
Submitted:
29 June 2023
Posted:
03 July 2023
You are already at the latest version
Abstract
(1) Background: Angiotensin II, a major culprit in cardiovascular disease, activates mediators that are also involved in pathological cardiac remodelling. We aimed at investigating the effects of two of them: aldosterone (Ald) and transforming growth factor-beta-1 (TGF-β1 ), on cardiac remodelling in an in vivo model. (2) Methods: Six-week-old male wild-type (WT) and TGF-β1 -overexpressing transgenic (TGF-β1 -TG) mice were infused with subhypertensive doses of Ald for 2 weeks, and/or treated orally with eplerenone from postnatal day 21. Heart’s ventricles were examined: by morphometry, immunoblotting to assess intracellular signalling pathways, RT qPCR to determine hypertrophy and fibrosis marker genes. (3) Results: TGF-β1 -TG mice spontaneously developed cardiac hypertrophy and interstitial fibrosis, exhibited higher baseline phosphorylation of p44/42 and p38 kinases, fibronectin and ANP mRNA expression. Ald induced a comparable increase in ventricular heart weight-to-body-weight ratio and cardiomyocyte diameter in both strains, but less pronounced increase in interstitial fibrosis in transgenic compared to WT mice (23.6% vs 80.9%, p<0.005). Ald increased phosphorylation of p44/42 and p38 in WT but not TGF-β1 -TG mice. While eplerenone-enriched chow partially prevented Ald-induced cardiac hypertrophy in both genotypes and interstitial fibrosis in WT controls, it completely protected from additional fibrosis in transgenic mice. (4) Conclusions: Ald appears to induce cardiac hypertrophy independently of TGF-β1 , while in case of fibrosis downstream signalling pathways of these two factors probably converge
Keywords:
aldosterone
; transforming growth factor beta1
; ventricular cardiac remodelling
; transgenic mice
; mitogen-activated protein kinases
1. Introduction
Pathological cardiac remodelling (CR) comprises structural and functional changes of the heart due to injury and/or chronic diseases. Ventricular hypertrophy and fibrosis are two major alterations seen in CR. While hypertrophy may serve adaptive purposes at least initially and/or in some cases, it has been established as a risk factor for cardiovascular (CV) events. Pathological cardiac hypertrophy is accompanied by fibrotic response, which triggers both systolic and diastolic dysfunction, leading to heart failure [1]. Both aldosterone (Ald) and transforming growth factor beta-1 (TGF-β1) have been demonstrated to cause hypertrophy and fibrosis in the course of CR [2,3].
Ald is a member of the renin-angiotensin-aldosterone system (RAAS) that regulates blood pressure and fluid balance by increasing sodium and water reabsorption via its mineralocorticoid receptor (MR) present in renal tubules. Apart from this classic effect, Ald directly influences the CV system: MRs are present in vascular smooth muscle cells, fibroblasts and cardiomyocytes, among others. Adverse effects of Ald include cardiac hypertrophy and fibrosis, endothelial dysfunction, and inflammation [2]. Recent research in humans indicates that the prevalence of primary hyperaldosteronism (PHA) as a cause of hypertension may be as high as 20-30%, but remains largely undiagnosed. This is unsatisfactory, since PHA patients suffer from much higher CV mortality and morbidity than those with primary hypertension, despite the availability of anti-MR targeted pharmacotherapy [4,5,6].
TGF-β1 is recognized as a fundamental mediator of fibrotic processes in kidney, lung, liver and heart diseases [7]. Elevated TGF-β1 expression has been shown in idiopathic hypertrophic cardiomyopathy, dilatative myopathy, and transition from stable hypertrophy to heart failure, all of which are associated with cardiac hypertrophy and fibrosis. At the molecular level, TGF-β1 acts as a local, para- and autocrine mediator as well as a secondary messenger in response to angiotensin II, Ald and beta-adrenergic signalling [3,8,9,10,11,12]. Upon binding to its receptor, TGF-β1′s intracellular signal transduction pathway follows through the Smad transcriptional activators; additionally, mitogen-activated protein kinases (MAPKs), i.e. Erk, p38 and c-Jun N-terminal kinase, are activated through TGF-Β1-activated kinase 1.
In this study we sought to analyze the individual effects of Ald and TGF-β1 on cardiac hypertrophy and fibrosis as well as downstream signalling pathways in a murine model.
2. Materials and methods
2.1. Treatment of animals
Male wild type (WT), i.e. C57Bl/J6 (Harlan and Winkelmann), and TGF-Β1-overexpressing transgenic mice (TGF-β1-TG or TG) mice were used in this study.
TGF-β1-TG mice were originally generated by Sanderson et al. using murine albumin promoter and enhancer linked to a porcine TGF-β1 construct and the 3‘- region of the human growth hormone gene, containing a polyadenylation signal [13]. Preferential secretion of mature TGF-β1 resulted from cysteine/serine substitutions at amino acid residues 223 and 225 in the TGF-β1 c-DNA. Line 25 mice were used here, in which solely males are transgenic and exhibit a 10-fold increased plasma TGF-β1 concentration compared with aged-matched controls [14]. The line has been maintained by continued backcrosses to WT F1 mice (which served as controls).
All investigations were performed in accordance with the National Institute of Health ‘Guide for the Care and Use of Laboratory Animals’ and Institutional Animal Care and Use Guidelines.
Eight study groups (7 to 14 mice per each group) were investigated, i.e. four WT and four TGF-β1-TG: control with regular chow diet and no Ald infusion (WT, TG), Ald-infused (WT+Ald, TG+Ald), eplerenone (Ep)-treated (WT+Ep, TG+Ep), and both Ep-enriched chow-fed and Ald-infused (WT+Ald+Ep, TG+Ald+Ep).
Selective MR antagonist Ep was provided from Pfizer in powder form and enriched to mouse chow by Harlan Laboratories (1.5 mg of Ep per gram of chow). A daily dosage of approximately 200-300 mg per kg body weight (BW) was obtained. Ald (ordered from Sigma) was dissolved in 20% ethanol to a concentration of 2 mg/ml and stored at 4°C. Dilution with 0.9% NaCl for a final 1 mg/ml concentration took place immediately prior to filling the solution into miniature infusion pumps (Alzet, model 1002). Manipulations were performed in sterile conditions.
Treatment with Ep started immediately after weaning (at 3 weeks of age), by providing drug-enriched chow, and lasted 5 weeks (until the age of 8 weeks), while Ald was infused via a mini-pump between day 42 and 56 (Figure 1).
Infusion pumps with Ald were implanted subcutaneously at the age of 6 weeks during isoflurane anaesthesia. 100 microliters of Ald solution was infused continuously for 14 days with an appropriate flow moderator, equivalent to a circa 0.35 mg/kg BW/day dose, i.e. a subhypertensive one [15]. Three control mice received pumps containing 0.9% NaCl to verify, whether pump implantation had any effect on cardiac morphology, which was not the case.
2.2. Organ extraction and sample preparation
At the age of 8 weeks animals’ organs were collected. Following isoflurane anaesthesia, BW were measured, mice were sacrificed by cervical dislocation; hearts were excised, perfused with cold saline to remove blood. Biventricular heart weight (vHW) was recorded after excising the atria. Hearts were sliced to obtain samples for immunohistochemistry (heart base cut off at organ’s equator, stored in 4% formaldehyde solution overnight), immunoblotting (liquid nitrogen snap-frozen and stored at -80°C), and mRNA quantification (RNA-Later containing tube and stored at 4°C). Had a mini-pump been implanted, it was excised and weighted after organ extraction; the BW of the mouse was reduced accordingly.—
2.3. Morphometric analysis
Cardiac hypertrophy was assessed by determining vHW to BW ratio (vHW/BW) (in milligrams per grams) as well as mean cardiomyocyte diameter for every mouse. The latter was acquired by measuring at least 100 cell diameters per animal. Horizontal sections at the equator of the heart were stained with haematoxylin-eosin. Ten sections were analyzed for each mouse. For each section the diameter from at least ten cardiomyocytes was determined and the averages were recorded. To ensure consistency, only cells with nuclei at their circumference were chosen. Here the shortest myocyte diameter was measured.
