerve [467,468]. Moreover, a deformation of the skull at the optic canal, and more specifically at its intracranial end, was reported also to be responsible of optic nerve injuries in indirect traumas [469]. The most reported causes of TON are vehicle accidents, bicycle accidents, falls, assaults, and sport injuries [24]. Indirect TON is more common than direct forms and occurs in 0.5-5% of all closed head injuries and 2.5% of all midfacial fractures [22,470,471]. In terms of diagnosis, a brain MRI scan is typically conducted[465]. Therapeutically, reported options for the treatment of traumatic optic neuropathy (TON) include high-dose corticosteroids or surgical decompressions[465]. However, both of these treatment approaches are accompanied by controversies due to the pharmacological side effects of steroids or the potential complications associated with surgery[465,472]. Consequently, exploring neuroprotective strategies to prevent optic nerve damage represents a promising curative option with substantial potential for TON [465].
5.1.2. Pathogenesis of Traumatic Optic Neuropathies: Role of oxidative stress and Ca²⁺
In murine models, traumatic optic neuropathy (TON) has been shown to be associated with RGC loss, axonal degeneration, and visual impairment[473]. The pathomechanisms potentially involved in RGC death include the disruption of the blood-brain barrier, leading to the infiltration of activated immune cells such as macrophages and neutrophils. These immune cells produce a significant amount of reactive oxygen species (ROS) and cytokines, further activating microglia and causing damage to mitochondria and endoplasmic reticulum[474,475,476]. Numerous studies on murine and rodent models of TON have demonstrated an overproduction of ROS [477,478,479], as well as a decrease in the antioxidant activity of SOD [478], and CAT [480,481]. Additionally, mitochondrial anomalies[482] and disturbances in Ca²⁺ fluxes, which can occur concomitantly with ROS formation, have been described [481]. Notably, the progression of damage in TON has been associated with intracellular Ca²⁺ influx, observed after stretching axonal myelinated fibers of the optic nerve, resulting in the dissolution of the axonal cytoskeleton[483]. An insightful review discussed the interplay between endoplasmic reticulum (ER) stress and oxidative stress in the pathophysiology of TON, highlighting the involvement of these two pathogenic factors in optic nerve trauma-induced RGC loss[484]. In this context, a study by Hu et al. on optic nerve crush models revealed an upregulation of the ER stress mediator protein CHOP as a consequence of optic nerve injury, further demonstrating that its deletion can prevent RGC death [485]. Consistent with these findings, other investigations on models of traumatic brain injury have reported the overexpression of CHOP and induction of ER stress, contributing to neurodegenerative processes following neurotrauma [486,487,488]. In summary, in line with existing literature, after an optic nerve insult, disruption of the blood-optic nerve barrier may allow the infiltration of activated immune cells, resulting in the generation of high concentrations of ROS and proinflammatory mediators. These events ultimately lead to axonal degeneration, myelin damage, and culminate in mitochondrial and ER stress, further exacerbating inflammation and triggering apoptotic RGC death.
5.1.3. Potential Antioxidants for Traumatic Optic Neuropathies
Numerous naturally occurring compounds have been tested for their antioxidant properties in models of traumatic optic neuropathy (TON). For example, the ethanol extract of Echium amoenum L. demonstrated antioxidant and anti-inflammatory effects in optic nerve crush models. It was shown to inhibit glutamate-induced ROS formation, reduce NF-kB activity, and decrease microglial activation and optic nerve damage[489]. Similarly, the ethanol extract of Lithospermum erythrorhizon in optic nerve crush models was found to decrease ROS generation and lower the levels of proapoptotic proteins such as caspase-3 [490]. In experimental neurotrauma murine models, a high-Vitamin E diet exhibited neuroprotective properties by preventing RGC death, reducing ROS levels, and suppressing inflammasome activation [480]. Neuroglobin, a mammalian heme protein with antioxidant features, was reported to enhance RGC survival and promote optic axon regeneration in TON murine models [491,492,493].
Moreover, synthetic compounds have also been investigated in TON models. Carvedilol, a nonselective β-adrenoreceptor blocker, was found to inhibit iNOS expression, ASK-1, and the p38/MAPK pathways, subsequently reducing RGC apoptosis [494]. Polydopamine is a polymer synthetically obtained through oxidation of the neurotransmitter dopamine [495], and also a major pigment of the naturally occurring eumelanin [496]. Polydopamine-based nanoreactors were demonstrated to scavenge ROS in RGCs after optic nerve injury in mice, as well as to mitigate microglial activation and to suppress ROS-related RGC-apoptosis [497]. Galantamine is an FDA-licensed compound used in the treatment of Alzheimer’s disease, which primarily acts as acetylcholinesterase inhibitor, but has also an antioxidant [498,499], and an anti-inflammatory feature [500]. This molecule was reported to preserve from visual function deficiency, attenuating oxidative stress, inflammation and inhibiting axonal degeneration in TON models [501]. Finally, the before reported resveratrol, which can activate SIRT1 and subsequently Nrf2 [262,263], appears to possess curative effects also in TON models. In fact, this compound showed protective effects on RGC in optic nerve crush models, attenuating oxidative stress [502]. In line with these findings, a recent investigation revealed that a regulation of the SIRT1/mTORC1 axis in the microglia has the potential of mitigating the optic nerve crush-induced RGC-death [503].
Taken together, many publications gathered evidence on promising preclinical results in animal models of optic nerve trauma. However, an important factor to be considered for practical uses of antioxidants after optic nerve trauma in humans is the prompt administration of the drug after diagnosis [474]. A delay in this context may equivale to unsuccessful effect of the medication, due to early activation of intracellular downstream transductions [474]. Thus, a combination of antioxidants and other medications with possibly different targets, may enhance the probability of effectiveness in contrasting damages after optic nerve injuries [474].
5.2. Compressive and Strecth Optic Neuropathies
5.2.1. General features
Graves orbitopathy (GO) can cause dysthyroid optic neuropathy (DON), which is due to optic nerve compression (found in over 90% of cases) through extension of extraocular muscles, orbital fat expansion and interstitial edema [504], or alternatively to optic nerve stretching, without compression (described in less than 10% of cases) [505]. DON is recognized as the most common form of compressive optic neuropathy, and its main reported risk factors comprise advanced age, male gender, smoking and diabetes mellitus [505]. This disorder clinically manifests with central vision defects, altered color perception, relative afferent pupillary defect, and mostly and typically through protrusio bulbi, also known as exophthalmos, due to widening of extraocular muscles, which lead to restricted ocular motility [505]. Diagnostic features include: (1) fundoscopy, which can evidence compression-induced disc edema; (2) perimetry, which help detect central scotomas; (3) OCT, which can show RNFL-thinning. Further, cranial MRI- or CT-scan may help to differentiate compressive from stretch conditions [506]. Gold standard therapy in DON caused by compression is a high intravenous dose of methylprednisolone (0.5-1 g for 3 days), eventually followed by surgical decompression in case of insufficient corticosteroid response [507]. In case of DON caused by stretching, the surgical option becomes the first-line treatment [505]. Other reported therapeutic options are biological drugs such as teprotumumab and tocilizumab, and also orbital radiotherapy [507]. Although first-line therapies are largely assumed to be effective to control symptomatic and to prevent vision loss in DON, deterioration of optic nerve function, relapses of DON and corticosteroid side effects or contraindications, induce to search for new curative options [507].
5.2.2. Oxidative Stress in The Pathogenesis of Dysthyroid Optic Neuropathy
DON is a consequence of the GO, an autoimmune inflammatory disorder, in which through stimulation of autoantibodies and interactions with activated T-cells, orbital fibroblasts are triggered and induce diverse intracellular transductions such as the MAPK-pathway with downstream NF-kB activation, as well as the PI3K/Akt axis, collectively inducing abnormal generation of pro-inflammatory cytokines such as IL-1α, IL-1β, IL-6, IL-8, macrophage chemoattractant protein-1 (MCP-1), transforming growth factor (TGF)-β [504,508]. Moreover, orbital fibroblasts differentiate into adipocytes and myofibroblasts, further producing glycosaminoglycan, a component of connective tissues, responsible, through its deposition, of the muscle enlargement, together with hyaluronic acid [509]. Investigations demonstrated also that indicators of oxidative damages are increased in blood, urine, as well as in orbital fibroblasts of patients with GO. In blood of patients affected by GO were demonstrated elevated levels of H₂O₂ as well as of lipid hydroperoxides (ROOH), and further enhanced activities of SOD and CAT, while GPX activity was reduced [510]. Biomarkers of oxidative stress such as 8-OHdG and MDA, as well as intracellular ROS were elevated in orbital fibroblasts of patients with GO compared to healthy controls [511]. High concentrations of MDA and 8-OHdG were also found in tears of patients with GO, particularly in active phases of the disease [512]. Another study reported that urinary 8-OHdG positive correlates with the disease activity and that smoking as risk factor has a higher influence on elevation of 8-OHdG [513]. In this regard, an in vitro investigation on orbital fibroblasts of patients affected by GO, exposed to cigarette smoke extracts, showed that fibroblasts react to cigarette smoke components through aberrant inducement of oxidative stress, as well as through elevation of TGF-β and IL-1 [514]. These studies globally evidenced that oxidative stress is present in GO and possibly indicate that ROS have a role in the pathophysiology of this disorder. In this context, hypotheses on the impact of ROS in the pathogenesis of GO were formulated and proven in some studies. For example, O2•− was suggested to trigger a retroocular fibroblast proliferation in patients with GO [515]. In addition, H₂O₂ was showed to stimulate the production of TGF-β and IL-1 in GO orbital fibroblasts [516].
Altogether, from the existing literature we can extrapolate that ROS, IL-1 and TGF-β play critical roles in the stimulation and progression of the inflammation, in the fibroblastic differentiation and in the deposition of connective tissue components in GO, which collectively lead to remodeling events that cause compression of the optic nerve in DON [517].
5.2.3. Antioxidant Candidates in Dysthyroid Optic Neuropathy
Some molecules with antioxidant characteristics showed encouraging results in dedicated investigations for mild stages of GO [504]. A relevant example is represented by selenium, a natural compound with antioxidant and anti-inflammatory properties [518], present among others in nuts, shrimps, eggs, meat, cereals [519]. This molecule was assessed in orbital fibroblasts of patients with GO, to inhibit fibroblastic proliferation, release of pro-inflammatory cytokines such as TNF-α, and also of the ROS-induced production of hyaluronic acid [520]. In line with these findings, another in vitro investigation on orbital fibroblasts affected by GO confirmed a suppressing action of selenium against ROS-formation, hyaluronan production and release of inflammatory mediators [521]. A dedicated trial on 159 patients with mild GO tested potential curative effects of selenium supplementations, and proved an improvement of symptomatic as well as quality of life, together with a slowed down disease progression, after selenium-based therapy, ultimately recommending a six-month treatment through selenium supplementation in patients with mild GO of short duration [522,523]. However, administration of selenium should be considered in dependence of the selenium-intake from the diet, which in turn varies in accordance to different geographical locations [519]. Hence, a detection of selenium concentrations in serum should occur preliminary to prevent high selenium-intake, which also may produce side effects [517,524].
Quercitin, an antioxidant flavonoid found for example in fruits and vegetables and used in the traditional Chinese medicine [525], was reported to decrease ROS-formation as well as to inhibit adipogenesis in primary cultured orbital fibroblasts obtained from GO patients exposed to cigarette-smoke extracts [526]. In addition to the demonstrated antioxidative effect, quercitin was also described as significant anti-inflammatory molecule in orbital fibroblasts affected by GO, suppressing the IL-1 related inflammation as well as hyaluronan formation and adipogenetic processes [527].
Other natural compounds which also showed in preclinical studies an effective antioxidant activity in orbital fibroblasts from patients with GO are ascorbic acid in combination with N-acetyl-L-cysteine and melatonin [528] and β-carotene [529]. Furthermore, pentoxifylline were tested in preclinical [530] and in clinical investigations [531,532], showing in both promising effects contrasting the glycosaminoglycan production from orbital fibroblasts. However, the same trial which confirmed curative effects of selenium in 159 patients with GO, tested also pentoxifylline, here not concluding an equivalent effectiveness for this class of molecules in GO [522].
The before mentioned resveratrol was also investigated in fibroblasts with GO, showing to decrease ROS levels and suppressing adipogenesis in vitro [533].
Additionally, also synthetic existing compounds were tested as potential antioxidants in GO. In this regard, allopurinol in combination with nicotinamide showed promising results in vitro, reducing ROS-formation in fibroblasts [534], as well as in clinical trial, positive impacting on the severity of the disorder [535]. Enalapril, a widespread anti-hypertensive drug, possesses also antioxidant features [536,537], and in vitro displayed decreased cell proliferation and reduced hyaluronic acid levels in both orbital fibroblasts affected by GO and control fibroblasts [538]. A dedicated trial confirmed the encouraging preclinical results in 12 patients with mild GO treated with enalapril for 6 months, concluding a beneficial impact on clinical course and disease progression [539].
Overviewing existing publications on this theme appears reasonable, that antioxidants may be considered as a promising additional therapy in combination with corticosteroids, to improve quality of life of patients, slow down the disease progression, as well as to reduce the severity of the inflammation in GO [517].
5.3. Infiltrative Optic Neuropathies
Infiltrative processes of the optic nerve likewise generate RGC degeneration. An example is the leukemic optic neuropathy, mainly associated with acute lymphoblastic leukemia [540,541,542]. Involvement of the optic nerve in leukemic cases remains anyway a rare circumstance, which may mostly appear in relapses and late in the disease course [543,544]. Optic nerve head affected by infiltration may dangerously mimic a disc edema, a pathological sign detected by a large variety of different optic neuropathies [545]. Fundoscopic, ONH can show irregular and nodular conformations [545]. Visual impairment is typically due to neoplastic infiltration of the ONH, which causes nerve fibers and vascular compressions [546]. Diagnostically, an MRI-scan of the brain is essential for early detections, to possibly rapid begin an appropriate therapy [547]. Pathogenetically, in relation to a possible direct involvement of oxidative stress in the pathophysiology of these pathologies, the current literature presents no dedicated studies on the argument.
5.4. Congenital Anomalies of the Optic Nerve
Optic nerve congenital anomalies also constitute a subgroup of optic neuropathies and include for example optic nerve hypoplasia and optic disc colobomas [548,549], this latter with a reported prevalence in children of ~8.9 in 100,000 [550]. Visual impairment in this large group of disorders may appear isolated or as part of a systemic malformation syndrome [551]. Congenital visual defects or visual loss are commonly associated with this devastating class of disorders [551]. No studies were performed to analyze pathogenetic roles of ROS in these diseases.
5.5. Nutritional and Toxic Optic Neuropathies
Nutritional deficits, such as in case of vitamin B12 (cyanocobalamin), B1 (thiamine) or B9 (folic acid) deficiency, as well as intoxications, caused for example by ethambutol, amiodarone, or also antibiotics such as chloramphenicol [552,553,554], may cause optic nerve disorders. Prevalence of these pathologies is variable and typically depends on social, economic, geographical and historical factors [31,32]. Some examples are the optic neuropathy in prisoners of the Japanese during World War II [555], the Cuban epidemic optic neuropathy [556] and the Tanzanian epidemic optic neuropathy [557], all of them caused by metabolic deficiencies. Interestingly, nutritional optic neuropathies are currently becoming increasingly common in consequence of bariatric surgery as well as strict vegetarian and vegan diets, thereby awaking scientific interests [32]. Nutritional optic neuropathies are usually caused by deficiency of molecules, which are crucial for the normal functionality of the mitochondria such as diverse subtypes of vitamin B [558]. Both intoxications or vitamin deficiencies usually manifest with optic disc pallor and anomalies in the papillomacular nerve fiber bundle, and symptomatically through reduced color perception and detectable visual field defects, corresponding to central or cecocentral scotomas, as observed in cases of LHON [558,559]. For example, toxic optic neuropathies caused by a chronic intake of chloramphenicol, can mimic an acute stage of LHON [554]. Chloramphenicol can suppress a mitochondrial protein synthesis, inducing alteration in the mitochondrial structure and subsequently dysfunction, with decreasing ATP and increasing ROS, which collectively reflect in a LHON-like symptomatic [560].
Possible treatments in these disorders obviously depend on the etiology. Indeed, nutritional optic neuropathies can be treated through vitamin B supplementations [561,562]. Toxic optic neuropathies should be treated rapidly after diagnosis through stopping of the drug therapy in case of pharmacological etiology, stopping smoking or alcohol consumption [563]. In this context, dedicated studies also demonstrated the effectiveness of erythropoietin in improving visual acuities in patients affected by methanol-related toxic optic neuropathies [564,565].
6. Conclusions and Future Directions
Optic nerve disorders pose significant challenges due to their prevalence, severity, and economic implications for patients and the healthcare system. The development and proposal of new effective curative strategies are crucial in addressing these conditions. The field of antioxidative research has garnered increasing scientific interest, with numerous studies demonstrating promising preclinical results. Oxidative stress has emerged as a potential therapeutic target in animal models of glaucoma, LHON, AION and ON. Clinical translations of these findings have already begun, as evidenced by the licensing of idebenone for LHON. However, considering the relative novelty of these potential antioxidants and the unknown aspects of their tolerability in humans, careful planning of clinical studies is necessary. The high prevalence of glaucoma worldwide suggests that cost-effective use of new antioxidant medications is feasible. Nonetheless, large-scale trials are imperative to ensure the applicability and long-term safety of these treatments for a large population of patients. The severity and rarity of conditions such as LHON may prompt exploration of new pharmacological avenues, including the antioxidant approach, as demonstrated by the development of idebenone [338]. Similarly, the lack of effective drugs for AION highlights the need to explore new directions, including the “antioxidant opportunity”.
Despite several positive findings in preclinical investigations, many potential antioxidant drugs have failed to progress beyond phase II in corresponding clinical trials [566,567,568]. Currently, only a limited number of clinical studies are planned in the antioxidant field. Recent studies have examined the reasons for these failures, which include low bioavailability of antioxidants, limited tissue targeting, poor antioxidant capacity, and high drug dosing that may be toxic for human use [569]. Additionally, some clinical investigations may have been limited by slow disease progression, such as in the case of glaucoma, resulting in long follow-up periods and a lack of sensitive biomarkers [566]. Significant efforts are currently being made to develop efficient drug delivery systems to enhance the translational success of potential antioxidants [453,454]. Alternative clinical trial designs, such as adaptive clinical trials, offer flexibility by allowing modifications based on predefined criteria [570,571].
In conclusion, the findings of our review have compiled significant preclinical evidence supporting the potential of targeting oxidative stress as a therapeutic approach in animal models of optic nerve diseases. These results highlight the importance of further advancing clinical trials in this research area, as they hold immense potential to bridge the current gap between preclinical and translational applications. By implementing and refining clinical trials, we can move closer to harnessing the benefits of oxidative stress modulation for the treatment of optic nerve diseases, ultimately improving patient outcomes and advancing therapeutic strategies in this field.
