
Submitted:
04 July 2023
Posted:
04 July 2023
You are already at the latest version
Abstract
Genome-wide sequencing data play an important role in evaluating the genomic level differences between superior and poor-quality crop plants and improving our understanding of molecular association with desired traits. We analyzed the obtained 92,921,066 raw reads from genome-wide resequencing of Cucumis sativus var. hardwickii through in-silico approaches and mapped to the reference genome of Cucumis sativus to identify the genome-wide single nucleotide polymorphisms (SNPs) and Single nucleotide variations (SNV). Here, we report 19, 74,213 candidate SNPs including 1,33,468 insertions and 1,43,237 deletions and 75 Indels genome-wide. A total of 2228224 identified variants were classified into four classes including 0.01% sequence alteration, 5.94% insertion, 6.37% deletion and 87.66% SNV respectively. These variations can be a major source of phenotypic diversity and sequence variation within the species. During the present study these variants were also utilized to resolve orthologous relationships among the genomes of C. melo and C. sativus var. hardwickii. Overall, the discovery of SNPs and genomic variants may help predict the plant response to certain environmental factors and can be utilized to improve crop plants' economically important traits.
Keywords:
Cucumis sativus var hardwickii
; VEP
; Genomic variation
; SNP
; SNV
1. Introduction
Cucumis sativus var. hardwickii is one of the wild relatives of cucumbers. It belongs to the Cucurbitaceae family, primarily found in forests and mountain slopes, at altitudes of 700- 2000 m, in China, Myanmar, NE India, Nepal and Thailand. The origin of the cucumber is known to be India, where the wild ancestor C. sativus var. hardwickii round, and bitter fruit grows [1]. It is also found in South India, particularly in the state of Kerala. C. sativus var. hardwickii (wild cucumber) is a self-compatible, insect-pollinated taxon with a stable genome (2n = 14) and high reproductive output (85%) as compared to cultivated cucumber (73.3%). It is primarily composed of two commercially available vegetables: cucumber (C. sativus L. var. sativus; 2n=2x=14) and melon (C. melo L; 2n=2x=24) [2,3]. From India to China, the plant seems to have spread eastward, and from China westward, to Asia Minor, North Africa, and Southern Europe. As such, it represents an extreme of C. sativus variation and has the potential to expand the genetic diversity available for commercial cucumber breeding [4].
It possesses a number of advantageous characteristics, including a sequential multiple fruiting habit, multiple lateral branching, and a high overall fruit weight per plant [5], dark green-coloured fruits, early maturation, and fruit production during the rainy season. It includes several critical genes for the evolution of cultivated cucumber but is under represented in the world's central gene banks [6,7]. Genetic markers (morphological, biochemical, and molecular) have been used to characterize the genetic variation found in cucumber [8,9]. Thus, by analyzing a more robust selection of this species, we can better understand the variation present in this closely related population used in plant improvement programs [10].
Agricultural lands have been compromised by salination, air pollution, and global warming [11]. Studies of single nucleotide polymorphisms (SNPs) and significant advances in the use of genomic methods have created new, quick mapping procedures for identifying features and determining their causes. To expand multidisciplinary science and it is necessary to maintain traditional practices in the field of molecular genetics. Because of their low costs and high performance, microarrays and next-generation sequencing are commonly used [12]. Recent microarrays have been applied to biological samples.
Although Northern or quantitative PCR has previously been used in gene studies and the ability to assess a more significant number of genes in a single run improved sensitivity and resolution. The primary goal of next-generation sequencing is to read hundreds of thousands or millions of DNA molecules.
Comparative genomics serves as an approach to predict shared genes among different species and to uncover genes that are unique and novel to specific species. Homologous genes, including orthologues and paralogues, play a crucial role in tracing the evolutionary patterns of organisms, especially in relation to speciation or duplication events. Additionally, inferring orthology provides the most accurate method for describing similarities and differences in a genome [13]. Orthologous genes typically exhibit a syntenic relationship with closely related species and can be traced back to a common ancestor during speciation events. The presence of similar orthologous sequences in different species indicates their similar biological function and contrary if sequence show greater divergence then other species may perform distinct function in particular species [14]. In this study, we employed a graph-based approach using the OrthoVenn 3 tool [15] to conduct synteny analysis, comparing the genomes of C. melo and C. sativus var. hardwickii.
The most common type of DNA sequence variation between alleles is single nucleotide polymorphisms (SNPs), which can be revealed by using advanced technologies for genome analysis. SNPs are single base-pair differences (substitutions or deletions) among chromosome sequences that result from point mutations and account for much of the genetic variation in the genome. The binding affinity between a regulatory protein and a regulatory DNA binding site may change if there is a SNP in that site [16,17]. SNP markers have become increasingly popular in plant molecular genetics due to their abundance in the genome and their ease of detection at ultra-high-throughput. Despite these benefits, GBS continues to face challenges due to a lack of quick, reliable data imputation algorithms and powerful computers capable of processing and storing large amounts of sequencing data [18]. Even in the face of such disadvantages or threats, genomic and other next-generation advances increase the pace and benefits of plant breeding. SNPs are the first step toward unravelling evolutionary and genetic relationships and elucidating agronomic characteristics and plant diseases [19]. Therefore, we performed the genome-wide in-silico mining of SNPs and genomic variants from C. sativus var. hardwickii and predicted their effects in detail.
