
Submitted:
30 June 2023
Posted:
30 June 2023
You are already at the latest version
Abstract
Bibliometric studies on inherited metabolic diseases(IMDs) didn't exist in the literature.Our research aims to conduct a bibliometric study to determine the current status,trending topics,and missing points of publications on IMDs.Between 1968-2023, we conducted a literature search with keyword "inherited metabolic disease" in the SCOPUS-database.We included research articles in medicine written in English,published in the final section.We created our data pool using VoSviewer, SciMAT, and Rstudio software programs for bibliometric parameters of the articles that met the inclusion criteria.We performed bibliometric analysis of the data with R-package "bibliometrix" and BibExcel programs.We included 2702 research articles published on IMDs.Top three countries that have written the most articles in this field are; USA(n= 501), United Kingdom(n=182), and China(n= 172).The most preferred keywords by the authors were;newborn screening(n=54), mutation(n=43), phenylketonuria(n=42), children(n=35), genetics(n= 34) and maple syrup urine disease(n=32).Trending topics were osteoporosis, computed tomography, bone marrow transplantation in the early years of the study, chronic kidney disease, urea cycle disorders, next-generation sequencing, newborn screening, and familial hypercholesterolemia in the final years of the study. This study provides clinicians with a new perspective showing that molecular and genetic studies in inherited metabolic diseases will play an essential role in diagnosis and treatment in the future.
Keywords:
Inherited metabolic disease
; bibliometric study
; newborn screening
; gene therapy
; metabolomics
; molecular genetic analysis
; phenylketonuria
1. Introduction
Inherited metabolic diseases are rare diseases caused by a genetic mutation and have unique biochemical, clinical, and pathophysiological consequences depending on the specific biochemical pathway affected [1,2]. Because of the advances in genetics and biochemistry, clinicians have more information about disease pathophysiology, early diagnosis, and treatment of inherited metabolic diseases [3,4,5]. Owing to recent studies, the positive contribution of early diagnosis and treatment on survival and morbidity in inherited metabolic diseases has led to the introduction of prenatal diagnosis methods and postnatal screening programs to diagnose these diseases [6,7,8,9]. Sir Archibald Garrod described the first inherited metabolic disease in 1908 as alkaptonuria [10]. He discovered the disease’s metabolic pathway and its relationship with Mendelian inheritance [10]. With these, the importance of biochemical analysis and genetics in medicine and inherited metabolic diseases was recognized for the first time. Then, in 1934, Ivar Asbjörn Følling discovered phenylketonuria, one of the most prevalent inherited metabolic diseases [11]. In the 1960s, Robert Guthrie found a diagnostic test with his name, and newborn screening started for phenylketonuria [12]. With the success of preventing mental retardation with dietary treatment, newborn screening for phenylketonuria has become widespread worldwide [13]. In the 1990s, with the introduction of tandem mass spectrometry, newborn screening for more than 50 inherited metabolic diseases have been started to be screened in certain places in the world [14]. In recent years, in some places, lysosomal storage diseases like Krabbe, Pompe, Fabry, Gaucher, Niemann-Pick, and Mucopolysaccharidosis, type I and II and also X-linked adrenoleukodystrophy a peroxisomal disease were included in the newborn screening program [15]. There is an increase in the number of metabolic diseases, which are diagnosed by developments in biochemistry and genetics, and in parallel, there are expansions in the newborn screening program. In the 1960s, 80 inherited metabolic diseases were mentioned, but this number increased to over 1000 in the 2020s [10]. This situation shows the development of inherited metabolic diseases over the years and the importance of following the literature on this subject. Evaluating the literature from a broad perspective regarding the trending topics over the years and the least mentioned topics that perhaps show the need to fill the lack of knowledge in this field is essential.
Bibliometric studies are one of the literature review methods that reveal the current status of publications in a particular research field and evaluate the quantitative and qualitative characteristics of these publications [16]. Bibliometric studies contribute to the development of scientific research by presenting the latest analysis of the publications in a particular research field, the latest current approaches, and the gaps that need to be filled in the literature [17]. Because of the bibliometric analysis, we can know about the trending topics, keywords, authors, journals, countries, etc. [18]. Bibliometric analysis programs such as CiteSpace [19], VoSviewer [20], R package “bibliometrix” [21], and HistCite [22] are used to visualize bibliometric analyses. Many bibliometric analysis studies have been carried out in different medical fields in recent years [23,24,25,26]. However, we found no bibliometric studies on inherited metabolic diseases when we examined the literature.
The study aims to make bibliometric analysis of published literature on inherited metabolic diseases. In this way, it is desired to illuminate the latest status, current issues, and missing points in the literature in this field.
2. Materials and Methods
2.1. Study Design and Search Strategy
We conducted this study retrospectively and cross-sectionally. Before starting the research, we received ethical approval from the ethics committee of Selcuk University Faculty of Medicine (Date 06.06.2023 Decision No: 2023/292). All authors participating in the study read and signed the Declaration of Helsinki. We carried out the study between 07.06.2023-23.06.2023. We conducted a literature search with the keyword “inherited metabolic disease” in the SCOPUS database between 1 January 1968 and 1 May 2023. We created our data pool using VoSviewer, SciMAT, and Rstudio software programs for the bibliometric parameters of the articles that met the inclusion criteria. We performed the bibliometric analysis of the data by creating mapping and table visuals with the R package “bibliometrix” and BibExcel programs.
2.2. Inclusion and Exclusion Criteria
We included articles in the field of medicine written in English, published in the final section, and not in the type of book or book series. We excluded from the study; the articles published in non-medicine fields (e.g., Pharmacology, Toxicology and Pharmaceutics, Nursing, Chemical Engineering, etc.), publications of books and book series, publications of non-research articles (e.g., review, case report, letter to the editor, etc.), articles in the “article in press” stage of which the final part was not published, and the articles published non-English language.
2.3. Data Collection and Statistics
In this study, we used bibliometric parameters, including the article title, publication year, authors, country of the corresponding author (first author of the study), the name of the journal in which it was published, keywords, and the number of citations to the article. We categorized the study data we obtained with a computer software program and expressed them numerically without the need for any statistical program.
3. Results
3.1. Analysis of Data
3.1.1. General Analysis of Published Articles on Inherited Metabolic Diseases
We included 2702 research articles published on inherited metabolic diseases within the specified dates and meeting the inclusion criteria. The flowchart of inclusion and exclusion is shown in detail in Figure 1. In these publications to which 14782 authors contributed, the annual growth rate was 3.9%, and the average citations per doc were 26.4%. General information about the study is given in Table 1.
