
Submitted:
28 June 2023
Posted:
29 June 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
It is important to be able to detect and differentiate between distinct porcine enteric coronaviruses that can cause similar diseases. However, the existence of naturally occurring recombinant coronaviruses such as swine enteric coronavirus (SeCoV) can give misleading results with currently used diagnostic methods. Therefore, we have developed and validated three duplex real-time quantitative RT-PCR assays for the simultaneous detection of, and differentiation between, porcine epidemic diarrhea virus (PEDV) and SeCoV. Transmissible gastroenteritis virus (TGEV) is also detected by two out of these three assays. In addition, a novel triplex assay was set up that was able to detect and differentiate between these alphacoronaviruses and the porcine deltacoronavirus (PDCoV). The validated assays have low limits of detection, close to 100% efficiency and were able to correctly identify the presence of PEDV and SeCoV in 55 field samples, while 20 samples of other pathogens did not give a positive result. Implementing one or more of these multiplex assays into the routine diagnostic surveillance for PEDV will ensure that the presence of SeCoV, TGEV and PDCoV will not go unnoticed.
Keywords:
Virus diagnosis
; RNA
; recombinant virus
; alphacoronavirus
; deltacoronavirus
; porcine epidemic diarrhea virus (PEDV)
; transmissible gastroenteritis virus (TGEV)
; swine enteric coronavirus (SeCoV)
; porcine deltacoronavirus (PDCoV)
1. Introduction
Enteric pathogens contribute to significant losses in pig production worldwide; young pigs are especially vulnerable to disease caused by gastrointestinal infections [1]. Important pathogens include porcine epidemic diarrhea virus (PEDV), an Alphacoronavirus, which caused the death of about 8 million newborn piglets within 1 year, following its first introduction into the USA in 2013 [2]. Historically, PEDV is the second enteric coronavirus that has been described in pigs after the initial identification of transmissible gastroenteritis virus (TGEV). The latter was responsible for epidemic outbreaks of virus enteritis in pigs, before the spike (S) protein gene deletion variant of TGEV, termed porcine respiratory coronavirus (PRCV), became widely prevalent and probably provided cross-protection against TGEV [3].
In recent years, new enteric porcine coronaviruses have been discovered including swine enteric alphacoronavirus (SeACoV or swine acute diarrhea syndrome coronavirus, SADS-CoV), which has only been detected in Asia so far [4]. In addition, a porcine deltacoronavirus (PDCoV) has been identified [5]; it has been found in China, USA and Canada, but not yet in Europe [6]. In Europe, several reports have described the detection of a diarrhea-associated, naturally occurring, recombinant virus termed swine enteric coronavirus (SeCoV) that is derived from PEDV and TGEV/PRCV [7,8,9,10,11] The major part of the genome of SeCoV consists of a TGEV/PRCV backbone, including the nucleocapsid gene (N), while the S protein gene is most closely related to PEDV, with 91-92% nucleotide identity to the S gene from known PEDVs [8]. Interestingly, other recombinant PEDV strains have been characterized from Slovenia, Hungary, Italy and Spain, where a part of the S gene (around 400 nucleotides from the 5’-end) is derived from SeCoV, while the rest of the genome sequence is derived from the European so-called INDEL type PEDV [10,12,13,14]. These recombinant strains seem to have outcompeted earlier European PEDV strains, and have become established as the most prevalent PEDVs in Northern Italy since 2017 [12]. Similarly, in Spain, it has been found that most PEDVs detected since 2017 are of this subgroup (termed SP4) [10].
Veterinary diagnosis of enteric viral pathogens typically relies on detection of the viruses by testing nucleic acids extracted from fecal samples using real-time quantitative PCR assays [15] and for RNA viruses these assays include a reverse transcriptase (RT) step, thus the assays are commonly referred to as RT-qPCR assays. Although the clinical pictures caused by these various enteric coronaviruses associated with severe diarrhea are indistinguishable [1], typically, only PEDV or TGEV would be suspected and investigated. RT-qPCR assays often used in Europe for PEDV and TGEV rely on detection of specific targets, normally within the N or S genes [16,17]. Only after a negative result would other coronaviruses be suspected, unless prior knowledge exists about the potential involvement of other viruses, e.g. the presence of SeCoV in Italy [9]. To differentiate this recombinant virus from its parental viruses, further testing would be needed. Furthermore, it is important to maintain surveillance in order to identify the entry of other swine enteric coronaviruses such as PDCoV.
The purpose of this study was to develop easy to use duplex diagnostic assays with combined detection of both PEDV and SeCoV from suspect field samples that build on methods currently used in European veterinary diagnostic laboratories. In addition, a triplex assay that detects PDCoV, as well as the targeted porcine enteric alphacoronaviruses, has been set up and characterized.
2. Materials and Methods
2.1. Nucleic acids from field samples and isolates
Sample materials from virus or bacterial isolates and field samples were mixed 1:10 with minimal enriched medium (MEM, internal production IZSLER, Brescia, Italy) and centrifuged for 5 min at 5000 RPM at 4°C. Sample supernatants (100µl), and a further 100µl of MEM plus the positive and negative controls were transferred to a sample plate and processed by the KingFisher™ Flex Purification System (ThermoFisher Scientific, Waltham, Massachusetts, USA) with the NucleoMag Vet kit (Macherey-Nagel, Düren, Germany) as previously described [16]. Extracted nucleic acid samples were stored at -80°C. Previously extracted nucleic acids from isolates of TGEV Purdue and PEDV CV777 (IZSLER variant, GenBank accession number LT905451) were used for the primer-probe optimizations and as positive controls in the duplex RT-qPCR assays. In vitro synthesized PDCoV, TGEV, PEDV RNA transcripts, described below, were used for the primer-probe optimizations and as positive controls in the triplex assay. Field samples, were selected for use in final sensitivity testing of the multiplex assays while viral and bacterial isolates were used for assessment of specificity.
2.2. RNA standards for testing and validation
In vitro transcribed RNA standards were generated corresponding to partial gene sequences of the S and N genes of SeCoV and PEDV, the N gene of TGEV, and the RNA-dependent RNA polymerase (RdRp) of PDCoV. In vitro transcribed RNA containing the PEDV ORF N gene was kindly provided by Beatrice Grasland, ANSES-France [18]. The PEDV S gene transcripts were produced as described [16]. Briefly, nucleic acids extracted from field samples (SeCoV strain Italy/77590/2019, accession number MT821905) or viral isolates (PEDV CV777 and TGEV Purdue) were reverse-transcribed, using SuperScript IV (Invitrogen, ThermoFisher Scientific) and gene specific primers (Supplementary Table S1) according to the manufacturer’s protocol, to generate cDNA. PCR amplicons of 651 bp (PEDV and SeCoV S) and 388 bp (TGEV and SeCoV N) were generated from these templates using GoTaq® G2 Flexi DNA polymerase (Promega, Madison, Wisconsin, USA), gel purified and inserted into the pCR®2.1-TOPO vector (Invitrogen), which was used for transformation of E.coli cells. A synthetic fragment (218 bp) corresponding to part of the PDCoV RdRp gene (KJ481931) was inserted into the plasmid pEX-A2 (Eurofins Genomics Srl, Milano, Italy), and then transferred into the pCR®2.1-TOPO vector as described above. Following identification of the required plasmids, linearized and purified DNAs were in vitro transcribed with HiScribe T7 RNA polymerase (New England Biolabs, Ipswich, Massachusetts, USA) according to the manufacturer’s protocol. The concentration of RNA was measured using an Infinite® 200 NanoQuant spectrophotometer (Tecan, Männedorf, Switzerland), and the copy number of RNA molecules per 1 μl was calculated using the formula:
In this formula, the number of plasmid-derived nucleotides (nucleotidesplasmid) in the final in vitro transcribed RNA was 128 for SeCoV N and S while NA is the Avogadro constant (approximately 6.02 × 1023 mol-1). The in vitro transcribed RNA was serially diluted with TE buffer to 107 molecules/µl, aliquoted and stored at -80°C.
