
Submitted:
30 June 2023
Posted:
04 July 2023
You are already at the latest version
Abstract
The identification of cancer predisposition syndromes (CPSs) plays a crucial role in understanding the etiology of pediatric cancers. CPSs are genetic mutations that increase the risk of developing cancer at an earlier age compared to the risk for the general population. This article aims to provide a comprehensive analysis of three unique cases involving pediatric patients with CPS who were diagnosed with multiple simultaneous or metachronous cancers. The first case involves a child with embryonal rhabdomyosarcoma, nephroblastoma, glioma, and subsequent medulloblastoma. Genetic analysis identified two pathogenic variants in the BRCA2 gene. The second case involves a child with alveolar rhabdomyosarcoma, Xanthogranuloma Juvenile, gliomas, and subsequent JMML/MDS/MPS. A pathogenic variant in the NF1 gene was identified. The third case involves a child with pleuropulmonary blastoma and pediatric cystic nephroma/nefroblastoma in whom a pathogenic variant in the DICER1 gene was identified. Multiple simultaneous and metachronous cancers in pediatric patients with CPSs are a rare but significant phenomenon. Comprehensive analysis and genetic testing play crucial roles in understanding the underlying mechanisms and guiding treatment strategies for these unique cases. Early detection and targeted interventions are crucial for improving outcomes in these individuals.
Keywords:
cancer predisposition syndromes
; genetic abnormalities
; neoplasms
; simultaneous occurrence
; pediatric cancers
; personalized treatment
; tumor suppressor genes
1. Introduction
Cancer incidence is increasing in all age groups in the general population, but neoplasms in children and adolescents are still considered rare diseases [1,2,3]. The global incidence of childhood cancer is estimated to be nearly 400,000 cases per year [4]. Multiple diagnoses are extremely rare. While the exact causes of oncological diseases remain largely unknown, the role of genetic factors and the identification of syndromes that predispose individuals to cancer are gradually being identified and understood with the advancements in genetics and genomics [5,6,7,8,9]. With medical advancements, targeted treatments, anticipatory screening, and prevention of subsequent cancers are possible, which is especially important for individuals with cancer predisposition syndromes (CPSs) [10,11,12,13]. Despite widespread prevention efforts, early detection of cancer is not fully effective. As early as 1889, Theodore Bilroth suggested that the possibility of developing a second cancer should not be excluded even if the first cancer has been completely removed through surgery. The increased risk of developing a second primary cancer among cancer survivors is well-known, but the simultaneous occurrence of multiple cancers remains unclear. Depending on the timing of the onset of multiple cancers, they can be classified as concurrent, synchronous, or metachronous. Most authors consider a time frame of two months or less for calling a cancer synchronous. For simultaneous cancers, some authors refer to them as a subset of lesions or use the term "metachronous" cancer [14].
Given the rarity of CPS, the main aim of this article is to identify cases with concomitant or metachronous multiple neoplasms of a Polish population in a single-centre study and to present a comprehensive genetic, phenotypic and a clinical analysis on disease progression and treatment.
2. Materials and Methods
Out of the 2,387 patients who were newly diagnosed with cancer and hospitalized at the Karol Jonscher Clinical Hospital of the Karol Marcinkowski Medical University in Poznan between 2000 and 2021, 182 were identified as having CPSs. These syndromes were defined based on the presence of characteristic phenotypic abnormalities, such as Down syndrome, NF1, and others, or through confirmation of mutations in genetic tests. Among them cases with simultaneous or metachronous multiple cancers were recognized. Targeted next-generation sequencing (NGS) was utilized for the identification of pathogenic variants in these patients (performed at the Medical University of Lodz). The study received a positive opinion of the Bioethics Committee of the Poznan University of Medical Sciences (Decision of the Bioethics Committee No. 574/23 issued on June 21, 2023). Written informed consent has been obtained from the parents to publish this paper, including consent to process the child's image for scientific purposes (Case 1; the date of approval January 1, 2021).
3. Results
Despite the relatively large number of patients included in the analysis, only three children were found to have simultaneous or metachronous multiple cancers.
3.1. Pathogenic variants of the BRCA2 gene - Case 1
The first patient is the firstborn child of a mother with hypothyroidism. The parents are unrelated, and the father has no history of chronic diseases. There is no family history of cancer. The girl was born at full term (at 40 weeks' gestation) with a birth weight of 2,950 g and scored 10 on the Apgar scale. Soon after birth, extensive café-au-lait spots were observed, primarily on the right thigh, which resembled those seen in the mother. Starting from the second month of life, the child exhibited left hemiplegic hypertrophy (Figure 1) and a slight delay in psychomotor development.
At 7 months of age, the child was admitted to the hospital due to isolated swelling of the left upper limb. During this hospitalization, additional abnormalities were detected, including a horseshoe kidney and an accessory spleen. At 14 months of age, the parents noticed a nodule in the child's left scapular region, which was subsequently diagnosed as embryonal rhabdomyosarcoma (ERMS) following its removal. On ultrasound, this tumor measured 2.5 x 1.8 x 1.0 cm and showed a probable connection to the muscle. Further tests revealed a horseshoe-shaped tumor in the kidney, measuring 8.0 x 5.7 x 8.7 cm, as identified by abdominal ultrasound, and confirmed by computed tomography (CT) scan. Biopsy results confirmed it to be a nephroblastoma (Wilms tumor, WT) of mixed, intermediate-risk, G3 type. Furthermore, an optic nerve glioma was detected during magnetic resonance imaging (MRI). To validate the histopathological findings, multiple diagnostic centers in Poland (The Children’s Memorial Health Institute in Warsaw) and Germany (at the University of Bonn and the Heidelberg University) were consulted, all of which confirmed the diagnoses of ERMS and Wilms tumor.
Given the concurrence of these three cancers, additional NGS testing was conducted. This revealed two distinct heterozygous pathogenic variants in the BRCA2 gene, namely c.1773_1776delTTAT (p.Ile591MetfsTer22; dbSNP rs80359304) inherited from the mother, and 886delGT(c.658_659delGT; p.Val220fs; dbSNP rs80359604) inherited from the father.
