
Submitted:
03 July 2023
Posted:
04 July 2023
Read the latest preprint version here
Abstract
While classical NOD-like receptor pyrin domain containing protein 1 (NLRP1) and NLRP3 inflammasomal proteins have been extensively investigated, the contribution of NLRP2 is still ill-defined in the nervous system. Given the putative significance of NLRP2 in orchestrating neuroinflammation, further inquiry is needed to gain a better understanding of its connectome, hence its specific targeting may hold a promising therapeutic implication. Therefore, bioinformatical approach for extracting information, specifically in the context of neuropathologies, is also undoubtedly preferred. To the best of our knowledge, there is no review study selectively targeting only NLRP2. Increasing, but still fragmentary evidence should encourage researchers to thoroughly investigate this inflammasome in various animal- and human models. Taken together, herein we aimed to review the current literature focusing on the role of NLRP2 inflammasome in the nervous system and more importantly, we provide an algorythm-based protein network of human NLRP2 for elucidating potentially valuable molecular partnerships.
Keywords:
inflammasome
; NLRP2
; neurological disorders
; connectome
; STRING
1. Introduction
Acute inflammation is a physiological defense mechanism in response to pathogens and cell damages; chronic inflammation however, is considered as a dysregulated maladaptive clinical phenomenon without any recuperative benefits [1]. Chronic inflammation has been associated with many neurological disorders, [2] thus studying inflammasomes is of particular importance. Classically, the currently known canonical inflammasomes (NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRP9, NLRP12, absent in melanoma 2 and pyrin inflammasomes) recruite caspase-1 enzyme, cleaving the zymogen interleukin-1β (IL-1β), IL-18 or IL-37 to induce lytic pyroptotic cell death and subsequent inflammatory downstream signaling [3,4,5,6,7]. NLRP2 (alternate names: NALP2, PYPAF2, NBS1, PAN1, CLR19.9) inflammasome is an intracellular multimer protein signaling hub assembled by three main elements: (i) a sensory component, termed NLRP, involved in the recognition of Pathogen-Associated Molecular patterns (PAMPs) and Damage-Associated Molecular Patterns (DAMPs), (ii) an adaptor unit, termed Apoptosis-associated Speck-like protein containing a caspase-activation and recruitment domain (ASC, CARD) as well as the (iii) effector caspase-1 enzyme [3,8,9,10,11]. NLRP2 activation recruites „function to find” (FIIND) and CARD-containing protein Cardinal that interacts with caspase-1 [12].
Of note, NLRP2 was earlier shown to govern inflammasome signaling by inhibiting nuclear factor kappa B (NF-κB) transcription factor [13,14]. Recent data have indicated that NLRP2 robustly increases the amount of NF-κB regulated cytokines in cystinosis [15]. Moreover, several lines of evidence support the critical regulator role of NLRP2 in the reproductive system, hence NLRP2 gene was reported as one of the mammalian maternal effect genes associated with murine embryogenesis, age-related maternal fertility and idiopathic recurrent miscarriage, respectively [16,17,18]. Besides, NLRP2 expression was also proven to associate with arsenic-induced skin lesion, chromosomal damages and respiratory disorders as well [19].
Intriguingly, despite the aforementioned past research on non-neural tissues, our gap of knowledge regarding NLRP2 and its connectome in neuropathological context has still yet to be improved (Figure 1 and Figure 2). First, the present study focused on the experiments related to NLRP2 inflammasome in human and rodent models by providing a state-of- the-art of the literature. Secondly, a publicly available cutting-edge biomedical database called Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, ver.11.5; string-db.org) [20] was employed to envisage potential protein interactions of NLRP2. Following the results of STRING analysis and Web of Science filtration we selected and described 15 proteins that are involved in neuropathologies, but not experimentally associated with NLRP2 inflammasome.
2. NLRP2 expression in the nervous system
2.1. Human NLRP2
For many years it remained elusive whether NLRP2 could be validated as a functional inflammasome in the human nervous system. The pioneering milestone was achieved by Julia Minkiewicz et al. in 2013 [7] by verifying the significance of NLRP2 inflammasome in human cortical astrocyte cultures. They proposed that NLRP2 interacted with ionotropic purinergic receptor X7 (P2X7) receptor and pannexin 1 (PNX1), hence mimicking inflammation with ATP stimulation rapidly resulted in caspase-1 activation and subsequent IL-1β release. Conversely, pharmacological blockade of astrocytic PNX1 and P2X7 receptor with probenecid and Brilliant Blue G (BBG) diminished the ATP-induced NLRP2 activation. In addition, siRNA-based silencing of NLRP2 was also found to disrupt caspase-1 activation, emphasizing its pivotal role in astrocyte immune responses. NLRP2 gene dysregulation was shown to be involved in human fetal brain development indicating a robust difference between cases of bipolar disorder and healthy individuals [21,22]. In line with this, Truvé et al. [23] in 2020 found considerable increase of NLRP2 expression in neural stem and mature cells of patients suffering from bipolar disorder.
2.2. Rodent NLRP2
To date, still more NLRP2 related experimental data are accessible from rodent models. Several rodent models are continously used in drug development pipelines, but regrettably the limitations of these concepts are major hindrances to translation attempts from bench to bedside. Few years previously Sun et al. in 2016 [24] have already determined NLRP2 distribution at specific areas of the murine central nervous system. Low constitutive NLRP2 levels were observed in the cortex, hippocampus and striatum, which were significantly elevated following occlusion of middle cerebral artery mimicking ischemic stroke. Similar results were obtained in primary astrocyte cultures upon oxygen-glucose deprivation. These findings were further corroborated by Cheon et al. in 2018 [25], who searched the interaction of apoptosis signal-regulating kinase 1 (ASK1) protein with astroglial NLRP2 inflammasome after ischemic stroke. ASK1 inhibition effectively downregulated NLRP2, as well as proinflammatory cytokine release in murine model and cultured astrocytes. In former studies, NLRP1 and NLRP3 inflammasomes were recognised in neuropathic models and spinal injury [26,27,28,29], but only scanty data were provided about NLRP2 protein following peripheral inflammation. Matsuoka and his colleagues in their study [30] characterised NLRP2 inflammasome in NeuN- and peripherin positive dorsal root ganglia (DRG) cells of C-fibers implying its significant role in nociceptive processing. They found that both siRNA gene silencing of NLRP2 expression and pharmacological blockade of caspase-1 prevented nociceptive hypersensitivity, measured in two peripheral inflammatory models using either complete Freund adjuvant (CFA)- or ceramide. Their data did not support possible activation of matrix metalloproteinase enzyme 9 or other inflammasome types except NLRP2, therefore they hypothesized that caspase-1 enzyme in DRG neurons worked presumably in NLRP2-dependent manner. Enigmatically, inhibition of caspase-1 and siRNA silencing of NLRP2 attenuated mechanical, but not thermal hypersensitivity upon inflammation. This finding can be explained by the low NLRP2 level in thermal sensor transient receptor potential vanilloid subtype-1 positive neurons.
Our workgroup also investigated the putative changes of NLRP2 expression in CFA- induced inflammatory pain model within the spinal dorsal horn [31]. Although, NLRP1, NLRP2 and NLRP3 inflammasomes have been all detected in astrocytes of different brain areas [32], our results showed that dorsal horn astrocytes mainly expressed NLRP2, and colocalisation was entirely lacking between NLRP1 and astrocytes. With protein blot analysis we also verified a robust increase of NLRP2 protein in spinal cord tissue lysates upon CFA injection. Not unexpectedly, the role of astroglial NLRP2 has also been reported in chronic mild stress induced depression [33], where they introduced an intraperitoneal injection of tryptophane derived metabolite kynurenine (Kyn), which upregulated NLRP2 in hippocampal primary astrocytes cultures. Simultaneously, Kyn treatment aided nuclear translocation of NFκB to promote NLRP2 transcription, eventually leading to inflammasome activation. These findings also illustrated that Kyn elicited depressive behaviour was eliminated following astrocytic NLRP2 knockdown, proposing a crucial role for NLRP2 in depressive behaviours.
Recently, the role of NLRP2 inflammasome has been elucidated in murine Alzheimer’s disease [34]. In this study, glucagon-like peptide-1 (GLP-1) receptor agonist exenatide attenuated neuroinflammation in piriform cortex resulting in cognitive improvement of 5x FAD transgenic mice. Additionally, upon exendine-4 treatment of cultured astrocytes, lower amyloid β1-42 level was detected confirming the reduced inflammation and oxidative stress presumably via downregulation of NLRP2 inflammasome.
3. Human NLRP2 connectome with STRING database
STRING (version 11.5; string-db.org) is a freely accessible online biological web resource. The query service of the platform was initiated with the following input: “NLRP2”, species: “Homo sapiens”. NLRP2 connnectome was generated by our parameters that were as follows: full-STRING network (containing both functional and physical interactions), evidence-based network edges (types of interactions are shown with different line colors) with a cut-off interaction confidence score: 0.4. For data visualisation all the interaction sources were collected: textmining, experiments, databases, coexpression, neighbourhood, gene fusion, cooccurence.
Based on our settings the network statistics of NLRP2 connectome involve 76 nodes (with an average node degree: 13.3), 504 edges, with a PPI enrichment p-value < 1.0e-16 (Figure 3). From the direct interactions of NLRP2 (45 nodes, Table 1), due to spatial limitations, possible functional partners with relevant role in neurological disorders (15 nodes), are introduced (in the order of their interaction score) in the Supplementary information (3.1.-3.15, Table S1). Here, we focused on those items that were either not experimentally investigated earlier with NLRP2 protein or not reviewed here.
4. Discussion and future prospects
To date, neurodegenerative disorders lack effective therapeutic agents or programs to prevent, efficiently influence or at least decelerate disease progrediation. Moreover, the rise in absolute numbers of people inflicted far and wide proposes that management of main neurological disorders is globally inadequate and these people pose a significant socioeconomic burden owing to disability, illness and premature death [191,192].
Unquestionably, inflammasome signaling is affected in several neuroinflammation-based neurodegenerative disorders including AD, PD or HD that attract more increasing attention in studies investigating their place in clinical practice. Specifically, the NLRP3 inflammasome has been meticulously investigated in this context [193,194], but recently, role of other inflammasomes as pathological drivers in brain diseases was also conceptualised by Anna Chiarini et al. [22]. This attempt markedly indicates that there is an urgent need to extend our comprehension regarding neurodegenerative mechanisms associated with inflammasomes other than NLRP3.
First, in this study we attempted to encompass the literature focusing on the expression and distribution of NLRP2 inflammasome in human and rodent neuropathological disorders (Figure 4.). Taken together, in these models major NLRP2 expression was upregulated in primary astrocyte cultures [7,24,25,33,34], as well as in astrocytes of rodent spinal dorsal horn, respectively [31]. Beyond the astrocytes the role of NLRP2 was also verified in NeuN- and peripherin positive mechanical, but not thermal sensor subsets of DRG cells by Matsuoka et al. [30]. Furthermore, tissue distribution of NLRP2 protein was observed not only in adult cortex, hippocampus, striatum or spinal cord [24,31], but in neural stem cells as well [23].
Regarding activatory stimuli, particular attention is given to purinergic signaling in human NLRP2 inflammasome activation, hence ATP is a well-known and adeptly characterised DAMP that facilitates inflammasome activation upon trauma [195]. Indeed, exogenous stimulation with ATP activates NLRP2 inflammasome resulting in caspase-1 mediated production of mature IL-1β [7]. According to all indications, ATP acts on P2X7 receptor that cooperates with PNX1, hence application of P2X7 receptor inhibitor BBG and PNX inhibitor probenecid diminishes NLRP2 activation. Nevertheless, it is important to underline that P2X4 receptor has been found to be functionally coupled with P2X7 receptor and pannexin-1 in NLRP3 inflammasome of gingival epithelial cells [196,197]. Overall, these observations are in accordance with findings reported by another group who found that heme, released following hemolysis or cell damage, activates NLRP3 inflammasome in macrophages via P2X7 and P2X4 signaling [198]. We speculate that this easily might be the case with NLRP2 inflammasome as well, however there are no experimental data that would confirm this hypothesis. In our earlier article we came up with the idea that overexpression of astroglial NLRP2 in spinal dorsal horn might be associated with their significant P2X4 upregulation as a consequence of intraplantar CFA injection [31,199]. Other triggering stimulus of NLRP2 activation is ischemic stroke, evoked experimentally by occlusion of middle cerebral artery mimicked in astrocyte cultures by oxygen-glucose deprivation [24]. Unsurprisingly, mounting evidence suggests that astrocyte-mediated inflammation may be potentially involved in the pathogenesis of mental disorders such as depression [200,201]. In their recent study, Zhang et al. [33] found that tryptophan metabolite Kyn, that was deemed as a specific biomarker of depressive behaviours, upregulated NLRP2 inflammasome in astrocytes, which was supported by other observations also reporting significantly elevated levels of proinflammatory cytokines [202]. In bipolar disorder, neuroinflammatory biomarkers of cerebrospinal fluid were found to associate with cognitive decline. Moreover, persistent cognitive impairment is increasingly recognised in people suffering from bipolar disorder, suggesting a link between neuroinflammation, neurodegenerative states and mood abnormalities [203], which may be significantly modulated by NLRP2 inflammasome [21,22,23].