3.1.2. Distribution of Main Authors
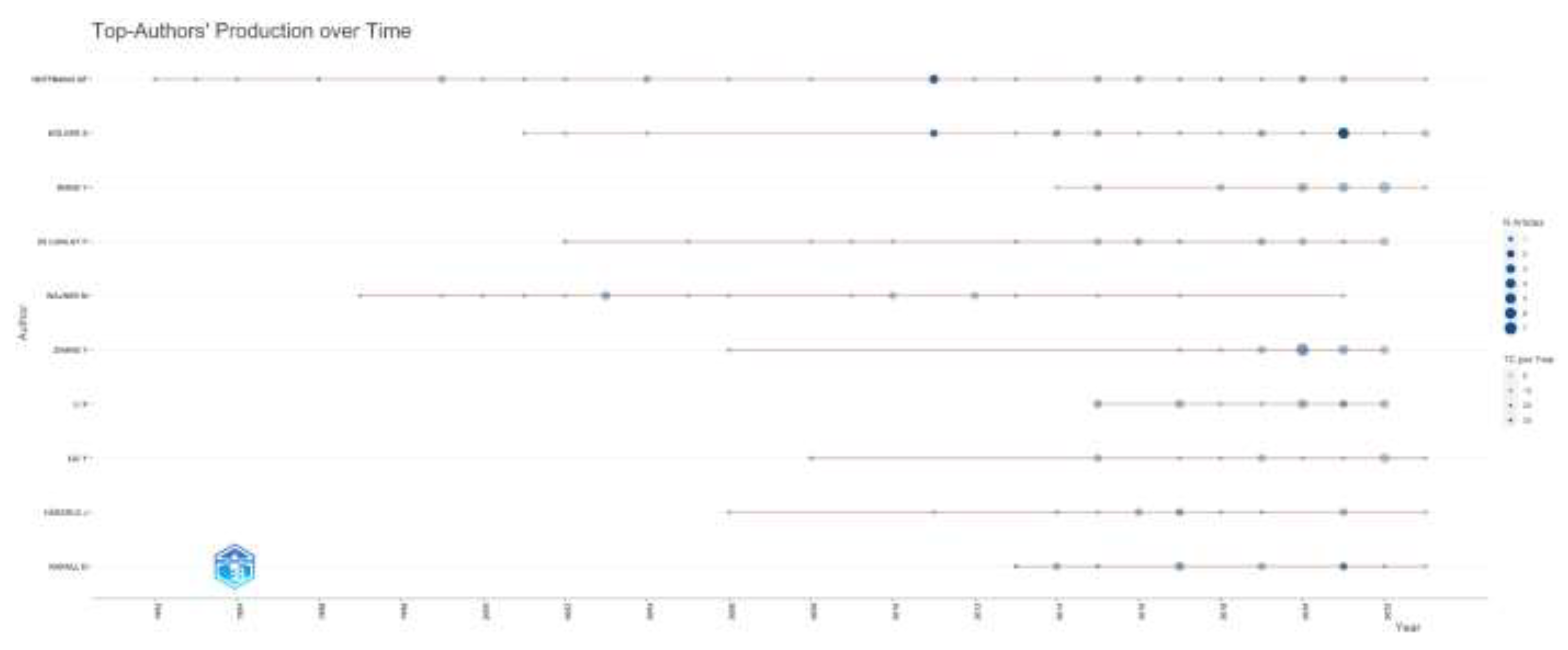
According to the number of articles on inherited metabolic diseases, Hoffmann GF. (n=30), Kölker S. (n=24), and Wang Y. (n=20) were the authors who wrote the most articles. According to their impacts, Hoffman GF. (H-index=22), Kolker S. (H-index=14), and Wagner M. (H-index=13) were the most influential authors. When the productivity of the authors according to the years is examined, it is seen as high, especially in the last ten years. The number of articles, the influence of the top ten authors who have publications in this field, and the distribution of the authors’ article productivity by years are given in Figure 2.
Figure 2a.
Most relevant authors’ number of articles are shown. N;number.
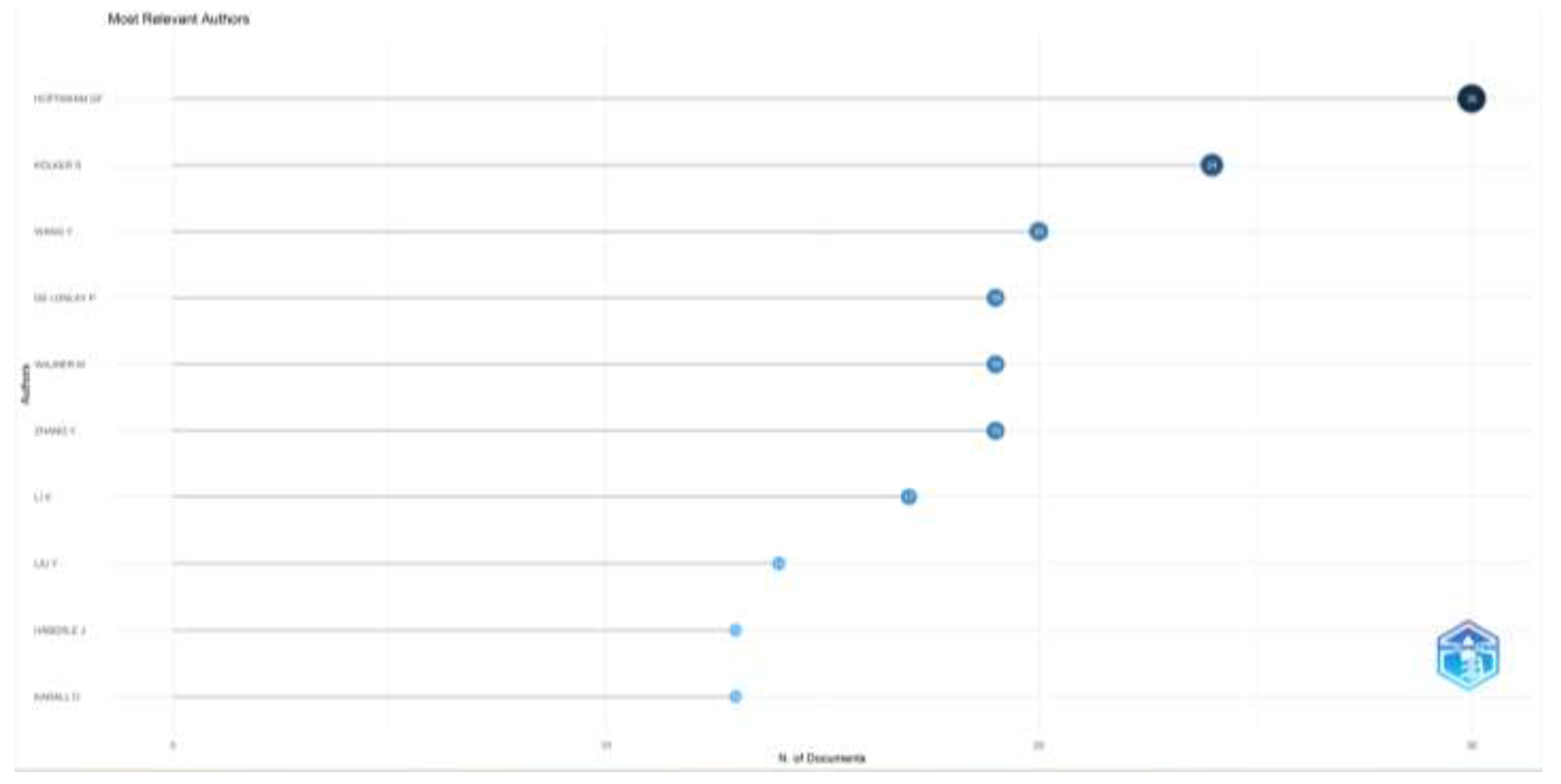
Figure 2b.
Author impact by H-index are shown.
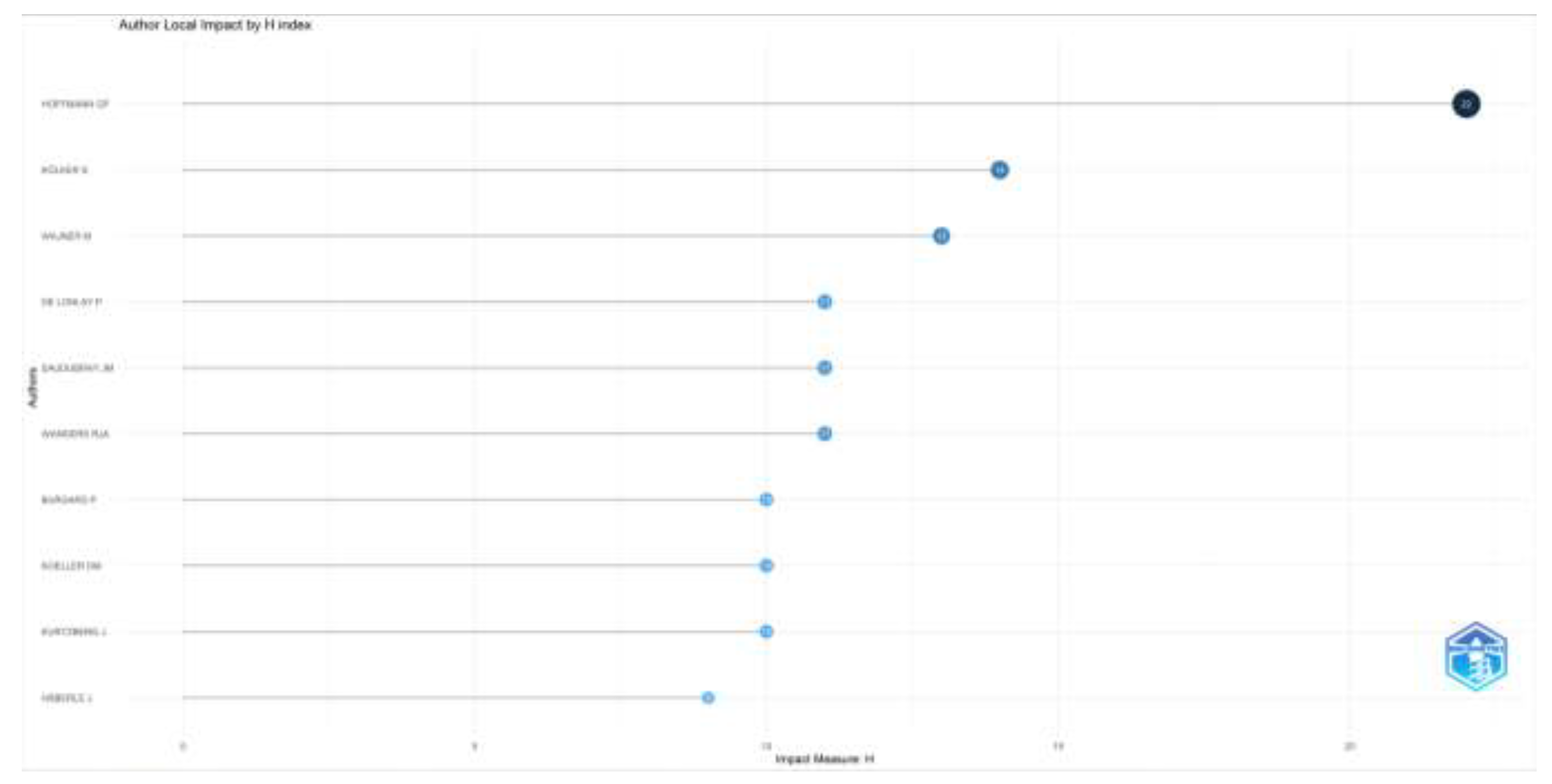
Figure 2c.