As a result of the hemihypertrophy, chromosome 11 methylation abnormality tests were conducted, which did not identify any large deletions or duplications in the 11p15 region through MLPA (Centogene, Germany). Utilizing Illumina TruSight One Expanded Sequencing Panel and Illumina NexSeq 550 instrument (Illumina Inc, San Diego, California), NGS analysis was performed, focusing on genes with documented clinical relevance to oncologic disorders. The analysis, conducted using Variant Studio v.3.0 (Illumina Inc, San Diego, California) and IGV v.2.3 (Broad Institute California) software, did not reveal the presence of any other pathogenic changes. Additionally, imprinted DNA methylation analysis was carried out at various loci, including DIRAS3 (1p31); PLAGL1 (6q24); GRB10 (7p12); PEG1/MEST (7q32); KCNQ1OT1/H19/IGF2 DMRO (11p15); DLK1 (14q32); SNRPN (15q11), PEG3 (19q32); and NESPAS/GNAS (20q13). Chemotherapy was initiated based on the Cooperative Weichteilsarkomstudiengruppe (CWS) protocol, and following confirmation of Wilms tumor through histopathological examination, treatment continued according to the UMBRELLA protocol of the International Society of Pediatric Oncology Renal Tumour Study Group (SIOP-RTSG). Two and a half years after the original diagnosis, an MRI revealed the presence of a medulloblastoma tumor (classic type, central nervous system (CNS) WHO G4) in the left cerebellar hemisphere. In light of the outcome of the non-radical surgery and tumor progression during chemotherapy, the child necessitated not only surgical intervention but also chemotherapy and radiation therapy. Considering the presence of BRCA2 pathogenic variants, posing a risk of Fanconi anemia, and anticipating bone marrow aplasia as a result of treatment, anticipatory allogeneic hematopoietic stem cell transplantation (HSCT) was performed. Figure 2 provides a timeline of the patient's disease progression.
3.2. Pathogenic variant of the NF1 gene - Case 2
The second patient is the first-born child of unrelated parents, both of whom are healthy with no family history of chronic diseases. However, each parent has more than 5 café-au-lait spots. The patient was born prematurely at 36 weeks' gestation due to premature rupture of the fetal membranes, with a birth weight of 3,315 grams and an Apgar score of 10. Café-au-lait spots were initially observed on the patient's skin after birth, and their number increased over time. The patient now has multiple café-au-lait spots distributed throughout the body. At 12 months of age, the patient's mother noticed blood in her diaper, which prompted her to seek medical attention. Abdominal ultrasound revealed a pelvic tumor measuring over 10.0 cm in diameter. Subsequent MRI confirmed a vaginal tumor measuring 11.0 x 7.7 x 5.8 cm (Figure 3).
Histopathological examination of the biopsy led to a diagnosis of alveolar rhabdomyosarcoma (ARMS). Simultaneous MRI of the CNS revealed eight gliomas, including one involving the optic nerve, measuring up to 1.5 x 1.4 x 1.2 cm. Additionally, three yellow-brown skin lesions were found in the craniofacial region, which were confirmed to be Xanthogranuloma Juvenile through histopathological examination. The patient underwent treatment following the CWS protocol, which included chemotherapy and postponed radical surgery. After treatment, complete remission of ARMS was achieved, and the Xanthogranuloma Juvenile lesions also resolved. The results of NGS genetic analysis with a TruSight One Expanded Sequencing Panel revealed the presence of a heterozygous pathogenic mutation in the NF1 gene (c.574C>T; p.Arg192Ter; dbSNP rs397514641).
After 2.5 years from the initial diagnosis of malignancy, a follow-up blood examination showed thrombocytopenia, which progressed to bicytopenia, with the addition of transfusion-dependent anemia. Further studies were conducted on peripheral blood and bone marrow smears (Figure 4 and Figure 5, respectively). Flow cytometry studies revealed the presence of immature myeloid cells with the immunophenotype CD33+CD38+CD31+CD11b+/-CD11c+CD64+CD19-CD10-CD13+CD34-CD117-HLADR-CD20-CD22-CD3-CD5-CD7-CD65+CD15+CD123+MPO+TdT- FSC mid, SSC high, identified by flow cytometry. In the bone marrow, these cells accounted for 77% of the total cell population, with 13% of them being myeloblasts.
Several additional tests were performed. Fluorescence in-situ hybridization (FISH) analysis showed that 85% of the cells exhibited chromosome 5 monosomy. Molecular analyses were also conducted, which revealed NF1 loss of heterozygosity (LOH) but no somatic mutations typically associated with juvenile myelomonocytic leukemia (JMML), including no mutations in PTPN11, NRAS, KRAS, CBL, JAK2 exon 12, CALR exon 9, MPL exon 10, or ASXL1 exon 13. These additional analyses were conducted by the EWOG MDS group in Freiburg.
Although NF1 mutations are risk factors for JMML, the presence of monosomy of chromosome 5, an atypical methylation pattern characterized by a high methylation profile, and low levels of fetal hemoglobin (HbF) were more indicative of secondary myelodysplastic/myeloproliferative syndrome (MDS/MPS). Following three courses of treatment with azacitidine, the patient underwent allogeneic hematopoietic stem cell transplantation (allo-HSCT). The disease course timeline for the second patient is depicted in Figure 6.
3.3. Pathogenic variant of the DICER1 gene - Case 3
The third patient is a second-born child. His parents are unrelated, with no family history of chronic diseases other than polycystic thyroid goiter (in the mother), which is also present in the child's grandmother and aunt. He was born at 38 weeks’ gestation with a birth weight of 4,220 g and a 10-point Apgar score. At the age of 6 months, he was admitted to our hospital due to suspected lung abscess. CT scan revealed a 4.6 x 4.5 x 5.5 cm lesion in the right lung, and ultrasound showed a 2.7 x 1.9 x 3.0 cm lithocystic lesion in the left kidney.
Considering the family history of multinodular thyroid goiter and the presence of a lung and kidney tumor in the boy, there was a suspicion of pleuropulmonary blastoma in the right lung and nephroma in the left kidney. Following the removal of the lung tumor, histopathological examination confirmed pleuropulmonary blastoma type II (PPB II). Due to the incomplete removal of the tumor during surgery (a non-radical surgery), chemotherapy was administered according to the Cooperative Weichteilsarkomstudiengruppe (CWS) protocol. After three months of chemotherapy, a follow-up abdominal ultrasound revealed progression of the left kidney tumor, leading to the decision to perform a nephrectomy. Histopathological examination confirmed pediatric cystic nephroma/nefroblastoma (Wilms tumor, WT). The boy continued chemotherapy as per the CWS protocol, and a resection of the residual lung tumor was also performed. The child successfully completed the treatment and remained in complete remission.