Midventricular sections were stained with Massons’-trichrome to evaluate the amount of fibrous tissue in the muscle. Cardiac interstitial fibrosis was quantified in 9 to 12 randomly chosen areas for each mouse heart (from a single heart section) in a blinded manner. 20x magnification was used. Regions with blood vessels as well as pericardium or endocardium were omitted. For every chosen area percentage of fibrous tissue was computed using a colour analysis method with Olympus’ Cell^P software. Blue- and grey-stained fibrotic elements were discriminated from white, artefactual, intracellular areas and black nuclei by setting colour thresholds. White artefacts were subtracted from the frame area set for each region to acquire an accurate percentage of fibrotic material. Perivascular fibrosis was assessed in a semi-quantitative way of randomly labelled Masson’s-trichrome stains. Coronary vessels were photographed—at least 5 for each animal—and scored on a 0 to 4 scale (with “0” representing no fibrosis, “1” minimal and “4” massive fibrosis). Average cardiac perivascular fibrosis score was calculated for each mouse.
2.4. Western blotting
The following antibodies were provided by Cell Signalling technology: phospho-p44/42 MAP Kinase (Thr202/Tyr204) Antibody (product number 9101), p44/42 MAP Kinase Antibody (product number 9101), phospho-p38 MAP Kinase (Thr180/Tyr182) Antibody (product number 9211), p38 MAP Kinase Antibody (product number 9212). Anti-rabbit monoclonal antibody was supplied from Sigma. Anti-Ras-Gap antibody was kindly supplied by professor A. Kazlauskas from Harvard Medical School.
Ventricular heart lysates were prepared from liquid nitrogen snap frozen pieces by homogenization in 1 ml RIPA proteinase inhibiting buffer at 4°C. Subsequently, homogenates were incubated for 2 hours at 4°C on gentle agitation and next centrifuged for 20 minutes at 10000 g. The supernatant was collected, aliquoted and stored at -80°C. For Western blotting, samples were thawed on ice. Protein concentration was quantified by NanoDrop (Thermo Scientific) and ranged between 14 and 27 mg/ml. Aliquots were diluted with RIPA to acquire equal sample concentrations. Homogenates were suspended in 4×SDS sample buffer. Samples with equal protein amounts were run on SDS–PAGE and transferred to PVDF membranes. Blots were probed with appropriate antibodies to reveal protein bands on hyperfilm, which were quantified using ImageJ software as described previously [16].
2.5. Real-time qPCR
Cardiac expression of mRNAs of two marker genes was investigated: arial natriuretic peptide (ANP) for cardiac hypertrophy and fibronectin (FBN) for fibrosis.
Rodent glyceraldehyde 3-phosphate dehydrogenase (GAPDH) primers and probe with VIC reporter dye were supplied by Applied Biosystems (product number 4308313). ANP and FBN primers and FAM reporter dye probes were ordered from Eurofins MWG Operon: ANP probe 5’-TCGCTGGCCCTCGGAGCCTAC-3’; forward primer 5’-GAAAAGCAAACTGAGGGCTCTG-3’; reverse primer 5’-CCCCGAAGCAGCTGGAT TGC-3’; FBN probe 5’-TCGGAGCCATTTGTTCCTGCACGT-3’, forward primer 5′-TGT-AGGAGAACAGTGGCAGAAAGA-3′, reverse primer 5′-CCGCTGGCCTCCGAA-3′.
Total RNA was extracted from heart tissues stored in RNAlater using the TRIzol (Life Technologies) RNA extraction method [17]. Equal amounts of isolated RNA were subsequently transcribed into cDNA using high-capacity cDNA reverse transcription kit (Applied Biosystems) as per manufacturer’s instructions. The iQ SYBR Green Supermix (Bio-Rad) kit was applied to perform qPCR. mRNA expression was analyzed with the ΔCt method. Ct values of target genes (ANP and FBN) were normalized to that of GAPDH (reference gene) using the equation ΔCt=Ct(reference)−Ct(target), and expressed as ΔCt.
2.6. Statistical Analysis
Control WT and TG animals were tested for differences with Student’s t-test or its non-parametric variant, the Mann-Whitney U test, depending on data distribution, which was verified with Shapiro-Wilk’s test. For each strain, parameters in four groups (control, Ald-infused, Ald-infused and Ep-treated, and Ep-treated) were tested for differences using parametric (with Tukey’s post-hoc test) or non-parametric (Kruskal Wallis) ANOVAs depending on data distribution. Results were presented as mean±standard deviation (SD) or median (interquartile range, IQR); graphs present mean and SD values. Statistical significance was set at 0.05.his section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
3. Results
3.1. Baseline characteristics of TGF-β1 mice
At baseline, mice with constitutive expression of the TGF-β1 transgene exhibited cardiac hypertrophy, as indicated by higher mean vWH/BW (+28.6%, p<10-4) and cardiomyocyte diameter (+24.2%, p<10-4) compared to the WT strain (Table 1 and Figure 2A–C). Furthermore, ANP mRNA expression was 89.7% higher in TG compared to WT mice, p=0.041 (Figure 2D).
Concerning cardiac fibrosis, the area occupied by interstitial fibrous tissue in the myocardium of TG animals was almost twice that of the WT mice: 1.03±0.2 vs 1.91±0.27%, p<10-4 (Figure 3A,C). In contrast, perivascular fibrosis score was similar for both strains (Figure 3B). Moreover, TGF-β1-TG mice showed a trend toward higher FBN mRNA expression in ventricular tissue compared to WT animals (p=0.15) (Figure 3D).
Constitutive higher Erk (p44/42) and p38 kinase phosphorylation in ventricular heart tissues samples from TGF-β1-TG compared to WT mice was recorded, which was 4.1 (3.1-5.1)- and 7.4 (3.5-12.1)-fold, respectively (Figure 4A).
3.2. Effects of Ald and Ep on cardiac hypertrophy
A 14-day Ald infusion lead to a 9.8±2.5% increase in the vHW/BW ratio of WT mice (p<10-4), while in TGF-β1-TG animals the increase was insignificant (p=0.31), and the change in vHW/BW due to Ald was comparable between strains at p=0.57 (Figure 5A). The increase in cardiomyocyte diameter was more pronounced in WT than TG mice: 36±13% versus 24.6±2.7%, p=0.026 (Figure 5B). In contrast, ANP mRNA expression did not differ between respective untreated and Ald-infused mice of each strain (Figure 2D).
Under basal conditions, Ep had no effect on the vHW/BW ratios or cardiomyocyte size in either mouse strain (Figure 2A–C). Accordingly, no differences were recorded in ANP mRNA expression between control and Ep-enriched chow groups (Figure 2D).
In Ald-infused animals, the vHW/BW ratio was not significantly affected by concurrent administration of Ald and its receptor antagonist in mice of either genotype, although a trend toward hypertrophy attenuation could be observed in TGF-β1-TG animals (Figure 2A). Ep administration with concurrent Ald infusion led to a 13.5% decrease in cardiomycyte diameter in WT mice versus those only treated with Ald, but not in TGF-β1-overexpressing mice (Figure 2B). No significant differences in ANP mRNA expression were found between groups infused with Ald only and those treated with both Ald and Ep (Figure 2D).
3.3. Effects of Ald and Ep on cardiac fibrosis
While both strains exhibited more interstitial fibrosis in the ventricles upon completion of a two-week Ald infusion, the increase was substantially higher in WT mice: 80.9±32% versus 23.6±30%, p=0.002 (Figure 5C). These morphometric observations were supported at the molecular level, as Ald induced a significant increase (+47.7%) in cardiac FBN mRNA expression only in WT mice (p=0.029, Figure 3D).
Importantly, the increase in cardiac interstitial fibrosis recorded in the WT strain was significantly higher than the increases in vHW/BW and cardiomyocyte diameter (ANOVA, p<0.0001, Figure 5). This was not the case in transgenic animals (p=0.175).
As expected, Ep treatment itself had no gross morphological effect on cardiac interstitial and perivascular fibrosis in either WT or TGF-β1-TG mice (Figure 3A–C). However, a trend was recorded toward lower myocardial expression of FBN mRNA in Ep-treated groups compared to their respective controls in both genotypes (Figure 3D).