2.3. Selection of primers and probes
Primers and probes from already established and validated assays for singleplex detection of PEDV and TGEV were selected to detect and differentiate between PEDV and SeCoV [17,19,20,21,22]. Primers and a probe for the detection of PDCoV were newly designed in this study. The primer and probe sequences are listed in Table 1. Alignments were made using partial genome sequences of PEDV and SeCoV samples tested in Italy and other publicly available sequences of these viruses. The primer and probe binding sites were then evaluated for mismatches. Three different duplex assays with different combinations of the primers and probes and one triplex assay were chosen for testing and validation. The expected properties of each assay are indicated in Table 2.
2.4. RT-qPCR protocol
The AgPath-ID One-Step RT-PCR kit (Applied Biosystems, ThermoFisher Scientific) was used for all singleplex format and duplex versions of the three different duplex assays while for the singleplex format and triplex versions of the triplex assays, the One Step PrimeScript ™ III RT-qPCR Mix (Takara Bio inc., Kusatsu, Shiga, Japan) system was used.
For the duplex assays, mastermixes of 20 µl were made of primers and probes in different concentrations (see Table 3 for final concentrations), 2x RT-PCR buffer and 25x enzyme mix (Applied Biosystems) were added according to the manufacturer’s protocol and the volume adjusted with RNase-free water. The mastermixes were aliquoted into clear plastic PCR plates and sealed with adhesive plastic foil or clear plastic strip lids. Aliquots (5μl) of template RNA were added using a separate laboratory bench, the contents were spun down and the PCR tubes incubated in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, California, USA). The RT-PCR program consisted of reverse transcription of RNA for 10 min at 48°C, activation of the DNA polymerase at 95°C for 10 min followed by 45 amplification cycles of denaturation at 95°C for 15 sec and annealing and elongation at 60°C for 45 sec. Fluorescence data was collected for the FAM and Cy5 channels.
For the triplex assays, the RNA was reverse transcribed at 52°C for 5 min, followed by Taq polymerase activation at 95°C for 10 sec. Amplification consisted of 45 cycles of 95°C for 5 sec and 60°C for 30 sec. Fluorescence data for the triplex assay was collected for the FAM, Texas Red and Cy5 channels.
The output files were analyzed using the program Bio-Rad CFX Maestro version1.1 (BioRad Laboratories).
2.5. Optimization of primer and probe concentrations
Primer and probe concentrations were optimized for all singleplex assays individually by testing a few samples with a range of different primer and probe concentrations in the same run. Following the singleplex optimizations, selected samples with high and low RNA concentrations were tested in parallel in the singleplex, duplex and triplex assays. The optimized primer and probe concentrations were used for each type of assay to assess whether the performance of the tests was similar in the multiplex and singleplex formats.
2.6. Standard curves
In vitro transcribed RNA samples were 10-fold serially diluted to be in the range of 106 or 107 to 10-1 copies/5µl and TGEV Purdue isolate nucleic acid extract was similarly 10-fold serially diluted (from undiluted to 10-5). The dilutions were tested with optimized primer and probe concentrations in three replicate samples. For the singleplex assays targeting the PEDV S and TGEV N coding sequences, in vitro transcribed RNA of SeCoV was used and for assays targeting the TGEV S coding sequence, the RNA extracted from the TGEV Purdue isolate was used. For other assays, in vitro transcribed RNA corresponding to the relevant regions of the PEDV S and N, TGEV N, and PDCoV RdRp genes were used for testing. From the data obtained, threshold limits were decided within the log phase of amplification and RNA standards within the linear dynamic range were selected to generate the standard curves. Standard curves were used to calculate efficiency (E = 10-1/slope – 1) and RNA copy numbers in these and other runs. The TGEV Purdue isolate RNA concentration was determined by testing a 10-4 dilution of the RNA in the TGEV N singleplex assay together with TGEV N in vitro transcribed RNA of previously determined concentrations using the standard curve.
2.7. Validation steps
Our aim was to validate the multiplex assays for qualitative (semi-quantitative) rather than fully quantitative use. Therefore, the assays were assessed with reference to the Minimum Information for Publication of Quantitative real-time PCR experiments (MIQE guidelines) [23] and also with reference to guidelines specific for qualitative assays [24].
RNA samples for validation were 10-fold serially diluted, as for the standard curves, but other dilutions were also used to be able to determine, more accurately, the limits of detection (LOD). The LOD were determined as the minimal number of genome copies/5μl in six replicates for each singleplex assay and for their combinations in the multiplex assays.
Repeatability or intra-assay variation was assessed by testing three samples in six replicates of high, intermediate and low concentrations of template, i.e. 106, 105/104 and 102 copies/5μl for all duplex assays, and 106, 104 and 103/102 copies/5μl for the triplex assay. Concentrations were calculated for each replicate and mean values and standard deviations calculated from these. The coefficient of variation (CV) was obtained by expressing the relative standard deviation (standard deviation divided by the mean) as a percentage value.
Reproducibility or inter-assay variation was assessed in a similar way by testing three samples of high, intermediate and low concentrations of template, i.e. 106, 104 and 102 copies/5μl for all duplex assays, and 106, 104 and 103/102 copies/5μl for the triplex assay. The samples were tested in three replicates in four separate runs. The mean concentration was calculated for each run of each sample, and the standard deviation of the four means was calculated for each sample.
For assessment of specificity and sensitivity, single replicate testing was done using panels of previously extracted RNA from field samples, bacteria and virus isolates (as described below, see Supplementary Tables S2 and S3).
3. Results
The primers and probes selected for use in the duplex and triplex assays for PEDV, TGEV, and PDCoV (see Table 1), were original or modified versions of oligonucleotides already in use for RT-qPCR assays for porcine coronaviruses in veterinary diagnostic laboratories [3,17,19,21,22], or newly designed oligonucleotides. The duplex and triplex combinations of these primers and probes are shown in Table 3 along with the optimal final concentrations determined experimentally. Expected characteristics of the selected assays are shown in Table 2. While each of the 3 duplex assays (termed Duplex 1, 2 and 3) were expected to be able to detect both PEDV and SeCoV, Duplex 3 was not expected to distinguish between them. Furthermore, TGEV should be detected by Duplex 2 and Duplex 3. The latter was intended to be specific for TGEV, whereas Duplex 2 was designed to detect PRCV as well, but without distinguishing between the two virus variants. Separately, the triplex assay was designed to detect PEDV, SeCoV, TGEV, PRCV and PDCoV; this assay was expected to distinguish PEDV from SeCoV but without distinguishing amongst SeCoV, TGEV and PRCV.
Standard curves and derived efficiencies are shown in Figure 1 for the four individual single targets used in the three duplex assays. Similarly, the standard curves for the combined assays, when run in the three duplex assays are shown in Figure 2.
It was found that both the singleplex and duplex assays performed well, with close to 100% efficiencies and the slopes of the curves were close to the optimal value of -3.32 (slope = - 1/log10(E+1), i.e. when E equals 100% (or 1.00 in the formula), then the slope equals - 3.32). In the Duplex 1 assay, the detection of PEDV N RNA had a slightly less than optimal efficiency of 98.9% and a slope of -3.348 was observed. The R2 value was close to 1.00 for all of the assays, but in the Duplex 3 and singleplex PEDV S assays, R2 was 0.998 and in the Duplex 3, TGEV S it was 0.995. It is considered that preferably the efficiency should in the range of 90-110% while the linearity R2 values should be ≥0.98 [24]; thus each of the duplex assays meet these criteria. The linear dynamic ranges of the duplex assays, which were also included in the standard curves, were 102 to 107 RNA copies/5µl (Figure 2) except for TGEV S in Duplex 3, where the linear range was 102 to 106 RNA copies/5µl. Similarly, the RT-qPCRs in the triplex assay had a high efficiency (99.8-103%) and a R2 value close to 1.00 (0.997-0.999) for each target within both singleplex and triplex formats (see Figure 3). The assays had a linear dynamic range of between 102-107 copies for the PDCoV and PEDV (singleplex) and PEDV (triplex) assays while it was between 103–107 copies for the TGEV (singleplex) and for PDCoV and TGEV in the triplex assays.