NGS genetic analysis using the same TruSight One Expanded Sequencing Panel, identified a pathogenic heterozygous variant in the DICER1 gene (c.4930T>G, p.Leu1573Arg). Genetic counseling was extended to the entire maternal family. Figure 7 illustrates the disease course timeline of the patient.
4. Discussion
Although the incidence of cancer increases with age, cancers in the pediatric population remain in the group of rare diseases, and their multiple diagnoses – especially within 3 years – represent an even rare phenomenon. [15]. The nomenclature of multiple neoplasm seems clear, however in each case at least one difficulty must be mentioned. Due to the time each disease takes to develop to reach the picture presented in examination studies, it is not known whether the first lesion occurred within two months as another when both are seen at the same time. Although malignant tumors imply a shorter expansion time, it still depends on the individual and accurate diagnosis.
Depending on the type of mutation, the presence of a genetic background is associated with the onset of the disease (sometimes indicating a better or worse prognosis) or enables its prevention [5,8,11,16]. The clinical cases reported in this paper exemplify the links identified between specific mutations in the genome and the induction of cancers in infancy and early childhood (at 14, 12 and 6 months of age at diagnosis, respectively). The simultaneously diagnosed tumors in the first reported case were embryonal rhabdomyosarcoma (ERMS), nephroblastoma (Wilms' tumor, WT), and glioma, which (after 2.5 years from the first oncological diagnosis) progressed to medulloblastoma classical CNS type WHO G4, which can be classified as metachronous tumor. What is more, this individual suffered Fanconi anemia. This complex phenotype was diagnosed in a carrier of two different heterozygous variants in BRCA2 gene. These variants were previously associated with hereditary CPSs [17,18]. The second clinical case involved an NF1 gene mutation carrier concurrently diagnosed with alveolar rhabdomyosarcoma (ARMS), Xanthogranuloma Juvenile and gliomas located in the CNS, including in the optic nerve. Approximately 2.5 years from the first oncological diagnosis, the metachronous disease - JMML/MDS/MPS was found, but it remains in full remission of previous neoplasms. The observed variant was previously associated a number of times with hereditary CPSs, and based on that, assigned using Ambry Genetics® General Variant Classification Scheme as pathogenic. The third patient (with a history of polycystic thyroid goiter in the female line in his mother's family members) was simultaneously diagnosed with pleumopulmonary blastoma type II (PPB II) and pediatric cystic nephroma/nefroblastoma related to mutations in the DICER1 gene. After 1.5 years of follow-up, he remains in full remission. The identified varianthad been previously shown to be associated with pleuropulmonary blastoma [19], and may also be involved in other cancers [20]. Table 2 presents the characteristics of genes with variants associated with CPS in children studied.
Awareness of CPS, manifested by increased susceptibility to cancer formation should lead to adjustments in therapy, insofar as proposed procedures may increase the possibility of neogenesis. However, the indications are very often ambiguous due to the benefit/risk ratio, which clinicians must keep in mind and consider. In fact, also avoiding high-risk courses of treatment leads to progression of the current disease - cancer - and may lead to the patient's death before developing another cancer due to the procedure under consideration. These topics are indirectly related to medical futility [30], an awareness of which should be an ethical challenge for everyone, as it brings more suffering than long-term or even short-term benefit to the patient.
Given the current knowledge of CPS, which is augmented by case-studies, defining standards for treatment modification is crucial for the transparency of medical procedures with oncology protocols. The Priority must be placed upon the quality of life of the patients and their families not only during but also after the treatment offered, and minimizing the risk of adverse events with the development of another metachronous cancer as one of the most important side effects to improve healthcare delivery [31,32,33]. Very often, patients with CPS are at higher risk of developing a number of cancers, not just one particular type [1,16], so preventive management is essential to avoid increasing the already elevated risk of cancer in these cases. The question "How to guide treatment?" remains open as long as we discuss the very or even extremely rare genetic disorders that cause CPS. There is currently one well-known genetic disorder, Down syndrome (DS), for which distinct diagnostic and treatment pathways have been developed, leading to early diagnosis and reduced mortality among these patients [5]. Such supportive care efforts should also be undertaken in other cases of CPS and lead to the establishment of dedicated procedures for such patients.
As suggested above, diagnostic, and therapeutic management should not further increase the risk of developing secondary cancers in patients with CPS. Radiation used not only in CT scans and other imaging procedures, but especially in therapy, should be avoided if proven to cause carcinogenesis, even when considering long-term risks measured in decades, not just a few years [34,35,36]. Moreover, awareness of the implications of CPS should prompt therapists to be more vigilant in monitoring in order to detect cancers at an early stage. The open question remains to what extent? The lack of developed standards should motivate most specialist centers to introduce such management algorithms in the face of the growing number of patients with CPS and the detection of new mutations that cause carcinogenesis. The validity of anticipatory treatment can also be considered, but further studies calculating the benefit/risk ratio would be needed to fully discuss and answer this thesis. So far, there is no targeted treatment, however further dynamic development in this field will generate it.
Author Contributions
Conceptualization, G. T., E. S., P. S.-S., and D. J.-L.; methodology, G. T., E. S., P. S.-S., and D. J.-L.; software, G. T., E. S., P. S.-S., and D. J.-L.; validation, E. S. and D. J.-L.; formal analysis, E. S. and D. J.-L.; investigation, G. T., P. S.-S., M. H. and D. J.-L.; resources, D. J.-L.; data curation, G. T., M. H., and D. J.-L.; writing—original draft preparation, G. T., E. S., M. H., D. J.-L.; writing—review and editing, P. S.-S.; visualization, G. T., D. J.-L.; supervision, D. J.-L.; project administration, G. T., and D. J.-L.; funding acquisition, D. J.-L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Ethical review and approval were waived for this study due to the retrospective and non-invasive nature of the study. These rules are compliant with the guidelines of the Bioethical Commission of Poznan University of Medical Science, Poland. The decision of the Bioethics Committee number 574/23; date of decision June 21, 2023.
Informed Consent Statement
Written informed consent has been obtained from the parents to publish this paper, including consent to process the child's image for scientific purposes (Case 1; the date of approval January 1, 2021).