Further, Ep effectively decreased Ald-induced cardiac interstitial fibrosis in TGF-β1-TG mice by 28% (TG+Ald+Ep versus TG+Ald). A similar trend was seen in WT animals, although it failed to reach statistical significance (Figure 3A). Importantly, Ep treatment nearly completely abolished Ald-induced upregulation of FBN mRNA in both WT and TGF-β1-TG mice (Figure 3D). These results indicate that Ald-induced cardiac interstitial fibrosis is mainly mediated via the MR and can be effectively attenuated by receptor antagonism.
3.4. Effects of Ald and Ep on MAPK-signalling
Since MAPK signalling is an important pathway mediating cardiac hypertrophy, we analysed phosphorylation of the main MAPKs, Erk (p44/42) and p38, in ventricular heart tissues of WT and TGF-β1-TG mice. In WT animals both signalling pathways were activated with Ald infusion (Figure 4B). In TGF-β1-TG mice, Erk and p38 phosphorylation had already been higher at baseline and did not further increase in response to Ald (Figure 4A,C).
Ep attenuated Erk (p44/42) and p38 phosphorylation in ventricular heart tissues of WT but not TG mice (Figure 4B,C). In addition, phosphorylation of these MAPKs in the WT+Ep group was also reduced compared to controls, indicating these pathways are mainly regulated by MR activation. In contrast to the WT strain, Ep had no statistically significant effect on either Erk (p44/42) or p38 phosphorylation in any of the TG groups, suggesting that activation of these pathways by TGF-β1 occurs independently of the MR.
4. Discussion
A To our knowledge, this study is the first to examine the effects of continuous Ald infusion and oral Ep treatment on cardiac remodelling in transgenic mice overexpressing TGF-β1. Therefore, direct comparisons with previous research are not possible. Our main finding is that the effect of Ald on cardiac hypertrophy is additive to that of TGF-β1, while with respect to interstitial fibrosis, downstream signalling mechanisms of these two mediators likely converge. The differential effects can be deduced from a comparison between WT and TG animals: in the former interstitial fibrosis increased significantly more than cardiac hypertrophy parameters in response to Ald infusion, whereas the change in all morphometric parameters was comparable in the setting of TGF-β1 excess (Figure 5). Furthermore, Ep-treatment alleviated the profibrotic effect of Ald infusion in TG animals, while only a partial reduction was observed for cardiomyocyte diameter. Concerning Erk and p38 activation, no significant alterations were observed here in case of TGF-β1-overexpressing mice across all groups: control, Ald and/or Ep treated, while in WT animals the MAPKs were activated by Ald.
Previous studies concerning the effects of both mediators on cardiac remodelling are scarce. Both molecules were investigated in vivo in mice with graded TGF-β1expression to reveal that its excess inhibited adrenal steroid synthesis including Ald. However, cardiac morphology was not reported [18]. Conversely, in a study by Nishioka et al., four-week Ald infusion along with 1% NaCl drinking water induced TGF-β1 mRNA expression and marked cardiac fibrosis [19]. Next, Ald stimulated TGF-β1 expression in cardiac fibroblasts and rat mesangial cells [20,21]. Still, a difficulty arises in discriminating the effect of Ald and Ang II on downstream signal transduction via TGF-β1 in light of interactions within the RAAS [22,23,24,25]. While Ang II has been established as a TGF-β1-inducing molecule, its antagonism has not been effective in abolishing TGF-β1′s cardiac effects [26].
A larger body of data has been generated with respect to TGF-β1′s role in cardiac remodelling alone. Previous studies using the same transgenic model also revealed cardiac hypertrophy and fibrosis [13,27,28]. Methodological differences and possible less homogenous expression of the TGF-β1 transgene years after the cited reports (apparent in greater variation of vHW/BW) may account for lower hypertrophy indices, while findings concerning ANP mRNA expression and interstitial fibrosis are comparable [27,28,29]. Data from studies with other animal models echo ours. Nakajima et al. showed TGF-1¬ overexpression increased heart-weight-to-tibia-length ratios as well as cardiomyocyte size, and, although a pro-fibrotic effect was seen only in the atria, this discrepancy had been partially explained by distinct approaches to TGF-β1 overexpression as well as low activity of the factor [27,28,30]. Further, TGF-β1 -/- and Rag1 -/- double-knockout mice did not develop cardiac hypertrophy induced by subpressor Ang II doses, as was the case for controls [10]. Moreover, the mediator’s role in age-associated myocardial fibrosis was evidenced by its amelioration in single-knockout TGF-β1 +/- mutant mice [31]. Finally, neutralizing TGF-β1 with an antibody prevented diastolic dysfunction in pressure-overloaded rats [32].
Analogically to morphometric findings, ample research exists supporting higher MAPK phosphorylation due to TGF-β1, which induces both Erk 1/2 [33,34,35] and p38 phosphrylation (particularly via the noncanonical, i.e. non-Smad-related, TAK1 pathway) [3,36,37]. Evidence has been accumulating for the role of p38 in the development of cardiac fibrosis, and Erk primarily in hypertrophy [38,39,40,41].
In discussing cardiac remodelling effects of Ald, the current study contrasts with most preceding ones in that neither nephrectomy nor salt-loading was applied. Also, here, Ald was infused at doses known not to induce hypertension (although lack of blood pressure measurements is a limitation of our data). In this setting, Ald’s actions could be extracted from the influence of other factors. With respect to MR antagonist, Ep was initiated three weeks prior to Ald infusion, while other researchers have applied simultaneous start of treatment.
Bearing these in mind, in a study similar to our, Yoshida et al. applied Ald infusion at a rate of 0.75 µg/h for 14 days in male Sprague-Dawley rats. A minor blood pressure increase was successfully reduced by an anti-oxidant without preventing cardiac hypertrophy (revealed by echocardiography, and a 34% increase in cardiomyocyte cross-sectional area). Spironolactone effectively attenuated Ald-induced cardiac hypertrophy [42]. Similar methodology, yet with 1% NaCl instead of drinking water, was applied by Nakano et al. to demonstrate higher LV weight/BW ratio with Ald infusion, and prevention thereof by spironolactone [43]. In another model, Ep ameliorated cardiac hypertrophy and fibrosis exhibited by mice overexpressing 11-beta-hydroxysteroid dehydrogenase 2 (which promotes Ald-MR binding) [44]. In contrast, Iglarz et al. did not record macroscopic cardiac hypertrophy after a 6-week Ald infusion in rats [45]. As for cardiac interstitial fibrous, an approximately 80% increase and higher FBN mRNA expression were recorded here in Ald-infused WT mice compared to untreated controls, which is in line with previous studies: that of Iglarz et al. mentioned above and another—by Johar et al., who also demonstrated higher FBN expression in Ald-induced rodents [45,46]. Both Ep and spironolactone were effective in fibrosis prevention [19,47,48].
Prior in vitro research also supports a trend toward higher Erk and p38 phosphorylation in response to Ald as was recorded here, e.g. [49]. In vivo data are scarce, yet, results by Nakano et al. mentioned above include this observation for Erk [43].
In the current study, Ep only partially prevented Ald-induced cardiac hypotrophy (significant difference for cardiomyocyte diameter, but not vHW/BW), and interstitial fibrosis (WT+Ald+Ep not significantly different from WT and WT+Ald groups), which contrasts with other reports [42,43,44,50]. Possibly, compensatory mechanisms to Ep treatment developed in postnatal weeks 3-6, which rendered MR antagonism ineffective once Ald was administered in weeks 6-8. These might include higher MR expression, Ald secretion, and altered Ang II signalling. The hypothesis of Ald escape is supported further with data of kidney-weight-to-BW ratios of the ‘+Ald+Ep’ group, which were higher those of untreated controls (Table 1).
5. Conclusions
A In the current and previous studies, TGF-β1 has been shown a vital cardiac pro-fibrotic and pro-hypertrophic mediator. Data obtained here also suggest that in the myocardium the pro-fibrotic actions of TGF-β1 and Ald overlap, while they are at least in part divergent in case of cardiac hypertrophy. Further research is necessary to elucidate the cross-talk between all components of the RAAS and TGF-β1.