Author Contributions
Conceptualization, F.B. and A.G.; writing—original draft preparation, F.B.; writing—review and editing, A.G., E.W.B. and N.P.; visualization, F.B.; supervision, A.G.; All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Riordan-Eva, P. Clinical assessment of optic nerve disorders. Eye (Lond) 2004, 18, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Van Stavern, G.P.; Newman, N.J. Optic neuropathies. An overview. Ophthalmol Clin North Am 2001, 14, 61–71. [Google Scholar] [PubMed]
- Sanz-Morello, B.; Ahmadi, H.; Vohra, R.; Saruhanian, S.; Freude, K.K.; Hamann, S.; Kolko, M. Oxidative Stress in Optic Neuropathies. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Stingl, J.V.; Wagner, F.M.; Liebezeit, S.; Baumgartner, R.; Spät, H.; Schuster, A.K.; Prokosch, V.; Grehn, F.; Hoffmann, E.M. Long-Term Efficacy and Safety of Modified Canaloplasty Versus Trabeculectomy in Open-Angle Glaucoma. Life (Basel) 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Wang, J.; Li, Y.; Jiang, B. Prevalence of primary open angle glaucoma in the last 20 years: a meta-analysis and systematic review. Sci Rep 2021, 11, 13762. [Google Scholar] [CrossRef] [PubMed]
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990-2020: a systematic review and meta-analysis. Lancet Glob Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [PubMed]
- Mascialino, B.; Leinonen, M.; Meier, T. Meta-analysis of the prevalence of Leber hereditary optic neuropathy mtDNA mutations in Europe. European Journal of Ophthalmology 2012, 22, 461–465. [Google Scholar] [CrossRef]
- Puomila, A.; Hämäläinen, P.; Kivioja, S.; Savontaus, M.L.; Koivumäki, S.; Huoponen, K.; Nikoskelainen, E. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet 2007, 15, 1079–1089. [Google Scholar] [CrossRef]
- Rosenberg, T.; Nørby, S.; Schwartz, M.; Saillard, J.; Magalhães, P.J.; Leroy, D.; Kann, E.C.; Duno, M. Prevalence and Genetics of Leber Hereditary Optic Neuropathy in the Danish Population. Investigative Ophthalmology & Visual Science 2016, 57, 1370–1375. [Google Scholar] [CrossRef]
- Spruijt, L.; Kolbach, D.N.; de Coo, R.F.; Plomp, A.S.; Bauer, N.J.; Smeets, H.J.; de Die-Smulders, C.E. Influence of mutation type on clinical expression of Leber hereditary optic neuropathy. Am J Ophthalmol 2006, 141, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Brown, D.T.; Howell, N.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of Leber hereditary optic neuropathy in the North East of England. Am J Hum Genet 2003, 72, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.C.; Davis, R.L.; Ravishankar, S.; Copty, J.; Kummerfeld, S.; Sue, C.M. Low disease risk and penetrance in Leber hereditary optic neuropathy. Am J Hum Genet 2023, 110, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Mackey, D.A.; Ong, J.S.; MacGregor, S.; Whiteman, D.C.; Craig, J.E.; Lopez Sanchez, M.I.G.; Kearns, L.S.; Staffieri, S.E.; Clarke, L.; McGuinness, M.B.; et al. Is the disease risk and penetrance in Leber hereditary optic neuropathy actually low? Am J Hum Genet 2023, 110, 170–176. [Google Scholar] [CrossRef]
- Li, K.J.; Semenov, D.; Turk, M.; Pope, J. A meta-analysis of the epidemiology of giant cell arteritis across time and space. Arthritis Research & Therapy 2021, 23, 82. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, K.A.; Oh, S.Y. Prevalence and incidence of non-arteritic anterior ischaemic optic neuropathy in South Korea: a nationwide population-based study. Br J Ophthalmol 2018, 102, 936–941. [Google Scholar] [CrossRef]
- Toosy, A.T.; Mason, D.F.; Miller, D.H. Optic neuritis. Lancet Neurol 2014, 13, 83–99. [Google Scholar] [CrossRef]
- Braithwaite, T.; Subramanian, A.; Petzold, A.; Galloway, J.; Adderley, N.J.; Mollan, S.P.; Plant, G.T.; Nirantharakumar, K.; Denniston, A.K. Trends in Optic Neuritis Incidence and Prevalence in the UK and Association With Systemic and Neurologic Disease. JAMA Neurol 2020, 77, 1514–1523. [Google Scholar] [CrossRef]
- Rodriguez, M.; Siva, A.; Cross, S.A.; O'Brien, P.C.; Kurland, L.T. Optic neuritis: a population-based study in Olmsted County, Minnesota. Neurology 1995, 45, 244–250. [Google Scholar] [CrossRef]
- Percy, A.K.; Nobrega, F.T.; Kurland, L.T. Optic neuritis and multiple sclerosis. An epidemiologic study. Arch Ophthalmol 1972, 87, 135–139. [Google Scholar] [CrossRef]
- Cockerham, G.C.; Goodrich, G.L.; Weichel, E.D.; Orcutt, J.C.; Rizzo, J.F.; Bower, K.S.; Schuchard, R.A. Eye and visual function in traumatic brain injury. J Rehabil Res Dev 2009, 46, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Karimi, S.; Arabi, A.; Ansari, I.; Shahraki, T.; Safi, S. A Systematic Literature Review on Traumatic Optic Neuropathy. J Ophthalmol 2021, 2021, 5553885. [Google Scholar] [CrossRef] [PubMed]
- Pirouzmand, F. Epidemiological trends of traumatic optic nerve injuries in the largest Canadian adult trauma center. J Craniofac Surg 2012, 23, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Miller, N.R. Traumatic Optic Neuropathy. J Neurol Surg B Skull Base 2021, 82, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Blandford, A.D.; Zhang, D.; Chundury, R.V.; Perry, J.D. Dysthyroid optic neuropathy: update on pathogenesis, diagnosis, and management. Expert Rev Ophthalmol 2017, 12, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Neigel, J.M.; Rootman, J.; Belkin, R.I.; Nugent, R.A.; Drance, S.M.; Beattie, C.W.; Spinelli, J.A. Dysthyroid optic neuropathy. The crowded orbital apex syndrome. Ophthalmology 1988, 95, 1515–1521. [Google Scholar] [CrossRef]
- Bartalena, L.; Piantanida, E.; Gallo, D.; Lai, A.; Tanda, M.L. Epidemiology, Natural History, Risk Factors, and Prevention of Graves’ Orbitopathy. Frontiers in Endocrinology 2020, 11. [Google Scholar] [CrossRef]
- Kincaid, M.C.; Green, W.R. Ocular and orbital involvement in leukemia. Surv Ophthalmol 1983, 27, 211–232. [Google Scholar] [CrossRef]
- Patel, L.; McNally, R.J.; Harrison, E.; Lloyd, I.C.; Clayton, P.E. Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J Pediatr 2006, 148, 85–88. [Google Scholar] [CrossRef]
- Tear Fahnehjelm, K.; Dahl, S.; Martin, L.; Ek, U. Optic nerve hypoplasia in children and adolescents; prevalence, ocular characteristics and behavioural problems. Acta Ophthalmol 2014, 92, 563–570. [Google Scholar] [CrossRef]
- Jefferis, J.M.; Hickman, S.J. Treatment and Outcomes in Nutritional Optic Neuropathy. Curr Treat Options Neurol 2019, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Roda, M.; di Geronimo, N.; Pellegrini, M.; Schiavi, C. Nutritional Optic Neuropathies: State of the Art and Emerging Evidences. Nutrients 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Levin, L.A. Neuroprotection in Optic Neuropathy. Asia Pac J Ophthalmol (Phila) 2018, 7, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Casson, R.J.; Chidlow, G.; Wood, J.P.; Crowston, J.G.; Goldberg, I. Definition of glaucoma: clinical and experimental concepts. Clin Exp Ophthalmol 2012, 40, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.K.; Su, E.-N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Progress in Retinal and Eye Research 2013, 36, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Jiang, S.; Musayeva, A.; Gericke, A. Oxidative Stress and Vascular Dysfunction in the Retina: Therapeutic Strategies. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res 2006, 25, 490–513. [Google Scholar] [CrossRef]
- Mozaffarieh, M.; Flammer, J. New insights in the pathogenesis and treatment of normal tension glaucoma. Curr Opin Pharmacol 2013, 13, 43–49. [Google Scholar] [CrossRef]
- Aslan, M.; Dogan, S.; Kucuksayan, E. Oxidative stress and potential applications of free radical scavengers in glaucoma. Redox Rep 2013, 18, 76–87. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol 2013, 13, 12–15. [Google Scholar] [CrossRef]
- Saccà, S.C.; Izzotti, A. Focus on molecular events in the anterior chamber leading to glaucoma. Cell Mol Life Sci 2014, 71, 2197–2218. [Google Scholar] [CrossRef] [PubMed]
- Langbøl, M.; Saruhanian, S.; Baskaran, T.; Tiedemann, D.; Mouhammad, Z.A.; Toft-Kehler, A.K.; Jun, B.; Vohra, R.; Bazan, N.G.; Kolko, M. Increased Antioxidant Capacity and Pro-Homeostatic Lipid Mediators in Ocular Hypertension—A Human Experimental Model. Journal of Clinical Medicine 2020, 9, 2979. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxidative Medicine and Cellular Longevity 2017, 2017, 9208489. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.Y.; Liu, P.K.; Wen, Y.T.; Quinn, P.M.J.; Levi, S.R.; Wang, N.K.; Tsai, R.K. Role of Oxidative Stress in Ocular Diseases Associated with Retinal Ganglion Cells Degeneration. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.P.; Sladen, P.E.; Yu-Wai-Man, P.; Cheetham, M.E. Induced Pluripotent Stem Cells for Inherited Optic Neuropathies-Disease Modeling and Therapeutic Development. J Neuroophthalmol 2022, 42, 35–44. [Google Scholar] [CrossRef]
- Freddi, T.d.A.L.; Ottaiano, A.C. The Optic Nerve: Anatomy and Pathology. Seminars in Ultrasound, CT and MRI 2022, 43, 378–388. [Google Scholar] [CrossRef]
- Yu, D.Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.K.; Su, E.N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog Retin Eye Res 2013, 36, 217–246. [Google Scholar] [CrossRef]
- De Moraes, C.G. Anatomy of the visual pathways. J Glaucoma 2013, 22 Suppl 5, S2–7. [Google Scholar] [CrossRef]
- Anderson, D.R. Vascular supply to the optic nerve of primates. Am J Ophthalmol 1970, 70, 341–351. [Google Scholar] [CrossRef]
- Linsenmeier, R.A.; Zhang, H.F. Retinal oxygen: from animals to humans. Prog Retin Eye Res 2017, 58, 115–151. [Google Scholar] [CrossRef]
- Arden, G.B.; Wolf, J.E.; Tsang, Y. Does dark adaptation exacerbate diabetic retinopathy?: Evidence and a linking hypothesis. Vision Research 1998, 38, 1723–1729. [Google Scholar] [CrossRef]
- Selhorst, J.B.; Chen, Y. The optic nerve. Semin Neurol 2009, 29, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Galetta, S.L. Chapter 1 - Anatomy and physiology of the afferent visual system. In Handbook of Clinical Neurology, Kennard, C., Leigh, R.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 102, pp. 3–19. [Google Scholar]
- Almazroa, A.; Burman, R.; Raahemifar, K.; Lakshminarayanan, V. Optic Disc and Optic Cup Segmentation Methodologies for Glaucoma Image Detection: A Survey. Journal of Ophthalmology 2015, 2015, 180972. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Mukherjee, R.; Dutta, K.; Sen, A. Deep learning based framework for automatic diagnosis of glaucoma based on analysis of focal notching in the optic nerve head. arXiv arXiv:2112.05748 2021.
- Wilczek, M. THE LAMINA CRIBROSA AND ITS NATURE. Br J Ophthalmol 1947, 31, 551–565. [Google Scholar] [CrossRef]
- Emery, J.M.; Landis, D.; Paton, D.; Boniuk, M.; Craig, J.M. The lamina cribrosa in normal and glaucomatous human eyes. Trans Am Acad Ophthalmol Otolaryngol 1974, 78, Op290–Op297. [Google Scholar]
- Quigley, H.A.; Hohman, R.M.; Addicks, E.M.; Massof, R.W.; Green, W.R. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am J Ophthalmol 1983, 95, 673–691. [Google Scholar] [CrossRef]
- Hayreh, S.S. Orbital vascular anatomy. Eye (Lond) 2006, 20, 1130–1144. [Google Scholar] [CrossRef]
- McCaa, C.S. The eye and visual nervous system: anatomy, physiology and toxicology. Environ Health Perspect 1982, 44, 1–8. [Google Scholar] [CrossRef]
- Juan, J.S.; Ana, I.R.; Rosa De, H.; Elena, S.-G.; Pilar, R.; José, A.F.-A.; Inés, L.-C.; Blanca, R.; Alberto, T.; José, M.R. Anatomy of the Human Optic Nerve: Structure and Function. In Optic Nerve; Felicia, M.F., Ed.; IntechOpen: Rijeka, 2018; Ch. 2. [Google Scholar] [CrossRef]
- Hattar, S.; Liao, H.W.; Takao, M.; Berson, D.M.; Yau, K.W. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science 2002, 295, 1065–1070. [Google Scholar] [CrossRef]
- Wang, L.; Dong, J.; Cull, G.; Fortune, B.; Cioffi, G.A. Varicosities of intraretinal ganglion cell axons in human and nonhuman primates. Investigative ophthalmology & visual science 2003, 44, 2–9. [Google Scholar]
- Bristow, E.A.; Griffiths, P.G.; Andrews, R.M.; Johnson, M.A.; Turnbull, D.M. The Distribution of Mitochondrial Activity in Relation to Optic Nerve Structure. Archives of Ophthalmology 2002, 120, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Serasinghe, M.N.; Chipuk, J.E. Mitochondrial Fission in Human Diseases. Handb Exp Pharmacol 2017, 240, 159–188. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 2000, 29, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biology 2015, 13, 89. [Google Scholar] [CrossRef]
- Walsh, C.; Barrow, S.; Voronina, S.; Chvanov, M.; Petersen, O.H.; Tepikin, A. Modulation of calcium signalling by mitochondria. Biochim Biophys Acta 2009, 1787, 1374–1382. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol 2012, 4, a006783. [Google Scholar] [CrossRef]
- Elkholi, R.; Renault, T.T.; Serasinghe, M.N.; Chipuk, J.E. Putting the pieces together: How is the mitochondrial pathway of apoptosis regulated in cancer and chemotherapy? Cancer Metab 2014, 2, 16. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 2010, 35, 505–513. [Google Scholar] [CrossRef]
- Shu, D.Y.; Chaudhary, S.; Cho, K.-S.; Lennikov, A.; Miller, W.P.; Thorn, D.C.; Yang, M.; McKay, T.B. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites 2023, 13, 187. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J Physiol 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int J Mol Med 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid Med Cell Longev 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. The International Journal of Biochemistry & Cell Biology 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Taurone, S.; Ralli, M.; Artico, M.; Madia, V.N.; Scarpa, S.; Nottola, S.A.; Maconi, A.; Betti, M.; Familiari, P.; Nebbioso, M.; et al. Oxidative stress and visual system: a review. EXCLI J 2022, 21, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: regulation and function. Eur Heart J 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Kannenkeril, D.; Bosch, A.; Kolwelter, J.; Jung, S.; Striepe, K.; Ott, C.; Delles, C.; Schmieder, R.E. Dependency of flow-mediated vasodilatation from basal nitric oxide activity. Clin Physiol Funct Imaging 2021, 41, 310–316. [Google Scholar] [CrossRef]
- Tibballs, J. The role of nitric oxide (formerly endothelium-derived relaxing factor-EDRF) in vasodilatation and vasodilator therapy. Anaesth Intensive Care 1993, 21, 759–773. [Google Scholar] [CrossRef]
- Simonsen, U.; Rodriguez-Rodriguez, R.; Dalsgaard, T.; Buus, N.H.; Stankevicius, E. Novel approaches to improving endothelium-dependent nitric oxide-mediated vasodilatation. Pharmacol Rep 2009, 61, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Sanders, K.M.; Ward, S.M. Nitric oxide and its role as a non-adrenergic, non-cholinergic inhibitory neurotransmitter in the gastrointestinal tract. Br J Pharmacol 2019, 176, 212–227. [Google Scholar] [CrossRef]
- Rand, M.J.; Li, C.G. Nitric oxide as a neurotransmitter in peripheral nerves: nature of transmitter and mechanism of transmission. Annu Rev Physiol 1995, 57, 659–682. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.R. Nitric oxide: a radical neurotransmitter in the central nervous system. Prog Neurobiol 1994, 42, 129–160. [Google Scholar] [CrossRef] [PubMed]
- Falak, N.; Imran, Q.M.; Hussain, A.; Yun, B.W. Transcription Factors as the "Blitzkrieg" of Plant Defense: A Pragmatic View of Nitric Oxide's Role in Gene Regulation. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Harari, O.; Liao, J.K. Inhibition of MHC II gene transcription by nitric oxide and antioxidants. Curr Pharm Des 2004, 10, 893–898. [Google Scholar] [CrossRef]
- Campbell, S.C.; Richardson, H.; Ferris, W.F.; Butler, C.S.; Macfarlane, W.M. Nitric oxide stimulates insulin gene transcription in pancreatic beta-cells. Biochem Biophys Res Commun 2007, 353, 1011–1016. [Google Scholar] [CrossRef]
- Gunnett, C.A.; Lund, D.D.; Chu, Y.; Brooks, R.M., 2nd; Faraci, F.M.; Heistad, D.D. NO-dependent vasorelaxation is impaired after gene transfer of inducible NO-synthase. Arterioscler Thromb Vasc Biol 2001, 21, 1281–1287. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Huie, R.E.; Padmaja, S. The reaction of no with superoxide. Free Radic Res Commun 1993, 18, 195–199. [Google Scholar] [CrossRef]
- Villamena, F.A. Chapter 2 - Chemistry of Reactive Species. In Reactive Species Detection in Biology; Villamena, F.A., Ed.; Elsevier: Boston, 2017; pp. 13–64. [Google Scholar] [CrossRef]
- Chronopoulos, P.; Manicam, C.; Zadeh, J.K.; Laspas, P.; Unkrig, J.C.; Göbel, M.L.; Musayeva, A.; Pfeiffer, N.; Oelze, M.; Daiber, A.; et al. Effects of Resveratrol on Vascular Function in Retinal Ischemia-Reperfusion Injury. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Gao, Y.; Song, M.; Cao, W.; Sun, X. Peroxynitrite is a novel risk factor and treatment target of glaucoma. Nitric Oxide 2020, 99, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Cantó, A.; Olivar, T.; Romero, F.J.; Miranda, M. Nitrosative Stress in Retinal Pathologies: Review. Antioxidants (Basel) 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Roos, D. Chronic Granulomatous Disease. Methods Mol Biol 2019, 1982, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Agita, A.; Alsagaff, M.T. Inflammation, Immunity, and Hypertension. Acta Med Indones 2017, 49, 158–165. [Google Scholar]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev 2014, 94, 329–354. [Google Scholar] [CrossRef]
- Hsueh, Y.J.; Chen, Y.N.; Tsao, Y.T.; Cheng, C.M.; Wu, W.C.; Chen, H.C. The Pathomechanism, Antioxidant Biomarkers, and Treatment of Oxidative Stress-Related Eye Diseases. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat Rev Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr Atheroscler Rep 2017, 19, 42. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free Radic Biol Med 2018, 125, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Chemistry of ROS-mediated oxidation to the guanine base in DNA and its biological consequences. Int J Radiat Biol 2022, 98, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.M.; Burrows, C.J. Interplay of Guanine Oxidation and G-Quadruplex Folding in Gene Promoters. J Am Chem Soc 2020, 142, 1115–1136. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Zhao, X.M.; Jiang, Z.S.; Wang, G.X.; Zhang, D.W. Protein tyrosine nitration in atherosclerotic endothelial dysfunction. Clin Chim Acta 2022, 529, 34–41. [Google Scholar] [CrossRef]
- Xiao, H.; Jedrychowski, M.P.; Schweppe, D.K.; Huttlin, E.L.; Yu, Q.; Heppner, D.E.; Li, J.; Long, J.; Mills, E.L.; Szpyt, J.; et al. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 2020, 180, 968–983. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys 1991, 288, 481–487. [Google Scholar] [CrossRef]
- Hiona, A.; Leeuwenburgh, C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp Gerontol 2008, 43, 24–33. [Google Scholar] [CrossRef]
- Sanz, A.; Caro, P.; Gómez, J.; Barja, G. Testing the vicious cycle theory of mitochondrial ROS production: effects of H2O2 and cumene hydroperoxide treatment on heart mitochondria. J Bioenerg Biomembr 2006, 38, 121–127. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef]
- Yuan, L.Q.; Wang, C.; Lu, D.F.; Zhao, X.D.; Tan, L.H.; Chen, X. Induction of apoptosis and ferroptosis by a tumor suppressing magnetic field through ROS-mediated DNA damage. Aging (Albany NY) 2020, 12, 3662–3681. [Google Scholar] [CrossRef]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell Mol Neurobiol 2015, 35, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Thompson, J.A. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Arch Biochem Biophys 1999, 366, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Kapoor, R.; Felts, P.A. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol 1999, 9, 69–92. [Google Scholar] [CrossRef]
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid Med Cell Longev 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Di Gioia, M.; Zanoni, I. Dooming Phagocyte Responses: Inflammatory Effects of Endogenous Oxidized Phospholipids. Front Endocrinol (Lausanne) 2021, 12, 626842. [Google Scholar] [CrossRef]
- Greig, F.H.; Kennedy, S.; Spickett, C.M. Physiological effects of oxidized phospholipids and their cellular signaling mechanisms in inflammation. Free Radic Biol Med 2012, 52, 266–280. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu Rev Biochem 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Ali, S.S.; Ahsan, H.; Zia, M.K.; Siddiqui, T.; Khan, F.H. Understanding oxidants and antioxidants: Classical team with new players. J Food Biochem 2020, 44, e13145. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J 2016, 15, 71. [Google Scholar] [CrossRef]
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awasthi, S. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol Appl Pharmacol 2015, 289, 361–370. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. J Neurochem 2009, 110, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Overview on Peroxiredoxin. Mol Cells 2016, 39, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic Biol Med 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res 2008, 52 Suppl 1, S128–138. [Google Scholar] [CrossRef]
- Engwa, G.A. Free Radicals and the Role of Plant Phytochemicals as Antioxidants Against Oxidative Stress-Related Diseases. Phytochemicals - Source of Antioxidants and Role in Disease Prevention, 2018. [Google Scholar]
- Yadav, A.; Mishra, P.C. Modeling the activity of glutathione as a hydroxyl radical scavenger considering its neutral non-zwitterionic form. J Mol Model 2013, 19, 767–777. [Google Scholar] [CrossRef]
- Stahl, W.; Sies, H. Antioxidant activity of carotenoids. Mol Aspects Med 2003, 24, 345–351. [Google Scholar] [CrossRef]
- Gęgotek, A.; Skrzydlewska, E. Ascorbic acid as antioxidant. Vitam Horm 2023, 121, 247–270. [Google Scholar] [CrossRef]
- Lee, G.Y.; Han, S.N. The Role of Vitamin E in Immunity. Nutrients 2018, 10. [Google Scholar] [CrossRef]
- Rochette, L.; Ghibu, S.; Richard, C.; Zeller, M.; Cottin, Y.; Vergely, C. Direct and indirect antioxidant properties of α -lipoic acid. Molecular nutrition & food research 2013, 57, 114–125. [Google Scholar] [CrossRef]
- Xia, N.; Daiber, A.; Förstermann, U.; Li, H. Antioxidant effects of resveratrol in the cardiovascular system. Br J Pharmacol 2017, 174, 1633–1646. [Google Scholar] [CrossRef] [PubMed]
- Musayeva, A.; Unkrig, J.C.; Zhutdieva, M.B.; Manicam, C.; Ruan, Y.; Laspas, P.; Chronopoulos, P.; Göbel, M.L.; Pfeiffer, N.; Brochhausen, C.; et al. Betulinic Acid Protects from Ischemia-Reperfusion Injury in the Mouse Retina. Cells 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.A.; Di Polo, A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Joyal, J.S.; Gantner, M.L.; Smith, L.E.H. Retinal energy demands control vascular supply of the retina in development and disease: The role of neuronal lipid and glucose metabolism. Prog Retin Eye Res 2018, 64, 131–156. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal energy metabolism in health and glaucoma. Progress in Retinal and Eye Research 2021, 81, 100881. [Google Scholar] [CrossRef] [PubMed]
- Wangsa-Wirawan, N.D.; Linsenmeier, R.A. Retinal Oxygen: Fundamental and Clinical Aspects. Archives of Ophthalmology 2003, 121, 547–557. [Google Scholar] [CrossRef]
- Cohen, L. Relationships between visual function and metabolism. Biochemistry of the Eye 1965, 36–50. [Google Scholar]
- ANDERSON, B.; SALTZMAN, H.A. Retinal oxygen utilization measured by hyperbaric blackout. Archives of ophthalmology 1964, 72, 792–795. [Google Scholar] [CrossRef]
- Ames III, A. Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: a commentary based on studies on retina. Canadian journal of physiology and pharmacology 1992, 70, S158–S164. [Google Scholar] [CrossRef]
- Birk, M.; Baum, E.; Zadeh, J.K.; Manicam, C.; Pfeiffer, N.; Patzak, A.; Helmstadter, J.; Steven, S.; Kuntic, M.; Daiber, A.; et al. Angiotensin II Induces Oxidative Stress and Endothelial Dysfunction in Mouse Ophthalmic Arteries via Involvement of AT1 Receptors and NOX2. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Gericke, A.; Mann, C.; Zadeh, J.K.; Musayeva, A.; Wolff, I.; Wang, M.; Pfeiffer, N.; Daiber, A.; Li, H.; Xia, N.; et al. Elevated Intraocular Pressure Causes Abnormal Reactivity of Mouse Retinal Arterioles. Oxid Med Cell Longev 2019, 2019, 9736047. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, H.; Xia, N.; Li, H.; van Beers, T.; Gericke, A.; Prokosch, V. Intraocular Pressure-Induced Endothelial Dysfunction of Retinal Blood Vessels Is Persistent, but Does Not Trigger Retinal Ganglion Cell Loss. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Zadeh, J.K.; Zhutdieva, M.B.; Laspas, P.; Yuksel, C.; Musayeva, A.; Pfeiffer, N.; Brochhausen, C.; Oelze, M.; Daiber, A.; Xia, N.; et al. Apolipoprotein E Deficiency Causes Endothelial Dysfunction in the Mouse Retina. Oxid Med Cell Longev 2019, 2019, 5181429. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, J.K.; Garcia-Bardon, A.; Hartmann, E.K.; Pfeiffer, N.; Omran, W.; Ludwig, M.; Patzak, A.; Xia, N.; Li, H.; Gericke, A. Short-Time Ocular Ischemia Induces Vascular Endothelial Dysfunction and Ganglion Cell Loss in the Pig Retina. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol 2015, 6, 524–551. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Scorziello, A.; Duchen, M.R. Three Distinct Mechanisms Generate Oxygen Free Radicals in Neurons and Contribute to Cell Death during Anoxia and Reoxygenation. The Journal of Neuroscience 2007, 27, 1129–1138. [Google Scholar] [CrossRef]
- Ono, T.; Tsuruta, R.; Fujita, M.; Aki, H.S.; Kutsuna, S.; Kawamura, Y.; Wakatsuki, J.; Aoki, T.; Kobayashi, C.; Kasaoka, S.; et al. Xanthine oxidase is one of the major sources of superoxide anion radicals in blood after reperfusion in rats with forebrain ischemia/reperfusion. Brain Research 2009, 1305, 158–167. [Google Scholar] [CrossRef]
- Dauth, A.; Breborowicz, A.; Ruan, Y.; Tang, Q.; Zadeh, J.K.; Bohm, E.W.; Pfeiffer, N.; Khedkar, P.H.; Patzak, A.; Vujacic-Mirski, K.; et al. Sulodexide Prevents Hyperglycemia-Induced Endothelial Dysfunction and Oxidative Stress in Porcine Retinal Arterioles. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Giurdanella, G.; Lazzara, F.; Caporarello, N.; Lupo, G.; Anfuso, C.D.; Eandi, C.M.; Leggio, G.M.; Drago, F.; Bucolo, C.; Salomone, S. Sulodexide prevents activation of the PLA2/COX-2/VEGF inflammatory pathway in human retinal endothelial cells by blocking the effect of AGE/RAGE. Biochem Pharmacol 2017, 142, 145–154. [Google Scholar] [CrossRef]
- Hein, T.W.; Xu, W.; Xu, X.; Kuo, L. Acute and Chronic Hyperglycemia Elicit JIP1/JNK-Mediated Endothelial Vasodilator Dysfunction of Retinal Arterioles. Invest Ophthalmol Vis Sci 2016, 57, 4333–4340. [Google Scholar] [CrossRef]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 1994, 269, 9889–9897. [Google Scholar] [CrossRef] [PubMed]
- Dehdashtian, E.; Mehrzadi, S.; Yousefi, B.; Hosseinzadeh, A.; Reiter, R.J.; Safa, M.; Ghaznavi, H.; Naseripour, M. Diabetic retinopathy pathogenesis and the ameliorating effects of melatonin; involvement of autophagy, inflammation and oxidative stress. Life Sci 2018, 193, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Pang, B.; Li, M.; Song, J.; Li, Q.W.; Wang, J.; Di, S.; Tong, X.L.; Ni, Q. Luo Tong formula attenuates retinal inflammation in diabetic rats via inhibition of the p38MAPK/NF-κB pathway. Chin Med 2020, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Lazzara, F.; Fidilio, A.; Platania, C.B.M.; Giurdanella, G.; Salomone, S.; Leggio, G.M.; Tarallo, V.; Cicatiello, V.; De Falco, S.; Eandi, C.M.; et al. Aflibercept regulates retinal inflammation elicited by high glucose via the PlGF/ERK pathway. Biochem Pharmacol 2019, 168, 341–351. [Google Scholar] [CrossRef]
- Tarr, J.M.; Kaul, K.; Chopra, M.; Kohner, E.M.; Chibber, R. Pathophysiology of diabetic retinopathy. ISRN Ophthalmol 2013, 2013, 343560. [Google Scholar] [CrossRef]
- Lee, S.R.; An, E.J.; Kim, J.; Bae, Y.S. Function of NADPH Oxidases in Diabetic Nephropathy and Development of Nox Inhibitors. Biomol Ther (Seoul) 2020, 28, 25–33. [Google Scholar] [CrossRef]
- Nakazawa, T.; Fukuchi, T. What is glaucomatous optic neuropathy? Jpn J Ophthalmol 2020, 64, 243–249. [Google Scholar] [CrossRef]
- Fortune, B.; Grzybowski, A. Glaucomatous or Non-glaucomatous Optic Neuropathy-It Is a Question? Am J Ophthalmol 2022, 234, A5–a7. [Google Scholar] [CrossRef]
- Burgoyne, C. The morphological difference between glaucoma and other optic neuropathies. J Neuroophthalmol 2015, 35 Suppl 1, S8–s21. [Google Scholar] [CrossRef]
- Anderson, D.R.; Patella, V.M. Automated static perimetry, 2nd ed.Mosby: Saint Louis (Mo.), 1999. [Google Scholar]
- Foster, P.J.; Buhrmann, R.; Quigley, H.A.; Johnson, G.J. The definition and classification of glaucoma in prevalence surveys. Br J Ophthalmol 2002, 86, 238–242. [Google Scholar] [CrossRef]
- McMonnies, C. Reactive oxygen species, oxidative stress, glaucoma and hyperbaric oxygen therapy. J Optom 2018, 11, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid Med Cell Longev 2016, 2016, 3164734. [Google Scholar] [CrossRef] [PubMed]
- Kroese, M.; Burton, H. Primary open angle glaucoma. The need for a consensus case definition. J Epidemiol Community Health 2003, 57, 752–754. [Google Scholar] [CrossRef]
- Khazaeni, B.; Khazaeni, L. Acute Closed Angle Glaucoma. In StatPearls, StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.: Treasure Island (FL), 2022.
