
Submitted:
29 June 2023
Posted:
04 July 2023
You are already at the latest version
Abstract
The classic, chronic Philadelphia chromosome-negative (Ph-) myeloproliferative neoplasms (MPN)- mainly essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF)— represent a heterogeneous group of stem cell disorders characterized by clonal proliferation of one or more hematopoietic cell lineages with organomegaly and constitutional symptoms. Several studies have shown that the presence of chronic inflammation and a dysregulated immune system play indisputable roles in the pathogenesis of these diseases. Lately, the treatment of cancer including hematological malignancy has progressed on the agents targeting the immune system, cytokine milieu, immunomodulatory agents, and targeted immune therapy. Immune checkpoints are the molecules that regulate T cells function in the tumor microenvironment (TME). The fully unraveled primary immune checkpoints are programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1), and cytotoxic T-lymphocyte antigen-4 (CTLA-4). Immune checkpoint inhibitor therapy (ICIT) is based on blocking the inhibitory pathways in T cells to promote anti-tumor immune responses and has revolutionized cancer treatment paradigms. Despite the impressive clinical success of ICIT, tumor intrinsic resistance remains a daunting challenge for oncologists leading to a low response rate in solid tumors and hematological malignancies. A phase II trial on Nivolumab for patients with Primary Myelofibrosis, post-Essential thrombocythemia myelofibrosis, or post- Polycythemia myelofibrosis has been performed (ClinicalTrials.gov Identifier: NCT02421354). This clinical trial, on the efficacy of PD-1 blockade using the monoclonal antibody Nivolumab, was prematurely terminated due to a lack of efficacy and adverse events. A multicenter, open-label, phase 2, single-arm study was conducted including pembrolizumab in patients with Dynamic International Prognostic Scoring System category of intermediate-2 or greater primary, post-essential thrombocythemia or post-polycythemia vera myelofibrosis that were ineligible for or were previously treated with Ruxolitinib. Pembrolizumab treatment was well tolerated, but there were no objective clinical responses, so the study closed after the first stage was completed. To permit more patients to benefit from immunotherapy, the focus has changed to targeting alternative novel immune checkpoints in the tumor microenvironment such as lymphocyte activation gene-3 (LAG-3), T cell immunoglobulin, and mucin domain 3 (TIM-3), V-domain immunoglobulin-containing suppressor of T-cell activation (VISTA), T cell immunoglobulin and ITIM domain (TIGIT) and human endogenous retrovirus-H long terminal repeat-associating protein 2 (HHLA2) forming the basis of next-generation ICIT. Our article aims at emphasizing and discovering the role of next-generation ICIT in MPN as targeted immunotherapy involving monoclonal antibodies, checkpoint inhibitors, or therapeutic vaccines against selected MPN epitopes that could further enhance tumor-specific immune responses. Immunotherapeutic approaches are expanding and hopefully will extend the therapeutic armamentarium in patients with myeloproliferative neoplasms. Preliminary studies from our laboratory showed over-expressed MDSC and over-expressed VISTA in MDSC, and in progenitor and immune cells directing the need for more clinical trials using next-generation ICI in the treatment of MPN.
Keywords:
Checkpoint Inhibitor
; Philadelphia Chromosome
; Myeloproliferative Neoplasm
Introduction
The MPN is defined by the clonal proliferation of one or more hematopoietic cell lineages [1]. As per International Consensus Calssification [2] (ICC) and World Health Organization (WHO), 5th edition [3] classic MPN comprises mainly myelofibrosis (MF), essential thrombocythemia (ET), and polycythemia vera (PV) [4]. Lately, the focus of the treatment of MPN is based on the agents targeting the immune system, cytokine environment, and immunotherapy agents. The pathogenesis of MPN is not very clear but studies have shown that TNF-α promotes the growth of JAK2V617F-positive MPN cells as compared to controls contributing to clonal formation of mutant copies during MPN progression [5]. Various driver mutations as studied by genetic sequencing and clonal protein expression showed that tyrosine kinase Janus Kinase 2 -JAK2V617F mutation was found in 95% of patients with PV and 50%- 60% of patients with ET and MF [6,7]. These mutations stimulate Janus Kinase and Signal Transducer and Activator of Transcription proteins (JAK-STAT) signaling path of thrombopoietin receptor and erythropoietin receptor [8]. Concurrently profound immune dysregulation and defective immune surveillance also have an important role in the pathogenesis of MPN [9]. The dysregulated genes related to the immune system and inflammation that are implicated in MPN are interferon-inducible gene [10], regulatory T cells (Tregs characterized as CD4 + CD25+ FOXP3+) [11], human leukocyte antigen (HLA) class I and II molecules, natural killer cells, β2-microglobulin, HLA I antigens (such as LMP7, LMP2, TAP1/2, tapasin) [10,12] and antioxidative stress genes (ATM, TP53, CYBA, NRF2, PTGS1, SIRT2) [13,14]. In addition, increased recruitment of suppressive cells, such as myeloid-derived suppressor cells (MDSC), leads to the escape of tumor cells from immune surveillance thus playing an important part in the etiopathogenesis of MPN [15]. The key events involved in the development of the neoplastic process are oncogenic transformation and immune escape allowing for uncontrolled proliferation and avoidance of apoptosis. Immune checkpoint inhibitory therapy (ICIT) is based on blocking the T cells inhibitory pathways thus promoting anti-tumor immune responses. Oncogenic JAK2 activation results in high expression of programmed death-ligand 1 (PD-L1) on the surface of megakaryocytes, monocytes, platelets, and MDSC which is mediated via the JAK2-STAT3 and JAK2-STAT5 axes [16]. Myeloid malignancies are found to have overexpressed PD-1 pathways and that has gained immense attention recently as pathbreaking therapeutic targets for immunotherapy. One such trial was: ClinicalTrials.gov Identifier: NCT02421354 where the safety and efficacy of nivolumab (PD-1 inhibitor) was tested in eight adult patients with myelofibrosis [17]. However, the study was discontinued due to failure to meet the efficacy endpoint. In 2020, at the annual meeting of American Society of Hematology (ASH), an open label, phase 2, multi-center, single-arm study of pembrolizumab was presented showing its use in patients with primary, post-essential thrombocythemia or post-polycythemia vera MF (NCT03065400) [18]. Nine cases were included, but none showed a clinical response.
The use of ICI in hematological malignancies brings a daunting challenge with a low response rate thus letting the oncologist/molecular physicians change their attention to focus deeply on the TME for novel therapeutic targets. To benefit more patients from immunotherapy, the paradigm has shifted to target alternative new immune checkpoints in the TME such as LAG-3, TIM-3, TIGIT, VISTA, and HHLA2 forming the basis of next-generation ICIT [19] as shown in Figure 1. Our review article aims at emphasizing and discovering the role of next-generation ICIT in MPN involving monoclonal antibodies as targeted immunotherapy or novel inhibitory checkpoints that would further broaden the horizon of tumor-specific immune responses and treatment.
LAG-3 targeted therapy and its role in hematological malignancies
LAG-3 (CD223) is a CD4-associated activation-induced cell surface inhibitory receptor that binds to major histocompatibility complex (MHC) class II molecules and negatively regulates T-cell effector functions [20]. Cells expressing LAG-3 are T cells, a few activated B cells, plasmacytoid dendritic cells (DCs), and neurons [21]. LAG-3 ligands are MHC class-II, galectin-3, and fibrinogen-like protein 1 (FGL1) with MHC-II being the main ligand [22]. LAG-3 attaches to MHC class II with higher affinity than CD4 inducing protein phosphorylation of phospholipase Cgamma2 (PLCgamma2) and p72syk as well as activation of phosphatidyl inositol 3-kinase/Akt, p42/44 extracellular protein kinase, and p38 mitogen-activated protein kinase pathways [23]. Galectin-3 is expressed on activated T cells and tumor cells that are needed for CD8/T-cell and plasmacytoid DC suppression [22]. FGL1 is highly produced by human cancer cells and binding of LAG-3 with FGL1 contributes to resistance /poor response to anti-PD-1/anti PD-L1 immunotherapies [24,25]. This mechanism forms the basis of therapies involving simultaneous blockade of PD-1 and LAG-3 responsible for several T-cell antitumor activities [26,27,28].
Currently, sixteen LAG-3 targeted immunotherapies are being tested at approximately 97 clinical trials by Bristol-Myers Squibb (BMS-986016), Regeneron Pharmaceuticals (REGN3767 and 89Zr-DFO-REGN3767), Merck (MK-4280), Novartis (LAG525), Tesaro (GSK) (TSR-033), Symphogen (Sym022), GlaxoSmith (GSK2831781), Incyte Biosciences International Sàrl (INCAGN02385), Prima BioMed/Immutep (IMP321), MacroGenics (MGD013), F-Star (FS118), Hoffmann-La Roche (RO7247669), Shanghai EpimAb Biotherapeutics (EMB-02), Xencor (XmAb841) and Innovent Biologics (IBI323) [29]. LAG-3 targeted therapies are divided into three categories namely monoclonal antibodies, LAG-3 –immunoglobulin fusion proteins, and anti-LAG-3 bispecific drugs [29]. Most trials are phase I/II with two of them reaching phase III including BMS-986016 (NCT05002569) [30] and MK-4280 drugs (NCT05064059) [31]. Table 1 demonstrates the use of LAG-3 agents in hematological malignancies in the current clinical trials.
- 1.
- Use of 89Zr-DFO-REGN3767 in PET Scans in people with diffuse large B Cell lymphoma (DLBCL) was the pilot study (NCT04566978) [32] undertaken at Memorial Sloan Kettering Hospital in 2022 with the main purpose of the study is looking at the way the body absorbs, distributes, and gets rid of 89Zr-DFO-REGN3767 [33]. 89Zr-DFO-REGN3767 is comprised of the anti-LAG-3 antibody, REGN3767 labeled with the positron-emitter zirconium-89 (89Zr) through the chelator-linker DFO and REGN3767 is an investigational monoclonal antibody that targets LAG-3 receptors. This study is a diagnostic research study determining the optimal time for imaging and tumor uptake post 89Zr-DFO-REGN3767 administration. However, it can help evaluate tumor uptake of 89Zr-DFO-REGN3767 and correlate with expression of LAG-3 by immunohistochemistry (IHC) in tumors that will be subsequently compared with other biomarkers of TME characterized in biopsies, such as IHC score (LAG-3 and/or other immune cell markers).
- 2.
- A safety and efficacy trial of JCAR017 (lisocabtagene maraleucel, also known as liso-cel) (a CD19-targeted chimeric antigen receptor CART-cell therapy) combinations in subjects with relapsed / refractory B-cell malignancies (PLATFORM) (NCT03310619) [34] was done. Relatlimab, BMS-986016 is an anti-LAG-3 fully human monoclonal IgG4-κ antibody that binds human LAG-3 with high affinity and inhibits its binding to MHC-II [35]. This trial was a global, open-label, multi-arm, parallel multi-cohort, multi-center, Phase 1/2 study to determine the safety, tolerability, pharmacokinetics, efficacy, and patient-reported quality of life of JCAR017 in combination with various agents including relatlimab, durvalumab, avadomide, iberdomide, ibrutinib, and nivolumab. The trial was completed, and the studied tumors were diffuse large B-cell lymphoma (DLBCL), non-Hodgkin lymphoma (NHL), and Follicular lymphoma (FL). The objective of the study during Phase 1 was to open different paths to test JCAR017 in combination with other agents in adult patients with R/R aggressive B-cell NHL. Different doses and schedules of JCAR017 were used in several arms and the combination agents were tested in several cohorts per arm. Phase 2 of the study involved the expansion of any dose level and schedule for any arm maintaining safety. All patients from Phase 1 and Phase 2 will then be followed for 24 months for adverse effects, survival, relapse, viral vector safety, and long term toxicity as per guidelines.
- 3.
- A similar trial was also designed with relatlimab by Bristol-Myers Squibb, NCT02061761 [36] administered alone or in combination with nivolumab to subjects with relapsed or refractory B-cell malignancies (relapsed or refractory Hodgkin lymphoma (HL) and relapsed or refractory DLBCL and to study its safety, tolerability, dose-limiting toxicities and maximum tolerated dose. The trial completed and studied hematological malignancies including chronic lymphocytic leukemia (CLL), HL, NHL, and Multiple Myeloma (MM). A detailed description of dose-related adverse events was studied and was +displayed in the result section of the trial.
- 4.
- Favezelimab (MK-4280) is another LAG-3 antibody that is studied in combination with pembrolizumab (MK-3475) in the clinical trial NCT03598608 [37] that was started in July 2018 to study and evaluate the safety and efficacy of these agents in hematologic malignancies. ). It included classical HL, DLBCL, and indolent HL. No results have been posted till the writing of this article. This study will also evaluate the safety and efficacy of pembrolizumab or favezelimab administered as monotherapy in participants with classical HL using a 1:1 randomized study design.
- 5.
- Relapsed or refractory acute myeloid leukemia (AML) and newly diagnosed older AML are included in the ClinicalTrials.gov Identifier: NCT04913922 [38] to study the combination of relatlimab with nivolumab and 5-azacytidine. No results have been posted yet.
All the above trials included LAG-3 as an ICI agent in the above-mentioned hematological malignancies, however, no trials have been done in the field of MPN. We unfold the mechanism of action of LAG-3 to provide a better understanding of its potential use in the future as depicted in Figure 2.