Secondly, the present study collects the connectome of NLRP2, envisaged with STRING platform. The obtained set of proteins were further filtered to 15 possible protein interactors involved in the disorders of the nervous system; their pathophysiological roles are discussed in details in Supplementary Information, Table S1. Many of these potential partnerships have not been earlier experimentally investigated. Beside, signaling of NLRP2 inflammasome also lacking from the collection of the Kyoto Encyclopedia of Genes and Genomes (KEGG) database Figure S1.
Concluding the revised literature and STRING based matches we hypothesize that the filtered NLRP2 connectome influences a plethora of molecular processes in neurons, such as glutamatergic excitotoxicity, apoptosis/ survival signaling and neuroinflammation.
Glutamatergic excitotoxicity - The integrity of glutamatergic signaling is essential for preserving neuronal homeostasis and evade neurodegeneration. SLA1 prevents glutamatergic excitotoxicity by degrading excessive NMDA receptors [35]. As a synergist, apelin also disrupts NMDA receptor-mediated excitotoxicity in rat hippocampus through survival kinases AKT and Raf/ERK-1/2 [143]. Furthermore, the role of DLGAPs is to anchor glutamate receptors in the postsynaptic membrane and to link them with other proteins including other glutamate receptors, signaling- and cytoskeletal factors while regulating both ionotropic and metabotropic glutamate receptors via synaptic scaling [119,120,121,122]. Low DLGAP2 expression was detected in age-related cognitive decline and AD [131], which may relate to other findings, such as low spine density or reduced excitatory postsynaptic current, revealed in orbitofrontal cortex of DLGAP2 knockout mice [126,128]. Directed expression bHLH transcription factor NEUROD1 stimulates neuronal maturation and integration as well as ameliorates deficits of dendritic spine density in hippocampal neurons of APPxPS1 model of AD [95]. Besides, ENT1 inhibitor J4 mitigates the damages of long-term potentiation, excitatory synaptic expression as well as GSK-3ß or PKA signaling of neuronal plasticity and alleviate memory deficits in APP/PS1 model of AD [165].
Apoptosis/survival signaling- It is of particular importance to gain a better understanding on the signaling of neuronal apoptosis/survival in neurodegenerative states. Impairment of autophagy via reduction of BECN1 level results in overproduction of microglial IL-1β and IL-18 proinflammatory cytokines in AD patients. Deficits of BECN1 mediated phagocytosis causes dysfunctional recruitment of phagocytic receptors CD36 and Trem2 that may associate with extracellular accumulation of Aβ plaques and other cellular debris. Recently, in early phase of HD BECN1 administration has successfully cleared mutant HTT accumulation and reversed progrediation [42,43,44,45,46]. Apelin exerts positive impacts on redox homeostasis and prevents mitochondrial cytochrome c release and caspase-3 activation in cultured murine cortical neurons [142]. Furthermore, EPS15 participates in the EGF-AKT pathway mediated pro-survival cell signaling reduced in dopaminergic neurodegeneration [82,83]. CCDC50 autophagy receptor inhibits inflammatory responses by disrupting NLRP3 [186,187] and prevents EGFR downregulation as well as regulate NF-κB, Fas and interferon signaling. As exposure, in contrast, triggers apoptosis of cerebellar neurons via activation of JNK and p38MAPK signaling [171] as well as upregulates Bax and decreases Bcl-2 factor [181]. A rescue factor SUGT1, functioning as a chaperone protein/heat shock protein, was also recognised to counteract pathological aggregation of α-synuclein and neurotoxicity in PD. SUGT1 mRNA was highly elevated in fronto-temporal cortex in human PD [74,75]. Its significance has been earlier described in AD, hence decreased SUGT1 immunopositivity was found in degenerating neurons of AD patients [76].
Dysregulation of aforementioned GSK-3 signaling, inhibited by PDK1/AKT kinase [204,205], contributes to the hyperphosphorylation of tau protein as well as Aβ-induced cell death in AD pathogenesis. Abundant GSK-3 level was found in postmortem brain of AD patients, moreover large body of evidence support that GSK-3 activator lysophosphatidic acid associates with AD biomarkers Aß, total tau and phospho-tau [56,58]. In contrast, GSK-3 inhibits ER stress sensor γ-TXLN, promoting apoptosis- and autophagy emphasized by the fact that blockade of γ-TXLN alone may lead to tau hyperphosphorylation in AD [116].
Neuroinflammation- Several key mechanims have been identified in neurodegenerative diseases as summarized above, also including neuroinflammation, which evokes a great challenge to clinical practice. Neuroinflammation has been recognised in dementia and is typically linked to cognitive decline with elevated levels of proinflammatory markers (IL-1, IL-6, IL-8, C-reactive protein) in patients suffering from dementia [206,207,208]. However, recent data suggest that inflammatory proteins may express both pro- and anti-inflammatory actions making the interpretation even more difficult in complex neurodegenerative states [209]. In fact, the versatility of postmortem samples as well as challenges of appropiate resolution in detection of cytokines taken from patient samples may all lead to controversial conclusions [210,211]. Indeed, neuroinflammation is still regarded as one of the crucial molecular processes of dementia, but we still do not know whether this mechanism is consequential or causative, in regards of neurodegenerative progrediation. Of note, it receives growing evidence that neuroinflammation may appear as an early temporal red flag event that may link to other mechanisms in contributing to neuropathologies [212,213,214].
In fact, neuroinflammation and generally the therapeutic potential of targeting inflammasomes has been increasingly recognised in neurodegenerative conditions since 2013, when Heneka et al. [215] proved the significance of NLRP3 inflammasome with nlrp3 -/- mice in AD. Since then many efforts have been made to seek selective and potent NLRP3 inhibitors, because the currently US Food and Drug Administration (FDA) approved inhibitors of multiple inflammatory diseases include only canakinumab, anakinra and rilonacept. Nevertheless, these inhibitors do not cross efficiently the blood-brain barrier and lack proper pharmacokinetic properties [216,217].
A great achievement was reached when a potent diarylsulfonylurea compound MCC950 (also termed as CRID3, CP-456773), endowed with NLRP3 selectivity, showed therapeutic improvement in several preclinical models such as experimental autoimmune encephalomyelitis, AD and PD, respectively [218,219,220,221]. In the search for new inhibitors Stavudine (d4T), acting as an inhibitor of nucleoside reverse transcriptase, has been published recently to downregulate NLRP3 activation in AD also suppressing caspase-1 activity [222]. Furthermore, Gastaldi et al. [223] by applying the pharmacophore-hybridization method synthesised and screened several benzo[d]imidazole-2-one derivatives to test their inhibitory effect on NLRP3 evoked pyroptosis and IL-1β production.
However, it is increasingly becoming clear that NLRP3 is not the only inflammasome involved in neurodegenerative states. Notably, Kaushal et al. [224] has already reported elevated mRNA level of NLRP1 and highlighted the causative role of NLRP1-caspase 1-caspase-6 signaling in the accumulation of Aβ42 deposits in AD.
In contrast, as NLRP3 and NLRP1 “steal” the show from NLRP2 although it has already been justified in human neuropathologies, till today no effective and specific pharmacological blockers have been designed. Hopefully, in the coming years focus will be also placed on the development of NLRP2 inhibitors by the representatives of industry and academia.
5. Conclusions
Taken together, NLRP2 inflammasome based on STRING analysis may cooperate with at least 15 proteins earlier described in neurological disorders, which are associated with the derangement of excitoxicity, celllular apoptosis/survival and neuroinflammation. Thus, the context of this filtered connectome in the nervous system requires an experimental approach both in rodent and human models. In the future, complete understanding of inflammasome signaling including NLRP2 mediated processes may open up new prospects for efficient inhibitors and/or therapeutic agents, which are currently lacking from our armamentarium.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization: LD Manuscript writing, data visualisation, editing and supervision: LD, BG. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by the Department of Anatomy, Histology and Embryology, University of Debrecen, Hungary and received no other external funding.
Data Availability Statement
The data sets and materials supporting the conclusions of this study are included within the article.
Acknowledgement
Not applicable
Conflicts of Interest
The authors declare no competing interests.
Ethical Approval
Not applicable.
References
- Yi Y. S. (2020). Functional crosstalk between non-canonical caspase-11 and canonical NLRP3 inflammasomes during infection-mediated inflammation. Immunology, 159(2), 142–155. [CrossRef]
- Singh, N., Baby, D., Rajguru, J. P., Patil, P. B., Thakkannavar, S. S., & Pujari, V. B. (2019). Inflammation and cancer. Annals of African medicine, 18(3), 121–126. [CrossRef]
- Schroder, K., & Tschopp, J. (2010). The inflammasomes. Cell, 140(6), 821–832. [CrossRef]
- Ross, C., Chan, A. H., von Pein, J. B., Maddugoda, M. P., Boucher, D., & Schroder, K. (2022). Inflammatory Caspases: Toward a Unified Model for Caspase Activation by Inflammasomes. Annual review of immunology, 40, 249–269. [CrossRef]
- Kelley, N., Jeltema, D., Duan, Y., & He, Y. (2019). The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. International journal of molecular sciences, 20(13), 3328. [CrossRef]
- Akbal, A., Dernst, A., Lovotti, M., Mangan, M. S. J., McManus, R. M., & Latz, E. (2022). How location and cellular signaling combine to activate the NLRP3 inflammasome. Cellular & molecular immunology, 19(11), 1201–1214. [CrossRef]
- Minkiewicz, J., de Rivero Vaccari, J. P., & Keane, R. W. (2013). Human astrocytes express a novel NLRP2 inflammasome. Glia, 61(7), 1113–1121. [CrossRef]
- 8de Rivero Vaccari, J. P., Dietrich, W. D., & Keane, R. W. (2014). Activation and regulation of cellular inflammasomes: gaps in our knowledge for central nervous system injury. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, 34(3), 369–375. [CrossRef]
- Martinon, F., Burns, K., & Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell, 10(2), 417–426.