- Flores-Sánchez, B.C.; Tatham, A.J. Acute angle closure glaucoma. Br J Hosp Med (Lond) 2019, 80, C174–c179. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.L.; Tham, C.C. Normal-tension glaucoma: Current concepts and approaches-A review. Clin Exp Ophthalmol 2022, 50, 247–259. [Google Scholar] [CrossRef]
- Patel, K.; Patel, S. Angle-closure glaucoma. Disease-a-Month 2014, 60, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Tawfik, M.A.; Waisbourd, M.; Katz, L.J. Primary angle-closure glaucoma: an update. Acta Ophthalmol 2016, 94, 217–225. [Google Scholar] [CrossRef]
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 2006, 90, 262–267. [Google Scholar] [CrossRef]
- Quigley, H.A. Number of people with glaucoma worldwide. Br J Ophthalmol 1996, 80, 389–393. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: a review. Jama 2014, 311, 1901–1911. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Thomas, R.; Li, S.Z.; Wang, N.L. Development of angle closure and associated risk factors: The Handan eye study. Acta Ophthalmologica 2022, 100, e253–e261. [Google Scholar] [CrossRef] [PubMed]
- Schuster, A.K.; Erb, C.; Hoffmann, E.M.; Dietlein, T.; Pfeiffer, N. The Diagnosis and Treatment of Glaucoma. Dtsch Arztebl Int 2020, 117, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.V.; Ervin, A.M.; Friedman, D.S.; Jampel, H.D.; Hawkins, B.S.; Vollenweider, D.; Chelladurai, Y.; Ward, D.; Suarez-Cuervo, C.; Robinson, K.A. Comparative effectiveness of treatments for open-angle glaucoma: a systematic review for the U.S. Preventive Services Task Force. Ann Intern Med 2013, 158, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Orgül, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.-P.; Stefánsson, E. The impact of ocular blood flow in glaucoma. Progress in Retinal and Eye Research 2002, 21, 359–393. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.C.; Roberts, M.D.; Burgoyne, C.F. Mechanical environment of the optic nerve head in glaucoma. Optom Vis Sci 2008, 85, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Prum, B.E., Jr.; Rosenberg, L.F.; Gedde, S.J.; Mansberger, S.L.; Stein, J.D.; Moroi, S.E.; Herndon, L.W., Jr.; Lim, M.C.; Williams, R.D. Primary Open-Angle Glaucoma Preferred Practice Pattern(®) Guidelines. Ophthalmology 2016, 123, P41–p111. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.P.; Yuan, H.H.; Zhu, X.; Cui, Y.Y.; Li, H.; Feng, X.M.; Qiu, Y.; Chen, H.Z.; Zhou, W. Activation of muscarinic receptors protects against retinal neurons damage and optic nerve degeneration in vitro and in vivo models. CNS Neurosci Ther 2014, 20, 227–236. [Google Scholar] [CrossRef]
- Li, T.; Lindsley, K.; Rouse, B.; Hong, H.; Shi, Q.; Friedman, D.S.; Wormald, R.; Dickersin, K. Comparative Effectiveness of First-Line Medications for Primary Open-Angle Glaucoma: A Systematic Review and Network Meta-analysis. Ophthalmology 2016, 123, 129–140. [Google Scholar] [CrossRef]
- Rouland, J.F.; Berdeaux, G.; Lafuma, A. The economic burden of glaucoma and ocular hypertension: implications for patient management: a review. Drugs Aging 2005, 22, 315–321. [Google Scholar] [CrossRef]
- Dirani, M.; Crowston, J.G.; Taylor, P.S.; Moore, P.T.; Rogers, S.; Pezzullo, M.L.; Keeffe, J.E.; Taylor, H.R. Economic impact of primary open-angle glaucoma in Australia. Clin Exp Ophthalmol 2011, 39, 623–632. [Google Scholar] [CrossRef]
- McGinley, P.; Ansari, E.; Sandhu, H.; Dixon, T. The cost burden of falls in people with glaucoma in National Health Service Hospital Trusts in the UK. J Med Econ 2020, 23, 106–112. [Google Scholar] [CrossRef]
- Shih, V.; Parekh, M.; Multani, J.K.; McGuiness, C.B.; Chen, C.-C.; Campbell, J.H.; Miller-Ellis, E.; Olivier, M.M.G. Clinical and Economic Burden of Glaucoma by Disease Severity: A United States Claims-Based Analysis. Ophthalmology Glaucoma 2021, 4, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, K.; Wolfram, C.; Breitscheidel, L.; Shlaen, M.; Verboven, Y.; Pfeiffer, N. Direct cost and predictive factors for treatment in patients with ocular hypertension or early, moderate and advanced primary open-angle glaucoma: the CoGIS study in Germany. Graefes Arch Clin Exp Ophthalmol 2013, 251, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Varma, R.; Lee, P.P.; Goldberg, I.; Kotak, S. An assessment of the health and economic burdens of glaucoma. Am J Ophthalmol 2011, 152, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, K.; Dada, R.; Dada, T. Oxidative DNA damage and reduced expression of DNA repair genes: Role in primary open angle glaucoma (POAG). Ophthalmic Genetics 2017, 38, 446–450. [Google Scholar] [CrossRef]
- Izzotti, A.; Saccà, S.C.; Longobardi, M.; Cartiglia, C. Mitochondrial Damage in the Trabecular Meshwork of Patients With Glaucoma. Archives of Ophthalmology 2010, 128, 724–730. [Google Scholar] [CrossRef]
- Saccà, S.C.; Pascotto, A.; Camicione, P.; Capris, P.; Izzotti, A. Oxidative DNA Damage in the Human Trabecular Meshwork: Clinical Correlation in Patients With Primary Open-Angle Glaucoma. Archives of Ophthalmology 2005, 123, 458–463. [Google Scholar] [CrossRef]
- Nucci, C.; Di Pierro, D.; Varesi, C.; Ciuffoletti, E.; Russo, R.; Gentile, R.; Cedrone, C.; Pinazo Duran, M.D.; Coletta, M.; Mancino, R. Increased malondialdehyde concentration and reduced total antioxidant capacity in aqueous humor and blood samples from patients with glaucoma. Mol Vis 2013, 19, 1841–1846. [Google Scholar]
- Zanon-Moreno, V.; Garcia-Medina, J.J.; Gallego-Pinazo, R.; Vinuesa-Silva, I.; Moreno-Nadal, M.A.; Pinazo-Duran, M.D. Antioxidant Status Modifications by Topical Administration of Dorzolamide in Primary Open-Angle Glaucoma. European Journal of Ophthalmology 2009, 19, 565–571. [Google Scholar] [CrossRef]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Oxidative stress markers in aqueous humor of glaucoma patients. American Journal of Ophthalmology 2004, 137, 62–69. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Kondkar, A.A.; Mousa, A.; Osman, E.A.; Al-Obeidan, S.A. Decreased total antioxidants in patients with primary open angle glaucoma. Curr Eye Res 2013, 38, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Asano, Y.; Himori, N.; Kunikata, H.; Yamazaki, M.; Shiga, Y.; Omodaka, K.; Takahashi, H.; Nakazawa, T. Age- and sex-dependency of the association between systemic antioxidant potential and glaucomatous damage. Sci Rep 2017, 7, 8032. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Kaidzu, S.; Takai, Y.; Ohira, A. Association between systemic oxidative stress and visual field damage in open-angle glaucoma. Sci Rep 2016, 6, 25792. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Kaidzu, S.; Takai, Y.; Ohira, A. Correlation between Systemic Oxidative Stress and Intraocular Pressure Level. PLOS ONE 2015, 10, e0133582. [Google Scholar] [CrossRef]
- Benoist d'Azy, C.; Pereira, B.; Chiambaretta, F.; Dutheil, F. Oxidative and Anti-Oxidative Stress Markers in Chronic Glaucoma: A Systematic Review and Meta-Analysis. PLoS One 2016, 11, e0166915. [Google Scholar] [CrossRef]
- Tang, B.; Li, S.; Cao, W.; Sun, X. The Association of Oxidative Stress Status with Open-Angle Glaucoma and Exfoliation Glaucoma: A Systematic Review and Meta-Analysis. Journal of Ophthalmology 2019, 2019, 1803619. [Google Scholar] [CrossRef]
- Aboobakar, I.F.; Allingham, R.R. Genetics of exfoliation syndrome and glaucoma. Int Ophthalmol Clin 2014, 54, 43–56. [Google Scholar] [CrossRef]
- Ritch, R.; Schlötzer-Schrehardt, U. Exfoliation syndrome. Surv Ophthalmol 2001, 45, 265–315. [Google Scholar] [CrossRef]
- Tsai, D.-C.; Hsu, W.-M.; Chou, C.-K.; Chen, S.-J.; Peng, C.-H.; Chi, C.-W.; Ho, L.L.-T.; Liu, J.-H.; Chiou, S.-H. Significant Variation of the Elevated Nitric Oxide Levels in Aqueous Humor from Patients with Different Types of Glaucoma. Ophthalmologica 2002, 216, 346–350. [Google Scholar] [CrossRef]
- Fernández-Durango, R.; Fernández-Martínez, A.; García-Feijoo, J.n.; Castillo, A.; de la Casa, J.M.n.; García-Bueno, B.; Pérez-Nievas, B.G.; Fernández-Cruz, A.; Leza, J.C. Expression of Nitrotyrosine and Oxidative Consequences in the Trabecular Meshwork of Patients with Primary Open-Angle Glaucoma. Investigative Ophthalmology & Visual Science 2008, 49, 2506–2511. [Google Scholar] [CrossRef]
- Llobet, A.; Gasull, X.; Gual, A. Understanding trabecular meshwork physiology: a key to the control of intraocular pressure? News Physiol Sci 2003, 18, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Oxidative stress markers in aqueous humor of glaucoma patients. Am J Ophthalmol 2004, 137, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Sorkhabi, R.; Ghorbanihaghjo, A.; Javadzadeh, A.; Rashtchizadeh, N.; Moharrery, M. Oxidative DNA damage and total antioxidant status in glaucoma patients. Mol Vis 2011, 17, 41–46. [Google Scholar]
- Saccà, S.C.; Izzotti, A.; Rossi, P.; Traverso, C. Glaucomatous outflow pathway and oxidative stress. Exp Eye Res 2007, 84, 389–399. [Google Scholar] [CrossRef]
- Hogg, P.; Calthorpe, M.; Batterbury, M.; Grierson, I. Aqueous humor stimulates the migration of human trabecular meshwork cells in vitro. Invest Ophthalmol Vis Sci 2000, 41, 1091–1098. [Google Scholar] [PubMed]
- Zhou, L.; Li, Y.; Yue, B.Y. Oxidative stress affects cytoskeletal structure and cell-matrix interactions in cells from an ocular tissue: the trabecular meshwork. J Cell Physiol 1999, 180, 182–189. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Current Opinion in Pharmacology 2013, 13, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Wax, M.B. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci 2000, 20, 8693–8700. [Google Scholar] [CrossRef]
- Tezel, G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog Brain Res 2008, 173, 409–421. [Google Scholar] [CrossRef]
- Tezel, G. Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets. Prog Retin Eye Res 2022, 87, 100998. [Google Scholar] [CrossRef]
- Ju, W.K.; Kim, K.Y.; Lindsey, J.D.; Angert, M.; Duong-Polk, K.X.; Scott, R.T.; Kim, J.J.; Kukhmazov, I.; Ellisman, M.H.; Perkins, G.A.; et al. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Invest Ophthalmol Vis Sci 2008, 49, 4903–4911. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G.; Fourth, A.P.O.R.I.C.W.G. The role of glia, mitochondria, and the immune system in glaucoma. Invest Ophthalmol Vis Sci 2009, 50, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Levkovitch-Verbin, H.; Valenta, D.; Baumrind, L.; Pease, M.E.; Quigley, H.A. Retinal glutamate transporter changes in experimental glaucoma and after optic nerve transection in the rat. Invest Ophthalmol Vis Sci 2002, 43, 2236–2243. [Google Scholar] [PubMed]
- Mantzaris, M.D.; Bellou, S.; Skiada, V.; Kitsati, N.; Fotsis, T.; Galaris, D. Intracellular labile iron determines H2O2-induced apoptotic signaling via sustained activation of ASK1/JNK-p38 axis. Free Radical Biology and Medicine 2016, 97, 454–465. [Google Scholar] [CrossRef]
- Harada, C.; Nakamura, K.; Namekata, K.; Okumura, A.; Mitamura, Y.; Iizuka, Y.; Kashiwagi, K.; Yoshida, K.; Ohno, S.; Matsuzawa, A.; et al. Role of apoptosis signal-regulating kinase 1 in stress-induced neural cell apoptosis in vivo. Am J Pathol 2006, 168, 261–269. [Google Scholar] [CrossRef]
- Kitsati, N.; Mantzaris, M.D.; Galaris, D. Hydroxytyrosol inhibits hydrogen peroxide-induced apoptotic signaling via labile iron chelation. Redox Biology 2016, 10, 233–242. [Google Scholar] [CrossRef]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef]
- Chang, Y.-S.; Chang, Y.-C.; Chen, P.-H.; Li, C.-Y.; Wu, W.-C.; Kao, Y.-H. MicroRNA-100 Mediates Hydrogen Peroxide-Induced Apoptosis of Human Retinal Pigment Epithelium ARPE-19 Cells. Pharmaceuticals 2021, 14, 314. [Google Scholar] [CrossRef]
- The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Collaborative Normal-Tension Glaucoma Study Group. Am J Ophthalmol 1998, 126, 498–505. [Google Scholar] [CrossRef]
- Killer, H.E.; Pircher, A. Normal tension glaucoma: review of current understanding and mechanisms of the pathogenesis. Eye (Lond) 2018, 32, 924–930. [Google Scholar] [CrossRef]
- Stroman, G.A.; Stewart, W.C.; Golnik, K.C.; Curé, J.K.; Olinger, R.E. Magnetic resonance imaging in patients with low-tension glaucoma. Arch Ophthalmol 1995, 113, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.; Farinelli, A.; Billson, F.; Houang, M.; Stern, M. Comparative study of brain magnetic resonance imaging findings in patients with low-tension glaucoma and control subjects. Ophthalmology 1995, 102, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Tomidokoro, A.; Araie, M.; Tomita, G.; Yamagami, J.; Okubo, T.; Masumoto, T. Visual field damage in normal-tension glaucoma patients with or without ischemic changes in cerebral magnetic resonance imaging. Jpn J Ophthalmol 2004, 48, 340–344. [Google Scholar] [CrossRef]
- Rupin, A.; Paysant, J.; Sansilvestri-Morel, P.; Lembrez, N.; Lacoste, J.M.; Cordi, A.; Verbeuren, T.J. Role of NADPH oxidase-mediated superoxide production in the regulation of E-selectin expression by endothelial cells subjected to anoxia/reoxygenation. Cardiovasc Res 2004, 63, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; König, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Kleikers, P.W.; Wingler, K.; Hermans, J.J.; Diebold, I.; Altenhöfer, S.; Radermacher, K.A.; Janssen, B.; Görlach, A.; Schmidt, H.H. NADPH oxidases as a source of oxidative stress and molecular target in ischemia/reperfusion injury. J Mol Med (Berl) 2012, 90, 1391–1406. [Google Scholar] [CrossRef]
- Kietzmann, T.; Görlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol 2005, 16, 474–486. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. Hypoxia-inducible factor 1alpha in the glaucomatous retina and optic nerve head. Arch Ophthalmol 2004, 122, 1348–1356. [Google Scholar] [CrossRef]
- Banasiak, K.J.; Xia, Y.; Haddad, G.G. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol 2000, 62, 215–249. [Google Scholar] [CrossRef]
- Yokota, H.; Narayanan, S.P.; Zhang, W.; Liu, H.; Rojas, M.; Xu, Z.; Lemtalsi, T.; Nagaoka, T.; Yoshida, A.; Brooks, S.E.; et al. Neuroprotection from retinal ischemia/reperfusion injury by NOX2 NADPH oxidase deletion. Invest Ophthalmol Vis Sci 2011, 52, 8123–8131. [Google Scholar] [CrossRef]
- Rieger, J.M.; Shah, A.R.; Gidday, J.M. Ischemia-reperfusion injury of retinal endothelium by cyclooxygenase- and xanthine oxidase-derived superoxide. Exp Eye Res 2002, 74, 493–501. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, X. Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci 2004, 45, 4049–4059. [Google Scholar] [CrossRef]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.-R.; Chae, H.-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Experimental & Molecular Medicine 2021, 53, 151–167. [Google Scholar] [CrossRef]
- Kroeger, H.; Chiang, W.-C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. The FEBS Journal 2019, 286, 399–412. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am J Pathol 2015, 185, 1800–1808. [Google Scholar] [CrossRef]
- Hurley, D.J.; Normile, C.; Irnaten, M.; O'Brien, C. The Intertwined Roles of Oxidative Stress and Endoplasmic Reticulum Stress in Glaucoma. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annual Review of Pathology: Mechanisms of Disease 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem Sci 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Chen, A.C.-H.; Burr, L.; McGuckin, M.A. Oxidative and endoplasmic reticulum stress in respiratory disease. Clinical & Translational Immunology 2018, 7, e1019. [Google Scholar] [CrossRef]
- Kaneko, M.; Niinuma, Y.; Nomura, Y. Activation Signal of Nuclear Factor-κB in Response to Endoplasmic Reticulum Stress is Transduced <i>via</i> IRE1 and Tumor Necrosis Factor Receptor-Associated Factor 2. Biological and Pharmaceutical Bulletin 2003, 26, 931–935. [Google Scholar] [CrossRef]
- Song, B.; Scheuner, D.; Ron, D.; Pennathur, S.; Kaufman, R.J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 2008, 118, 3378–3389. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Zhai, P.; Shao, D.; Zablocki, D.; Nagarajan, N.; Terada, L.S.; Volpe, M.; Sadoshima, J. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2α/activating transcription factor 4 pathway. Circ Res 2013, 113, 1253–1264. [Google Scholar] [CrossRef]
- Pahl, H.L.; Baeuerle, P.A. The ER-overload response: activation of NF-κB. Trends in Biochemical Sciences 1997, 22, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.; Xue, R.; Yang, Y.; Zhang, S.X.; Xiao, H.; Zhu, H.; Li, J.; Chen, G.; Ye, Y.; Yu, M.; et al. Activation of ATF4 triggers trabecular meshwork cell dysfunction and apoptosis in POAG. Aging (Albany NY) 2021, 13, 8628–8642. [Google Scholar] [CrossRef]
- Kasetti, R.B.; Patel, P.D.; Maddineni, P.; Patil, S.; Kiehlbauch, C.; Millar, J.C.; Searby, C.C.; Raghunathan, V.; Sheffield, V.C.; Zode, G.S. ATF4 leads to glaucoma by promoting protein synthesis and ER client protein load. Nature Communications 2020, 11, 5594. [Google Scholar] [CrossRef]
- Peters, J.C.; Bhattacharya, S.; Clark, A.F.; Zode, G.S. Increased Endoplasmic Reticulum Stress in Human Glaucomatous Trabecular Meshwork Cells and Tissues. Investigative Ophthalmology & Visual Science 2015, 56, 3860–3868. [Google Scholar] [CrossRef]
- Chai, F.; Yan, H.; Zhao, X.; Li, J.; Pei, C. The role of GRP78 in oxidative stress induced by tunicamycin in trabecular meshwork cells. Acta Biochim Pol 2022, 69, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Doh, S.H.; Kim, J.H.; Lee, K.M.; Park, H.Y.; Park, C.K. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res 2010, 1308, 158–166. [Google Scholar] [CrossRef]
- Marola, O.J.; Syc-Mazurek, S.B.; Libby, R.T. DDIT3 (CHOP) contributes to retinal ganglion cell somal loss but not axonal degeneration in DBA/2J mice. Cell Death Discov 2019, 5, 140. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Sato, T.; Ohno-Oishi, M.; Ozawa, M.; Maekawa, S.; Shiga, Y.; Yabana, T.; Yasuda, M.; Himori, N.; Omodaka, K.; et al. CHOP deletion and anti-neuroinflammation treatment with hesperidin synergistically attenuate NMDA retinal injury in mice. Exp Eye Res 2021, 213, 108826. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Zhang, X.; Xu, X. Nerve Growth Factor Protects Retinal Ganglion Cells Related to Inhibiting Endoplasmic Reticulum Stress by Inhibiting IRE1-JNK-CHOP Signaling Pathway. Ocul Immunol Inflamm 2022, 30, 1341–1346. [Google Scholar] [CrossRef]
- Gao, Z.; Li, M.; Yao, F.; Xia, X.; Duan, T.; Meng, J.; Huang, Y.; He, Y.; Saro, A.; Huang, J. Valdecoxib Protects against Cell Apoptosis Induced by Endoplasmic Reticulum Stress via the Inhibition of PERK-ATF4-CHOP Pathway in Experimental Glaucoma. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Galiniak, S.; Aebisher, D.; Bartusik-Aebisher, D. Health benefits of resveratrol administration. Acta Biochim Pol 2019, 66, 13–21. [Google Scholar] [CrossRef]
- Avotri, S.; Eatman, D.; Russell-Randall, K. Effects of Resveratrol on Inflammatory Biomarkers in Glaucomatous Human Trabecular Meshwork Cells. Nutrients 2019, 11. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of Human SIRT1 Activation by Resveratrol*. Journal of Biological Chemistry 2005, 280, 17187–17195. [Google Scholar] [CrossRef]
- Ciccone, L.; Piragine, E.; Brogi, S.; Camodeca, C.; Fucci, R.; Calderone, V.; Nencetti, S.; Martelli, A.; Orlandini, E. Resveratrol-like Compounds as SIRT1 Activators. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Pirhan, D.; Yüksel, N.; Emre, E.; Cengiz, A.; Kürşat Yıldız, D. Riluzole- and Resveratrol-Induced Delay of Retinal Ganglion Cell Death in an Experimental Model of Glaucoma. Curr Eye Res 2016, 41, 59–69. [Google Scholar] [CrossRef]
- Ye, M.J.; Meng, N. Resveratrol acts via the mitogen-activated protein kinase (MAPK) pathway to protect retinal ganglion cells from apoptosis induced by hydrogen peroxide. Bioengineered 2021, 12, 4878–4886. [Google Scholar] [CrossRef]
- Yue, Y.K.; Mo, B.; Zhao, J.; Yu, Y.J.; Liu, L.; Yue, C.L.; Liu, W. Neuroprotective effect of curcumin against oxidative damage in BV-2 microglia and high intraocular pressure animal model. J Ocul Pharmacol Ther 2014, 30, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Buccarello, L.; Dragotto, J.; Hassanzadeh, K.; Maccarone, R.; Corbo, M.; Feligioni, M. Retinal ganglion cell loss in an ex vivo mouse model of optic nerve cut is prevented by curcumin treatment. Cell Death Discov 2021, 7, 394. [Google Scholar] [CrossRef] [PubMed]
- Noro, T.; Namekata, K.; Azuchi, Y.; Kimura, A.; Guo, X.; Harada, C.; Nakano, T.; Tsuneoka, H.; Harada, T. Spermidine Ameliorates Neurodegeneration in a Mouse Model of Normal Tension Glaucoma. Invest Ophthalmol Vis Sci 2015, 56, 5012–5019. [Google Scholar] [CrossRef]
- Noro, T.; Namekata, K.; Kimura, A.; Guo, X.; Azuchi, Y.; Harada, C.; Nakano, T.; Tsuneoka, H.; Harada, T. Spermidine promotes retinal ganglion cell survival and optic nerve regeneration in adult mice following optic nerve injury. Cell Death Dis 2015, 6, e1720. [Google Scholar] [CrossRef]
- Liu, X.G.; Wu, S.Q.; Li, P.; Yang, H. Advancement in the chemical analysis and quality control of flavonoid in Ginkgo biloba. J Pharm Biomed Anal 2015, 113, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Dong, L.-H.; Zhang, Y.; Liu, Q. A network pharmacology-based strategy for predicting the protective mechanism of Ginkgo biloba on damaged retinal ganglion cells. Chinese Journal of Natural Medicines 2022, 20, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Anne-Marie, B.; Beatriz, V.; Ana Isabel, J.; Covadonga, P. Managing Intraocular Pressure: Innovation in Glaucoma Management. In Glaucoma; Parul, I., Ed.; IntechOpen: Rijeka, 2016; Ch. 6. [Google Scholar] [CrossRef]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress-mediated mitochondrial alteration in a mouse model of glaucoma. Invest Ophthalmol Vis Sci 2014, 55, 993–1005. [Google Scholar] [CrossRef]