Mechanism of action of LAG-3
LAG-3 was discovered in 1990, by Triebel and colleagues, as a new 498-amino acid type I transmembrane protein present on activated natural killer (NK) and T cell lines [39]. The LAG-3 gene is found close to CD4 on chromosome 12 in humans (chromosome 6 in mice) displaying structural homology to CD4 with extracellular immunoglobulin superfamily (IgSF)-like domains namely D1–D4 [40]. The structural motifs are conserved between LAG-3 and CD4, translating to the same extracellular folding patterns as a result of which LAG-3 can bind with greater affinity to MHC class II than CD4 [41]. LAG-3 was speculated to be spatially related to the T-cell receptor TCR: CD3 complex present in microdomains of lipid raft promoting clustering of signaling molecules and the development of the immunological synapse however the exact mechanism is still unclear [42]. The cytoplasmic tail for the tyrosine kinase p56Lck, lacks a binding site for LAG-3, which is normally used by CD4 to promote downstream signal transduction of the T cell receptor (TCR) [41]. Conversely, the LAG-3 cytoplasmic domain has three well-defined motifs namely serine-based motif acting as a PKC substrate, repetitive “EP” motif comprising of a series of glutamic acid-proline dipeptide repeats, and relatively unique “KIEELE” motif, highlighted by an essential lysine residue [43,44]. LAG-3 cytoplasmic tailless mutants neither mediate the inhibitory effects of LAG-3 nor compete with CD4, emphasizing the importance of the function of this domain needed for the transmission of an inhibitory signal [20]. Expression of MHC class II molecules by human melanoma cells is correlated with poor prognosis thus, LAG-3 ligation with MHC-II class, which is seen on melanoma-infiltrating T cells, may facilitate their clonal exhaustion [45]. In vitro, the demonstration showed that such an interaction may help tumor cells to adopt an escape mechanism giving them protection against apoptosis, with a recent study showing that MHC class II-expressing melanoma cells causes infiltration of tumor-specific CD4+ T cells, mediated by interaction with LAG-3, which in turn negatively regulates CD8+ T cell responses [46,47.] Galectin-3 is a ligand, that is expressed by several cells within the TME but not the tumor itself, facilitating interaction with LAG-3 (present on tumor-specific CD8+ T cells) that may regulate anti-tumor immune activities [48]. Liver sinusoidal endothelial cell lectin (LSECL) is present in the liver as well as identified in melanoma tumor cells where it stimulates growth by inhibiting anti-tumor T-cell dependent responses [49]. The interaction between LSECL in melanoma cells and LAG-3 inhibited IFNγ production, mediated by effector T cells (antigen-specific), altering the TME [49]. Continuous T cell activation in an inflammatory state, specifically in a tumor, results in persistent co-expression of LAG-3 on T cells along with additional inhibitory receptors (IR) such as PD1, TIGIT, TIM3, CD160, 2B4 leading to T cell dysfunction [50]. Several hematopoietic cell types, including CD11clow B220+ PDCA-1+ plasmacytoid dendritic cells (pDCs) constitutively express LAG-3 [51] however it is not expressed on any myeloid or lymphoid DC subset. In vitro, MHC class II-expressing melanoma cells could stimulate LAG-3 positive pDCs to mature and produce IL-6 which was later confirmed in vivo as well with LAG-3 positive pDCs showing increased IL-6 production and an activated phenotype similar to melanoma cells [52]. Bo Huang et al showed that increased IL-6 promotes the release of CCL2 by monocytes in vitro, which then may recruit MDSCs thus forming the hypothesis that LAG-3 positive pDCs may indirectly mediate MDSC-related immunosuppression by engaging MHC class II+ melanoma cells [53]. LAG-3 functions are regulated by cell surface cleavage mainly ADAM10 and ADAM17 disintegrin /metalloproteases, although in mice soluble LAG-3 seems to have no biological function [54].
V-domain immunoglobulin suppressor of T cell activation (VISTA) targeted therapy and its role in hematological malignancies
VISTA (also known as B7-H5, PD-1H, DD1α, c10orf54, VSIR, SISP1, Gi24, and Dies1) is primarily expressed in myeloid cells mainly microglia, and neutrophils followed by macrophages, monocytes, and dendritic cells [55,56]. Additionally, it is highly expressed on new CD4+ and Foxp3+ regulatory T cells [57]. VISTA is a type I transmembrane protein consisting of a single N-terminal immunoglobulin V-domain that has the greatest homology with PD-L1 [58]. The exact function and role of VISTA in regulating the immune system are still complex and not very clear. It works both as a ligand expressed on antigen-presenting cells and as a receptor on T cells [59]. To date, various studies have described the inhibitory effect of VISTA on the immune system and the ability of VISTA-deficiency or anti-VISTA treatment to upregulate immune responses [60]. Due to its predominant expression on macrophages, VISTA is implicated as a potential immunotherapeutic target in melanoma [61]. Studies claim that melanoma survival correlates with PD-L1/VISTA expressions [62,63]. Furthermore, tumor cell expression of VISTA, which is regulated by factor forkhead box D3 (FOXD3), encourages tumorigenesis and promotes PD-L1 expression on tumor-infiltrating macrophages in vivo along with increased intra-tumoral T regulatory cells [62]. VISTA is expressed on MDSCs in the peripheral circulation, with a strong positive association between MDSC expression of VISTA and T cell expression of PD-1 in acute myeloid leukemia (AML) patients, although there is no evidence of direct regulation [64,65]. MDSCs are myeloid cells that are defined into subsets namely monocytic MDSCs (CD15-) and granulocytic MDSCs (CD15+) [66]. Patients with AML displayed increased expression of VISTA on MDSCs highlighting the role of VISTA in MDSC-mediated CD8 T cell response [64]. There is conflicting evidence with some studies supporting that VISTA is an immune checkpoint marker expressed on tumor-infiltrating T lymphocytes and myeloid cells, causing suppression of T cell activation, proliferation, and cytokine production [67,68] whereas other studies have shown that VISTA is overexpressed in tumor cells and may functions as a co-stimulatory molecule [69,70].
Currently, clinical trials of VISTA-targeted cancer immunotherapy are in progress namely ClinicalTrials.gov Identifier: NCT02671955 [71] and ClinicalTrials.gov Identifier: NCT02812875 [72]. JNJ-61610588 (CI-8993) [71] is a human monoclonal antibody against VISTA with negative checkpoint regulatory and antitumor activities that is being studied in advanced cancer patients. No study results have yet been posted. Meanwhile, a study of CA-170 [72], an inhibitory molecule that selectively aims for PD-L1 and VISTA, is still currently being conducted in advanced solid tumors or lymphomas, although the trial is not recruiting any more subjects and the last update was posted on May 6, 2019. There are pre-clinical trials of VISTA mentioned in hematological cancer and solid tumors involving IGN-381 (mAbs by Ingenica Biotherapeutics) and HMBD-002 (mABs by Hummingbird Bioscience) [73]. HMBD-002 exerted significant inhibitory effects on tumor progression and its combination with anti-PD-L1 was found to be more effective in tumors that showed abundant MDSC infiltration [74]. Table 2 summarizes the potential clinical trials of VISTA in hematological malignancies.
Tumor and Immune System Interaction Database (TISIDB) [69,75] analyzed the potential relevance of VISTA in cancer immunity across 30 different cancer types and the outcomes were: 1) Almost all types of TILs, with tumor-suppressing or tumor-promoting functions across 30 types of cancers, correlated positively with VISTA expression, including activated CD8 T cells, NK cells, MDSC, and Tregs cells 2) VISTA expression levels correlated positively with almost all significant immunomodulators including immune inhibitors, immunostimulators, or MHCs, including but not limited to, the critical immune checkpoints such as PD-L1, PD-1, CD80, and CD86. 3) Additionally, VISTA expression correlated positively with almost all well-known chemokines and their receptors, including but not limited to CXCL1, CXCL8, CXCL10, and CXCR3. VISTA can function as a receptor as well as a ligand interacting with distinct partners modulating immune response. VISTA modulator is a promising target, and its mechanism is worthy of further investigation specifically in hematological cancers including MPN. Targeting VISTA may promote releasing suppression by myeloid cells leading to improve T cell-focused therapies like anti-PD1 and anti-CTLA4 especially when monotherapy resistance of other ICIT appears.
Role of T cell immunoglobulin and mucin domain 3 (TIM-3) as next-generation ICI in hematological malignancies
TIM-3 is a type I transmembrane protein that was discovered on CD4+ type 1 helper T cells (TH1 cells) and CD8+ cytotoxic T cells (CTLs) [76]. Subsequently, other T-cell subtypes also expressed TIM-3 along with other immune cells including DCs, NK cells, macrophages, monocytes, mast cells, and some malignant cells [77,78,79,80]. TIM-3 is pertinently expressed on DCs and macrophages in both humans and mice, specifically in humans where it suppresses IL-12 expression [81,82]. TIM-3 inhibits DCs cell activation and maturation by blocking NF-κB signaling via a Btk-c-Src signaling-dependent mechanism, interfering with the ability of cytoplasmic toll-like receptors (TLRs) to sense immunogenicity and thereby suppressing anti-tumor immunity [83]. There are four known ligands for TIM-3 namely galectin-9 [84] - which induces apoptosis in TH1 cells, High-mobility group protein B1 (HMGB1) [85] – also called “alarmin”, released from damaged cells and activates phagocytes, phosphatidylserine (PtdSer) [86]- “eat-me” signal induction molecule and carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1) [87] – known for both cis and trans interactions with TIM-3. Studies claim that interactions between TIM-3 with its ligands (galectin-9 and Ceacam-1) lead to phosphorylation of tyrosine residues namely Y256 and Y263, stimulating the release of HLA-B associated transcript 3 (Bat3) from the tail, thereby enhancing T cell inhibitory function by allowing binding of SH2 domain-containing Src kinases and subsequent regulation of TCR signaling [88,89]. Studies have reported that a higher expression of TIM-3 poses a higher risk for myelodysplastic syndrome (MDS) transformation to leukemia as increased levels of TIM-3 and Gal-9 are reported on bone marrow cells and MDSCs from MDS patients [90,91]. This highlights the role of the TIM-3/Gal-9 axis in the blast proliferation, induction of immune escape, and T cell exhaustion supporting disease progression [92]. Bruck et al reported TIM-3 overexpression on exhausted CD4+ and CD8+ T cells in untreated chronic myeloid leukemia (CML) patients and observed a correlation between PD-1 positive TIM-3 CD8+ T cells along with a poor response to Tyrosine kinase inhibitors (TKIs) [93]. Dysfunctional immunity plays a major role in malignancy formation, but many more clinical studies are required to investigate the role of TIM-3 in MPN pathogenesis and establish its role in the formation, therapy resistance, relapse, and immune scoring of this malignancy. Several clinical trials involving co-blockade of TIM-3 and PD-1, have demonstrated promising preliminary results against solid tumors namely HBV-related hepatocellular carcinoma [94,95,96]. TIM-3 is highly expressed in peripheral blood and bone marrow exhausted T cells in various hematological malignancies, including acute lymphoblastic leukemia (ALL), chronic lymphocytic leukemia (CLL), and multiple myeloma (MM) however, few reports have demonstrated its clinical significance as monotherapy with TIM-3 inhibitors alone [97,98,99].
Further studies are required to evaluate the efficacy of TIM-3 inhibitors in different types and stages of leukemias and MPNs with emphasis on TME. Currently, the TIM-3 inhibitors used in clinical trials include MBG453 (also known as sabatolimab), TSR-022 (Tesaro), BMS-986258, LY3321367, SYM023, BGB-A425, and SHR-1702 [100,101]. Currently, sabatolimab (high-affinity IgG4 mAb) is the only anti–TIM-3 mAb being investigated in MDS and AML with preliminary safety and efficacy data. ClinicalTrials.gov Identifier: NCT03066648 is an active phase I trial of TIM-3 involving the study of PDR001 and/or MBG453 in combination with decitabine or azacitidine in patients with AML or high-risk MDS [102]. It includes AML, MDS, chronic myelomonocytic leukemia, and bone marrow diseases. No result was posted till the writing of this article, but preliminary results reported that the combination of sabatolimab plus HMA (either decitabine or azacitidine) was associated with mostly grade 1 or 2 adverse events and showed preliminary evidence of antitumor activity with a durable response. As per preliminary follow-up, overall response rates (ORRs) in patients with HR-MDS with sabatolimab plus decitabine and sabatolimab plus azacitidine were 61.1% and 57.1%, respectively, with complete response (CR) rates of 33.3% and 7.1% [100]. TIM-3 is relatively higher expressed on leukemic stem cells than non-tumor stem cells, often with other surface antigens such as CD33, CD123, and CLL, thus targeting TIM3 might be a novel approach in cancer treatment in future [103]. Targeting TIM-3 along with other checkpoint inhibitors or combining TIM-3 inhibition with new immunotherapeutic modalities that activate cancer-specific T-cell stimulatory molecules have immense potential for developing therapies with durable clinical benefits.
Role of T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory domain (TIGIT) as a target for next-generation ICI in hematological malignancies
TIGIT belongs to a family of PVR-like proteins, discovered in 2009, composed of one extracellular immunoglobulin variable domain (a type I transmembrane domain) and a short intracellular domain with one immunoreceptor tyrosine-based inhibitory motif (ITIM) and one immunoglobulin tyrosine tail (ITT)-like motif [104,105]. TIGIT (also called Washington University cell adhesion molecule, WUCAM) along with DNAX accessory molecule-1 (DNAM-1) and CD96 are expressed on NK cells and T cells and share CD155 [polio virus receptors (PVR), nectin and nectin-like (NECL) NECL-5] as a ligand [106,107]. The immunoglobulin variable domain is homologous with other members of the PVR-like family, including DNAM-1, CD96, CD111, CD155, CD112, CD113, and PVRL4 [104]. In both humans and mice, CD155 serves as a ligand of TIGIT, and comparatively, it binds with lower affinity with CD112 and CD113 [105,108]. CD155 is mainly expressed on DCs, B cells, T cells, macrophages, kidneys, nervous system, and intestines [109], CD112 has a wide expression in bone marrow, pancreas, kidney, and lung [110], and CD113 is limited to non-hematopoietic tissues, including placenta, kidneys, testis, liver, and lung [111]. The mechanism of action of TIGIT involves an extrinsic pathway, as a ligand for CD155 [104] or a cell-intrinsic pathway by interfering with DNAM-1 co-stimulation [112,113] or by directly delivering inhibitory signals to the effector cell [105]. The interaction of TIGIT with CD155 sends signals to human monocyte-derived DCs leading to increased secretion of IL-10 and decreased secretion of IL-12 thus promoting tolerogenic DCs that down-regulate T cell responses [106]. For the cell-intrinsic mechanism of action, it was postulated that the high affinity of TIGIT for CD155 out-numbers DNAM-1 for the binding of CD155 leading to T-cell inhibition. This was first observed that TIGIT knock-down in CD4+ T cells increased their expression of T-bet and IFN-γ, and this could be overcome by DNAM-1 blockade [112,113]. Several malignancies, including melanoma, breast cancer, non-small-cell lung carcinoma (NSCLC), colon adenocarcinoma, gastric cancer, multiple myeloma (MM), and AML have shown increased expression of TIGIT thus paving the path for anti-TIGIT therapies [114,115,116,117,118].