Data Availability Statement
Data available on request due to restrictions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA: a cancer journal for clinicians 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A. International incidence of childhood cancer, 2001-10: a population-based registry study. The Lancet. Oncology 2017, 18, 719–731. [Google Scholar] [CrossRef]
- Kentsis, A. Why do young people get cancer? Pediatr Blood Cancer 2020, 67, e28335. [Google Scholar] [CrossRef]
- Ward, Z.J.; Yeh, J.M.; Bhakta, N.; Frazier, A.L.; Atun, R. Estimating the total incidence of global childhood cancer: a simulation-based analysis. The Lancet. Oncology 2019, 20, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Shahani, S.A.; Marcotte, E.L. Landscape of germline cancer predisposition mutations testing and management in pediatrics: Implications for research and clinical care. Frontiers in pediatrics 2022, 10, 1011873. [Google Scholar] [CrossRef]
- Newman, S.; Nakitandwe, J.; Kesserwan, C.A.; Azzato, E.M.; Wheeler, D.A.; Rusch, M.; Shurtleff, S.; Hedges, D.J.; Hamilton, K.V.; Foy, S.G.; et al. Genomes for Kids: The Scope of Pathogenic Mutations in Pediatric Cancer Revealed by Comprehensive DNA and RNA Sequencing. Cancer Discov 2021, 11, 3008–3027. [Google Scholar] [CrossRef] [PubMed]
- Kratz, C.P.; Jongmans, M.C.; Cavé, H.; Wimmer, K.; Behjati, S.; Guerrini-Rousseau, L.; Milde, T.; Pajtler, K.W.; Golmard, L.; Gauthier-Villars, M.; et al. Predisposition to cancer in children and adolescents. Lancet Child Adolesc Health 2021, 5, 142–154. [Google Scholar] [CrossRef]
- Benton, M.L.; Abraham, A.; LaBella, A.L.; Abbot, P.; Rokas, A.; Capra, J.A. The influence of evolutionary history on human health and disease. Nature Reviews Genetics 2021, 22, 269–283. [Google Scholar] [CrossRef]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med 2015, 373, 2336–2346. [Google Scholar] [CrossRef]
- Fair, D.; Potter, S.L.; Venkatramani, R. Challenges and solutions to the study of rare childhood tumors. Current opinion in pediatrics 2020, 32, 7–12. [Google Scholar] [CrossRef]
- Scollon, S.; Anglin, A.K.; Thomas, M.; Turner, J.T.; Wolfe Schneider, K. A Comprehensive Review of Pediatric Tumors and Associated Cancer Predisposition Syndromes. Journal of genetic counseling 2017, 26, 387–434. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Nichols, K.E.; Schiffman, J.D.; Plon, S.E.; Brodeur, G.M. The Future of Surveillance in the Context of Cancer Predisposition: Through the Murky Looking Glass. Clin Cancer Res 2017, 23, e133–e137. [Google Scholar] [CrossRef]
- Druker, H.; Zelley, K.; McGee, R.B.; Scollon, S.R.; Kohlmann, W.K.; Schneider, K.A.; Wolfe Schneider, K. Genetic Counselor Recommendations for Cancer Predisposition Evaluation and Surveillance in the Pediatric Oncology Patient. Clin Cancer Res 2017, 23, e91–e97. [Google Scholar] [CrossRef]
- Krueger, H.; McLean, D.; Williams, D. The Prevention of Second Primary Cancers: A Resource for Clinicians and Health Managers; S.Karger AG; 2008. [Google Scholar]
- Zahnreich, S.; Schmidberger, H. Childhood Cancer: Occurrence, Treatment and Risk of Second Primary Malignancies. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Bakhuizen, J.J.; Hopman, S.M.J.; Bosscha, M.I.; Dommering, C.J.; van den Heuvel-Eibrink, M.M.; Hol, J.A.; Kester, L.A.; Koudijs, M.J.; Langenberg, K.P.S.; Loeffen, J.L.C.; et al. Assessment of Cancer Predisposition Syndromes in a National Cohort of Children With a Neoplasm. JAMA network open 2023, 6, e2254157. [Google Scholar] [CrossRef]
- Duzkale, N.; Kandemir, O. The Relationship of Mutation Carriage of BRCA1/2 and Family History in Triple-Negative Breast Cancer: Experience from a Diagnostic Center in Turkey. Eur J Breast Health 2021, 17, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Offit, K.; Levran, O.; Mullaney, B.; Mah, K.; Nafa, K.; Batish, S.D.; Diotti, R.; Schneider, H.; Deffenbaugh, A.; Scholl, T.; et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J Natl Cancer Inst 2003, 95, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.A.; Ivanovich, J.; Priest, J.R.; Gurnett, C.A.; Dehner, L.P.; Desruisseau, D.; Jarzembowski, J.A.; Wikenheiser-Brokamp, K.A.; Suarez, B.K.; Whelan, A.J.; et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 2009, 325, 965. [Google Scholar] [CrossRef]
- Hata, A.; Kashima, R. Dysregulation of microRNA biogenesis machinery in cancer. Crit Rev Biochem Mol Biol 2016, 51, 121–134. [Google Scholar] [CrossRef]