Top-Authors’ Production over time is shown.
3.1.3. Distribution of Main Journals
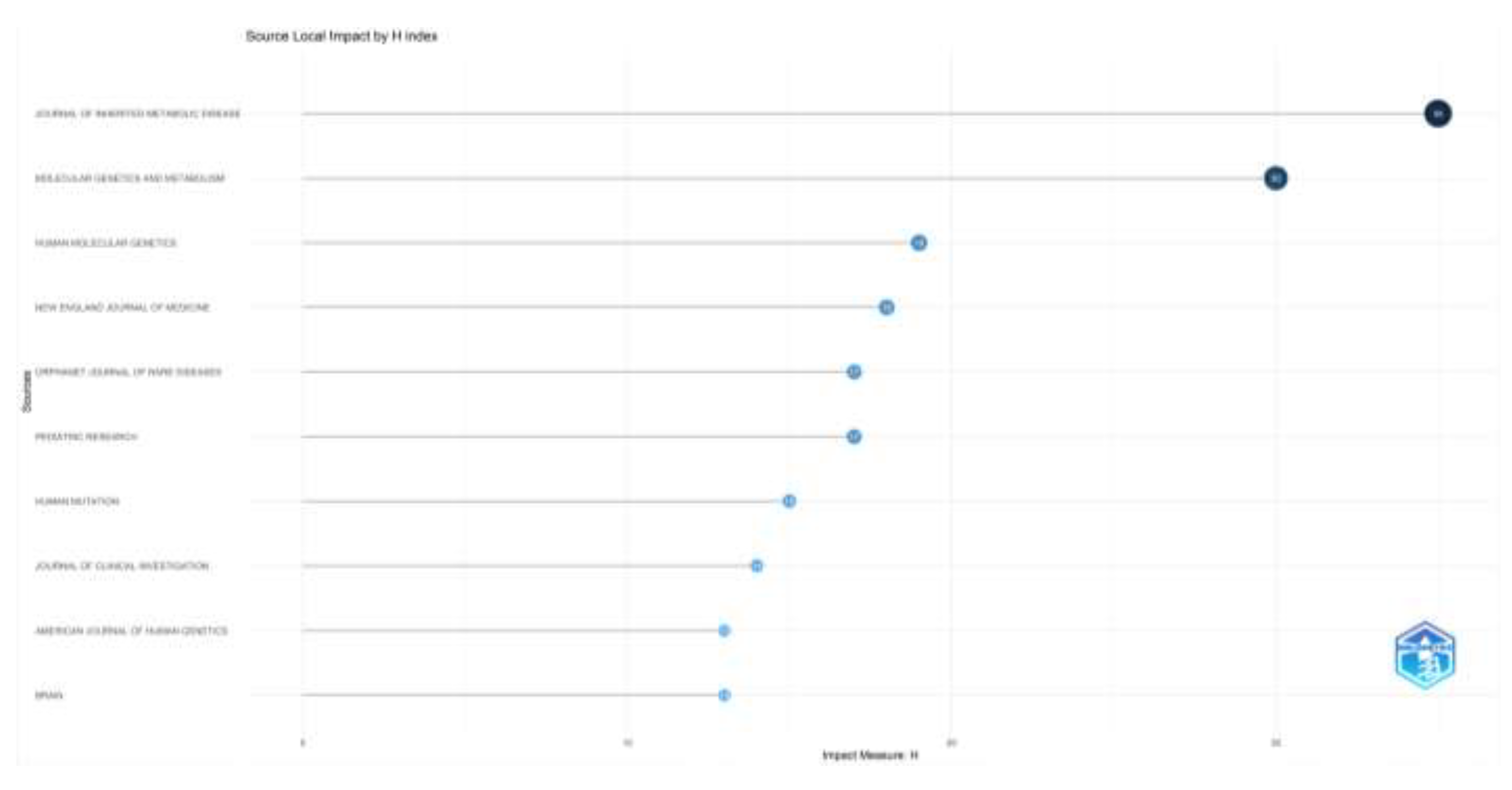
When the number of articles and the impact factor of the journals that publish articles on inherited metabolic diseases are examined, the Journal of Inherited Metabolic Disease (n=169), Molecular Genetics and Metabolism (n=90), Orphanet Journal of Rare Diseases (n=64) are the first three journals to publish the most articles; Journal of Inherited Metabolic Disease (H-index= 35), Molecular Genetics and Metabolism (H-index=30), and Human Molecular Genetics (H-index=19) were the most influential journals. The number of articles and the impact factor of the top ten journals that publish articles in this field are given in Figure 3.
Figure 3a.
Most relevant Journals’ number of documents are shown.
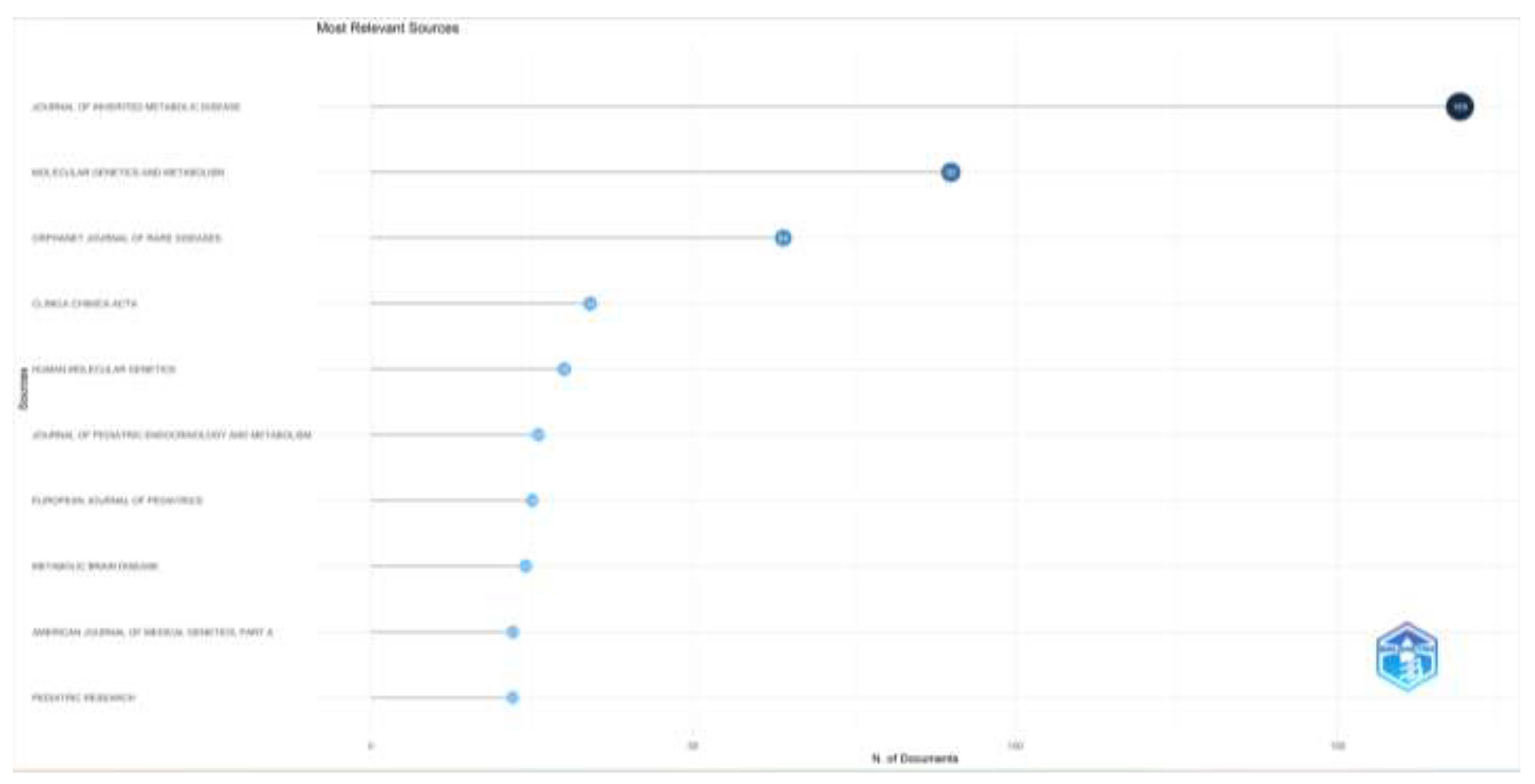
Figure 3b.