- Quaranta, L.; Riva, I.; Biagioli, E.; Rulli, E.; Rulli, E.; Poli, D.; Legramandi, L. Evaluating the Effects of an Ophthalmic Solution of Coenzyme Q10 and Vitamin E in Open-Angle Glaucoma Patients: A Study Protocol. Adv Ther 2019, 36, 2506–2514. [Google Scholar] [CrossRef]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. α-Lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS One 2013, 8, e65389. [Google Scholar] [CrossRef]
- Sanz-González, S.M.; Raga-Cervera, J.; Aguirre Lipperheide, M.; Zanón-Moreno, V.; Chiner, V.; Ramírez, A.I.; Pinazo-Durán, M.D. Effect of an oral supplementation with a formula containing R-lipoic acid in glaucoma patients. Arch Soc Esp Oftalmol (Engl Ed) 2020, 95, 120–129. [Google Scholar] [CrossRef]
- Kamat, J.P.; Devasagayam, T.P. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox Rep 1999, 4, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.I.; Kim, Y.C.; Park, C.K. Dietary Niacin and Open-Angle Glaucoma: The Korean National Health and Nutrition Examination Survey. Nutrients 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; John, S.W.M. Glaucoma as a Metabolic Optic Neuropathy: Making the Case for Nicotinamide Treatment in Glaucoma. J Glaucoma 2017, 26, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin Exp Ophthalmol 2020, 48, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Fan Gaskin, J.C.; Shah, M.H.; Chan, E.C. Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Deliyanti, D.; Wilkinson-Berka, J.L. Inhibition of NOX1/4 with GKT137831: a potential novel treatment to attenuate neuroglial cell inflammation in the retina. J Neuroinflammation 2015, 12, 136. [Google Scholar] [CrossRef]
- Dionysopoulou, S.; Wikström, P.; Walum, E.; Thermos, K. Effect of NADPH oxidase inhibitors in an experimental retinal model of excitotoxicity. Exp Eye Res 2020, 200, 108232. [Google Scholar] [CrossRef]
- Goetz, R.K.; Irnaten, M.; O'Brien, C.J. TGF-β induces NOX4 and fibrotic genes in trabecular meshwork cells: role in glaucoma. Investigative Ophthalmology & Visual Science 2019, 60, 3800–3800. [Google Scholar]
- Wan, P.; Su, W.; Zhang, Y.; Li, Z.; Deng, C.; Zhuo, Y. Trimetazidine protects retinal ganglion cells from acute glaucoma via the Nrf2/Ho-1 pathway. Clin Sci (Lond) 2017, 131, 2363–2375. [Google Scholar] [CrossRef]
- Yuan, J.P.; Peng, J.; Yin, K.; Wang, J.H. Potential health-promoting effects of astaxanthin: a high-value carotenoid mostly from microalgae. Mol Nutr Food Res 2011, 55, 150–165. [Google Scholar] [CrossRef]
- Kumar, S.; Kumar, R.; Kumari, A.; Panwar, A. Astaxanthin: A super antioxidant from microalgae and its therapeutic potential. J Basic Microbiol 2022, 62, 1064–1082. [Google Scholar] [CrossRef] [PubMed]
- Cort, A.; Ozturk, N.; Akpinar, D.; Unal, M.; Yucel, G.; Ciftcioglu, A.; Yargicoglu, P.; Aslan, M. Suppressive effect of astaxanthin on retinal injury induced by elevated intraocular pressure. Regul Toxicol Pharmacol 2010, 58, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Dong, Z.; Shinmei, Y.; Murata, M.; Kanda, A.; Noda, K.; Harada, T.; Ishida, S. Cytoprotective Effect of Astaxanthin in a Model of Normal Intraocular Pressure Glaucoma. J Ophthalmol 2020, 2020, 9539681. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Q.; Chu, C.; Liu, S. Astaxanthin protects retinal ganglion cells from acute glaucoma via the Nrf2/HO-1 pathway. J Chem Neuroanat 2020, 110, 101876. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Xing, Y.; Xiong, L.; Wang, J. Sestrin2 overexpression alleviates hydrogen peroxide-induced apoptosis and oxidative stress in retinal ganglion cells by enhancing Nrf2 activation via Keap1 downregulation. Chem Biol Interact 2020, 324, 109086. [Google Scholar] [CrossRef]
- Li, X.; Leng, Y.; Jiang, Q.; Wang, Z.; Luo, P.; Zhang, C.; Chen, L.; Wang, Y.; Wang, H.; Yue, X.; et al. Eye Drops of Metformin Prevents Fibrosis After Glaucoma Filtration Surgery in Rats via Activating AMPK/Nrf2 Signaling Pathway. Front Pharmacol 2020, 11, 1038. [Google Scholar] [CrossRef]
- Naguib, S.; DeJulius, C.R.; Backstrom, J.R.; Haider, A.A.; Ang, J.M.; Boal, A.M.; Calkins, D.J.; Duvall, C.L.; Rex, T.S. Intraocular Sustained Release of EPO-R76E Mitigates Glaucoma Pathogenesis by Activating the NRF2/ARE Pathway. Antioxidants (Basel) 2023, 12. [Google Scholar] [CrossRef]
- Li, G.; Lee, C.; Read, A.T.; Wang, K.; Ha, J.; Kuhn, M.; Navarro, I.; Cui, J.; Young, K.; Gorijavolu, R.; et al. Anti-fibrotic activity of a rho-kinase inhibitor restores outflow function and intraocular pressure homeostasis. Elife 2021, 10. [Google Scholar] [CrossRef]
- Batra, M.; Gupta, S.; Nair, A.B.; Dhanawat, M.; Sandal, S.; Morsy, M.A. Netarsudil: A new ophthalmic drug in the treatment of chronic primary open angle glaucoma and ocular hypertension. Eur J Ophthalmol 2021, 31, 2237–2244. [Google Scholar] [CrossRef]
- Fujimoto, T.; Inoue, T.; Ohira, S.; Awai-Kasaoka, N.; Kameda, T.; Inoue-Mochita, M.; Tanihara, H. Inhibition of Rho Kinase Induces Antioxidative Molecules and Suppresses Reactive Oxidative Species in Trabecular Meshwork Cells. Journal of Ophthalmology 2017, 2017, 7598140. [Google Scholar] [CrossRef]
- Chen, W.; Yang, X.; Fang, J.; Zhang, Y.; Zhu, W.; Yang, X. Rho-Associated Protein Kinase Inhibitor Treatment Promotes Proliferation and Phagocytosis in Trabecular Meshwork Cells. Front Pharmacol 2020, 11, 302. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, T.; Omae, T.; Nakabayashi, S.; Takahashi, K.; Tanner, A.; Yoshida, A. Effect of Rho Kinase Inhibitor Ripasudil (K-115) on Isolated Porcine Retinal Arterioles. J Ocul Pharmacol Ther 2021, 37, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Feillet, F.; Leonard, J.V. Alternative pathway therapy for urea cycle disorders. J Inherit Metab Dis 1998, 21 Suppl 1, 101–111. [Google Scholar] [CrossRef]
- Brown, C.R.; Hong-Brown, L.Q.; Biwersi, J.; Verkman, A.S.; Welch, W.J. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1996, 1, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Ghosh, A.; Jana, A.; Liu, X.; Brahmachari, S.; Gendelman, H.E.; Pahan, K. Sodium phenylbutyrate controls neuroinflammatory and antioxidant activities and protects dopaminergic neurons in mouse models of Parkinson's disease. PLoS One 2012, 7, e38113. [Google Scholar] [CrossRef]
- Dong, Y.; Li, L.; Xia, T.; Wang, L.; Xiao, L.; Ding, N.; Wu, Y.; Lu, K. Oxidative Stress Can Be Attenuated by 4-PBA Caused by High-Fat or Ammonia Nitrogen in Cultured Spotted Seabass: The Mechanism Is Related to Endoplasmic Reticulum Stress. Antioxidants (Basel) 2022, 11. [Google Scholar] [CrossRef]
- Zode, G.S.; Kuehn, M.H.; Nishimura, D.Y.; Searby, C.C.; Mohan, K.; Grozdanic, S.D.; Bugge, K.; Anderson, M.G.; Clark, A.F.; Stone, E.M.; et al. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. J Clin Invest 2011, 121, 3542–3553. [Google Scholar] [CrossRef]
- Maddineni, P.; Kasetti, R.B.; Kodati, B.; Yacoub, S.; Zode, G.S. Sodium 4-Phenylbutyrate Reduces Ocular Hypertension by Degrading Extracellular Matrix Deposition via Activation of MMP9. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Lapchak, P.A. A critical assessment of edaravone acute ischemic stroke efficacy trials: is edaravone an effective neuroprotective therapy? Expert Opin Pharmacother 2010, 11, 1753–1763. [Google Scholar] [CrossRef]
- Masuda, T.; Shimazawa, M.; Hara, H. Retinal Diseases Associated with Oxidative Stress and the Effects of a Free Radical Scavenger (Edaravone). Oxid Med Cell Longev 2017, 2017, 9208489. [Google Scholar] [CrossRef]
- Akaiwa, K.; Namekata, K.; Azuchi, Y.; Guo, X.; Kimura, A.; Harada, C.; Mitamura, Y.; Harada, T. Edaravone suppresses retinal ganglion cell death in a mouse model of normal tension glaucoma. Cell Death Dis 2017, 8, e2934. [Google Scholar] [CrossRef] [PubMed]
- Aksar, A.T.; Yuksel, N.; Gok, M.; Cekmen, M.; Caglar, Y. Neuroprotective effect of edaravone in experimental glaucoma model in rats: a immunofluorescence and biochemical analysis. Int J Ophthalmol 2015, 8, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qin, X.; Zhao, X.; Tong, N.; Gong, Y.; Zhang, W.; Wu, X. Valproic acid regulates antioxidant enzymes and prevents ischemia/reperfusion injury in the rat retina. Curr Eye Res 2012, 37, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Guo, X.; Noro, T.; Harada, C.; Tanaka, K.; Namekata, K.; Harada, T. Valproic acid prevents retinal degeneration in a murine model of normal tension glaucoma. Neurosci Lett 2015, 588, 108–113. [Google Scholar] [CrossRef]
- Tribble, J.R.; Kastanaki, E.; Uslular, A.B.; Rutigliani, C.; Enz, T.J.; Williams, P.A. Valproic Acid Reduces Neuroinflammation to Provide Retinal Ganglion Cell Neuroprotection in the Retina Axotomy Model. Front Cell Dev Biol 2022, 10, 903436. [Google Scholar] [CrossRef]
- Mahalingam, K.; Chaurasia, A.K.; Gowtham, L.; Gupta, S.; Somarajan, B.I.; Velpandian, T.; Sihota, R.; Gupta, V. Therapeutic potential of valproic acid in advanced glaucoma: A pilot study. Indian J Ophthalmol 2018, 66, 1104–1108. [Google Scholar] [CrossRef]
- Harada, C.; Noro, T.; Kimura, A.; Guo, X.; Namekata, K.; Nakano, T.; Harada, T. Suppression of Oxidative Stress as Potential Therapeutic Approach for Normal Tension Glaucoma. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Ozdemir, G.; Tolun, F.I.; Gul, M.; Imrek, S. Retinal Oxidative Stress Induced by Intraocular Hypertension in Rats May be Ameliorated by Brimonidine Treatment and N-acetyl Cysteine Supplementation. Journal of Glaucoma 2009, 18, 662–665. [Google Scholar] [CrossRef]
- Yang, L.; Tan, P.; Zhou, W.; Zhu, X.; Cui, Y.; Zhu, L.; Feng, X.; Qi, H.; Zheng, J.; Gu, P.; et al. N-acetylcysteine protects against hypoxia mimetic-induced autophagy by targeting the HIF-1α pathway in retinal ganglion cells. Cell Mol Neurobiol 2012, 32, 1275–1285. [Google Scholar] [CrossRef]
- Sano, H.; Namekata, K.; Kimura, A.; Shitara, H.; Guo, X.; Harada, C.; Mitamura, Y.; Harada, T. Differential effects of N-acetylcysteine on retinal degeneration in two mouse models of normal tension glaucoma. Cell Death Dis 2019, 10, 75. [Google Scholar] [CrossRef]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin Protects against Neuron Death in In Vitro andIn Vivo Models of Parkinson's Disease. Journal of Neuroscience 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Deng, J.J.; Bai, Y.; Thornton, F.B.; Oddo, S. Rapamycin rescues TDP-43 mislocalization and the associated low molecular mass neurofilament instability. Journal of Biological Chemistry 2009, 284, 27416–27424. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Li, Z.; Jia, Y.; Zhuo, Y. Rapamycin is neuroprotective in a rat chronic hypertensive glaucoma model. PLoS One 2014, 9, e99719. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Kwon, Y.W.; Kondo, N.; Bai, J.; Masutani, H.; Nakamura, H.; Fujii, J.; Ohira, A.; Yodoi, J. Cytoprotective effects of geranylgeranylacetone against retinal photooxidative damage. J Neurosci 2005, 25, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shinmei, Y.; Dong, Y.; Inafuku, S.; Fukuhara, J.; Ando, R.; Kitaichi, N.; Kanda, A.; Tanaka, K.; Noda, K.; et al. Effect of geranylgeranylacetone on the protection of retinal ganglion cells in a mouse model of normal tension glaucoma. Heliyon 2016, 2, e00191. [Google Scholar] [CrossRef] [PubMed]
- Zibold, J.; von Livonius, B.; Kolarova, H.; Rudolph, G.; Priglinger, C.S.; Klopstock, T.; Catarino, C.B. Vitamin B12 in Leber hereditary optic neuropathy mutation carriers: a prospective cohort study. Orphanet J Rare Dis 2022, 17, 310. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Turnbull, D.M.; Chinnery, P.F. Leber hereditary optic neuropathy. J Med Genet 2002, 39, 162–169. [Google Scholar] [CrossRef]
- Mashima, Y.; Yamada, K.; Wakakura, M.; Kigasawa, K.; Kudoh, J.; Shimizu, N.; Oguchi, Y. Spectrum of pathogenic mitochondrial DNA mutations and clinical features in Japanese families with Leber's hereditary optic neuropathy. Current Eye Research 1998, 17, 403–408. [Google Scholar] [CrossRef]
- Mackey, D.A.; Oostra, R.J.; Rosenberg, T.; Nikoskelainen, E.; Bronte-Stewart, J.; Poulton, J.; Harding, A.E.; Govan, G.; Bolhuis, P.A.; Norby, S. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet 1996, 59, 481–485. [Google Scholar]
- Macmillan, C.; Johns, T.A.; Fu, K.; Shoubridge, E.A. Predominance of the T14484C Mutation in French-Canadian Families with Leber Hereditary Optic Neuropathy Is Due to a Founder Effect. The American Journal of Human Genetics 2000, 66, 332–335. [Google Scholar] [CrossRef]
- Laberge, A.-M.; Jomphe, M.; Houde, L.; Vézina, H.; Tremblay, M.; Desjardins, B.; Labuda, D.; St-Hilaire, M.; Macmillan, C.; Shoubridge, E.A.; et al. A “Fille du Roy” Introduced the T14484C Leber Hereditary Optic Neuropathy Mutation in French Canadians. The American Journal of Human Genetics 2005, 77, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Macmillan, C.; Kirkham, T.; Fu, K.; Allison, V.; Andermann, E.; Chitayat, D.; Fortier, D.; Gans, M.; Hare, H.; Quercia, N.; et al. Pedigree analysis of French Canadian families with T14484C Leber's hereditary optic neuropathy. Neurology 1998, 50, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Hage, R.; Vignal-Clermont, C. Leber Hereditary Optic Neuropathy: Review of Treatment and Management. Front Neurol 2021, 12, 651639. [Google Scholar] [CrossRef] [PubMed]
- Man, P.Y.W.; Turnbull, D.M.; Chinnery, P.F. Leber hereditary optic neuropathy. Journal of Medical Genetics 2002, 39, 162–169. [Google Scholar] [CrossRef]
- Hudson, G.; Keers, S.; Yu-Wai-Man, P.; Griffiths, P.; Huoponen, K.; Savontaus, M.L.; Nikoskelainen, E.; Zeviani, M.; Carrara, F.; Horvath, R.; et al. Identification of an X-chromosomal locus and haplotype modulating the phenotype of a mitochondrial DNA disorder. Am J Hum Genet 2005, 77, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.P.; Fingert, J.H.; Carelli, V.; Valentino, M.L.; King, T.M.; Daiger, S.P.; Salomao, S.R.; Berezovsky, A.; Belfort, R., Jr.; Braun, T.A.; et al. Evidence for a novel x-linked modifier locus for leber hereditary optic neuropathy. Ophthalmic Genet 2008, 29, 17–24. [Google Scholar] [CrossRef]
- Rabenstein, A.; Catarino, C.B.; Rampeltshammer, V.; Schindler, D.; Gallenmüller, C.; Priglinger, C.; Pogarell, O.; Rüther, T.; Klopstock, T. Smoking and alcohol, health-related quality of life and psychiatric comorbidities in Leber's Hereditary Optic Neuropathy mutation carriers: a prospective cohort study. Orphanet J Rare Dis 2021, 16, 127. [Google Scholar] [CrossRef]
- Pott, J.W.; Wong, K.H. Leber's hereditary optic neuropathy and vitamin B12 deficiency. Graefes Arch Clin Exp Ophthalmol 2006, 244, 1357–1359. [Google Scholar] [CrossRef]
- Chen, B.S.; Holzinger, E.; Taiel, M.; Yu-Wai-Man, P. The Impact of Leber Hereditary Optic Neuropathy on the Quality of Life of Patients and Their Relatives: A Qualitative Study. Journal of Neuro-Ophthalmology 2022, 42, 316–322. [Google Scholar] [CrossRef]
- Gong, J.; Cheung, S.; Fasso-Opie, A.; Galvin, O.; Moniz, L.S.; Earle, D.; Durham, T.; Menzo, J.; Li, N.; Duffy, S.; et al. The Impact of Inherited Retinal Diseases in the United States of America (US) and Canada from a Cost-of-Illness Perspective. Clin Ophthalmol 2021, 15, 2855–2866. [Google Scholar] [CrossRef]
- Amore, G.; Romagnoli, M.; Carbonelli, M.; Barboni, P.; Carelli, V.; La Morgia, C. Therapeutic Options in Hereditary Optic Neuropathies. Drugs 2021, 81, 57–86. [Google Scholar] [CrossRef]
- Carelli, V.; Carbonelli, M.; de Coo, I.F.; Kawasaki, A.; Klopstock, T.; Lagrèze, W.A.; La Morgia, C.; Newman, N.J.; Orssaud, C.; Pott, J.W.R.; et al. International Consensus Statement on the Clinical and Therapeutic Management of Leber Hereditary Optic Neuropathy. J Neuroophthalmol 2017, 37, 371–381. [Google Scholar] [CrossRef]
- Stramkauskaitė, A.; Povilaitytė, I.; Glebauskienė, B.; Liutkevičienė, R. Clinical Overview of Leber Hereditary Optic Neuropathy. Acta Med Litu 2022, 29, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Nikoskelainen, E.; Hoyt, W.F.; Nummelin, K. Ophthalmoscopic findings in Leber's hereditary optic neuropathy. II. The fundus findings in the affected family members. Arch Ophthalmol 1983, 101, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Lyseng-Williamson, K.A. Idebenone: A Review in Leber's Hereditary Optic Neuropathy. Drugs 2016, 76, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Jaber, S.; Polster, B.M. Idebenone and neuroprotection: antioxidant, pro-oxidant, or electron carrier? J Bioenerg Biomembr 2015, 47, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Gueven, N.; Ravishankar, P.; Eri, R.; Rybalka, E. Idebenone: When an antioxidant is not an antioxidant. Redox Biol 2021, 38, 101812. [Google Scholar] [CrossRef]
- Nagai, Y.; Yoshida, K.; Narumi, S.; Tanayama, S.; Nagaoka, A. Brain distribution of idebenone and its effect on local cerebral glucose utilization in rats. Arch Gerontol Geriatr 1989, 8, 257–272. [Google Scholar] [CrossRef]
- Torii, H.; Yoshida, K.; Kobayashi, T.; Tsukamoto, T.; Tanayama, S. Disposition of idebenone (CV-2619), a new cerebral metabolism improving agent, in rats and dogs. J Pharmacobiodyn 1985, 8, 457–467. [Google Scholar] [CrossRef]
- Heitz, F.D.; Erb, M.; Anklin, C.; Robay, D.; Pernet, V.; Gueven, N. Idebenone protects against retinal damage and loss of vision in a mouse model of Leber's hereditary optic neuropathy. PLoS One 2012, 7, e45182. [Google Scholar] [CrossRef]
- Montenegro, L.; Turnaturi, R.; Parenti, C.; Pasquinucci, L. Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials (Basel) 2018, 8. [Google Scholar] [CrossRef]
- Giorgio, V.; Schiavone, M.; Galber, C.; Carini, M.; Da Ros, T.; Petronilli, V.; Argenton, F.; Carelli, V.; Acosta Lopez, M.J.; Salviati, L.; et al. The idebenone metabolite QS10 restores electron transfer in complex I and coenzyme Q defects. Biochim Biophys Acta Bioenerg 2018, 1859, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.-Y.; Wang, A.-G.; Wei, Y.-H. Leber's hereditary optic neuropathy: A multifactorial disease. Progress in Retinal and Eye Research 2006, 25, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Trounce, I.A.; Jun, A.S.; Allen, J.C.; Wallace, D.C. Functional Analysis of Lymphoblast and Cybrid Mitochondria Containing the 3460, 11778, or 14484 Leber's Hereditary Optic Neuropathy Mitochondrial DNA Mutation*. Journal of Biological Chemistry 2000, 275, 39831–39836. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Rugolo, M.; Sgarbi, G.; Ghelli, A.; Zanna, C.; Baracca, A.; Lenaz, G.; Napoli, E.; Martinuzzi, A.; Solaini, G. Bioenergetics shapes cellular death pathways in Leber's hereditary optic neuropathy: a model of mitochondrial neurodegeneration. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2004, 1658, 172–179. [Google Scholar] [CrossRef]
- Yen, M.Y.; Kao, S.H.; Wang, A.G.; Wei, Y.H. Increased 8-hydroxy-2'-deoxyguanosine in leukocyte DNA in Leber's hereditary optic neuropathy. Invest Ophthalmol Vis Sci 2004, 45, 1688–1691. [Google Scholar] [CrossRef] [PubMed]
- Ghelli, A.; Porcelli, A.M.; Zanna, C.; Martinuzzi, A.; Carelli, V.; Rugolo, M. Protection against oxidant-induced apoptosis by exogenous glutathione in Leber hereditary optic neuropathy cybrids. Invest Ophthalmol Vis Sci 2008, 49, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Floreani, M.; Napoli, E.; Martinuzzi, A.; Pantano, G.; De Riva, V.; Trevisan, R.; Bisetto, E.; Valente, L.; Carelli, V.; Dabbeni-Sala, F. Antioxidant defences in cybrids harboring mtDNA mutations associated with Leber's hereditary optic neuropathy. Febs j 2005, 272, 1124–1135. [Google Scholar] [CrossRef]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc Natl Acad Sci U S A 2012, 109, 20065–20070. [Google Scholar] [CrossRef]
- Qi, X.; Sun, L.; Lewin, A.S.; Hauswirth, W.W.; Guy, J. The mutant human ND4 subunit of complex I induces optic neuropathy in the mouse. Invest Ophthalmol Vis Sci 2007, 48, 1–10. [Google Scholar] [CrossRef]
- Porcelli, A.M.; Angelin, A.; Ghelli, A.; Mariani, E.; Martinuzzi, A.; Carelli, V.; Petronilli, V.; Bernardi, P.; Rugolo, M. Respiratory complex I dysfunction due to mitochondrial DNA mutations shifts the voltage threshold for opening of the permeability transition pore toward resting levels. J Biol Chem 2009, 284, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- Barrientos, A.; Moraes, C.T. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem 1999, 274, 16188–16197. [Google Scholar] [CrossRef] [PubMed]
- Battisti, C.; Formichi, P.; Cardaioli, E.; Bianchi, S.; Mangiavacchi, P.; Tripodi, S.A.; Tosi, P.; Federico, A. Cell response to oxidative stress induced apoptosis in patients with Leber's hereditary optic neuropathy. J Neurol Neurosurg Psychiatry 2004, 75, 1731–1736. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, G.; Cortopassi, G. Oxidative stress in inherited mitochondrial diseases. Free Radic Biol Med 2015, 88, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Ghelli, A.; Zanna, C.; Porcelli, A.M.; Schapira, A.H.; Martinuzzi, A.; Carelli, V.; Rugolo, M. Leber's hereditary optic neuropathy (LHON) pathogenic mutations induce mitochondrial-dependent apoptotic death in transmitochondrial cells incubated with galactose medium. J Biol Chem 2003, 278, 4145–4150. [Google Scholar] [CrossRef]
- Danielson, S.R.; Wong, A.; Carelli, V.; Martinuzzi, A.; Schapira, A.H.; Cortopassi, G.A. Cells bearing mutations causing Leber's hereditary optic neuropathy are sensitized to Fas-Induced apoptosis. J Biol Chem 2002, 277, 5810–5815. [Google Scholar] [CrossRef]