- Meyer, S.; Tischkowitz, M.; Chandler, K.; Gillespie, A.; Birch, J.M.; Evans, D.G. Fanconi anaemia, BRCA2 mutations and childhood cancer: a developmental perspective from clinical and epidemiological observations with implications for genetic counselling. Journal of medical genetics 2014, 51, 71–75. [Google Scholar] [CrossRef]
- Rosenbaum, T.; Wimmer, K. Neurofibromatosis type 1 (NF1) and associated tumors. Klinische Padiatrie 2014, 226, 309–315. [Google Scholar] [CrossRef]
- Robertson, J.C.; Jorcyk, C.L.; Oxford, J.T. DICER1 syndrome: DICER1 mutations in rare cancers. Cancers 2018, 10, 143. [Google Scholar] [CrossRef]
- González, I.A.; Stewart, D.R.; Schultz, K.A.P.; Field, A.P.; Hill, D.A.; Dehner, L.P. DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma. Modern Pathology 2022, 35, 4–22. [Google Scholar] [CrossRef]
- Grant, C.N.; Rhee, D.; Tracy, E.T.; Aldrink, J.H.; Baertschiger, R.M.; Lautz, T.B.; Glick, R.D.; Rodeberg, D.A.; Ehrlich, P.F.; Christison-Lagay, E. Pediatric solid tumors and associated cancer predisposition syndromes: Workup, management, and surveillance. A summary from the APSA Cancer Committee. J Pediatr Surg 2022, 57, 430–442. [Google Scholar] [CrossRef]
- Patil, P.; Pencheva, B.B.; Patil, V.M.; Fangusaro, J. Nervous system (NS) Tumors in Cancer Predisposition Syndromes. Neurotherapeutics 2022, 19, 1752–1771. [Google Scholar] [CrossRef]
- Woodward, E.R.; Meyer, S. Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef]
- Radtke, H.B.; Berger, A.; Skelton, T.; Goetsch Weisman, A. Neurofibromatosis Type 1 (NF1): Addressing the Transition from Pediatric to Adult Care. Pediatric Health Med Ther 2023, 14, 19–32. [Google Scholar] [CrossRef]
- Caroleo, A.M.; De Ioris, M.A.; Boccuto, L.; Alessi, I.; Del Baldo, G.; Cacchione, A.; Agolini, E.; Rinelli, M.; Serra, A.; Carai, A.; et al. DICER1 Syndrome and Cancer Predisposition: From a Rare Pediatric Tumor to Lifetime Risk. Front Oncol 2020, 10, 614541. [Google Scholar] [CrossRef]
- Ferdynus, M.P. Why the term ‘persistent therapy’ is not worse than the term ‘medical futility’. Journal of Medical Ethics 2022, 350–352. [Google Scholar] [CrossRef]
- Rossini, L.; Durante, C.; Bresolin, S.; Opocher, E.; Marzollo, A.; Biffi, A. Diagnostic Strategies and Algorithms for Investigating Cancer Predisposition Syndromes in Children Presenting with Malignancy. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Al-Sarhani, H.; Gottumukkala, R.V.; Grasparil, A.D.S., 2nd; Tung, E.L.; Gee, M.S.; Greer, M.C. Screening of cancer predisposition syndromes. Pediatr Radiol 2022, 52, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Schimmel, M.; Dunstheimer, D.; Nemes, K.; Richter, M.; Streble, J.; Vollert, K.; Walden, U.; Frühwald, M.C.; Kuhlen, M. The need for tumor surveillance of children and adolescents with cancer predisposition syndromes: a retrospective cohort study in a tertiary-care children's hospital. Eur J Pediatr 2022, 181, 1585–1596. [Google Scholar] [CrossRef]
- Khanna, L.; Prasad, S.R.; Yedururi, S.; Parameswaran, A.M.; Marcal, L.P.; Sandrasegaran, K.; Tirumani, S.H.; Menias, C.O.; Katabathina, V.S. Second Malignancies after Radiation Therapy: Update on Pathogenesis and Cross-sectional Imaging Findings; Radiographics: a review publication of the Radiological Society of North America, Inc., 2021; pp. 876–894. [Google Scholar]
- Tanjak, P.; Suktitipat, B.; Vorasan, N.; Juengwiwattanakitti, P.; Thiengtrong, B.; Songjang, C.; Therasakvichya, S.; Laiteerapong, S.; Chinswangwatanakul, V. Risks and cancer associations of metachronous and synchronous multiple primary cancers: a 25-year retrospective study. BMC Cancer 2021, 21, 1045. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Chen, Y.; Wong, F.L.; Hageman, L.; Smith, K.; Korf, B.; Cannon, A.; Leidy, D.J.; Paz, A.; Andress, J.E.; et al. Subsequent Neoplasms After a Primary Tumor in Individuals With Neurofibromatosis Type 1. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2019, 37, 3050–3058. [Google Scholar] [CrossRef]
Figure 1.
Photographs of the first patient, taken in January 2021, showing left-sided hemihypertrophy (a) and café-au-lait spots on the right thigh (b).
Figure 1.
Photographs of the first patient, taken in January 2021, showing left-sided hemihypertrophy (a) and café-au-lait spots on the right thigh (b).
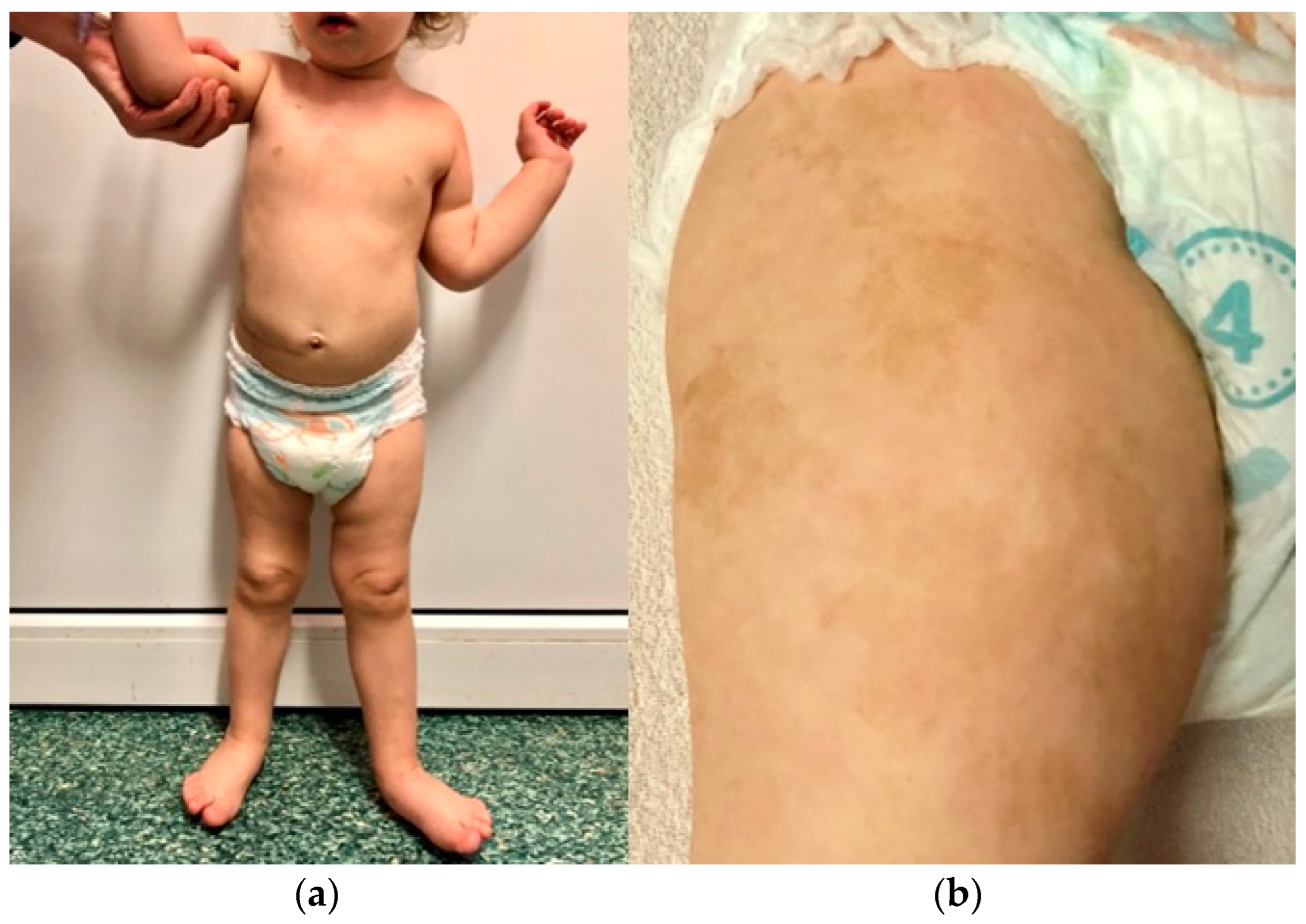
Figure 2.