Author Contributions
conceptualization, EC; methodology, PK, EC, MO; formal analysis, SR, EC, PK; investigation, EC, PK.; resources, SR; data curation, PK; writing—original draft preparation, PK; writing—review and editing, EC, SR, PK, MO; supervision, EC, SR; funding acquisition, SR. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The animal study protocol was approved by the North Rhine-Westphalia State Agency for Nature, Environment and Consumer protection – file number 9.93.2.10.31.07.189.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Buffolo, F.; Tetti, M.; Mulatero, P.; Monticone, S. Aldosterone as a Mediator of Cardiovascular Damage. Hypertension 2022, 79, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Xiao H, Zhang YY. Understanding the role of transforming growth factor-beta signalling in the heart: overview of studies using genetic mouse models. Clin Exp Pharmacol Physiol. 2008, 35, 335–341. [CrossRef]
- Brown JM, Siddiqui M, Calhoun DA, Carey RM, Hopkins PN, Williams GH, et al. The Unrecognized Prevalence of Primary Aldosteronism: A Cross-sectional Study. Annals of Internal Medicine. 2020, 173, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Hundemer, G.L.; Nanba, K.; Parksook, W.W.; Brown, J.M. Primary Aldosteronism: State-of-the-Art Review. Am. J. Hypertens. 2022, 35, 967–988. [Google Scholar] [CrossRef]
- Funder JW, Carey RM. Primary Aldosteronism: Where Are We Now? Where to From Here? Hypertension. 2022, 79, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Rosenkranz, S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004, 63, 423–432. [Google Scholar] [CrossRef]
- Frangogiannis, NG. Cardiac fibrosis. Cardiovasc Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Schultz JEJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. 2002, 109, 787–796. [Google Scholar] [CrossRef]
- Scaglione R, Argano C, Di Chiara T, Parrinello G, Colomba D, Avellone G, et al. Effect of dual blockade of renin-angiotensin system on TGFbeta1 and left ventricular structure and function in hypertensive patients. J Hum Hypertens. 2007, 21, 307–315. [Google Scholar] [CrossRef]
- Schluter, K.D.; Zhou, X.J.; Piper, H.M. Induction of hypertrophic responsiveness to isoproterenol by TGF-beta in adult rat cardiomyocytes. Am. J. Physiol. Physiol. 1995, 269, C1311–C1316. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, N.; Factor, V.; Nagy, P.; Kopp, J.; Kondaiah, P.; Wakefield, L.; Roberts, A.B.; Sporn, M.B.; Thorgeirsson, S.S. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. USA 1995, 92, 2572–2576. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.; Hurta, R.; Kondaiah, P.; Khalil, N.; Turley, E.; Wright, J.; Greenberg, A. Autocrine induction of tumor protease production and invasion by a metallothionein-regulated TGF-beta 1 (Ser223, 225). EMBO J. 1992, 11, 1599–1605. [Google Scholar] [CrossRef]
- Bruder-Nascimento, T.; Ferreira, N.S.; Zanotto, C.Z.; Ramalho, F.; Pequeno, I.O.; Olivon, V.C.; Neves, K.B.; Alves-Lopes, R.; Campos, E.; Silva, C.A.A.; et al. NLRP3 Inflammasome Mediates Aldosterone-Induced Vascular Damage. Circulation 2016, 134, 1866–1880. [Google Scholar] [CrossRef] [PubMed]
- Stael, S.; Miller, L.P. ; Fernández-Fernández, D.; Van Breusegem, F. Detection of Damage-Activated Metacaspase Activity by Western Blot in Plants. 2022, 2447, 127–137. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Purification of RNA Using TRIzol (TRI Reagent). Cold Spring Harb. Protoc. 2010, 2010, pdb–prot5439. [Google Scholar] [CrossRef] [PubMed]
- Kakoki, M.; Pochynyuk, O.M.; Hathaway, C.M.; Tomita, H.; Hagaman, J.R.; Kim, H.-S.; Zaika, O.L.; Mamenko, M.; Kayashima, Y.; Matsuki, K.; et al. Primary aldosteronism and impaired natriuresis in mice underexpressing TGFβ1. Proc. Natl. Acad. Sci. 2013, 110, 5600–5605. [Google Scholar] [CrossRef]
- Nishioka, T.; Suzuki, M.; Onishi, K.; Takakura, N.; Inada, H.; Yoshida, T.; Hiroe, M.; Imanaka-Yoshida, K. Eplerenone Attenuates Myocardial Fibrosis in the Angiotensin II-Induced Hypertensive Mouse: Involvement of Tenascin-C Induced by Aldosterone-Mediated Inflammation. J. Cardiovasc. Pharmacol. 2007, 49, 261–268. [Google Scholar] [CrossRef]
- Tsybouleva, N.; Zhang, L.; Chen, S.; Patel, R.; Lutucuta, S.; Nemoto, S.; DeFreitas, G.; Entman, M.; Carabello, B.A.; Roberts, R.; et al. Aldosterone, Through Novel Signaling Proteins, Is a Fundamental Molecular Bridge Between the Genetic Defect and the Cardiac Phenotype of Hypertrophic Cardiomyopathy. Circulation 2004, 109, 1284–1291. [Google Scholar] [CrossRef]
- Han JS, Choi BS, Yang CW, Kim YS. Aldosterone-induced TGF-beta1 expression is regulated by mitogen-activated protein kinases and activator protein-1 in mesangial cells. J Korean Med Sci. 2009, 24, S195–203. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, A.; Takeda, Y.; Karashima, S.; Kometani, M.; Aono, D.; Demura, M.; Higashitani, T.; Konishi, S.; Yoneda, T.; Takeda, Y. Impact of mineralocorticoid receptor blockade with direct renin inhibition in angiotensin II-dependent hypertensive mice. Hypertens. Res. 2020, 43, 1099–1104. [Google Scholar] [CrossRef]
- Zhang Y, Shao L, Ma A, Guan G, Wang J, Wang Y, et al. Telmisartan delays myocardial fibrosis in rats with hypertensive left ventricular hypertrophy by TGF-β1/Smad signal pathway. Hypertens Res. 2014, 37, 43–9. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-F.; Yang, S.-F.; Chu, H.-J.; Ueng, K.-C. Cross-talk between mineralocorticoid receptor/angiotensin II type 1 receptor and mitogen-activated protein kinase pathways underlies aldosterone-induced atrial fibrotic responses in HL-1 cardiomyocytes. Int. J. Cardiol. 2013, 169, 17–28. [Google Scholar] [CrossRef]
- Matsuki, K.; Hathaway, C.K.; Chang, A.S.; Smithies, O.; Kakoki, M. Transforming growth factor beta1 and aldosterone. Curr. Opin. Nephrol. Hypertens. 2015, 24, 139–144. [Google Scholar] [CrossRef]
- Khan, R. Examining Potential Therapies Targeting Myocardial Fibrosis through the Inhibition of Transforming Growth Factor-Beta 1. Cardiology 2007, 108, 368–380. [Google Scholar] [CrossRef]
- Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol. 2002, 283, H1253–1262. [Google Scholar] [CrossRef]
- Seeland U, Haeuseler C, Hinrichs R, Rosenkranz S, Pfitzner T, Scharffetter-Kochanek K, et al. Myocardial fibrosis in transforming growth factor-beta(1) (TGF-beta(1)) transgenic mice is associated with inhibition of interstitial collagenase. Eur J Clin Invest. 2002, 32, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Seeland U, Schäffer A, Selejan S, Hohl M, Reil JC, Müller P, et al. Effects of AT1- and beta-adrenergic receptor antagonists on TGF-beta1-induced fibrosis in transgenic mice. Eur J Clin Invest. 2009, 39, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Nakajima H, Nakajima HO, Salcher O, Dittiè AS, Dembowsky K, Jing S, et al. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res. 2000, 86, 571–579. [Google Scholar] [CrossRef]
- Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J Mol Cell Cardiol. 2000, 32, 187–195. [Google Scholar] [CrossRef]
- Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, et al. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002, 106, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Burch ML, Yang SNY, Ballinger ML, Getachew R, Osman N, Little PJ. TGF-beta stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad2. Cell Mol Life Sci. 2010, 67, 2077–2790. [Google Scholar] [CrossRef]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Liu X, Sun SQ, Hassid A, Ostrom RS. cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol Pharmacol. 2006, 70, 1992–2003. [Google Scholar] [CrossRef]
- Muslin, A.J. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin. Sci. 2008, 115, 203–218. [Google Scholar] [CrossRef]
- Zhang, D.; Gaussin, V.; Taffet, G.E.; Belaguli, N.S.; Yamada, M.; Schwartz, R.J.; Michael, L.H.; Overbeek, P.A.; Schneider, M.D. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat. Med. 2000, 6, 556–563. [Google Scholar] [CrossRef]
- Romero-Becerra, R.; Santamans, A.M.; Folgueira, C.; Sabio, G. p38 MAPK Pathway in the Heart: New Insights in Health and Disease. Int. J. Mol. Sci. 2020, 21, 7412. [Google Scholar] [CrossRef] [PubMed]
- Gallo, S.; Vitacolonna, A.; Bonzano, A.; Comoglio, P.; Crepaldi, T. ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 2164. [Google Scholar] [CrossRef]
- Duangrat, R.; Parichatikanond, W.; Morales, N.P.; Pinthong, D.; Mangmool, S. Sustained AT1R stimulation induces upregulation of growth factors in human cardiac fibroblasts via Gαq/TGF-β/ERK signaling that influences myocyte hypertrophy. Eur. J. Pharmacol. 2022, 937, 175384. [Google Scholar] [CrossRef]
- Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000, 19, 6341–6350. [Google Scholar] [CrossRef]
- Yoshida, K.; Kim-Mitsuyama, S.; Wake, R.; Izumiya, Y.; Izumi, Y.; Yukimura, T.; Ueda, M.; Yoshiyama, M.; Iwao, H. Excess Aldosterone under Normal Salt Diet Induces Cardiac Hypertrophy and Infiltration via Oxidative Stress. Hypertens. Res. 2005, 28, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Kobayashi, N.; Yoshida, K.; Ohno, T.; Matsuoka, H. Cardioprotective Mechanisms of Spironolactone Associated with the Angiotensin-Converting Enzyme/Epidermal Growth Factor Receptor/Extracellular Signal-Regulated Kinases, NAD(P)H Oxidase/Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1, and Rho-Kinase Pathways in Aldosterone/Salt-Induced Hypertensive Rats. Hypertens. Res. 2005, 28, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Rudolph, A.E.; Bond, B.R.; Rocha, R.; Blomme, E.A.; Goellner, J.J.; Funder, J.W.; McMahon, E.G.; K, T.; R, W.; et al. Transgenic Model of Aldosterone-Driven Cardiac Hypertrophy and Heart Failure. Circ. Res. 2003, 93, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Iglarz M, Touyz RM, Viel EC, Amiri F, Schiffrin EL. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction wtih the renin-angiotension system. Am J Hypertens. 2004, 17, 597–603. [Google Scholar] [CrossRef]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006, 20, 1546–1548. [Google Scholar] [CrossRef]
- Brilla, C.G. Aldosterone and myocardial fibrosis in heart failure. Herz 2000, 25, 299–306. [Google Scholar] [CrossRef]
- Tsukamoto, O.; Minamino, T.; Sanada, S.; Okada, K.-I.; Hirata, A.; Fujita, M.; Shintani, Y.; Yulin, L.; Asano, Y.; Takashima, S.; et al. The Antagonism of Aldosterone Receptor Prevents the Development of Hypertensive Heart Failure Induced by Chronic Inhibition of Nitric Oxide Synthesis in Rats. Cardiovasc. Drugs Ther. 2006, 20, 93–102. [Google Scholar] [CrossRef]
- Okoshi, M.P.; Yan, X.; Okoshi, K.; Nakayama, M.; Schuldt, A.J.; O'Connell, T.D.; Simpson, P.C.; Lorell, B.H. Aldosterone directly stimulates cardiac myocyte hypertrophy. J. Card. Fail. 2004, 10, 511–518. [Google Scholar] [CrossRef]
- Nehme, J.; Mercier, N.; Labat, C.; Benetos, A.; E Safar, M.; Delcayre, C.; Lacolley, P. Differences Between Cardiac and Arterial Fibrosis and Stiffness in Aldosterone-Salt Rats: Effect of Eplerenone. J. Renin-Angiotensin-Aldosterone Syst. 2006, 7, 31–39. [Google Scholar] [CrossRef]
Figure 1.
Mice treatment scheme. There were four study groups for both WT and TG mice. Regular or eplerenone-enriched chow was given from day 21 to 56, while Ald infused subcutaneously via a mini-pump from day 42 to 56. Ald – aldosterone-infused groups; Ald+Ep – both Ald-infused and Ep-enriched chow fed groups; Con – control groups; Ep—eplerenone-enriched chow fed groups.
Figure 1.
Mice treatment scheme. There were four study groups for both WT and TG mice. Regular or eplerenone-enriched chow was given from day 21 to 56, while Ald infused subcutaneously via a mini-pump from day 42 to 56. Ald – aldosterone-infused groups; Ald+Ep – both Ald-infused and Ep-enriched chow fed groups; Con – control groups; Ep—eplerenone-enriched chow fed groups.
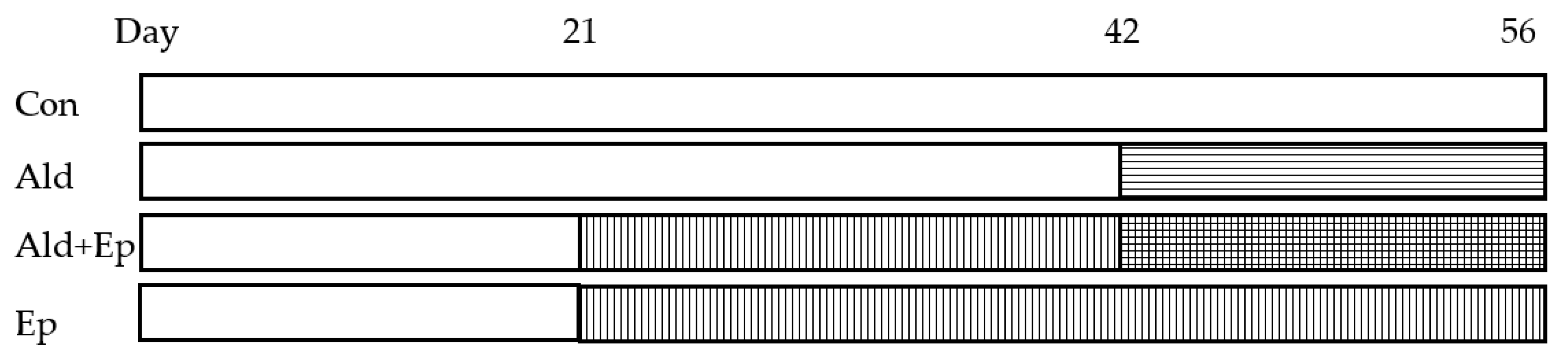
Figure 2.
Cardiac hypertrophy morphometric and ANP mRNA expression data. Significance level for the WT vs TG comparison is given; symbols annotate respective comparisons with: control (*), ‘+Ald’ (#), and ‘+Ald+Ep’ ($) groups; 2A—ventricular heart weight/body weight; 2B—mean diameters of ventricular cardiomyocytes; 2C—ANP mRNA expression in ventricular heart tissues normalised to GAPDH; 2D—representative microphotographs of midventricular cross sections; at the same magnification (200x) differences in cardiomyocytes’ diameters can be noticed. HE staining; 3A—cardiac interstitial fibrosis under basal and experimental conditions; 3B—fibronectin mRNA expression in ventricular heart tissues normalised to GAPDH mRNA; 3C—representative microphotographs, at the same magnification (400x) the amount of fibrotic material can be noticed. Masson’s trichrome staining; 3D—a scoring system was used to assess perivascular fibrosis as described in ‘Methods.
Figure 2.
Cardiac hypertrophy morphometric and ANP mRNA expression data. Significance level for the WT vs TG comparison is given; symbols annotate respective comparisons with: control (*), ‘+Ald’ (#), and ‘+Ald+Ep’ ($) groups; 2A—ventricular heart weight/body weight; 2B—mean diameters of ventricular cardiomyocytes; 2C—ANP mRNA expression in ventricular heart tissues normalised to GAPDH; 2D—representative microphotographs of midventricular cross sections; at the same magnification (200x) differences in cardiomyocytes’ diameters can be noticed. HE staining; 3A—cardiac interstitial fibrosis under basal and experimental conditions; 3B—fibronectin mRNA expression in ventricular heart tissues normalised to GAPDH mRNA; 3C—representative microphotographs, at the same magnification (400x) the amount of fibrotic material can be noticed. Masson’s trichrome staining; 3D—a scoring system was used to assess perivascular fibrosis as described in ‘Methods.