- Ghelli, A.; Zanna, C.; Porcelli, A.M.; Schapira, A.H.V.; Martinuzzi, A.; Carelli, V.; Rugolo, M. Leber's Hereditary Optic Neuropathy (LHON) Pathogenic Mutations Induce Mitochondrial-dependent Apoptotic Death in Transmitochondrial Cells Incubated with Galactose Medium*. Journal of Biological Chemistry 2003, 278, 4145–4150. [Google Scholar] [CrossRef]
- Danielson, S.R.; Wong, A.; Carelli, V.; Martinuzzi, A.; Schapira, A.H.V.; Cortopassi, G.A. Cells Bearing Mutations Causing Leber's Hereditary Optic Neuropathy Are Sensitized to Fas-induced Apoptosis*. Journal of Biological Chemistry 2002, 277, 5810–5815. [Google Scholar] [CrossRef]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Martinuzzi, A.; Carelli, V.; Rugolo, M. Caspase-independent death of Leber’s hereditary optic neuropathy cybrids is driven by energetic failure and mediated by AIF and Endonuclease G. Apoptosis 2005, 10, 997–1007. [Google Scholar] [CrossRef]
- Ng, W.S.V.; Trigano, M.; Freeman, T.; Varrichio, C.; Kandaswamy, D.K.; Newland, B.; Brancale, A.; Rozanowska, M.; Votruba, M. New avenues for therapy in mitochondrial optic neuropathies. Therapeutic Advances in Rare Disease 2021, 2, 26330040211029037. [Google Scholar] [CrossRef]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab 2012, 105, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Sadun, A.A.; Chicani, C.F.; Ross-Cisneros, F.N.; Barboni, P.; Thoolen, M.; Shrader, W.D.; Kubis, K.; Carelli, V.; Miller, G. Effect of EPI-743 on the clinical course of the mitochondrial disease Leber hereditary optic neuropathy. Arch Neurol 2012, 69, 331–338. [Google Scholar] [CrossRef]
- Chicani, C.; Chu, E.; Ross-Cisneros, F.; Rockwell, S.; Murase, K.; Thoolen, M.; Miller, G.; Sadun, A. Treatment of Leber's hereditary optic neuropathy (LHON): Results using a novel quinone, EPI-743. Investigative Ophthalmology & Visual Science 2013, 54, 4574–4574. [Google Scholar]
- Pitceathly, R.D.S.; Keshavan, N.; Rahman, J.; Rahman, S. Moving towards clinical trials for mitochondrial diseases. J Inherit Metab Dis 2021, 44, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, B.; Ma, J.; Ge, J.; Wang, K. Protective effect of mitochondria-targeted peptide MTP-131 against oxidative stress-induced apoptosis in RGC-5 cells. Mol Med Rep 2017, 15, 2179–2185. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.S.; Kim, J.H.; Min, K.N.; Moon, J.A.; Roh, T.C.; Lee, M.J.; Lee, K.W.; Min, J.E.; Lee, Y.M. KL1333, a Novel NAD(+) Modulator, Improves Energy Metabolism and Mitochondrial Dysfunction in MELAS Fibroblasts. Front Neurol 2018, 9, 552. [Google Scholar] [CrossRef]
- Patel, H.R.; Margo, C.E. Pathology of Ischemic Optic Neuropathy. Arch Pathol Lab Med 2017, 141, 162–166. [Google Scholar] [CrossRef]
- Hayreh, S.S. Ischemic optic neuropathy. Prog Retin Eye Res 2009, 28, 34–62. [Google Scholar] [CrossRef]
- Hattenhauer, M.G.; Leavitt, J.A.; Hodge, D.O.; Grill, R.; Gray, D.T. Incidence of Nonarteritic Anteripr Ischemic Optic Neuropathy. American Journal of Ophthalmology 1997, 123, 103–107. [Google Scholar] [CrossRef]
- Chen, T.-W.; Wu, P.-Y.; Wen, Y.-T.; Desai, T.D.; Huang, C.-T.; Liu, P.-K.; Tsai, R.-K. Vitamin B3 Provides Neuroprotection via Antioxidative Stress in a Rat Model of Anterior Ischemic Optic Neuropathy. Antioxidants 2022, 11, 2422. [Google Scholar] [CrossRef]
- Winkler, A.; True, D. Giant Cell Arteritis: 2018 Review. Mo Med 2018, 115, 468–470. [Google Scholar] [PubMed]
- Bilton, E.J.; Mollan, S.P. Giant cell arteritis: reviewing the advancing diagnostics and management. Eye (Lond) 2023, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Smit, E.; Palmer, A.J.; Hewitt, A.W. Projected worldwide disease burden from giant cell arteritis by 2050. J Rheumatol 2015, 42, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Mackie, S.L.; Dejaco, C.; Appenzeller, S.; Camellino, D.; Duftner, C.; Gonzalez-Chiappe, S.; Mahr, A.; Mukhtyar, C.; Reynolds, G.; de Souza, A.W.S.; et al. British Society for Rheumatology guideline on diagnosis and treatment of giant cell arteritis. Rheumatology (Oxford) 2020, 59, e1–e23. [Google Scholar] [CrossRef]
- Danesh-Meyer, H.; Savino, P.J.; Gamble, G.G. Poor prognosis of visual outcome after visual loss from giant cell arteritis. Ophthalmology 2005, 112, 1098–1103. [Google Scholar] [CrossRef]
- Dinkin, M.; Johnson, E. One Giant Step for Giant Cell Arteritis: Updates in Diagnosis and Treatment. Curr Treat Options Neurol 2021, 23, 6. [Google Scholar] [CrossRef]
- Lyons, H.S.; Quick, V.; Sinclair, A.J.; Nagaraju, S.; Mollan, S.P. A new era for giant cell arteritis. Eye 2020, 34, 1013–1026. [Google Scholar] [CrossRef]
- Genovese, M.C.; McKay, J.D.; Nasonov, E.L.; Mysler, E.F.; da Silva, N.A.; Alecock, E.; Woodworth, T.; Gomez-Reino, J.J. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis & Rheumatism: Official Journal of the American College of Rheumatology 2008, 58, 2968–2980. [Google Scholar]
- Jones, G.; Sebba, A.; Gu, J.; Lowenstein, M.B.; Calvo, A.; Gomez-Reino, J.J.; Siri, D.A.; Tomšič, M.; Alecock, E.; Woodworth, T. Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Annals of the rheumatic diseases 2010, 69, 88–96. [Google Scholar] [CrossRef]
- Hellmich, B.; Águeda, A.F.; Monti, S.; Luqmani, R. Treatment of Giant Cell Arteritis and Takayasu Arteritis—Current and Future. Current Rheumatology Reports 2020, 22, 84. [Google Scholar] [CrossRef]
- Liu, B.; Yu, Y.; Liu, W.; Deng, T.; Xiang, D. Risk Factors for Non-arteritic Anterior Ischemic Optic Neuropathy: A Large Scale Meta-Analysis. Front Med (Lausanne) 2021, 8, 618353. [Google Scholar] [CrossRef]
- Hayreh, S.S.; Podhajsky, P.A.; Zimmerman, B. Nonarteritic Anterior Ischemic Optic Neuropathy: Time of Onset of Visual Loss. American Journal of Ophthalmology 1997, 124, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Hayreh, S.S.; Zimmerman, B. Visual Field Abnormalities in Nonarteritic Anterior Ischemic Optic Neuropathy: Their Pattern and Prevalence at Initial Examination. Archives of Ophthalmology 2005, 123, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.D.; Biousse, V.; Newman, N.J. Ischemic Optic Neuropathies: Current Concepts. Ann Indian Acad Neurol 2022, 25, S54–s58. [Google Scholar] [CrossRef] [PubMed]
- Hayreh, S.S.; Zimmerman, M.B. Non-arteritic anterior ischemic optic neuropathy: role of systemic corticosteroid therapy. Graefes Arch Clin Exp Ophthalmol 2008, 246, 1029–1046. [Google Scholar] [CrossRef]
- Berry, S.; Lin, W.V.; Sadaka, A.; Lee, A.G. Nonarteritic anterior ischemic optic neuropathy: cause, effect, and management. Eye Brain 2017, 9, 23–28. [Google Scholar] [CrossRef]
- Katz, D.M.; Trobe, J.D. Is there treatment for nonarteritic anterior ischemic optic neuropathy. Curr Opin Ophthalmol 2015, 26, 458–463. [Google Scholar] [CrossRef]
- Saxena, R.; Singh, D.; Sharma, M.; James, M.; Sharma, P.; Menon, V. Steroids versus No Steroids in Nonarteritic Anterior Ischemic Optic Neuropathy: A Randomized Controlled Trial. Ophthalmology 2018, 125, 1623–1627. [Google Scholar] [CrossRef]
- Goldenberg-Cohen, N.; Dadon-Bar-El, S.; Hasanreisoglu, M.; Avraham-Lubin, B.C.; Dratviman-Storobinsky, O.; Cohen, Y.; Weinberger, D. Possible neuroprotective effect of brimonidine in a mouse model of ischaemic optic neuropathy. Clin Exp Ophthalmol 2009, 37, 718–729. [Google Scholar] [CrossRef]
- Danylkova, N.O.; Alcala, S.R.; Pomeranz, H.D.; McLoon, L.K. Neuroprotective effects of brimonidine treatment in a rodent model of ischemic optic neuropathy. Exp Eye Res 2007, 84, 293–301. [Google Scholar] [CrossRef]
- Aktaş, Z.; Gürelik, G.; Akyürek, N.; Onol, M.; Hasanreisoğlu, B. Neuroprotective effect of topically applied brimonidine tartrate 0.2% in endothelin-1-induced optic nerve ischaemia model. Clin Exp Ophthalmol 2007, 35, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Liao, Y.J.; Goronzy, J.J. The Immunopathology of Giant Cell Arteritis: Diagnostic and Therapeutic Implications. Journal of Neuro-Ophthalmology 2012, 32, 259–265. [Google Scholar] [CrossRef]
- Bilton, E.J.; Mollan, S.P. Giant cell arteritis: reviewing the advancing diagnostics and management. Eye 2023. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Hashimoto, M. Aging-Related Vascular Inflammation: Giant Cell Arteritis and Neurological Disorders. Frontiers in Aging Neuroscience 2022, 14. [Google Scholar] [CrossRef]
- Cid, M.C.; Font, C.; Coll-Vinent, B.; Grau, J.M. Large vessel vasculitides. Current Opinion in Rheumatology 1998, 10. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.J.; Blin, M.G.; Song, J.; Wood, S.C.; Zhang, M.; Beard, D.A.; Goldstein, D.R. Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circulation Research 2020, 126, 298–314. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circulation Research 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Wang, L.; Ai, Z.; Khoyratty, T.; Zec, K.; Eames, H.L.; van Grinsven, E.; Hudak, A.; Morris, S.; Ahern, D.; Monaco, C.; et al. ROS-producing immature neutrophils in giant cell arteritis are linked to vascular pathologies. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Ianni, A.; Kumari, P.; Tarighi, S.; Argento, F.R.; Fini, E.; Emmi, G.; Bettiol, A.; Braun, T.; Prisco, D.; Fiorillo, C.; et al. An Insight into Giant Cell Arteritis Pathogenesis: Evidence for Oxidative Stress and SIRT1 Downregulation. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef]
- Ophir, A.; Berenshtein, E.; Kitrossky, N.; Berman, E.R.; Photiou, S.; Rothman, Z.; Chevion, M. Hydroxyl radical generation in the cat retina during reperfusion following ischemia. Exp Eye Res 1993, 57, 351–357. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Zhang, J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke 1996, 27, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Osborne, N.N.; Casson, R.J.; Wood, J.P.M.; Chidlow, G.; Graham, M.; Melena, J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Progress in Retinal and Eye Research 2004, 23, 91–147. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol Cell Physiol 2002, 282, C227–C241. [Google Scholar] [CrossRef] [PubMed]
- Aghai, Z.H.; Kumar, S.; Farhath, S.; Kumar, M.A.; Saslow, J.; Nakhla, T.; Eydelman, R.; Strande, L.; Stahl, G.; Hewitt, C.; et al. Dexamethasone Suppresses Expression of Nuclear Factor-kappaB in the Cells of Tracheobronchial Lavage Fluid in Premature Neonates with Respiratory Distress. Pediatric Research 2006, 59, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Birer, S.; Arda, H.; Kilic, D.; Baskol, G. Systemic oxidative stress in non-arteritic anterior ischemic optic neuropathy. Eye (Lond) 2019, 33, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, X.; Wu, X.; Jiang, L.; Ahsan, A.; Ma, S.; Xiao, Z.; Han, F.; Qin, Z.H.; Hu, W.; et al. Somatic autophagy of axonal mitochondria in ischemic neurons. J Cell Biol 2019, 218, 1891–1907. [Google Scholar] [CrossRef]
- Wan, W.; Peng, T.; Jin, X.; Li, Q.; Zhang, F.; Zheng, G.; Lv, Y.; Wan, G.; Zhu, Y. Glutathione-S-Transferase Deletions and Non-arteritic Anterior Ischemic Optic Neuropathy. Molecular Neurobiology 2016, 53, 2361–2367. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Milcarek, B.; Bosley, T.M. GSTM1 and GSTT1 deletion genotypes in various spontaneous optic neuropathies in Arabs. British Journal of Ophthalmology 2009, 93, 1101. [Google Scholar] [CrossRef]
- Bosley, T.M.; Abu-Amero, K.K.; Ozand, P.T. Mitochondrial DNA nucleotide changes in non-arteritic ischemic optic neuropathy. Neurology 2004, 63, 1305–1308. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, X.; Zhang, L. Negative regulation of inflammation by SIRT1. Pharmacol Res 2013, 67, 60–67. [Google Scholar] [CrossRef]
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid Redox Signal 2018, 28, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.-T.; Huang, C.-W.; Liu, C.-P.; Chen, C.-H.; Tu, C.-M.; Hwang, C.-S.; Chen, Y.-H.; Chen, W.-R.; Lin, K.-L.; Ho, Y.-C.; et al. Inhibition of Retinal Ganglion Cell Loss By a Novel ROCK Inhibitor (E212) in Ischemic Optic Nerve Injury Via Antioxidative and Anti-Inflammatory Actions. Investigative Ophthalmology & Visual Science 2021, 62, 21–21. [Google Scholar] [CrossRef]
- Chien, J.Y.; Lin, S.F.; Chou, Y.Y.; Huang, C.F.; Huang, S.P. Protective Effects of Oroxylin A on Retinal Ganglion Cells in Experimental Model of Anterior Ischemic Optic Neuropathy. Antioxidants (Basel) 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Nakamura, K.; Guo, X.; Kitaichi, N.; Mitamura, Y.; Yoshida, K.; Ohno, S.; Yoshida, H.; Harada, T. Neuroprotective effect of geranylgeranylacetone against ischemia-induced retinal injury. Mol Vis 2007, 13, 1601–1607. [Google Scholar] [PubMed]
- Zhang, L.; Xue, K.; Fan, P.; Chen, C.; Hu, J.; Huang, J.; Lu, W.; Xu, J.; Xu, S.; Ran, J.; et al. Geranylgeranylacetone-induced heat shock protein70 expression reduces retinal ischemia-reperfusion injury through PI3K/AKT/mTOR signaling. Exp Eye Res 2023, 229, 109416. [Google Scholar] [CrossRef]
- Lin, W.N.; Kapupara, K.; Wen, Y.T.; Chen, Y.H.; Pan, I.H.; Tsai, R.K. Haematococcus pluvialis-Derived Astaxanthin Is a Potential Neuroprotective Agent against Optic Nerve Ischemia. Mar Drugs 2020, 18. [Google Scholar] [CrossRef]
- Ji, K.; Li, Z.; Lei, Y.; Xu, W.; Ouyang, L.; He, T.; Xing, Y. Resveratrol attenuates retinal ganglion cell loss in a mouse model of retinal ischemia reperfusion injury via multiple pathways. Exp Eye Res 2021, 209, 108683. [Google Scholar] [CrossRef]
- Pang, Y.; Qin, M.; Hu, P.; Ji, K.; Xiao, R.; Sun, N.; Pan, X.; Zhang, X. Resveratrol protects retinal ganglion cells against ischemia induced damage by increasing Opa1 expression. Int J Mol Med 2020, 46, 1707–1720. [Google Scholar] [CrossRef]
- Ross, A.G.; Chaqour, B.; McDougald, D.S.; Dine, K.E.; Duong, T.T.; Shindler, R.E.; Yue, J.; Liu, T.; Shindler, K.S. Selective Upregulation of SIRT1 Expression in Retinal Ganglion Cells by AAV-Mediated Gene Delivery Increases Neuronal Cell Survival and Alleviates Axon Demyelination Associated with Optic Neuritis. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Abel, A.; McClelland, C.; Lee, M.S. Critical review: Typical and atypical optic neuritis. Surv Ophthalmol 2019, 64, 770–779. [Google Scholar] [CrossRef]
- Pau, D.; Al Zubidi, N.; Yalamanchili, S.; Plant, G.T.; Lee, A.G. Optic neuritis. Eye (Lond) 2011, 25, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol 2008, 65, 727–732. [CrossRef]
- Ebers, G.C. Optic neuritis and multiple sclerosis. Arch Neurol 1985, 42, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.L. Optic Neuritis. Continuum (Minneap Minn) 2019, 25, 1236–1264. [Google Scholar] [CrossRef] [PubMed]
- Aranda, M.L.; Narvaez, O.; Altschuler, F.; Calanni, J.S.; González Fleitas, M.F.; Sande, P.H.; Dorfman, D.; Concha, L.; Rosenstein, R.E. Chronobiotic effect of melatonin in experimental optic neuritis. Neuropharmacology 2021, 182, 108401. [Google Scholar] [CrossRef]
- Söderström, M.; Link, H.; Sun, J.B.; Fredrikson, S.; Wang, Z.Y.; Huang, W.X. Autoimmune T cell repertoire in optic neuritis and multiple sclerosis: T cells recognising multiple myelin proteins are accumulated in cerebrospinal fluid. J Neurol Neurosurg Psychiatry 1994, 57, 544–551. [Google Scholar] [CrossRef]
- YOUL, B.D.; TURANO, G.; MILLER, D.H.; TOWELL, A.D.; MACMANUS, D.G.; MOORE, S.G.; JONES, S.J.; BARRETT, G.; KENDALL, B.E.; MOSELEY, I.F.; et al. THE PATHOPHYSIOLOGY OF ACUTE OPTIC NEURITIS: AN ASSOCIATION OF GADOLINIUM LEAKAGE WITH CLINICAL AND ELECTROPHYSIOLOGICAL DEFICITS. Brain 1991, 114, 2437–2450. [Google Scholar] [CrossRef]
- Balcer, L.J. Clinical practice. Optic neuritis. N Engl J Med 2006, 354, 1273–1280. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Noro, T.; Harada, C.; Harada, T. Targeting Oxidative Stress for Treatment of Glaucoma and Optic Neuritis. Oxid Med Cell Longev 2017, 2017, 2817252. [Google Scholar] [CrossRef]
- Trip, S.A.; Schlottmann, P.G.; Jones, S.J.; Altmann, D.R.; Garway-Heath, D.F.; Thompson, A.J.; Plant, G.T.; Miller, D.H. Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann Neurol 2005, 58, 383–391. [Google Scholar] [CrossRef]
- Costello, F.; Coupland, S.; Hodge, W.; Lorello, G.R.; Koroluk, J.; Pan, Y.I.; Freedman, M.S.; Zackon, D.H.; Kardon, R.H. Quantifying axonal loss after optic neuritis with optical coherence tomography. Ann Neurol 2006, 59, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Keltner, J.L.; Johnson, C.A.; Cello, K.E.; Dontchev, M.; Gal, R.L.; Beck, R.W. Visual field profile of optic neuritis: a final follow-up report from the optic neuritis treatment trial from baseline through 15 years. Arch Ophthalmol 2010, 128, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.W.; Cleary, P.A.; Anderson, M.M., Jr.; Keltner, J.L.; Shults, W.T.; Kaufman, D.I.; Buckley, E.G.; Corbett, J.J.; Kupersmith, M.J.; Miller, N.R.; et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med 1992, 326, 581–588. [Google Scholar] [CrossRef]
- Gal, R.L.; Vedula, S.S.; Beck, R. Corticosteroids for treating optic neuritis. Cochrane Database Syst Rev 2015, 2015, Cd001430. [Google Scholar] [CrossRef]
- Tselis, A.; Perumal, J.; Caon, C.; Hreha, S.; Ching, W.; Din, M.; Van Stavern, G.; Khan, O. Treatment of corticosteroid refractory optic neuritis in multiple sclerosis patients with intravenous immunoglobulin. Eur J Neurol 2008, 15, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Roed, H.G.; Langkilde, A.; Sellebjerg, F.; Lauritzen, M.; Bang, P.; Mørup, A.; Frederiksen, J.L. A double-blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology 2005, 64, 804–810. [Google Scholar] [CrossRef]
- Noseworthy, J.H.; O'Brien, P.C.; Petterson, T.M.; Weis, J.; Stevens, L.; Peterson, W.K.; Sneve, D.; Cross, S.A.; Leavitt, J.A.; Auger, R.G.; et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology 2001, 56, 1514–1522. [Google Scholar] [CrossRef]
- Guy, J.; Ellis, E.A.; Hope, G.M.; Rao, N.A. Antioxidant enzyme suppression of demyelination in experimental optic neuritis. Current Eye Research 1989, 8, 467–477. [Google Scholar] [CrossRef]
- Guy, J.; Ellis, E.A.; Rao, N.A. Hydrogen Peroxide Localization in Experimental Optic Neuritis. Archives of Ophthalmology 1990, 108, 1614–1621. [Google Scholar] [CrossRef]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. Suppression of Mitochondrial Oxidative Stress Provides Long-term Neuroprotection in Experimental Optic Neuritis. Investigative Ophthalmology & Visual Science 2007, 48, 681–691. [Google Scholar] [CrossRef]
- Qi, X.; Sun, L.; Lewin, A.S.; Hauswirth, W.W.; Guy, J. Long-term Suppression of Neurodegeneration in Chronic Experimental Optic Neuritis: Antioxidant Gene Therapy. Investigative Ophthalmology & Visual Science 2007, 48, 5360–5370. [Google Scholar] [CrossRef]
- Vural, G.; Gümüşyayla, Ş.; Deniz, O.; Neşelioğlu, S.; Erel, Ö. Relationship between thiol-disulphide homeostasis and visual evoked potentials in patients with multiple sclerosis. Neurological Sciences 2019, 40, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Liang, X.M.; Zhang, X.L.; Ling, S.Q.; Yang, T.T.; Li, M.; Peng, F.H. Relationship between serum bilirubin levels and optic neuritis. Chin Med J (Engl) 2013, 126, 3307–3310. [Google Scholar] [PubMed]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid Med Cell Longev 2016, 2016, 5698931. [Google Scholar] [CrossRef]
- Plant, G.T.; Sibtain, N.A.; Thomas, D. Hyperacute corticosteroid treatment of optic neuritis at the onset of pain may prevent visual loss: a case series. Mult Scler Int 2011, 2011, 815068. [Google Scholar] [CrossRef]
- Wang, L.; Liu, K.; Tan, X.; Zhou, L.; Zhang, Y.; Liu, X.; Fu, Y.; Qiu, W.; Yang, H. Remedial Effect of Intravenous Cyclophosphamide in Corticosteroid-Refractory Patients in the Acute Phase of Neuromyelitis Optica Spectrum Disorder-Related Optic Neuritis. Front Neurol 2020, 11, 612097. [Google Scholar] [CrossRef]
- Zyla, K.; Larabee, C.M.; Georgescu, C.; Berkley, C.; Reyna, T.; Plafker, S.M. Dimethyl fumarate mitigates optic neuritis. Mol Vis 2019, 25, 446–461. [Google Scholar]
- Alhasani, R.H.; Biswas, L.; Tohari, A.M.; Zhou, X.; Reilly, J.; He, J.F.; Shu, X. Gypenosides protect retinal pigment epithelium cells from oxidative stress. Food Chem Toxicol 2018, 112, 76–85. [Google Scholar] [CrossRef]
- Zhang, H.-K.; Ye, Y.; Li, K.-J.; Zhao, Z.-n.; He, J.-F. Gypenosides Prevent H2O2-Induced Retinal Ganglion Cell Apoptosis by Concurrently Suppressing the Neuronal Oxidative Stress and Inflammatory Response. Journal of Molecular Neuroscience 2020, 70, 618–630. [Google Scholar] [CrossRef]
- Dietrich, M.; Helling, N.; Hilla, A.; Heskamp, A.; Issberner, A.; Hildebrandt, T.; Kohne, Z.; Küry, P.; Berndt, C.; Aktas, O.; et al. Early alpha-lipoic acid therapy protects from degeneration of the inner retinal layers and vision loss in an experimental autoimmune encephalomyelitis-optic neuritis model. J Neuroinflammation 2018, 15, 71. [Google Scholar] [CrossRef]
- Sarezky, D.; Raquib, A.R.; Dunaief, J.L.; Kim, B.J. Tolerability in the elderly population of high-dose alpha lipoic acid: a potential antioxidant therapy for the eye. Clinical Ophthalmology 2016, 10, 1899–1903. [Google Scholar] [CrossRef]
- Huntemann, N.; Rolfes, L.; Pawlitzki, M.; Ruck, T.; Pfeuffer, S.; Wiendl, H.; Meuth, S.G. Failed, Interrupted, or Inconclusive Trials on Neuroprotective and Neuroregenerative Treatment Strategies in Multiple Sclerosis: Update 2015-2020. Drugs 2021, 81, 1031–1063. [Google Scholar] [CrossRef]