Journals’ impact by H-index are shown.
3.1.4. Analysis of Corresponding Authors’ Countries
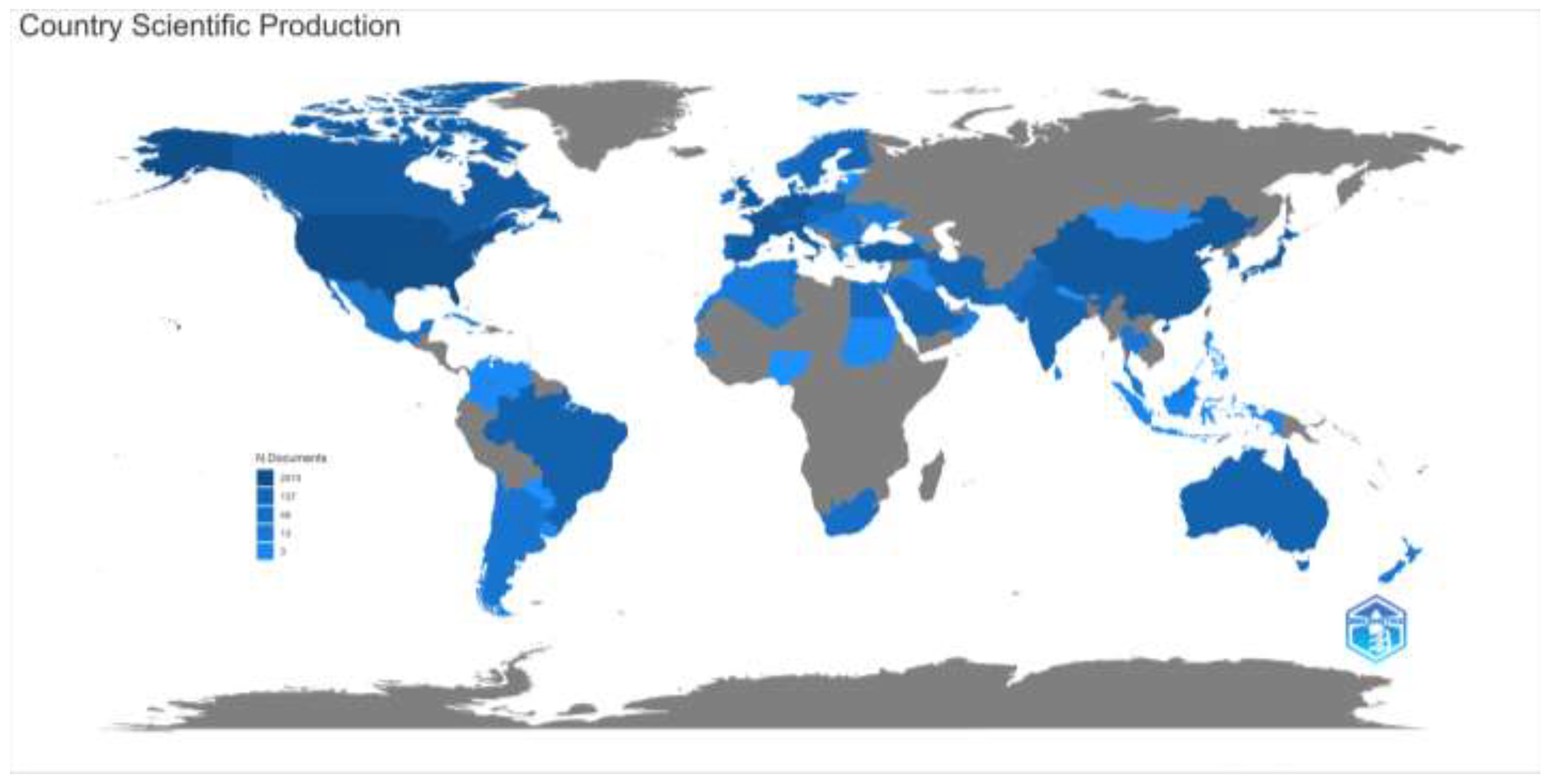
When the distribution of the articles on inherited metabolic diseases according to the countries of the corresponding authors is examined, the USA (n= 501), the United Kingdom (n=182), and China (n= 172) were in the top three places. We found that studies in China, Turkey, and Japan are mainly carried out in a single center, while multicenter studies are mainly carried out in the USA and European countries. Again, when the distribution of countries on the world map according to the article productivity is examined, we found that productivity is high in developed countries such as the USA, European countries, and Australia and developing countries such as China. However, the production of articles in this field is low in Russia and the least developed countries in Africa. The distribution of the articles according to the countries on the world map, the number of articles in the top ten countries according to the countries of the corresponding authors, and the number of single or multi-centered studies are given in Figure 4.
Figure 4a.
Number of documents of corresponding authors’ country and also single country or multiple country publications are shown.
Figure 4a.
Number of documents of corresponding authors’ country and also single country or multiple country publications are shown.