Timeline illustrating the disease course of Case 1.
Figure 3.
Pelvic MRI scan illustrating a large vaginal tumor (indicated by arrow) in the second patient presented.
Figure 3.
Pelvic MRI scan illustrating a large vaginal tumor (indicated by arrow) in the second patient presented.
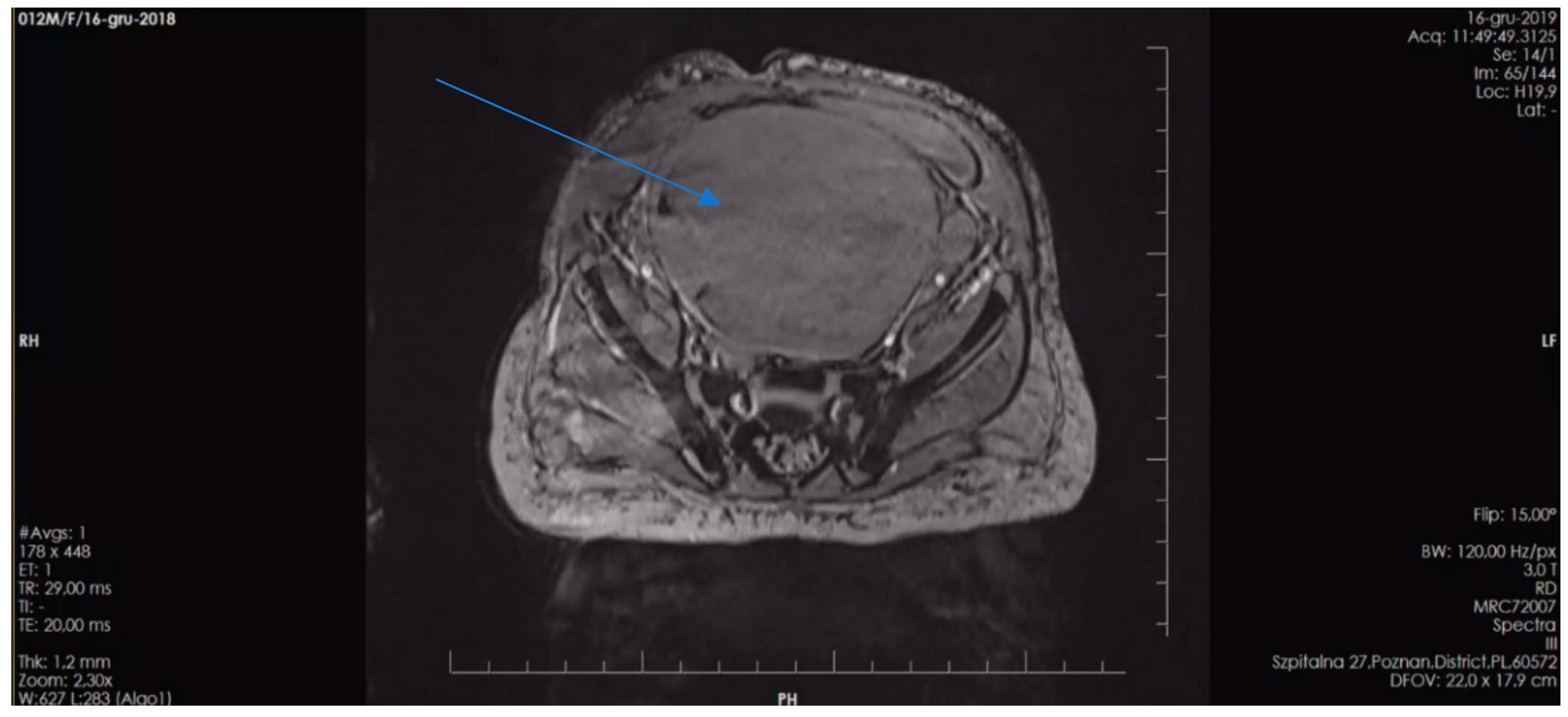
Figure 4.
Peripheral blood smear showing leukocytosis with monocytosis , left-shifted neutrophils and immature cells, dysplastic granulocytes with pseudo Pelger cells and pycnotic nuclei, and 6% blasts.
Figure 4.
Peripheral blood smear showing leukocytosis with monocytosis , left-shifted neutrophils and immature cells, dysplastic granulocytes with pseudo Pelger cells and pycnotic nuclei, and 6% blasts.