2. Materials and Methods
2.1. Raw reads and Mapping
In-Silico investigation has been done with the raw SRA data available in the public domain at NCBI (National Center for Biotechnology Information). The whole genomic data was used for the SNPs and variants calling using the whole genome sequencing data. Raw reads from the whole genome sequence of C. sativus var hardwickii were downloaded from the NCBI, SRA with an accession number SRR2096458 using the Illumina platform 92,921,066 raw reads were generated. These reads were mapped to the C. sativus genome using BWA, and the version of the Cucumber genome was ASM407v2 (var. sativus, cv. Chinese Long, inbred line 9930) [20].
2.2. Genome-Wide SNPs Calling
BWA-mem (v0.7.17) was used with its default parameters. The VCFstats feature of RTG-tools version 3.12 was used to calculate mapping statistics such as total mapped, unmapped, and paired mapped reads and perform additional analysis on the alignment files. We used Unified Genotyper from GATK (v.3.8-0) to call SNPs and indels for variant analysis. We separated the raw variant calling data into SNPs and indels using Select Variants from GATK (v. 4.0.1.2). Then we applied hard-filtering to these raw SNP calls using Variant Filtration from GATK (v. 4.0.1.2) based on these threshold criteria: Mapping Quality Rank Sum of < -12.5, QUAL < 30, QD < 3.0, FS > 30.0, MQ < 30.0, DP < 200, and genotype-filter-expression DP < 15. We then choose bi-allelic variants using VCFtools (version 0.1.15) [21,22].
2.3. Variant Effect Analysis
The mapped data was used for Variant effect prediction using Variant Effect Predictor (VEP) software (version (API), 102) to verify the effect of variants [23]. The script for Ensembl Variant Effect Predictor is available at (http://www.ensembl.org/info/docs/tools/vep/script/index.html) [24].
2.4. Synteny Analysis
In order to demonstrate the synteny relationship between the orthologous genes derived from C. melo and C. sativus var. hardwickii, syntenic analysis maps were generated using the OrthoVenn3 toolkit program, utilizing the default parameters.
3. Results
3.1. Genome-Wide Identification of SNPs
We identified a total 19, 74,213 polymorphic SNPs in C. sativus var. hardwickii in relation to the reference genome of cucumber with MNPs (0.00), Insertions (1, 33,468), deletions (1,43,237), Indels (75) from all the processed reads and 1,62,673 genotypes were missing (Table 1). Furthermore, the heterozygous/homozygous ratio was also predicted for SNPs (0.20%), insertion (0.99%), deletion (0.85%) with total Het/Hom ratio (0.26%) and not detectable for MNPs and indels (Table 1).
3.2. Genome-Wide Variant Analysis Using Ensembl Variant Effect Predictor
VEP analysis was performed to determine the effect of variants on genes, mRNA transcripts, protein-coding, and regulatory sequences that may help evaluate the genetic diversity within the species.
3.2.1. General Statistics Data Description of Analyzed Variants
A total of 22,28,224 variants were processed out of 6404727 input reads along with 23,777 number of overlapped genes and similar for overlapped transcripts with zero novel variants and overlapped regulatory features (Table 2).
3.2.2. Variants Classes
Single-nucleotide variants (SNV) are the most commonly present variants genome wide and detected with great value in present study. Total identified variants in present study with computational analysis are segregated mainly into four classes of genomic variants which includes 0.01% Sequence alteration (275), 5.94% Insertion (1,32,531), 6.37% Deletion (1,42,040) and 87.66% SNV (19,53,378) respectively (Table 3).
3.2.3. All Consequences and Most Severe Consequences of Variants
Most severe consequences were detected in 21 classes with significant difference in number, highest for upstream gene variants i.e., 1,029,734 and lowest for Start retained variant i. e., 02 only. Some major classes are frame shift variants (1,114), in frame insertion (588), in frame deletion (712), missense variant (38,641), protein altering variant (29), synonymous variant (42,823), coding sequence variant (21), 5’ prime UTR variant (20,079), 3’ prime UTR variant (27,691), intron variant (3,04,952), downstream gene variant (3,66,513) and intergenic variant (3,87,161) were also detected for most severe consequences. Similarly, all consequences were detected in 21 classes with great value change, highest for upstream gene variants i.e., 1,547,272 and lowest for start retained variant (13) (Table 4).
3.2.4. Coding Consequences and Variant Location on Chromosomes
A total of 86435 variants were recorded for Coding consequences (a variant that makes changes in coding sequence) and classified into 12 classes with variable count range and highly detected 50.55% (43,701) in Synonymous variant (A sequence variant changes one base of terminator codon but does not impact on encoded amino acids, the terminator remains), followed by 44.86% (38,779) Missense variant (A variant that make changes in one base of the transcript first codon) and least detected for 0.01% (13) Start retained variant class (Table 5). A total of 22,28,224 variants were detected by Ensembl VEP, although variants were identified in all chromosomes, the number of detected variants on chromosomes varied considerably. These variants distributed on all seven chromosomes of C. sativus var. hardwickii with high number 18.03% (401,920) of total detected variants on the chromosome 3 followed by 15.84% (352,961) chromosome 5, 15.46% (344,544) chromosome 1, 14.72% (328,039) chromosome 6, 12.92% (287,924) chromosome 2, 12.13% (270,449) chromosome 4 while less 10.87% (242,387) on chromosome 7 (Table 6).