For the targets of the duplex assays, testing and validation showed that the LODs in the singleplex assays were in the range of 10-50 copies/5µl with the exception of the PEDV N assay which gave variable and higher values for the LOD (up to 1000 copies/5µl) (Table 4) while the PEDV S assay had a lower LOD (just 10 copies/5µl). In the duplex assays, consistent detection of the PEDV S gene derived RNA down to 25 copies/5µl template was achieved, while, the N gene of PEDV was detected down to 50 copies/5µl template and the TGEV S and N genes down to 100 copies/5µl template in the three assays (Table 4). In the triplex assay, the singleplex reactions showed a low LOD (10-100 copies/5µl) (Table 4). The LOD was also low in the triplex assay for TGEV N gene (25 copies/5µl) but higher for PDCoV RdRp and PEDV N genes (1000 and 25 copies/5µl, respectively).
For the targets of the duplex assays, testing and validation showed that the LODs in the singleplex assays were in the range of 10-50 copies/5µl with the exception of the PEDV N assay which gave variable and higher values for the LOD (up to 1000 copies/5µl) (Table 4) while the PEDV S assay had a lower LOD (just 10 copies/5µl). In the duplex assays, consistent detection of the PEDV S gene derived RNA down to 25 copies/5µl template was achieved, while, the N gene of PEDV was detected down to 50 copies/5µl template and the TGEV S and N genes down to 100 copies/5µl template in the three assays (Table 4). In the triplex assay, the singleplex reactions showed a low LOD (10-100 copies/5µl) (Table 4). The LOD was also low in the triplex assay for TGEV N gene (25 copies/5µl) but higher for PDCoV RdRp and PEDV N genes (1000 and 25 copies/5µl, respectively).
The intra-assay variation was low (coefficient of variation 3-16%) for samples with high and intermediate concentrations of template for all the duplex and triplex assays (Table 5). However, they did not perform as well for the low concentration samples, as may be expected. Here, the coefficient of variation for detection of PEDV was 11-31% and for TGEV 13-110%. It should be noted that the concentration of this sample was close to the approximate LOD for TGEV. The inter-assay variation was not dependent on the concentration of template and showed coefficients of variation between 4 and 43% (mean 23%) (Table 5).
Specificity testing using a panel of twenty samples of different swine pathogens (viruses and bacteria), other than PEDV, TGEV and SeCoV, (Supplementary Table S2) showed no false positive samples in any of the duplex assays.
For sensitivity testing, a panel of fifty-five field samples of PEDV and SeCoV including recombinant PEDV/SeCoV, as described previously [12], resulted in the expected positive tests for nearly all the samples with just one exception (a summary of the results is shown in Table 6, while the full set of results, including Ct values, are in Supplementary Table S3). Out of 24 PEDV samples tested, a single PEDV sample (number 20) was not detected by either the PEDV N test in Duplex 1 or the triplex assay, but it was detected by all of the PEDV S gene tests in the Duplex 2 and Duplex 3 assays. The assays correctly differentiated between PEDV and TGEV (Duplex 2 and 3 and triplex assays). Furthermore, the assays gave distinctive patterns of results for SeCoV (negative for PEDV N and TGEV S but positive for PEDV S and TGEV N genes) compared to PEDV and the recombinant SeCoV/PEDV samples (negative for TGEV N and TGEV S but positive for PEDV N and PEDV S genes). These assays do not discriminate between PEDV and the recombinant SeCoV/PEDV genomes (Table 6, Supplementary Table S3). The triplex assay showed the same ability to detect all the tested samples except for two recombinant SeCoV/PEDV samples (numbers 25 and 42) which gave high Ct values (about 30 or above) in the duplex assays.
4. Discussion
The purpose of developing a duplex assay was to be able to detect both PEDVs and SeCoVs and to distinguish between them within a single assay. Different combinations of existing assays were possible and thus three different duplex assays were designed in order to assess which assay performed the best before selecting one for diagnostic implementation. All three duplex assays included the PEDV S gene targeted assay that was originally developed after the outbreaks in the USA [16,19]. However, an assay targeting the S protein gene, which is under evolutionary selection pressure, could be challenged by accumulating genetic changes. Therefore, it is important to continue to monitor and sequence circulating PEDV and SeCoV strains to identify mismatches in the primer/probe-binding regions of the S gene. When trying to document the absence of a pathogen, it is important to have a low limit of detection. In this study, we found that the LOD for the PEDV S gene RNA in the three duplexes was 25 copies/5µl, which was a bit higher than in the singleplex assay (10 copies/5µl), but still sufficient for diagnostic use. Since the PEDV S assay is able to detect both SeCoV and PEDV, which were the pathogens of interest in this study, we consider that it is acceptable for the other gene targets included in the duplex assays to have higher LODs. Apart from the PEDV S target detected using the FAM-labelled probe, each of the three duplex assays in this study included one additional target detected using a Cy5-labelled probe. In Duplex 1, a PEDV N gene targeted assay was included, which was already in use for diagnostic testing of PEDV suspect samples in Denmark. When tested in the duplex format, this assay had a LOD of 50 copies/5µl, which was better than the LOD of 100 copies/5µl observed for TGEV N and TGEV S in Duplex 2 and 3. The TGEV S of Duplex 3 was only validated using the RNA of a single TGEV isolate, and therefore the validation parameters such as linear dynamic range, repeatability and sensitivity could possibly be improved by testing more TGEV samples and by using in vitro transcribed RNA of TGEV of known concentrations. As two of the duplex assays will detect the same target, albeit using two sets of primers and probes (PEDV in Duplex 1 and SeCoV in Duplex 2), it is possible that there will be competition for reagents in the reaction, although this did not seem to affect the performance during testing.
Overall, the assays performed well, and selection of one over the others would depend on the specific purpose as each of the three duplex assays have their own specific properties (Table 3). Duplex 1 has a good likelihood of detecting PEDV, as two different targets are recognized. Furthermore, SeCoV is detected and distinguished from PEDV in this assay. Apart from detection of PEDV and SeCoV, Duplex 2 and 3 are also able to detect TGEV, which could be beneficial, as this is an important differential diagnosis. However, Duplex 2 is not able to distinguish between TGEV and PRCV, which could be problematic, as PRCV is prevalent in many swine producing countries, which are free from TGEV [3]. It should be noted, however, that although PRCV has a predominant tissue tropism for the respiratory tract, virus is also shed via the feces [25]. This means that positive TGEV N results in Duplex 2 would have to be followed-up by further testing, e.g. using the TGEV S singleplex assay, this would enable the important discrimination between the presence of TGEV and PRCV. No testing for PRCV is necessary when using Duplex 3, but positive PEDV S results should be further examined to distinguish PEDV from SeCoV.
As previously mentioned, other porcine enteric pathogens, apart from TGEV and SeCoV, are clinically indistinguishable from PEDV. Therefore, it made sense to develop further multiplex assays capable of detecting and distinguishing between these and other relevant viruses (e.g. PDCoV and SeACoV), as shown in other studies [26,27,28]. However, none of these previously described assays specifically recognize SeCoV.
In addition, PDCoV detection might be important, even in Europe. In fact, while PDCoV was initially identified in Asia and North America [6], it is essential to recognize that infectious diseases can cross geographical boundaries. Given the global nature of the swine industry and the extensive international trade, Europe remains susceptible to the introduction and spread of PDCoV. Integrating PDCoV detection alongside other porcine coronaviruses in diagnostic protocols enables early identification, surveillance, and monitoring of this emerging pathogen. This proactive approach provides valuable insights into the epidemiology of PDCoV, enhances preparedness for potential outbreaks and supports the implementation of effective control measures to safeguard the European swine population. However, the triplex assay developed in this study, is not able to distinguish between SeCoV and TGEV/PRCV. Further testing would be needed in cases of non- discriminative results.