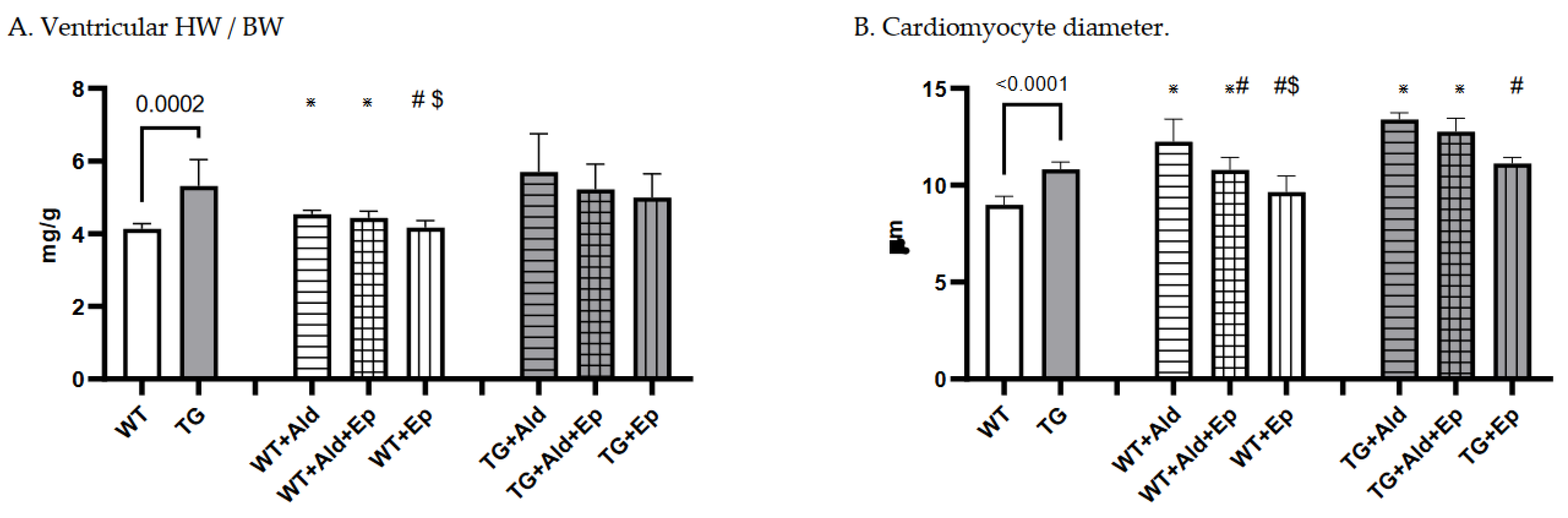
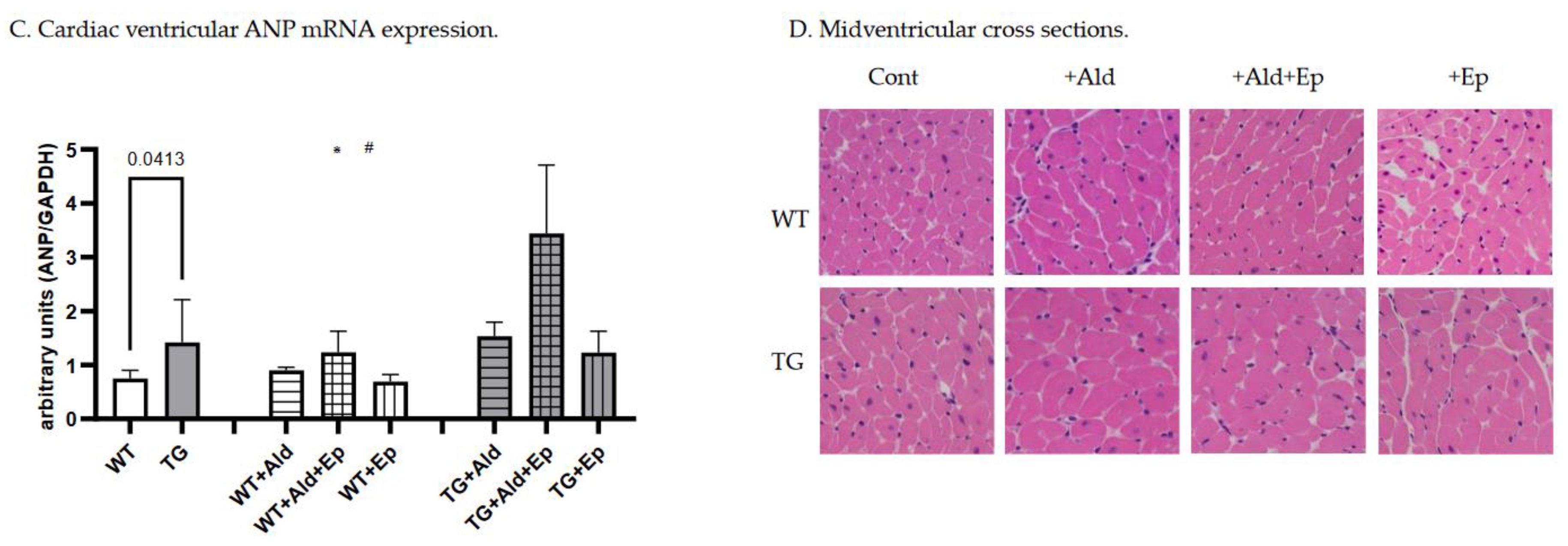
Figure 3.
Cardiac fibrosis morphometric and fibronectin mRNA expression data. Significance level for the WT vs TG comparison is given; symbols annotate respective comparisons with: control (*), ‘+Ald’ (#), and ‘+Ald+Ep’ ($) groups; 2A—ventricular heart weight/body weight; 2B—mean diameters of ventricular cardiomyocytes; 2C—ANP mRNA expression in ventricular heart tissues normalised to GAPDH; 2D—representative microphotographs of midventricular cross sections; at the same magnification (200x) differences in cardiomyocytes’ diameters can be noticed. HE staining; 3A—cardiac interstitial fibrosis under basal and experimental conditions; 3B—fibronectin mRNA expression in ventricular heart tissues normalised to GAPDH mRNA; 3C—representative microphotographs, at the same magnification (400x) the amount of fibrotic material can be noticed. Masson’s trichrome staining; 3D—a scoring system was used to assess perivascular fibrosis as described in ‘Methods.
Figure 3.
Cardiac fibrosis morphometric and fibronectin mRNA expression data. Significance level for the WT vs TG comparison is given; symbols annotate respective comparisons with: control (*), ‘+Ald’ (#), and ‘+Ald+Ep’ ($) groups; 2A—ventricular heart weight/body weight; 2B—mean diameters of ventricular cardiomyocytes; 2C—ANP mRNA expression in ventricular heart tissues normalised to GAPDH; 2D—representative microphotographs of midventricular cross sections; at the same magnification (200x) differences in cardiomyocytes’ diameters can be noticed. HE staining; 3A—cardiac interstitial fibrosis under basal and experimental conditions; 3B—fibronectin mRNA expression in ventricular heart tissues normalised to GAPDH mRNA; 3C—representative microphotographs, at the same magnification (400x) the amount of fibrotic material can be noticed. Masson’s trichrome staining; 3D—a scoring system was used to assess perivascular fibrosis as described in ‘Methods.