3.3. Synteny Analysis
A synteny relationship can provide insight into the evolutionary history of a genome. To further investigate the synteny of genes in rep-presentative crop species, comparative syntenic maps were constructed for C. melo and C. sativus var. hardwickii genes (Figure 1). Interestingly, our findings indicate a significant correlation between the majority of genes on chromosome 10 and 11 of C. melo, and chromosome 5 of C. sativus var. hardwickii. Additionally, a majority of genes on chromosome 12 of C. melo exhibit synteny with chromosome 6 of C. sativus var. hardwickii. Similarly, it is observed that the majority of genes on chromosome 13 of C. melo display synteny with chromosome 1 of C. sativus var. hardwickii. In contrast, the genes located on chromosome 2 of C. melo did not exhibit clear synteny with C. sativus var. hardwickii. Instead, they were observed to be distributed across chromosomes 1, 2, 3, 4, 5, and 6 of C. sativus var. hardwickii.
According to the predicted summarized clustering result, the species are represented in the columns, and each cell in the row represents an orthologous cluster group. The program's prediction indicated the presence of a total of four cluster groups, with two in each Cucumis species, and the absence of a cluster group in two cells (Figure 2). The number of protein sequences predicted to be part of the cluster group varied across each Cucumis species, ranging from 183 to 15,677 in cumulative count (bar plot stacked at right in Figure 2). The C. melo and C. sativus var. hardwickii revealed to have 15677 number of protein sequences involved in cluster group.
4. Discussion
Cucumber is a vegetable crop, the economic importance of which varies according to the region of the world. Cucumber is widely considered an excellent model for studying sex determination in monoecious plants [25]. Cucumber plant breeding can benefit from research into the phenotypic diversity found in cucumber genotypes. Superior genotypes for fruit yield and component production could be used to increase fruit yield, and they are also recommended for hybridization in cucumber cultivation [13]. Next-generation sequencing (NGS) offers a new perspective on the genetic code, revealing new genes and regulatory sequences, as well as a wealth of molecular markers. When it comes to traits that are influenced by a large number of factors, expression studies provide breeders with a wealth of knowledge about the molecular basis of these traits [26]. Advances in next-generation sequencing technology have facilitated the widespread use of SNPs in crops. Computational approaches dominate SNP discovery methods because increasing sequence awareness in public databases makes it easier to identify insightful SNPs computationally; however, complex genomes complicate SNP discovery significantly more, necessitating the use of alternative strategies for some crops. Plant genomes, in their entirety, have been shown to be repetitive [27].
Genome-wide sequencing analysis for SNPs and Variants prediction, along with their genome-wide distribution, may help to improve our understanding of genetic diversity, evolutionary studies and superior economically important traits of crop plants [28]. In this study, we conducted genome-wide SNPs identification and variants analysis from whole-genome sequencing data of C. sativus var. hardwickii. SNPs are very useful for studying genetic diversity because they can reveal the connections between different varieties. SNPs have been used for several years to measure diversity in specific genes or genomic regions, and the results are used to infer phylogenetic relationships between species [29]. The successful mapping of 92.9M raw reads with the reference genome revealed 19, 74,213 SNPs and 22, 28,224 variants genome-wide. SNPs allowed us to better understand genetic diversity and agricultural improvement, which will result in the enhancement of critical crop traits.
Variants calling in cucumber mapped sequencing data resulted in four variant classes with high number of SNV (87.66%) followed by deletion (6.37%), insertion (5.94%) and negligible for sequence alteration (0.01%). Here we recorded 21 variants classes for the most severe consequences with a total count of 2228224, highest for upstream gene variants (1,029,734), which located in 2 Kb 5’ of a gene followed by Intergenic variant found in intergenic regions (387161) and lowest for Start retained variant (02). Similarly, 21 variant classes for all consequences type with total number 3741865, highly detected for upstream gene variants, i.e., 1,547,27, followed by Downstream gene variant, i.e., 1352690 and lowest for Start retained variant, i.e., 13. Based on the variant location in the protein-coding and non-coding regions, including non-synonymous, missense, nonsense or Frameshift variants [30].
Impact of variants for coding consequences category revealed high change is synonymous (50.55%) where one base of stop codon changed. Still, terminator remains and does not affect the encoding amino acid peptide, missense (44.86%), which change at least one base in start codon and affect the encoding protein, followed by frame shift (1.32%) which resulting in disruption of translating reading frame as insertion or deletion is not multiple of three and stop gained (1.00%) (Liu et al., 2019). The discovered Variants were distributed across all 7 chromosomes. Overall, 18.03% (401920) of the total discovered variants (2228224) were on chromosome 3, followed by 15.84% (352961) on chromosome 5 and at least 10.87% (242387) on chromosome 7. The above genome-wide analyzed SNPs and Variants may improve our understanding of genetic evolution and diversity in Cucumis sativus and significantly increase our knowledge about the variants' association with genomic diversity and plants' response to environmental factors.