- Xue, Y., Enosi Tuipulotu, D., Tan, W. H., Kay, C., & Man, S. M. (2019). Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends in immunology, 40(11), 1035–1052. [CrossRef]
- Sonnessa, M., Cioffi, A., Brunetti, O., Silvestris, N., Zito, F. A., Saponaro, C., & Mangia, A. (2020). NLRP3 Inflammasome From Bench to Bedside: New Perspectives for Triple Negative Breast Cancer. Frontiers in oncology, 10, 1587. [CrossRef]
- Khare, S., Luc, N., Dorfleutner, A., & Stehlik, C. (2010). Inflammasomes and their activation. Critical reviews in immunology, 30(5), 463–487. [CrossRef]
- Bruey, J. M., Bruey-Sedano, N., Newman, R., Chandler, S., Stehlik, C., & Reed, J. C. (2004). PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. The Journal of biological chemistry, 279(50), 51897–51907. [CrossRef]
- Fontalba, A., Gutierrez, O., & Fernandez-Luna, J. L. (2007). NLRP2, an inhibitor of the NF-kappaB pathway, is transcriptionally activated by NF-kappaB and exhibits a nonfunctional allelic variant. Journal of immunology (Baltimore, Md. : 1950), 179(12), 8519–8524. [CrossRef]
- Rossi, M. N., Pascarella, A., Licursi, V., Caiello, I., Taranta, A., Rega, L. R., Levtchenko, E., Emma, F., De Benedetti, F., & Prencipe, G. (2019). NLRP2 Regulates Proinflammatory and Antiapoptotic Responses in Proximal Tubular Epithelial Cells. Frontiers in cell and developmental biology, 7, 252. [CrossRef]
- Peng, H., Chang, B., Lu, C., Su, J., Wu, Y., Lv, P., Wang, Y., Liu, J., Zhang, B., Quan, F., Guo, Z., & Zhang, Y. (2012). Nlrp2, a maternal effect gene required for early embryonic development in the mouse. PloS one, 7(1), e30344. [CrossRef]
- Kuchmiy, A. A., D'Hont, J., Hochepied, T., & Lamkanfi, M. (2016). NLRP2 controls age-associated maternal fertility. The Journal of experimental medicine, 213(13), 2851–2860. [CrossRef]
- Huang, J. Y., Su, M., Lin, S. H., & Kuo, P. L. (2013). A genetic association study of NLRP2 and NLRP7 genes in idiopathic recurrent miscarriage. Human reproduction (Oxford, England), 28(4), 1127–1134. [CrossRef]
- Bhattacharjee, P., Das, N., Chatterjee, D., Banerjee, A., Das, J. K., Basu, S., Banerjee, S., Majumder, P., Goswami, P., & Giri, A. K. (2013). Association of NALP2 polymorphism with arsenic induced skin lesions and other health effects. Mutation research, 755(1), 1–5. [CrossRef]
- Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., Simonovic, M., Doncheva, N. T., Morris, J. H., Bork, P., Jensen, L. J., & Mering, C. V. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic acids research, 47(D1), D607–D613. [CrossRef]
- Vizlin-Hodzic, D., Zhai, Q., Illes, S., Södersten, K., Truvé, K., Parris, T. Z., Sobhan, P. K., Salmela, S., Kosalai, S. T., Kanduri, C., Strandberg, J., Seth, H., Bontell, T. O., Hanse, E., Ågren, H., & Funa, K. (2017). Early onset of inflammation during ontogeny of bipolar disorder: the NLRP2 inflammasome gene distinctly differentiates between patients and healthy controls in the transition between iPS cell and neural stem cell stages. Translational psychiatry, 7(1), e1010. [CrossRef]
- Chiarini, A., Armato, U., Gui, L., & Dal Prà, I. (2022). "Other Than NLRP3" Inflammasomes: Multiple Roles in Brain Disease. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry, 10738584221106114. Advance online publication. [CrossRef]
- Truvé, K., Parris, T. Z., Vizlin-Hodzic, D., Salmela, S., Berger, E., Ågren, H., & Funa, K. (2020). Identification of candidate genetic variants and altered protein expression in neural stem and mature neural cells support altered microtubule function to be an essential component in bipolar disorder. Translational psychiatry, 10(1), 390. [CrossRef]
- Sun, X., Song, X., Zhang, L., Sun, J., Wei, X., Meng, L., & An, J. (2016). NLRP2 is highly expressed in a mouse model of ischemic stroke. Biochemical and biophysical research communications, 479(4), 656–662. [CrossRef]
- Cheon, S. Y., Kim, E. J., Kim, S. Y., Kim, J. M., Kam, E. H., Park, J. K., & Koo, B. N. (2018). Apoptosis Signal-regulating Kinase 1 Silencing on Astroglial Inflammasomes in an Experimental Model of Ischemic Stroke. Neuroscience, 390, 218–230. [CrossRef]
- Li, Q., Tian, Y., Wang, Z. F., Liu, S. B., Mi, W. L., Ma, H. J., Wu, G. C., Wang, J., Yu, J., & Wang, Y. Q. (2013). Involvement of the spinal NALP1 inflammasome in neuropathic pain and aspirin-triggered-15-epi-lipoxin A4 induced analgesia. Neuroscience, 254, 230–240. [CrossRef]
- Liu, S., Li, Q., Zhang, M. T., Mao-Ying, Q. L., Hu, L. Y., Wu, G. C., Mi, W. L., & Wang, Y. Q. (2016). Curcumin ameliorates neuropathic pain by down-regulating spinal IL-1β via suppressing astroglial NALP1 inflammasome and JAK2-STAT3 signalling. Scientific reports, 6, 28956. [CrossRef]
- He, W., Long, T., Pan, Q., Zhang, S., Zhang, Y., Zhang, D., Qin, G., Chen, L., & Zhou, J. (2019). Microglial NLRP3 inflammasome activation mediates IL-1β release and contributes to central sensitization in a recurrent nitroglycerin-induced migraine model. Journal of neuroinflammation, 16(1), 78. [CrossRef]
- Grace, P. M., Strand, K. A., Galer, E. L., Urban, D. J., Wang, X., Baratta, M. V., Fabisiak, T. J., Anderson, N. D., Cheng, K., Greene, L. I., Berkelhammer, D., Zhang, Y., Ellis, A. L., Yin, H. H., Campeau, S., Rice, K. C., Roth, B. L., Maier, S. F., & Watkins, L. R. (2016). Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proceedings of the National Academy of Sciences of the United States of America, 113(24), E3441–E3450. [CrossRef]
- Matsuoka, Y., Yamashita, A., Matsuda, M., Kawai, K., Sawa, T., & Amaya, F. (2019). NLRP2 inflammasome in dorsal root ganglion as a novel molecular platform that produces inflammatory pain hypersensitivity. Pain, 160(9), 2149–2160. [CrossRef]
- Ducza, L., Szücs, P., Hegedűs, K., Bakk, E., Gajtkó, A., Wéber, I., & Holló, K. (2021). NLRP2 Is Overexpressed in Spinal Astrocytes at the Peak of Mechanical Pain Sensitivity during Complete Freund Adjuvant-Induced Persistent Pain. International journal of molecular sciences, 22(21), 11408. [CrossRef]
- Downer, E. J., Johnston, D. G., & Lynch, M. A. (2013). Differential role of Dok1 and Dok2 in TLR2-induced inflammatory signaling in glia. Molecular and cellular neurosciences, 56, 148–158. [CrossRef]
- Zhang, Q., Sun, Y., He, Z., Xu, Y., Li, X., Ding, J., Lu, M., & Hu, G. (2020). Kynurenine regulates NLRP2 inflammasome in astrocytes and its implications in depression. Brain, behavior, and immunity, 88, 471–481. [CrossRef]
- Zhang, M., Wu, Y., Gao, R., Chen, X., Chen, R., & Chen, Z. (2022). Glucagon-like peptide-1 analogs mitigate neuroinflammation in Alzheimer's disease by suppressing NLRP2 activation in astrocytes. Molecular and cellular endocrinology, 542, 111529. [CrossRef]
- Marton, N., Baricza, E., Érsek, B., Buzás, E. I., & Nagy, G. (2015). The Emerging and Diverse Roles of Src-Like Adaptor Proteins in Health and Disease. Mediators of inflammation, 2015, 952536. [CrossRef]
- Ge, M. M., Zhou, Y. Q., Tian, X. B., Manyande, A., Tian, Y. K., Ye, D. W., & Yang, H. (2020). Src-family protein tyrosine kinases: A promising target for treating chronic pain. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie, 125, 110017. [CrossRef]
- Yang, H., Wang, L., Zang, C., Wang, Y., Shang, J., Zhang, Z., Liu, H., Bao, X., Wang, X., & Zhang, D. (2020). Src Inhibition Attenuates Neuroinflammation and Protects Dopaminergic Neurons in Parkinson's Disease Models. Frontiers in neuroscience, 14, 45. [CrossRef]
- Giraud, F., Pereira, E., Anizon, F., & Moreau, P. (2021). Recent Advances in Pain Management: Relevant Protein Kinases and Their Inhibitors. Molecules (Basel, Switzerland), 26(9), 2696. [CrossRef]
- Tran, S., Fairlie, W. D., & Lee, E. F. (2021). BECLIN1: Protein Structure, Function and Regulation. Cells, 10(6), 1522. [CrossRef]
- Kang, R., Zeh, H. J., Lotze, M. T., & Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell death and differentiation, 18(4), 571–580. [CrossRef]
- Zhang, D., Wang, W., Sun, X., Xu, D., Wang, C., Zhang, Q., Wang, H., Luo, W., Chen, Y., Chen, H., & Liu, Z. (2016). AMPK regulates autophagy by phosphorylating BECN1 at threonine 388. Autophagy, 12(9), 1447–1459. [CrossRef]
- Lucin, K. M., O'Brien, C. E., Bieri, G., Czirr, E., Mosher, K. I., Abbey, R. J., Mastroeni, D. F., Rogers, J., Spencer, B., Masliah, E., & Wyss-Coray, T. (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron, 79(5),873–886. [CrossRef]
- Heppner, F. L., Ransohoff, R. M., & Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nature reviews. Neuroscience, 16(6), 358–372. [CrossRef]
- Houtman, J., Freitag, K., Gimber, N., Schmoranzer, J., Heppner, F. L., & Jendrach, M. (2019). Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. The EMBO journal, 38(4), e99430. [CrossRef]
- Lucin, K. M., O'Brien, C. E., Bieri, G., Czirr, E., Mosher, K. I., Abbey, R. J., Mastroeni, D. F., Rogers, J., Spencer, B., Masliah, E., & Wyss-Coray, T. (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron, 79(5), 873–886. [CrossRef]
- Brattås, P. L., Hersbach, B. A., Madsen, S., Petri, R., Jakobsson, J., & Pircs, K. (2021). Impact of differential and time-dependent autophagy activation on therapeutic efficacy in a model of Huntington disease. Autophagy, 17(6), 1316–1329. [CrossRef]
- Peek, S. L., Mah, K. M., & Weiner, J. A. (2017). Regulation of neural circuit formation by protocadherins. Cellular and molecular life sciences : CMLS, 74(22), 4133–4157. [CrossRef]
- Langfelder, P., Cantle, J. P., Chatzopoulou, D., Wang, N., Gao, F., Al-Ramahi, I., Lu, X. H., Ramos, E. M., El-Zein, K., Zhao, Y., Deverasetty, S., Tebbe, A., Schaab, C., Lavery, D. J., Howland, D., Kwak, S., Botas, J., Aaronson, J. S., Rosinski, J., Coppola, G., … Yang, X. W. (2016). Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nature neuroscience, 19(4), 623–633. [CrossRef]
- Carrasquillo, M. M., Zou, F., Pankratz, V. S., Wilcox, S. L., Ma, L., Walker, L. P., Younkin, S. G., Younkin, C. S., Younkin, L. H., Bisceglio, G. D., Ertekin-Taner, N., Crook, J. E., Dickson, D. W., Petersen, R. C., Graff-Radford, N. R., & Younkin, S. G. (2009). Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nature genetics, 41(2), 192–198. [CrossRef]
- Beecham, G. W., Naj, A. C., Gilbert, J. R., Haines, J. L., Buxbaum, J. D., & Pericak-Vance, M. A. (2010). PCDH11X variation is not associated with late-onset Alzheimer disease susceptibility. Psychiatric genetics, 20(6), 321–324. [CrossRef]
- Miar, A., Alvarez, V., Corao, A. I., Alonso, B., Díaz, M., Menéndez, M., Martínez, C., Calatayud, M., Morís, G., & Coto, E. (2011). Lack of association between protocadherin 11-X/Y (PCDH11X and PCDH11Y) polymorphisms and late onset Alzheimer's disease. Brain research, 1383, 252–256. [CrossRef]
- Marr, L., Biswas, D., Daly, L. A., Browning, C., Vial, S. C. M., Maskell, D. P., Hudson, C., Bertrand, J. A., Pollard, J., Ranson, N. A., Khatter, H., Eyers, C. E., Sakamoto, K., & Zeqiraj, E. (2022). Mechanism of glycogen synthase inactivation and interaction with glycogenin. Nature communications, 13(1), 3372. [CrossRef]