In mouse pre-clinical models and cancer patients, TIGIT expression on tumor-infiltrating CD8+ T cells often correlates with increased expression of other inhibitory receptors such as PD-1, LAG-3, TIM-3, and with decreased expression of DNAM-1 [115,119,120,121]. Similarly, a high TIGIT/DNAM-1 ratio on tumor-infiltrating Tregs was shown to correlate with poor clinical outcomes following ICB targeting PD-1 and/or CTLA-4 [122]. In the pre-clinical mice TIGIT negative mice bearing colon cancer (MC38 model), co-blockade of TIGIT and PD-1 was associated with enhanced effector cell functions of both CD4+ and CD8+ T cells compared to either therapy alone; and TIGIT/PD-1 co-blockade produced a 100% cure rate [123].
As explained earlier tumor cells create a microenvironment by either promoting secretion of immunosuppressive cytokines such as IL-10 and transforming growth factor (TGF)-β, or by recruiting regulatory cells including Tregs, MDSCs, and type 2 macrophages or by affecting immune cell metabolism [124,125]. However, most of these pathways comprise receptor-ligand pairs, and their interaction leading to suppression of the effector functions of T cells and NK cells and thereby impairing anti-tumor immunity [126]. However, despite the enormous success and popularity of ICIT, there is still a substantial number of patients who either do not respond to currently available immunotherapies or develop treatment-related toxicities termed ‘immune-related adverse events’ (irAEs), which sometimes led to fatalities [127,128]. Thus, there is great interest in discovering new immune checkpoints that can be targeted with safety without affecting the anti-tumor efficacy across various malignancies. TIGIT is a negative regulator of cytotoxic T cells and has emerged as a particularly attractive target for cancer therapy with possibly fewer irAEs than anti-PD-1 or anti-CTLA-4 mAbs [129,130].
Presently, six human clinical trials of anti-TIGIT-mAb of IgG1 isotype are undergoing including etigilimab (OMP-313M32), in phase I/II, either as monotherapy or combinations with PD-1/PD-L1 blockade, for the treatment of solid cancers [131,132,133,134,135,136].
TIGIT is highly expressed on tumor-infiltrating lymphocytes (TIL) in several hematological malignancies including follicular lymphoma, CLL, classic HL, AML, and relapsed MM, helping in tumor progression and poor outcome [137]. Research studies have shown the immense potential of anti-TIGIT therapy as reported by Catakovic's in vitro experiment showing reduce CLL viability by TIGIT blockade [138]. Anti-TIGIT treatment prevented T cell exhaustion and prolonged survival in MM mice [139]. Current clinical trials based on therapeutic strategies targeting TIGIT have encouraging efficacy in hematological malignancies [140,141,142,143,144,145,146,147,148]. Table 3 shows current clinical trials of anti-TIGIT antibodies in hematological malignancies.
The current and future role of ICI in the management of MPN
The management of MPNs is constantly evolving and highly individualized. Optimal management of MPN patients is based on considering specific disease types, complex decision-making, individualized prognosis, age, comorbidities, and the risks and benefits of available treatment. Patients with PV and ET use aspirin for thrombotic risk reduction as well as hydroxyurea (HU) or interferon-based therapy for cytoreduction [149,150]. As per the revised IPSET score, cytoreductive therapy is reserved for patients with high-risk factors including age > 60 years, previous thrombosis, and JAK2 mutation [151]. HU is associated with significant side effects and subsequently, 24% of patients with PV or ET develop resistance to primary therapy necessitating the need for second-line therapy [152]. Interferon is frequently used as a frontline or second-line therapy including a novel, mono-pegylated formulation called Ropeginterferon alfa-2b, the first and only approved treatment for PV independent of previous hydroxyurea exposure [150,153]. With the advances in molecular science, there is the discovery of the JAK2 V617F mutation and its role in JAK-STAT pathway dysregulation, which led to the development of the JAK inhibitor ruxolitinib, which currently represents the cornerstone of medical therapy in MF and hydroxyurea-refractory/intolerant PV [150]. Furthermore, the JAK1/2 inhibitor ruxolitinib is approved in intermediate to high-risk MF, as well as advanced PV after HU intolerance or failure [154]. JAK inhibitors are known to alleviate symptoms, improve performance status and disease-associated cachexia further adding the survival benefit of these drugs [155]. Long-term follow-up studies showed improved bone marrow morphology (up to 50% of patients might achieve some regression in marrow fibrosis after 60 months) [156] however complete molecular remissions are rare (3 and 6 patients in RESPONSE-I and COMFORT-I trials, respectively) [157,158]. The major limitations of the use of these agents are that they have debatable disease-modifying activities, there is the likelihood of losing response over time, development of treatment resistance, chronic anemia, and thrombocytopenia stemming from JAK2 inhibition frequently limiting their safety profile and dosing [159].
Ongoing research efforts are dedicated to improving the efficacy and safety profile of established treatment modalities as well as discovering novel therapeutic approaches, many of which target the immune system. Lately, the focus of the treatment of MPN is based on the agents targeting the immune system, cytokine environment, and immunomodulatory agents with targeted therapy. At the American Society of Hematology (ASH) annual meeting in 2020 Mascarenhas et al [18] presented an open-label, multi-center, phase 2, single-arm study of pembrolizumab in patients with primary, post-essential thrombocythemia or post-polycythemia vera myelofibrosis (MF) (NCT03065400). Nine case studies were done, but none had a clinical response. Wang et al published an article demonstrating that PD-1 and PD-L1 expressions were increased in MPN disease in immune cells, including CD4, CD8, monocyte, and CD34+ cells [160]. The potential stimulators of PD-L1 expression are interferon-gamma (IFN-ϒ), IL-10, VEGF, and hypoxia leading to activation of PD-L1 transcription [161,162]. Treg cells can stimulate B7-H1 expression in MDSCs thus enhancing each other’s immune suppression functions [163]. The role of MDSCs in the tumor microenvironment is getting defined day by day and they are implicated in inducing therapeutic resistance to ICI therapy [164,165]. Further studies summarized that in patients with advanced melanoma, non-small cell lung cancer, and breast cancer, there is an accumulation of MDSC that led to resistance to immunotherapy proven by the positive correlation between the MDSC percentage and neutrophil/lymphocyte rate (NLR) (a prognostic marker in both ipilimumab and nivolumab therapy) [166,167,168,169]. ICI targeting PD-1 stimulated circulating Treg levels but did not change Granulocyte-MDSC (G-MDSC) and Myeloid-MDSC (M-MDSC) levels. However, the partial response group had a higher baseline level of M-MDSCs, which showed a significant decrease after the first cycle of anti-PD-1 treatment [170]. Therefore, MDSC accumulation plays a significant role within the tumor microenvironment and is implicated in the failure of ICI.
There have been limited studies on the use of ICI in the treatment of MPN as described earlier in the article with three NCI-sponsored clinical trials related to combined immune- therapy (NCT03065400, NCT02421354, and NCT02871323) in 2021 [18,19,171].
We have collected preliminary data in our laboratory showing the expressions of VISTA, TIM-3, and LAG-3 on the progenitor, immune, and MDSC cells in MPN patients. We found that VISTA is the predominant next ICI receptor or ligand found in MPN patients. Other next-generation checkpoints including TIM-3, TIGIT, and LAG-3 were not different in expressions between controls and MPN patients as shown in Figure 3, Figure 4, Figure 5 and Figure 6. We had previously found MDSC over-expressed in cells including CD34+, CD14+, CD4+, and CD8+, and now our preliminary data suggest that VISTA (one of the next generation ICI) as compared to others like TIM-3, LAG-3, TIGIT could be the predominant ICI target in MPN.
Future directions and perspectives
There is now immense interest in integrating immunotherapy into the standard of care for various tumor types specifically hematological cancers, in large part due to the considerable progress made in discovering new immune checkpoint targets as part of next-generation ICIT. First, although combination immunotherapy has shown a ray of hope by yielding significant therapeutic improvement, there is substantial debate over the optimal types and dosage of these modalities. Second, IR blockade can produce remarkable tumor shrinkage and remission in only a proportion of patients, and it is critically important to understand why. While several known factors, such as IR ligand expression, the brevity, and the immunogenicity of neoantigen expression, could contribute, there may be many other factors that remain unknown. Determining these factors and identifying biomarkers that can predict responsiveness to each immunological modality will be critical. Third, while the novel immunotherapies tested in clinical trials represent a significant step forward, it remains important to continue the search for new targets that might be critical components of future combinatorial approaches. This is especially important in the future to promote new modalities with higher efficacy but reduced adverse events. It is also important to continue to identify new potential immunotherapeutic targets and mechanisms that can lay the foundation of new targeted approaches.
Our preliminary results showed that VISTA than others including TIM-3, LAG-3, and TIGIT, were the predominant next-generation ICI expressed on CD3+, CD14 +, and CD 34 + cells as measured by the percentage of positive 2nd G- ICI cells (Figure 3) and MFI (Figure 4). Also, we demonstrated that VISTA also wares the predominant 2nd G-ICI on both the G-MDSC and M-MDSC as measured by the percentage of positive cells (Figure 5) and MFI (Figure 6) respectively. This would lead to further clinical trials specifically involving VISTA with a possible combination anti-Vista and anti-PD-1 in MPN disease. This may lead to reviving the ICI therapy in MPN which ICI was found to be a negative trial in using anti-PD-1 only in the treatment of MPN.
References
- Passamonti F, Mora B, Maffioli M. New molecular genetics in the diagnosis and treatment of myeloproliferative neoplasms. Curr Opin Hematol. 2016;23(2):137-143. [CrossRef]
- Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140(11):1200-1228. [CrossRef]
- Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36(7):1703-1719. [CrossRef]
- Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667-679. [CrossRef]
- Fleischman AG, Aichberger KJ, Luty SB, et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood. 2011;118(24):6392-6398. [CrossRef]
- James C, Ugo V, Le Couédic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144-1148. [CrossRef]
- Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. The Lancet. 2005;365(9464):1054-1061. [CrossRef]
- Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387-397. [CrossRef]
- Barosi G. An Immune Dysregulation in MPN. Curr Hematol Malig Rep. 2014;9(4):331-339. [CrossRef]
- Skov V, Larsen TS, Thomassen M, et al. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leukemia Research. 2012;36(11):1387-1392. [CrossRef]
- Zhao W bo, Li Y, Liu X, Zhang L yan, Wang X. Involvement of CD4+CD25+ regulatory T cells in the pathogenesis of polycythaemia vera. Chin Med J (Engl). 2008;121(18):1781-1786.
- Skov V, Riley CH, Thomassen M, et al. Whole blood transcriptional profiling reveals significant down-regulation of human leukocyte antigen class I and II genes in essential thrombocythemia, polycythemia vera and myelofibrosis. Leukemia & Lymphoma. 2013;54(10):2269-2273. [CrossRef]
- Marty C, Lacout C, Droin N, et al. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia. 2013;27(11):2187-2195. [CrossRef]
- Bjørn ME, Hasselbalch HC. The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms. Mediators of Inflammation. 2015;2015:e648090. [CrossRef]
- Wang JC, Kundra A, Andrei M, et al. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leukemia Research. 2016;43:39-43. [CrossRef]
- Marzec M, Zhang Q, Goradia A, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105(52):20852-20857. [CrossRef]
- Abou Dalle I, Kantarjian H, Daver N, et al. Phase II study of single-agent nivolumab in patients with myelofibrosis. Ann Hematol. 2021;100(12):2957-2960. [CrossRef]
- Hobbs G, Cimen Bozkus C, Moshier E, et al. PD-1 inhibition in advanced myeloproliferative neoplasms. Blood Adv. 2021;5(23):5086-5097. [CrossRef]
- Long L, Zhang X, Chen F, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer. 2018;9(5-6):176-189. [CrossRef]
- Workman CJ, Dugger KJ, Vignali DAA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. 2002;169(10):5392-5395. [CrossRef]
- Saleh R, Toor SM, Sasidharan Nair V, Elkord E. Role of Epigenetic Modifications in Inhibitory Immune Checkpoints in Cancer Development and Progression. Front Immunol. 2020;11:1469. [CrossRef]
- Kouo T, Huang L, Pucsek AB, et al. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res. 2015;3(4):412-423. [CrossRef]
- Andreae S, Buisson S, Triebel F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood. 2003;102(6):2130-2137. [CrossRef]
- Wang J, Sanmamed MF, Datar I, et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell. 2019;176(1-2):334-347.e12. [CrossRef]
- Zuazo M, Arasanz H, Fernández-Hinojal G, et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol Med. 2019;11(7):e10293. [CrossRef]
- Lichtenegger FS, Rothe M, Schnorfeil FM, et al. Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Front Immunol. 2018;9:385. [CrossRef]
- Jing W, Gershan JA, Weber J, et al. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J Immunother Cancer. 2015;3(1):2. [CrossRef]
- Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(17):7875-7880. [CrossRef]
- Chocarro L, Blanco E, Arasanz H, et al. Clinical landscape of LAG-3-targeted therapy. Immunooncol Technol. 2022;14:100079. [CrossRef]
- Bristol-Myers Squibb. A Phase 3, Randomized, Double-Blind Study of Adjuvant Immunotherapy With Nivolumab + Relatlimab Fixed-Dose Combination Versus Nivolumab Monotherapy After Complete Resection of Stage III-IV Melanoma. clinicaltrials.gov; 2023. Accessed March 9, 2023. https://clinicaltrials.gov/ct2/show/NCT05002569.