Comparative genetic mapping proves valuable in uncovering the syntenic relationships present among closely related plant species [31,32]. Gaining insights into syntenic relationships among species enables the exploration of genome evolution and dynamics [33,34]. Moreover, it facilitates the utilization of genetic information from related species in gene isolation and molecular tagging experiments [35-37]. Our study's identification of synteny aligns with the earlier discoveries of Huang et al. [38], Fukino et al. [39], and Li et al. (2011) [40]. They revealed that following divergence from C. melo, five out of cucumber's seven chromosomes emerged through the fusion of ten ancestral chromosomes. Comparative mapping has proven successful in establishing syntenic relationships among closely related plant species across various families. For instance, in the Solanaceae family (pepper, tomato, and potato), studies by [41-43] Park et al. [41], Wu et al. [42], and Wu and Tanksley [43] have utilized comparative mapping to define these relationships. Similarly, in Gramineae grasses, Byrne et al. [44], Rohner et al. [45], Zhu et al. [46], Khahani et al. [47] and Shariatipour et al. [48] have applied this approach. Comparative mapping has also been employed in Fabaceae legumes by Mudge et al. [33], Kalo et al. [50], Yan et al. [51], Cannon et al. [52], Phan et al. [53], and McClean et al. [54]. Additionally, Wang et al. [55] explored comparative mapping in Brassicaceae, while Dirlewanger et al. [56] and Jung et al. [57] employed it in the Rosaceae family.
5. Conclusion
Genome-wide sequencing data have become a powerful tool for exploring the genetic diversity and functional significance of SNP) and variants in various organisms. These molecular markers can reveal the evolutionary history, population structure, and adaptive responses of different species and cultivars. In this study, we performed a comprehensive analysis of SNPs and variants in the re-sequenced genome of C. sativus var. hardwickii, a wild relative of cultivated cucumber. We detected a total of 1974213 SNPs and 2228224 variants across the seven chromosomes of C. sativus var. hardwickii, with chromosome 3 having the highest density and chromosome 7 having the lowest density. These genomic variations reflect the high level of genetic diversity and divergence between C. sativus var. hardwickii and other cucurbit species.
Furthermore, we annotated the functional effects of these SNPs and variants on genes and regulatory elements involved in various biological processes related to plant growth, development, stress tolerance, and disease resistance. Our results provide valuable resources for future studies on the molecular basis and phenotypic consequences of genome-wide variation in C. sativus var. hardwickii and its potential applications for cucumber breeding.
Author Contributions
P.K. and A.K.D. conceived the research idea. S.R., and V.K.S. performed the analysis part. S.R., V.K.S., R.K. and M.S. contributed to the writing of the manuscript. P.S.D., A.Y. and P.K. edited and finalized the manuscript. The manuscript was read and revised by all authors and finalized by P.K.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lee, H.Y.; Kim, J.G.; Kang, B.C; Song, K. Assessment of the Genetic Diversity of the Breeding Lines and a Genome Wide Association Study of Three Horticultural Traits Using Worldwide Cucumber (Cucumis spp.) Germplasm Collection. Agronomy 2020, 10, 1736. [CrossRef]
- Datar, M.N.; Pathak, G; Ghate, H.V. A note on the occurrence of Cucumis sativus L. forma hardwickii (Royle) WJ De Wilde and Duyfjes (Cucurbitaceae) in peninsular India. J. Threa. Taxa 2013, 5, 5010-5012.
- Golabadi, M.; Golkar, P; Eghtedary, A.R. Assessment of genetic variation in cucumber (Cucumis sativus L.) genotypes. Eur. J. Exp. Biol 2012, 2, 1382-1388.
- Naegele, R.P.; Wehner, T.C.; Genetic resources of cucumber. In Genetics and genomics of Cucurbitaceae. Springer, Cham 2016, 61-86.
- Staub, J.E; Grumet, R. Selection for multiple disease resistance reduces cucumber yield potential. Euphytica, 1993, 67, 205-213. [CrossRef]
- Behera, T.K.; Behera, S.; Bharathi, L.K.; John, K.J.; Simon, P.W; Staub, J.E. 2 bitter-gourd: botany, horticulture, Breeding. Hort. reviews 2010, 37, 101.
- Mariod, A.A.; Mirghani, M.E.S; Hussein, I.H. Unconventional oilseeds and oil sources. Academic Press 2017.