- Falardeau, J.; Fryman, A.; Wanchu, R.; Marracci, G.H.; Mass, M.; Wooliscroft, L.; Bourdette, D.N.; Murchison, C.F.; Hills, W.L.; Yadav, V. Oral lipoic acid as a treatment for acute optic neuritis: a blinded, placebo controlled randomized trial. Mult Scler J Exp Transl Clin 2019, 5, 2055217319850193. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, M.; Zhao, S.; Tian, Y.; Zhang, C. Matrine promotes mitochondrial biosynthesis and reduces oxidative stress in experimental optic neuritis. Front Pharmacol 2022, 13, 936632. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Harada, C.; Namekata, K.; Matsuzawa, A.; Camps, M.; Ji, H.; Swinnen, D.; Jorand-Lebrun, C.; Muzerelle, M.; Vitte, P.A.; et al. Regulation of the severity of neuroinflammation and demyelination by TLR-ASK1-p38 pathway. EMBO Mol Med 2010, 2, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Harada, C.; Namekata, K.; Kimura, A.; Mitamura, Y.; Yoshida, H.; Matsumoto, Y.; Harada, T. Spermidine Alleviates Severity of Murine Experimental Autoimmune Encephalomyelitis. Investigative Ophthalmology & Visual Science 2011, 52, 2696–2703. [Google Scholar] [CrossRef]
- Luo, W.; Xu, H.; Xu, L.; Jiang, W.; Chen, C.; Chang, Y.; Liu, C.; Tian, Z.; Qiu, X.; Xie, C.; et al. Remyelination in neuromyelitis optica spectrum disorder is promoted by edaravone through mTORC1 signaling activation. Glia 2023, 71, 284–304. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, H.; Zhai, Q.; Li, H.; Wang, C.; Wang, Y. Traumatic optic neuropathy: a review of current studies. Neurosurg Rev 2022, 45, 1895–1913. [Google Scholar] [CrossRef]
- Sarkies, N. Traumatic optic neuropathy. Eye (Lond) 2004, 18, 1122–1125. [Google Scholar] [CrossRef]
- Atkins, E.J.; Newman, N.J.; Biousse, V. Post-traumatic visual loss. Rev Neurol Dis 2008, 5, 73–81. [Google Scholar]
- Walsh, F.B. Pathological-clinical correlations. I. Indirect trauma to the optic nerves and chiasm. II. Certain cerebral involvements associated with defective blood supply. Invest Ophthalmol 1966, 5, 433–449. [Google Scholar] [PubMed]
- Gross, C.E.; DeKock, J.R.; Panje, W.R.; Hershkowitz, N.; Newman, J. Evidence for orbital deformation that may contribute to monocular blindness following minor frontal head trauma. J Neurosurg 1981, 55, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Singman, E.; McCulley, T.; Wu, C.; Daphalapurkar, N. The Biomechanics of Indirect Traumatic Optic Neuropathy Using a Computational Head Model With a Biofidelic Orbit. Front Neurol 2020, 11, 346. [Google Scholar] [CrossRef] [PubMed]
- Carta, A.; Ferrigno, L.; Salvo, M.; Bianchi-Marzoli, S.; Boschi, A.; Carta, F. Visual prognosis after indirect traumatic optic neuropathy. J Neurol Neurosurg Psychiatry 2003, 74, 246–248. [Google Scholar] [CrossRef]
- Urolagin, S.B.; Kotrashetti, S.M.; Kale, T.P.; Balihallimath, L.J. Traumatic optic neuropathy after maxillofacial trauma: a review of 8 cases. J Oral Maxillofac Surg 2012, 70, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Steinsapir, K.D.; Goldberg, R.A. Traumatic Optic Neuropathy: An Evolving Understanding. American Journal of Ophthalmology 2011, 151, 928–933.e922. [Google Scholar] [CrossRef]
- Khan, R.S.; Ross, A.G.; Aravand, P.; Dine, K.; Selzer, E.B.; Shindler, K.S. RGC and Vision Loss From Traumatic Optic Neuropathy Induced by Repetitive Closed Head Trauma Is Dependent on Timing and Force of Impact. Transl Vis Sci Technol 2021, 10, 8. [Google Scholar] [CrossRef]
- Ryan, A.K.; Rich, W.; Reilly, M.A. Oxidative stress in the brain and retina after traumatic injury. Front Neurosci 2023, 17, 1021152. [Google Scholar] [CrossRef]
- Bond, W.S.; Rex, T.S. Evidence That Erythropoietin Modulates Neuroinflammation through Differential Action on Neurons, Astrocytes, and Microglia. Front Immunol 2014, 5, 523. [Google Scholar] [CrossRef]
- DeJulius, C.R.; Bernardo-Colón, A.; Naguib, S.; Backstrom, J.R.; Kavanaugh, T.; Gupta, M.K.; Duvall, C.L.; Rex, T.S. Microsphere antioxidant and sustained erythropoietin-R76E release functions cooperate to reduce traumatic optic neuropathy. J Control Release 2021, 329, 762–773. [Google Scholar] [CrossRef]
- Szymanski, C.R.; Chiha, W.; Morellini, N.; Cummins, N.; Bartlett, C.A.; O'Hare Doig, R.L.; Savigni, D.L.; Payne, S.C.; Harvey, A.R.; Dunlop, S.A.; et al. Paranode Abnormalities and Oxidative Stress in Optic Nerve Vulnerable to Secondary Degeneration: Modulation by 670 nm Light Treatment. PLoS One 2013, 8, e66448. [Google Scholar] [CrossRef] [PubMed]
- O'Hare Doig, R.L.; Bartlett, C.A.; Maghzal, G.J.; Lam, M.; Archer, M.; Stocker, R.; Fitzgerald, M. Reactive species and oxidative stress in optic nerve vulnerable to secondary degeneration. Exp Neurol 2014, 261, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Fatteh, N.; El-Sherbiny, N.M.; Naime, M.; Ibrahim, A.S.; El-Sherbini, A.M.; El-Shafey, S.A.; Khan, S.; Fulzele, S.; Gonzales, J.; et al. Potential role of A2A adenosine receptor in traumatic optic neuropathy. J Neuroimmunol 2013, 264, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Bernardo-Colón, A.; Vest, V.; Clark, A.; Cooper, M.L.; Calkins, D.J.; Harrison, F.E.; Rex, T.S. Antioxidants prevent inflammation and preserve the optic projection and visual function in experimental neurotrauma. Cell Death Dis 2018, 9, 1097. [Google Scholar] [CrossRef]
- Wells, J.; Kilburn, M.R.; Shaw, J.A.; Bartlett, C.A.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Early in vivo changes in calcium ions, oxidative stress markers, and ion channel immunoreactivity following partial injury to the optic nerve. J Neurosci Res 2012, 90, 606–618. [Google Scholar] [CrossRef]
- Cummins, N.; Bartlett, C.A.; Archer, M.; Bartlett, E.; Hemmi, J.M.; Harvey, A.R.; Dunlop, S.A.; Fitzgerald, M. Changes to mitochondrial ultrastructure in optic nerve vulnerable to secondary degeneration in vivo are limited by irradiation at 670 nm. BMC Neurosci 2013, 14, 98. [Google Scholar] [CrossRef]
- Maxwell, W.L.; McCreath, B.J.; Graham, D.I.; Gennarelli, T.A. Cytochemical evidence for redistribution of membrane pump calcium-ATPase and ecto-Ca-ATPase activity, and calcium influx in myelinated nerve fibres of the optic nerve after stretch injury. J Neurocytol 1995, 24, 925–942. [Google Scholar] [CrossRef]
- Cansler, S.M.; Evanson, N.K. Connecting endoplasmic reticulum and oxidative stress to retinal degeneration, TBI, and traumatic optic neuropathy. Journal of Neuroscience Research 2020, 98, 571–574. [Google Scholar] [CrossRef]
- Hu, Y.; Park, K.K.; Yang, L.; Wei, X.; Yang, Q.; Cho, K.S.; Thielen, P.; Lee, A.H.; Cartoni, R.; Glimcher, L.H.; et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron 2012, 73, 445–452. [Google Scholar] [CrossRef]
- Hylin, M.J.; Holden, R.C.; Smith, A.C.; Logsdon, A.F.; Qaiser, R.; Lucke-Wold, B.P. Juvenile Traumatic Brain Injury Results in Cognitive Deficits Associated with Impaired Endoplasmic Reticulum Stress and Early Tauopathy. Dev Neurosci 2018, 40, 175–188. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Nguyen, L.; Bailes, J.E.; Lee, J.M.; Robson, M.J.; Omalu, B.I.; Huber, J.D.; Rosen, C.L. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J Neurosurg 2016, 124, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, A.F.; Lucke-Wold, B.P.; Nguyen, L.; Matsumoto, R.R.; Turner, R.C.; Rosen, C.L.; Huber, J.D. Salubrinal reduces oxidative stress, neuroinflammation and impulsive-like behavior in a rodent model of traumatic brain injury. Brain Res 2016, 1643, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Behnammanesh, G.; Wu, Z.; Rong, R.; You, M.; Majid, A.S.A.; Ji, D. Echium amoenum L. Ethanol Extract Protects Retinal Ganglion Cell after Glutamate and Optic Nerve Crush Injury. Dis Markers 2022, 2022, 3631532. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.K.; Le, T.T.; Kim, K.A.; Kim, Y.J.; Lee, W.B.; Jung, S.H. Roots of Lithospermum erythrorhizon promotes retinal cell survival in optic nerve crush-induced retinal degeneration. Exp Eye Res 2021, 203, 108419. [Google Scholar] [CrossRef]
- Li, R.C.; Morris, M.W.; Lee, S.K.; Pouranfar, F.; Wang, Y.; Gozal, D. Neuroglobin protects PC12 cells against oxidative stress. Brain research 2008, 1190, 159–166. [Google Scholar] [CrossRef]
- Watanabe, S.; Takahashi, N.; Uchida, H.; Wakasugi, K. Human neuroglobin functions as an oxidative stress-responsive sensor for neuroprotection. Journal of Biological Chemistry 2012, 287, 30128–30138. [Google Scholar] [CrossRef]
- Sugitani, K.; Koriyama, Y.; Sera, M.; Arai, K.; Ogai, K.; Wakasugi, K. A novel function of neuroglobin for neuroregeneration in mice after optic nerve injury. Biochem Biophys Res Commun 2017, 493, 1254–1259. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Y.J. Carvedilol Promotes Retinal Ganglion Cell Survival Following Optic Nerve Injury via ASK1-p38 MAPK Pathway. CNS Neurol Disord Drug Targets 2019, 18, 695–704. [Google Scholar] [CrossRef]
- Liebscher, J. Chemistry of Polydopamine – Scope, Variation, and Limitation. European Journal of Organic Chemistry 2019, 2019, 4976–4994. [Google Scholar] [CrossRef]
- Mei, S.; Xu, X.; Priestley, R.D.; Lu, Y. Polydopamine-based nanoreactors: synthesis and applications in bioscience and energy materials. Chem Sci 2020, 11, 12269–12281. [Google Scholar] [CrossRef]
- Lou, X.; Hu, Y.; Zhang, H.; Liu, J.; Zhao, Y. Polydopamine nanoparticles attenuate retina ganglion cell degeneration and restore visual function after optic nerve injury. J Nanobiotechnology 2021, 19, 436. [Google Scholar] [CrossRef] [PubMed]
- Ezoulin, M.J.; Ombetta, J.-E.; Dutertre-Catella, H.; Warnet, J.-M.; Massicot, F. Antioxidative properties of galantamine on neuronal damage induced by hydrogen peroxide in SK–N–SH cells. Neurotoxicology 2008, 29, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Egea, J.; García, A.G.; López, M.G. Synergistic neuroprotective effect of combined low concentrations of galantamine and melatonin against oxidative stress in SH-SY5Y neuroblastoma cells. Journal of pineal research 2010, 49, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Yang, L.; Sameshima, H. Galantamine, an acetylcholinesterase inhibitor, reduces brain damage induced by hypoxia-ischemia in newborn rats. International Journal of Developmental Neuroscience 2014, 37, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Naguib, S.; Bernardo-Colón, A.; Cencer, C.; Gandra, N.; Rex, T.S. Galantamine protects against synaptic, axonal, and vision deficits in experimental neurotrauma. Neurobiol Dis 2020, 134, 104695. [Google Scholar] [CrossRef]
- Zuo, L.; Khan, R.S.; Lee, V.; Dine, K.; Wu, W.; Shindler, K.S. SIRT1 promotes RGC survival and delays loss of function following optic nerve crush. Invest Ophthalmol Vis Sci 2013, 54, 5097–5102. [Google Scholar] [CrossRef]
- Mou, Q.; Yao, K.; Ye, M.; Zhao, B.; Hu, Y.; Lou, X.; Li, H.; Zhang, H.; Zhao, Y. Modulation of Sirt1-mTORC1 Pathway in Microglia Attenuates Retinal Ganglion Cell Loss After Optic Nerve Injury. J Inflamm Res 2021, 14, 6857–6869. [Google Scholar] [CrossRef]
- Khong, J.J.; McNab, A.A.; Ebeling, P.R.; Craig, J.E.; Selva, D. Pathogenesis of thyroid eye disease: review and update on molecular mechanisms. Br J Ophthalmol 2016, 100, 142–150. [Google Scholar] [CrossRef]
- Dolman, P.J. Dysthyroid optic neuropathy: evaluation and management. J Endocrinol Invest 2021, 44, 421–429. [Google Scholar] [CrossRef]
- Giaconi, J.A.; Kazim, M.; Rho, T.; Pfaff, C. CT scan evidence of dysthyroid optic neuropathy. Ophthalmic Plast Reconstr Surg 2002, 18, 177–182. [Google Scholar] [CrossRef]
- Pelewicz-Sowa, M.; Miśkiewicz, P. Dysthyroid optic neuropathy: emerging treatment strategies. Journal of Endocrinological Investigation 2023, 46, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Khoo, T.K.; Bahn, R.S. Pathogenesis of Graves' ophthalmopathy: the role of autoantibodies. Thyroid 2007, 17, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Pappa, A.; Jackson, P.; Stone, J.; Munro, P.; Fells, P.; Pennock, C.; Lightman, S. An ultrastructural and systemic analysis of glycosaminoglycans in thyroid-associated ophthalmopathy. Eye (Lond) 1998, 12 ( Pt 2) Pt 2, 237–244. [Google Scholar] [CrossRef]
- Bednarek, J.; Wysocki, H.; Sowiński, J. Peripheral parameters of oxidative stress in patients with infiltrative Graves' ophthalmopathy treated with corticosteroids. Immunol Lett 2004, 93, 227–232. [Google Scholar] [CrossRef]
- Tsai, C.C.; Wu, S.B.; Cheng, C.Y.; Kao, S.C.; Kau, H.C.; Chiou, S.H.; Hsu, W.M.; Wei, Y.H. Increased oxidative DNA damage, lipid peroxidation, and reactive oxygen species in cultured orbital fibroblasts from patients with Graves' ophthalmopathy: evidence that oxidative stress has a role in this disorder. Eye (Lond) 2010, 24, 1520–1525. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.; Li, Y.; Ji, Y.S.; Yoon, K.C. Oxidative stress markers in tears of patients with Graves’ orbitopathy and their correlation with clinical activity score. BMC Ophthalmology 2018, 18, 303. [Google Scholar] [CrossRef]
- Tsai, C.C.; Cheng, C.Y.; Liu, C.Y.; Kao, S.C.; Kau, H.C.; Hsu, W.M.; Wei, Y.H. Oxidative stress in patients with Graves' ophthalmopathy: relationship between oxidative DNA damage and clinical evolution. Eye (Lond) 2009, 23, 1725–1730. [Google Scholar] [CrossRef]
- Kau, H.C.; Wu, S.B.; Tsai, C.C.; Liu, C.J.; Wei, Y.H. Cigarette Smoke Extract-Induced Oxidative Stress and Fibrosis-Related Genes Expression in Orbital Fibroblasts from Patients with Graves' Ophthalmopathy. Oxid Med Cell Longev 2016, 2016, 4676289. [Google Scholar] [CrossRef]
- Burch, H.B.; Lahiri, S.; Bahn, R.S.; Barnes, S. Superoxide radical production stimulates retroocular fibroblast proliferation in Graves' ophthalmopathy. Exp Eye Res 1997, 65, 311–316. [Google Scholar] [CrossRef]
- Tsai, C.C.; Wu, S.B.; Kao, S.C.; Kau, H.C.; Lee, F.L.; Wei, Y.H. The protective effect of antioxidants on orbital fibroblasts from patients with Graves' ophthalmopathy in response to oxidative stress. Mol Vis 2013, 19, 927–934. [Google Scholar]
- Hou, T.Y.; Wu, S.B.; Kau, H.C.; Tsai, C.C. The Role of Oxidative Stress and Therapeutic Potential of Antioxidants in Graves' Ophthalmopathy. Biomedicines 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Duntas, L.H. Selenium and inflammation: underlying anti-inflammatory mechanisms. Horm Metab Res 2009, 41, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Rotondo Dottore, G.; Leo, M.; Casini, G.; Latrofa, F.; Cestari, L.; Sellari-Franceschini, S.; Nardi, M.; Vitti, P.; Marcocci, C.; Marinò, M. Antioxidant Actions of Selenium in Orbital Fibroblasts: A Basis for the Effects of Selenium in Graves' Orbitopathy. Thyroid 2017, 27, 271–278. [Google Scholar] [CrossRef]
- Kim, B.Y.; Jang, S.Y.; Choi, D.H.; Jung, C.H.; Mok, J.O.; Kim, C.H. Anti-inflammatory and Antioxidant Effects of Selenium on Orbital Fibroblasts of Patients With Graves Ophthalmopathy. Ophthalmic Plast Reconstr Surg 2021, 37, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Marcocci, C.; Kahaly, G.J.; Krassas, G.E.; Bartalena, L.; Prummel, M.; Stahl, M.; Altea, M.A.; Nardi, M.; Pitz, S.; Boboridis, K.; et al. Selenium and the course of mild Graves' orbitopathy. N Engl J Med 2011, 364, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Bartalena, L.; Baldeschi, L.; Boboridis, K.; Eckstein, A.; Kahaly, G.J.; Marcocci, C.; Perros, P.; Salvi, M.; Wiersinga, W.M. The 2016 European Thyroid Association/European Group on Graves' Orbitopathy Guidelines for the Management of Graves' Orbitopathy. Eur Thyroid J 2016, 5, 9–26. [Google Scholar] [CrossRef]
- Lanzolla, G.; Marinò, M.; Marcocci, C. Selenium in the Treatment of Graves' Hyperthyroidism and Eye Disease. Front Endocrinol (Lausanne) 2020, 11, 608428. [Google Scholar] [CrossRef]
- Xu, D.; Hu, M.J.; Wang, Y.Q.; Cui, Y.L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24. [Google Scholar] [CrossRef]
- Yoon, J.S.; Lee, H.J.; Chae, M.K.; Lee, S.Y.; Lee, E.J. Cigarette smoke extract-induced adipogenesis in Graves' orbital fibroblasts is inhibited by quercetin via reduction in oxidative stress. J Endocrinol 2013, 216, 145–156. [Google Scholar] [CrossRef]
- Yoon, J.S.; Lee, H.J.; Choi, S.H.; Chang, E.J.; Lee, S.Y.; Lee, E.J. Quercetin inhibits IL-1β-induced inflammation, hyaluronan production and adipogenesis in orbital fibroblasts from Graves' orbitopathy. PLoS One 2011, 6, e26261. [Google Scholar] [CrossRef] [PubMed]
- Rotondo Dottore, G.; Ionni, I.; Menconi, F.; Casini, G.; Sellari-Franceschini, S.; Nardi, M.; Vitti, P.; Marcocci, C.; Marinò, M. Action of three bioavailable antioxidants in orbital fibroblasts from patients with Graves' orbitopathy (GO): a new frontier for GO treatment? J Endocrinol Invest 2018, 41, 193–201. [Google Scholar] [CrossRef]
- Rotondo Dottore, G.; Ionni, I.; Menconi, F.; Casini, G.; Sellari-Franceschini, S.; Nardi, M.; Vitti, P.; Marcocci, C.; Marinò, M. Antioxidant effects of β-carotene, but not of retinol and vitamin E, in orbital fibroblasts from patients with Graves' orbitopathy (GO). J Endocrinol Invest 2018, 41, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chang, T.C.; Kao, S.C.; Kuo, Y.F.; Chien, L.F. Pentoxifylline inhibits the proliferation and glycosaminoglycan synthesis of cultured fibroblasts derived from patients with Graves' ophthalmopathy and pretibial myxoedema. Acta Endocrinol (Copenh) 1993, 129, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Balazs, C.; Kiss, E.; Vamos, A.; Molnar, I.; Farid, N.R. Beneficial effect of pentoxifylline on thyroid associated ophthalmopathy (TAO)*: a pilot study. J Clin Endocrinol Metab 1997, 82, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Finamor, F.E.; Martins, J.R.; Nakanami, D.; Paiva, E.R.; Manso, P.G.; Furlanetto, R.P. Pentoxifylline (PTX)--an alternative treatment in Graves' ophthalmopathy (inactive phase): assessment by a disease specific quality of life questionnaire and by exophthalmometry in a prospective randomized trial. Eur J Ophthalmol 2004, 14, 277–283. [Google Scholar] [CrossRef]
- Kim, C.Y.; Lee, H.J.; Chae, M.K.; Byun, J.W.; Lee, E.J.; Yoon, J.S. Therapeutic Effect of Resveratrol on Oxidative Stress in Graves' Orbitopathy Orbital Fibroblasts. Invest Ophthalmol Vis Sci 2015, 56, 6352–6361. [Google Scholar] [CrossRef]
- Bartalena, L.; Martino, E.; Marcocci, C.; Bogazzi, F.; Panicucci, M.; Velluzzi, F.; Loviselli, A.; Pinchera, A. More on smoking habits and Graves' ophthalmopathy. J Endocrinol Invest 1989, 12, 733–737. [Google Scholar] [CrossRef]
- Bouzas, E.A.; Karadimas, P.; Mastorakos, G.; Koutras, D.A. Antioxidant agents in the treatment of Graves' ophthalmopathy. Am J Ophthalmol 2000, 129, 618–622. [Google Scholar] [CrossRef]
- Chandran, G.; Sirajudeen, K.N.; Yusoff, N.S.; Swamy, M.; Samarendra, M.S. Effect of the antihypertensive drug enalapril on oxidative stress markers and antioxidant enzymes in kidney of spontaneously hypertensive rat. Oxid Med Cell Longev 2014, 2014, 608512. [Google Scholar] [CrossRef]
- de Cavanagh, E.M.; Inserra, F.; Ferder, L.; Fraga, C.G. Enalapril and captopril enhance glutathione-dependent antioxidant defenses in mouse tissues. Am J Physiol Regul Integr Comp Physiol 2000, 278, R572–R577. [Google Scholar] [CrossRef] [PubMed]
- Botta, R.; Lisi, S.; Marcocci, C.; Sellari-Franceschini, S.; Rocchi, R.; Latrofa, F.; Menconi, F.; Altea, M.A.; Leo, M.; Sisti, E.; et al. Enalapril reduces proliferation and hyaluronic acid release in orbital fibroblasts. Thyroid 2013, 23, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Ataabadi, G.; Dabbaghmanesh, M.H.; Owji, N.; Bakhshayeshkaram, M.; Montazeri-Najafabady, N. Clinical Features of Graves' Ophthalmopathy and Impact of Enalapril on the Course of Mild Graves' Ophthalmopathy: A Pilot Study. Endocr Metab Immune Disord Drug Targets 2020, 20, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Rachwani Anil, R.; Rocha-de-Lossada, C.; Zamorano Martín, F.; Santos Ortega, A.; Luque Aranda, G.; España Contreras, M.; Escudero Gómez, J. Infiltrative optic neuropathy as a relapse of acute lymphoblastic leukemia. J Fr Ophtalmol 2021, 44, 293–295. [Google Scholar] [CrossRef]
- Kuan, H.C.; Mustapha, M.; Oli Mohamed, S.; Abdul Aziz, R.A.; Loh, C.K.; Mohammed, F.; Naffi, A.A.; Othman, O.; Nasaruddin, R.A.; Alias, H. Isolated Infiltrative Optic Neuropathy in an Acute Lymphoblastic Leukemia Relapse. Cureus 2022, 14, e25625. [Google Scholar] [CrossRef]
- Myers, K.A.; Nikolic, A.; Romanchuk, K.; Weis, E.; Brundler, M.A.; Lafay-Cousin, L.; Costello, F. Optic neuropathy in the context of leukemia or lymphoma: diagnostic approach to a neuro-oncologic emergency. Neurooncol Pract 2017, 4, 60–66. [Google Scholar] [CrossRef]
- Jain, R.; Trehan, A.; Singh, R.; Srinivasan, R.; Dogra, M.; Bansal, D.; Marwaha, R.K. Unusual sites of relapse in pre-B acute lymphoblastic leukemia. J Pediatr Hematol Oncol 2014, 36, e506–e508. [Google Scholar] [CrossRef]
- Currie, J.N.; Lessell, S.; Lessell, I.M.; Weiss, J.S.; Albert, D.M.; Benson, E.M. Optic neuropathy in chronic lymphocytic leukemia. Arch Ophthalmol 1988, 106, 654–660. [Google Scholar] [CrossRef]
- Takkar, A.; Naheed, D.; Dogra, M.; Goyal, M.K.; Singh, R.; Gupta, N.; Gupta, K.; Mittal, B.R.; Lal, V. Infiltrative Optic Neuropathies: Opening Doors to Sinister Pathologies. Neuroophthalmology 2017, 41, 279–283. [Google Scholar] [CrossRef]
- Takahashi, T.; Oda, Y.; Isayama, Y. Leukemic optic neuropathy. Ophthalmologica 1982, 185, 37–45. [Google Scholar] [CrossRef]
- Soares, M.d.F.; Braga, F.T.; Rocha, A.J.d.; Lederman, H.M. Optic nerve infiltration by acute lymphoblastic leukemia: MRI contribution. Pediatric Radiology 2005, 35, 799–802. [Google Scholar] [CrossRef]
- Vegunta, S.; Patel, B.C. Optic Nerve Coloboma. In StatPearls, StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.: Treasure Island (FL), 2022.