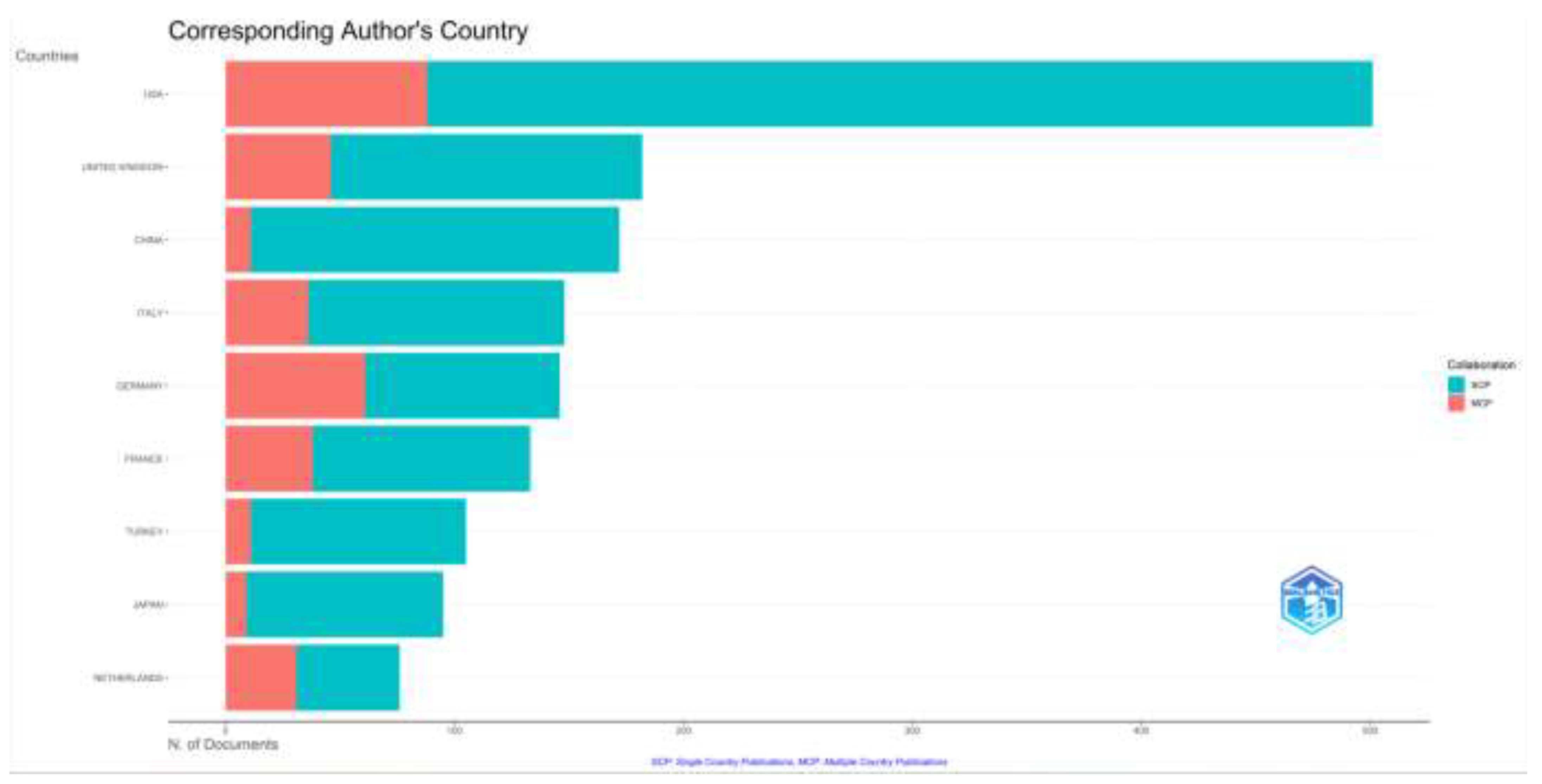
Figure 4b.
Distribution of countries by article productivity are shown.
3.1.5. Analysis of Keywords
In the evaluation of the keywords preferred by the authors in the research articles on inherited metabolic diseases, according to the number of articles, the most preferred six keywords were newborn screening (n=54), mutation (n=43), phenylketonuria (n=42), children (n=35), genetics (n= 34) and maple syrup urine disease (n=32) (Table 2). The distribution of the 100 most preferred keywords by the authors is given in Figure 5.
Figure 5.
The most frequent 100 keywords used by authors are shown. The most frequent keywords are shown bigger, and the less frequent keywords are shown smaller.
Figure 5.
The most frequent 100 keywords used by authors are shown. The most frequent keywords are shown bigger, and the less frequent keywords are shown smaller.

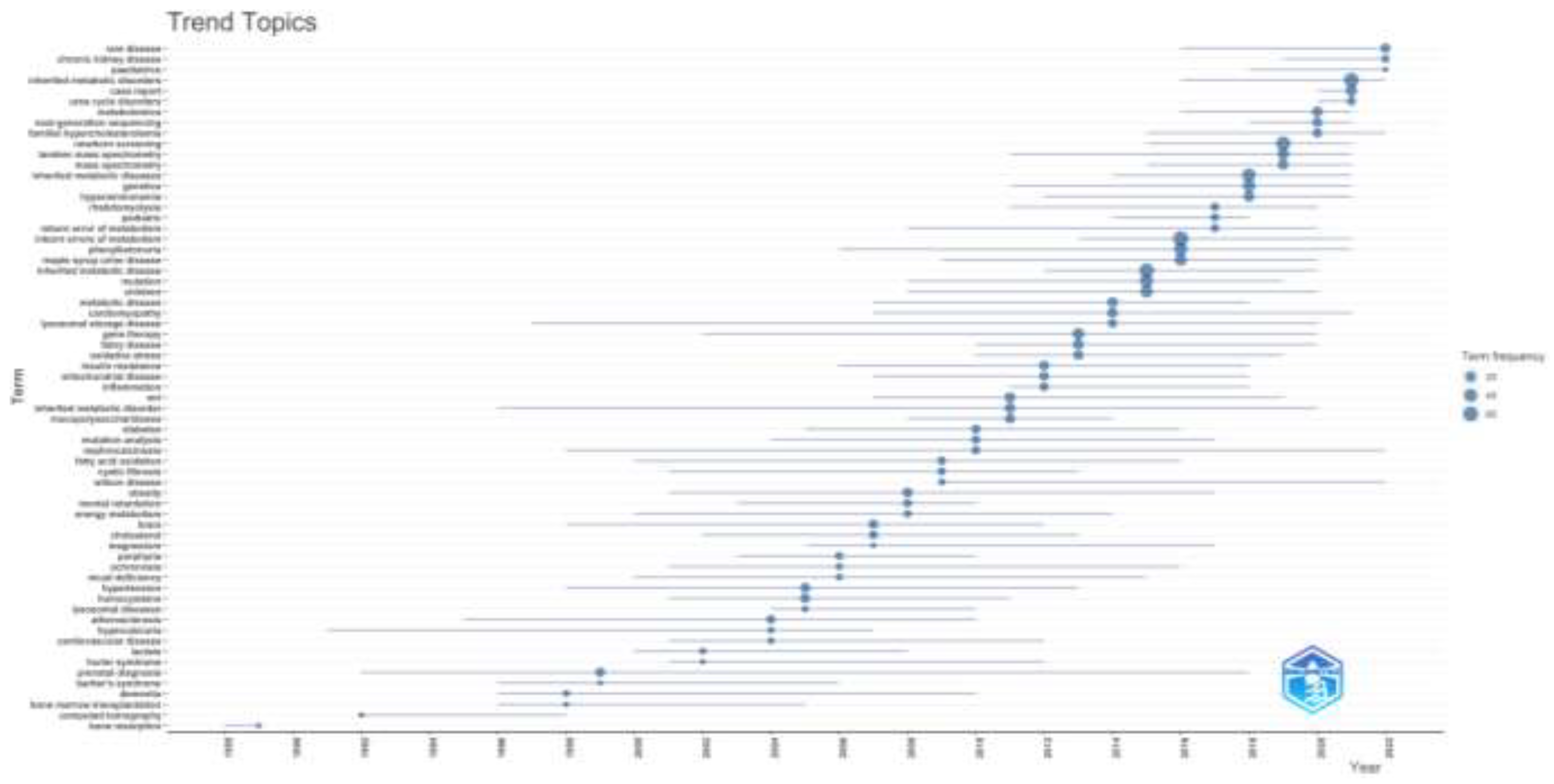
3.1.6. Trending Topics
When the trending topics in the field of inherited metabolic diseases are evaluated according to the years, it was seen that bone resorption, computed tomography, and bone marrow transplantation subjects were at the forefront in the first periods of the study. Chronic kidney disease, urea cycle disorders, next-generation sequencing, newborn screening, and familial hypercholesterolemia subjects were preferred in recent years. It is seen that prenatal diagnosis and cardiomyopathy trending topics have a wider distribution over the years. It is observed that the subjects of porphyria, hypercalciuria, ochronosis, and mucopolysaccharidoses have decreased in the last ten years, while studies on the subjects of Fabry disease, gene therapy, lysosomal storage disease, maple syrup urine disease, phenylketonuria have increased. In addition, physicians working on inherited metabolic diseases have recently focused on diagnostic methods such as newborn screening, next-generation sequencing, and tandem mass spectrometry. The distribution of trending topics by year is shown in Figure 6.
Figure 6.
The most frequent keywords used by authors according to the years are shown.
4. Discussion
In this bibliometric study, in which we evaluated research articles published on inherited metabolic diseases, we evaluated the latest status of publications on this subject, current issues, and missing points in the literature over a wide period of time. Our study is important as it is the first bibliometric study on inherited metabolic diseases.