- Pandey, S.; Ansari, W.A.; Mishra, V.K.; Singh, A.K; Singh, M. Genetic diversity in Indian cucumber based on microsatellite and morphological markers. Biochemical Systematics and Ecology, 2013, 51, 19-27. [CrossRef]
- Singh, D.K.; Tewari, R.; Singh, N.K; Singh, S.S. Genetic diversity cucumber using inter simple sequence repeats (ISSR). Transcriptomics 2016, 4, 1-4. [CrossRef]
- Kesh, H.; Kaushik, P. Advances in melon (Cucumis melo L.) breeding: An update. Scientia Horticulturae 282, 110045, 2021. [CrossRef]
- Arzani, A; Ashraf, M. Smart engineering of genetic resources for enhanced salinity tolerance in crop plants. Crit. Rev. Plant Sci 2016, 35, 146-189. [CrossRef]
- Nadeem, M.A.; Nawaz, M.A.; Shahid, M.Q.; Doğan, Y.; Comertpay, G.; Yıldız, M.; Hatipoğlu, R.; Ahmad, F.; Alsaleh, A.; Labhane, N.; Özkan, H. DNA molecular markers in plant breeding: current status and recent advancements in genomic selection and genome editing. Biotechnol. Biotechnol. Equip 2018, 32, 261-285. [CrossRef]
- Gabaldón, T.; Koonin, E.V. Functional and evolutionary implications of gene orthology. Nat Rev Genet, 2013, 14, 360-366. [CrossRef]
- Fitch, W.M. Homology a personal view on some of the problems. Trends Genet 2000, 16, 227-31. [CrossRef]
- Xu et al. OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species, Nucleic Acids Research, Volume 47, Issue W1, Pages 2019, W52–W58. [CrossRef]
- Fareed, M; Afzal, M. Single nucleotide polymorphism in genome-wide association of human population: a tool for broad spectrum service. Egypt. J. Med. Hum. Genet 2013, 14, 123-134. [CrossRef]
- Zaid, I.U.; Tang, W.; Liu, E.; Khan, S.U.; Wang, H.; Mawuli, E.W; Hong, D. Genome-wide single-nucleotide polymorphisms in CMS and restorer lines discovered by genotyping using sequencing and association with marker-combining ability for 12 yield- related traits in Oryza sativa L. subsp. japonica. Front. Plant Sci 2017. [CrossRef]
- Patel, D.A.; Zander, M.; Dalton-Morgan, J.; Batley, J. Advances in plant genotyping: where the future will take us, in: Plant Genotyping. Springer 2015, 1-11. [CrossRef]
- Rasheed, A.; Hao, Y.; Xia, X.; Khan, A.; Xu, Y.; Varshney, R.K.; He, Z. Crop breeding chips and genotyping platforms: progress, challenges, and perspectives. Mol. plant 2017, 10, 1047–1064. [CrossRef]
- Li, H; Durbin, R. Fast and accurate short read alignment with Burrows– Wheeler transform. Bioinformatics, 2009, 25, 1754-1760. [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del, G.; Rivas, M.A.; Hanna, M; McKenna, A. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genet 2011, 43, 491. [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T; McVean, G. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156-2158. [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P; Cunningham, F. The ensembl variant effect predictor. Genome biology 2016, 17, 1-14.
- Ensembl Variant Effect Predictor script http://www.ensembl.org/info/docs/tools/vep/scrip t/index.html. Accessed 17 Mar 2016.
- Pawelkowicz, M.E.; Skarzyńska, A.; Pląder, W; Przybecki, Z. Genetic and molecular bases of cucumber (Cucumis sativus L.) sex determination. Mole. breed 2019, 39, 1-27. [CrossRef]
- Pawełkowicz, M.; Zieliński, K.; Zielińska, D.; Pląder, W.; Yagi, K.; Wojcieszek, M.; Siedlecka, E.; Bartoszewski, G.; Skarzyńska, A; Przybecki, Z. Next generation sequencing and omics in cucumber (Cucumis sativus L.) breeding directed research. Plant sci, 2016, 242, 77-88. [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R; Kumpatla, S. SNP markers and their impact on plant breeding. Inter J plant genom 2012. [CrossRef]
- Cavagnaro, P.F.; Senalik, D.A.; Yang, L.; Simon, P.W.; Harkins, T.T.; Kodira, C.D.; Huang, S; Weng, Y. Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC genomics 2010, 11, 1-18. [CrossRef]
- Morgil, H.; Gercek, Y.C; Tulum, I. Single nucleotide polymorphisms (SNPs) in plant genetics and breeding. In the Recent Topics in Genetic Polymorphisms. Intech Open 2020. [CrossRef]
- Shameer, K.; Tripathi, L.P.; Kalari, K.R.; Dudley, J.T; Sowdhamini, R. Interpreting functional effects of coding variants: challenges in proteome-scale prediction, annotation and assessment. Brief. Bioinfo 2016, 17, 841-862. [CrossRef]
- Keller, B.; Feuillet, C. Colinearity and gene density in grass genomes. Trends Plant Sci 2000, 5, 246-251. [CrossRef]
- Paterson, A.; Bowers, J.; Burow, M.; Draye, X.; Eisik, C.; Jiang, C.; Katsar, C.; Land, T.; Lin, Y.; Ing, R.; Wright, R. Comparative genomics of plant chromosomes. Plant Cell 2000, 12, 1523-1539.
- Mudge, J.; Cannon, S.B.; Kalo, P.; Oldroyd, G.E.; Roe, B.A.; Town, C.D.; Young, N, D. Highly syntenic regions in the genomes of soybean, Medicago truncatula, and Arabidopsis thaliana. BMC Plant Biol 2005, 5, 15. [CrossRef]
- Tang, H.; Bowers, J.E.; Wang, X.; Ming, R.; Maqsudul, A.; Paterson, A.H. Synteny and collinearity in plant genomes. Science 2008, 320, 486-488. [CrossRef]
- Chen, M.; SanMiguel, P.; Bennetzen, J.L. Sequence organization and conservation in sh2/a1-homologous regions of sorghum and rice. Genetics 1998, 148, 435-443. [CrossRef]
- Ramakrishna, W.; Bennetzen, J.L. Genomic colinearity as a tool for plant gene isolation Methods. Mol. Biol 2003, 236, 109-122.
- Nieto, C.; Morales, M.; Orjeda, G.; Clepet, C.; Monfort, A.; Sturbois, B.; Puigdomenech, P.; Pitrat, M.; Caboche, M.; Dogimont, C.; Garcia-Mas, J.; Aranda, M.; Bendahmane, A. An eIF4E allele confers resistance to an uncapped and non-polyadenylated RNA virus in melon. Plant J 2006, 48, 452-462. [CrossRef]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.I.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; Ren, Y. The genome of the cucumber, Cucumis sativus L. Nature genet 2009, 41, 1275-1281. [CrossRef]
- Fukino, N.; Yoshioka, Y.; Sakata, Y.; Matsumoto, S.; Thies, J.A.; Kousik, S; Levi, A. Construction of an intervarietal genetic map of cucumber and its comparison with the melon genetic map. Cucurbitaceae 2010, 22-25.