- Saffren, B.D.; Yassin, S.H.; Geddie, B.E.; de Faber, J.; Blieden, L.S.; Bhate, M.; Gamio, S.; Rutar, T.; Levin, A.V. Optic Nerve Aplasia. J Neuroophthalmol 2022, 42, e140–e146. [Google Scholar] [CrossRef] [PubMed]
- Skriapa Manta, A.; Olsson, M.; Ek, U.; Wickström, R.; Teär Fahnehjelm, K. Optic Disc Coloboma in children - prevalence, clinical characteristics and associated morbidity. Acta Ophthalmol 2019, 97, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Amador-Patarroyo, M.J.; Pérez-Rueda, M.A.; Tellez, C.H. Congenital anomalies of the optic nerve. Saudi J Ophthalmol 2015, 29, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Margolin, E.; Blair, K.; Shemesh, A. Toxic and Nutritional Optic Neuropathy. In StatPearls, StatPearls Publishing Copyright © 2022, StatPearls Publishing LLC.: Treasure Island (FL), 2022.
- Grzybowski, A.; Zülsdorff, M.; Wilhelm, H.; Tonagel, F. Toxic optic neuropathies: an updated review. Acta Ophthalmol 2015, 93, 402–410. [Google Scholar] [CrossRef]
- Kovač, L.; Volk, M.; Šuštar Habjan, M.; Hawlina, M. Oxidative Stress in Antibiotic Toxic Optic Neuropathy Mimicking Acute LHON in a Patient with Exacerbation of Cystic Fibrosis. Stresses 2023, 3, 387–396. [Google Scholar] [CrossRef]
- Spillane, J.D. Nutritional disorders of the nervous system in the Middle East. Proc R Soc Med 1946, 39, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Epidemic Neuropathy—Cuba, 1991-1994. JAMA 1994, 271, 1154–1156. [CrossRef]
- Plant, G.T.; Dolin, P.; Mohamed, A.A.; Mlingi, N. Confirmation that neither cyanide intoxication nor mutations commonly associated with Leber's Hereditary Optic Neuropathy are implicated in Tanzanian Epidemic Optic Neuropathy. J Neurol Sci 1997, 152, 107–108. [Google Scholar] [CrossRef]
- Baj, J.; Forma, A.; Kobak, J.; Tyczyńska, M.; Dudek, I.; Maani, A.; Teresiński, G.; Buszewicz, G.; Januszewski, J.; Flieger, J. Toxic and Nutritional Optic Neuropathies-An Updated Mini-Review. Int J Environ Res Public Health 2022, 19. [Google Scholar] [CrossRef]
- Margolin, E.; Blair, K.; Shemesh, A. Toxic and Nutritional Optic Neuropathy. In StatPearls, StatPearls Publishing Copyright © 2023, StatPearls Publishing LLC.: Treasure Island (FL), 2023.
- Yu, J.J.; Lee, D.H.; Gallagher, S.P.; Kenney, M.C.; Boisvert, C.J. Mitochondrial Impairment in Antibiotic Induced Toxic Optic Neuropathies. Current Eye Research 2018, 43, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Devalia, V.; Hamilton, M.S.; Molloy, A.M. Guidelines for the diagnosis and treatment of cobalamin and folate disorders. Br J Haematol 2014, 166, 496–513. [Google Scholar] [CrossRef] [PubMed]
- Langan, R.C.; Goodbred, A.J. Vitamin B12 Deficiency: Recognition and Management. Am Fam Physician 2017, 96, 384–389. [Google Scholar] [PubMed]
- Grzybowski, A.; Zülsdorff, M.; Wilhelm, H.; Tonagel, F. Toxic optic neuropathies: an updated review. Acta Ophthalmologica 2015, 93, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Pakdel, F.; Sanjari, M.S.; Naderi, A.; Pirmarzdashti, N.; Haghighi, A.; Kashkouli, M.B. Erythropoietin in Treatment of Methanol Optic Neuropathy. J Neuroophthalmol 2018, 38, 167–171. [Google Scholar] [CrossRef]
- Pakravan, M.; Esfandiari, H.; Sanjari, N.; Ghahari, E. Erythropoietin as an adjunctive treatment for methanol-induced toxic optic neuropathy. Am J Drug Alcohol Abuse 2016, 42, 633–639. [Google Scholar] [CrossRef]
- Fang, C.E.H.; Guo, L.; Hill, D.; Yap, T.E.; Cordeiro, M.F. Neuroprotective Strategies in Glaucoma - Translation to Clinical Trials. OBM Neurobiology 2020, 04, 062. [Google Scholar] [CrossRef]
- Wubben, T.J.; Besirli, C.G.; Johnson, M.W.; Zacks, D.N. Retinal Neuroprotection: Overcoming the Translational Roadblocks. American Journal of Ophthalmology 2018, 192, xv–xxii. [Google Scholar] [CrossRef]
- Almasieh, M.; Levin, L.A. Neuroprotection in Glaucoma: Animal Models and Clinical Trials. Annual Review of Vision Science 2017, 3, 91–120. [Google Scholar] [CrossRef]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef]
- Normando, E.M.; Yap, T.E.; Maddison, J.; Miodragovic, S.; Bonetti, P.; Almonte, M.; Mohammad, N.G.; Ameen, S.; Crawley, L.; Ahmed, F.; et al. A CNN-aided method to predict glaucoma progression using DARC (Detection of Apoptosing Retinal Cells). Expert Review of Molecular Diagnostics 2020, 20, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, D.L.; Mehta, C. Adaptive Designs for Clinical Trials. New England Journal of Medicine 2016, 375, 65–74. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Prevalence (per 100,000) in some of the most frequent optic neuropathies. LHON: Leber’s hereditary optic neuropathy; ON: optic neuritis; NA-AION: non-arteritic anterior ischemic optic neuropathy; A-AION: arteritic anterior ischemic optic neuropathy; GCA: giant cell arteritis. *We used a y-axis break in consideration of the remarkably higher prevalence of glaucoma compared to all other optic nerve disorders. **LHON prevalence in case of incomplete penetrance is meaningfully higher than in complete penetrance, due to the high frequency of the variant mutant carriers.
Figure 1.
Prevalence (per 100,000) in some of the most frequent optic neuropathies. LHON: Leber’s hereditary optic neuropathy; ON: optic neuritis; NA-AION: non-arteritic anterior ischemic optic neuropathy; A-AION: arteritic anterior ischemic optic neuropathy; GCA: giant cell arteritis. *We used a y-axis break in consideration of the remarkably higher prevalence of glaucoma compared to all other optic nerve disorders. **LHON prevalence in case of incomplete penetrance is meaningfully higher than in complete penetrance, due to the high frequency of the variant mutant carriers.
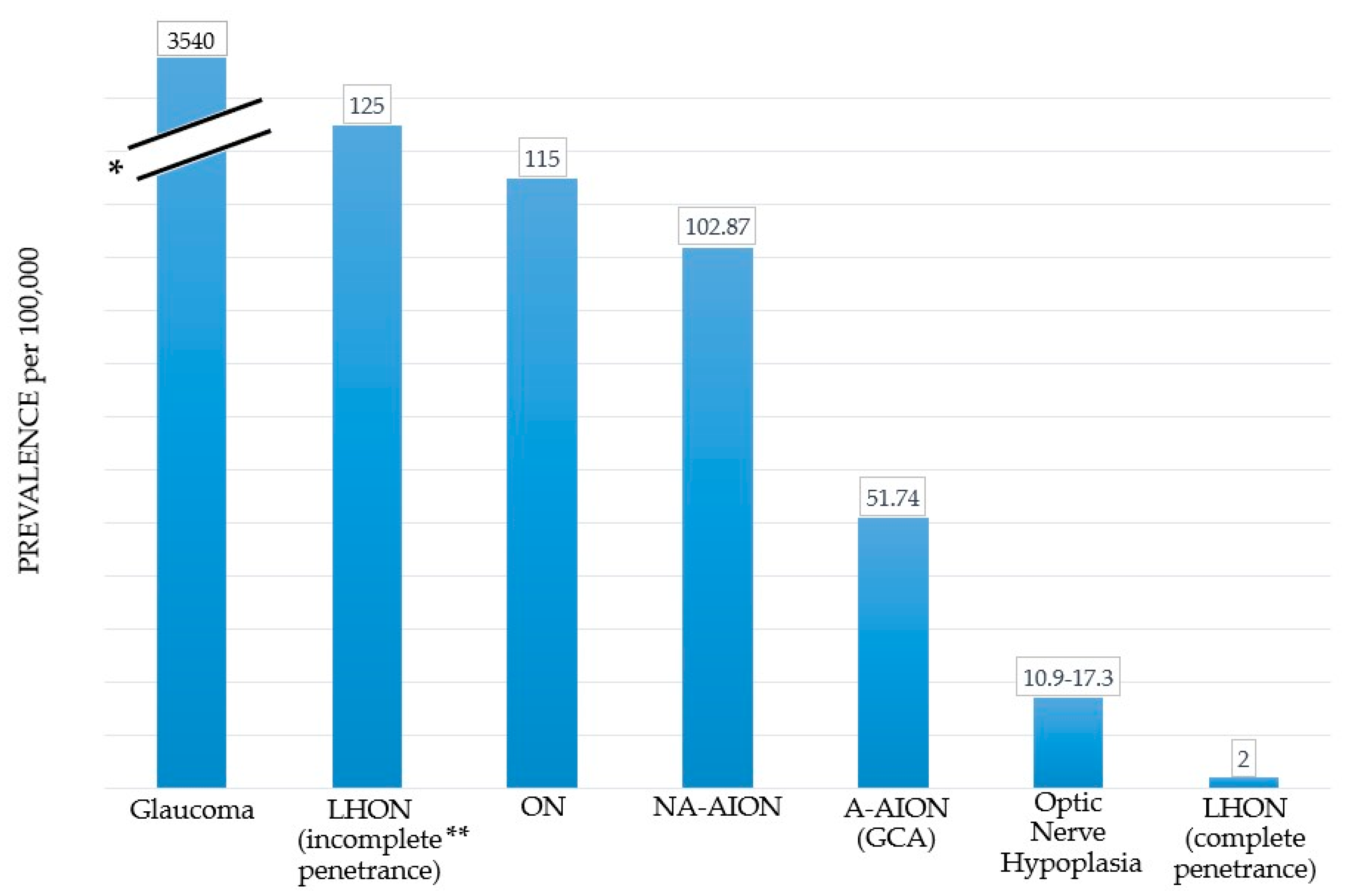
Figure 2.
Anatomy and perfusion of the visual pathway. LGN: lateral geniculate nucleus.
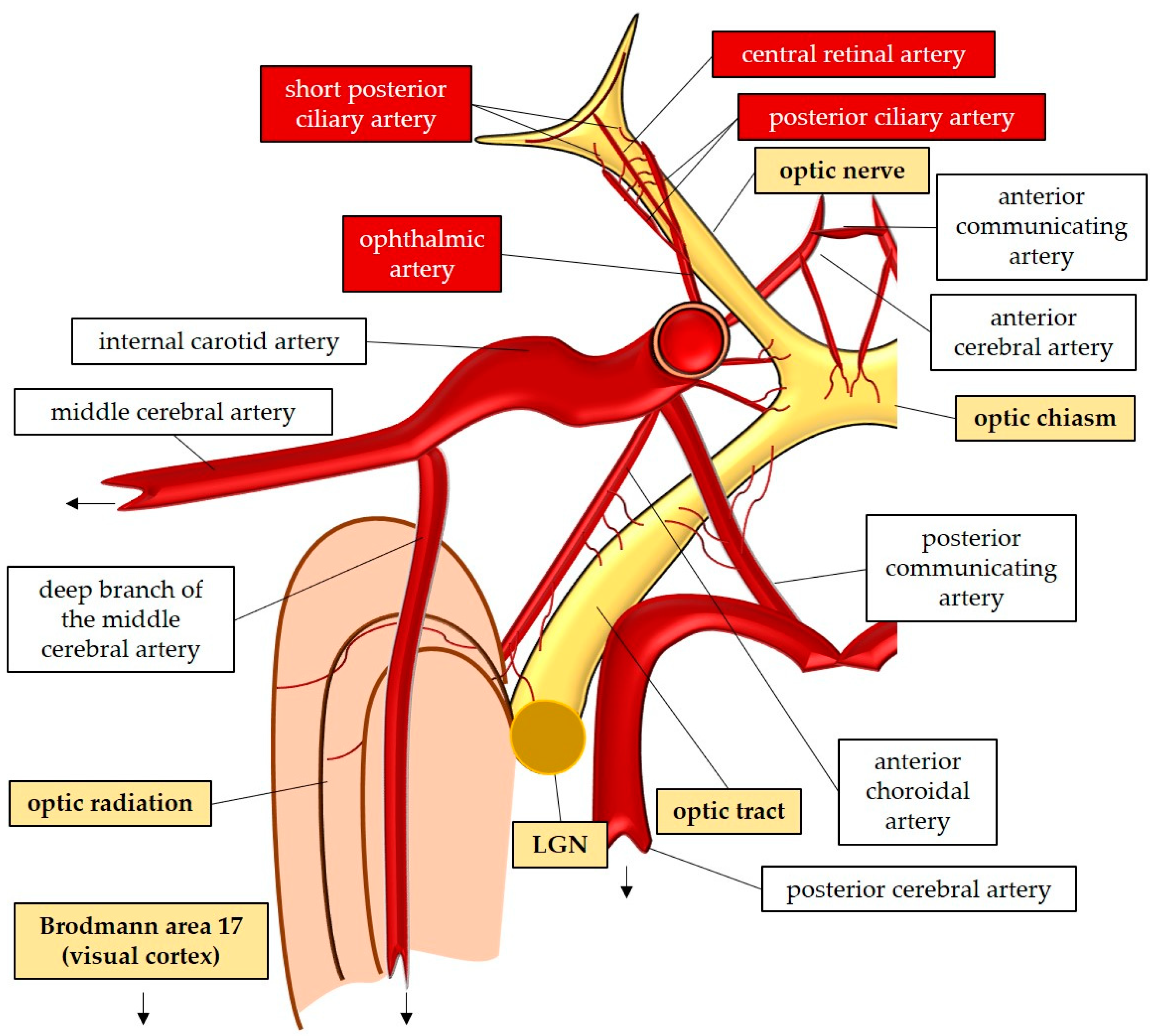
Figure 3.
Model representing the ROS-impact on the retina and on the optic nerve. ROS: reactive oxidative species; NOX2: NADPH Oxidase type 2; XO: Xanthine Oxidase; eNOS: endothelial nitric oxide synthase; RGC: retinal ganglion cell; RNFL: retinal nerve fiber layer; RPE: retinal pigment epithelium; BM: Brunch’s membrane.
Figure 3.
Model representing the ROS-impact on the retina and on the optic nerve. ROS: reactive oxidative species; NOX2: NADPH Oxidase type 2; XO: Xanthine Oxidase; eNOS: endothelial nitric oxide synthase; RGC: retinal ganglion cell; RNFL: retinal nerve fiber layer; RPE: retinal pigment epithelium; BM: Brunch’s membrane.
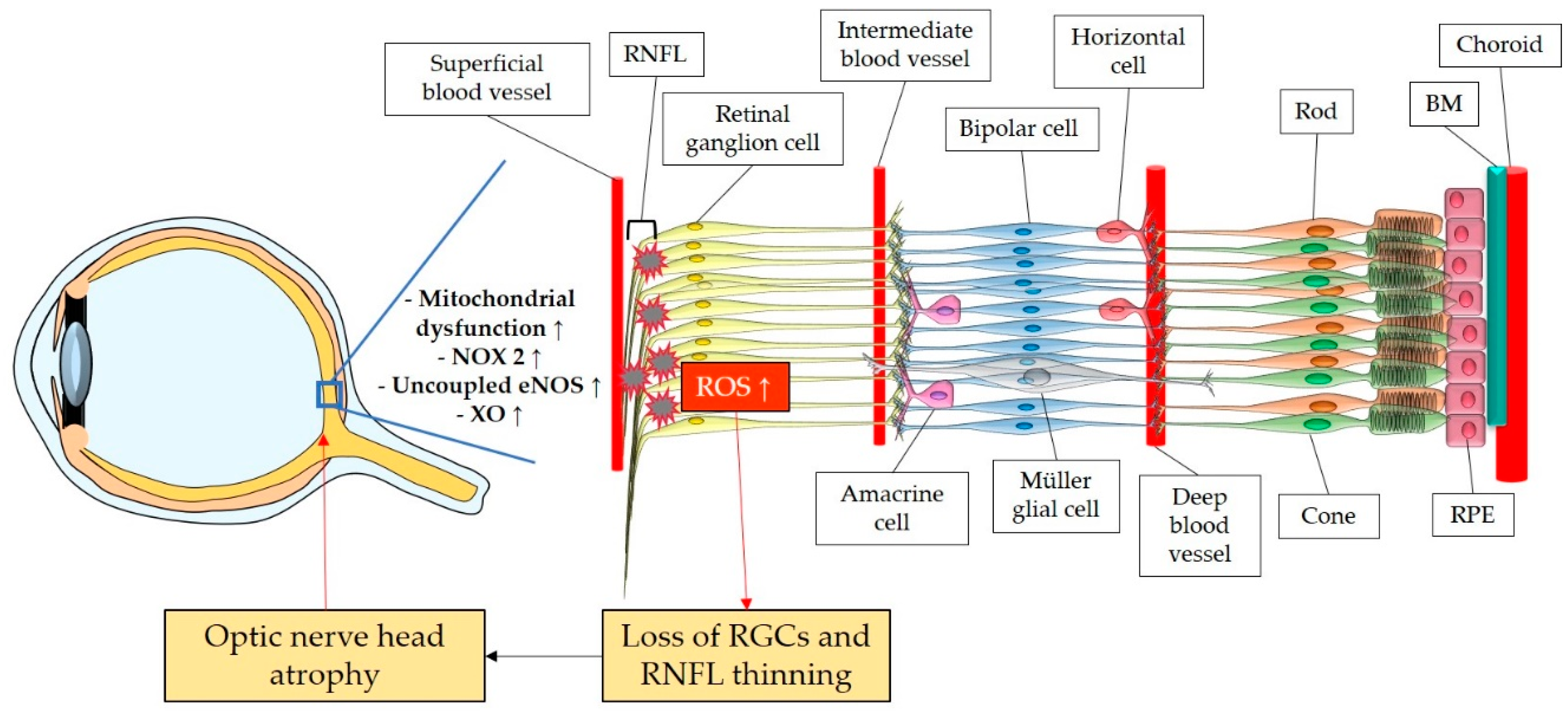
Figure 4.