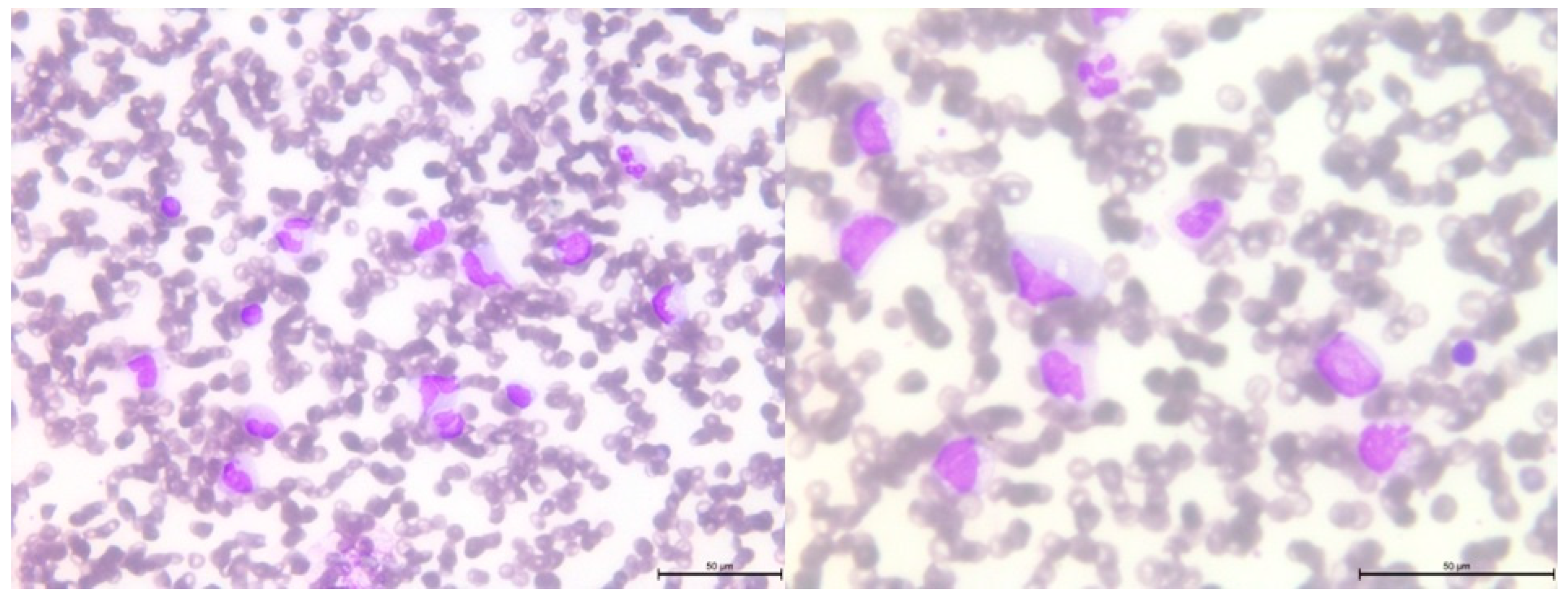
Figure 5.
Bone marrow smear showing reduced cell content, absence of megakaryocytes, monocytosis with dysplastic and immature monocytes, very dysplastic myelopoiesis with pseudo Pelger cells, and aplastic erythropoiesis. Additionally, 13% blasts are observed.
Figure 5.
Bone marrow smear showing reduced cell content, absence of megakaryocytes, monocytosis with dysplastic and immature monocytes, very dysplastic myelopoiesis with pseudo Pelger cells, and aplastic erythropoiesis. Additionally, 13% blasts are observed.
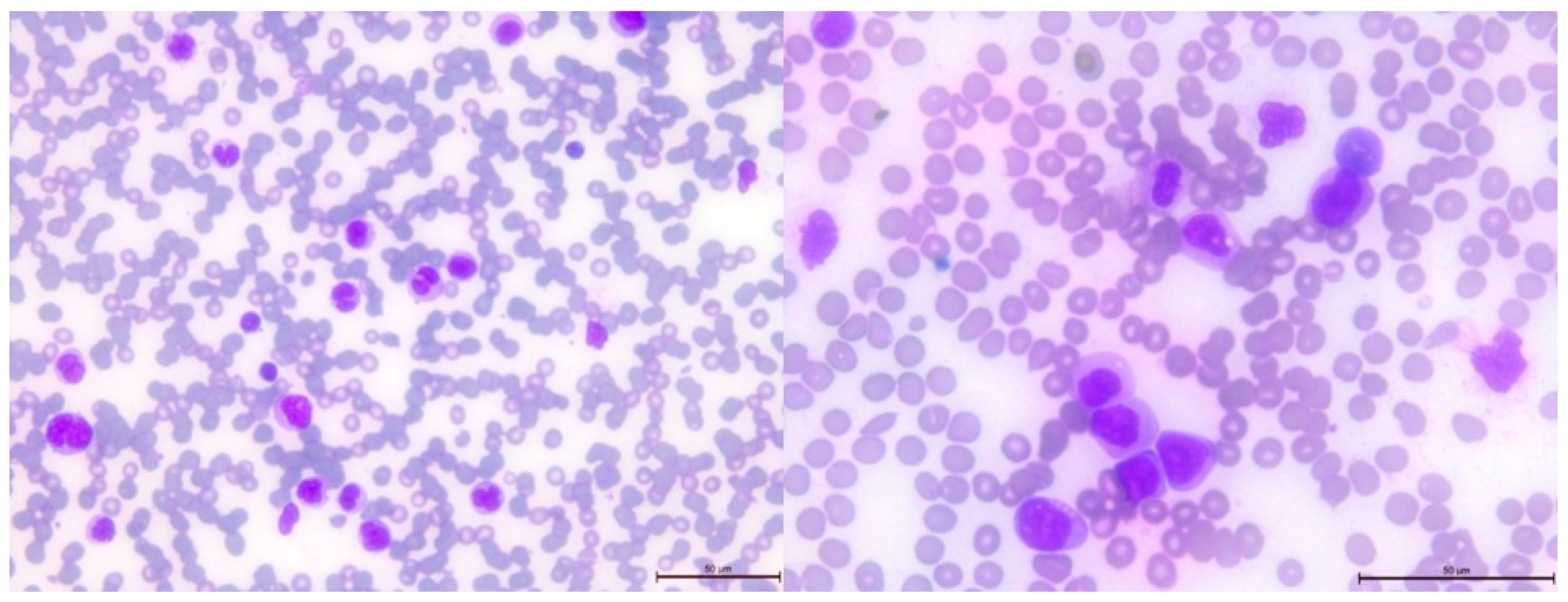
Figure 6.
Timeline illustrating the disease course of Case 2.