Most inherited metabolic diseases show autosomal recessive inheritance [27]. For this reason, it is expected to be more common in least developed and developing countries with a high level of consanguineous marriage. However, in our study, we found that authors from developed countries such as the USA and the United Kingdom published more articles on inherited metabolic disease. These results were similar to many bibliometric studies in other medical fields [28,29,30]. In developed countries, existing better data recording automation systems may be a reason for this situation. The novelty and reliability of information are among the publication criteria of journals in scientific research [31]. Robust data recording systems of developed countries may have contributed to the productivity of publications in developed countries by ensuring the reliability of the information. In addition, more funds and resources are provided in developed countries for research on the diagnosis and treatment of metabolic diseases may be another reason. The fact that the multicenter studies in our study are less in countries such as Turkey and China than in the USA and European countries also supports this situation. In addition, unlike in developed countries, clinicians dealing with inherited metabolic diseases in these countries are gathered in certain centers, and the inadequacy of the country-wide data recording system may cause studies to be conducted in a single center. The inadequacy of studies in developing countries does not mean that inherited metabolic diseases are less common in these countries. It is due to the difficulties in diagnosing for the reasons described above.
In our study, newborn screening was the main topic that clinicians researched on inherited metabolic diseases were most interested in. Newborn screening, first initiated in the 1960s to diagnose patients with phenylketonuria, has gradually become a part of countries’ national health policies as an important public health initiative to prevent disability and death [32]. Although the number of diseases investigated varies according to country, the scope of newborn screening has been expanded in recent years with the increase in the number of defined metabolic diseases [10,32]. For example, the issue of including lysosomal storage diseases treated with enzyme replacement therapy in the screening program has come to the fore. However, the presence of patients with borderline enzyme activity, the inability to adequately interpret new gene variants, whether the affected individuals need treatment in the pre-symptomatic stage, and the concerns that the balance between the cost and benefit of treatment cannot be achieved have led to debates about inclusion of these disease in newborn screening [10]. In a program organized in New York in 2006, the treatment results of five newborns diagnosed with Krabbe disease after screening 2 million newborns increased the debate on the search for lysosomal storage diseases in newborn screening [33]. Because there were concerns such as not knowing how many Krabbe patients would become symptomatic, not being able to distinguish between newborns affected and unaffected by the disease, undesired treatment results, treatment complications, and ethical concerns. The interest of clinicians working on inherited metabolic diseases in this field may be their desire to eliminate these uncertainties in newborn screening and to bring an international standard. Early diagnosis can reduce mortality and morbidity by providing early treatment for inherited metabolic diseases. For this reason, screening for the highest number of inherited metabolic diseases in newborns is essential, considering treatment efficacy, ethical issues, and cost-benefit balance. We believe that newborn screening will maintain its popularity in the future.
Phenylketonuria (PKU) was also the starting point for newborn screening as being one of the first treated inherited metabolic disease [32,34]. Patients diagnosed with PKU by newborn screening were treated with a low phenylalanine diet, preventing neurological damage [34]. However, over time, problems in patients’ adherence to diet and the presence of patients whose neurocognitive functions did not improve despite diet therapy brought up new ideas for developing new treatment strategies [34]. In addition to enzymatic treatments, clinical studies have been carried out in gene therapy in recent years [34,35]. The overprocessing of PKU in the studies published on inherited metabolic diseases may be because it is the first known disease among inherited metabolic diseases, and clinicians have more information about PKU because it is diagnosed more frequently than other diseases because of newborn screening programs. In addition, the recent search for genetic and molecular treatments may have led to increased PKU studies.
Inherited metabolic diseases are diseases that occur as a result of genetic disorders. Many different mutations can be seen in the same disease group. In addition, it may show clinical variability according to mutation [36,37,38,39]. Clinicians’ detection of the mutation type plays a role in identifying variants that cause symptomatic disease and may also be a guide in gene therapy [39]. While curable inherited metabolic diseases were diagnosed by newborn screening, many inherited metabolic diseases could not be diagnosed. Over time, with the addition of tandem mass spectrometry to newborn screening, the number of inherited metabolic diseases diagnosed has increased. Examining more genetic variants allows authors to notice different clinical findings in the same disease [40]. Because of this, genotype-phenotype relationships began to be investigated [40]. Detailed diagnostic examinations with whole exome sequencing became routine [40,41]. However, whether some variants of unknown significance caused the disease remained a question mark [40]. Furthermore, emerging metabolomics studies have enabled the discovery of new biomarkers in inherited metabolic diseases [40]. In addition, this metabolomics sheds light on whether mutations are responsible for the disease [40]. We see the journey of the diagnostic approach in inherited metabolic diseases from the detection of the accumulated substance to the enzymatic evaluation, molecular genetic studies, and metabolomics. We think this change parallels the change from dietary treatment and enzyme replacement therapy to gene therapy in the treatment. The increase in studies on genetics, the use of “next generation sequencing” and the use of “tandem mass spectrometry” in extended newborn screenings, and the discovery of “metabolomics” explains the fact that these three subjects are current and frequently researched topics in “trending topics” [42]. The most recent treatment approach in metabolic diseases is correcting for the primary genetic defect. In this approach, conventional gene therapy is applied by delivering a vector containing the correct coding DNA (cDNA) sequence of the defective gene to the host rather than resolving the endogenous genetic defect [43]. Gene therapy has been successfully applied in preclinical trials of inherited metabolic diseases such as glycogen storage type 1a, familial hypercholesterolemia, ornithine transcarbamylase deficiency, and hereditary tyrosinemia type 1 [39,44,45,46]. With the increasing mutation studies in inherited metabolic diseases, the clinical diversity of inherited metabolic diseases may have been expanded. At the same time, it may have led to increased genetic studies, such as gene therapy, especially in inherited metabolic diseases that are not treatable. However, in our study, we observed that clinicians’ willingness to research incurable or very rare diseases (peroxisomal disease, Farber disease, cerebrotendinous xanthomatosis, etc.) was weak. The intensive studies of clinicians on treatable inherited metabolic diseases such as glycogen storage disease type 1, methylmalonic acidemia, and Fabry disease have caused complications such as chronic renal failure due to the prolongation of life expectancy to be seen more frequently in these patients and have been over-reported in recent studies. We think that in the future, clinicians will follow the complication issue in inherited metabolic diseases more closely with the advances in molecular diagnostic methods and gene therapy.
4.1. Limitations
Evaluation of publications in a single database is a limitation of our study. Another limitation is searching with a single keyword. Although we created a data set that could evaluate the inherited metabolic diseases in our study, it cannot be said that we have thoroughly analyzed the studies for the subgroups of this. Because the inclusion of an article in our research was directly related to whether the keyword we preferred was also preferred by the author or not. In addition, although our aim in evaluating only research articles in our study was to determine the research trend in inherited metabolic diseases, another limitation is that we did not include case reports and case series publications that refer to individual differences in inherited metabolic diseases. Another limitation of the study is that we did not analyze the impact of publications.
5. Conclusions
As a result, the diagnostic process that started with newborn screening has increased the number and clinical diversity of inherited metabolic diseases over the years. Phenylketonuria, the first preventable disease diagnosed with newborn screening and its treatment, has been the subject of greatest interest to clinicians dealing with metabolism. We observed that the diagnosis of inherited metabolic diseases was expanded by newborn screening, then tandem mass spectrometry, next-generation sequencing, and metabolomics. With the development of molecular studies, we predict that treatment will change from enzymatic therapy to gene therapy in the future. There is a need for bibliometric studies that include more databases and evaluate more original articles on this subject.