It could also be worth including these pathogens into a high throughput platform such as the Fluidigm system, using high throughput microfluidic RT-qPCRs [29], or employing several duplex or multiplex assays simultaneously, as described from Spain [30] and here. An alternative approach is to use a broad screening strategy detecting all coronaviruses, e.g. using a pan-coronavirus real-time RT-PCR assay [31,32,33,34,35] followed by further characterization of positive samples by specific RT-qPCRs or by sequencing.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Sequences of the oligonucleotides used for amplifying fragments inserted into cloning vector to enable In vitro transcription of RNA standards; Table S2: Specificity panel and results from testing in the three separate duplex assays and in the triplex assay. Table S3: Specificity panel and results from testing in the three separate duplex assays and in the triplex assay.
Author Contributions
Conceptualization, M.B.B.; methodology, C.M.L.; A.P.; validation, C.M.L.; A.P..; formal analysis, C.M.L.; A.P.; investigation, C.M.L.; resources, A.B.; data curation, A.P..; writing—original draft preparation, C.M.L.; A.P.; writing—review and editing, All authors.; visualization, C.M.L.; A-P.; supervision, G.J.B, A.B.; T.B.R.; M.B.B; project administration, A.B.; funding acquisition, M.B.B.. All authors have read and agreed to the published version of the manuscript.
Funding
The research stay of CML at IZSLER was supported by scholarships received by CML from the EPIZONE short term mission program, Christian og Otillia Brorsons rejselegat and a grant from Otto Mønsteds Fond (reference no. 19-70-1185). The study was also partially funded by the Italian Ministry of Health, grant number PRC2014005. The APC was funded by internal resources of the University of Copenhagen.
Institutional Review Board Statement
“Not applicable” as the studies did not involve humans or animals.
Data Availability Statement
Relevant data are included in the paper.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Jung, K.; Saif, L.J.; Wang, Q. Porcine epidemic diarrhea virus (PEDV): An update on etiology, transmission, pathogenesis, and prevention and control. Virus Research 2020, 286, 198045. [Google Scholar] [CrossRef]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virology Journal 2015, 12, 1–16. [Google Scholar] [CrossRef]
- Chen, F.; Knutson, T.P.; Rossow, S.; Saif, L.J.; Marthaler, D.G. Decline of transmissible gastroenteritis virus and its complex evolutionary relationship with porcine respiratory coronavirus in the United States. Scientific Reports 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.; Lou, Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; Zheng, X.S.; Li, B.; Li, J.M.; Guo, H.; Pei, G.Q.; An, X.P.; Chen, J.W.; Zhou, L.; Mai, K.J.; … Ma, J.Y. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–259. [CrossRef]
- Woo, P.C. Y. ; Lau, S.K. P. ; Lam, C.S. F. ; Lau, C.C. Y. ; Tsang, A.K. L. ; Lau, J.H. N. ; Bai, R. ; Teng, J.L. L. ; Tsang, C.C. C. ; Wang, M. ; Zheng, B.-J. ; Chan, K.-H. ; Yuen, K.-Y. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavi. Journal of Virology 2012, 86, 3995–4008. [CrossRef]
- Wang, L.; Byrum, B.; Zhang, Y. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerging Infectious Diseases 2014, 20, 1227–1230. [Google Scholar] [CrossRef]
- Akimkin, V.; Beer, M.; Blome, S.; Hanke, D.; Höper, D.; Jenckel, M.; Pohlmann, A. New chimeric porcine coronavirus in Swine Feces, Germany, 2012. Emerging Infectious Diseases 2016, 22, 1314–1315. [Google Scholar] [CrossRef] [PubMed]
- Belsham, G.J.; Rasmussen, T.B.; Normann, P.; Vaclavek, P.; Strandbygaard, B.; Bøtner, A. Characterization of a Novel Chimeric Swine Enteric Coronavirus from Diseased Pigs in Central Eastern Europe in 2016. Transboundary and Emerging Diseases 2016, 63, 595–601. [Google Scholar] [CrossRef]
- Boniotti, M.B.; Papetti, A.; Lavazza, A.; Alborali, G.; Sozzi, E.; Chiapponi, C.; Faccini, S.; Bonilauri, P.; Cordioli, P.; Marthaler, D. Porcine epidemic diarrhea virus and discovery of a recombinant swine enteric coronavirus, Italy. Emerging Infectious Diseases 2016, 22, 83–87. [Google Scholar] [CrossRef]
- de Nova, P.J.G.; Cortey, M.; Díaz, I.; Puente, H.; Rubio, P.; Martín, M.; Carvajal, A. A retrospective study of porcine epidemic diarrhoea virus (PEDV) reveals the presence of swine enteric coronavirus (SeCoV) since 1993 and the recent introduction of a recombinant PEDV-SeCoV in Spain. Transboundary and Emerging Diseases 2020, 67, 2911–2922. [Google Scholar] [CrossRef]
- Papetti, A.; Bonilauri, P.; Chiapponi, C.; Baioni, L.; Boniotti, M.B. Complete Genome Sequence of an Italian Swine Enteric Coronavirus Strain 77590/2019. Microbiology Resource Announcements 2022, 15, e00386–22. [Google Scholar] [CrossRef]
- Boniotti, M.B.; Papetti, A.; Bertasio, C.; Giacomini, E.; Lazzaro, M.; Cerioli, M.; Faccini, S.; Bonilauri, P.; Vezzoli, F.; Lavazza, A.; Alborali, G.L. Porcine Epidemic Diarrhoea Virus in Italy: Disease spread and the role of transportation. Transboundary and Emerging Diseases 2018, 65, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Valkó, A.; Biksi, I.; Cságola, A.; Tuboly, T.; Kiss, K.; Ursu, K.; Dán, Á. Porcine epidemic diarrhoea virus with a recombinant S gene detected in Hungary, 2016. Acta Veterinaria Hungarica 2017, 65, 253–261. [Google Scholar] [CrossRef]
- Valkó, A.; Albert, E.; Cságola, A.; Varga, T.; Kiss, K.; Farkas, R.; Rónai, Z.; Biksi, I.; Dán, Á. Isolation and characterisation of porcine epidemic diarrhoea virus in Hungary. Acta Veterinaria Hungarica 2019, 67, 307–313. [Google Scholar] [CrossRef]
- Mackay, I.M.; Arden, K.E.; Nitsche, A. Real-time PCR in virology. Nucleic Acids Research 2002, 30, 1292–1305. [Google Scholar] [CrossRef]
- Bertasio, C.; Giacomini, E.; Lazzaro, M.; Perulli, S.; Papetti, A.; Lavazza, A.; Lelli, D.; Alborali, G.; Boniotti, M.B. Porcine epidemic diarrhea virus shedding and antibody response in swine farms: A longitudinal study. Frontiers in Microbiology 2016, 7, 2009. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, I.J.; Pyo, H.M.; Tark, D.S.; Song, J.Y.; Hyun, B.H. Multiplex real-time RT-PCR for the simultaneous detection and quantification of transmissible gastroenteritis virus and porcine epidemic diarrhea virus. Journal of Virological Methods 207, 146, 172–177. [Google Scholar] [CrossRef]