- Merck Sharp & Dohme LLC. A Phase 3 Study of MK-4280A (Coformulated Favezelimab [MK-4280] Plus Pembrolizumab [MK-3475]) Versus Standard of Care in Previously Treated Metastatic PD-L1 Positive Colorectal Cancer. clinicaltrials.gov; 2022. Accessed March 9, 2023. https://clinicaltrials.gov/ct2/show/NCT05064059.
- Tawbi HA, Schadendorf D, Lipson EJ, et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N Engl J Med. 2022;386(1):24-34. [CrossRef]
- Memorial Sloan Kettering Cancer Center. A Pilot Study of 89Zr-DFO-REGN3767 Anti LAG-3 Antibody Positron Emission Tomography in Patients With Relapsed/Refractory DLBCL. clinicaltrials.gov; 2022. Accessed March 12, 2023. https://clinicaltrials.gov/ct2/show/NCT04566978.
- Celgene. An Exploratory Phase 1/2 Trial To Evaluate The Safety And Efficacy Of JCAR017 Combinations In Subjects With Relapsed/Refractory B-Cell Malignancies (PLATFORM). clinicaltrials.gov; 2023. Accessed March 12, 2023. https://clinicaltrials.gov/ct2/show/NCT03310619.
- Albershardt TC, Parsons AJ, Reeves RS, et al. Therapeutic efficacy of PD1/PDL1 blockade in B16 melanoma is greatly enhanced by immunization with dendritic cell-targeting lentiviral vector and protein vaccine. Vaccine. 2020;38(17):3369-3377. [CrossRef]
- Bristol-Myers Squibb. A Phase 1/2a Dose Escalation and Cohort Expansion Study of the Safety, Tolerability, and Efficacy of Anti-LAG-3 Monoclonal Antibody (Relatlimab, BMS-986016) Administered Alone and in Combination With Anti-PD-1 Monoclonal Antibody (Nivolumab, BMS-936558) in Relapsed or Refractory B-Cell Malignancies. clinicaltrials.gov; 2022. Accessed March 12, 2023. https://clinicaltrials.gov/ct2/show/NCT02061761.
- Merck Sharp & Dohme LLC. A Phase 1/Phase 2 Clinical Study to Evaluate the Safety and Efficacy of a Combination of MK-4280 and Pembrolizumab (MK-3475) in Participants With Hematologic Malignancies. clinicaltrials.gov; 2022. Accessed March 13, 2023. https://clinicaltrials.gov/ct2/show/NCT03598608.
- MD VB. An Open-Label Phase II Study of Relatlimab (BMS-986016) With Nivolumab (BMS-936558) in Combination With 5-Azacytidine for the Treatment of Patients With Refractory/Relapsed Acute Myeloid Leukemia and Newly Diagnosed Older Acute Myeloid Leukemia Patients. clinicaltrials.gov; 2022. Accessed March 13, 2023. https://clinicaltrials.gov/ct2/show/NCT04913922.
- Triebel F, Jitsukawa S, Baixeras E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171(5):1393-1405. [CrossRef]
- Wang JH, Meijers R, Xiong Y, et al. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proc Natl Acad Sci U S A. 2001;98(19):10799-10804. [CrossRef]
- Huard B, Mastrangeli R, Prigent P, et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc Natl Acad Sci U S A. 1997;94(11):5744-5749. [CrossRef]
- Hannier S, Triebel F. The MHC class II ligand lymphocyte activation gene-3 is co-distributed with CD8 and CD3-TCR molecules after their engagement by mAb or peptide-MHC class I complexes. Int Immunol. 1999;11(11):1745-1752. [CrossRef]
- Mastrangeli R, Micangeli E, Donini S. Cloning of murine LAG-3 by magnetic bead bound homologous probes and PCR (gene-capture PCR). Anal Biochem. 1996;241(1):93-102. [CrossRef]
- Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a Cancer Immunotherapy Target. Immunol Rev. 2017;276(1):80-96. [CrossRef]
- Ruiter DJ, Mattijssen V, Broecker EB, Ferrone S. MHC antigens in human melanomas. Semin Cancer Biol. 1991;2(1):35-45.
- Hemon P, Jean-Louis F, Ramgolam K, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011;186(9):5173-5183. [CrossRef]
- Donia M, Andersen R, Kjeldsen JW, et al. Aberrant Expression of MHC Class II in Melanoma Attracts Inflammatory Tumor-Specific CD4+ T- Cells, Which Dampen CD8+ T-cell Antitumor Reactivity. Cancer Res. 2015;75(18):3747-3759. [CrossRef]
- Dumic J, Dabelic S, Flögel M. Galectin-3: an open-ended story. Biochim Biophys Acta. 2006;1760(4):616-635. [CrossRef]
- Xu F, Liu J, Liu D, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014;74(13):3418-3428. [CrossRef]
- Zarour HM. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clin Cancer Res. 2016;22(8):1856-1864. [CrossRef]
- Workman CJ, Wang Y, El Kasmi KC, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol. 2009;182(4):1885-1891. [CrossRef]
- Camisaschi C, De Filippo A, Beretta V, et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J Invest Dermatol. 2014;134(7):1893-1902. [CrossRef]
- Huang B, Lei Z, Zhao J, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett. 2007;252(1):86-92. [CrossRef]
- Li N, Wang Y, Forbes K, et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007;26(2):494-504. [CrossRef]
- Wang L, Rubinstein R, Lines JL, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208(3):577-592. [CrossRef]
- Borggrewe M, Grit C, Den Dunnen WFA, et al. VISTA expression by microglia decreases during inflammation and is differentially regulated in CNS diseases. Glia. 2018;66(12):2645-2658. [CrossRef]
- ElTanbouly MA, Croteau W, Noelle RJ, Lines JL. VISTA: a novel immunotherapy target for normalizing innate and adaptive immunity. Semin Immunol. 2019;42:101308. [CrossRef]
- Flies DB, Wang S, Xu H, Chen L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol. 2011;187(4):1537-1541. [CrossRef]
- Huang X, Zhang X, Li E, et al. VISTA: an immune regulatory protein checking tumor and immune cells in cancer immunotherapy. Journal of Hematology & Oncology. 2020;13(1):83. [CrossRef]
- ElTanbouly MA, Schaafsma E, Noelle RJ, Lines JL. VISTA: Coming of age as a multi-lineage immune checkpoint. Clin Exp Immunol. 2020;200(2):120-130. [CrossRef]
- Blando J, Sharma A, Higa MG, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci U S A. 2019;116(5):1692-1697. [CrossRef]
- Rosenbaum SR, Knecht M, Mollaee M, et al. FOXD3 Regulates VISTA Expression in Melanoma. Cell Rep. 2020;30(2):510-524.e6. [CrossRef]
- Kuklinski LF, Yan S, Li Z, et al. VISTA expression on tumor-infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease-specific survival. Cancer Immunol Immunother. 2018;67(7):1113-1121. [CrossRef]
- Wang L, Jia B, Claxton DF, et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology. 2018;7(9):e1469594. [CrossRef]
- Aru B, Pehlivanoğlu C, Dal Z, Dereli-Çalışkan NN, Gürlü E, Yanıkkaya-Demirel G. A potential area of use for immune checkpoint inhibitors: Targeting bone marrow microenvironment in acute myeloid leukemia. Frontiers in Immunology. 2023;14. Accessed March 19, 2023. [CrossRef]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253-268. [CrossRef]
- Lines JL, Sempere LF, Broughton T, Wang L, Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. 2014;2(6):510-517. [CrossRef]
- Le Mercier I, Chen W, Lines JL, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014;74(7):1933-1944. [CrossRef]
- Ru B, Wong CN, Tong Y, et al. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics. 2019;35(20):4200-4202. [CrossRef]
- Loeser H, Kraemer M, Gebauer F, et al. The expression of the immune checkpoint regulator VISTA correlates with improved overall survival in pT1/2 tumor stages in esophageal adenocarcinoma. Oncoimmunology. 2019;8(5):e1581546. [CrossRef]
- Janssen Research & Development, LLC. An Open-Label, First-in-Human, Phase 1 Study of the Safety, Pharmacokinetics, and Pharmacodynamics of JNJ-61610588, a Fully Human IgG1 Kappa Anti-VISTA (V-Domain Ig Suppressor of T-Cell Activation) Monoclonal Antibody, in Subjects With Advanced Cancer. clinicaltrials.gov; 2018. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT02671955.
- Curis, Inc. A Phase 1, Open-Label, Dose Escalation and Dose Expansion Trial Evaluating the Safety, Pharmacokinetics, Pharmacodynamics, and Clinical Effects of Orally Administered CA-170 in Patients With Advanced Tumors and Lymphomas. clinicaltrials.gov; 2020. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT02812875.
- Wu C, Cao X, Zhang X. VISTA inhibitors in cancer immunotherapy: a short perspective on recent progresses. RSC Med Chem. 12(10):1672-1679. [CrossRef]
- Ingram PJ, Thakkar D, Boyd-Kirkup JD. Abstract 587: HMBD002, a novel neutralizing antibody targeting a specific epitope on the co-inhibitory immune checkpoint receptor VISTA, displays potent anti-tumor effects in pre-clinical models. Cancer Research. 2017;77(13_Supplement):587. [CrossRef]
- Gou R, Zhu L, Zheng M, et al. Annexin A8 can serve as potential prognostic biomarker and therapeutic target for ovarian cancer: based on the comprehensive analysis of Annexins. J Transl Med. 2019;17(1):275. [CrossRef]
- Monney L, Sabatos CA, Gaglia JL, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536-541. [CrossRef]
- Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol. 2014;193(4):1525-1530. [CrossRef]
- Phong BL, Avery L, Sumpter TL, et al. Tim-3 enhances FcεRI-proximal signaling to modulate mast cell activation. J Exp Med. 2015;212(13):2289-2304. [CrossRef]
- Baitsch L, Baumgaertner P, Devêvre E, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350-2360. [CrossRef]
- Kikushige Y, Shima T, Takayanagi S ichiro, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708-717. [CrossRef]
- Anderson AC, Anderson DE, Bregoli L, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science. 2007;318(5853):1141-1143. [CrossRef]
- Zhang Y, Ma CJ, Wang JM, et al. Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS One. 2011;6(5):e19664. [CrossRef]
- Maurya N, Gujar R, Gupta M, Yadav V, Verma S, Sen P. Immunoregulation of dendritic cells by the receptor T cell Ig and mucin protein-3 via Bruton’s tyrosine kinase and c-Src. J Immunol. 2014;193(7):3417-3425. [CrossRef]
- Su EW, Bi S, Kane LP. Galectin-9 regulates T helper cell function independently of Tim-3. Glycobiology. 2011;21(10):1258-1265. [CrossRef]
- Chiba S, Baghdadi M, Akiba H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832-842. [CrossRef]
- DeKruyff RH, Bu X, Ballesteros A, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184(4):1918-1930. [CrossRef]
- Huang YH, Zhu C, Kondo Y, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517(7534):386-390. [CrossRef]
- Rangachari M, Zhu C, Sakuishi K, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat Med. 2012;18(9):1394-1400. [CrossRef]
- Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276(1):97-111. [CrossRef]
- Tao J lian, Li L juan, Fu R, et al. Elevated TIM3+ hematopoietic stem cells in untreated myelodysplastic syndrome displayed aberrant differentiation, overproliferation and decreased apoptosis. Leuk Res. 2014;38(6):714-721. [CrossRef]
- Tcvetkov N, Gusak A, Morozova E, et al. Immune checkpoints bone marrow expression as the predictor of clinical outcome in myelodysplastic syndrome. Leuk Res Rep. 2020;14:100215. [CrossRef]
- Asayama T, Tamura H, Ishibashi M, et al. Functional expression of Tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget. 2017;8(51):88904-88917. [CrossRef]
- Brück O, Blom S, Dufva O, et al. Immune cell contexture in the bone marrow tumor microenvironment impacts therapy response in CML. Leukemia. 2018;32(7):1643-1656. [CrossRef]
- Liu F, Zeng G, Zhou S, et al. Blocking Tim-3 or/and PD-1 reverses dysfunction of tumor-infiltrating lymphocytes in HBV-related hepatocellular carcinoma. Bull Cancer. 2018;105(5):493-501. [CrossRef]
- Schnell A, Bod L, Madi A, Kuchroo VK. The yin and yang of co-inhibitory receptors: toward anti-tumor immunity without autoimmunity. Cell Res. 2020;30(4):285-299. [CrossRef]
- Liu F, Liu Y, Chen Z. Tim-3 expression and its role in hepatocellular carcinoma. J Hematol Oncol. 2018;11(1):126. [CrossRef]
- Tan J, Huang S, Huang J, et al. Increasing Tim-3+CD244+, Tim-3+CD57+, and Tim-3+PD-1+ T cells in patients with acute myeloid leukemia. Asia Pac J Clin Oncol. 2020;16(3):137-141. [CrossRef]
- Hadadi L, Hafezi M, Amirzargar AA, Sharifian RA, Abediankenari S, Asgarian-Omran H. Dysregulated Expression of Tim-3 and NKp30 Receptors on NK Cells of Patients with Chronic Lymphocytic Leukemia. Oncol Res Treat. 2019;42(4):202-208. [CrossRef]
- Rezazadeh H, Astaneh M, Tehrani M, et al. Blockade of PD-1 and TIM-3 immune checkpoints fails to restore the function of exhausted CD8+ T cells in early clinical stages of chronic lymphocytic leukemia. Immunol Res. 2020;68(5):269-279. [CrossRef]
- Zeidan AM, Komrokji RS, Brunner AM. TIM-3 pathway dysregulation and targeting in cancer. Expert Rev Anticancer Ther. 2021;21(5):523-534. [CrossRef]
- Harding JJ, Moreno V, Bang YJ, et al. Blocking TIM-3 in Treatment-refractory Advanced Solid Tumors: A Phase Ia/b Study of LY3321367 with or without an Anti-PD-L1 Antibody. Clin Cancer Res. 2021;27(8):2168-2178. [CrossRef]
- Novartis Pharmaceuticals. Phase 1b, Multi-Arm, Open-Label Study of PDR001 and/or MBG453 in Combination With Decitabine in Patients With Acute Myeloid Leukemia or High Risk Myelodysplastic Syndrome. clinicaltrials.gov; 2023. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT03066648.