- Fastman, N. M., Liu, Y., Ramanan, V., Merritt, H., Ambing, E., DePaoli-Roach, A. A., Roach, P. J., Hurley, T. D., Mellem, K. T., Ullman, J. C., Green, E., Morgans, D., Jr, & Tzitzilonis, C. (2022). The structural mechanism of human glycogen synthesis by the GYS1-GYG1 complex. Cell reports, 40(1), 111041. [CrossRef]
- Imagawa, E., Osaka, H., Yamashita, A., Shiina, M., Takahashi, E., Sugie, H., Nakashima, M., Tsurusaki, Y., Saitsu, H., Ogata, K., Matsumoto, N., & Miyake, N. (2014). A hemizygous GYG2 mutation and Leigh syndrome: a possible link?. Human genetics, 133(2), 225–234. [CrossRef]
- Duran, J., Hervera, A., Markussen, K. H., Varea, O., López-Soldado, I., Sun, R. C., Del Río, J. A., Gentry, M. S., & Guinovart, J. J. (2021). Astrocytic glycogen accumulation drives the pathophysiology of neurodegeneration in Lafora disease. Brain : a journal of neurology, 144(8), 2349–2360. [CrossRef]
- Sayas, C. L., & Ávila, J. (2021). GSK-3 and Tau: A Key Duet in Alzheimer's Disease. Cells, 10(4), 721. [CrossRef]
- Leroy, K., Yilmaz, Z., & Brion, J. P. (2007). Increased level of active GSK-3beta in Alzheimer's disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathology and applied neurobiology, 33(1), 43–55. [CrossRef]
- Ahmad, S., Orellana, A., Kohler, I., Frölich, L., de Rojas, I., Gil, S., Boada, M., Hernández, I., Hausner, L., Bakker, M. H. M., Cabrera-Socorro, A., Amin, N., Ramírez, A., Ruiz, A., Hankemeier, T., & Van Duijn, C. M. (2020). Association of lysophosphatidic acids with cerebrospinal fluid biomarkers and progression to Alzheimer's disease. Alzheimer's research & therapy, 12(1), 124. [CrossRef]
- Madabhushi, R., Pan, L., & Tsai, L. H. (2014). DNA damage and its links to neurodegeneration. Neuron, 83(2), 266–282. [CrossRef]
- Kannan, A., Bhatia, K., Branzei, D., & Gangwani, L. (2018). Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic acids research, 46(16), 8326–8346. [CrossRef]
- Suberbielle, E., Sanchez, P. E., Kravitz, A. V., Wang, X., Ho, K., Eilertson, K., Devidze, N., Kreitzer, A. C., & Mucke, L. (2013). Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nature neuroscience, 16(5), 613–621. [CrossRef]
- Thadathil, N., Hori, R., Xiao, J., & Khan, M. M. (2019). DNA double-strand breaks: a potential therapeutic target for neurodegenerative diseases. Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology, 27(4), 345–364. [CrossRef]
- Lu, R., Zhang, H., Jiang, Y. N., Wang, Z. Q., Sun, L., & Zhou, Z. W. (2021). Post-Translational Modification of MRE11: Its Implication in DDR and Diseases. Genes, 12(8), 1158. [CrossRef]
- Miyamoto, R., Morino, H., Yoshizawa, A., Miyazaki, Y., Maruyama, H., Murakami, N., Fukada, K., Izumi, Y., Matsuura, S., Kaji, R., & Kawakami, H. (2014). Exome sequencing reveals a novel MRE11 mutation in a patient with progressive myoclonic ataxia. Journal of the neurological sciences, 337(1-2), 219–223. [CrossRef]
- Matsumoto, Y., Miyamoto, T., Sakamoto, H., Izumi, H., Nakazawa, Y., Ogi, T., Tahara, H., Oku, S., Hiramoto, A., Shiiki, T., Fujisawa, Y., Ohashi, H., Sakemi, Y., & Matsuura, S. (2011). Two unrelated patients with MRE11A mutations and Nijmegen breakage syndrome-like severe microcephaly. DNA repair, 10(3), 314–321. [CrossRef]
- Sedghi, M., Salari, M., Moslemi, A. R., Kariminejad, A., Davis, M., Goullée, H., Olsson, B., Laing, N., & Tajsharghi, H. (2018). Ataxia-telangiectasia-like disorder in a family deficient for MRE11A, caused by a MRE11 variant. Neurology. Genetics, 4(6), e295. [CrossRef]
- Ding, M., Qing, X., Zhang, G., Baade-Büttner, C., Gruber, R., Lu, H., Ferguson, D. O., Geis, C., Wang, Z. Q., & Zhou, Z. W. (2022). The Essential DNA Damage Response Complex MRN Is Dispensable for the Survival and Function of Purkinje Neurons. Frontiers in aging neuroscience, 13, 786199. [CrossRef]
- Jacobsen, E., Beach, T., Shen, Y., Li, R., & Chang, Y. (2004). Deficiency of the Mre11 DNA repair complex in Alzheimer's disease brains. Brain research. Molecular brain research, 128(1), 1–7. [CrossRef]
- Kitagawa, K., Skowyra, D., Elledge, S. J., Harper, J. W., & Hieter, P. (1999). SGT1 encodes an essential component of the yeast kinetochore assembly pathway and a novel subunit of the SCF ubiquitin ligase complex. Molecular cell, 4(1), 21–33. [CrossRef]
- Willhoft, O., Kerr, R., Patel, D., Zhang, W., Al-Jassar, C., Daviter, T., Millson, S. H., Thalassinos, K., & Vaughan, C. K. (2017). The crystal structure of the Sgt1-Skp1 complex: the link between Hsp90 and both SCF E3 ubiquitin ligases and kinetochores. Scientific reports, 7, 41626. [CrossRef]
- Lee, Y. T., Jacob, J., Michowski, W., Nowotny, M., Kuznicki, J., & Chazin, W. J. (2004). Human Sgt1 binds HSP90 through the CHORD-Sgt1 domain and not the tetratricopeptide repeat domain. The Journal of biological chemistry, 279(16), 16511–16517. [CrossRef]
- Eisele, F., Eisele-Bürger, A. M., Hao, X., Berglund, L. L., Höög, J. L., Liu, B., & Nyström, T. (2021). An Hsp90 co-chaperone links protein folding and degradation and is part of a conserved protein quality control. Cell reports, 35(13), 109328. [CrossRef]
- da Silva Correia, J., Miranda, Y., Leonard, N., & Ulevitch, R. (2007). SGT1 is essential for Nod1 activation. Proceedings of the National Academy of Sciences of the United States of America, 104(16), 6764–6769. [CrossRef]
- Bohush, A., Góral, A., Sierant, M., Nawrot, B., Leśniak, W., & Filipek, A. (2021). Sgt1 Regulates α-Synuclein Subcellular Localization and Expression of Parkinson's Disease Related Genes, PINK1 and PARK9. Biomolecules, 11(11), 1675. [CrossRef]
- Bohush, A., Niewiadomska, G., Weis, S., & Filipek, A. (2019). HSP90 and Its Novel Co-Chaperones, SGT1 and CHP-1, in Brain of Patients with Parkinson's Disease and Dementia with Lewy Bodies. Journal of Parkinson's disease, 9(1), 97–107. [CrossRef]
- Spiechowicz, M., Bernstein, H. G., Dobrowolny, H., Leśniak, W., Mawrin, C., Bogerts, B., Kuźnicki, J., & Filipek, A. (2006). Density of Sgt1-immunopositive neurons is decreased in the cerebral cortex of Alzheimer's disease brain. Neurochemistry international, 49(5), 487–493. [CrossRef]
- Fazioli, F., Minichiello, L., Matoskova, B., Wong, W. T., & Di Fiore, P. P. (1993). eps15, a novel tyrosine kinase substrate, exhibits transforming activity. Molecular and cellular biology, 13(9), 5814–5828. [CrossRef]
- Benmerah, A., Gagnon, J., Bègue, B., Mégarbané, B., Dautry-Varsat, A., & Cerf-Bensussan, N. (1995). The tyrosine kinase substrate eps15 is constitutively associated with the plasma membrane adaptor AP-2. The Journal of cell biology, 131(6 Pt 2), 1831–1838. [CrossRef]
- Carbone, R., Fré, S., Iannolo, G., Belleudi, F., Mancini, P., Pelicci, P. G., Torrisi, M. R., & Di Fiore, P. P. (1997). eps15 and eps15R are essential components of the endocytic pathway. Cancer research, 57(24), 5498–5504.
- Huang, F., Khvorova, A., Marshall, W., & Sorkin, A. (2004). Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. The Journal of biological chemistry, 279(16), 16657–16661. [CrossRef]
- van Bergen En Henegouwen P. M. (2009). Eps15: a multifunctional adaptor protein regulating intracellular trafficking. Cell communication and signaling : CCS, 7, 24. [CrossRef]
- Iwakura, Y., Piao, Y. S., Mizuno, M., Takei, N., Kakita, A., Takahashi, H., & Nawa, H. (2005). Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson's disease and its model: neurotrophic implication in nigrostriatal neurons. Journal of neurochemistry, 93(4), 974–983. [CrossRef]
- Atkin, G., & Paulson, H. (2014). Ubiquitin pathways in neurodegenerative disease. Frontiers in molecular neuroscience, 7, 63. [CrossRef]
- Conway, J. A., Kinsman, G., & Kramer, E. R. (2022). The Role of NEDD4 E3 Ubiquitin-Protein Ligases in Parkinson's Disease. Genes, 13(3), 513. [CrossRef]
- Dokucu, M. E., Zipursky, S. L., & Cagan, R. L. (1996). Atonal, rough and the resolution of proneural clusters in the developing Drosophila retina. Development (Cambridge, England), 122(12), 4139–4147. [CrossRef]
- Sommer, L., Ma, Q92., & Anderson, D. J. (1996). neurogenins, a novel family of atonal-related bHLH transcription factors, are putative mammalian neuronal determination genes that reveal progenitor cell heterogeneity in the developing CNS and PNS. Molecular and cellular neurosciences, 8(4), 221–241. [CrossRef]
- Bertrand, N., Castro, D. S., & Guillemot, F. (2002). Proneural genes and the specification of neural cell types. Nature reviews. Neuroscience, 3(7), 517–530. [CrossRef]
- Tutukova, S., Tarabykin, V., & Hernandez-Miranda, L. R. (2021). The Role of Neurod Genes in Brain Development, Function, and Disease. Frontiers in molecular neuroscience, 14, 662774. [CrossRef]
- Schwab, M. H., Druffel-Augustin, S., Gass, P., Jung, M., Klugmann, M., Bartholomae, A., Rossner, M. J., & Nave, K. A. (1998). Neuronal basic helix-loop-helix proteins (NEX, neuroD, NDRF): spatiotemporal expression and targeted disruption of the NEX gene in transgenic mice. The Journal of neuroscience : the official journal of the Society for Neuroscience, 18(4), 1408–1418. [CrossRef]
- Boulle, F., Massart, R., Stragier, E., Païzanis, E., Zaidan, L., Marday, S., Gabriel, C., Mocaer, E., Mongeau, R., & Lanfumey, L. (2014). Hippocampal and behavioral dysfunctions in a mouse model of environmental stress: normalization by agomelatine. Translational psychiatry, 4(11), e485. [CrossRef]
- Rubio-Cabezas, O., Minton, J. A., Kantor, I., Williams, D., Ellard, S., & Hattersley, A. T. (2010). Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes, 59(9), 2326–2331. [CrossRef]
- Liu, M. H., Li, W., Zheng, J. J., Xu, Y. G., He, Q., & Chen, G. (2020). Differential neuronal reprogramming induced by NeuroD1 from astrocytes in grey matter versus white matter. Neural regeneration research, 15(2), 342–351. [CrossRef]
- Fedele, V., Roybon, L., Nordström, U., Li, J. Y., & Brundin, P. (2011). Neurogenesis in the R6/2 mouse model of Huntington's disease is impaired at the level of NeuroD1. Neuroscience, 173, 76–81. [CrossRef]
- Satoh, J., Yamamoto, Y., Asahina, N., Kitano, S., & Kino, Y. (2014). RNA-Seq data mining: downregulation of NeuroD6 serves as a possible biomarker for alzheimer's disease brains. Disease markers, 2014, 123165. [CrossRef]
- Richetin, K., Leclerc, C., Toni, N., Gallopin, T., Pech, S., Roybon, L., & Rampon, C. (2015). Genetic manipulation of adult-born hippocampal neurons rescues memory in a mouse model of Alzheimer's disease. Brain : a journal of neurology, 138(Pt 2), 440–455. [CrossRef]
- Lee, T. Y., Cho, I. S., Bashyal, N., Naya, F. J., Tsai, M. J., Yoon, J. S., Choi, J. M., Park, C. H., Kim, S. S., & Suh-Kim, H. (2020). ERK Regulates NeuroD1-mediated Neurite Outgrowth via Proteasomal Degradation. Experimental neurobiology, 29(3), 189–206. [CrossRef]
- Pomeshchik, Y., Klementieva, O., Gil, J., Martinsson, I., Hansen, M. G., de Vries, T., Sancho-Balsells, A., Russ, K., Savchenko, E., Collin, A., Vaz, A. R., Bagnoli, S., Nacmias, B., Rampon, C., Sorbi, S., Brites, D., Marko-Varga, G., Kokaia, Z., Rezeli, M., Gouras, G. K., … Roybon, L. (2020). Human iPSC-Derived Hippocampal Spheroids: An Innovative Tool for Stratifying Alzheimer Disease Patient-Specific Cellular Phenotypes and Developing Therapies. Stem cell reports, 15(1), 256–273. [CrossRef]