Anatomic overview of aqueous humor outflow, with focus on the drainage system through the trabecular meshwork and the Schlemm’s canal, to the superficial veins.
Figure 4.
Anatomic overview of aqueous humor outflow, with focus on the drainage system through the trabecular meshwork and the Schlemm’s canal, to the superficial veins.
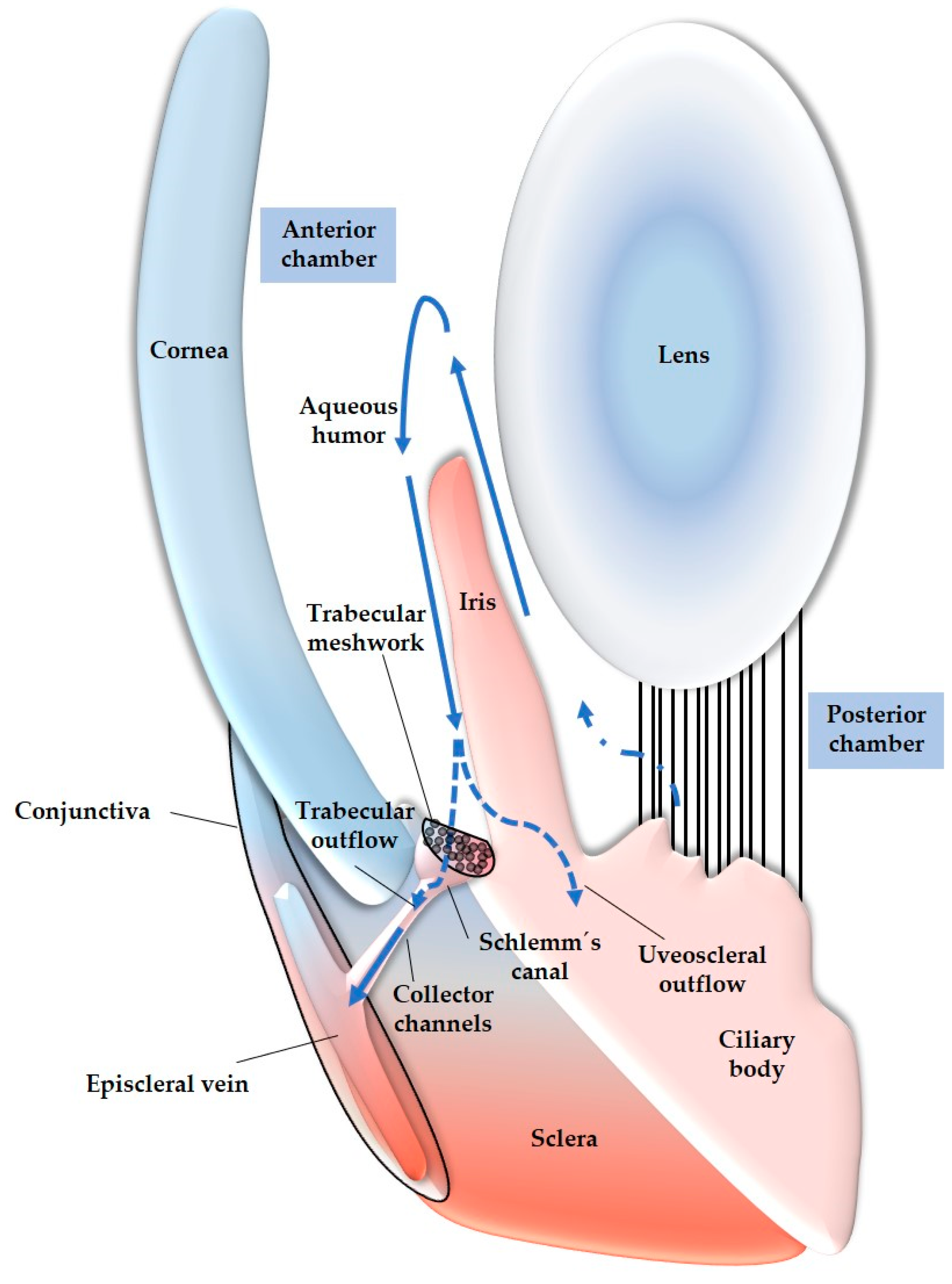
Figure 5.
Model of etiopathogenesis in glaucomatous optic neuropathies. AH: aqueous humor; TM: trabecular meshwork; TMC: trabecular meshwork cell; ONH: optic nerve head; OPA1: optic atrophy 1 gene; TNF-α:tumor necrosis factor alpha; NF-kB: nuclear factor ’kappa-light-chain-enhancer’ of activated B-cells; ATP: adenosintriphosphat; RGC: retinal ganglion cell; oxLDL: oxidized low density lipoprotein; AGE: advanced glycation end product; pJNK: c-Jun N-terminal kinase; pERK: extracellular-signal-regulated kinase.
Figure 5.
Model of etiopathogenesis in glaucomatous optic neuropathies. AH: aqueous humor; TM: trabecular meshwork; TMC: trabecular meshwork cell; ONH: optic nerve head; OPA1: optic atrophy 1 gene; TNF-α:tumor necrosis factor alpha; NF-kB: nuclear factor ’kappa-light-chain-enhancer’ of activated B-cells; ATP: adenosintriphosphat; RGC: retinal ganglion cell; oxLDL: oxidized low density lipoprotein; AGE: advanced glycation end product; pJNK: c-Jun N-terminal kinase; pERK: extracellular-signal-regulated kinase.
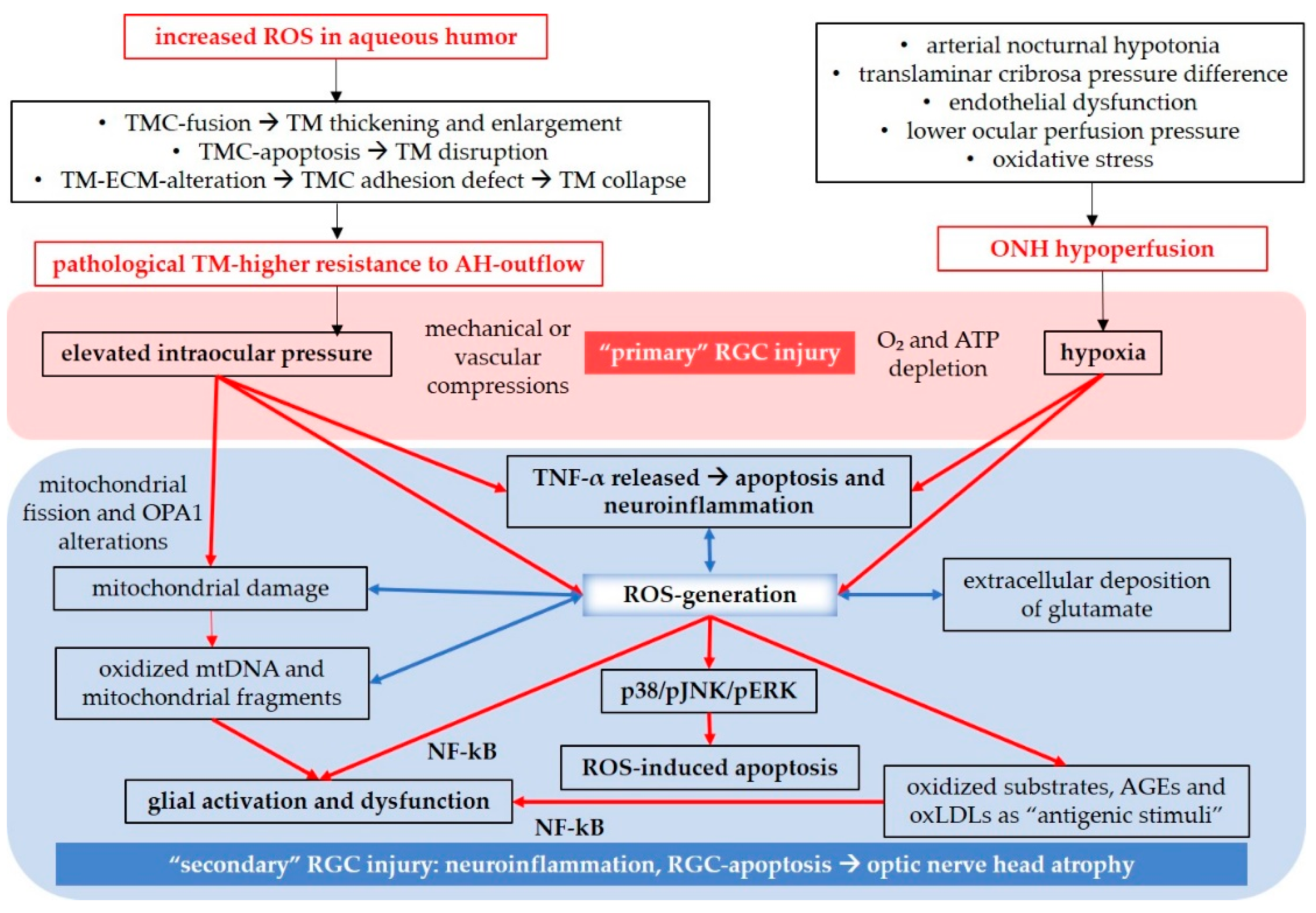
Figure 6.
Role of calcium and reactive oxygen species in the interplay between ER and Mitochondria. ER: endoplasmic reticulum; PERK: protein kinase RNA-like ER-kinase; ATF: activating transcription factor; IRE-1: inositol-requiring protein 1; CHOP: CCAAT-enhancer-binding protein homologous protein; eIF2α: Eukaryotic Initiation Factor 2α; sXBP1: spliced X-box binding protein-1; TNFR2: tumor necrosis factor alpha receptor 2; ASK-1: apoptosis signal-regulating kinase 1; JNK: c-Jun N-terminal kinase; ERAD: ER-associated degradation; NF-kB: nuclear factor ’kappa-light-chain-enhancer’ of activated B-cells.
Figure 6.
Role of calcium and reactive oxygen species in the interplay between ER and Mitochondria. ER: endoplasmic reticulum; PERK: protein kinase RNA-like ER-kinase; ATF: activating transcription factor; IRE-1: inositol-requiring protein 1; CHOP: CCAAT-enhancer-binding protein homologous protein; eIF2α: Eukaryotic Initiation Factor 2α; sXBP1: spliced X-box binding protein-1; TNFR2: tumor necrosis factor alpha receptor 2; ASK-1: apoptosis signal-regulating kinase 1; JNK: c-Jun N-terminal kinase; ERAD: ER-associated degradation; NF-kB: nuclear factor ’kappa-light-chain-enhancer’ of activated B-cells.
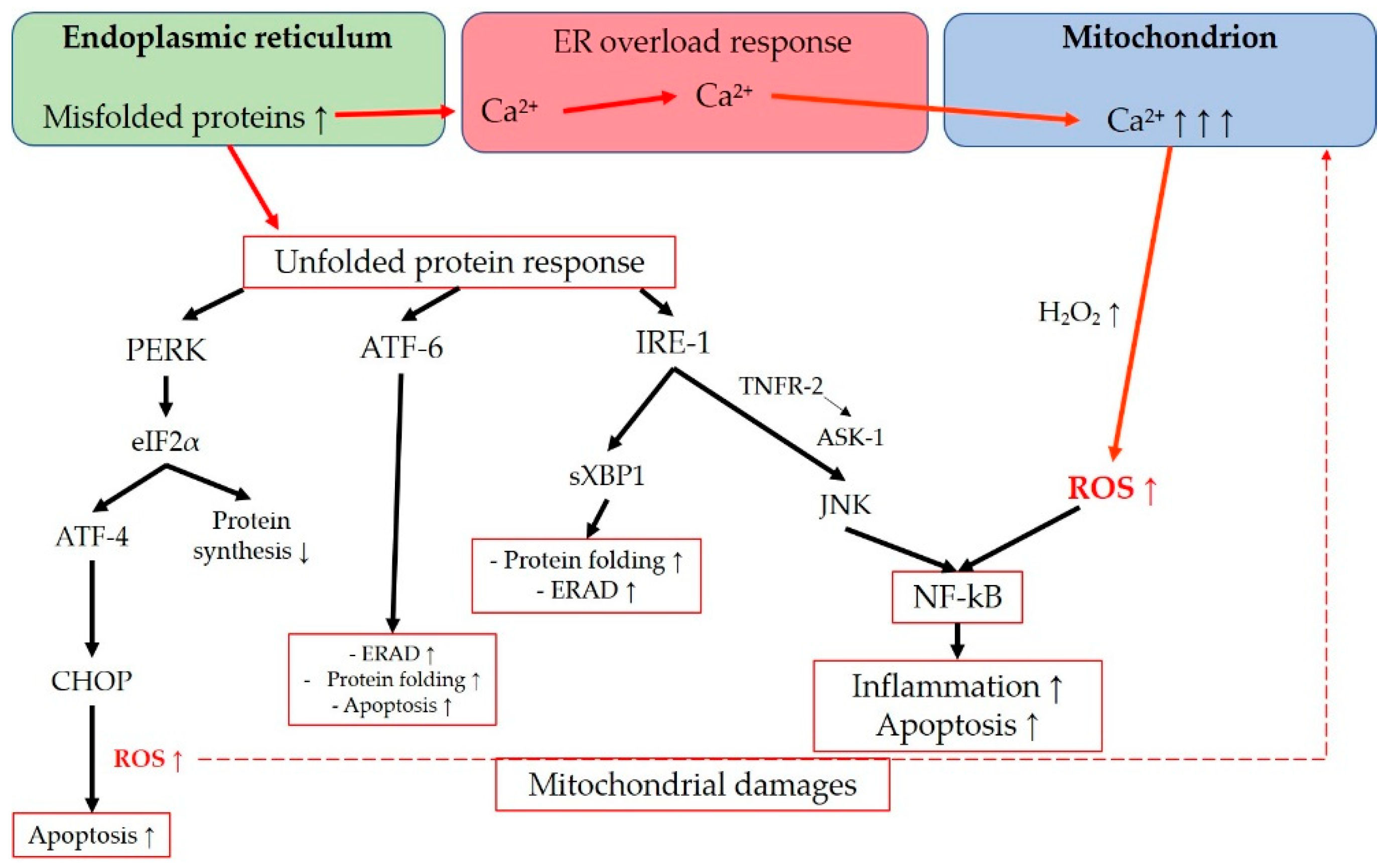
Figure 7.
Direct and indirect effect of idebenone on the mitochondrial oxidative metabolism. COQ₁₀: Cofactor Q 10; Cyt c: Cytochrom c; GSH: Gluthatione; SOD: Superoxide dismutase; GPX: Gluthatione peroxidase; NQO1: NAD(P)H quinone oxidoreductase 1; NOX2: nicotinamide adenine dinucleotide phosphate oxidase 2; Nrf2: nuclear factor erythroid-derived 2-related factor 2.
Figure 7.
Direct and indirect effect of idebenone on the mitochondrial oxidative metabolism. COQ₁₀: Cofactor Q 10; Cyt c: Cytochrom c; GSH: Gluthatione; SOD: Superoxide dismutase; GPX: Gluthatione peroxidase; NQO1: NAD(P)H quinone oxidoreductase 1; NOX2: nicotinamide adenine dinucleotide phosphate oxidase 2; Nrf2: nuclear factor erythroid-derived 2-related factor 2.
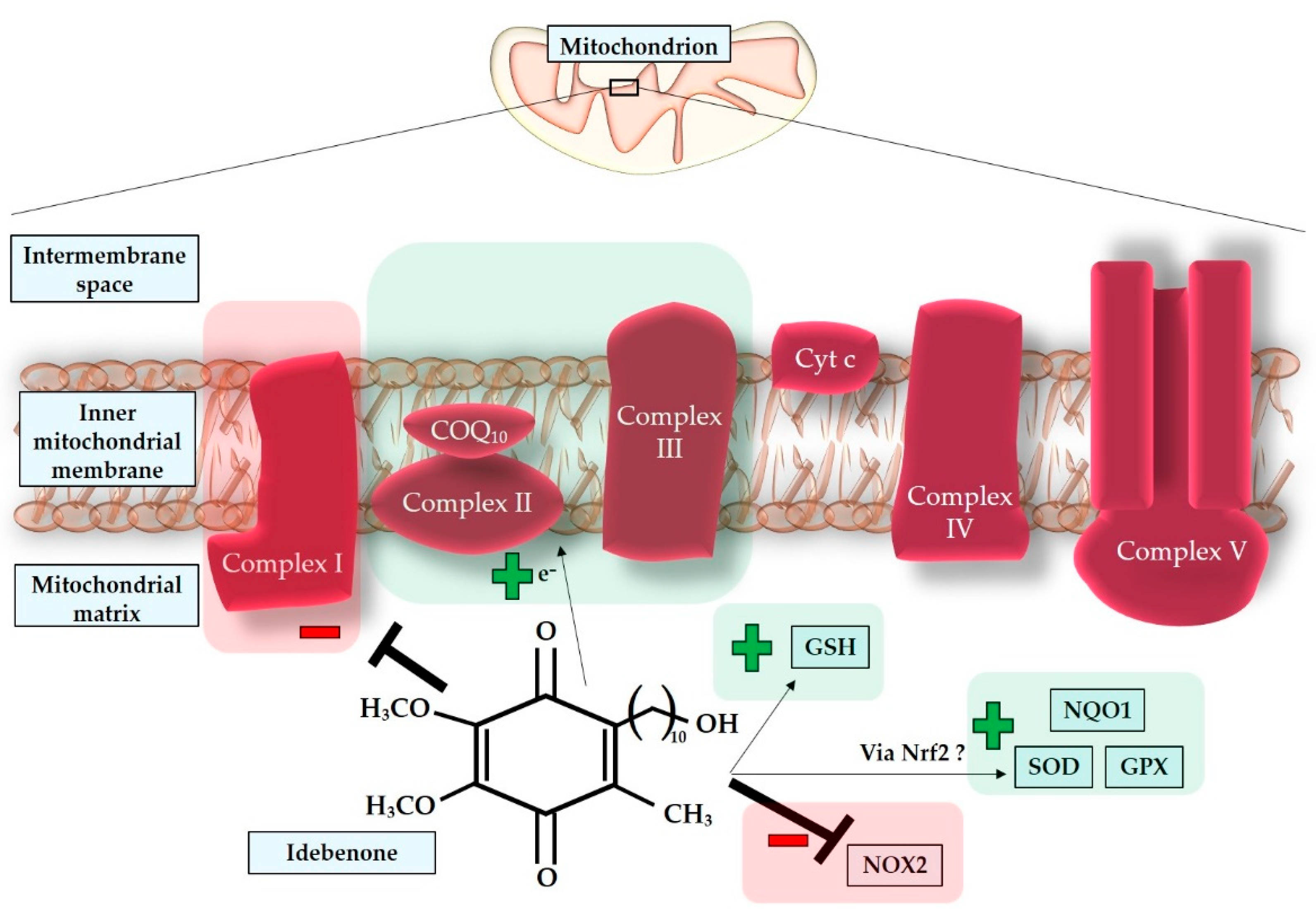
Figure 8.
Model of LHON-Pathogenesis. NAD: Nicotinamidedinucleotide; ETC: electron transport chain; ATP: Adenosintriphopshat; cyt c: cytochrom c; Bid: BH3 interacting-domain death agonist; Apaf-1: apoptotic protease-activating factor 1; PTP: permeability transition pore.
Figure 8.
Model of LHON-Pathogenesis. NAD: Nicotinamidedinucleotide; ETC: electron transport chain; ATP: Adenosintriphopshat; cyt c: cytochrom c; Bid: BH3 interacting-domain death agonist; Apaf-1: apoptotic protease-activating factor 1; PTP: permeability transition pore.
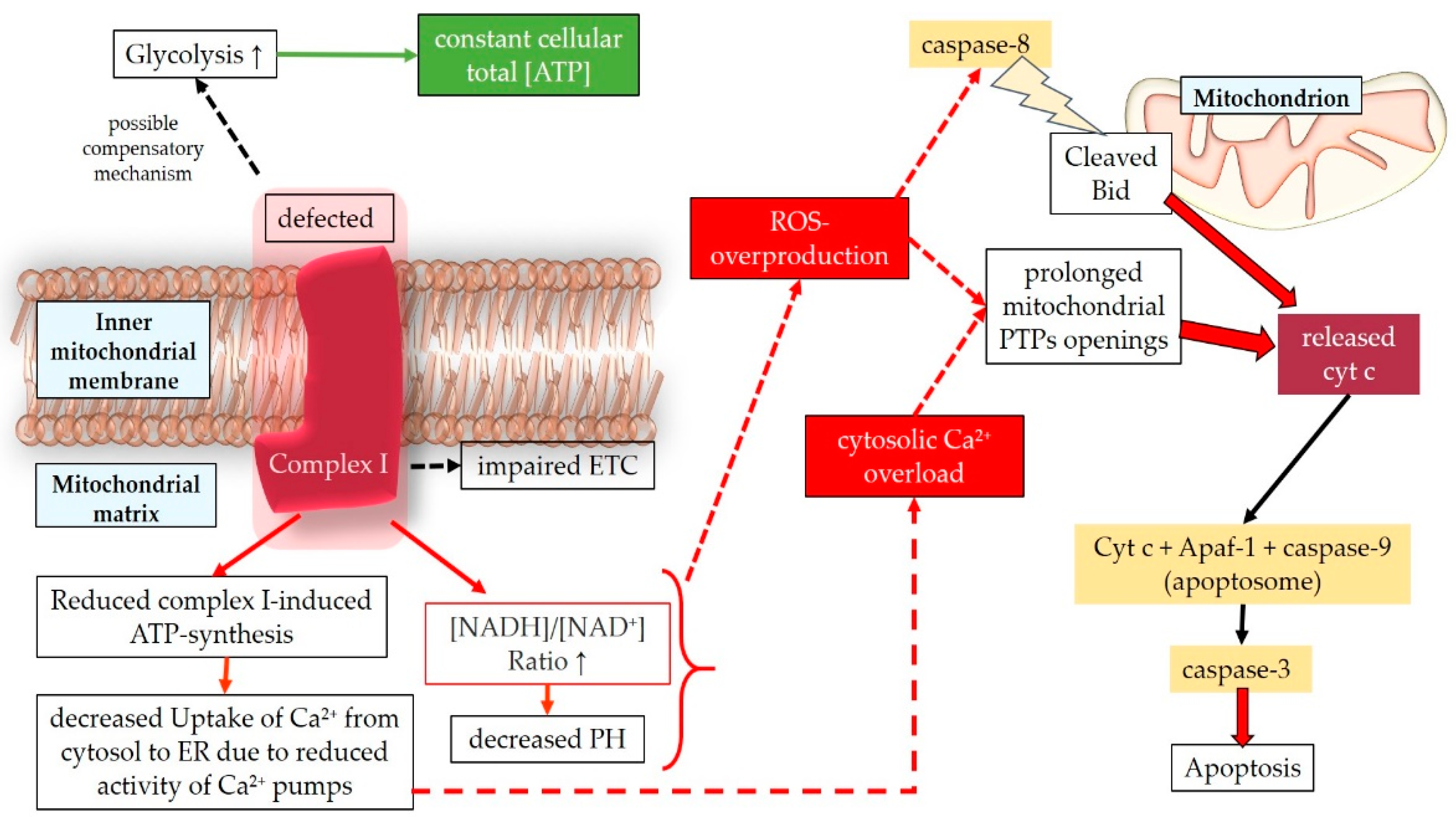
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