- Bigault, L.; Brown, P.; Bernard, C.; Blanchard, Y.; Grasland, B. Porcine epidemic diarrhea virus: Viral RNA detection and quantification using a validated one-step real time RT-PCR. Journal of Virological Methods 2020, 283, 113906. [Google Scholar] [CrossRef]
- Alonso, C.; Goede, D.P.; Morrison, R.B.; Davies, P.R.; Rovira, A.; Marthaler, D.G.; Torremorell, M. Evidence of infectivity of airborne porcine epidemic diarrhea virus and detection of airborne viral RNA at long distances from infected herds. Veterinary Research 2014, 45, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Li, G.; Stasko, J.; Thomas, J.T.; Stensland, W.R.; Pillatzki, A.E.; Gauger, P.C.; Schwartz, K.J.; Madson, D.; Yoon, K.J.; Stevenson, G.W.; Burrough, E.R.; Harmon, K.M.; Main, R.G.; Zhang, J. Isolation and characterization of porcine epidemic diarrhea viruses associated with the 2013 disease outbreak among swine in the united states. Journal of Clinical Microbiology 2014, 52, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Vemulapalli, R.; Gulani, J.; Santrich, C. A real-time TaqMan® RT-PCR assay with an internal amplification control for rapid detection of transmissible gastroenteritis virus in swine fecal samples. Journal of Virological Methods 2009, 162, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Liu, Y.; Chen, Y.; Xing, G.; Hao, H.; Wei, Q.; Liang, Y.; Xie, W.; Li, D.; Huang, H.; Deng, R.; Zhang, G. A novel duplex TaqMan probe-based real-time RT-qPCR for detecting and differentiating classical and variant porcine epidemic diarrhea viruses. Molecular and Cellular Probes 2018, 37, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; Vandesompele, J.; Wittwer, C.T. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Broeders, S.; Huber, I.; Grohmann, L.; Berben, G.; Taverniers, I.; Mazzara, M.; Roosens, N.; Morisset, D. Guidelines for validation of qualitative real-time PCR methods. Trends in Food Science and Technology 2014, 37, 115–126. [Google Scholar] [CrossRef]
- Costantini, V.; Lewis, P.; Alsop, J.; Templeton, C.; Saif, L.J. Respiratory and fecal shedding of Porcine respiratory coronavirus (PRCV) in sentinel weaned pigs and sequence of the partial S-gene of the PRCV isolates. Archives of Virology 2004, 149, 957–974. [Google Scholar] [CrossRef]
- Ding, G.; Fu, Y.; Li, B.; Chen, J.; Wang, J.; Yin, B.; Sha, W.; Liu, G. Development of a multiplex RT-PCR for the detection of major diarrhoeal viruses in pig herds in China. Transboundary and Emerging Diseases 2020, 67, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Tsuchiaka, S.; Ashiba, T.; Yamasato, H.; Fukunari, K.; Omatsu, T.; Furuya, T.; Shirai, J.; Mizutani, T.; Nagai, M. Development of one-step real-time reverse transcriptase-PCR-based assays for the rapid and simultaneous detection of four viruses causing porcine diarrhea. Japanese Journal of Veterinary Research 2016, 64, 5–14. [Google Scholar] [CrossRef]
- Si, G.; Niu, J.; Zhou, X.; Xie, Y.; Chen, Z.; Li, G.; Chen, R.; He, D. Use of dual priming oligonucleotide system-based multiplex RT-PCR assay to detect five diarrhea viruses in pig herds in South China. AMB Express 2021, 11. [Google Scholar] [CrossRef]
- Goecke, N.B.; Hjulsager, C.K.; Krog, J.S.; Skovgaard, K.; Larsen, L.E. Development of a high-throughput real-time PCR system for detection of enzootic pathogens in pigs. Journal of Veterinary Diagnostic Investigation 2020, 32, 51–64. [Google Scholar] [CrossRef]
- Puente, H.; Argüello, H.; Mencía-Ares, Ó.; Gómez-García, M.; Rubio, P.; Carvajal, A. Detection and Genetic Diversity of Porcine Coronavirus Involved in Diarrhea Outbreaks in Spain. Frontiers in Veterinary Science 2021, 8, 651999. [Google Scholar] [CrossRef]
- de Souza Luna, L.K.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; Drosten, C. Generic Detection of Coronaviruses and Differentiation at the Prototype Strain Level by Reverse Transcription-PCR and Nonfluorescent Low-Density Microarray. Journal of Clinical Microbiology 2007, 45, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Escutenaire, S.; Mohamed, N.; Isaksson, M.; Thorén, P.; Klingeborn, B.; Belák, S.; Berg, M.; Blomberg, J. SYBR Green Real-Time Reverse Transcription-Polymerase Chain Reaction Assay for the Generic Detection of Coronaviruses. Archives of Virology 2007, 152, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Jung, K.; Wang, Q.; Saif, L.J.; Vlasova, A.N. Development of a one-step RT-PCR assay for detection of pancoronaviruses (α-, β-, γ-, and δ-coronaviruses) using newly designed degenerate primers for porcine and avian fecal samples. Journal of Virological Methods 2018, 256, 116–122. [Google Scholar] [CrossRef]
- Vijgen, L.; Moës, E.; Keyaerts, E.; Li, S.; Van Ranst, M. A Pancoronavirus RT-PCR Assay for Detection of All Known Coronaviruses. Methods in Molecular Biology 2008, 454, 3–12. [Google Scholar] [CrossRef]
- Lazov, C.M.; Chriél, M.; Baagøe, H.J.; Fjederholt, E.; Deng, Y.; Kooi, E.A.; Belsham, G.J.; Bøtner, A.; Rasmussen, T.B. Detection and Characterization of Distinct Alphacoronaviruses in Five Different Bat Species in Denmark. Viruses 2018, 10, 486. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Song, D.S.; Park, B.K. Differential detection of transmissible gastroenteritis virus and porcine epidemic diarrhea virus by duplex RT-PCR. Journal of Veterinary Diagnostic Investigation 2001, 13, 516–520. [Google Scholar] [CrossRef]
Figure 1.
Singleplex standard curves generated on the basis of serially diluted RNA standards. In vitro transcribed RNA was used for the standards for PEDV N (from PEDV CV777), for TGEV N and for PEDV S (from SeCoV). For the TGEV S standard, RNA extracted from the virus isolate TGEV Purdue was used, and the genome copies were approximated for the run. Standard curves were generated for the data points within the linear dynamic range. Efficiency (E) was calculated automatically from the standard curve by the Bio-Rad CFX Maestro 1.1 program.
Figure 1.
Singleplex standard curves generated on the basis of serially diluted RNA standards. In vitro transcribed RNA was used for the standards for PEDV N (from PEDV CV777), for TGEV N and for PEDV S (from SeCoV). For the TGEV S standard, RNA extracted from the virus isolate TGEV Purdue was used, and the genome copies were approximated for the run. Standard curves were generated for the data points within the linear dynamic range. Efficiency (E) was calculated automatically from the standard curve by the Bio-Rad CFX Maestro 1.1 program.