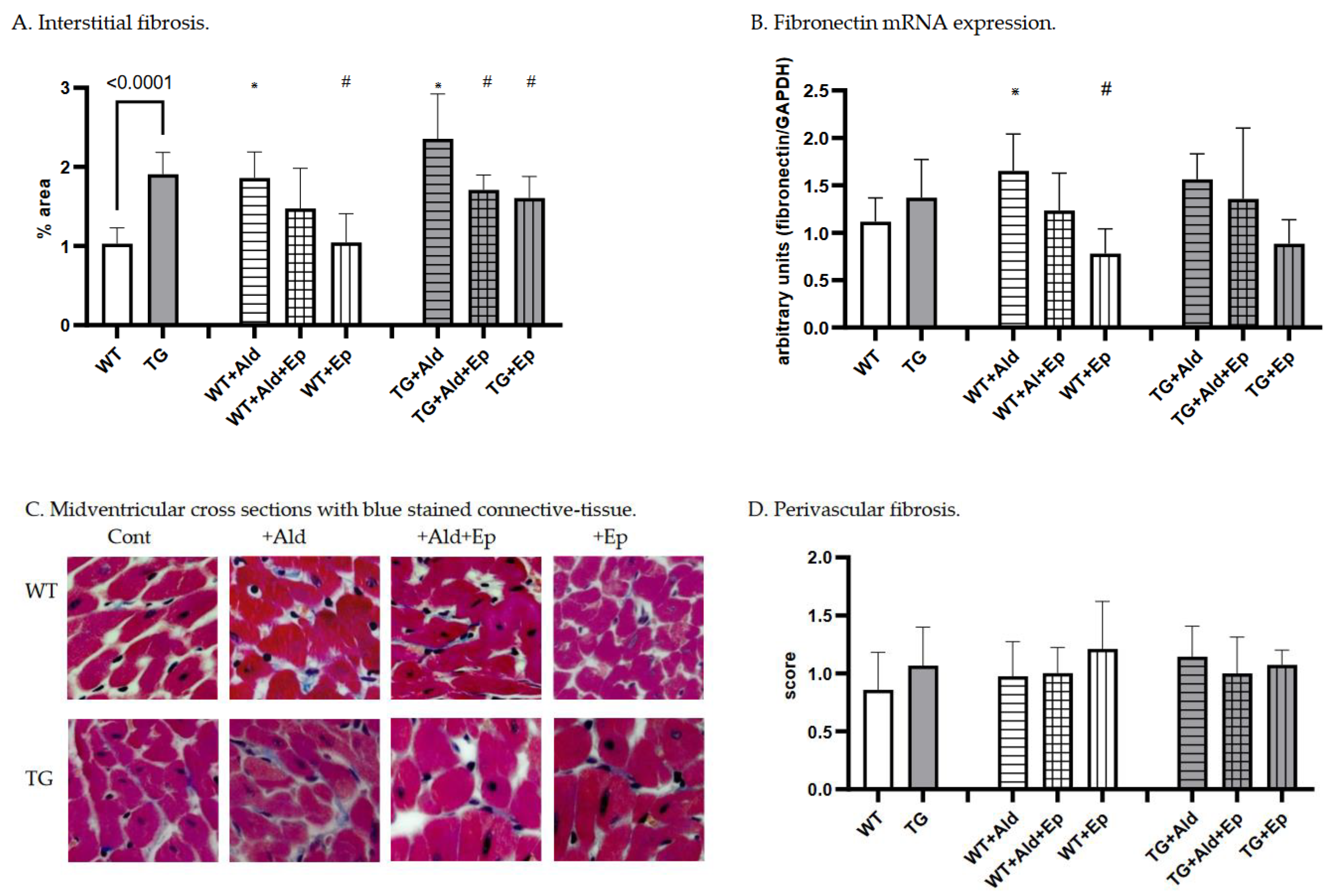
Figure 4.
Western blot analysis. Representative Western blot scans of p-Erk and p-p38 of WT and TG animals with RasGap loading control; A—control (untreated) WT and TG animals; B and C—untreated, Ald-infused (‘+Ald’), Ald-infused and Ep-treated (‘+Al+Ep), eplerenone-treated (‘+Ep) wild type (WT) and TGF-β1-transgenic (TG) mice. Three or four separate experiments were performed for each comparison.
Figure 4.
Western blot analysis. Representative Western blot scans of p-Erk and p-p38 of WT and TG animals with RasGap loading control; A—control (untreated) WT and TG animals; B and C—untreated, Ald-infused (‘+Ald’), Ald-infused and Ep-treated (‘+Al+Ep), eplerenone-treated (‘+Ep) wild type (WT) and TGF-β1-transgenic (TG) mice. Three or four separate experiments were performed for each comparison.
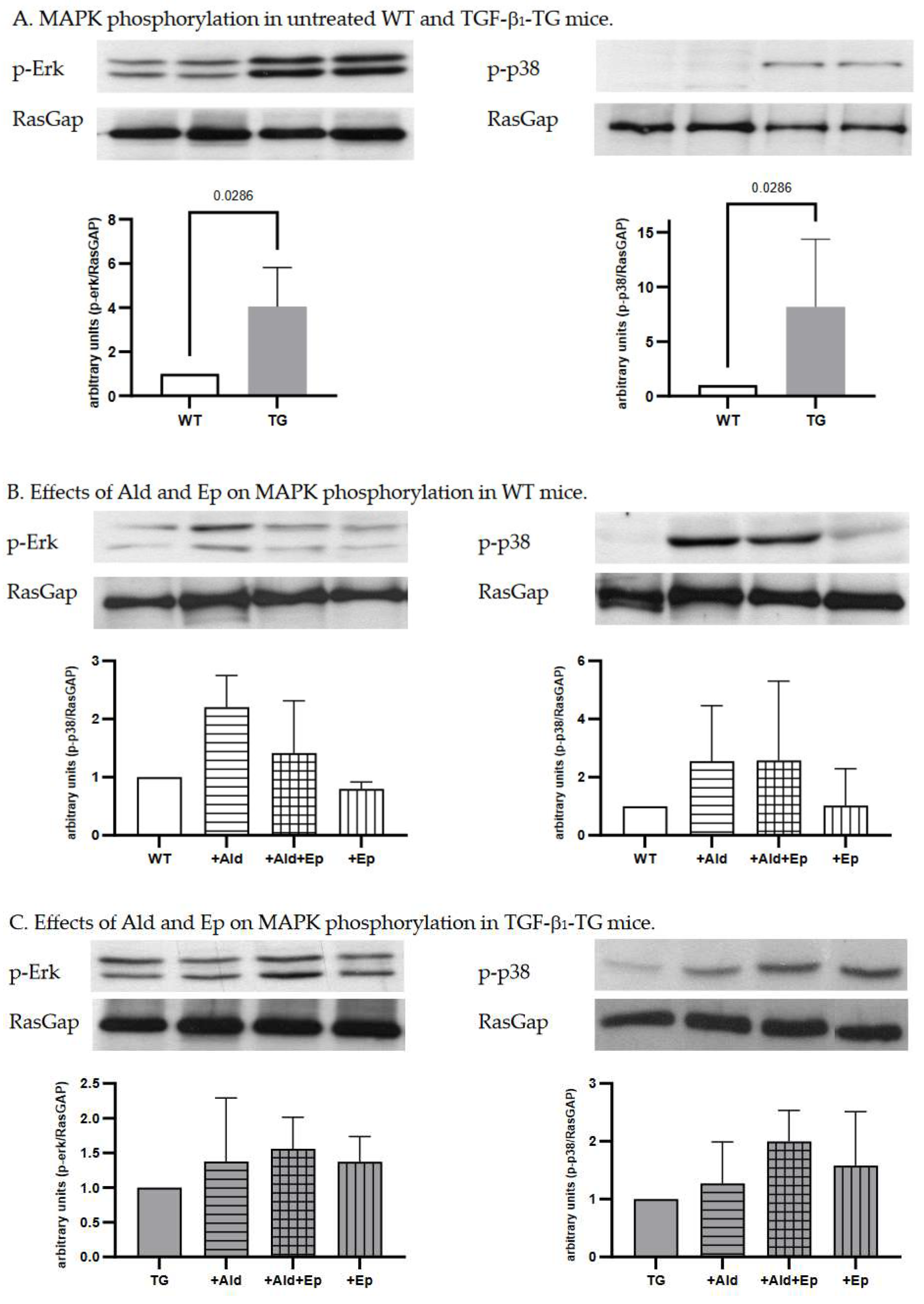
Figure 5.
Relative changes in morphometric parameters due to Ald infusion in WT and TG mice. TG—TGF-1-transgenic; vHW/BW—ventricular heart weight-to-body weight ratio; WT—wild type.
Figure 5.