- Guo, Z., Zhang, L., Wu, Z., Chen, Y., Wang, F., & Chen, G. (2014). In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer's disease model. Cell stem cell, 14(2), 188–202. [CrossRef]
- Ge, L. J., Yang, F. H., Li, W., Wang, T., Lin, Y., Feng, J., Chen, N. H., Jiang, M., Wang, J. H., Hu, X. T., & Chen, G. (2020). In vivo Neuroregeneration to Treat Ischemic Stroke Through NeuroD1 AAV-Based Gene Therapy in Adult Non-human Primates. Frontiers in cell and developmental biology, 8, 590008. [CrossRef]
- Sorimachi, H., Hata, S., & Ono, Y. (2011). Calpain chronicle--an enzyme family under multidisciplinary characterization. Proceedings of the Japan Academy. Series B, Physical and biological sciences, 87(6), 287–327. [CrossRef]
- Chen, L., Xiao, D., Tang, F., Gao, H., & Li, X. (2020). CAPN6 in disease: An emerging therapeutic target (Review). International journal of molecular medicine, 46(5), 1644–1652. [CrossRef]
- Dear, N., Matena, K., Vingron, M., & Boehm, T. (1997). A new subfamily of vertebrate calpains lacking a calmodulin-like domain: implications for calpain regulation and evolution. Genomics, 45(1), 175–184. [CrossRef]
- Dear, T. N., & Boehm, T. (1999). Diverse mRNA expression patterns of the mouse calpain genes Capn5, Capn6 and Capn11 during development. Mechanisms of development, 89(1-2), 201–209. [CrossRef]
- Moretti, D., Del Bello, B., Allavena, G., & Maellaro, E. (2014). Calpains and cancer: friends or enemies?. Archives of biochemistry and biophysics, 564, 26–36. [CrossRef]
- Ono, Y., Ojima, K., Shinkai-Ouchi, F., Hata, S., & Sorimachi, H. (2016). An eccentric calpain, CAPN3/p94/calpain-3. Biochimie, 122, 169–187. [CrossRef]
- Filali, H., Vidal, E., Bolea, R., Márquez, M., Marco, P., Vargas, A., Pumarola, M., Martin-Burriel, I., & Badiola, J. J. (2013). Gene and protein patterns of potential prion-related markers in the central nervous system of clinical and preclinical infected sheep. Veterinary research, 44(1), 14. [CrossRef]
- Su, X., Xiao, D., Huang, L., Li, S., Ying, J., Tong, Y., Ye, Q., Mu, D., & Qu, Y. (2019). MicroRNA Alteration in Developing Rat Oligodendrocyte Precursor Cells Induced by Hypoxia-Ischemia. Journal of neuropathology and experimental neurology, 78(10), 900–909. [CrossRef]
- Medeiros, R., Kitazawa, M., Chabrier, M. A., Cheng, D., Baglietto-Vargas, D., Kling, A., Moeller, A., Green, K. N., & LaFerla, F. M. (2012). Calpain inhibitor A-705253 mitigates Alzheimer's disease-like pathology and cognitive decline in aged 3xTgAD mice. The American journal of pathology, 181(2), 616–625. [CrossRef]
- Mahaman, Y. A. R., Huang, F., Kessete Afewerky, H., Maibouge, T. M. S., Ghose, B., & Wang, X. (2019). Involvement of calpain in the neuropathogenesis of Alzheimer's disease. Medicinal research reviews, 39(2), 608–630. [CrossRef]
- Kim, J. H., Kwon, S. J., Stankewich, M. C., Huh, G. Y., Glantz, S. B., & Morrow, J. S. (2016). Reactive protoplasmic and fibrous astrocytes contain high levels of calpain-cleaved alpha 2 spectrin. Experimental and molecular pathology, 100(1), 1–7. [CrossRef]
- Nogami, S., Satoh, S., Nakano, M., Shimizu, H., Fukushima, H., Maruyama, A., Terano, A., & Shirataki, H. (2003). Taxilin; a novel syntaxin-binding protein that is involved in Ca2+-dependent exocytosis in neuroendocrine cells. Genes to cells : devoted to molecular & cellular mechanisms, 8(1), 17–28. [CrossRef]
- Nogami, S., Satoh, S., Tanaka-Nakadate, S., Yoshida, K., Nakano, M., Terano, A., & Shirataki, H. (2004). Identification and characterization of taxilin isoforms. Biochemical and biophysical research communications, 319(3), 936–943. [CrossRef]
- Horii, Y., Sakane, H., Nogami, S., Ohtomo, N., Tomiya, T., & Shirataki, H. (2014). Expression of α-taxilin in the murine gastrointestinal tract: potential implication in cell proliferation. Histochemistry and cell biology, 141(2), 165–180. [CrossRef]
- Sakane, H., Makiyama, T., Nogami, S., Horii, Y., Akasaki, K., & Shirataki, H. (2016). β-Taxilin participates in differentiation of C2C12 myoblasts into myotubes. Experimental cell research, 345(2), 230–238. [CrossRef]
- Makiyama, T., Higashi, S., Sakane, H., Nogami, S., & Shirataki, H. (2018). γ-Taxilin temporally regulates centrosome disjunction in a Nek2A-dependent manner. Experimental cell research, 362(2), 412–423. [CrossRef]
- Hotokezaka, Y., Katayama, I., van Leyen, K., & Nakamura, T. (2015). GSK-3β-dependent downregulation of γ-taxilin and αNAC merge to regulate ER stress responses. Cell death & disease, 6(4), e1719. [CrossRef]
- Ma, D., Wang, F., Wang, R., Hu, Y., Chen, Z., Huang, N., Tian, Y., Xia, Y., Teng, J., & Chen, J. (2022). α-/γ-Taxilin are required for centriolar subdistal appendage assembly and microtubule organization. eLife, 11, e73252. [CrossRef]
- Higashi, S., Makiyama, T., Sakane, H., Nogami, S., & Shirataki, H. (2022). Regulation of Hook1-mediated endosomal sorting of clathrin-independent cargo by γ-taxilin. Journal of cell science, 135(1), jcs258849. [CrossRef]
- Kornau, H. C., Schenker, L. T., Kennedy, M. B., & Seeburg, P. H. (1995). Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science (New York, N.Y.), 269(5231), 1737–1740. [CrossRef]
- Naisbitt, S., Kim, E., Tu, J. C., Xiao, B., Sala, C., Valtschanoff, J., Weinberg, R. J., Worley, P. F., & Sheng, M. (1999). Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron, 23(3), 569–582. [CrossRef]
- Chen, L., Chetkovich, D. M., Petralia, R. S., Sweeney, N. T., Kawasaki, Y., Wenthold, R. J., Bredt, D. S., & Nicoll, R. A. (2000). Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature, 408(6815), 936–943. [CrossRef]
- Steiner, P., Higley, M. J., Xu, W., Czervionke, B. L., Malenka, R. C., & Sabatini, B. L. (2008). Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron, 60(5), 788–802. [CrossRef]
- Naisbitt, S., Kim, E., Weinberg, R. J., Rao, A., Yang, F. C., Craig, A. M., & Sheng, M. (1997). Characterization of guanylate kinase-associated protein, a postsynaptic density protein at excitatory synapses that interacts directly with postsynaptic density-95/synapse-associated protein 90. The Journal of neuroscience : the official journal of the Society for Neuroscience, 17(15), 5687–5696. [CrossRef]
- Welch, J. M., Wang, D., & Feng, G. (2004). Differential mRNA expression and protein localization of the SAP90/PSD-95-associated proteins (SAPAPs) in the nervous system of the mouse. The Journal of comparative neurology, 472(1), 24–39. [CrossRef]
- Yao, I., Iida, J., Nishimura, W., & Hata, Y. (2003). Synaptic localization of SAPAP1, a synaptic membrane-associated protein. Genes to cells : devoted to molecular & cellular mechanisms, 8(2), 121–129. [CrossRef]
- Rasmussen, A. H., Rasmussen, H. B., & Silahtaroglu, A. (2017). The DLGAP family: neuronal expression, function and role in brain disorders. Molecular brain, 10(1), 43. [CrossRef]
- Welch, J. M., Wang, D., & Feng, G. (2004). Differential mRNA expression and protein localization of the SAP90/PSD-95-associated proteins (SAPAPs) in the nervous system of the mouse. The Journal of comparative neurology, 472(1), 24–39. [CrossRef]
- Jiang-Xie, L. F., Liao, H. M., Chen, C. H., Chen, Y. T., Ho, S. Y., Lu, D. H., Lee, L. J., Liou, H. H., Fu, W. M., & Gau, S. S. (2014). Autism-associated gene Dlgap2 mutant mice demonstrate exacerbated aggressive behaviors and orbitofrontal cortex deficits. Molecular autism, 5, 32. [CrossRef]
- Li, J. M., Lu, C. L., Cheng, M. C., Luu, S. U., Hsu, S. H., Hu, T. M., Tsai, H. Y., & Chen, C. H. (2014). Role of the DLGAP2 gene encoding the SAP90/PSD-95-associated protein 2 in schizophrenia. PloS one, 9(1), e85373. [CrossRef]
- Gilbertson, M. W., Shenton, M. E., Ciszewski, A., Kasai, K., Lasko, N. B., Orr, S. P., & Pitman, R. K. (2002). Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nature neuroscience, 5(11), 1242–1247. [CrossRef]
- Ouellette, A. R., Neuner, S. M., Dumitrescu, L., Anderson, L. C., Gatti, D. M., Mahoney, E. R., Bubier, J. A., Churchill, G., Peters, L., Huentelman, M. J., Herskowitz, J. H., Yang, H. S., Smith, A. N., Reitz, C., Kunkle, B. W., White, C. C., De Jager, P. L., Schneider, J. A., Bennett, D. A., Seyfried, N. T., … Kaczorowski, C. C. (2020). Cross-Species Analyses Identify Dlgap2 as a Regulator of Age-Related Cognitive Decline and Alzheimer's Dementia. Cell reports, 32(9), 108091. [CrossRef]
- O'Dowd, B. F., Heiber, M., Chan, A., Heng, H. H., Tsui, L. C., Kennedy, J. L., Shi, X., Petronis, A., George, S. R., & Nguyen, T. (1993). A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene, 136(1-2), 355–360. [CrossRef]
- Hu, G., Wang, Z., Zhang, R., Sun, W., & Chen, X. (2021). The Role of Apelin/Apelin Receptor in Energy Metabolism and Water Homeostasis: A Comprehensive Narrative Review. Frontiers in physiology, 12, 632886. [CrossRef]
- Luo, H., Han, L., & Xu, J. (2020). Apelin/APJ system: A novel promising target for neurodegenerative diseases. Journal of cellular physiology, 235(2), 638–657. [CrossRef]
- Pi, J., Cheng, Y., Sun, H., Chen, X., Zhuang, T., Liu, J., Li, Y., Chang, H., Zhang, L., Zhang, Y., & Tao, T. (2017). Apln-CreERT:mT/mG reporter mice as a tool for sprouting angiogenesis study. BMC ophthalmology, 17(1), 163. [CrossRef]
- Mughal, A., & O'Rourke, S. T. (2018). Vascular effects of apelin: Mechanisms and therapeutic potential. Pharmacology & therapeutics, 190, 139–147. [CrossRef]
- Tatemoto, K., Takayama, K., Zou, M. X., Kumaki, I., Zhang, W., Kumano, K., & Fujimiya, M. (2001). The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regulatory peptides, 99(2-3), 87–92. [CrossRef]
- O'Carroll, A. M., Lolait, S. J., Harris, L. E., & Pope, G. R. (2013). The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. The Journal of endocrinology, 219(1), R13–R35. [CrossRef]
- Kazemi, F., & Zahediasl, S. (2018). Effects of exercise training on adipose tissue apelin expression in streptozotocin-nicotinamide induced diabetic rats. Gene, 662, 97–102. [CrossRef]
- Ashley, E., Chun, H. J., & Quertermous, T. (2006). Opposing cardiovascular roles for the angiotensin and apelin signaling pathways. Journal of molecular and cellular cardiology, 41(5), 778–781. [CrossRef]
- Taheri, S., Murphy, K., Cohen, M., Sujkovic, E., Kennedy, A., Dhillo, W., Dakin, C., Sajedi, A., Ghatei, M., & Bloom, S. (2002). The effects of centrally administered apelin-13 on food intake, water intake and pituitary hormone release in rats. Biochemical and biophysical research communications, 291(5), 1208–1212. [CrossRef]
- Zeng, X. J., Yu, S. P., Zhang, L., & Wei, L. (2010). Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Experimental cell research, 316(11), 1773–1783. [CrossRef]
- O'Donnell, L. A., Agrawal, A., Sabnekar, P., Dichter, M. A., Lynch, D. R., & Kolson, D. L. (2007). Apelin, an endogenous neuronal peptide, protects hippocampal neurons against excitotoxic injury. Journal of neurochemistry, 102(6), 1905–1917. [CrossRef]
- Yang, S., Li, H., Tang, L., Ge, G., Ma, J., Qiao, Z., Liu, H., & Fang, W. (2015). Apelin-13 protects the heart against ischemia-reperfusion injury through the RISK-GSK-3β-mPTP pathway. Archives of medical science : AMS, 11(5), 1065–1073.