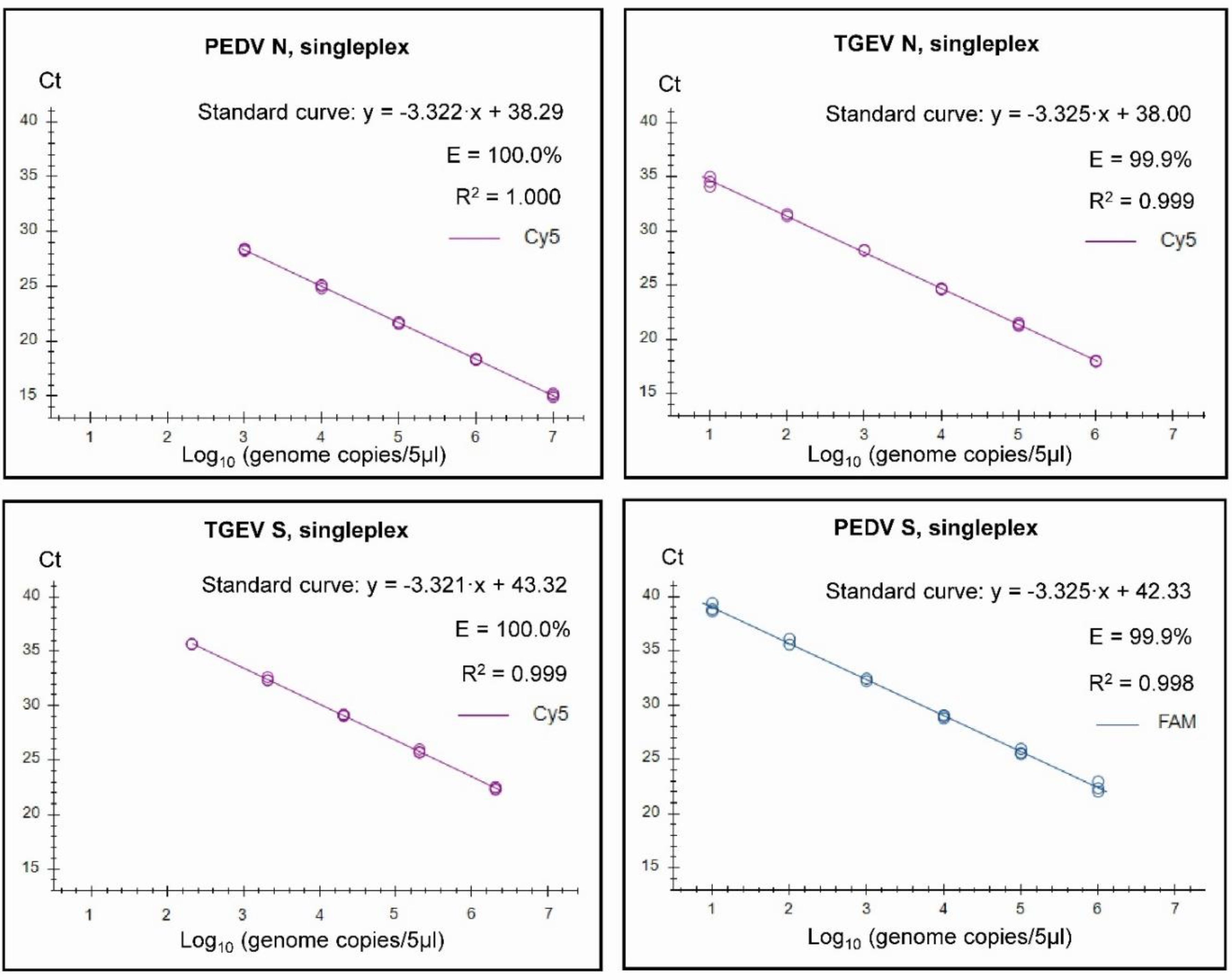
Figure 2.
Standard curves for the three duplex assays generated on the basis of testing serially diluted RNA standards in three replicates. Each tested sample contained a mix of two standards and fluorescence data was collected for the two channels (FAM and Cy5) as indicated.
Figure 2.
Standard curves for the three duplex assays generated on the basis of testing serially diluted RNA standards in three replicates. Each tested sample contained a mix of two standards and fluorescence data was collected for the two channels (FAM and Cy5) as indicated.
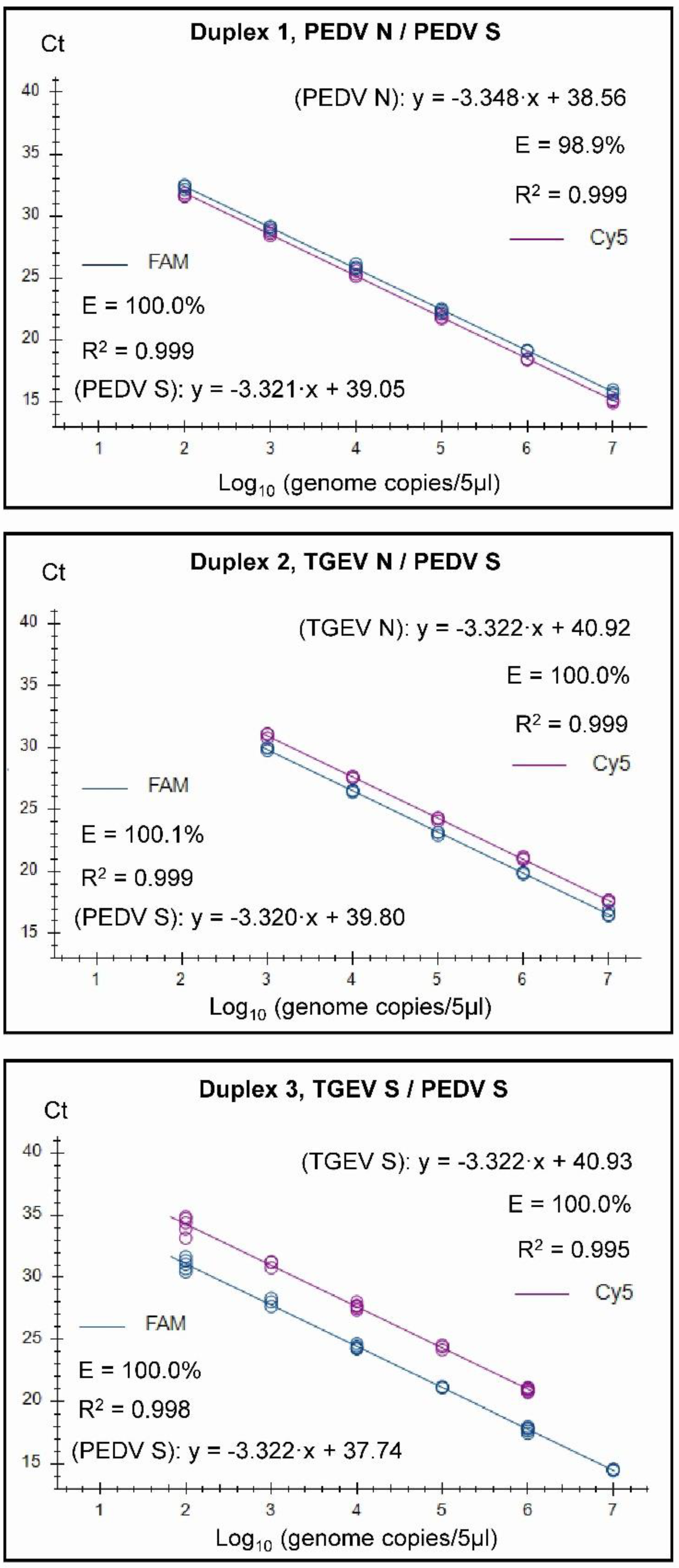
Figure 3.
Singleplex and triplex standard curves generated on the basis of serially diluted RNA standards in three replicates. In vitro transcribed RNA was used for the standards for PDCoV RdRp, for TGEV N and for PEDV N. Standard curves were generated for the data points within the linear dynamic range. Efficiency (E) was calculated automatically from the standard curve by the Bio-Rad CFX Maestro 1.1 program.
Figure 3.
Singleplex and triplex standard curves generated on the basis of serially diluted RNA standards in three replicates. In vitro transcribed RNA was used for the standards for PDCoV RdRp, for TGEV N and for PEDV N. Standard curves were generated for the data points within the linear dynamic range. Efficiency (E) was calculated automatically from the standard curve by the Bio-Rad CFX Maestro 1.1 program.