- Bewersdorf JP, Stahl M, Zeidan AM. Immune checkpoint-based therapy in myeloid malignancies: a promise yet to be fulfilled. Expert Review of Anticancer Therapy. 2019;19(5):393-404. [CrossRef]
- Yu X, Harden K, Gonzalez LC, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48-57. [CrossRef]
- Stanietsky N, Simic H, Arapovic J, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci U S A. 2009;106(42):17858-17863. [CrossRef]
- Harjunpää H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol. 2020;200(2):108-119. [CrossRef]
- Chan CJ, Andrews DM, Smyth MJ. Receptors that interact with nectin and nectin-like proteins in the immunosurveillance and immunotherapy of cancer. Curr Opin Immunol. 2012;24(2):246-251. [CrossRef]
- Boles KS, Vermi W, Facchetti F, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol. 2009;39(3):695-703. [CrossRef]
- Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56(5):855-865. [CrossRef]
- Eberlé F, Dubreuil P, Mattei MG, Devilard E, Lopez M. The human PRR2 gene, related to the human poliovirus receptor gene (PVR), is the true homolog of the murine MPH gene. Gene. 1995;159(2):267-272. [CrossRef]
- Reymond N, Borg JP, Lecocq E, et al. Human nectin3/PRR3: a novel member of the PVR/PRR/nectin family that interacts with afadin. Gene. 2000;255(2):347-355. [CrossRef]
- Johnston RJ, Comps-Agrar L, Hackney J, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26(6):923-937. [CrossRef]
- Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. 2012;188(8):3869-3875. [CrossRef]
- Kong Y, Zhu L, Schell TD, et al. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin Cancer Res. 2016;22(12):3057-3066. [CrossRef]
- Chauvin JM, Pagliano O, Fourcade J, et al. TIGIT and PD-1 impair tumor antigen-specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125(5):2046-2058. [CrossRef]
- Guillerey C, Harjunpää H, Carrié N, et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood. 2018;132(16):1689-1694. [CrossRef]
- Stålhammar G, Seregard S, Grossniklaus HE. Expression of immune checkpoint receptors Indoleamine 2,3-dioxygenase and T cell Ig and ITIM domain in metastatic versus nonmetastatic choroidal melanoma. Cancer Med. 2019;8(6):2784-2792. [CrossRef]
- O’Brien SM, Klampatsa A, Thompson JC, et al. Function of Human Tumor-Infiltrating Lymphocytes in Early-Stage Non-Small Cell Lung Cancer. Cancer Immunol Res. 2019;7(6):896-909. [CrossRef]
- Kurtulus S, Sakuishi K, Ngiow SF, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125(11):4053-4062. [CrossRef]
- Degos C, Heinemann M, Barrou J, et al. Endometrial Tumor Microenvironment Alters Human NK Cell Recruitment, and Resident NK Cell Phenotype and Function. Front Immunol. 2019;10:877. [CrossRef]
- Josefsson SE, Huse K, Kolstad A, et al. T Cells Expressing Checkpoint Receptor TIGIT Are Enriched in Follicular Lymphoma Tumors and Characterized by Reversible Suppression of T-cell Receptor Signaling. Clin Cancer Res. 2018;24(4):870-881. [CrossRef]
- Fourcade J, Sun Z, Chauvin JM, et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight. 2018;3(14):e121157, 121157. [CrossRef]
- Dixon KO, Schorer M, Nevin J, et al. Functional Anti-TIGIT Antibodies Regulate Development of Autoimmunity and Antitumor Immunity. J Immunol. 2018;200(8):3000-3007. [CrossRef]
- Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267-296. [CrossRef]
- Wu AA, Drake V, Huang HS, Chiu S, Zheng L. Reprogramming the tumor microenvironment: tumor-induced immunosuppressive factors paralyze T cells. Oncoimmunology. 2015;4(7):e1016700. [CrossRef]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252-264. [CrossRef]
- Martins F, Sofiya L, Sykiotis GP, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol. 2019;16(9):563-580. [CrossRef]
- Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):1-11. [CrossRef]
- Solomon BL, Garrido-Laguna I. TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother. 2018;67(11):1659-1667. [CrossRef]
- Harjunpää H, Blake SJ, Ahern E, et al. Deficiency of host CD96 and PD-1 or TIGIT enhances tumor immunity without significantly compromising immune homeostasis. Oncoimmunology. 2018;7(7):e1445949. [CrossRef]
- Genentech, Inc. A Phase II, Randomized, Blinded, Placebo-Controlled Study of Tiragolumab, An Anti-TIGIT Antibody, In Combination With Atezolizumab In Chemotherapy-Naïve Patients With Locally Advanced Or Metastatic Non-Small Cell Lung Cancer. clinicaltrials.gov; 2023. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT03563716.
- Arcus Biosciences, Inc. A Phase 1 Study to Evaluate the Safety and Tolerability of AB154 Monotherapy and Combination Therapy in Participants With Advanced Malignancies. clinicaltrials.gov; 2023. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT03628677.
- Merck Sharp & Dohme LLC. A Phase 1 Trial of MK-7684 as Monotherapy and in Combination With Pembrolizumab in Subjects With Advanced Solid Tumors. clinicaltrials.gov; 2022. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT02964013.
- Bristol-Myers Squibb. Phase 1/2a First-In-Human Study of BMS-986207 Monoclonal Antibody Alone and in Combination With Nivolumab or With Nivolumab and Ipilimumab in Advanced Solid Tumors. clinicaltrials.gov; 2023. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT02913313.
- Astellas Pharma Global Development, Inc. A Phase 1b Study of ASP8374, an Immune Checkpoint Inhibitor, as a Single Agent and in Combination With Pembrolizumab in Subjects With Advanced Solid Tumors. clinicaltrials.gov; 2022. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT03260322.
- Astellas Pharma Inc. A Phase 1, Open Label Study of ASP8374, an Immune Checkpoint Inhibitor, in Japanese Patients With Advanced Solid Tumors. clinicaltrials.gov; 2021. Accessed March 16, 2023. https://clinicaltrials.gov/ct2/show/NCT03945253.
- Jin S, Zhang Y, Zhou F, Chen X, Sheng J, Zhang J. TIGIT: A promising target to overcome the barrier of immunotherapy in hematological malignancies. Frontiers in Oncology. 2022;12. Accessed May 14, 2023. [CrossRef]
- Catakovic K, Gassner FJ, Ratswohl C, et al. TIGIT expressing CD4+T cells represent a tumor-supportive T cell subset in chronic lymphocytic leukemia. Oncoimmunology. 2017;7(1):e1371399. [CrossRef]
- Minnie SA, Kuns RD, Gartlan KH, et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood. 2018;132(16):1675-1688. [CrossRef]
- Hoffmann-La Roche. A Phase Ib/II Open-Label, Multicenter Study Evaluating the Safety, Efficacy, and Pharmacokinetics of Mosunetuzumab in Combination With Tiragolumab With or Without Atezolizumab in Patients With Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma. clinicaltrials.gov; 2023. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT05315713.
- Genentech, Inc. A Phase Ia/Ib Open-Label, Multicenter Study Evaluating the Safety and Pharmacokinetics of Tiragolumab as a Single Agent and in Combination With Atezolizumab and/or Daratumumab in Patients With Relapsed or Refractory Multiple Myeloma, and as a Single Agent and in Combination With Rituximab in Patients With Relapsed or Refractory B-Cell Non-Hodgkin Lymphoma. clinicaltrials.gov; 2023. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT04045028.
- BeiGene. A Phase 1b/2 Study Investigating the Safety, Tolerability, Pharmacokinetics, and Preliminary Antitumor Activity of Ociperlimab (BGB A1217) in Combination With Tislelizumab (BGB A317) or Rituximab in Patients With Relapsed or Refractory Diffuse Large B Cell Lymphoma. clinicaltrials.gov; 2022. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT05267054.
- Multiple Myeloma Research Consortium. A Phase I/II Assessment of Combination Immuno-Oncology Drugs Elotuzumab, Anti-LAG-3 (BMS-986016) and Anti-TIGIT (BMS-986207). clinicaltrials.gov; 2021. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT04150965.
- Merck Sharp & Dohme LLC. A Phase 2, Open-Label Study to Evaluate the Safety and Efficacy of MK-7684A (MK-7684 [Vibostolimab] With MK-3475 [Pembrolizumab] Coformulation) in Participants With Relapsed or Refractory Hematological Malignancies. clinicaltrials.gov; 2023. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT05005442.
- Compugen Ltd. A Phase 1 Study of The Safety and Tolerability of COM902 in Subjects With Advanced Malignancies. clinicaltrials.gov; 2023. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT04354246.
- Seagen Inc. A Phase 1 Study of SEA-TGT (SGN-TGT) in Subjects With Advanced Malignancies. clinicaltrials.gov; 2023. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT04254107.
- Arcus Biosciences, Inc. A Phase 1/1b Study to Evaluate the Safety and Tolerability of AB308 in Combination With AB122 in Participants With Advanced Malignancies. clinicaltrials.gov; 2023. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT04772989.
- iTeos Belgium SA. Study of EOS884448 Alone, and in Combination With Iberdomide With or Without Dexamethasone, in Participants With Relapsed or Refractory Multiple Myeloma. clinicaltrials.gov; 2022. Accessed May 12, 2023. https://clinicaltrials.gov/ct2/show/NCT05289492.
- Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761-770. [CrossRef]
- Masarova L, Bose P, Verstovsek S. The Rationale for Immunotherapy in Myeloproliferative Neoplasms. Curr Hematol Malig Rep. 2019;14(4):310-327. [CrossRef]
- Barbui T, Vannucchi AM, Buxhofer-Ausch V, et al. Practice-relevant revision of IPSET-thrombosis based on 1019 patients with WHO-defined essential thrombocythemia. Blood Cancer J. 2015;5(11):e369. [CrossRef]
- Alvarez-Larrán A, Garrote M, Ferrer-Marín F, et al. Real-world analysis of main clinical outcomes in patients with polycythemia vera treated with ruxolitinib or best available therapy after developing resistance/intolerance to hydroxyurea. Cancer. 2022;128(13):2441-2448. [CrossRef]
- Wagner SM, Melchardt T, Greil R. Ropeginterferon alfa-2b for the treatment of patients with polycythemia vera. Drugs Today (Barc). 2020;56(3):195-202. [CrossRef]
- Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus Standard Therapy for the Treatment of Polycythemia Vera. New England Journal of Medicine. 2015;372(5):426-435. [CrossRef]
- Mascarenhas J, Hoffman R. A comprehensive review and analysis of the effect of ruxolitinib therapy on the survival of patients with myelofibrosis. Blood. 2013;121(24):4832-4837. [CrossRef]
- Kvasnicka HM, Thiele J, Bueso-Ramos CE, et al. Long-term effects of ruxolitinib versus best available therapy on bone marrow fibrosis in patients with myelofibrosis. J Hematol Oncol. 2018;11(1):42. [CrossRef]
- Vannucchi AM, Verstovsek S, Guglielmelli P, et al. Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol. 2017;96(7):1113-1120. [CrossRef]
- Deininger M, Radich J, Burn TC, Huber R, Paranagama D, Verstovsek S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood. 2015;126(13):1551-1554. [CrossRef]
- Vannucchi AM, Kantarjian HM, Kiladjian JJ, et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. 2015;100(9):1139-1145. [CrossRef]
- Wang JC, Chen C, Kundra A, et al. Programmed Cell Death Receptor (PD-1) Ligand (PD-L1) expression in Philadelphia chromosome-negative myeloproliferative neoplasms. Leuk Res. 2019;79:52-59. [CrossRef]
- Fang W, Zhang J, Hong S, et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget. 2014;5(23):12189-12202. [CrossRef]
- Mandai M, Hamanishi J, Abiko K, Matsumura N, Baba T, Konishi I. Dual Faces of IFNγ in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clinical Cancer Research. 2016;22(10):2329-2334. [CrossRef]
- Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH. Regulatory T Cells Stimulate B7-H1 Expression in Myeloid-Derived Suppressor Cells in ret Melanomas. J Invest Dermatol. 2012;132(4):1239-1246. [CrossRef]
- Hou A, Hou K, Huang Q, Lei Y, Chen W. Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Frontiers in Immunology. 2020;11. Accessed March 28, 2023. [CrossRef]
- Weber R, Fleming V, Hu X, et al. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Frontiers in Immunology. 2018;9. Accessed March 28, 2023. [CrossRef]
- Meyer C, Cagnon L, Costa-Nunes CM, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247-257. [CrossRef]
- Martens A, Wistuba-Hamprecht K, Foppen MG, et al. Baseline Peripheral Blood Biomarkers Associated with Clinical Outcome of Advanced Melanoma Patients Treated with Ipilimumab. Clinical Cancer Research. 2016;22(12):2908-2918. [CrossRef]
- Weide B, Martens A, Zelba H, et al. Myeloid-Derived Suppressor Cells Predict Survival of Patients with Advanced Melanoma: Comparison with Regulatory T Cells and NY-ESO-1- or Melan-A–Specific T Cells. Clinical Cancer Research. 2014;20(6):1601-1609. [CrossRef]
- Soda H, Ogawara D, Fukuda Y, et al. Dynamics of blood neutrophil-related indices during nivolumab treatment may be associated with response to salvage chemotherapy for non-small cell lung cancer: A hypothesis-generating study. Thoracic Cancer. 2019;10(2):341-346. [CrossRef]
- Feng J, Chen S, Li S, et al. The association between monocytic myeloid-derived suppressor cells levels and the anti-tumor efficacy of anti-PD-1 therapy in NSCLC patients. Translational Oncology. 2020;13(12):100865. [CrossRef]
- Northwestern University. A Phase 1, Single Arm, Single Center Pilot Study of Medi4736, an Anti-Pdl1 Therapy, for Patients With Myelofibrosis. clinicaltrials.gov; 2019. Accessed March 28, 2023. https://clinicaltrials.gov/ct2/show/NCT02871323.