- Aminyavari, S., Zahmatkesh, M., Farahmandfar, M., Khodagholi, F., Dargahi, L., & Zarrindast, M. R. (2019). Protective role of Apelin-13 on amyloid β25-35-induced memory deficit; Involvement of autophagy and apoptosis process. Progress in neuro-psychopharmacology & biological psychiatry, 89, 322–334. [CrossRef]
- Agostinho, P., Cunha, R. A., & Oliveira, C. (2010). Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer's disease. Current pharmaceutical design, 16(25), 2766–2778. [CrossRef]
- Xin, Q., Cheng, B., Pan, Y., Liu, H., Yang, C., Chen, J., & Bai, B. (2015). Neuroprotective effects of apelin-13 on experimental ischemic stroke through suppression of inflammation. Peptides, 63, 55–62. [CrossRef]
- Yang, L., Wang, H., Liu, L., & Xie, A. (2018). The Role of Insulin/IGF-1/PI3K/Akt/GSK3β Signaling in Parkinson's Disease Dementia. Frontiers in neuroscience, 12, 73. [CrossRef]
- Roche, J., Ramé, C., Reverchon, M., Mellouk, N., Cornuau, M., Guerif, F., Froment, P., & Dupont, J. (2016). Apelin (APLN) and Apelin Receptor (APLNR) in Human Ovary: Expression, Signaling, and Regulation of Steroidogenesis in Primary Human Luteinized Granulosa Cells. Biology of reproduction, 95(5), 104. [CrossRef]
- Haghparast, E., Esmaeili-Mahani, S., Abbasnejad, M., & Sheibani, V. (2018). Apelin-13 ameliorates cognitive impairments in 6-hydroxydopamine-induced substantia nigra lesion in rats. Neuropeptides, 68, 28–35. [CrossRef]
- Pouresmaeili-Babaki, E., Esmaeili-Mahani, S., Abbasnejad, M., & Ravan, H. (2018). Protective Effect of Neuropeptide Apelin-13 on 6-Hydroxydopamine-Induced Neurotoxicity in SH-SY5Y Dopaminergic Cells: Involvement of Its Antioxidant and Antiapoptotic Properties. Rejuvenation research, 21(2), 162–167. [CrossRef]
- Chi, Y., Chai, J., Xu, C., Luo, H., & Zhang, Q. (2015). Apelin inhibits the activation of the nucleotide-binding domain and the leucine-rich, repeat-containing family, pyrin-containing 3 (NLRP3) inflammasome and ameliorates insulin resistance in severely burned rats. Surgery, 157(6), 1142–1152. [CrossRef]
- Fernández-Nogales, M., Santos-Galindo, M., Hernández, I. H., Cabrera, J. R., & Lucas, J. J. (2016). Faulty splicing and cytoskeleton abnormalities in Huntington's disease. Brain pathology (Zurich, Switzerland), 26(6), 772–778. [CrossRef]
- Proskura, A. L., Vechkapova, S. O., Zapara, T. A., & Ratushniak, A. S. (2017). Molekuliarnaia biologiia, 51(4), 734–742. [CrossRef]
- Li, Y., Bai, Y. J., Jiang, Y. R., Yu, W. Z., Shi, X., Chen, L., Feng, J., & Sun, G. B. (2018). Apelin-13 Is an Early Promoter of Cytoskeleton and Tight Junction in Diabetic Macular Edema via PI-3K/Akt and MAPK/Erk Signaling Pathways. BioMed research international, 2018, 3242574. [CrossRef]
- Bryan, M. R., & Bowman, A. B. (2017). Manganese and the Insulin-IGF Signaling Network in Huntington's Disease and Other Neurodegenerative Disorders. Advances in neurobiology, 18, 113–142. [CrossRef]
- Zhang, Y., Zhang, Y., Sun, K., Meng, Z., & Chen, L. (2019). The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. Journal of molecular cell biology, 11(1), 1–13. [CrossRef]
- César-Razquin, A., Snijder, B., Frappier-Brinton, T., Isserlin, R., Gyimesi, G., Bai, X., Reithmeier, R. A., Hepworth, D., Hediger, M. A., Edwards, A. M., & Superti-Furga, G. (2015). A Call for Systematic Research on Solute Carriers. Cell, 162(3), 478–487. [CrossRef]
- Brzica, H., Abdullahi, W., Ibbotson, K., & Ronaldson, P. T. (2017). Role of Transporters in Central Nervous System Drug Delivery and Blood-Brain Barrier Protection: Relevance to Treatment of Stroke. Journal of central nervous system disease, 9, 1179573517693802. [CrossRef]
- Türková, A., & Zdrazil, B. (2019). Current Advances in Studying Clinically Relevant Transporters of the Solute Carrier (SLC) Family by Connecting Computational Modeling and Data Science. Computational and structural biotechnology journal, 17, 390–405. [CrossRef]
- Hu, C., Tao, L., Cao, X., & Chen, L. (2020). The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian journal of pharmaceutical sciences, 15(2), 131–144. [CrossRef]
- Young, J. D., Yao, S. Y., Baldwin, J. M., Cass, C. E., & Baldwin, S. A. (2013). The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Molecular aspects of medicine, 34(2-3), 529–547. [CrossRef]
- Pastor-Anglada, M., Urtasun, N., & Pérez-Torras, S. (2018). Intestinal Nucleoside Transporters: Function, Expression, and Regulation. Comprehensive Physiology, 8(3), 1003–1017. [CrossRef]
- Kao, Y. H., Lin, M. S., Chen, C. M., Wu, Y. R., Chen, H. M., Lai, H. L., Chern, Y., & Lin, C. J. (2017). Targeting ENT1 and adenosine tone for the treatment of Huntington's disease. Human molecular genetics, 26(3), 467–478. [CrossRef]
- Lee, C. C., Chang, C. P., Lin, C. J., Lai, H. L., Kao, Y. H., Cheng, S. J., Chen, H. M., Liao, Y. P., Faivre, E., Buée, L., Blum, D., Fang, J. M., & Chern, Y. (2018). Adenosine Augmentation Evoked by an ENT1 Inhibitor Improves Memory Impairment and Neuronal Plasticity in the APP/PS1 Mouse Model of Alzheimer's Disease. Molecular neurobiology, 55(12), 8936–8952. [CrossRef]
- Chang, C. P., Chang, Y. G., Chuang, P. Y., Nguyen, T. N. A., Wu, K. C., Chou, F. Y., Cheng, S. J., Chen, H. M., Jin, L. W., Carvalho, K., Huin, V., Buée, L., Liao, Y. F., Lin, C. J., Blum, D., & Chern, Y. (2021). Equilibrative nucleoside transporter 1 inhibition rescues energy dysfunction and pathology in a model of tauopathy. Acta neuropathologica communications, 9(1), 112. [CrossRef]
- Chung, J. Y., Yu, S. D., & Hong, Y. S. (2014). Environmental source of arsenic exposure. Journal of preventive medicine and public health = Yebang Uihakhoe chi, 47(5), 253–257. [CrossRef]
- Ahmad, S. A., Khan, M. H., & Haque, M. (2018). Arsenic contamination in groundwater in Bangladesh: implications and challenges for healthcare policy. Risk management and healthcare policy, 11, 251–261. [CrossRef]
- Emadi, A., & Gore, S. D. (2010). Arsenic trioxide - An old drug rediscovered. Blood reviews, 24(4-5), 191–199. [CrossRef]
- Singh, A. P., Goel, R. K., & Kaur, T. (2011). Mechanisms pertaining to arsenic toxicity. Toxicology international, 18(2), 87–93. [CrossRef]
- Rahman, M. A., Hannan, M. A., Uddin, M. J., Rahman, M. S., Rashid, M. M., & Kim, B. (2021). Exposure to Environmental Arsenic and Emerging Risk of Alzheimer's Disease: Perspective Mechanisms, Management Strategy, and Future Directions. Toxics, 9(8), 188. [CrossRef]
- Chandravanshi, L. P., Gupta, R., & Shukla, R. K. (2018). Developmental Neurotoxicity of Arsenic: Involvement of Oxidative Stress and Mitochondrial Functions. Biological trace element research, 186(1), 185–198. [CrossRef]
- Hannan, M. A., Dash, R., Sohag, A. A. M., Haque, M. N., & Moon, I. S. (2020). Neuroprotection Against Oxidative Stress: Phytochemicals Targeting TrkB Signaling and the Nrf2-ARE Antioxidant System. Frontiers in molecular neuroscience, 13, 116. [CrossRef]
- Dwivedi, N., & Flora, S. J. (2011). Concomitant exposure to arsenic and organophosphates on tissue oxidative stress in rats. Food and chemical toxicology : an international journal published for the British Industrial Biological Research Association, 49(5), 1152–1159. [CrossRef]
- Dash, R., Mitra, S., Ali, M. C., Oktaviani, D. F., Hannan, M. A., Choi, S. M., & Moon, I. S. (2021). Phytosterols: Targeting Neuroinflammation in Neurodegeneration. Current pharmaceutical design, 27(3), 383–401. [CrossRef]
- Wisessaowapak, C., Visitnonthachai, D., Watcharasit, P., & Satayavivad, J. (2021). Prolonged arsenic exposure increases tau phosphorylation in differentiated SH-SY5Y cells: The contribution of GSK3 and ERK1/2. Environmental toxicology and pharmacology, 84, 103626. [CrossRef]
- Medda, N., Patra, R., Ghosh, T. K., & Maiti, S. (2020). Neurotoxic Mechanism of Arsenic: Synergistic Effect of Mitochondrial Instability, Oxidative Stress, and Hormonal-Neurotransmitter Impairment. Biological trace element research, 198(1), 8–15. [CrossRef]
- Prakash, C., Soni, M., & Kumar, V. (2016). Mitochondrial oxidative stress and dysfunction in arsenic neurotoxicity: A review. Journal of applied toxicology : JAT, 36(2), 179–188. [CrossRef]
- Prakash, C., Soni, M., & Kumar, V. (2015). Biochemical and Molecular Alterations Following Arsenic-Induced Oxidative Stress and Mitochondrial Dysfunction in Rat Brain. Biological trace element research, 167(1), 121–129. [CrossRef]
- Rahman, M. A., Rahman, M. S., Uddin, M. J., Mamum-Or-Rashid, A. N. M., Pang, M. G., & Rhim, H. (2020). Emerging risk of environmental factors: insight mechanisms of Alzheimer's diseases. Environmental science and pollution research international, 27(36), 44659–44672. [CrossRef]
- Zhang, W., Cui, X., Gao, Y., Sun, L., Wang, J., Yang, Y., Liu, X., Li, Y., Guo, X., & Sun, D. (2019). Role of pigment epithelium-derived factor (PEDF) on arsenic-induced neuronal apoptosis. Chemosphere, 215, 925–931. [CrossRef]
- Dash, R., Jahan, I., Ali, M. C., Mitra, S., Munni, Y. A., Timalsina, B., Hannan, M. A., & Moon, I. S. (2021). Potential roles of natural products in the targeting of proteinopathic neurodegenerative diseases. Neurochemistry international, 145, 105011. [CrossRef]
- Dash, R.; Ali, M.C.; Jahan, I.; Munni, Y.A.; Mitra, S.; Hannan, M.A.; Timalsina, B.; Oktaviani, D.F.; Choi, H.J.; Moon, I.S. Emerging potential of cannabidiol in reversing proteinopathies. Ageing Res.