Author Contributions
Conceptualization, B.K.Y.; methodology, B.K.Y. and A.H.A.; software, A.H.A.; validation, B.K.Y. and A.H.A.; formal analysis, A.H.A.; investigation, B.K.Y.; data curation, A.H.A.; writing—original draft preparation, B.K.Y.; writing—review and editing, B.K.Y. and A.H.A.; visualization, B.K.Y.; supervision, B.K.Y.; project administration, B.K.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was conducted according to the guidelines of the Declaration of Helsinki and approved by the ethics committee of Selcuk University Faculty of Medicine (Decision No: 2023/292), ethical date is 06 June 2023.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Acknowledgments
We want to acknowledge Muhammet Akgül for providing technical support in creating the database with bibliometric data and checking the accuracy of the data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Li, Y.-J.; Kan, X. Recent research on inherited metabolic diseases in children. . 2022, 24, 326–331. [Google Scholar] [PubMed]
- Ferreira, C.R.; Rahman, S.; Keller, M.; Zschocke, J. ; ICIMD Advisory Group An international classification of inherited metabolic disorders (ICIMD). J. Inherit. Metab. Dis. 2020, 44, 164–177. [Google Scholar] [CrossRef]
- Schweitzer-Krantz, S. Early diagnosis of inherited metabolic disorders towards improving outcome: the controversial issue of galactosaemia. Eur. J. Pediatr. 2003, 162, S50–S53. [Google Scholar] [CrossRef] [PubMed]
- van Konijnenburg, E.M.M.H.; Wortmann, S.B.; Koelewijn, M.J.; Tseng, L.A.; Houben, R.; Stöckler-Ipsiroglu, S.; Ferreira, C.R.; van Karnebeek, C.D.M. Treatable inherited metabolic disorders causing intellectual disability: 2021 review and digital app. Orphanet J. Rare Dis. 2021, 16, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Lenzini, L.; Carraro, G.; Avogaro, A.; Vitturi, N. Genetic Diagnosis in a Cohort of Adult Patients with Inherited Metabolic Diseases: A Single-Center Experience. Biomolecules 2022, 12, 920. [Google Scholar] [CrossRef]
- Wasim M, Khan HN, Ayesha H, Awan FR. Need and Challenges in Establishing Newborn Screening Programs for Inherited Metabolic Disorders in Developing Countries [published online ahead of print, 2023 Apr 5]. Adv Biol (Weinh) 2023, e2200318. [CrossRef]
- Villoria, J.G.; Pajares, S.; López, R.M.; Marin, J.L.; Ribes, A. Neonatal Screening for Inherited Metabolic Diseases in 2016. Semin. Pediatr. Neurol. 2016, 23, 257–272. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, Q.; Zheng, T.; Zheng, Z.; Lin, W.; Fu, Q. Expanded newborn screening for inherited metabolic disorders and genetic characteristics in a southern Chinese population. Clin. Chim. Acta 2019, 494, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Thomas, D.C.; Sharma, S.; Jhingan, G.; Saxena, R.; Kohli, S.; Puri, R.D.; Bijarnia, S.; Verma, I.C. Inherited metabolic disorders: prenatal diagnosis of lysosomal storage disorders. Prenat. Diagn. 2015, 35, 1137–1147. [Google Scholar] [CrossRef]
- Arnold, G.L.; L. , G. Inborn errors of metabolism in the 21st century: past to present. Ann. Transl. Med. 2018, 6, 467–467. [Google Scholar] [CrossRef]
- Gonzalez, J. , & Willis, M. S. Ivar Asbjörn FøllingDiscovered Phenylketonuria (PKU). Laboratory Medicine 2010, 41, 118–119. [Google Scholar]
- Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet 2010, 376, 1417–1427.
- Scriver, CR. The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat. 2007, 28, 831–845. [Google Scholar] [CrossRef]
- Chace, D.H.; A Kalas, T.; Naylor, E.W. Use of Tandem Mass Spectrometry for Multianalyte Screening of Dried Blood Specimens from Newborns. Clin. Chem. 2003, 49, 1797–1817. [Google Scholar] [CrossRef]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef]
- Wu, F.; Gao, J.; Kang, J.; Wang, X.; Niu, Q.; Liu, J.; Zhang, L. Knowledge Mapping of Exosomes in Autoimmune Diseases: A Bibliometric Analysis (2002–2021). Front. Immunol. 2022, 13, 939433. [Google Scholar] [CrossRef]
- De Oliveira, O.J.; Da Silva, F.F.; Juliani, F.; Barbosa, L.C.F.M.; Nunhes, T.V. Bibliometric Method for Mapping the State-of-the-Art and Identifying Research Gaps and Trends in Literature: An Essential Instrument to Support the Development of Scientific Projects; Intech: London, UK, 2019. [Google Scholar] [CrossRef]
- Ke, L.; Lu, C.; Shen, R.; Lu, T.; Ma, B.; Hua, Y. Knowledge Mapping of Drug-Induced Liver Injury: A Scientometric Investigation (2010–2019). Front. Pharmacol. 2020, 11, 551617. [Google Scholar] [CrossRef]
- Synnestvedt, M.B.; Chen, C.; Holmes, J.H. CiteSpace II: visualization and knowledge discovery in bibliographic databases. AMIA. Annu. Symp. Proc. AMIA Symp. 2005, 2005, 724–728. [Google Scholar]
- Yeung, A.W.K.; Tzvetkov, N.T.; Balacheva, A.A.; Georgieva, M.G.; Gan, R.-Y.; Jozwik, A.; Pyzel, B.; Horbańczuk, J.O.; Novellino, E.; Durazzo, A.; et al. Lignans: Quantitative Analysis of the Research Literature. Front. Pharmacol. 2020, 11, 37. [Google Scholar] [CrossRef]
- Li, C.; Ojeda-Thies, C.; Renz, N.; Margaryan, D.; Perka, C.; Trampuz, A. The global state of clinical research and trends in periprosthetic joint infection: A bibliometric analysis. Int. J. Infect. Dis. 2020, 96, 696–709. [Google Scholar] [CrossRef]
- Lu, C.; Liu, M.; Shang, W.; Yuan, Y.; Li, M.; Deng, X.; Li, H.; Yang, K. Knowledge Mapping of Angelica sinensis (Oliv.) Diels (Danggui) Research: A Scientometric Study. Front. Pharmacol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Wilson, M.; Sampson, M.; Barrowman, N.; Doja, A. Bibliometric Analysis of Neurology Articles Published in General Medicine Journals. JAMA Netw. Open 2021, 4, e215840–e215840. [Google Scholar] [CrossRef]
- Liu B, He X, Wang Y; et al. Bibliometric Analysis of γδ T Cells as Immune Regulators in Cancer Prognosis. Front Immunol 2022, 13, 874640. [CrossRef]
- Li, C.; Wang, L.; Perka, C.; Trampuz, A. Clinical application of robotic orthopedic surgery: a bibliometric study. BMC Musculoskelet. Disord. 2021, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Vural, S.; Kaya, H.B.; Çoşkun, F. A bibliometric study on the publication errors in emergency medicine journals from 2000 to 2020. Am. J. Emerg. Med. 2022, 60, 140–144. [Google Scholar] [CrossRef]
- Bharadwaj, A.; Wahi, N.; Saxena, A. Occurrence of Inborn Errors of Metabolism in Newborns, Diagnosis and Prophylaxis. Endocrine, Metab. Immune Disord.—Drug Targets 2021, 21, 592–616. [Google Scholar] [CrossRef]
- Wang, H.; Shi, J.; Shi, S.; Bo, R.; Zhang, X.; Hu, Y. Bibliometric Analysis on the Progress of Chronic Heart Failure. Curr. Probl. Cardiol. 2022, 47, 101213. [Google Scholar] [CrossRef]
- Akmal, M.; Hasnain, N.; Rehan, A.; Iqbal, U.; Hashmi, S.; Fatima, K.; Farooq, M.Z.; Khosa, F.; Siddiqi, J.; Khan, M.K. Glioblastome Multiforme: A Bibliometric Analysis. World Neurosurg. 2020, 136, 270–282. [Google Scholar] [CrossRef]
- Koo, M. Systemic Lupus Erythematosus Research: A Bibliometric Analysis over a 50-Year Period. Int. J. Environ. Res. Public Heal. 2021, 18, 7095. [Google Scholar] [CrossRef]
- de Diego, I.M.; González-Fernández, C.; Fernández-Isabel, A.; Fernández, R.R.; Cabezas, J. System for evaluating the reliability and novelty of medical scientific papers. J. Inf. 2021, 15, 101188. [Google Scholar] [CrossRef]
- Rajabi, F. Updates in Newborn Screening. Pediatr. Ann. 2018, 47, e187–e190. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Andriola, M.; Arnold, G.; Aron, A.; Duffner, P.; Erbe, R.W.; Escolar, M.L.; Estrella, L.; Galvin-Parton, P.; Iglesias, A.; et al. Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State. Anesthesia Analg. 2016, 18, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen FJ, Enns GM. Future treatment strategies in phenylketonuria. Mol Genet Metab 2010, 99 (Suppl 1), 90–95.