Relative changes in morphometric parameters due to Ald infusion in WT and TG mice. TG—TGF-1-transgenic; vHW/BW—ventricular heart weight-to-body weight ratio; WT—wild type.
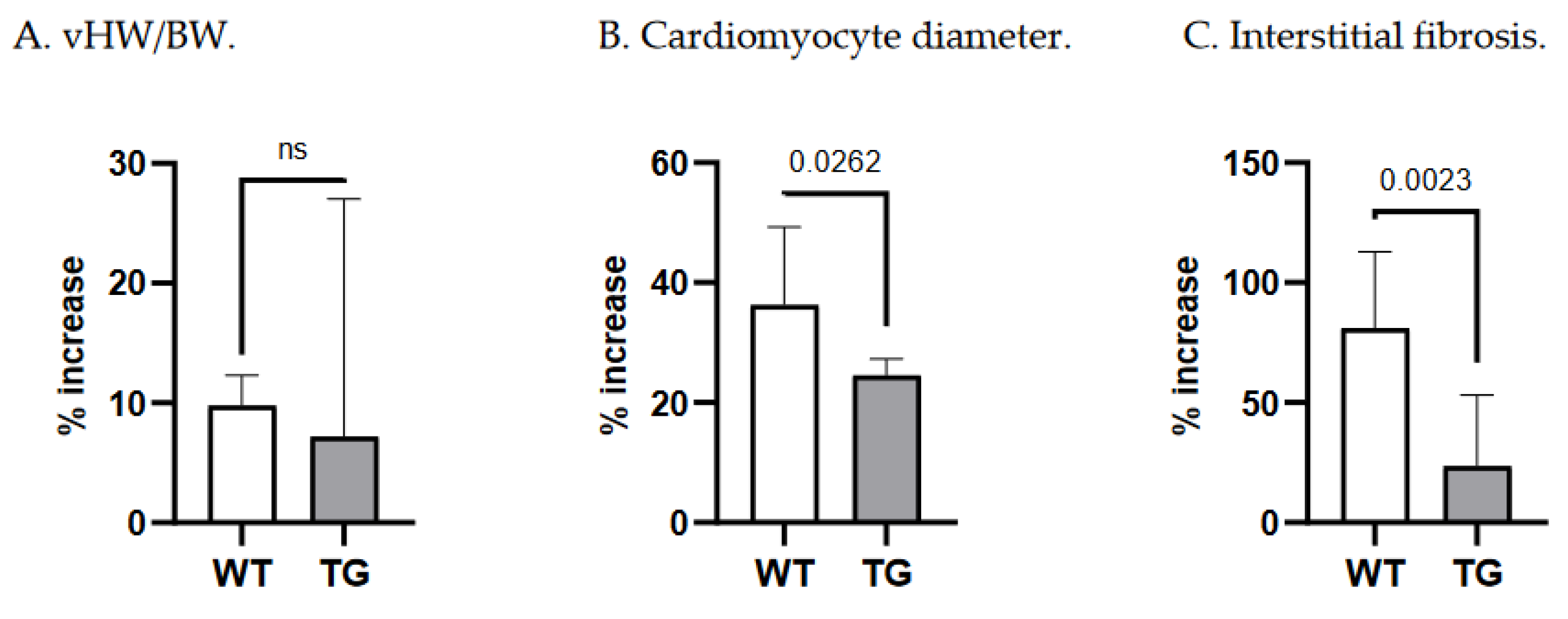

Table 1.
Morphometric and qPCR data of wild-type and TGF-β1-TG mice.
| WT | TG |
p |
WT+Ald | WT+Ald+Ep |
WT+Ep |
p (WT) |
TG+Ald | TG+Ald+Ep |
TG+Ep | p (TG) |
||
| Ventricular HW /BW |
n | 8 | 14 |
<10-3 |
7 | 7 | 8 |
<10-4 |
9 | 9 | 9 |
0.31 |
| mg/g | 4.1±0.1 | 5.3±0.7 | 4.5±0.1* | 4.4±0.2* | 4.2±0.2#$ | 5.7±1.1 | 5.2±0.7 | 5±0.6 | ||||
| Cardiomyocyte diameter | n | 8 | 9 |
<10-4 |
7 | 7 | 8 |
<10-4 |
9 | 9 | 9 |
<10-4 |
| µm | 8.8(0.8) | 10.9(0.3) | 12.3±1.2* | 10.8±0.6*# | 9.6±0.8#$ | 13.4(0.5)* | 12.8(1.1)* | 11(0.5)# | ||||
| ANP/GAPDH mRNA |
n | 7 | 14 |
0.041 |
6 | 7 | 6 |
10-3 |
7 | 7 | 6 |
<10-4 |
| - | 0.8±0.2 | 1.4±0.8 | 0.9±0.06 | 1.2±0.4* | 0.7±0.1$ | 1.5±0.3 | 3.4±1.3*# | 1.2±0.4$ | ||||
| Interstitial fibrosis |
n | 8 | 9 |
<10-4 |
7 | 7 | 8 |
<10-3 |
9 | 9 | 10 |
<10-3 |
| % | 1±0.1 | 1.9±0.1 | 1.9±0.1* | 1.5±0.2 | 1.05±0.1# | 2.4±0.2* | 1.7±0.1# | 1.61±0.1# | ||||
| Perivascular fibrosis |
n | 8 | 9 |
0.21 |
7 | 6 | 6 |
0.27 |
9 | 9 | 10 |
0.73 |
| score | 0.9±0.3 | 1.1±0.3 | 1±0.3 | 1±0.22 | 1.2± 0.4 | 1.2±0.26 | 1±0.3 | 1.1±0.1 | ||||
| FBN/GAPDH mRNA |
n | 7 | 15 |
0.15 |
7 | 7 | 7 |
<10-3 |
7 | 9 | 7 |
0.06 |
| - | 1.1±0.3 | 1.4±0.4 | 1.7±0.4* | 1.2±0.4 | 0.8±0.3# | 1.6±0.3 | 1.4±0.8 | 0.9±0.3 | ||||
| Kidney W /BW |
n | 8 | 14 |
0.15 |
7 | 10 | 8 |
<10-4 |
9 | 9 | 9 |
<10-4 |
| mg/g | 11.9±0.3 | 12.8±1.6 | 14.9±1.6* | 13.7±1.2* | 11.4±1.5#$ | 15.9±1.5* | 13±0.8# | 12.3±1.8# | ||||
Legend: p for comparisons of WT and TG controls, and four-group (each genotype) ANOVAs/Kruskall-Wallis test was given; significant differences in Tukey’s/Dunn’s post-hoc tests are denoted with * vs controls, # vs ‘+Ald’, $ vs ‘+Ald+Ep’: * for vHW/BW vs WT+Ald p=3x10-4, WT+Ald+Ep p=6x10-3; for cardiomyocyte diameter vs WT+Ald p<10-4, WT+Ald+Ep p=10-3, TG+Ald p<10-4, and TG+Ald+Ep p=0.006; for ANP/GAPDH mRNA vs WT+Ald+Ep p=0.004, and TG+Ald+Ep p<10-4; for interstitial fibrosis vs WT+Ald p<10-3, and TG+Ald p=0.0498; for FBN/GAPDH mRNA vs WT+Ald p=0.029; for kidney W/BW vs WT+Ald p<10-3, TG+Ald p<10-4; # for vHW/BW vs WT+Ep p=8x10-4, for cardiomyocyte diameter vs WT+Ald+Ep p=0.01, WT+Ep p<10-4, and TG+Ep p=6x10-4; for interstitial fibrosis vs: WT+Ep p=10-3, TG+Ald p=0.003, and TG+Ep p=3x10-4; for FBN/GAPDH mRNA vs WT+Ep p=3x10-4; for kidney W/BW vs WT+Ep p<10-4, TG+Ald+Ep p=10-3, and TG+Ep p<10-4; $ for vHB/BW vs WT+Ep p=0.019; for cardiomyocyte diameter vs WT+Ep p=0.049; for ANP/GAPDH mRNA vs WT+Ep p=0.002; for kidney W/BW vs WT+Ep p=0.003; Ald—aldosterone; BW—body weight; Ep—eplerenone; HW—heart weight; TG –transgenic; W—weight; WT—wild-type.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