- Mizushima, N., & Komatsu, M. (2011). Autophagy: renovation of cells and tissues. Cell, 147(4), 728–741. [CrossRef]
- Levine, B., Mizushima, N., & Virgin, H. W. (2011). Autophagy in immunity and inflammation. Nature, 469(7330), 323–335. [CrossRef]
- Vazza, G., Picelli, S., Bozzato, A., & Mostacciuolo, M. L. (2003). Identification and characterization of C3orf6, a new conserved human gene mapping to chromosome 3q28. Gene, 314, 113–120. [CrossRef]
- Lin, Y., Li, Z., Wang, Y., Tian, T., Jia, P., Ye, Y., He, M., Yang, Z., Li, C., Guo, D., & Hou, P. (2022). CCDC50 suppresses NLRP3 inflammasome activity by mediating autophagic degradation of NLRP3. EMBO reports, 23(5), e54453. [CrossRef]
- Farfsing, A., Engel, F., Seiffert, M., Hartmann, E., Ott, G., Rosenwald, A., Stilgenbauer, S., Döhner, H., Boutros, M., Lichter, P., & Pscherer, A. (2009). Gene knockdown studies revealed CCDC50 as a candidate gene in mantle cell lymphoma and chronic lymphocytic leukemia. Leukemia, 23(11), 2018–2026. [CrossRef]
- Hou, P., Yang, K., Jia, P., Liu, L., Lin, Y., Li, Z., Li, J., Chen, S., Guo, S., Pan, J., Wu, J., Peng, H., Zeng, W., Li, C., Liu, Y., & Guo, D. (2021). A novel selective autophagy receptor, CCDC50, delivers K63 polyubiquitination-activated RIG-I/MDA5 for degradation during viral infection. Cell research, 31(1), 62–79. [CrossRef]
- Cruchaga, C., Kauwe, J. S., Harari, O., Jin, S. C., Cai, Y., Karch, C. M., Benitez, B. A., Jeng, A. T., Skorupa, T., Carrell, D., Bertelsen, S., Bailey, M., McKean, D., Shulman, J. M., De Jager, P. L., Chibnik, L., Bennett, D. A., Arnold, S. E., Harold, D., Sims, R., … Goate, A. M. (2013). GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron, 78(2), 256–268. [CrossRef]
- Skaria A. P. (2022). The economic and societal burden of Alzheimer disease: managed care considerations. The American journal of managed care, 28(10 Suppl), S188–S196. [CrossRef]
- Wong W. (2020). Economic burden of Alzheimer disease and managed care considerations. The American journal of managed care, 26(8 Suppl), S177–S183. [CrossRef]
- Holbrook, J. A., Jarosz-Griffiths, H. H., Caseley, E., Lara-Reyna, S., Poulter, J. A., Williams-Gray, C. H., Peckham, D., & McDermott, M. F. (2021). Neurodegenerative Disease and the NLRP3 Inflammasome. Frontiers in pharmacology, 12, 643254. [CrossRef]
- Anderson, F. L., Biggs, K. E., Rankin, B. E., & Havrda, M. C. (2023). NLRP3 inflammasome in neurodegenerative disease. Translational research : the journal of laboratory and clinical medicine, 252, 21–33. [CrossRef]
- Chakraborty, S., Kaushik, D. K., Gupta, M., & Basu, A. (2010). Inflammasome signaling at the heart of central nervous system pathology. Journal of neuroscience research, 88(8), 1615–1631. [CrossRef]
- Hung, S. C., Choi, C. H., Said-Sadier, N., Johnson, L., Atanasova, K. R., Sellami, H., Yilmaz, Ö., & Ojcius, D. M. (2013). P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PloS one, 8(7), e70210. [CrossRef]
- Kanellopoulos, J. M., Almeida-da-Silva, C. L. C., Rüütel Boudinot, S., & Ojcius, D. M. (2021). Structural and Functional Features of the P2X4 Receptor: An Immunological Perspective. Frontiers in immunology, 12, 645834. [CrossRef]
- Li, Q., Fu, W., Yao, J., Ji, Z., Wang, Y., Zhou, Z., Yan, J., & Li, W. (2014). Heme induces IL-1β secretion through activating NLRP3 in kidney inflammation. Cell biochemistry and biophysics, 69(3), 495–502. [CrossRef]
- Ducza, L., Gajtkó, A., Hegedűs, K., Bakk, E., Kis, G., Gaál, B., Takács, R., Szücs, P., Matesz, K., & Holló, K. (2023). Neuronal P2X4 receptor may contribute to peripheral inflammatory pain in rat spinal dorsal horn. Frontiers in molecular neuroscience, 16, 1115685. [CrossRef]
- Du, R. H., Wu, F. F., Lu, M., Shu, X. D., Ding, J. H., Wu, G., & Hu, G. (2016). Uncoupling protein 2 modulation of the NLRP3 inflammasome in astrocytes and its implications in depression. Redox biology, 9, 178–187. [CrossRef]
- Zhou, X., Xiao, Q., Xie, L., Yang, F., Wang, L., & Tu, J. (2019). Astrocyte, a Promising Target for Mood Disorder Interventions. Frontiers in molecular neuroscience, 12, 136. [CrossRef]
- Zang, X., Zheng, X., Hou, Y., Hu, M., Wang, H., Bao, X., Zhou, F., Wang, G., & Hao, H. (2018). Regulation of proinflammatory monocyte activation by the kynurenine-AhR axis underlies immunometabolic control of depressive behavior in mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 32(4), 1944–1956. [CrossRef]
- Rolstad, S., Jakobsson, J., Sellgren, C., Isgren, A., Ekman, C. J., Bjerke, M., Blennow, K., Zetterberg, H., Pålsson, E., & Landén, M. (2015). CSF neuroinflammatory biomarkers in bipolar disorder are associated with cognitive impairment. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology, 25(8), 1091–1098. [CrossRef]
- Tsai, P. J., Lai, Y. H., Manne, R. K., Tsai, Y. S., Sarbassov, D., & Lin, H. K. (2022). Akt: a key transducer in cancer. Journal of biomedical science, 29(1), 76. [CrossRef]
- Peng, S., Gu, J. H., Dai, C. L., Iqbal, K., Liu, F., & Gong, C. X. (2022). AKT/GSK-3β signaling is altered through downregulation of mTOR during cerebral Ischemia/Reperfusion injury. Molecular biology reports, 49(5), 3955–3964. [CrossRef]
- Stefaniak, J., & O'Brien, J. (2016). Imaging of neuroinflammation in dementia: a review. Journal of neurology, neurosurgery, and psychiatry, 87(1), 21–28. [CrossRef]
- Passamonti, L., Tsvetanov, K. A., Jones, P. S., Bevan-Jones, W. R., Arnold, R., Borchert, R. J., Mak, E., Su, L., O'Brien, J. T., & Rowe, J. B. (2019). Neuroinflammation and Functional Connectivity in Alzheimer's Disease: Interactive Influences on Cognitive Performance. The Journal of neuroscience : the official journal of the Society for Neuroscience, 39(36), 7218–7226. [CrossRef]
- Ng, A., Tam, W. W., Zhang, M. W., Ho, C. S., Husain, S. F., McIntyre, R. S., & Ho, R. C. (2018). IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer's disease: Systematic Review and Meta-Analysis. Scientific reports, 8(1), 12050. [CrossRef]
- Cuello A. C. (2017). Early and Late CNS Inflammation in Alzheimer's Disease: Two Extremes of a Continuum?. Trends in pharmacological sciences, 38(11), 956–966. [CrossRef]
- Aziz N. (2015). Measurement of Circulating Cytokines and Immune-Activation Markers by Multiplex Technology in the Clinical Setting: What Are We Really Measuring?. Forum on immunopathological diseases and therapeutics, 6(1-2), 19–22. [CrossRef]
- Koelman, L., Pivovarova-Ramich, O., Pfeiffer, A. F. H., Grune, T., & Aleksandrova, K. (2019). Cytokines for evaluation of chronic inflammatory status in ageing research: reliability and phenotypic characterisation. Immunity & ageing : I & A, 16, 11. [CrossRef]
- Zuliani, G., Ranzini, M., Guerra, G., Rossi, L., Munari, M. R., Zurlo, A., Volpato, S., Atti, A. R., Blè, A., & Fellin, R. (2007). Plasma cytokines profile in older subjects with late onset Alzheimer's disease or vascular dementia. Journal of psychiatric research, 41(8), 686–693. [CrossRef]
- Belkhelfa, M., Beder, N., Mouhoub, D., Amri, M., Hayet, R., Tighilt, N., Bakheti, S., Laimouche, S., Azzouz, D., Belhadj, R., & Touil-Boukoffa, C. (2018). The involvement of neuroinflammation and necroptosis in the hippocampus during vascular dementia. Journal of neuroimmunology, 320, 48–57. [CrossRef]
- Engelhart, M. J., Geerlings, M. I., Meijer, J., Kiliaan, A., Ruitenberg, A., van Swieten, J. C., Stijnen, T., Hofman, A., Witteman, J. C., & Breteler, M. M. (2004). Inflammatory proteins in plasma and the risk of dementia: the rotterdam study. Archives of neurology, 61(5), 668–672. [CrossRef]
- Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., Griep, A., Axt, D., Remus, A., Tzeng, T. C., Gelpi, E., Halle, A., Korte, M., Latz, E., & Golenbock, D. T. (2013). NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature, 493(7434), 674–678. [CrossRef]
- Baldwin, A. G., Brough, D., & Freeman, S. (2016). Inhibiting the Inflammasome: A Chemical Perspective. Journal of medicinal chemistry, 59(5), 1691–1710. [CrossRef]
- Bertinaria M. (2021). Inflammasome Inhibitors. Molecules (Basel, Switzerland), 26(22), 6912. [CrossRef]
- Coll, R. C., Robertson, A. A., Chae, J. J., Higgins, S. C., Muñoz-Planillo, R., Inserra, M. C., Vetter, I., Dungan, L. S., Monks, B. G., Stutz, A., Croker, D. E., Butler, M. S., Haneklaus, M., Sutton, C. E., Núñez, G., Latz, E., Kastner, D. L., Mills, K. H., Masters, S. L., Schroder, K., … O'Neill, L. A. (2015). A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nature medicine, 21(3), 248–255. [CrossRef]
- Dempsey, C., Rubio Araiz, A., Bryson, K. J., Finucane, O., Larkin, C., Mills, E. L., Robertson, A. A. B., Cooper, M. A., O'Neill, L. A. J., & Lynch, M. A. (2017). Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain, behavior, and immunity, 61, 306–316. [CrossRef]
- Gordon, R., Albornoz, E. A., Christie, D. C., Langley, M. R., Kumar, V., Mantovani, S., Robertson, A. A. B., Butler, M. S., Rowe, D. B., O'Neill, L. A., Kanthasamy, A. G., Schroder, K., Cooper, M. A., & Woodruff, T. M. (2018). Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Science translational medicine, 10(465), eaah4066. [CrossRef]
- Piancone, F., La Rosa, F., Marventano, I., Saresella, M., & Clerici, M. (2021). The Role of the Inflammasome in Neurodegenerative Diseases. Molecules (Basel, Switzerland), 26(4), 953. [CrossRef]
- La Rosa, F., Saresella, M., Marventano, I., Piancone, F., Ripamonti, E., Al-Daghri, N., Bazzini, C., Zoia, C. P., Conti, E., Ferrarese, C., & Clerici, M. (2019). Stavudine Reduces NLRP3 Inflammasome Activation and Modulates Amyloid-β Autophagy. Journal of Alzheimer's disease : JAD, 72(2), 401–412. [CrossRef]
- Gastaldi, S., Boscaro, V., Gianquinto, E., Sandall, C. F., Giorgis, M., Marini, E., Blua, F., Gallicchio, M., Spyrakis, F., MacDonald, J. A., & Bertinaria, M. (2021). Chemical Modulation of the 1-(Piperidin-4-yl)-1,3-dihydro-2H-benzo[d]imidazole-2-one Scaffold as a Novel NLRP3 Inhibitor. Molecules (Basel, Switzerland), 26(13), 3975. [CrossRef]
- Kaushal, V., Dye, R., Pakavathkumar, P., Foveau, B., Flores, J., Hyman, B., Ghetti, B., Koller, B. H., & LeBlanc, A. C. (2015). Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell death and differentiation, 22(10), 1676–1686. [CrossRef]
Figure 1.
Litmaps visualisation of original research articles targeting NLRP2 inflammasome in the nervous system Articles are shown in display mode “ Momentum”. Calculation is based on "Cited By" count, weighed by recency.
Figure 1.
Litmaps visualisation of original research articles targeting NLRP2 inflammasome in the nervous system Articles are shown in display mode “ Momentum”. Calculation is based on "Cited By" count, weighed by recency.
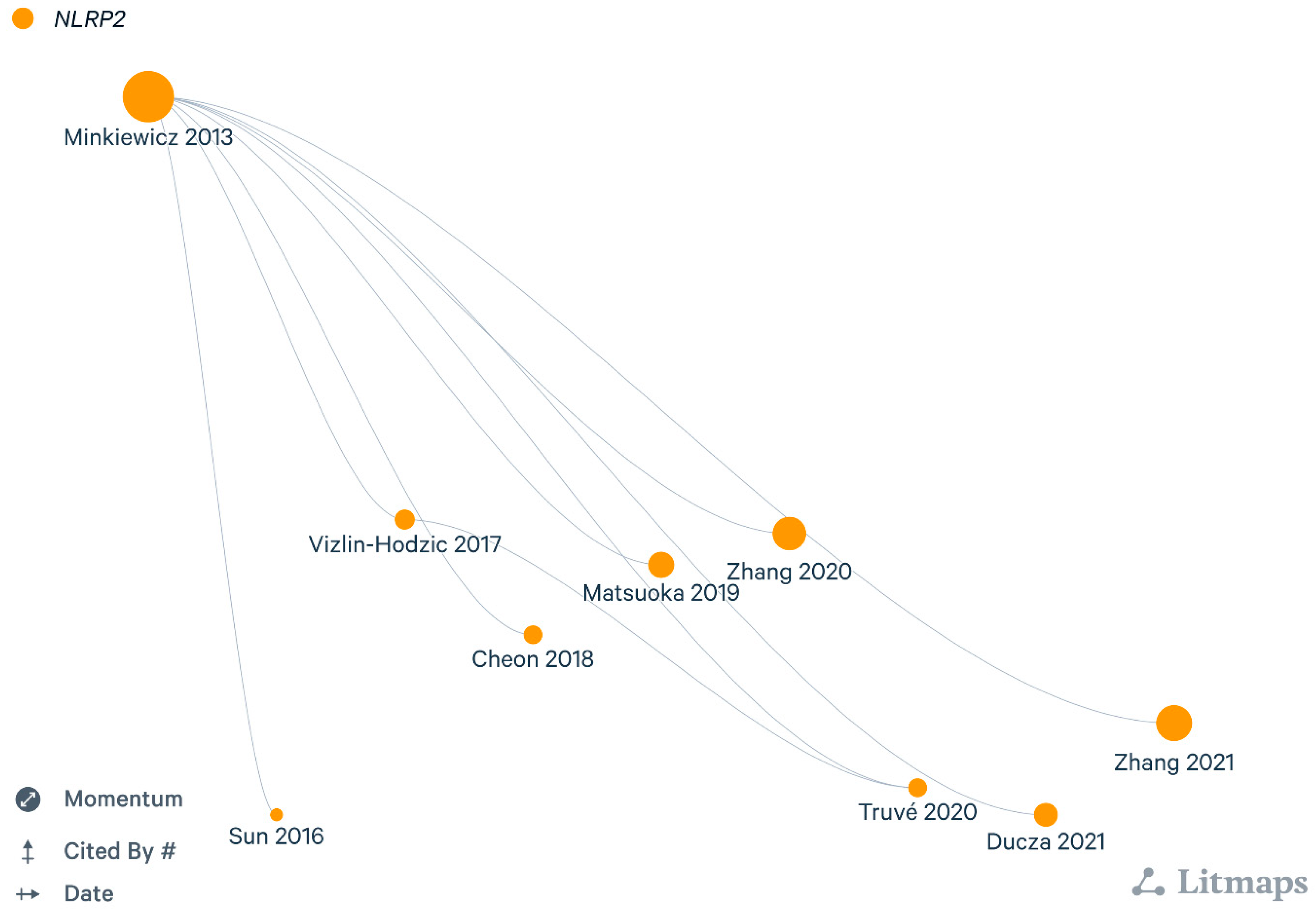
Figure 2.
Number of publications and citations related to NLRP2 inflammasome in the nervous system (2005-2023).
Figure 2.
Number of publications and citations related to NLRP2 inflammasome in the nervous system (2005-2023).
Figure 3.