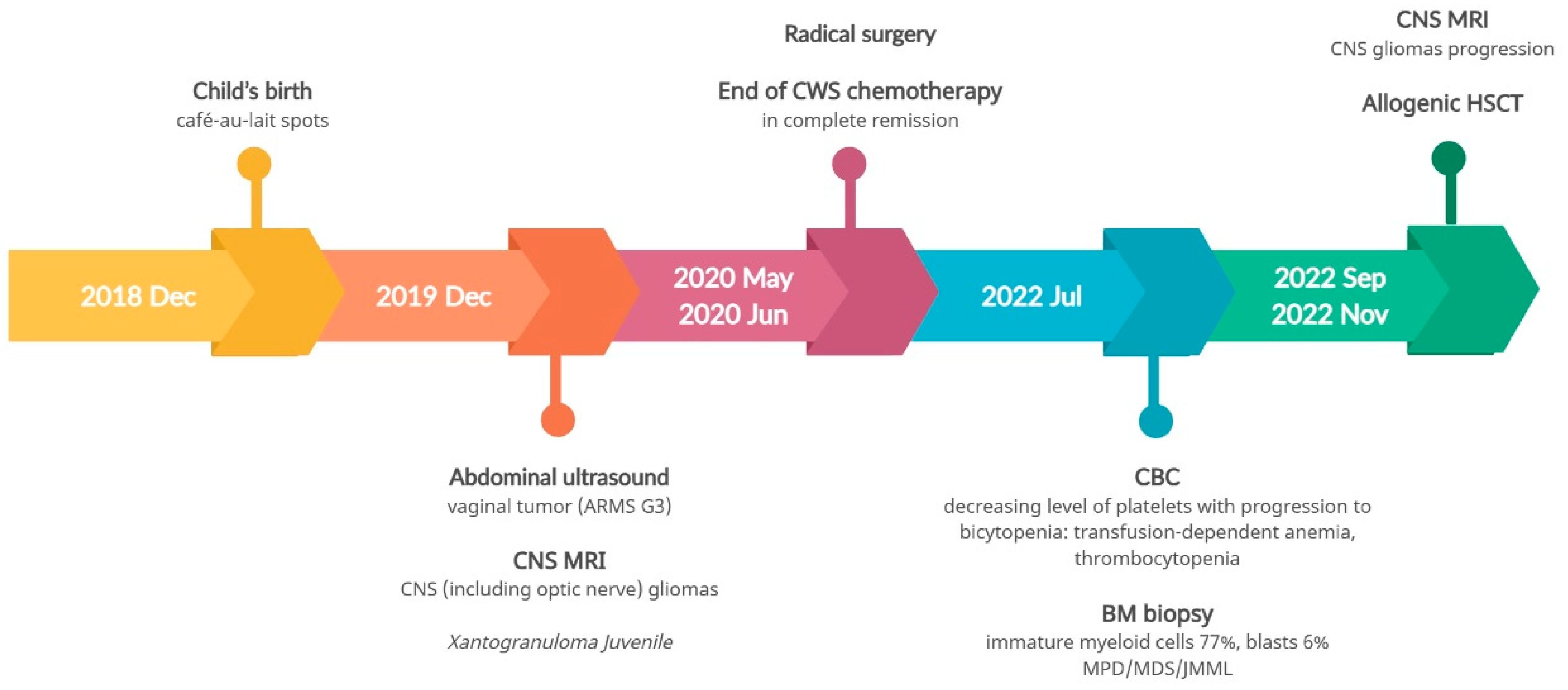
Figure 7.
Timeline illustrating the disease course of Case 3.
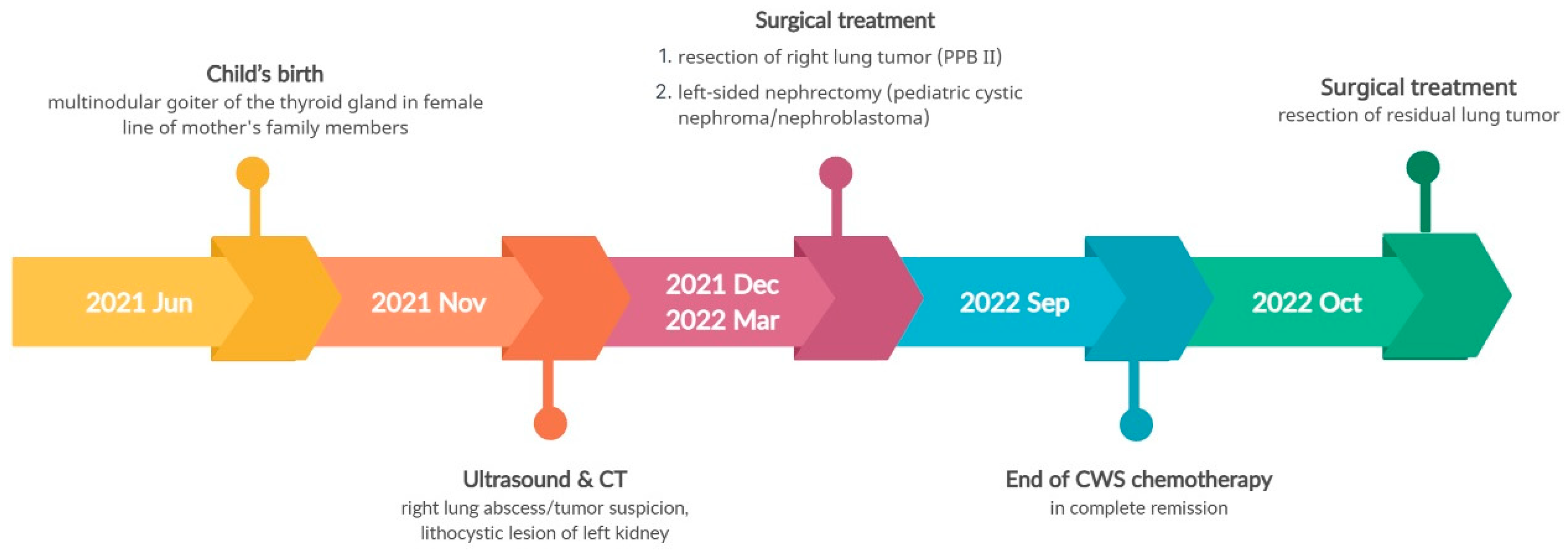

Table 1.
Characterization of BRCA2, NF1, and DICER1 genes in terms of the function of the proteins they encode and their impact on tumorigenesis (based on previous data [7,11,21,22,23,24,25,26,27,28,29]).
| Gene | Function | Mutation consequences | Cancerous manifestations of gene mutation |
| BRCA2 |
|
The protein activity loss as the cause of increased sensitivity to DNA-damaging factors leading to neogenesis induction |
|
| NF1 |
|
Neogenesis induction – proliferation and cell division promoting, increased susceptibility to harmful UV effects – as results of the protein activity loss |
|
| DICER1 |
|
Suppressor genes function loss or proto-oncogene activity enhancement leading to neogenesis induction |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