Figure 1.
Immune checkpoints in a tumor microenvironment (TME). APCs present tumor antigens to naïve T cells inducing T cell activation. Through the MHC and TCR signaling pathway a first signal for T-cell activation is provided whereas co-inhibitory immune checkpoints suppress T-cell activation in TME. Immune checkpoints are expressed on T-cells, ligands are present on APCs, tumor cells, and stromal cells like CAFs and MDSCs. Abbreviations: APCs - antigen presenting cells; MHC – major histocompatibility complex; TCR – T-cell receptor; TME – tumor microenvironment; MDSCs – myeloid-derived suppressor cells; PD-1 – programmed death 1; PD-L2 – programmed cell death ligand-2; VISTA – V-domain immunoglobulincontaining suppressor T-cell activation; HHLA2 – human endogenous retrovirus-H long terminal repeatassociated protein 2; TIM-3 – T-cell immunoglobulin and mucin domain 3; Gal-9 – Galedctin-9; CAFs – cancer associated fibroblasts; LAG-3 – lymphocyte activated gene-3; CTLA-4 – cytotoxic T-lymphocyte antigen-.
Figure 1.
Immune checkpoints in a tumor microenvironment (TME). APCs present tumor antigens to naïve T cells inducing T cell activation. Through the MHC and TCR signaling pathway a first signal for T-cell activation is provided whereas co-inhibitory immune checkpoints suppress T-cell activation in TME. Immune checkpoints are expressed on T-cells, ligands are present on APCs, tumor cells, and stromal cells like CAFs and MDSCs. Abbreviations: APCs - antigen presenting cells; MHC – major histocompatibility complex; TCR – T-cell receptor; TME – tumor microenvironment; MDSCs – myeloid-derived suppressor cells; PD-1 – programmed death 1; PD-L2 – programmed cell death ligand-2; VISTA – V-domain immunoglobulincontaining suppressor T-cell activation; HHLA2 – human endogenous retrovirus-H long terminal repeatassociated protein 2; TIM-3 – T-cell immunoglobulin and mucin domain 3; Gal-9 – Galedctin-9; CAFs – cancer associated fibroblasts; LAG-3 – lymphocyte activated gene-3; CTLA-4 – cytotoxic T-lymphocyte antigen-.
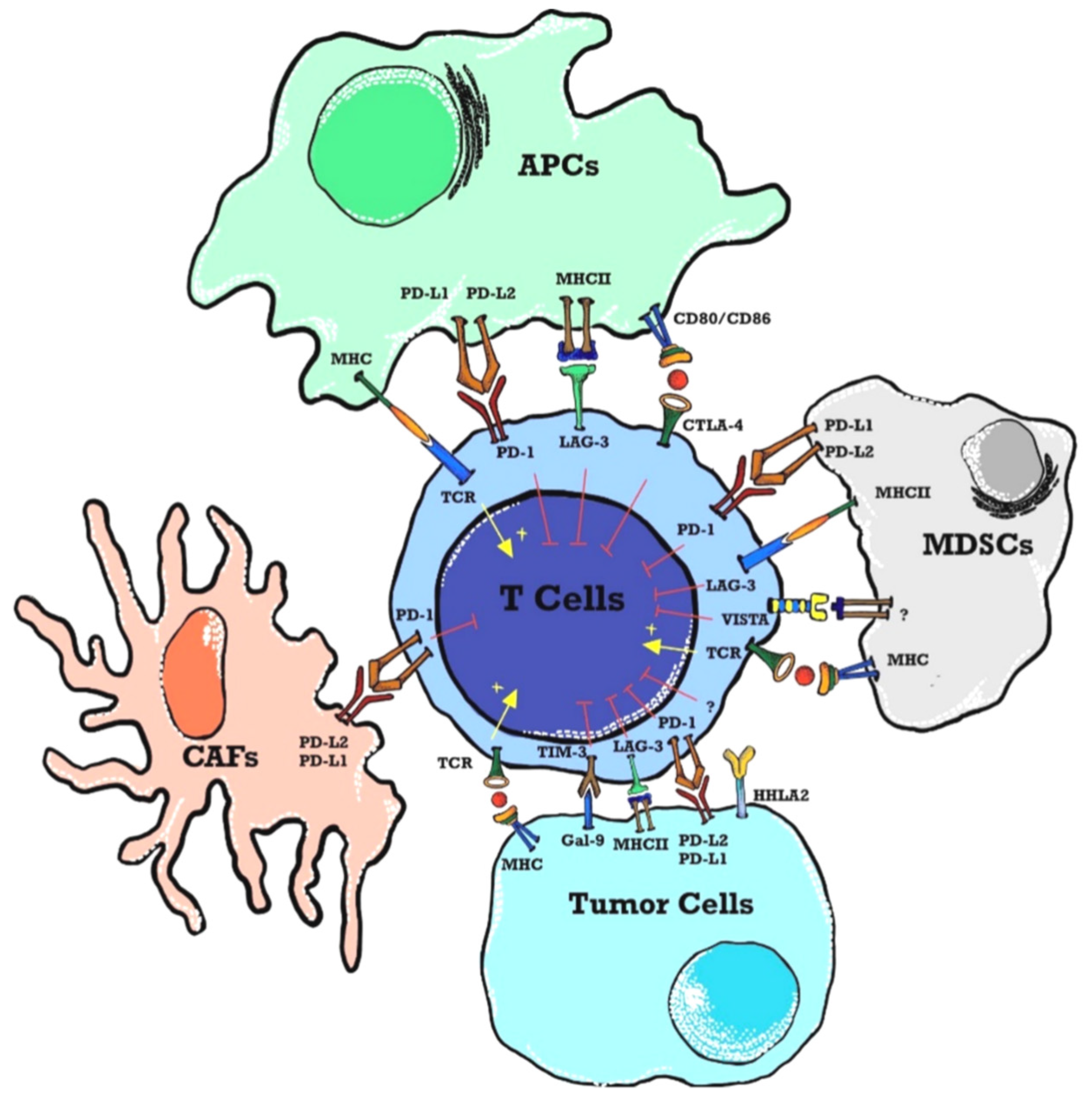
Figure 2.
Structural similarities between LAG-3 and CD4. The LAG-3 gene is predicted to be highly structurally homologous to CD4 with four extracellular immunoglobulin superfamily (IgSF)-like domains (D1-D4). LAG-3 binds to MHC class II with high affinity. LAG-3 cytoplasmic domain appears to have three well-defined motifs namely serine-based motif which could act as a PKC substrate, repetitive “EP” motif consisting of a series of glutamic acid-proline dipeptide repeats and relatively unique “KIEELE” motif, highlighted by an essential lysine residue. LAG3 has two additional ligands namely LSECtin expressed on melanoma cells and Galectin-3 expressed on stromal cells and CD8+T cells in TME. Abbreviations: TCR -Toll like recepto.
Figure 2.
Structural similarities between LAG-3 and CD4. The LAG-3 gene is predicted to be highly structurally homologous to CD4 with four extracellular immunoglobulin superfamily (IgSF)-like domains (D1-D4). LAG-3 binds to MHC class II with high affinity. LAG-3 cytoplasmic domain appears to have three well-defined motifs namely serine-based motif which could act as a PKC substrate, repetitive “EP” motif consisting of a series of glutamic acid-proline dipeptide repeats and relatively unique “KIEELE” motif, highlighted by an essential lysine residue. LAG3 has two additional ligands namely LSECtin expressed on melanoma cells and Galectin-3 expressed on stromal cells and CD8+T cells in TME. Abbreviations: TCR -Toll like recepto.
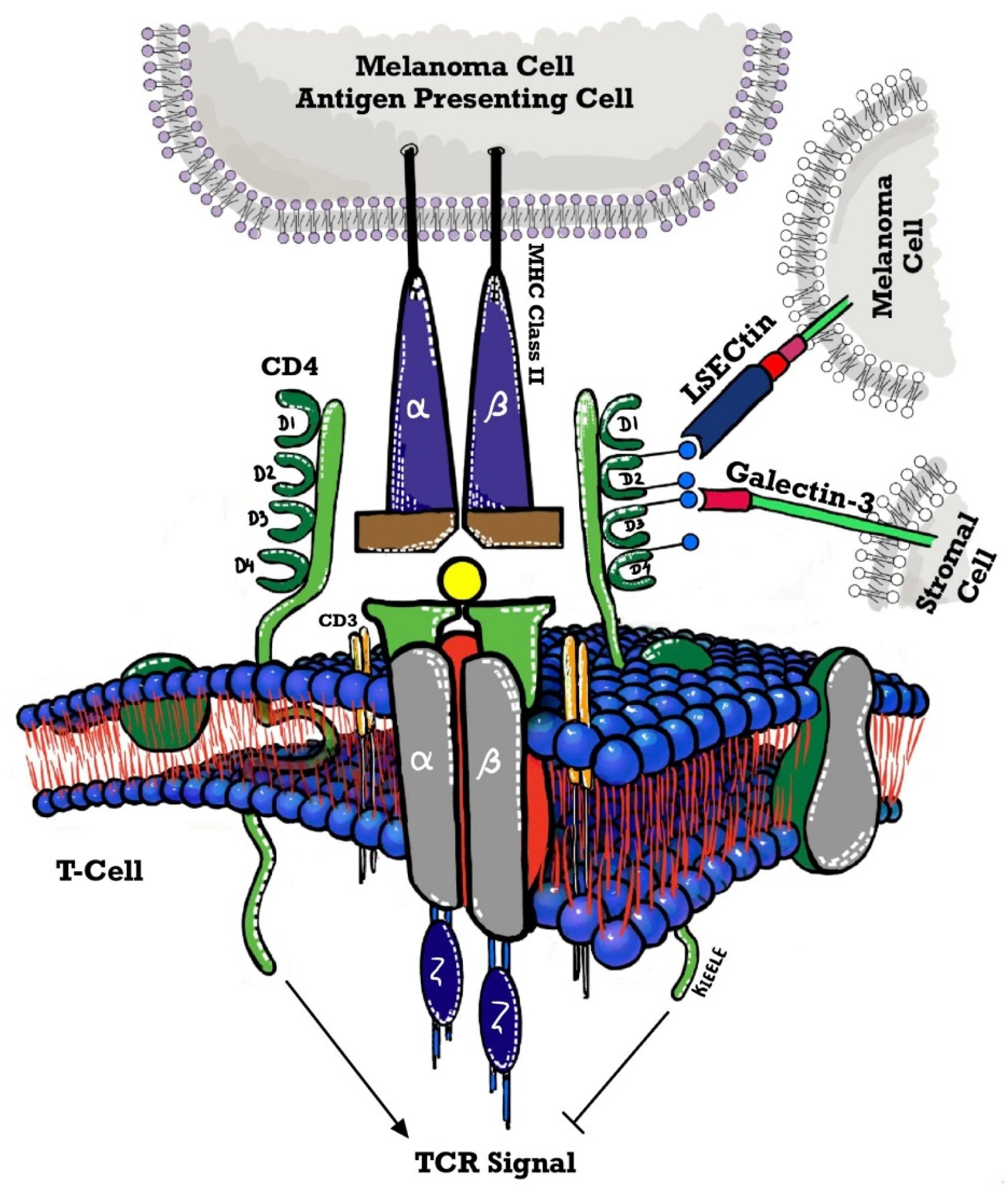
Figure 3.
2nd generation of ICI expressions (% of cells) on the different cell populations were done on MPN patients and controls. The results showed that there were no difference on the LAG-3, TIGIT, and TIM-3 on the different cell population between MPN and controls, but there was a significance of the VISTA expression (between MPN and controls) ( mean + SE) on the CD3 ( 20.4 + 5.94 Vs 0.91 + 0.44 , P< 0.05) CD14 (38.86 + 6.12 Vs 0.79 + 0.43, P =0.003) , CD 34 (2.30 + 1.26 Vs 2.30 + 1.28 , P < 0.05 ), other ICI marker of LAG3 and TIM3 were of no significant difference.
Figure 3.
2nd generation of ICI expressions (% of cells) on the different cell populations were done on MPN patients and controls. The results showed that there were no difference on the LAG-3, TIGIT, and TIM-3 on the different cell population between MPN and controls, but there was a significance of the VISTA expression (between MPN and controls) ( mean + SE) on the CD3 ( 20.4 + 5.94 Vs 0.91 + 0.44 , P< 0.05) CD14 (38.86 + 6.12 Vs 0.79 + 0.43, P =0.003) , CD 34 (2.30 + 1.26 Vs 2.30 + 1.28 , P < 0.05 ), other ICI marker of LAG3 and TIM3 were of no significant difference.