The extended STRING connectome of NLRP2 protein. Color code is as follows: known interactions: light blue, from curated databases; magenta, experimentally determined; predicted interactions: green, gene neighbourhood; dark blue, gene co-occurrence; others: lime, text-mining; black, coexpression; purple, protein homology. Red colour indicates the selected 15 nodes potentially involved in neuropathologies.
Figure 3.
The extended STRING connectome of NLRP2 protein. Color code is as follows: known interactions: light blue, from curated databases; magenta, experimentally determined; predicted interactions: green, gene neighbourhood; dark blue, gene co-occurrence; others: lime, text-mining; black, coexpression; purple, protein homology. Red colour indicates the selected 15 nodes potentially involved in neuropathologies.
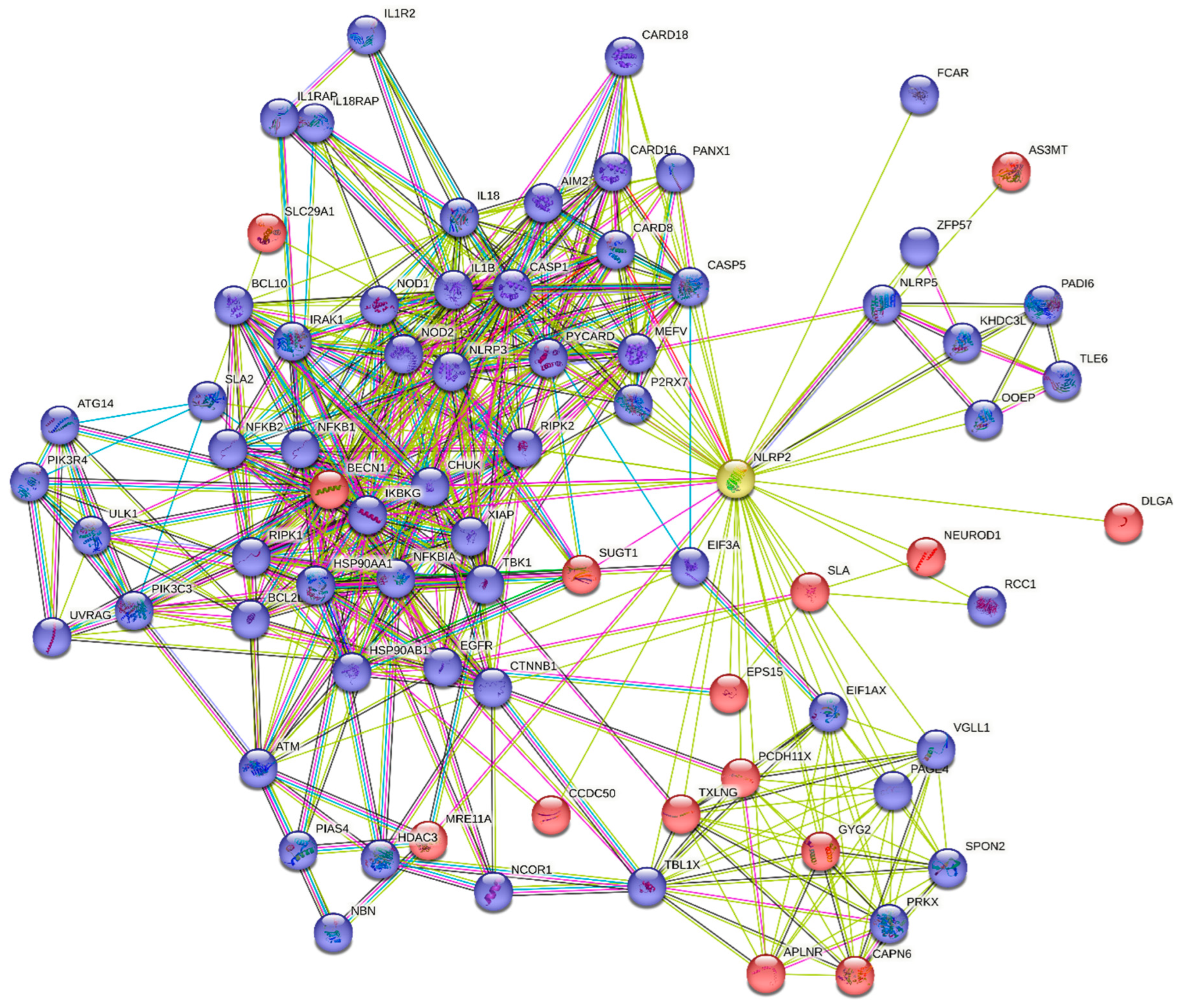
Figure 4.
State-of-the-art in relation to NLRP2 protein in neuropathologies. ASC: Apoptosis-associated Speck-like protein containing a caspase-activation and recruitment domain, ASK1: Apoptosis signal-regulating kinase 1, BBG: Brilliant Blue G, GLP-1: Glucagon-like peptide-1, NFκB: Nuclear factor kappa B, P2X4, P2X7- P2X purinoreceptor X4,7, PNX1-Pannexin-1, ROS-reactive oxygen species
Figure 4.
State-of-the-art in relation to NLRP2 protein in neuropathologies. ASC: Apoptosis-associated Speck-like protein containing a caspase-activation and recruitment domain, ASK1: Apoptosis signal-regulating kinase 1, BBG: Brilliant Blue G, GLP-1: Glucagon-like peptide-1, NFκB: Nuclear factor kappa B, P2X4, P2X7- P2X purinoreceptor X4,7, PNX1-Pannexin-1, ROS-reactive oxygen species
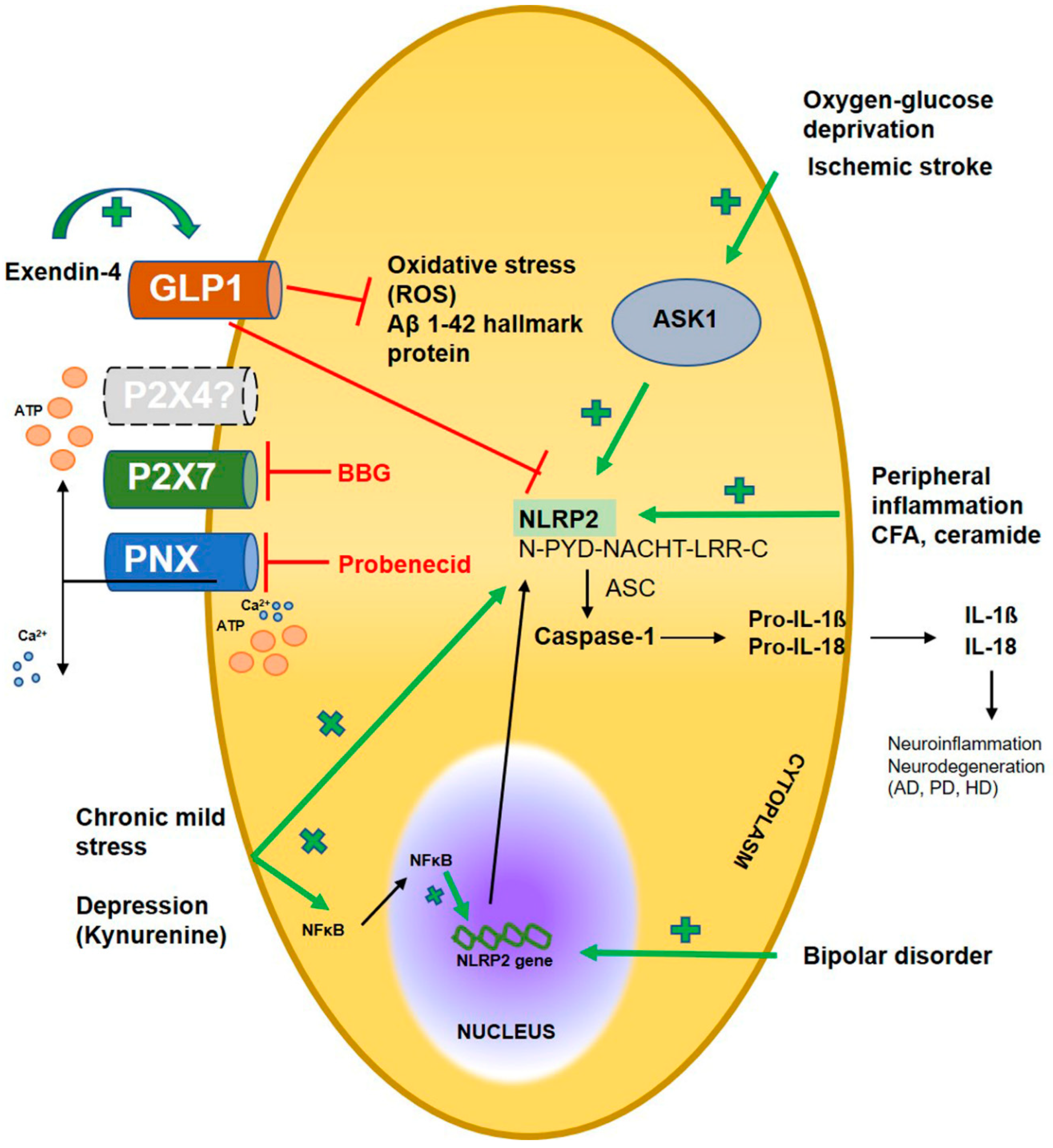

Table 1.
List of direct interactions of NLRP2

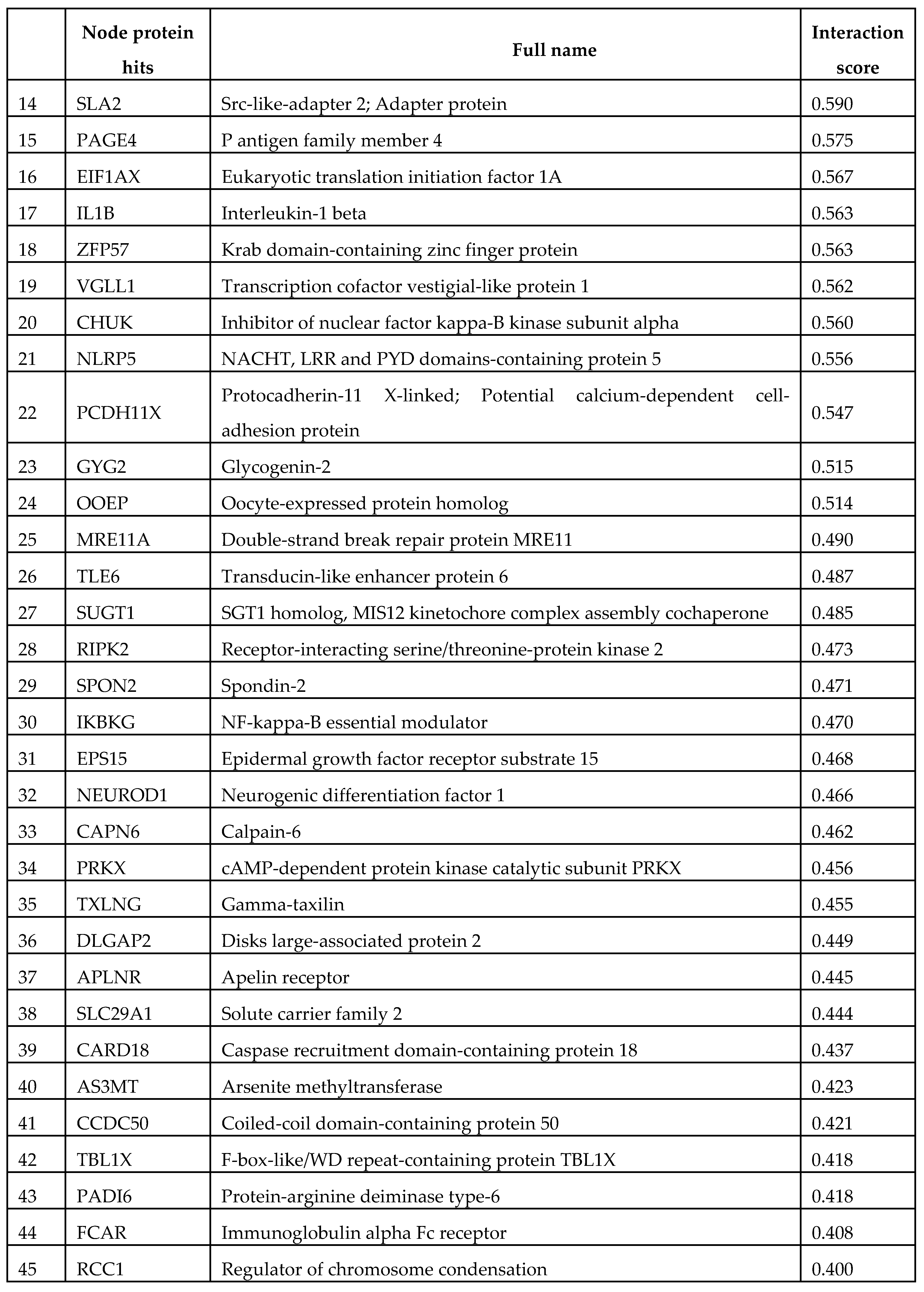
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