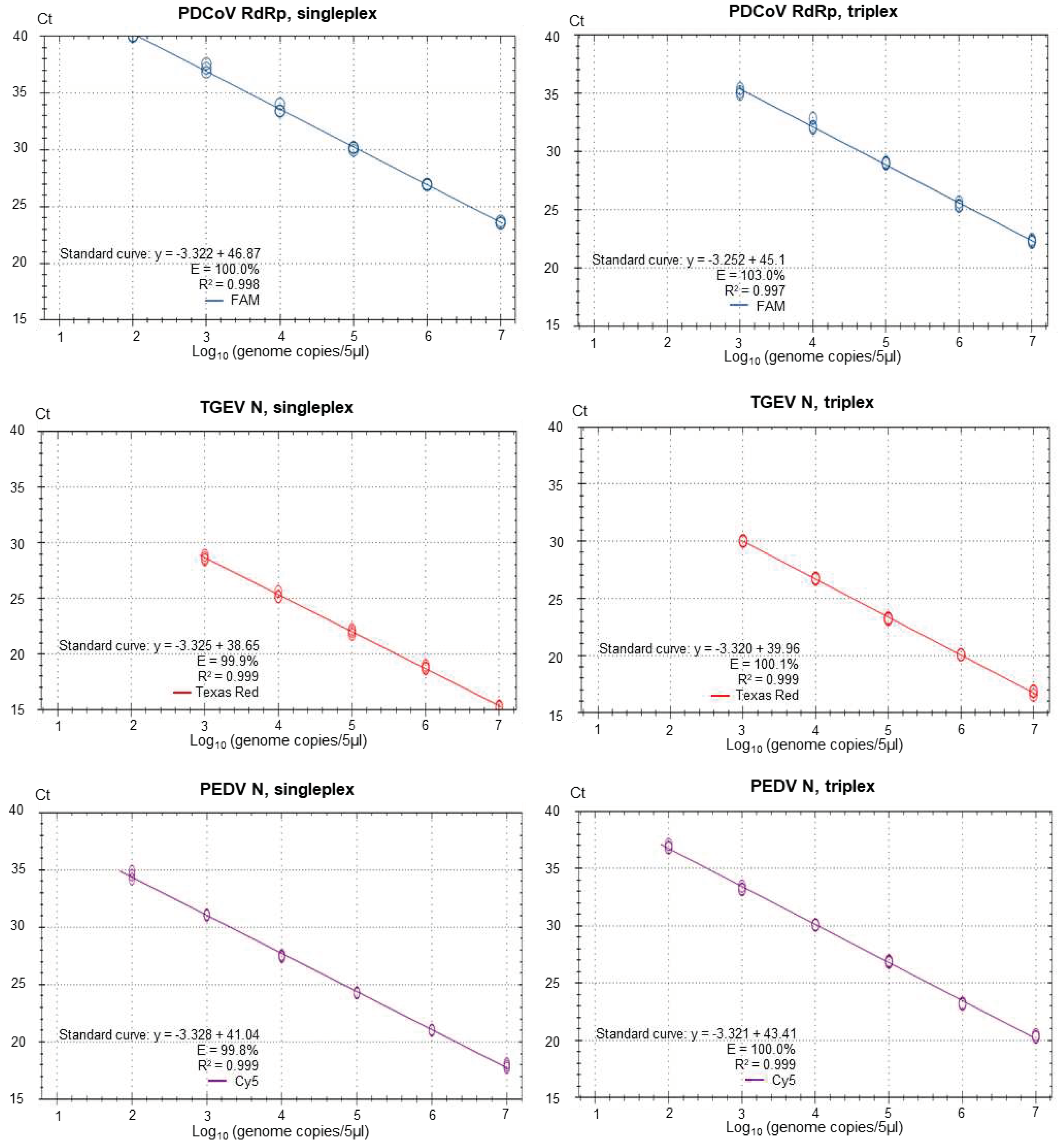

Table 1.
The sequences of the primers and probes used for single and multiplex assays. The forward and reverse primers are indicated, respectively, by the F and R suffix, while probes are indicated by the P suffix. The duplex assay targeting the PEDV N gene included two forward (F) primers that varied in their last three nucleotides at the 3´-end (underlined). Changes were made to some of the oligonucleotides compared to the originally published sequences: the duplex PEDV N probe (PEDV-N-P) as well as the duplex primers of the TGEV N assay included a single degenerate base R (purine, marked in bold) corresponding to the nucleotides A and G. The forward primer of TGEV N gene is identical in both the duplex and triplex assays. The duplex PEDV-N-P was a mixture of the two already published probe sequences. The triplex PEDV R primer was modified to include genetic diversity among recently circulating PEDV strains. For the duplex and triplex TGEV N assays, these were new modifications to accommodate variations seen in recent SeCoV genomes detected in Italy.
Table 1.
The sequences of the primers and probes used for single and multiplex assays. The forward and reverse primers are indicated, respectively, by the F and R suffix, while probes are indicated by the P suffix. The duplex assay targeting the PEDV N gene included two forward (F) primers that varied in their last three nucleotides at the 3´-end (underlined). Changes were made to some of the oligonucleotides compared to the originally published sequences: the duplex PEDV N probe (PEDV-N-P) as well as the duplex primers of the TGEV N assay included a single degenerate base R (purine, marked in bold) corresponding to the nucleotides A and G. The forward primer of TGEV N gene is identical in both the duplex and triplex assays. The duplex PEDV-N-P was a mixture of the two already published probe sequences. The triplex PEDV R primer was modified to include genetic diversity among recently circulating PEDV strains. For the duplex and triplex TGEV N assays, these were new modifications to accommodate variations seen in recent SeCoV genomes detected in Italy.
| Oligo | Sequence 5´ - 3´ | Location** | References | |
|---|---|---|---|---|
| Duplex assays | PEDV-N-R | TTGCCTCTGTTGTTACTTGGAGAT | 26,853-26,876 | [17] |
| PEDV-N-F | CGCAAAGACTGAACCCACTAATTT | 26,679-26,702 | ||
| PEDV-N-US-F | CGCAAAGACTGAACCCACTAACCT | 26,679-26,702 | [17,20] | |
| PEDV-N-P | TGTTGCCATTRCCACGACTCCTGC | 26,819-26,842 | ||
| TGEV-N-F | GCAGGTARAGGTGATGTGACAA | 27,637-27,658 | [17] | |
| TGEV-N-R | ACATTCAGCCARTTGTGGGTAA | 27,735-27,756 | ||
| TGEV-N-P | TGGCACTGCTCCCATTGGCAACGA | 27,707-27,730 | ||
| PEDV-S-F | ACGTCCCTTTACTTTCAATTCACA | 22,474-22,497 | [19] | |
| PEDV-S-R | TATACTTGGTACACACATCCAGAGTCA | 22,559-22,585 | ||
| PEDV-S-P | TGAGTTGATTACTGGCACGCCTAAACCAC | 22,503-22,531 | ||
| TGEV-S-F | TCTGCTGAAGGTGCTATTATATGC | 20,734-20,757 | [21] | |
| TGEV-S-R | CCACAATTTGCCTCTGAATTAGAA* | 20,856-20,879 | ||
| TGEV-S-P | *AAGGGCTCACCACCTACTACCACCA | 20,764-20,788 | ||
| Triplex assay | PDCoV-RdRp-F | AACTGACATGAATGTTGGCCCT | 13,777-13,798 | This study |
| PDCoV-RdRp-R | CATGCACCCAGAATGCGAGA | 13,874-13,893 | ||
| PDCoV-RdRp-P | AGCATACTGTGTTAGCAGAGCATGATGGT | 13,815-13,843 | ||
| TGEV-N-F | GCAGGTARAGGTGATGTGACAA | 27,637-27,658 | [17] | |
| Triplex-TGEV-N-R | TGCTRGACACAGATGGAACACA | 27,754-27,775 | ||
| Triplex-TGEV-N-P | GGAGCAGTGCCAAGCATTACCCACAA | 27,719-27,744 | ||
| Triplex-PEDV-N-F | CGCAAAGACTGAACCCACTAAC | 26,679-26,699 | [22] | |
| Triplex-PEDV-N-R | TGGTTRTTGCCTCTGTTGTTACT | 26,860-26,882 | ||
| Triplex-PEDV-N-P | TGTTGCCATTGCCACGACTCCTGC | 26,819-26,842 |
*In the TGEV S assay, a G at the 3´-end of the R primer as well as the C/T degenerate nucleotide at the 5´-end of the P were removed compared to the published assay. ** Locations of the oligonucleotides in the genome are indicated relative to reference sequences: PEDV strain CV777 (Acc. no. NC_003436), TGEV strain Purdue (Acc. no. NC_038861) and PDCoV strain USA/Illinois121/2014 (Acc. no. KJ481931), respectively.
Table 2.
Expected characteristics of the three duplex and triplex real-time RT-PCR assays with highlighted limitations (in bold font) for detection and differentiation between selected porcine coronaviruses. N/A= not applicable.
Table 2.