- Li, D.; Cuevas, H.E.; Yang, L.; Li, Y.; Garcia-Mas, J.; Zalapa, J.; Staub, J.E.; Luan, F.; Reddy, U.; He, X.; Gong, Z. Syntenic relationships between cucumber (Cucumis sativus L.) and melon (C. melo L.) chromosomes as revealed by comparative genetic mapping. BMC genom 2011, 12,1-14. [CrossRef]
- Park, M.; Jo, S.H.; Kwon, J.K.; Park, J.; Ahn, J.H.; Kim, S.; Lee, Y.H.; Yang, T.J.; Hur, C.G.; Kang, B.C.; Kim, S.D.; Choi, D. Comparative analysis of pepper and tomato reveals euchromatin expansion of pepper genome caused by differential accumulation of Ty3/Gypsy-like elements. BMC Genomics 2011, 12, 85. [CrossRef]
- Wu, F.; Eannetta, N.T.; Xu, Y.; Durrett, R.; Mazourek, M.; Jahn, M.M; Tanksley, S.D. A COSII genetic map of the pepper genome provides a detailed picture of synteny with tomato and new insights into recent chromosome evolution in the genus Capsicum. Theor. appl. Genet 2009, 118, 1279-1293. [CrossRef]
- Wu, F.N.; Tanksley, S.D. Chromosomal evolution in the plant family Solanaceae. BMC Genomics 2010, 11, 182. [CrossRef]
- Byrne, S.L.; Nagy, I.; Pfeifer, M.; Armstead, I.; Swain, S.; Studer, B.; Mayer, K.; Campbell, J.D.; Czaban, A.; Hentrup, S; Panitz, F. A synteny-based draft genome sequence of the forage grass Lolium perenne. The Plant Journal, 2015, 84, 816-826. [CrossRef]
- Rohner, M.; Manzanares, C.; Yates, S.; Thorogood, D.; Copetti, D.; Lübberstedt, T.; Asp, T.; Studer, B. Fine-mapping and comparative genomic analysis reveal the gene composition at the S and Z self-incompatibility loci in grasses. Mole. Biol. Evol 2023, 40, 259. [CrossRef]
- Zhu, S.; Liu, C.; Gong, S.; Chen, Z.; Chen, R.; Liu, T.; Liu, R.; Du, H.; Guo, R.; Li, G; Li, M. Orthologous genes Pm12 and Pm21 from two wild relatives of wheat show evolutionary conservation but divergent powdery mildew resistance. Plant Commun, 2023. [CrossRef]
- Khahani, B.; Tavakol, E.; Shariati, V; Fornara, F. Genome wide screening and comparative genome analysis for Meta-QTLs, ortho-MQTLs and candidate genes controlling yield and yield-related traits in rice. BMC genom, 2020, 21, 1-24. [CrossRef]
- Shariatipour, N.; Heidari, B.; Tahmasebi, A.; Richards, C. Comparative genomic analysis of quantitative trait loci associated with micronutrient contents, grain quality, and agronomic traits in wheat (Triticum aestivum L.). Front. Plant Sci 2021, 2142. [CrossRef]
- Kalo, P.; Seres, A.; Taylor, S.A.; Jakab, J.; Kevei, Z.; Kereszt, A.; Endre, G.; Ellis, T.H.; Kiss, G.B. Comparative mapping between Medicago sativa and Pisum sativum. Mole. Genet. Genom 2004, 272, 235-246.. [CrossRef]
- Yan, H.H.; Mudge, J.; Kim, D.J.; Shoemaker, R.C.; Cook, D.R.; Young, N.D. Comparative physical mapping reveals features of microsynteny between Glycine max, Medicago truncatula, and Arabidopsis thaliana. Genome 2004, 47, 141-55. [CrossRef]
- Cannon, S.B.; Sterck, L.; Rombauts, S.; Sato, S.; Cheung, F.; Gouzy, J.; Wang, X.; et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci 2006, 103, 14959-14964. [CrossRef]
- Phan, H.T.; Ellwood, S.R.; Hane, J.K.; Ford, R.; Materne, M.; Oliver, R.P. Extensive macrosynteny between Medicago truncatula and Lens culinaris ssp. culinaris. Theor. Appl. Genet 2007, 114, 549-558. [CrossRef]
- McClean, P.E.; Mamidi, S.; McConnell, M.; Chikara, S.; Lee, R. Synteny mapping between common bean and soybean reveals extensive blocks of shared loci. BMC Genom 2010, 11, 184. [CrossRef]
- Wang, J.; Lydiate, D.J.; Parkin, I.A.P.; Falentin, C.; Delourme, R.; Carion, P.W.C.; King, G.J. Integration of linkage maps for the amphidiploid Brassica napus and comparative mapping with Arabidopsis and Brassica rapa. BMC Genom 2011, 12, 101. [CrossRef]
- Dirlewanger, E.; Graziano, E.; Joobeur, T.; Garriga-Calderé, F.; Cosson, P.; Howad, W.; Arús, P. Comparative mapping and marker-assisted selection in Rosaceae fruit crops. Pro. Natl. Acad. Sci 2004, 101, 9891-9896. [CrossRef]
- Jung, S.; Jiwan, D.; Cho, I.; Lee, T.; Abbott, A.; Sosinski, B.; Main, D. Synteny of Prunus and other model plant species. BMC Genom 2009, 10, 76. [CrossRef]
Figure 1.