- Wiedemann A, Oussalah A, Jeannesson É, Guéant JL, Feillet F. La phénylcétonurie—De la diététique à la thérapie génique [Phenylketonuria, from diet to gene therapy]. Med Sci (Paris) 2020, 36(8-9), 725-734. [CrossRef]
- Dursun, A.; Henneke, M.; Özgül, K.; Gartner, J.; Coşkun, T.; Tokatli, A.; Kalkanoğlu, H.S.; Demirkol, M.; Wendel, U.; Özalp, I. Maple syrup urine disease: Mutation analysis in Turkish patients. J. Inherit. Metab. Dis. 2002, 25, 89–97. [Google Scholar] [CrossRef]
- Carrillo-Carrasco, N.; Chandler, R.J.; Venditti, C.P. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J. Inherit. Metab. Dis. 2011, 35, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Yu Y, Ling S, Shuai R; et al. Clinical features and outcomes of patients with cblC type methylmalonic acidemia carrying gene c.609G>A mutation. Zhejiang Da Xue Xue Bao Yi Xue Ban 2021, 50, 436–443. [CrossRef]
- Yilmaz, B.S.; Gurung, S.; Perocheau, D.; Counsell, J.; Baruteau, J. Gene Therapy for Inherited Metabolic Diseases. 24, 64. [CrossRef]
- Coene, K.L.M.; Kluijtmans, L.A.J.; van der Heeft, E.; Engelke, U.F.H.; de Boer, S.; Hoegen, B.; Kwast, H.J.T.; van de Vorst, M.; Huigen, M.; Keularts, I.; et al. Next-generation metabolic screening: Targeted and untargeted metabolomics for the diagnosis of inborn errors of metabolism in individual patients. J. Inherit. Metab. Dis. 2018, 41, 337–353. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Heiles, S. Advanced tandem mass spectrometry in metabolomics and lipidomics—methods and applications. Anal. Bioanal. Chem. 2021, 413, 5927–5948. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef]
- Brooks, E.D.; Landau, D.J.; Everitt, J.I.; Brown, T.T.; Grady, K.M.; Waskowicz, L.; Bass, C.R.; D’Angelo, J.; Asfaw, Y.G.; Williams, K.; et al. Long-term complications of glycogen storage disease type Ia in the canine model treated with gene replacement therapy. J. Inherit. Metab. Dis. 2018, 41, 965–976. [Google Scholar] [CrossRef]
- Bryson, T.E.; Anglin, C.M.; Bridges, P.H.; Cottle, R.N. Nuclease-Mediated Gene Therapies for Inherited Metabolic Diseases of the Liver. Yale J. Biol. Med. 2017, 90, 553–566. [Google Scholar] [PubMed]
- Lee, Y.M.; Conlon, T.J.; Specht, A.; Coleman, K.E.; Brown, L.M.; Estrella, A.M.; Dambska, M.; Dahlberg, K.R.; Weinstein, D.A. Long-term safety and efficacy of AAV gene therapy in the canine model of glycogen storage disease type Ia. J. Inherit. Metab. Dis. 2018, 41, 977–984. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The screening flowchart of articles including the keyword “inherited metabolic disease”. IMD; inherited metabolic disease, n; number.
Figure 1.
The screening flowchart of articles including the keyword “inherited metabolic disease”. IMD; inherited metabolic disease, n; number.
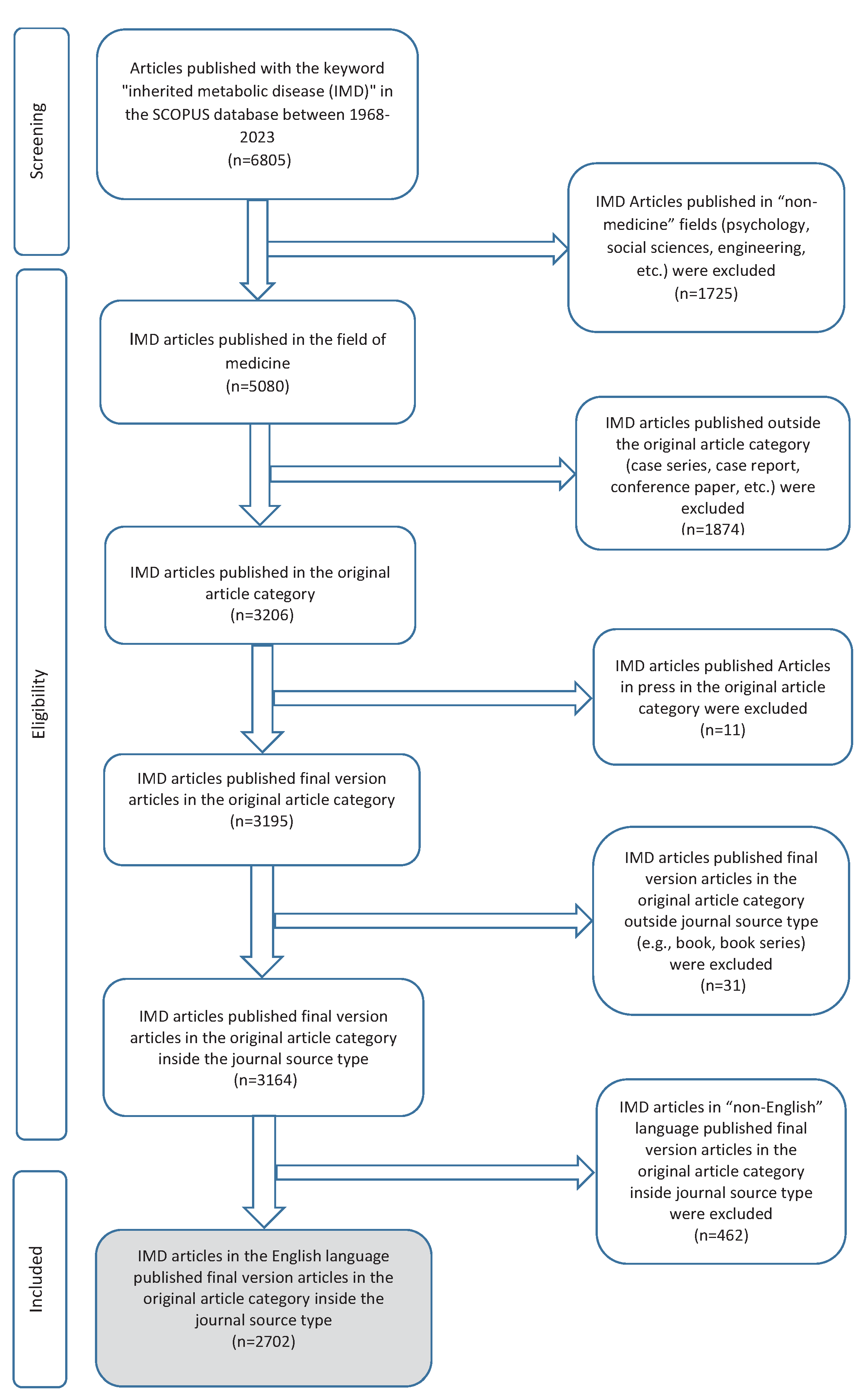

Table 1.
General information of the study.
| Description | Results |
|---|---|
| MAIN INFORMATION ABOUT DATA | |
| Timespan | 1968:2023 |
| Sources (Journals, Books, etc) | 1012 |
| Documents | 2702 |
| Annual Growth Rate % | 3,69 |
| Document Average Age | 14,2 |
| Average citations per doc | 26,4 |
| References | 88136 |
| DOCUMENT CONTENTS | |
| Keywords Plus (ID) | 16542 |
| Author’s Keywords (DE) | 5380 |
| AUTHORS COLLABORATION | |
| Authors | 14782 |
| Single-authored docs | 207 |
| Co-Authors per Doc | 6,97 |
| International co-authorships % | 21,06 |
| DOCUMENT TYPES | |
| Research article | 2702 |
Table 2.
Top 15 most commonly used keywords in IMD publications.
| Keywords | Number of articles |
|---|---|
| newborn screening | 54 |
| Mutation | 43 |
| Phenylketonuria | 42 |
| Children | 35 |
| Genetics | 34 |
| maple syrup urine disease | 32 |
| gene therapy | 31 |
| case report | 28 |
| tandem mass spectrometry | 28 |
| glycogen storage disease | 27 |
| Mucopolysaccharidosis | 27 |
| liver transplantation | 25 |
| Mitochondria | 25 |
| Hyperammonemia | 24 |
| enzyme replacement therapy | 21 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