Expected characteristics of the three duplex and triplex real-time RT-PCR assays with highlighted limitations (in bold font) for detection and differentiation between selected porcine coronaviruses. N/A= not applicable.
| Expected assay characteristics | Duplex 1 | Duplex 2 | Duplex 3 | Triplex |
|---|---|---|---|---|
| Target genes | PEDV N + PEDV S |
TGEV N + PEDV S |
TGEV S + PEDV S |
PDCoV + TGEV N + PEDV N |
| Detects PEDV | Yes | Yes | Yes | Yes |
| Detects SeCoV | Yes | Yes | Yes | Yes |
| Detects TGEV | No | Yes | Yes | Yes |
| Detects PRCV | No | Yes | No | Yes |
| Detects PDCoV | No | No | No | Yes |
| Distinguishes PEDV from SeCoV | Yes | Yes | No | Yes |
| Distinguishes SeCoV from TGEV/PRCV | N/A | Yes | Yes | No |
| Distinguishes TGEV from PRCV | N/A | No | N/A | No |
Table 3.
The conditions for the three duplex and triplex real-time RT-PCR assays, with primer and probe final concentrations and their gene targets viruses.
Table 3.
The conditions for the three duplex and triplex real-time RT-PCR assays, with primer and probe final concentrations and their gene targets viruses.
| Assay | Gene target (Virus detected) | Forward primer | Conc. | Reverse primer | Conc. | Probe | Conc. |
|---|---|---|---|---|---|---|---|
| Duplex 1 |
PEDV N (PEDV) |
PEDV-N-F | 400 nM | PEDV-N-R | 800 nM | PEDV-N-probe (Cy5) |
240 nM |
| PEDV-N-US-F | 400 nM | ||||||
|
PEDV S (PEDV + SeCoV) |
PEDV-S-F | 900 nM | PEDV-S-R | 900 nM | PEDV-S-probe (FAM) |
200 nM | |
| Duplex 2 |
TGEV N (TGEV/PRCV + SeCoV) |
TGEV-N-F | 700 nM | TGEV-N-R | 700 nM | TGEV-N-probe (Cy5) |
200 nM |
|
PEDV S (PEDV + SeCoV) |
PEDV-S-F | 900 nM |
PEDV-S-R | 900 nM | PEDV-S-probe (FAM) |
200 nM | |
| Duplex 3 |
TGEV S (TGEV) |
TGEV-S-F | 900 nM | TGEV-S-R | 900 nM | TGEV-S-probe (Cy5) |
200 nM |
|
PEDV S (PEDV + SeCoV) |
PEDV-S-F | 900 nM | PEDV-S-R | 900 nM | PEDV-S-probe (FAM) |
200 nM | |
| Triplex | PDCoV RdRp(PDCoV) | PDCoV-RdRp-F | 900 nM | PDCoV-RdRp-R | 900 nM | PDCoV-RdRp-P (FAM) | 200 nM |
|
TGEV N (TGEV/PRCV + SeCoV) |
TGEV-N-F | 900 nM | TGEV-N-R | 900 nM | TGEV-N-P (Texas Red) |
200 nM | |
|
PEDV N (PEDV) |
PEDV-N-F | 700 nM | PEDV-N-R | 700 nM | PEDV-N-P (Cy5) |
200 nM |
Table 4.
Summary of the limits of detection (LOD) (expressed as RNA copies/5µl) for each of the indicated assays in the singleplex and multiplex formats. The assays were performed using the primers and probes shown in Table 1.
Table 4.
Summary of the limits of detection (LOD) (expressed as RNA copies/5µl) for each of the indicated assays in the singleplex and multiplex formats. The assays were performed using the primers and probes shown in Table 1.
| LOD of duplex assays | LOD of triplex assay | |||||
|---|---|---|---|---|---|---|
| Assay | Singleplex | Duplex 1 | Duplex 2 | Duplex 3 | Singleplex | Triplex |
| PEDV N | 1000* | 50 | - | - | ||
| PEDV S | 10 | 25 | 25 | 25 | ||
| TGEV N | 25 | - | 100 | - | ||
| TGEV S** | 50 | - | - | 100 | ||
| PDCoV RdRp | 100 | 1000 | ||||
| TGEV N | 25 | 25 | ||||
| PEDV N | 10 | 25 | ||||
*No consistent detection of the target in the singleplex PEDV N assay was obtained in concentrations below 1000 copies/5µl although individual replicates were positive in concentrations down to 200 copies/5µl. **Note that the TGEV LOD was determined based on an estimated concentration for the TGEV Purdue isolate RNA as described in Materials and Methods.
Table 5.
Summary of intra-assay and inter-assay variation expressed as coefficient of variation (CV) from tests of three samples of high, intermediate and low concentrations (conc.) of template, i.e. 106, 105/104 and 102 copies/5μl for the duplex assays and 106, 104 and 103/102 copies/5μl for the triplex assay. The intra-assay variation was tested in six replicates, while the inter-assay was calculated in three replicates for four runs.
Table 5.
Summary of intra-assay and inter-assay variation expressed as coefficient of variation (CV) from tests of three samples of high, intermediate and low concentrations (conc.) of template, i.e. 106, 105/104 and 102 copies/5μl for the duplex assays and 106, 104 and 103/102 copies/5μl for the triplex assay. The intra-assay variation was tested in six replicates, while the inter-assay was calculated in three replicates for four runs.
| Tested sample for repeatability | Tested sample for reproducibility | ||||||
|---|---|---|---|---|---|---|---|
| Assay | High conc. | Intermediate conc. | Low Conc | High conc. |
Intermediate conc. | Low conc. | |
| Duplex 1 | PEDV N | 4% | 6% | 17% | 27% | 27% | 16% |
| PEDV S | 7% | 8% | 31% | 29% | 34% | 22% | |
| Duplex 2 | TGEV N | 4% | 6% | 43% | 27% | 27% | 24% |
| PEDV S | 6% | 7% | 19% | 29% | 33% | 43% | |
| Duplex 3 | TGEV S | 7% | 11% | 110% | 19% | 21% | *37% |
| PEDV S | 4% | 3% | 11% | 32% | 34% | 35% | |
| Triplex | PDCoV RdRp | 10% | 16% | 9% | 14% | 16% | 4% |
| TGEV N | 5% | 7% | 13% | 10% | 6% | 32% | |
| PEDV N | 5% | 8% | 22% | 5% | 7% | 16% | |
* Five replicates were negative and excluded from the calculation.
Table 6.
Summary of sensitivity testing for porcine alphacoronaviruses using the duplex and triplex assays. A panel of 55 field samples of PEDV and SeCoV, including recombinant PEDV/SeCoVs, were tested in each of the multiplex assays. The number and percentages of tested samples testing positive in each assay are shown. Negative results are highlighted in grey. The full set of results (including Ct values) are shown in Supplementary Table S2.
Table 6.
Summary of sensitivity testing for porcine alphacoronaviruses using the duplex and triplex assays. A panel of 55 field samples of PEDV and SeCoV, including recombinant PEDV/SeCoVs, were tested in each of the multiplex assays. The number and percentages of tested samples testing positive in each assay are shown. Negative results are highlighted in grey. The full set of results (including Ct values) are shown in Supplementary Table S2.
| Duplex 1 | Duplex 2 | Duplex 3 | Triplex | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pathogen samples | No. tested | PEDV N positive (%) | PEDV S positive (%) | TGEV N positive (%) | PEDV S positive (%) | TGEV S positive (%) | PEDV S positive (%) | PDCoV RdRp positive (%) | TGEV N positive (%) | PEDV N positive (%) |
| PEDV | 24 | 23 (96) | 24 (100) | 0 (0) | 24 (100) | 0 (0) | 24 (100) | 0 (0) | 0 (0) | 23 (96) |
| Rec. SeCoV/PEDV | 22 | 22 (100) | 22 (100) | 0 (0) | 22 (100) | 0 (0) | 22 (100) | 0 (0) | 0 (0) | 20 (91) |
| SeCoV | 9 | 0 (0) | 9 (100) | 9 (100) | 9 (100) | 0 (0) | 9 (100) | 0 (0) | 9 (100) | 0 (0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