Synteny analysis of genes between Cucumis melo and Cucumis sativus var. hardwickii.
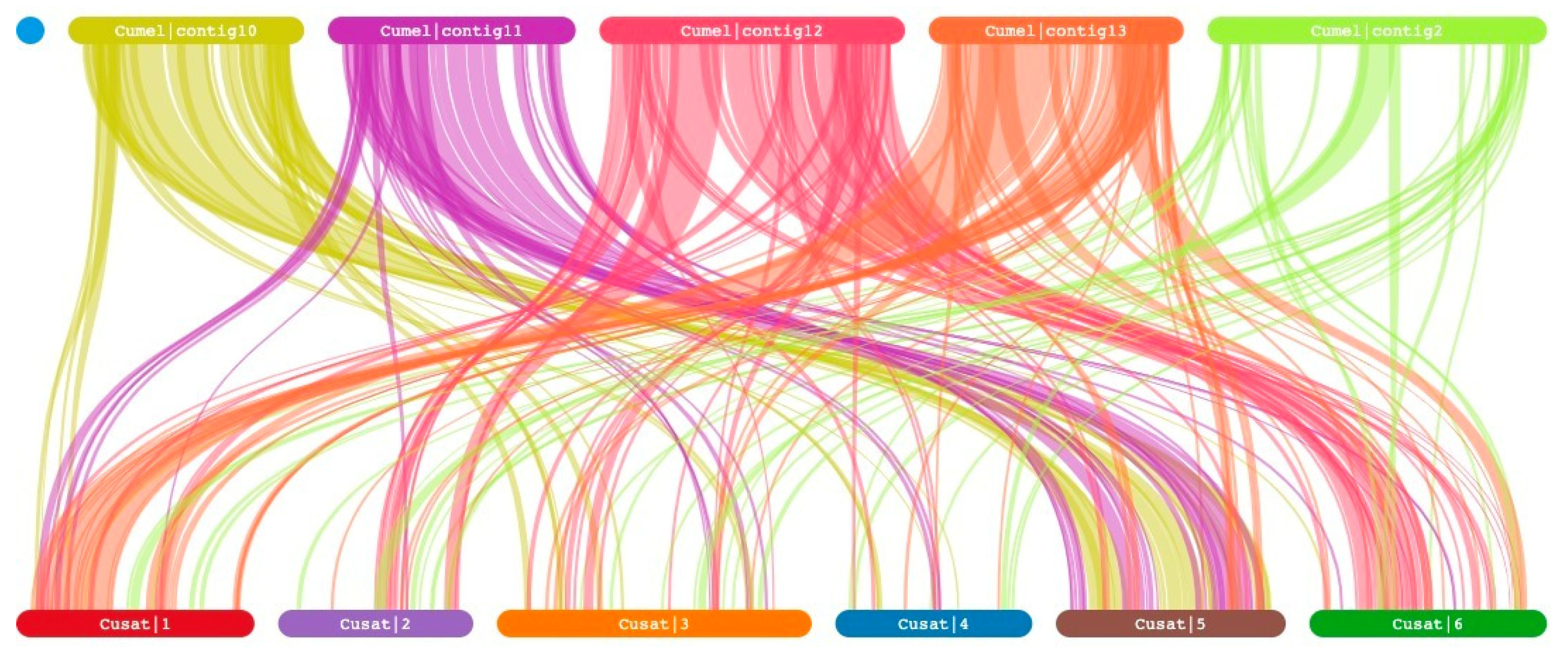
Figure 2.
The occurrence table displays the patterns of shared orthologous groups between C. melo and C. sativus var. hardwickii. The left pattern reveals the Cucumis species involved in the clusters, the number of shared clusters among them, and the protein count within the shared clusters.
Figure 2.
The occurrence table displays the patterns of shared orthologous groups between C. melo and C. sativus var. hardwickii. The left pattern reveals the Cucumis species involved in the clusters, the number of shared clusters among them, and the protein count within the shared clusters.
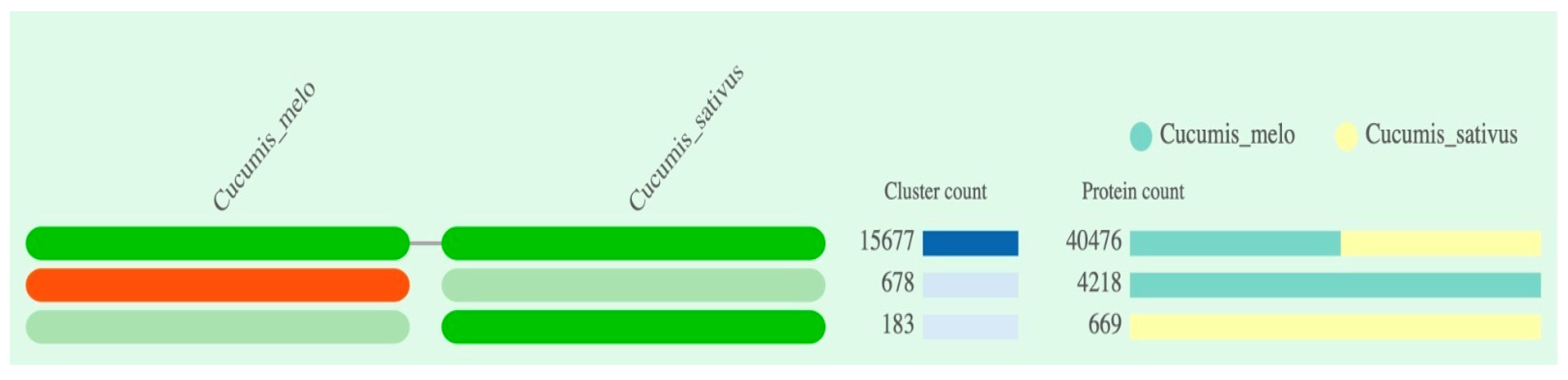

Table 1.