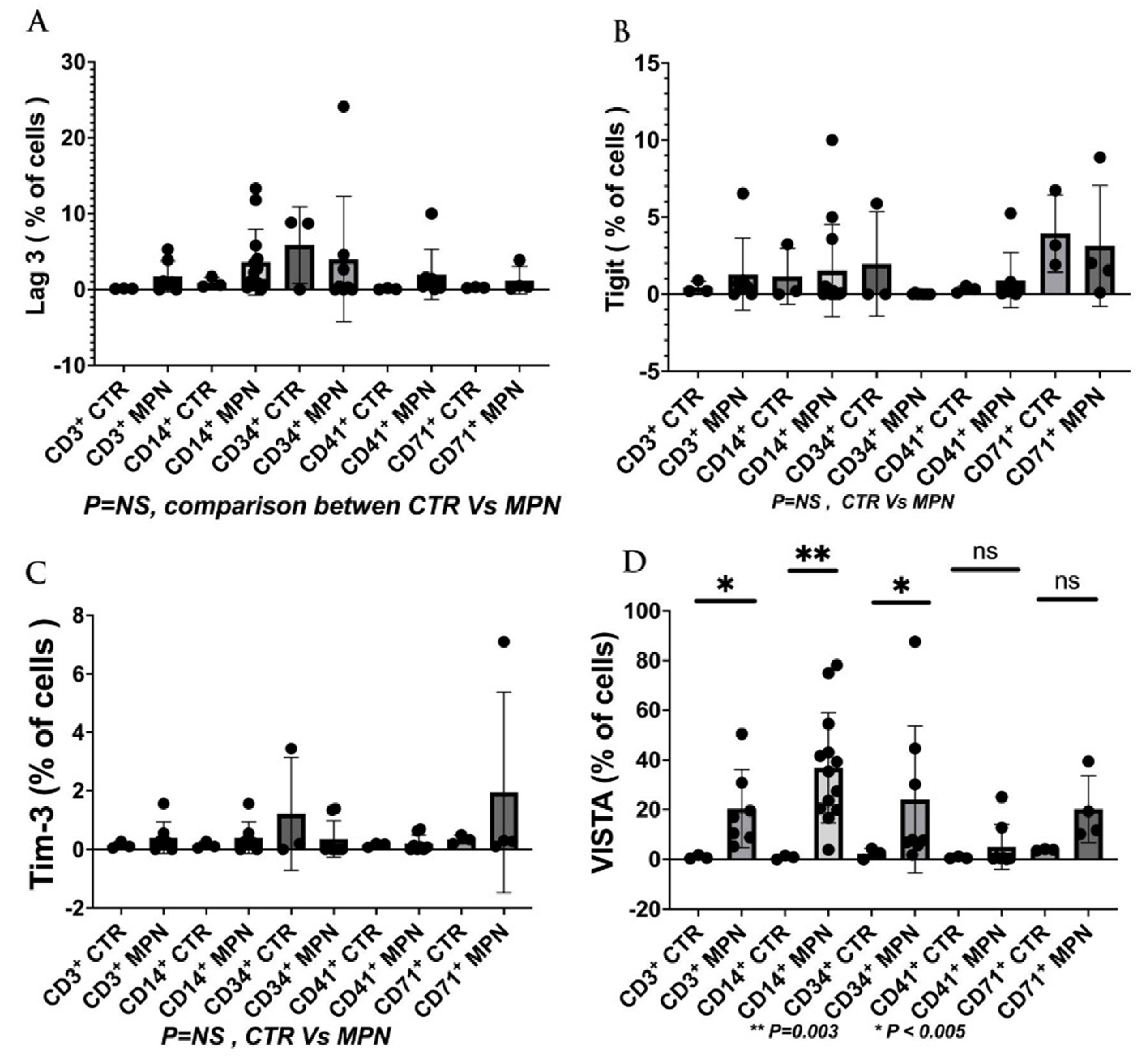
Figure 4.
2nd generation of ICI expression (MFI) on different cell populations on MPN patients and controls. The results showed there were no differences of the LAG-3, TIGIT, and TIM-3 on the different cell population between MPN and controls, but there was a significance of the VISTA expression (between MPN and controls) ( mean + SE) on the CD3 ( 3257 + 673.4 Vs 457,0 + 59.0 , P< 0.05) CD14 (5399 + 994.3 Vs 879.3 + 325.2, P =0.003) , CD 34 (2300 + 1262 Vs 24.0 + 10.4 , P < 0.05 ).
Figure 4.
2nd generation of ICI expression (MFI) on different cell populations on MPN patients and controls. The results showed there were no differences of the LAG-3, TIGIT, and TIM-3 on the different cell population between MPN and controls, but there was a significance of the VISTA expression (between MPN and controls) ( mean + SE) on the CD3 ( 3257 + 673.4 Vs 457,0 + 59.0 , P< 0.05) CD14 (5399 + 994.3 Vs 879.3 + 325.2, P =0.003) , CD 34 (2300 + 1262 Vs 24.0 + 10.4 , P < 0.05 ).
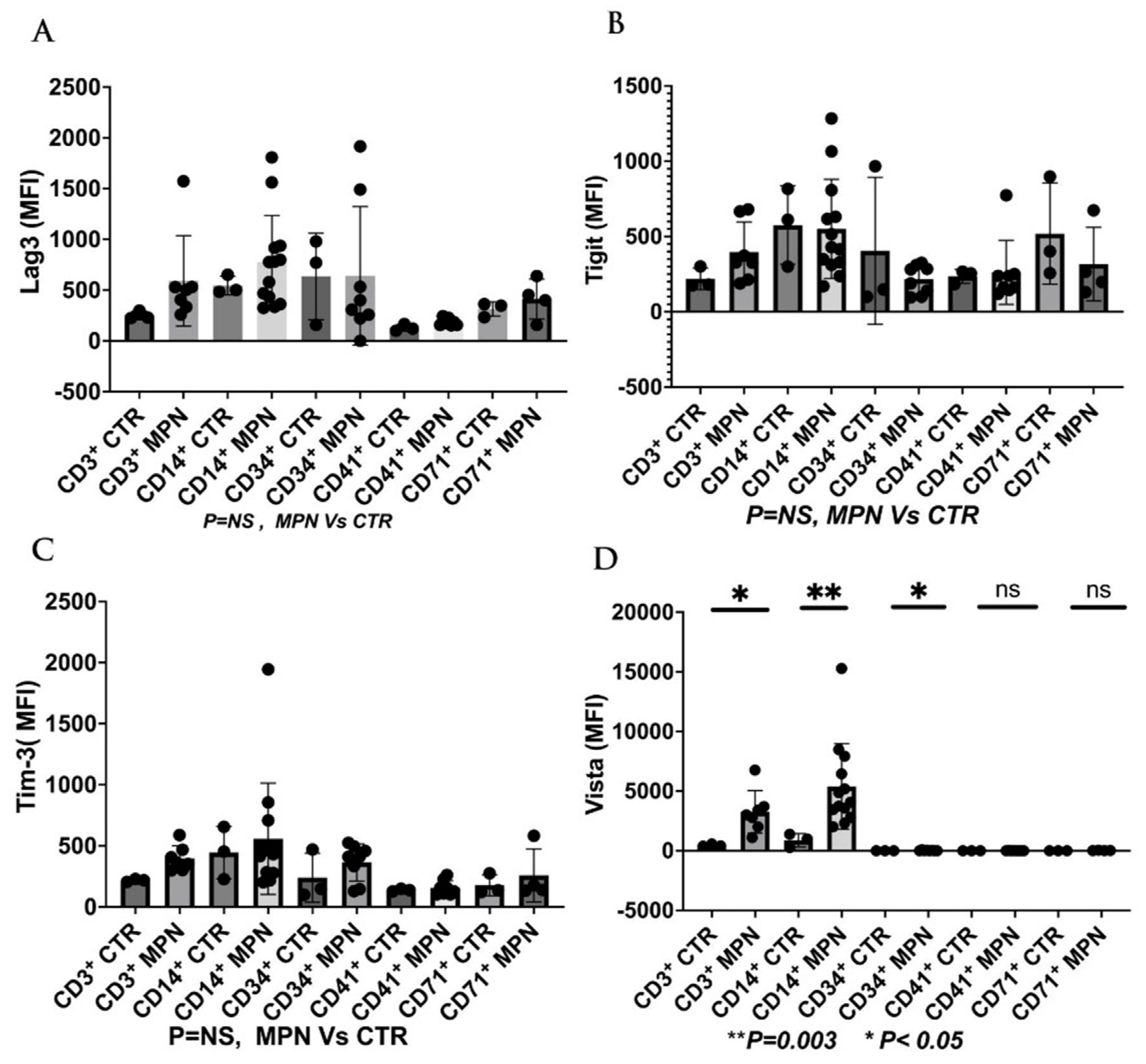
Figure 5.
Expression of 2nd generation of ICI (% of positive cells) on the G- MDSC, and M-MDSC in patients with MPN and controls. The results showed there was no difference of the LAG-3, TIGIT, and TIM-3 on G- MDSC or M-MDSC between MPN and controls, but there was a significance of the VISTA expression (between MPN and controls) ( mean + SE) on the G-MDSC (23.9 + 6.8 Vs 0.00 + 0.0 , P< 0.003) , and M-MDSC ( 31.5 + 6.6 Vs 1.47 + 1.47, P =0.003) respectively .
Figure 5.
Expression of 2nd generation of ICI (% of positive cells) on the G- MDSC, and M-MDSC in patients with MPN and controls. The results showed there was no difference of the LAG-3, TIGIT, and TIM-3 on G- MDSC or M-MDSC between MPN and controls, but there was a significance of the VISTA expression (between MPN and controls) ( mean + SE) on the G-MDSC (23.9 + 6.8 Vs 0.00 + 0.0 , P< 0.003) , and M-MDSC ( 31.5 + 6.6 Vs 1.47 + 1.47, P =0.003) respectively .
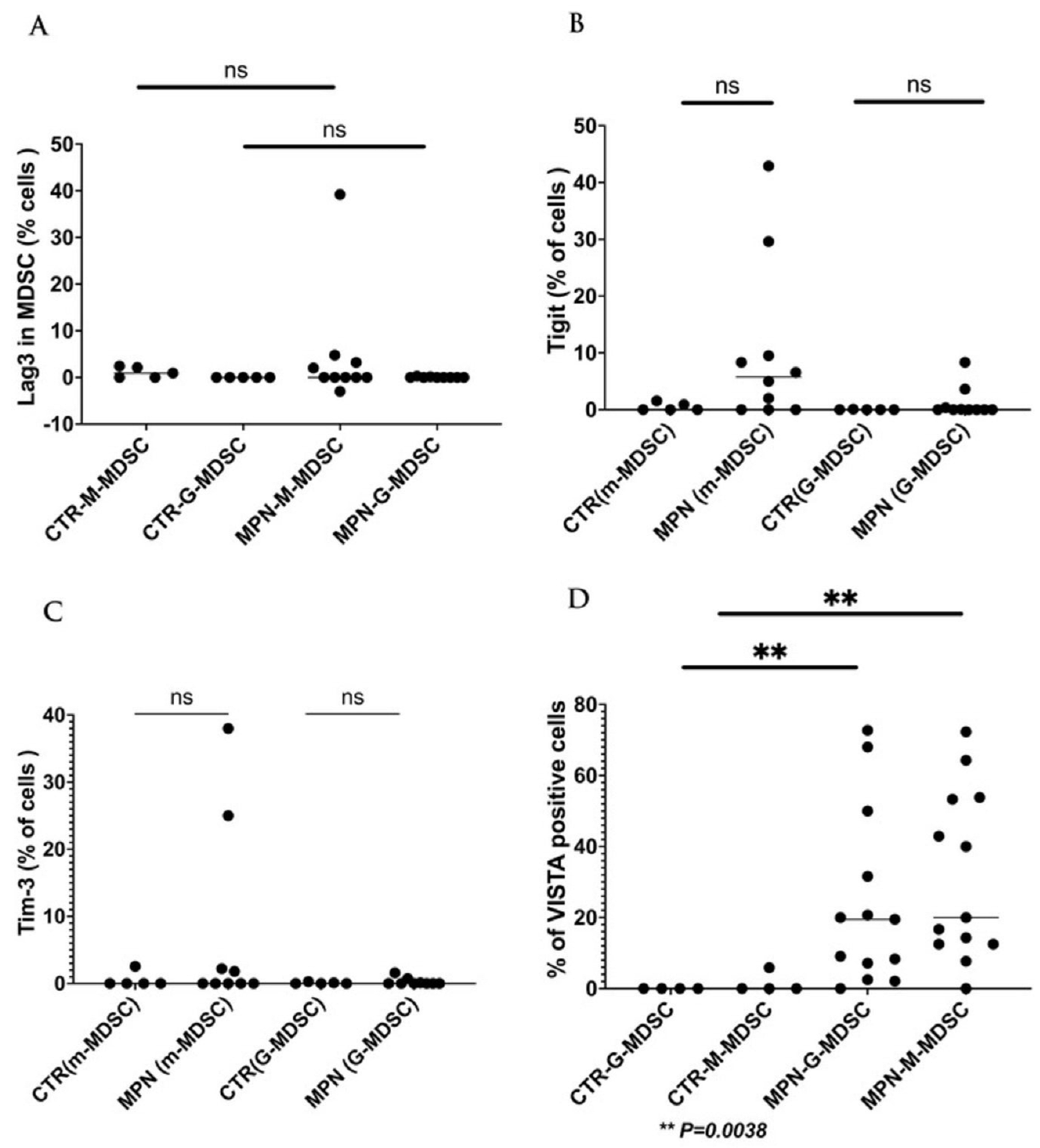
Figure 6.