Summary of SNPs identified in C. sativus var. hardwickii.
| Analyzed Factors | Values |
| Failed Filters | 0 |
| Passed Filters | 6404727 |
| SNPs | 1974213 |
| MNPs | 0 |
| Insertions | 133468 |
| Deletions | 143237 |
| Indels | 75 |
| Same as reference | 3991061 |
| Missing Genotype | 162673 |
| SNP Transitions/Transversions | 1.53 (2189498/1432901) |
| Total Het/Hom ratio | 0.26 (459863/1791130) |
| SNP Het/Hom ratio | 0.20 (327665/1646548) |
| MNP Het/Hom ratio | - (0/0) |
| Insertion Het/Hom ratio | 0.99 (66254/67214) |
| Deletion Het/Hom ratio | 0.85 (65869/77368) |
| Indel Het/Hom ratio | - (75/0) |
| Insertion/Deletion ratio | 0.93 (133468/143237) |
| Indel/SNP+MNP ratio | 0.14 (276780/1974213) |
Table 2.
Statistics of processed variants in C. sativus var. hardwickii.
| Lines of input read | 6404727 |
| Variants processed | 2228224 |
| Variants filtered out | 0 |
| Novel / existing variants | - |
| Overlapped genes | 23777 |
| Overlapped transcripts | 23777 |
| Overlapped regulatory features | - |
Table 3.
Summary of predicted variant classes in C. sativus var. hardwickii.
| Variant class | Count | % |
| Sequence alteration | 275 | 0.01 |
| Insertion | 132,531 | 5.94 |
| Deletion | 142,040 | 6.37 |
| SNV | 1,953,378 | 87.66 |
| Total | 2228224 |
Table 4.
Most severe consequences and all consequences type data in C. sativus var. hardwickii.
| Most severe Consequence type | Count | All Consequence type | Count |
| Splice donor variant | 220 | Splice donor variant | 220 |
| Splice acceptor variant | 235 | Splice acceptor variant | 235 |
| Stop gained | 869 | Stop gained | 869 |
| Frameshift variant | 1,114 | Frameshift variant | 1,147 |
| Stop lost | 174 | Stop lost | 191 |
| Start lost | 192 | Start lost | 220 |
| Inframe insertion | 588 | Inframe insertion | 602 |
| Inframe deletion | 712 | Inframe deletion | 719 |
| Missense variant | 38,641 | Protein altering variant | 30 |
| Protein altering variant | 29 | Missense variant | 38,779 |
| Splice region variant | 6,386 | Splice region variant | 7,034 |
| Start retained variant | 2 | Start retained variant | 13 |
| Synonymous variant | 42,823 | Synonymous variant | 43,701 |
| Stop retained variant | 88 | Stop retained variant | 112 |
| Coding sequence variant | 21 | Coding sequence variant | 44 |
| 5’ Prime UTR variant | 20,079 | 5’ Prime UTR variant | 20,418 |
| 3’ Prime UTR variant | 27,691 | 3’ Prime UTR variant | 28,389 |
| Intron variant | 304,952 | Intron variant | 311,414 |
| Upstream gene variant | 1,029,734 | Upstream gene variant | 1,547,272 |
| Downstream gene variant | 366,513 | Downstream gene variant | 1,352,690 |
| Intergenic variant | 387,161 | Intergenic variant | 387,766 |
| Total | 2228224 | Total | 3741865 |
Table 5.
Coding consequences identified in C. sativus var. hardwickii.
| Consequence type | Count | % |
| Stop gained | 869 | 1.00 |
| Frameshift variant | 1,147 | 1.32 |
| Stop lost | 191 | 0.22 |
| Start lost | 220 | 0.25 |
| Inframe insertion | 602 | 0.69 |
| Inframe deletion | 719 | 0.83 |
| Protein altering variant | 30 | 0.03 |
| Missense variant | 38,779 | 44.86 |
| Start retained variant | 13 | 0.01 |
| Stop retained variant | 112 | 0.12 |
| Synonymous variant | 43,701 | 50.55 |
| Coding sequence variant | 44 | 0.05 |
| Total | 86435 |
Table 6.
Chromosomal distribution of predicted variants in the Cucumis sativus var. hardwickii genome.
Table 6.
Chromosomal distribution of predicted variants in the Cucumis sativus var. hardwickii genome.
| Chromosome | Count | % |
| 1 | 344,544 | 15.46 |
| 2 | 287,924 | 12.92 |
| 3 | 401,920 | 18.03 |
| 4 | 270,449 | 12.13 |
| 5 | 352,961 | 15.84 |
| 6 | 328,039 | 14.72 |
| 7 | 242,387 | 10.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