Expression of 2nd generation of ICI (MFI ) on the G- MDSC, and M-MDSC in patients with MPN and controls. The results showed there were no difference of the LAG-3, TIGIT, and TIM-3 on GMDSC or M-MDSC between MPN and controls, but there was a significance of the VISTA expression (between MPN and controls) ( mean + SE) on the G-MDSC (3085 + 763.6 Vs 179.7 + 64.6 , P= 0.008) , and M-MDSC ( 4241 + 617.7 Vs 159.7 + 31.29, P =0.0004) respectively.
Figure 6.
Expression of 2nd generation of ICI (MFI ) on the G- MDSC, and M-MDSC in patients with MPN and controls. The results showed there were no difference of the LAG-3, TIGIT, and TIM-3 on GMDSC or M-MDSC between MPN and controls, but there was a significance of the VISTA expression (between MPN and controls) ( mean + SE) on the G-MDSC (3085 + 763.6 Vs 179.7 + 64.6 , P= 0.008) , and M-MDSC ( 4241 + 617.7 Vs 159.7 + 31.29, P =0.0004) respectively.
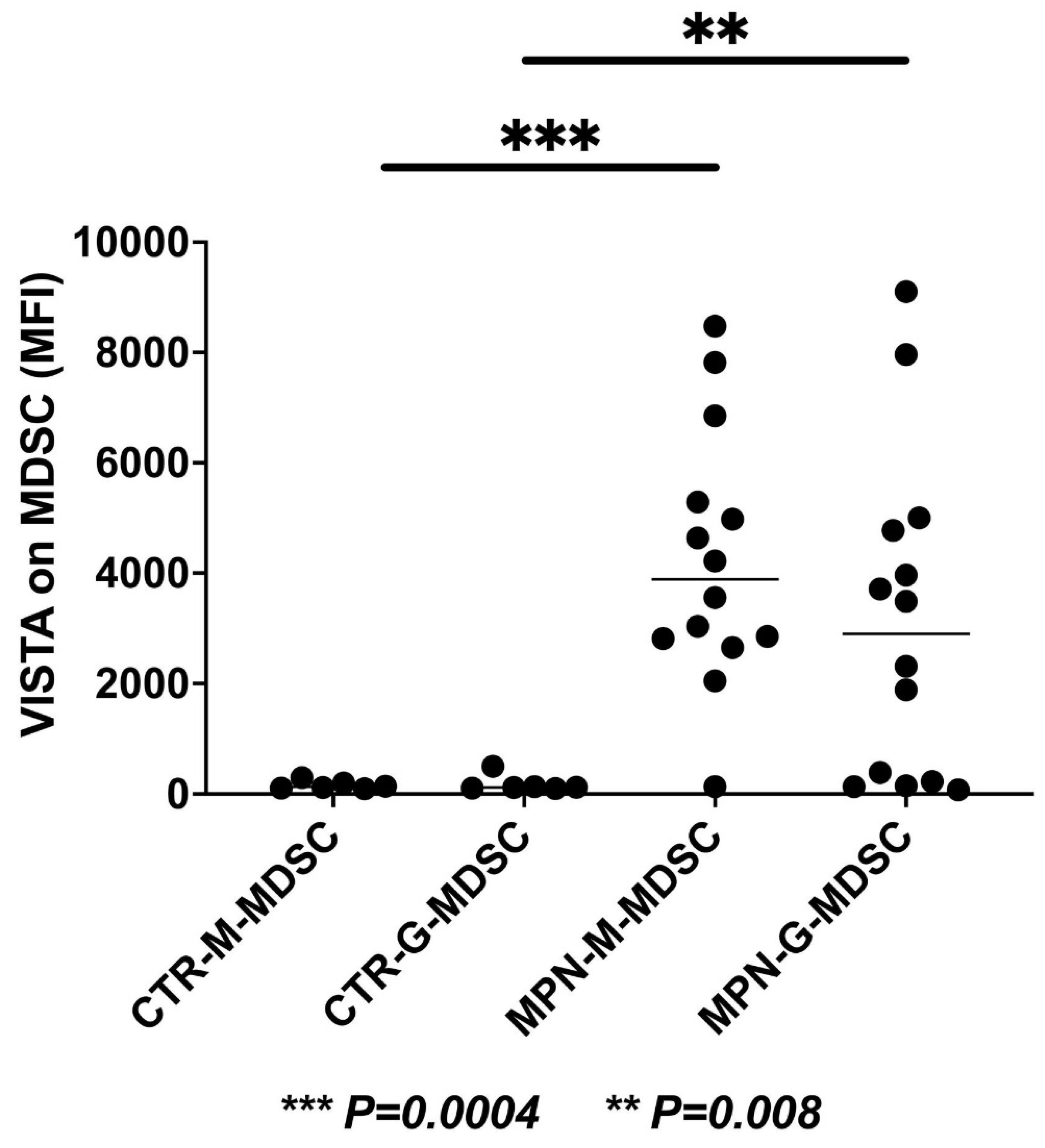

Table 1.
Summarizes all the clinical trials of LAG-3 therapy being used in hematological or related malignancies.
Table 1.
Summarizes all the clinical trials of LAG-3 therapy being used in hematological or related malignancies.
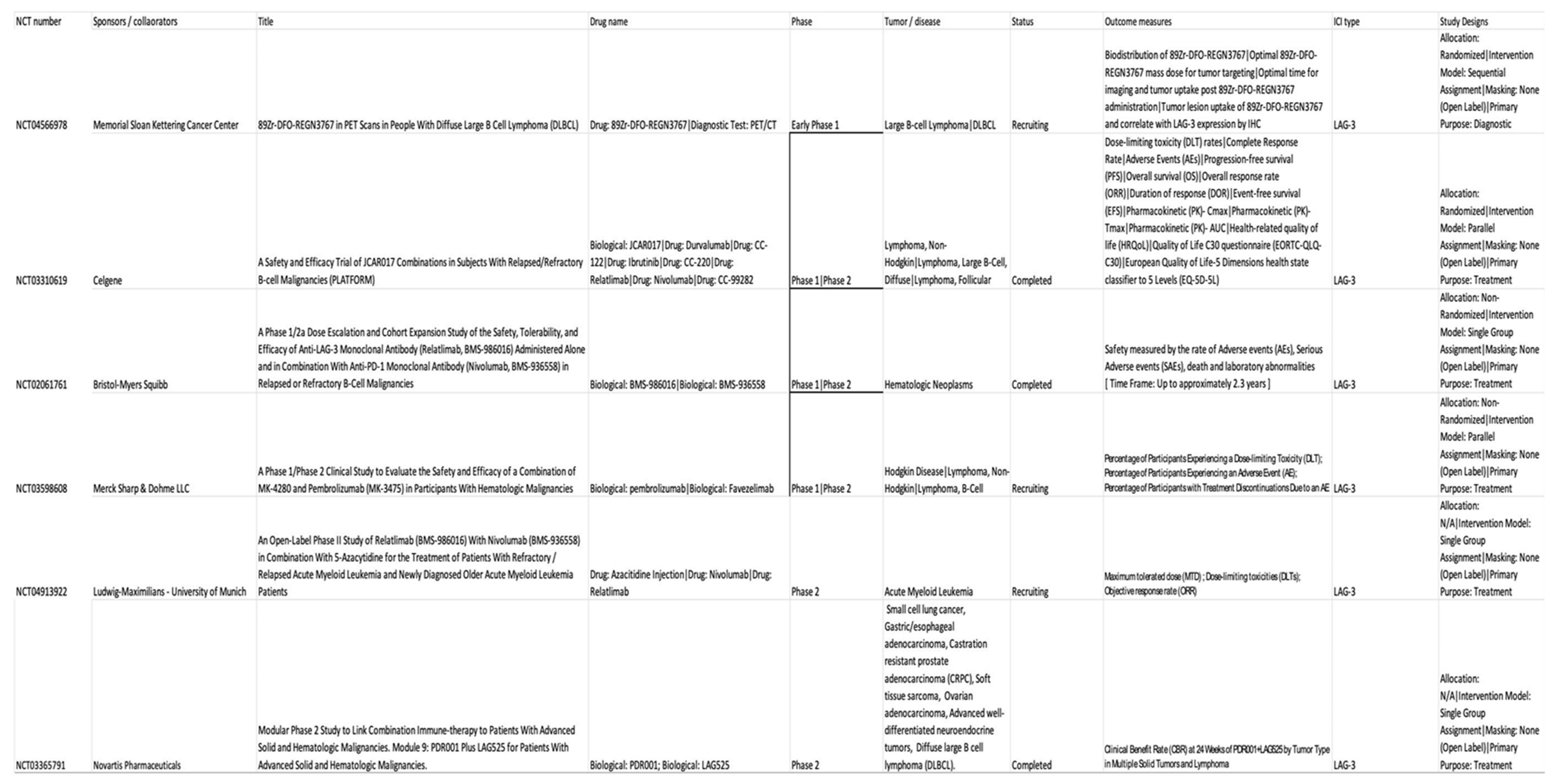
Table 2.
Summarizes the potential clinical trials of VISTA in hematological malignancies.
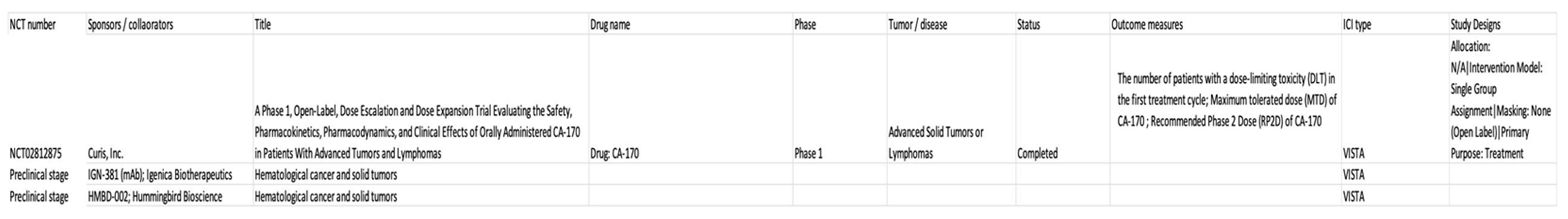
Table 3.
Shows current clinical trials of anti-TIGIT antibodies in hematological malignancies.
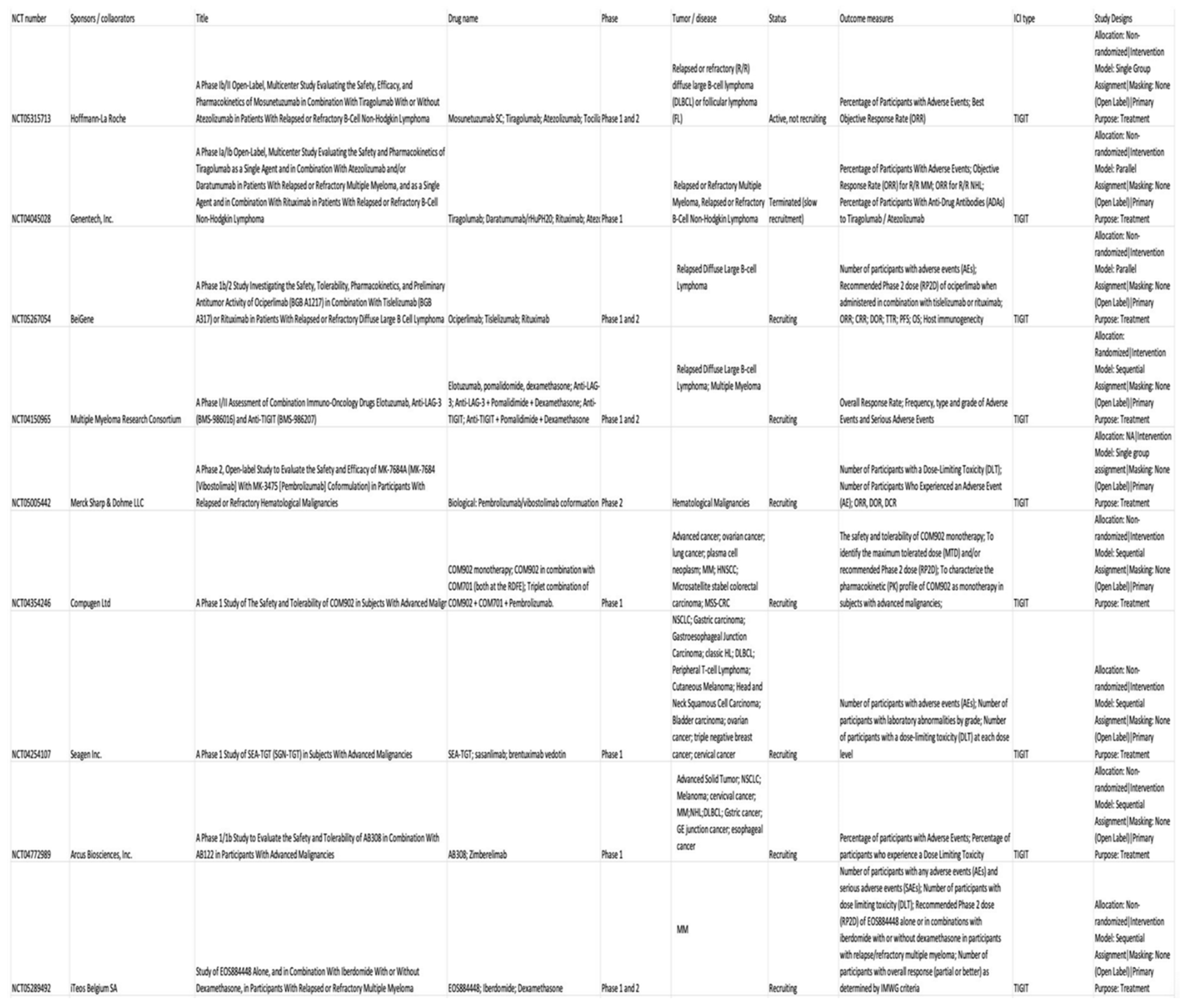
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



